 Jayanth Kumar Palanichamy
Jayanth Kumar Palanichamy Dinesh S. Rao
Dinesh S. Rao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 18 March 2014
Sec. RNA
Volume 5 - 2014 | https://doi.org/10.3389/fgene.2014.00054
This article is part of the Research Topic Micro-RNAS and Diseases: Beyond expression arrays View all 9 articles
It is now well known that gene expression is intricately regulated inside each cell especially in mammals. There are multiple layers of gene regulation active inside a cell at a given point of time. Gene expression is regulated post-transcriptionally by microRNAs and other factors. Mechanistically, microRNAs are known to bind to the 3’ UTR of mRNAs and cause repression of gene expression and the number of known microRNAs continues to increase every day. Dysregulated microRNA signatures in different types of cancer are being uncovered consistently implying their importance in cellular homeostasis. However when studied in isolation in mouse models, clear-cut cellular and molecular mechanisms have been described only for a select few microRNAs. What is the reason behind this discrepancy? Are microRNAs small players in gene regulation helping only to fine tune gene expression? Or are their roles tissue and cell type-specific with single-cell level effects on mRNA expression and microRNA threshold levels? Or does it all come down to the technical limitations of high-throughput techniques, resulting in false positive results? In this review, we will assess the challenges facing the field and potential avenues for resolving the cellular and molecular mechanisms of these small but important regulators of gene expression.
microRNAs (miRNAs) are short single stranded RNAs that regulate mRNA expression at the post-transcriptional level. The very first miRNA:mRNA target pair was discovered in the labs of Victor Ambros and Gary Ruvkun (lin-4 and lin-14, respectively) who were studying the regulation of developmental timing in C. elegans (Lee et al., 1993; Wightman et al., 1993). The mature miRNA is a single stranded ~22 nucleotide RNA that is sequentially processed from a primary transcript (pri-miRNA) and the resulting stem cell loop structure (pre-miRNA) by the proteins Drosha and Dicer respectively (Kim et al., 2009). The basic principle of miRNA mediated regulation- that miRNAs target mRNAs for repression by binding to the latter’s 3’UTR- was recognized in these early studies, but the pervasiveness of this mechanism in gene regulation was not recognized until much later. The number of miRNAs in humans and mice is constantly being revised, and it is likely that much of the genome is under the control of miRNA-mediated gene expression regulation (Kim et al., 2009).
Dysregulation of miRNAs has been observed in a number of diseases, especially hematopoietic malignancies like chronic lymphocytic leukemia (CLL) and epithelial malignancies such as lung cancers and breast carcinomas (Calin and Croce, 2006). However, the exact cause-effect mechanism has not been established for many miRNAs and diseases despite significant efforts into such research. Indeed, while numerous cancer-causing or promoting miRNAs have been proposed on the basis of their dysregulation in disease states, few have been established as truly oncogenic or tumor suppressive. Implicit in this observation is the question of whether we should be interpreting the roles of miRNAs in disease states in the same way we evaluate the role of proteins in oncogenesis, and what molecular and cell biological mechanisms distinguish miRNAs as novel factors in disease causation and progression. In this review, we will evaluate these questions critically, using hematologic cancers as an example. We speculate that the paucity of definitive experimental evidence is likely attributable to a combination of several factors- including, but not limited to, the inherent genetic redundancy of miRNAs, non-genetic heterogeneity of cells and our technical limitations in small RNA profiling. We begin with a brief primer on the complexity of miRNA based regulation and then examine research findings with the aim of understanding the limitations of current strategies to study miRNAs and exploring alternatives for the future.
miRNA biogenesis and mechanism of action have been extensively studied and detailed mechanisms are reviewed elsewhere, but we review some of the mechanisms whereby complexity in gene regulation may be generated by miRNAs (Kim et al., 2009; Krol et al., 2010). The gene structure of miRNAs is the first source of complexity. Briefly, miRNAs can be encoded within an exon of a unique transcript, can occur as “polycistronic” miRNAs, or can be found within the introns of protein-coding or non-coding genes. The primary transcript undergoes endoribonucleolytic cleavage by Drosha/DGCR8 in the nucleus, and then in the cytoplasm. Intronic microRNAs (mirtrons) have also shown to be generated by a non-Drosha/DGCR8 mediated, Dicer only dependent mechanism (Castellano and Stebbing, 2013). Only one of the strands (the guide strand) thus generated is incorporated into the RNA induced silencing complex (RISC). Although the passenger strand is usually degraded, there are now reports emerging that show an important role for the passenger strand in gene regulation (Almeida et al., 2012; Epis et al., 2012; Uchino et al., 2013). The two strands of the microRNA are termed 5p and 3p depending on their orientation. Recent reports suggest that incorporation of either strand into the RISC complex is possible. Examples include miR-142 and miR-17 where incorporation of both the strands into the RISC complex occurs, leading to the presence of both 5p as well as 3p transcripts inside the cell (Kasashima et al., 2004; Yang et al., 2013). This introduces yet another layer of regulation since the 5p and 3p microRNAs would target different genes because of the difference in their seed sequence. This is the case with miR-28 where overexpression of miR-28-5p and miR-28-3p caused different effects in colorectal cancer cells (Almeida et al., 2012). Interestingly, miR-17-5p as well as miR-17-3p were shown to target the same gene TIMP3 and induced prostate cancer growth (Yang et al., 2013). All of these steps are subject to regulation and can therefore create intricate patterns of miRNA expression.
Once the miRNA is incorporated into RISC, it causes repression of target gene expression. miRNAs usually target the 3’UTR (untranslated region) of the mRNA. Perfect complementarity between the miRNA and its target leads to destruction of the RNA by the “slicer” activity in the Argonaute-2 protein which is part of the RISC (Meister, 2013). This type of repression is called RNAi and is very rare in mammals but more commonly seen in plants. More commonly, incomplete complementarity between the miRNA and its target is seen leading to translational repression. Proposed mechanisms include interference of the miRNA with the formation of a looped mRNA structure required for optimal translation, but further study is needed (Kim et al., 2009). Nonetheless, the net effect of miRNA expression is thought to be repression of the target gene. The repression is thought to be limited in extent (i.e., the gene is partially repressed) because miRNAs are induced and degraded over time.
Although binding to the 3’UTR and repression of the gene is considered to be the current canonical pathway for miRNA mediated gene regulation, non-canonical binding and targeting has also been described for microRNAs. Some microRNAs have been shown to target the promoters of genes and cause either gene activation (miR-373, miR-744, miR-1186) or gene repression (miR-320, miR-373). Gene activation was accompanied by the presence of active histone marks (H3K4 trimethylation) while gene repression was caused by a closed chromatin structure due to H3K9 dimethylation (Huang and Li, 2012). Recently, CLASH (cross-linking, ligation, and sequencing of hybrids) was used along with overexpression of an Ago1 fusion protein and high-throughput sequencing to map the miRNA-interactome. This revealed that even though the majority of the miRNA binding sites were in the 3’UTRs, microRNAs also bound to 5’UTRs, gene bodies, long non-coding RNAs (lncRNAs) as well as introns. There was also a class of microRNAs whose mRNA interactions did not involve the seed region (miR-92a), or so-called “seedless binding” (Helwak et al., 2013).
The temporal patterns of miRNA expression add significantly to the complexity of the problem. Some miRNAs are strongly induced by stimuli such as LPS (miR-155; O’Connell et al., 2009) whereas others are expressed at baseline levels and seem to maintain fairly constant levels during developmental processes (Pauli et al., 2011; Guan et al., 2013). In addition to their biogenesis, which can be regulated transcriptionally and post-transcriptionally at the level of RNA processing (van Kouwenhove et al., 2011), their degradation can be regulated both by proteins, as is the case with Xrn2 (Grosshans and Chatterjee, 2010), and by cell divisions, which dilute out the pool of miRNAs if their rate of production is less than the sum of dilution and degradation. As one considers disease-related changes in miRNA expression, understanding the temporal expression pattern, particularly whether phenotypes may be induced by various biological stimuli, impacts the interpretation of phenotypic changes. As an example, miR-155-deficient mice showed normal baseline levels of B-cells and T-cells, but these mice clearly had major defects in immune responses (Thai et al., 2007; O’Connell et al., 2010b; Huffaker et al., 2012). It is clear that miR-155 has major functions when induced, and that these functions have importance in the context of oncogenesis.
Adding to the complexity of miRNA mediated repression is computational and experimental evidence that a single miRNA can target hundreds or potentially thousands of mRNA molecules. The stoichiometry of such inhibition is incompletely understood, and it would seem that any two targets could be inhibited to different degrees by a given miRNA. Given computational algorithms that are imperfect and the lack of a methodology to predict the magnitude of repression, both the identity of and extent of effect on target gene expression have to be defined in an individual fashion by experimentation. Hence, it would be extremely difficult to predict the effects of miRNA expression on gene expression at the global level.
Post-transcriptional gene regulation by miRNAs is further modulated by RNA binding proteins (RBPs). Numerous RBPs have recently been characterized recently and many play a role in mRNA stability. A target mRNA can be competitively bound by a RBP and a miRNA, leading to a reduction in repression levels by the miRNA. RBP binding can also cause a switch in mRNA secondary structure, causing steric hindrance to miRNA binding. One example is the binding of IGF2BP1 (Insulin like growth factor 2 binding protein 1) to the BTRC and MITF mRNAs, preventing the binding of microRNAs 183 and 340 respectively. This leads to de-repression of these genes and subsequent downstream effects (Elcheva et al., 2009; Goswami et al., 2010; van Kouwenhove et al., 2011).
Lastly, there is a consideration about miRNA-based regulation that seems obvious but has important implications. miRNAs can only target transcripts that have been induced in a particular developmental or pathological context; they cannot, a priori, cause a gene to be expressed. Hence, the set of genes that a miRNA regulates in a given cell is highly dependent on the transcriptional program of that particular cell. Finding functional targets in one cell type does not necessarily implicate the same targets in a different cell type. Similarly, signaling pathways implicated in one cell type may not be operant in a different cell type. This has been highlighted by the germline knockout of miR-146a, which demonstrated a phenotype that was almost exclusively restricted to the hematopoietic system (Boldin et al., 2011; Zhao et al., 2011). This has important implications for how one predicts the effects of miRNA manipulation across different tissue types.. This also emphasizes the fact that microRNA targets will most probably be tissue type specific. So microRNA: mRNA interaction studies need to be performed specific to cell types and their results interpreted according to the tissue of origin.
After the recognition of miRNAs as being widespread in the genome of humans, there was a keen interest to identify dysregulated miRNA profiles in a number of diseases, especially cancer. Altered miRNA profiles were indeed found in many cancers (Calin and Croce, 2006). For example miR-21 was found to be up regulated in a number of malignancies like glioblastomas, breast, colon and pancreatic cancer and has been anointed as an oncogenic miRNA (Garzon et al., 2009). The miR 17–92 cluster was up regulated in lymphomas and in breast, lung, colon, stomach, and pancreatic cancers (Calin and Croce, 2009; Garzon et al., 2009).MiR-155 was found to be highly expressed in certain types of B-cell lymphomas, including CLL with an aggressive course and diffuse large B-cell lymphoma (Calin et al., 2004; Eis et al., 2005; Chen et al., 2008). Numerous other miRNAs have also been identified in patient-based samples as being dysregulated in disease states.
Similarly, some miRNAs are down regulated in tumors and are therefore classified as tumor suppressor miRNAs. The classic examples include miR-15a and miR-16-1 loss in CLL and multiple myeloma, let-7 miRNA loss in lung and breast cancers (Garzon et al., 2009). miR-34 is a p53 responsive miRNA family and its members, notably miR-34a, have been observed to be lost in pancreatic, colon, breast and liver cancers, and is a predictor of poor prognosis in CLL (Chim et al., 2010; Christoffersen et al., 2010; Corney et al., 2010; Hermeking, 2010; Choi et al., 2011; Fabbri et al., 2011). In myelodysplastic syndrome, miR-146a was downregulated, and at least part of the time, this occurs as a consequence of deletion of the long arm of chromosome 5 in the malignant hematopoietic progenitor cells (Starczynowski et al., 2010; Boldin et al., 2011; Zhao et al., 2013).
The last 10 years have seen a proliferation of miRNA profiling studies, and while many miRNAs are dysregulated at the level of their expression, it is unclear how miRNAs become downregulated in specific cancers. Until recently, the chromosomal deletion of miR-15a/16 (chromosome 12p) and miR-146a (chromosome 5q) comprised the only two recurrent genetic abnormalities that can be traced as a direct cause for miRNA downregulation in the hematopoietic system and as noted below, in cancer pathogenesis. Earlier last year, whole genome sequencing of de novo acute myeloid leukemia (AML) revealed that miR-142 undergoes point mutations, potentially resulting in loss of targeting by the 3p form (Cancer Genome Atlas Research Network, 2013). Further work has shown that miR-142 mutations in AML are localized to the seed region of miR-142-3p, leading to reduced expression of miR-142-5p. It is an interesting case where the stability of one form of the miRNA is dependent on the other form (Trissal et al., 2013). Other possible mechanisms include dysregulated miRNA expression resulting downstream of mutations in transcription factors. Since epigenetic studies of gene expression regulation have largely focused on protein-coding genes (where rules for histone modifications are better understood), annotations of miRNA promoters and enhancers have lagged behind those in the protein-coding genes. Therefore, further work in elucidating mechanisms of miRNA dysregulation and their integration into large scale genetic networks that are dysregulated in cancer is required.
As expected, altered miRNA profiles have been observed in numerous malignancies. However, there are only a few malignancies where a causal relationship has been directly established for the dysregulation of the miRNA and tumor development. A general role for the miRNA pathway in oncogenesis was established by the observation that mice heterozygous for Dicer developed an increased incidence of tumors (Kumar et al., 2009; Ravi et al., 2012). Hence, global abnormalities in miRNA expression are indeed causally linked to an increased incidence of tumors.
Among the well-known oncomiRs, overexpression of miR-21 in mice caused pre-B cell leukemia/lymphoma (Medina et al., 2010). In this study, miR-21 was expressed in a Nestin-Cre-inducible manner, surprisingly producing B-cell tumors. miR-155 overexpression led to a lymphoproliferative disorder in B-cells and eventually acute lymphoblastic lymphoma/leukemia (ALL), and myeloproliferative disease when overexpressed in myeloid cells (Costinean et al., 2006, 2009; O’Connell et al., 2011; Babar et al., 2012). Recently, overexpression of miR-22 in hematopoietic cells has been shown to lead to MDS (Song et al., 2013). Ectopic expression of miR-29a (Han et al., 2010) and enforced expression of miR-125b in hematopoietic stem cells (HSCs) led to a myeloproliferative disorder which progressed to AML (O’Connell et al., 2010a; Chaudhuri et al., 2012). Overexpression of the miR-17-19b cluster (five miRNAs) in the bone marrow of Eμ-Myc transgenic mice led to an acceleration of lymphoma development (He et al., 2005). Recently, B-cell specific overexpression of the miR 17–92 cluster led to lymphoma development, establishing its role as an independent oncogene (Jin et al., 2013). Other miRNAs which are highly expressed in tumors however, have failed to recapitulate the full tumor phenotype. For example, overexpression of the putative oncogenic miR-25-106b-93 cluster in mice showed no tumor development.
Deletions of tumor suppressor miRNA in mice have to date yielded phenotypes with a long latency. Deletion of the tumor suppressor miRNA cluster, encoding miR-15a and miR-16-1 led to a CLL like disease in mouse models, albeit at around 18 months of age and only in ~30% of the involved mice (Aqeilan et al., 2010; Klein et al., 2010). Deletion of the miRNA-146a led to a myeloproliferative phenotype and hematologic malignancies, again late in life (~18 months; Boldin et al., 2011; Zhao et al., 2011). Mice with targeted deletions of the following putative tumor suppressor miRNAs did not show development of an overt malignancy: miR-145, miR-223, miR-133, and miR-206 (Park et al., 2010). Although the overexpression of miR-29 had been shown to reduce liver cancer growth in mice (which is contrary to the role that it plays in the hematopoietic system), knockout of the miR-29 locus led to liver fibrosis and not any overt malignancy (Xiong et al., 2010; Roderburg et al., 2011). Perhaps the most surprising example of a knockout lacking a phenotype is the miR-34a knockout mouse, which did not develop any overt malignancies despite being downstream of arguably the most important tumor suppressor in the human cell, p53. Indeed, the triple knockout of all three members of miRNA family did not lead to an increase in cancer incidence (Concepcion et al., 2012). This is despite the fact that miR-34a targets numerous transcripts for proteins important in cell cycle progression, cell cycle progression, and oncogenesis (Siemens et al., 2013).
From a therapeutic standpoint, overexpression and in vivo delivery of miRNAs/ modified small nucleic acid molecules has met with some success. The overexpression of tumor suppressor miRNAs have been discovered to be successful in delaying tumor development or reducing the tumor burden in mouse models. Examples include miR-34a (lung adenocarcinoma), miR-145 and miR-33 (colon carcinoma) and miR-15 and 16-1 (colon carcinoma). Systemic delivery of the tumor suppressor let-7 increased the latency to lung cancer in mouse models (Croce, 2009; Nana-Sinkam and Croce, 2013).There are also some in vivo studies where knock down of oncomiRs has helped in attenuating tumorigenesis. In a mouse model of Ras-driven lung cancer, miR-21 inhibition slowed tumor progression (Hatley et al., 2010). Administration of antagomiRs in mice to block miR-10b and miR-223 resulted in significant antitumor activity against breast carcinoma and hepatocellular carcinoma respectively (Garzon et al., 2010).
In essence, not all dysregulated miRNAs discovered by expression profiling of tumors are pathogenetic and many do not have a clear mechanism of action in oncogenesis. In general, cancer phenotypes driven by overexpression seem to be more severe than those generated by loss-of-function, and the latter seem to be highly age dependent. These findings may be explained by the fact that miRNAs can only act on a “pre-existing” transcriptional program. Hence, miRNA gain of function in a cell type could lead to aberrant functions by targeting genes that are not normally repressed, while loss of function would only affect cells where the appropriate target genes and the miRNA of interest are concurrently expressed. This again brings forward the idea that miRNA gain and loss-of-function should be studied in the appropriate system and the appropriate biological context. One should also bear in mind the fact that microRNA overexpression or knockdown phenotypes have little utility in the absence of human disease specific dysregulation. Hence animal models and human studies should be used together to unravel the mechanism behind dysregulated miRNAs.
Profiling of tumors provides patient-based data on miRNA expression patterns, which is undeniably important, but this presents its own set of problems. Centrally, it is very difficult to control for variability in collection of tumor cells between patients, as opposed to the ease of collecting high-quality controlled samples under experimental conditions. While it seems that miRNAs are better preserved between samples, as small RNAs demonstrate more stability than long RNAs, the techniques used to detect miRNAs also engender some variability. The common techniques for high-throughput profiling include microarrays, RNA-Seq and qPCR arrays. Cost and ease of data analysis remain the two main criteria which determine the type of platform utilized. The gold standard for individual miRNA detection remains the Northern blot and for samples with a low RNA yield (patient samples), reverse transcriptase (RT)-qPCR is the technique of choice for miRNA quantitation (Pritchard et al., 2012). However, interpretation of fold changes obtained from RT-qPCR experiments should always consider absolute levels of miRNA expression in control cells. For example, the biological impact of a 10-fold increase of a low expressed miRNA (1 molecule per cell increases to 10 molecules per cell) may be less significant than a 1.5-fold increase of a highly expressed miRNA (50,000 molecules per cell to 75,000 molecules per cell), depending on the relative expression levels of their mRNA targets. A last consideration in qPCR is the use of appropriate normalization techniques, investigators should consider the use of multiple reference genes or mean expression value of all expressed microRNAs in a sample as a normalization factor (Mestdagh et al., 2009).
Technically, both the RT step and the amount of RNA can be a source of significant bias during RNA profiling. A study comparing the efficiency of different types of RT enzymes showed around a 100-fold difference between enzymes for some genes (Stahlberg et al., 2004). This effect was particularly pronounced for mRNAs whose expression is low. Introduction of errors due to secondary structure, differences in priming efficiency and properties of the enzyme itself (like RNAse H activity) can all influence the product yield from the RT reaction. Also some miRNAs are GC rich and have a higher Tm which is a reason for reduced reverse priming efficiency and consequently smaller representation in the cDNA library. This effect will be much more pronounced when the quantity and quality of the RNA is low (for example, paraffin embedded samples). Similar to the RT reaction, each reaction in the sample preparation workflow may lead to variations including the adaptor ligation step using RNA ligase, linear amplification step as well as the fluorochrome labeling step for microarrays (Stahlberg et al., 2004; Baker, 2010).
With so many commercial options available for miRNA profiling, it is somewhat alarming that different groups have reported poor reproducibility across these different platforms. To avoid this problem, it has been suggested that once a certain profiling method has been standardized in the lab, maintaining the same technique is advisable to eliminate bias across methods (Git et al., 2010). Earlier quality control studies showed a big variation in the profiling of miRNAs by high-throughput sequencing especially when the quantity of transcripts was not abundant. However, encouragingly, recent reports on quality control of RNA-seq using newer kits suggests that current platforms are reproducible and comparable across each other (Adiconis et al., 2013).In addition to the range of platforms, data analysis can differ significantly between studies as well as labs, particularly in the stringency of false discovery rates and in the numerical level of difference between the samples. This too could contribute to the seemingly disparate results obtained by different groups.
Apart from these technical considerations, other factors may also lead to difficulty in fully understanding miRNA expression data. For example, gene duplications have led to miRNAs originating from different chromosomal loci, and each locus is subject to unique regulation. At present, we can only quantify the total amount of mature miRNA being produced because routine profiling studies do not distinguish which locus they originated from. Moreover, researchers have identified so-called IsomiRNAs, which are miRNAs that differ from their cognate miRNA by a few nucleotides either at the 3′ or even the 5′ end. The presence of IsomiRNAs could certainly cause a bias in mature miRNA quantitation by these high-throughput methods (Vickers et al., 2013). Hence, factors intrinsic to the roles of miRNAs in cells can cause variability in data generated by high-throughput studies.
Several proposals have been made to improve technical issues with miRNA based profiling techniques. Some laboratories have advocated the use of external reference samples for the exact quantitation of miRNAs (Git et al., 2010). But these reference samples do not mimic conditions inside the human body: most importantly, total RNA samples probably contain less than 0.01% miRNA (Git et al., 2010). There is ribosomal RNA, tRNA and mRNA that could compete for binding with preparative enzymes and thereby could potentially affect results. This is being negated to a certain extent now by removing the abundant ribosomal RNA before using the RNA for profiling (Git et al., 2010). It would be ideal if guidelines similar to the MIQE guidelines (minimum information for publication of quantitative real-time PCR experiments) were made available for miRNA profiling techniques (Bustin et al., 2013). This would ensure better experimental practice allowing more reliable comparative analysis of miRNA profiling data (Witwer, 2013). Hence, having a carefully curated database using uniform analytic techniques would go a long way toward removing obstacles that confound efforts to jump from profiling studies to functional analyses characterizing dysregulated miRNAs in cancer states.
Apart from high-throughput RNA sequencing, other novel techniques are also being employed to delineate targets of microRNAs. One of these is cross-linking immunoprecipitation-sequencing (CLIP-seq), where small RNAs and mRNAs bound to one of the proteins in the RISC, usually Ago-1, are cross-linked by ultraviolet radiation, pulled down using an anti-Ago1 antibody and then sequenced (Mittal and Zavolan, 2014). RISC-seq is a method similar to CLIP-seq using anti-Argonaute antibody pull-down followed by reverse transcription and sequencing. The difference here is the absence of any cross-linking (Matkovich et al., 2011). As mentioned previously, CLASH utilizes a similar cross-linking technique but ligates the miRNA and the mRNA using RNA ligase. While the specificity of CLASH is high, its sensitivity is reported to be very low (~2%; Helwak et al., 2013). The usage of these techniques in parallel with gene expression analyses in the setting of miRNA gain- or loss-of-function will likely increase the specificity of the observed microRNA targets.
Here, we turn to some possible reasons as to why we see small phenotypes in miRNA deficient mice. The first of these relates to redundancy of both miRNAs and targeting of particular mRNAs by different miRNAs. In addition to there being multiple genomic loci producing the same miRNA, many miRNAs are part of miRNA “families,” which target the same seed sequence. Hence, there is a great deal of redundancy at the genomic level. Because the seed sequence is small, a single miRNA can target a large number of mRNAs (Thomson et al., 2011). The opposite is also true, that a single mRNA can be targeted by numerous miRNAs, including those from different families.
There are a number of miRNA target prediction tools available online including but not limited to TargetScan, PicTar, and Miranda (Ritchie et al., 2013). Confirmation of direct miRNA targeting of mRNA is usually done by a luciferase reporter-3′UTR of the mRNA construct and overexpressing the miRNA. However, not all of the predicted mRNA targets are actual targets of the miRNA. So what factors decide whether a miRNA will target a particular mRNA? There are many factors which likely play a role beyond seed matching and binding of complementary RNA strands. These may include structural accessibility of the 3′UTR of the mRNA to miRNAs, epigenetic modifications in targeted region preventing miRNA binding and the presence of miRNA sponges called competing endogenous RNAs (ceRNAs) which preferentially bind to miRNAs thereby de-repressing their mRNA targets (Karreth and Pandolfi, 2013). The prediction of miRNA-deficient phenotypes suffers from this lack of predictability of target repression.
Knocking out a single genetic locus encoding a miRNA may not be sufficient to fully understand the function of a given miRNA. Conceptually, this is similar to the situation of genetic redundancy seen with protein-coding genes, but it is different because miRNAs are most commonly found as families, and the majority of miRNAs share seed sequences with other microRNAs. Another important thing to note is the unperturbed levels of the miRNA in a particular tissue type. For example, miR-122 has been shown to account for nearly 50% of the total hepatocyte miRNA pool. In this situation, one could imagine that a knockout would have significant effects on gene expression (Szabo and Bala, 2013).
An ideal loss-of-function study will concurrently target all redundant miRNAs for deletion. This requires a large amount of resources to generate conventional knockout mice, which can be prohibitive to undertake such a project. However, with the advent of novel knockdown technologies like ZFNs (zinc finger nucleases), CRISPRs (clustered regularly interspaced short palindromic repeats) and TALENs (transcription activator-like effector nucleases), these complicated knockout animals may now be more feasible to create and study (Gaj et al., 2013). Alternative and novel approaches, such as designing ceRNAs that can target entire families of miRNAs, should also be considered and carefully examined. This approach could provide a viable method to knock down a large number of miRNAs at the same time. Lastly, the role of specific miRNA-target interactions should be carefully studied in carefully selected systems. In certain cases and in certain developmental/ pathologic contexts, a single miRNA-target pair may be of paramount importance (Xiao et al., 2007; O’Connell et al., 2009; Xiao and Rajewsky, 2009; Rao et al., 2010). Such interactions should be further studied using targeted mutations of the miRNA binding site in 3’UTRs, as described for miR-155 and AID (Borchert et al., 2011).
In most currently utilized techniques, the extent of overexpression is usually higher than physiologically attainable limits. Such overexpression may overwhelm the miRNA processing machinery and cause off-target effects (Singh et al., 2011; Karlsen and Brinchmann, 2013). Hence, some of the phenotypes that have been elaborated with overexpression may be more striking simply because of the extent of overexpression. Usage of endogenous promoters which are weaker than the conventional ones or using an inducible promoter which follows linear kinetics would be ideal, but this remains technically challenging. Yet, many of these experiments have revealed the central importance of miRNAs in driving oncogenesis.
Recent studies have examined the role of variability of gene expression in the generation of different cell types during differentiation, and presumably similar mechanisms are operant in cancer causation and progression. Even when a tissue contains cells of the same type, the expression of a particular protein can show stochastic cell-to-cell as well as temporal fluctuations within the same cell. Sometimes these fluctuations were up to ~1000-fold between the low expressing and high expressing cells at one point of time (Chang et al., 2008). These differing gene expression levels might be purely stochastic or represent phenotypic variants within the same population (Mukherji et al., 2011). Recent reports suggest that such non-genetic heterogeneity might be generated by miRNA-mediated effects on gene expression, and that this may depend on the number of miRNA binding sites in the 3’UTR, the level of mRNA in the cell as well as the level of miRNA in the cell.
It has been demonstrated that at a constant level of miRNA, there is a particular threshold of mRNA below which the repression by the miRNA is dramatically increased (Mukherji et al., 2011). On the opposite end of the spectrum, past a certain concentration of mRNA, miRNA-mediated repression is virtually ineffective. Similarly when the number of binding sites in the 3’UTR for the miRNA was increased from 1 to 7 in the UTR, the repression increased by 40-fold (Chang et al., 2008). This threshold effect between the miRNA and the mRNA is reminiscent of biochemical precipitation reactions involving antigen- antibody interactions at the optimum concentration of both the antigen as well as the antibody. The precipitin line forms only at this zone of equivalence and neither in the region of antigen excess nor in antibody excess (Hornbeck, 2001). Hence, there are several relationships between the miRNA and mRNA that can profoundly change the distribution of gene expression within a population of cells.
It is implied that a cell requires a particular level of mRNA and miRNA to coexist for repression to occur. The net effect of these threshold effects is that our conventional thinking about miRNA- that they partially repress gene expression- may only hold for intermediate concentrations of the miRNA and the target mRNA. The implications for experimental science are important: we have to be cognizant of the level of the target mRNA in the cells that are being targeted for perturbation of the miRNA. In experiments with miRNA overexpression or knock down, most groups usually collect the RNA from an entire population of cells, revealing a snapshot of the average gene expression in the cells. This certainly doesn’t tell us the entire story- different cells with differing unperturbed target mRNA levels would have different levels of change, depending on the mRNA threshold (Figure 1). Therefore, single-cell expression studies are necessary to study the cell to cell variation in response to miRNAs. Recent reports have shown that miRNA expression can be studied at the single-cell level using microfluidics (Wu et al., 2013). This, coupled with lineage tracing studies may help us understand how phenotypes develop in miRNA-deficient or overexpressing cells, both in development and in disease. The threshold effect is an important consideration in miRNA therapeutics- particularly against cancers. Overexpression of miRNAs might be highly efficient at repressing the expression of a critical target mRNA in cells that express the target at the optimum level (Figure 2). However cells with extremely high levels of the target mRNA would escape repression, if the above-proposed mechanisms are correct, and may even be selected for. In theory, this could lead to the elaboration of a more aggressive cancer following an initial remission. Hence, targeted therapy utilizing small RNA molecules requires careful consideration of ranges of target gene expression in the cancer cells.
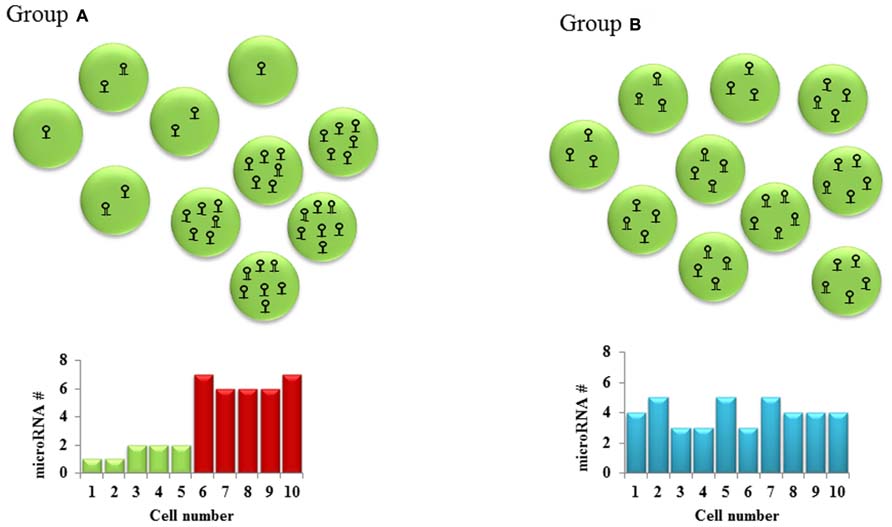
FIGURE 1. Cellular heterogeneity in microRNA levels: Hypothetical cell groups (A,B) have the same average number of a particular microRNA per cell (average = 4). SNA extraction and qPCR of both these groups of cells would give the same microRNA expression in both groups. However, a closer look at the two groups shows a bimodal type of microRNA expression pattern in group A and a homogenous expression pattern in group B. This cell to cell difference in microRNA levels which might lead to different downstream gene expression is often overlooked in the absence of single-cell studies.
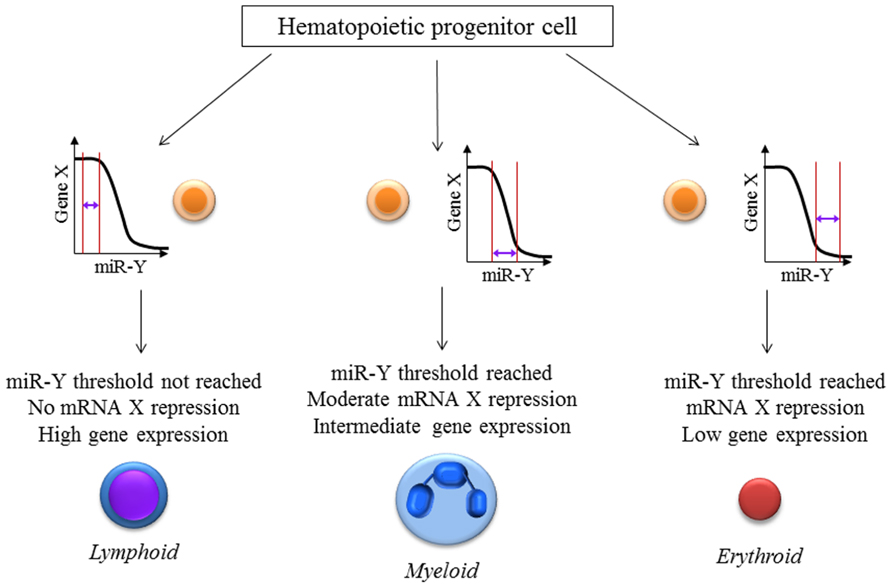
FIGURE 2. miRNA:mRNA interaction-the threshold effect: The level of repression of gene expression is dependent on the levels of both the mRNA as well as the microRNA present inside a particular cell. This hypothetical figure assumes that the expression of mRNA X is important for hematopoietic cell lineage determination with high, intermediate and low levels leading to lymphoid, myeloid and erythroid lineage differentiation respectively. Gene X is a known target of the microRNA miR-Y. These effects would be missed if single-cell measurements of X and miR-Y were not performed which reiterates the necessity for newer techniques and analyses especially at the single-cell level.
The plethora of profiling experiments has to date yielded important data about miRNA expression in primary human disease states, but we suggest that these constitute a starting point for disease-relevant studies. A major obstacle to understanding these miRNAs in vivo is the resource set required to individually target these miRNAs and miRNA-target pairs. However, it seems that such detailed studies will be required to understand the roles of these small RNAs in disease pathogenesis and for their utilization as therapeutic interventions.
We have tried to provide an overview of some of the factors that we believe to be obstacles in moving miRNA research from the bench to the bedside (or vice-versa). These include technical difficulties and variations in high-throughput profiling studies between labs and across platforms. These could be standardized in the future with rigorous quality control, better kits and standard guidelines for sample preparation and data analysis. The question of genetic redundancy in miRNAs calls for more complex mouse models which are also foreseeable in the future. Non-genetic heterogeneity presents a totally different question altogether. Sophisticated single-cell expression studies of mRNA, miRNA and proteins are required to pave the way to decipher the actual role of non-genetic variants present within what many researchers have previously presumed to be homogeneous populations. With the advent of more robust and reproducible profiling techniques, more sophisticated genetic manipulations in model systems, and single-cell analytical advances, we anticipate a clearer understanding of miRNA roles in biology and pathogenesis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank members of the Rao lab for helpful discussions regarding this review. This work was supported by an R01 CA166450-01 and Career Development Award K08CA133521 from the National Institutes of Health (Dinesh S. Rao). Dinesh S. Rao was a Sidney Kimmel Scholar supported by the Sidney Kimmel Foundation for Cancer Research (Translational Award SKF-11-013).
Adiconis, X., Borges-Rivera, D., Satija, R., DeLuca, D. S., Busby, M. A., Berlin, A. M., et al. (2013). Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629. doi: 10.1038/nmeth.2483
Almeida, M. I., Nicoloso, M. S., Zeng, L., Ivan, C., Spizzo, R., Gafà, R., et al. (2012). Strand-specific miR-28-5p and miR-28-3p have distinct effects in colorectal cancer cells. Gastroenterology 142, 886–896. doi: 10.1053/j.gastro.2011.12.047
Aqeilan, R. I., Calin, G. A., and Croce, C. M. (2010). miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 17, 215–220. doi: 10.1038/cdd.2009.69
Babar, I. A., Cheng, C. J., Booth, C. J., Liang, X., Weidhaas, J. B., Saltzman, W. M., et al. (2012). Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. U.S.A. 109, E1695–E1704. doi: 10.1073/pnas.1201516109
Baker, M. (2010). microRNA profiling: separating signal from noise. Nat. Methods 7, 687–692. doi: 10.1038/nmeth0910-687
Boldin, M. P., Taganov, K. D., Rao, D. S., Yang, L., Zhao, J. L., Kalwani, M., et al. (2011). miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 208, 1189–1201. doi: 10.1084/jem.20101823
Borchert, G. M., Holton, N. W., and Larson, E. D. (2011). Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer 11:347. doi: 10.1186/1471-2407-11-347
Bustin, S. A., Benes, V., Garson, J., Hellemans, J., Huggett, J., Kubista, M., et al. (2013). The need for transparency and good practices in the qPCR literature. Nat. Methods 10, 1063–1067. doi: 10.1038/nmeth.2697
Calin, G. A., and Croce, C. M. (2006). microRNA signatures in human cancers. Nat. Rev. Cancer 6, 857–866. doi: 10.1038/nrc1997
Calin, G. A., and Croce, C. M. (2009). Chronic lymphocytic leukemia: interplay between noncoding RNAs and protein-coding genes. Blood 114, 4761–4770. doi: 10.1182/blood-2009-07-192740
Calin, G. A., Liu, C. G., Sevignani, C., Ferracin, M., Felli, N., Dumitru, C. D., et al. (2004). microRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc. Natl. Acad. Sci. U.S.A. 101, 11755–11760. doi: 10.1073/pnas.0404432101
Cancer Genome Atlas Research Network. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074. doi: 10.1056/NEJMoa1301689
Castellano, L., and Stebbing, J. (2013). Deep sequencing of small RNAs identifies canonical and non-canonical miRNA and endogenous siRNAs in mammalian somatic tissues. Nucleic Acids Res. 41, 3339–3351. doi: 10.1093/nar/gks1474
Chang, H. H., Hemberg, M., Barahona, M., Ingber, D. E., and Huang, S. (2008). Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453, 544–547. doi: 10.1038/nature06965
Chaudhuri, A. A., So, A. Y., Mehta, A., Minisandram, A., Sinha, N., Jonsson, V. D., et al. (2012). Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc. Natl. Acad. Sci. U.S.A. 109, 4233–4238. doi: 10.1073/pnas.1200677109
Chen, L., Cui, B., Zhang, S., Chen, G., Croce, C. M., and Kipps, T. J. (2008). Association between the proficiency of B-cell receptor signaling and the relative expression levels of ZAP-70, SHIP-1, and Mir-155 in chronic lymphocytic leukemia. Blood 112, 3155.
Chim, C. S., Wong, K. Y., Qi, Y., Loong, F., Lam, W. L., Wong, L. G., et al. (2010). Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 31, 745–750. doi: 10.1093/carcin/bgq033
Choi, Y. J., Lin, C. P., Ho, J. J., He, X., Okada, N., Bu, P., et al. (2011). miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol. 13, 1353–1360. doi: 10.1038/ncb2366
Christoffersen, N. R., Shalgi, R., Frankel, L. B., Leucci, E., Lees, M., Klausen, M., et al. (2010). p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 17, 236–245. doi: 10.1038/cdd.2009.109
Concepcion, C. P., Han, Y. C., Mu, P., Bonetti, C., Yao, E., D’Andrea, A., et al. (2012). Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet. 8:e1002797. doi: 10.1371/journal.pgen.1002797
Corney, D. C., Hwang, C. I., Matoso, A., Vogt, M., Flesken-Nikitin, A., Godwin, A. K., et al. (2010). Frequent downregulation of miR-34 family in human ovarian cancers. Clin. Cancer Res. 16, 1119–1128. doi: 10.1158/1078-0432.CCR-09-2642
Costinean, S., Sandhu, S. K., Pedersen, I. M., Tili, E., Trotta, R., Perrotti, D., et al. (2009). Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood 114, 1374–1382. doi: 10.1182/blood-2009-05-220814
Costinean, S., Zanesi, N., Pekarsky, Y., Tili, E., Volinia, S., Heerema, N., et al. (2006). Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 103, 7024–7029. doi: 10.1073/pnas.0602266103
Croce, C. M. (2009). Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 10, 704–714. doi: 10.1038/nrg2634
Eis, P. S., Tam, W., Sun, L., Chadburn, A., Li, Z., Gomez, M. F., et al. (2005). Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc. Natl. Acad. Sci. U.S.A. 102, 3627–3632. doi: 10.1073/pnas.0500613102
Elcheva, I., Goswami, S., Noubissi, F. K., and Spiegelman, V. S. (2009). CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 35, 240–246. doi: 10.1016/j.molcel.2009.06.007
Epis, M. R., Giles, K. M., Kalinowski, F. C., Barker, A., Cohen, R. J., and Leedman, P. J. (2012). Regulation of expression of deoxyhypusine hydroxylase (DOHH), the enzyme that catalyzes the activation of eIF5A, by miR-331-3p and miR-642-5p in prostate cancer cells. J. Biol. Chem. 287, 35251–35259. doi: 10.1074/jbc.M112.374686
Fabbri, M., Bottoni, A., Shimizu, M., Spizzo, R., Nicoloso, M. S., Rossi, S., et al. (2011). Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA 305, 59–67. doi: 10.1001/jama.2010.1919
Gaj, T., Gersbach, C. A., and Barbas, C. F. III. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004
Garzon, R., Calin, G. A., and Croce, C. M. (2009). microRNAs in Cancer. Annu. Rev. Med. 60, 167–179. doi: 10.1146/annurev.med.59.053006.104707
Garzon, R., Marcucci, G., and Croce, C. M. (2010). Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discov. 9, 775–789. doi: 10.1038/nrd3179
Git, A., Dvinge, H., Salmon-Divon, M., Osborne, M., Kutter, C., Hadfield, J., et al. (2010). Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 16, 991–1006. doi: 10.1261/rna.1947110
Goswami, S., Tarapore, R. S., Teslaa, J. J., Grinblat, Y., Setaluri, V., and Spiegelman, V. S. (2010). microRNA-340-mediated degradation of microphthalmia-associated transcription factor mRNA is inhibited by the coding region determinant-binding protein. J. Biol. Chem. 285, 20532–20540. doi: 10.1074/jbc.M110.109298
Grosshans, H., and Chatterjee, S. (2010). microRNAses and the regulated degradation of mature animal miRNAs. Adv. Exp. Med. Biol. 700, 140–155. doi: 10.1007/978-1-4419-7823-3_12
Guan, D., Zhang, W., Zhang, W., Liu, G. H., and Belmonte, J. C. (2013). Switching cell fate, ncRNAs coming to play. Cell Death Dis. 4:e464. doi: 10.1038/cddis.2012.196
Han, Y. C., Park, C. Y., Bhagat, G., Zhang, J., Wang, Y., Fan, J. B., et al. (2010). microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med. 207, 475–489. doi: 10.1084/jem.20090831
Hatley, M. E., Patrick, D. M., Garcia, M. R., Richardson, J. A., Bassel-Duby, R., van Rooij, E., et al. (2010). Modulation of K-Ras-dependent lung tumorigenesis by microRNA-21. Cancer Cell 18, 282–293. doi: 10.1016/j.ccr.2010.08.013
He, L., Thomson, J. M., Hemann, M. T., Hernando-Monge, E., Mu, D., Goodson, S., et al. (2005). A microRNA polycistron as a potential human oncogene. Nature 435, 828–833. doi: 10.1038/nature03552
Helwak, A., Kudla, G., Dudnakova, T., and Tollervey, D. (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. doi: 10.1016/j.cell.2013.03.043
Hermeking, H. (2010). The miR-34 family in cancer and apoptosis. Cell Death Differ. 17, 193–199. doi: 10.1038/cdd.2009.56
Hornbeck, P. (2001). Double-immunodiffusion assay for detecting specific antibodies. Curr. Protoc. Immunol. Chap. 2:Unit 2.3. doi: 10.1002/0471142735.im0203s00
Huffaker, T. B., Hu, R., Runtsch, M. C., Bake, E., Chen, X., Zhao, J., et al. (2012). Epistasis between microRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell Rep. 2, 1697–1709. doi: 10.1016/j.celrep.2012.10.025
Jin, H. Y., Oda, H., Lai, M., Skalsky, R. L., Bethel, K., Shepherd, J., et al. (2013). microRNA-17~92 plays a causative role in lymphomagenesis by coordinating multiple oncogenic pathways. EMBO J. 32, 2377–2391. doi: 10.1038/emboj.2013.178
Karlsen, T. A., and Brinchmann, J. E. (2013). Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol. Ther. 21, 1169–1181. doi: 10.1038/mt.2013.55
Karreth, F. A., and Pandolfi, P. P. (2013). ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 3, 1113–1121. doi: 10.1158/2159-8290.CD-13-0202
Kasashima, K., Nakamura, Y., and Kozu, T. (2004). Altered expression profiles of microRNAs during TPA-induced differentiation of HL-60 cells. Biochem. Biophys. Res. Commun. 322, 403–410. doi: 10.1016/j.bbrc.2004.07.130
Kim, V. N., Han, J., and Siomi, M. C. (2009). Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139. doi: 10.1038/nrm2632
Klein, U., Lia, M., Crespo, M., Siegel, R., Shen, Q., Mo, T., et al. (2010). The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17, 28–40. doi: 10.1016/j.ccr.2009.11.019
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. doi: 10.1038/nrg2843
Kumar, M. S., Pester, R. E., Chen, C. Y., Lane, K., Chin, C., Lu, J., et al. (2009). Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 23, 2700–2704. doi: 10.1101/gad.1848209
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Matkovich, S. J., Van Booven, D. J., Eschenbacher, W. H., and Dorn, G. W. II. (2011). RISC RNA sequencing for context-specific identification of in vivo microRNA targets. Circ. Res. 108, 18–26. doi: 10.1161/CIRCRESAHA.110.233528
Medina, P. P., Nolde, M., and Slack, F. J. (2010). OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 467, 86–90. doi: 10.1038/nature09284
Meister, G. (2013). Argonaute proteins: functional insights and emerging roles. Nat. Rev. Genet. 14, 447–459. doi: 10.1038/nrg3462
Mestdagh, P., Van Vlierberghe, P., De Weer, A., Muth, D., Westermann, F., Speleman, F., et al. (2009). A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 10, R64. doi: 10.1186/gb-2009-10-6-r64
Mittal, N., and Zavolan, M. (2014). Seq and CLIP through the miRNA world. Genome Biol. 15, 202. doi: 10.1186/gb4151
Mukherji, S., Ebert, M. S., Zheng, G. X., Tsang, J. S., Sharp, P. A., and van Oudenaarden, A. (2011). microRNAs can generate thresholds in target gene expression. Nat. Genet. 43, 854–859. doi: 10.1038/ng.905
Nana-Sinkam, S. P., and Croce, C. M. (2013). Clinical applications for microRNAs in cancer. Clin. Pharmacol. Ther. 93, 98–104. doi: 10.1038/clpt.2012.192
O’Connell, R. M., Chaudhuri, A. A., Rao, D. S., and Baltimore, D. (2009). Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. U.S.A. 106, 7113–7118. doi: 10.1073/pnas.0902636106
O’Connell, R. M., Chaudhuri, A. A., Rao, D. S., Gibson, W. S., Balazs, A. B., and Baltimore, D. (2010a). microRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc. Natl. Acad. Sci. U.S.A 107, 14235–14240. doi: 10.1073/pnas.1009798107
O’Connell, R. M., Kahn, D., Gibson, W. S., Round, J. L., Scholz, R. L., Chaudhuri, A. A., et al. (2010b). microRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 33, 607–619. doi: 10.1016/j.immuni.2010.09.009
O’Connell, R. M., Zhao, J. L., and Rao, D. S. (2011). microRNA function in myeloid biology. Blood 118, 2960–2969. doi: 10.1182/blood-2011-03-291971
Park, C. Y., Choi, Y. S., and McManus, M. T. (2010). Analysis of microRNA knockouts in mice. Hum. Mol. Genet. 19, R169–R175. doi: 10.1093/hmg/ddq367
Pauli, A., Rinn, J. L., and Schier, A. F. (2011). Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 12, 136–149. doi: 10.1038/nrg2904
Pritchard, C. C., Cheng, H. H., and Tewari, M. (2012). microRNA profiling: approaches and considerations. Nat. Rev. Genet. 13, 358–369. doi: 10.1038/nrg3198
Rao, D. S., O’Connell, R. M., Chaudhuri, A. A., Garcia-Flores, Y., Geiger, T. L., and Baltimore, D. (2010). microRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 33, 48–59. doi: 10.1016/j.immuni.2010.06.013
Ravi, A., Gurtan, A. M., Kumar, M. S., Bhutkar, A., Chin, C., Lu, V., et al. (2012). Proliferation and tumorigenesis of a murine sarcoma cell line in the absence of DICER1. Cancer Cell 21, 848–855. doi: 10.1016/j.ccr.2012.04.037
Ritchie, W., Rasko, J. E., and Flamant, S. (2013). microRNA target prediction and validation. Adv. Exp. Med. Biol. 774, 39–53. doi: 10.1007/978-94-007-5590-1_3
Roderburg, C., Urban, G. W., Bettermann, K., Vucur, M., Zimmermann, H., Schmidt, S., et al. (2011). micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218. doi: 10.1002/hep.23922
Siemens, H., Jackstadt, R., Kaller, M., and Hermeking, H. (2013). Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget 4, 1399–1415.
Singh, S., Narang, A. S., and Mahato, R. I. (2011). Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm. Res. 28, 2996–3015. doi: 10.1007/s11095-011-0608-1
Song, S. J., Ito, K., Ala, U., Kats, L., Webster, K., Sun, S. M., et al. (2013). The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 13, 87–101. doi: 10.1016/j.stem.2013.06.003
Stahlberg, A., Kubista, M., and Pfaffl, M. (2004). Comparison of reverse transcriptases in gene expression analysis. Clin. Chem. 50, 1678–1680. doi: 10.1373/clinchem.2004.035469
Starczynowski, D. T., Kuchenbauer, F., Argiropoulos, B., Sung, S., Morin, R., Muranyi, A., et al. (2010). Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nat. Med. 16, 49–58. doi: 10.1038/nm.2054
Szabo, G., and Bala, S. (2013). microRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol. 10, 542–552. doi: 10.1038/nrgastro.2013.87
Thai, T. H., Calado, D. P., Casola, S., Ansel, K. M., Xiao, C., Xue, Y., et al. (2007). Regulation of the germinal center response by microRNA-155. Science 316, 604–608. doi: 10.1126/science.1141229
Thomson, D. W., Bracken, C. P., and Goodall G. J. (2011). Experimental strategies for microRNA target identification. Nucleic Acids Res. 39, 6845–6853. doi: 10.1093/nar/gkr330
Trissal, M., Silva, J., Wylie, T., Hundal, J., McGrath, S., Magrini, V., et al. (2013). Dysregulation and recurrent mutation of miRNA-142 in de novo AML. Blood 122, 472.
Uchino, K., Takeshita, F., Takahashi, R. U., Kosaka1, N., Fujiwara, K., Naruoka, H., et al. (2013). Therapeutic effects of microRNA-582-5p and -3p on the inhibition of bladder cancer progression. Mol. Ther. 21, 610–619. doi: 10.1038/mt.2012.269
van Kouwenhove, M., Kedde, M., and Agami, R. (2011). microRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 11, 644–656. doi: 10.1038/nrc3107
Vickers, K. C., Sethupathy, P., Baran-Gale, J., and Remaley, A. T. (2013). Complexity of microRNA function and the role of isomiRs in lipid homeostasis. J. Lipid Res. 54, 1182–1191. doi: 10.1194/jlr.R034801
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. doi: 10.1016/0092-8674(93)90530-4
Witwer, K. W. (2013). Data submission and quality in microarray-based microRNA profiling. Clin. Chem. 59, 392–400. doi: 10.1373/clinchem.2012.193813
Wu, M., Piccini, M., Koh, C. Y., Lam, K. S., and Singh, A. K. (2013). Single cell microRNA analysis using microfluidic flow cytometry. PLoS ONE 8:e55044. doi: 10.1371/journal.pone.0055044
Xiao, C., and Rajewsky, K. (2009). microRNA control in the immune system: basic principles. Cell 136, 26–36. doi: 10.1016/j.cell.2008.12.027
Xiao, C., Calado, D. P., Galler, G., Thai, T. H., Patterson, H. C., Wang, J., et al. (2007). MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131, 146–159. doi: 10.1016/j.cell.2007.07.021
Xiong, Y., Fang, J. H., Yun, J. P., Yang, J., Zhang, Y., Jia, W. H., et al. (2010). Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 51, 836–845. doi: 10.1002/hep.23380
Yang, X., Du, W. W., Li, H., Liu, F., Khorshidi, A., Rutnam, Z. J., et al. (2013). Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 41, 9688–9704. doi: 10.1093/nar/gkt680
Zhao, J. L., Rao, D. S., Boldin, M. P., Taganov, K. D., O’Connell, R. M., and Baltimore, D. (2011). NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. U.S.A. 108, 9184–9189. doi: 10.1073/pnas.1105398108
Keywords: microRNAs, non-genetic heterogeneity, genetic redundancy, microRNA profiling, single-cell studies
Citation: Palanichamy JK and Rao DS (2014) MiRNA dysregulation in cancer: towards a mechanistic understanding. Front. Genet. 5:54. doi: 10.3389/fgene.2014.00054
Received: 17 January 2014; Paper pending published: 07 February 2014;
Accepted: 02 March 2014; Published online: 18 March 2014.
Edited by:
Ryan Michael O’Connell, University of Utah, USAReviewed by:
Todd A. Fehniger, Washington University School of Medicine, USACopyright © 2014 Palanichamy and Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dinesh S. Rao, Department of Pathology and Laboratory Medicine, University of California, Los Angeles, CA 90095, USA
e-mail:ZHJhb0BtZWRuZXQudWNsYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.