 Brendan M. Olynik1,2
Brendan M. Olynik1,2 Mojgan Rastegar1,2*
Mojgan Rastegar1,2*- 1 Regenerative Medicine Program, Faculty of Medicine, University of Manitoba, Winnipeg, MB, Canada
- 2 Department of Biochemistry and Medical Genetics, University of Manitoba, Winnipeg, MB, Canada
Epigenetic changes occur throughout life from embryonic development into adulthood. This results in the timely expression of developmentally important genes, determining the morphology and identity of different cell types and tissues within the body. Epigenetics regulate gene expression and cellular morphology through multiple mechanisms without alteration in the underlying DNA sequences. Different epigenetic mechanisms include chromatin condensation, post-translational modification of histone proteins, DNA cytosine marks, and the activity of non-coding RNA molecules. Epigenetics play key roles in development, stem cell differentiation, and have high impact in human disease. In this review, we will discuss our current knowledge about these epigenetic mechanisms, with a focus on histone and DNA marks. We will then talk about the genetics and epigenetics of embryonic stem cell self-renewal and differentiation into neural stem cells, and further into specific neuronal cell types.
Introduction
Life begins upon fertilization of a haploid egg and a haploid sperm. The resulting zygote will subsequently undergo many cellular divisions in a tightly regulated manner, as part of a complex developmental journey. The underlying genetic code remains the same in all cells within an individual, yet an enormously diverse population of cell types will be generated, which will constitute a multicellular organism. By controlling gene expression, epigenetic mechanisms instruct cellular identity and morphology, greatly contributing to the vast assortment of cellular phenotypes (Barros and Offenbacher, 2009; Delcuve et al., 2009; Rastegar et al., 2011). The term “epigenetics” was used for the first time by Waddington (1942) as a way of “genotype” controlling “phenotype” during development. Since then, we have come a long way and the field of epigenetics has been revolutionized substantially. The word “epigenetics” is named after the Greek word, “epi” that means “on,” or “above” the “genetics.” Epigenetics are known as inheritable programming in gene expression that control cellular morphology and identity, without changing the corresponding DNA sequences (Delcuve et al., 2009; Rastegar et al., 2011). Accumulation of all epigenetic marks define the epigenome of any given cell, similarly in that all genetic information pertains to the genome, and protein components to the proteome.
Epigenetics are profoundly involved in mammalian development (Barber and Rastegar, 2010), human disease (Choo, 2011; Zachariah and Rastegar, 2012) and embryonic stem cell (ESC) differentiation (Delcuve et al., 2009; Rastegar et al., 2011). For the first time in 1981, ESCs were isolated and expanded in vitro from the inner cell mass (ICM) of the developing mouse blastocysts (Martin, 1981). Mouse and human ESC are pluripotent stem cells that are capable of indefinite self-renewal and differentiation into all three germ layers (Martin, 1981; Pan and Thomson, 2007). The discovery of these properties presented ESC as a perfect tool for investigating early development and modeling human disease. In addition, ESC self-renewal and differentiation properties provide a great promise as an inexhaustible resource for cell-based therapies in human degenerative diseases. These unique characteristics have brought ESCs into the forefront of today’s research. ESC differentiation into somatic nerve cells involves a cascade of molecular events, and signaling pathways from the extracellular microenvironment, to the transcriptional activities within the nucleus. In many neurological disorders, we need to direct ESCs toward specific type of neurons to be suitable for transplantation strategies and in regenerative medicine. Such comprehensive knowledge of gene expression profiles and cell fate commitment of stem cells are essential for future stem cell-based therapy applications and for proper in vitro modeling of neurological disorders. This information are particularly essential for most applications in cell-based therapy strategies and in regenerative medical approaches, where pure populations of differentiated cell types are required for successful transplantation. Although our scientific community has made great advancement toward directing ESC to differentiate into particular cell types and lineages, even with our best available protocols, we still end up generating heterogeneous populations of differentiated cells. The challenge of our today’s research is therefore to understand how ESCs execute gene expression profiles to undergo such extensive genomic, proteomic, and epigenetic changes during differentiation. Eventually, we would be able to benefit from this knowledge to control the cell fate commitment of ESCs toward any desired cell type.
In this review, we will first describe epigenetic programming, with a focus on chromatin structure, histone post-translational modifications (PTM), and DNA marks. We will then discuss ESC self-renewal and differentiation followed by the current state of knowledge about the underlying genetics and epigenetic mechanisms in neural development.
Epigenetic Mechanisms
Chromatin Structure
The epigenetic information is mainly carried within the “chromatin” structure, referred to the combination of DNA and DNA-bound histones (Delcuve et al., 2009; Rastegar et al., 2011). The repetitive fundamental unit of the chromatin is termed the “nucleosome,” which is a combination of 146 bp of DNA molecules wrapped around a histone octamer (Kornberg, 1974). The histone octamer is composed of four nucleosomal histones, including two of each histone H3 and histone H4 (one tetramer of H3–2H4) and two dimers of histones H2A–H2B. Nucleosomes are further joined by the linker DNA that is bound to the fifth histone, namely the linker histone H1, and can be visualized as “beads-on-a-string” structure (Olins and Olins, 1974). At higher level of organization, nucleosomes are assembled into a fiber-like arrangement, with a diameter of 10 or 30 nm, depending on the compaction state of the chromatin (Fussner et al., 2011a). However, recent evidence support the idea that the 30-nm fiber does not exist in vivo (Lieberman-Aiden et al., 2009), and in case of higher order chromatin structure what actually exists is the densely compacted 10 nm fiber. A chromosome is ultimately comprised of these “fibers” and their associated proteins, which allow the chromatin structure to condense, as can be seen throughout cellular division. The compaction state of chromatin can be segregated into tightly packed, “heterochromatin” and lightly packed “euchromatic” regions; with the latter being accessible to gene transcription pertaining to the expressed genome (Horvath et al., 2001; Ellis et al., 2007; Bassett et al., 2009). As a result, the division of chromatin into euchromatin (actively transcribed genes) is marked by histone acetylation, and the heterochromatin (inactive non-transcribed genes) by particular histone methylation with or without DNA methylation. Different heterochromatin regions are characterized by specific epigenetic marks on the inactive repressed genes (referred to as the constitutive heterochromatin) and inactive silenced genes (referred to as the facultative heterochromatin). The euchromatin and heterochromatin compartments are usually dynamic and the chromatin structure can transition between “opened” and “closed” states by carrying a variety of chemical groups to histone proteins or directly to the cytosine nucleotides; in response to extrinsic factors (Delcuve et al., 2009; Rastegar et al., 2011). In general, epigenetic marks are inheritable and can be passed on through cellular division from the mother cells into the daughter cells, but they are also dynamic and reversible in response to stimuli and signaling pathways. Recently, there is an emerging focus on the molecular mechanisms of extracellular signals translating into epigenetic changes, bringing excitement, and introducing new concepts. This is particularly important toward the future application of epigenetics in novel therapeutic strategies for diseases that currently have no treatment and epigenetic disorders that are caused by environmental insults. In addition to modification in chromatin structure, histone variants, histone PTM, and non-coding RNAs (ncRNA) can further decrease or inhibit gene expression, adding to the diversity of features, which constitute the epigenome (Delcuve et al., 2009; Malecova and Morris, 2010; Rastegar et al., 2011; Faust et al., 2012).
Histone Variants and Post-Translational Modifications
Each nucleosome constitutes of 146 bp of DNA wrapped around a histone octamer, with an extra 20–50 bp stretch of DNA called the linker region, which associates with the linker histone H1 (Delcuve et al., 2009). Besides histone H4, histone variants are identified for all other histones and they can influence nuclear chromatin both structurally and functionally (Talbert and Henikoff, 2010). These histone variants are expressed either at the time of DNA replication in S phase (H3.1 and H2A.1) or throughout the cell cycle (H2AZ and H3.3; Mizuguchi et al., 2004; Boulard et al., 2007; Elsaesser et al., 2010; Goldberg et al., 2010; Szenker et al., 2011). In addition to the existence of histone variants, the epigenetic information is further transferred through post-translational modification of histone proteins. Histone PTM include: acetylation, phosphorylation, methylation, ubiquitination, sumoylation, and ADP-ribosylation (Kouzarides, 2007; Delcuve et al., 2009). In general, histone PTM take place on the N-terminal tail of histones, yet in limited cases, they have been found within the core sequence of histones (Delcuve et al., 2009; Barber and Rastegar, 2010). Such examples are the H3K56 acetylation (Tjeertes et al., 2009) and H3Y41 phosphorylation (Dawson et al., 2009), which can occur within the core of histone H3. The existence of the particular covalently bound small chemical molecules on histones or DNA molecules, will determine the epigenetic code at any given locus. Overall, the epigenetic proteins include “writer” molecules, which will deposit these covalent marks; the “erasers” that will remove these marks; and “readers” that interpret these marks and will intermediate or facilitate the intended function. The cumulative action of these three types of epigenetic proteins will eventually determine the gene accessibility state for transcription.
Depending on the specific amino acid residues that histone PTM are attached to, the outcome may be a tighter association of histone proteins with DNA and repression of transcriptional activities; or looser association, allowing transcriptional accessibility of RNA polymerase and affiliated transcriptional machinery (Jenuwein and Allis, 2001; Delcuve et al., 2009; Rastegar et al., 2011). Additions of acetyl groups to the lysines 9 (K9) and 14 (K14) of histone H3 or lysine 16 of histone H4 (H4K16) are performed by K-histone acetyl transferases (KAT), and correspond with open chromatin state (Roth et al., 2001). The lysine residues have a positive charge that leads to their tight association with the negatively charged DNA. The addition of an acetyl group to lysines will eliminate this positive charge leading to a more relaxed association with DNA molecules and as a result the corresponding genomic loci will become more accessible for transcription. Removal of acetylation marks on histones is accomplished by histone de-acetylases (HDAC; Zhang et al., 2002; de Ruijter et al., 2003; Brandl et al., 2009; Delcuve et al., 2009; Rastegar et al., 2011). Phosphorylation of histones usually occurs by addition of a phosphate group onto a hydroxyl group on serine (S) or threonine (T) causing a negative charge, leading to general relaxation and chromatin de-condensation. A well-known example is histone H3S10 phosphorylation that is associated with transcriptional initiation in many genes (Delcuve et al., 2009). Histone methylation is catalyzed by the activity of lysine methyl transferases (KMT), characterized by the existence of a SET domain. Histone methylation can occur at the lysine (K) and arginine (R) residues of histones, and depending to the degree of methylation and the amino acid residues that become methylated, the outcome would be different. For example, H3K4me3 is associated with active genes, H3K9me3 is a hallmark of constitutive heterochromatin and H3K27me3 marks facultative heterochromatin. Interestingly, H3K9me2 is not associated with constitutive heterochromatin, but rather it marks the silenced genes within the facultative compartment of the heterochromatin. Using specific antibodies raised against each of these particular histone marks, chromatin immunoprecipitation (ChIP) techniques can be used to identify the association of particular genomic loci with each of these epigenetic marks. There are some histone lysine demethylation (KDM) enzymes that can erase the histone methyl marks from active or repressed genes, which would also depend on the amino acid residues that are methylated.
In addition to these histone marks, other modifications are found at the lysine residues such as ubiquitination, which has been found on the histones H2A (K119) and H2B (K120; Delcuve et al., 2009; Rastegar et al., 2011). The molecular mechanism of histone ubiquitination is still not fully understood, but it is likely that it functions in keeping chromatin at the open/active state due to its large molecular size (Calvanese and Fraga, 2012). Another identified histone PTM is the sumoylation that occurs in the core region of the four nucleosomal histones, and it happens at the exact same lysines that can become acetylated or ubiquitinated, in an exclusive manner. Other histone PTM include proline isomerization and ADP-ribosylation, both of which are involved in transcriptional gene regulation. It should be noted that the existence of histone marks on the same amino acids are exclusive and they may not occur at an individual residue, simultaneously. This is one of several forms of histone crosstalks, and here the existence of one histone PTM would prevent the deposition of another form to happen at the same residue. Histone crosstalks are not limited to the same histone molecules; rather, modification of an amino acid on one histone can also influence deposition of a different form of PTM on another histone molecule. In addition to the histone PTM crosstalks, these covalent modifications can direct gene expression through crosstalks with chemical groups on DNA molecules such as DNA methylation or 5-hydroxymethylation (Delcuve et al., 2009; Calvanese and Fraga, 2012).
DNA Marks
In addition to methylation of lysine residues within the histone proteins, methylation can also proceed directly on DNA molecules. DNA methylation occurs on the 5′ carbon of the cytosine nucleotides (5mC), next to guanine (CpG dinucleotides). DNA methylation is a hallmark of transcriptionally inactive genes and usually correlates with gene repression and silencing (Suzuki and Bird, 2008; Delcuve et al., 2009), genomic imprinting (Li et al., 1993), and X-chromosome inactivation (Mohandas et al., 1981). De-regulated DNA methylation leads to human disease, mental retardation, and neurological disorders, such as Rett Syndrome (Warren, 2007; Jin et al., 2008; Delcuve et al., 2009). Epigenetic re-programming of primordial germ cells is crucial for erasing inherited epigenetic marks; and in the establishment of totipotency (Reik et al., 2001). Shortly after fertilization, the male pro-nucleus within the zygote will go through a process of genome-wide active and passive demethylation events, followed by de novo methylation (Santos et al., 2002; Delcuve et al., 2009). How cytosine methylation exerts a gene silencing effect is contributed to a couple of scenarios. Firstly, transcription factors may have difficulties in binding nearby methylated DNA, due to the structural hindrances associated with methylated CpG sites (Watt and Molloy, 1988; Tate and Bird, 1993). Secondly, repressor protein complexes and methyl-CpG-binding proteins (MBD) can directly bind to methylated cytosines, preventing transcription initiation (Hendrich and Bird, 1998; Wade, 2001; Zachariah and Rastegar, 2012). The primary identified member of the MBD proteins is the methyl-CpG-binding protein 2 (MeCP2; Meehan et al., 1992), mutations in which lead to Rett Syndrome (Amir et al., 1999). MeCP2 is an interesting protein, as it can bind to both methylated and non-methylated DNA and act as either a transcriptional repressor or an activator (Chahrour et al., 2008; Delcuve et al., 2009; Zachariah and Rastegar, 2012). In the brain, direct association of MeCP2 with constitutive and facultative heterochromatin marks depends on DNA methylation (Thambirajah et al., 2011), and its genomic localization follows DNA methylation (Skene et al., 2010). While MeCP2 is a widely expressed nuclear protein and can be found in multiple tissues and cell types, it is particularly important in the maturation of the central nervous system (CNS) and plays key roles in neuronal maturation and dendrite formation (Rastegar et al., 2009; Squillaro et al., 2012; Zachariah and Rastegar, 2012). MeCP2 is involved in many cellular pathways, acting both as an activator or a repressor, depending on the specific proteins with which it may interact (Chahrour et al., 2008). Its direct and indirect interacting protein partners include HDAC1, HDAC2, the transcriptional repressors mSin3A, and CoREST (Jones et al., 1998; Nan et al., 1998; Ballas et al., 2005). The enzymes responsible for 5mC are DNA methyltransferases (DNMTs), which consist of DNMT1, acting during replication, and DNMT3a and DNMT3b in de novo circumstances (Okano et al., 1999; Bestor, 2000; Delcuve et al., 2009; Zachariah and Rastegar, 2012). There are pivotal crosstalks between 5mC and histone PTM (Fuks, 2005; Cedar and Bergman, 2009), which for example, are evident in heterochromatin with high levels of H3K9me3 equating with 5mC (Stancheva, 2005). Also, the recruitment of HDAC to CpG sites augments the repression of genomic loci (Dobosy and Selker, 2001). These epigenetic conjunctions are apparent throughout the epigenome, and consequently, should be taken into consideration when investigating the epigenetic regulation of any given gene.
First discovered and reported in phage DNA over half a century ago (Wyatt and Cohen, 1953), the 5-hydroxymethylcytosine (5hmC) is recognized as the “sixth base” of the genome. The 5hmC is an epigenetic mark found directly on DNA, with specific tissue (Nestor et al., 2012) and unique distribution pattern during mammalian development (Tahiliani et al., 2009; Ruzov et al., 2011). Within the paternal pro-nucleus of the post-fertilized zygote, the global loss of 5mC, coincides with genome-wide gain of 5hmC (Ruzov et al., 2011). Through the action of TET (10–11 Translocation) proteins, 5mC is converted into 5hmC (Ito et al., 2010). Although the role of 5hmC is still not fully clear, it is reported to have relatively higher expression in pluripotent stem cells, multipotent stem cells, and within the CNS (Ruzov et al., 2011). In the cerebellum, 5hmC is more enriched in the Purkinje cells compared to the granular cells, being reported to be at about 40% of the 5mC marks highlighting a cell type-specific role for 5hmC (Kriaucionis and Heintz, 2009). Nuclear proteins that bind to 5mC, such as MeCP2 or MBD do not interact with 5hmC (Huang et al., 2010), and the bisulfite sequencing technique that has been used commonly for the determination of DNA CpG methylation does not discern between 5hmC and 5mC (Munzel et al., 2011). As a result, this may reinforce a re-validation of our current knowledge about known methylated sites attributed with specific genomic localities obtained with classical bisulfate sequencing. It is recently shown that the TET enzymes not only convert 5mC into 5hmC, but they are also capable of further oxidization into formylcytosine (5fC) and 5 carboxylcytosine (5caC; Ito et al., 2011). The presence of 5fC and 5caC have been seen in murine ESC (Ito et al., 2011; Pfaffeneder et al., 2011), however, the functional role of these novel cytosine marks have not been fully uncovered yet.
Micro RNA
Apart from the role of chromatin structure, histone PTM and DNA marks, another form of epigenetic regulation involves RNA molecules that are affiliated with epigenetic memory (Morris, 2009). MicroRNAs (miRNAs) are ∼22 nucleotide-long Non-coding RNA molecules that can regulate gene expression via binding to the messenger RNA with anti-sense sequence homology, leading to transcript degradation or translational inhibition, thereby adversely impacting protein expression (Bartel, 2004). MicroRNAs are critical in the epigenetic regulation and they have been shown to participate in the establishment of de novo DNA methylation in murine ESC (Sinkkonen et al., 2008), with capability to modulate chromatin structure and key histone modifiers (Chuang and Jones, 2007). MicroRNAs have been elucidated to play regulatory roles in stem cell proliferation and differentiation (Dubois-Dauphin et al., 1991) and loss of components in the miRNA pathway, such as Dicer, can alter these processes (Bernstein et al., 2003). It is the coordinated pattern of DNA marks and histone modifications, along with miRNA activities, which allows the proper differentiation of fully functional cell types in the CNS, and throughout the body.
Central Nervous System Development; the Role of Homeobox Genes
As embryonic development proceeds, the blastocyst will form three main layers namely: the mesoderm, the ectoderm, and the endoderm. It is from the ectoderm that nervous tissue arises, through a tightly regulated process. The early embryo consists of the neural tube, which will eventually become the brain and the spinal cord with full development of the CNS. In each tissue lineage, multipotent stem cells exist which have a more restricted range of cells to become; in the nervous system, these would be neural stem cells (NSC; Temple, 1989; Merkle and Alvarez-Buylla, 2006; Figure 1). Within the CNS, there is an emerging focus on molecular mechanisms of extracellular signals translating into “epigenetic” modifications. In this regard, the activities of homeobox transcription factors exemplify the intricately coordinated network of developmental control.
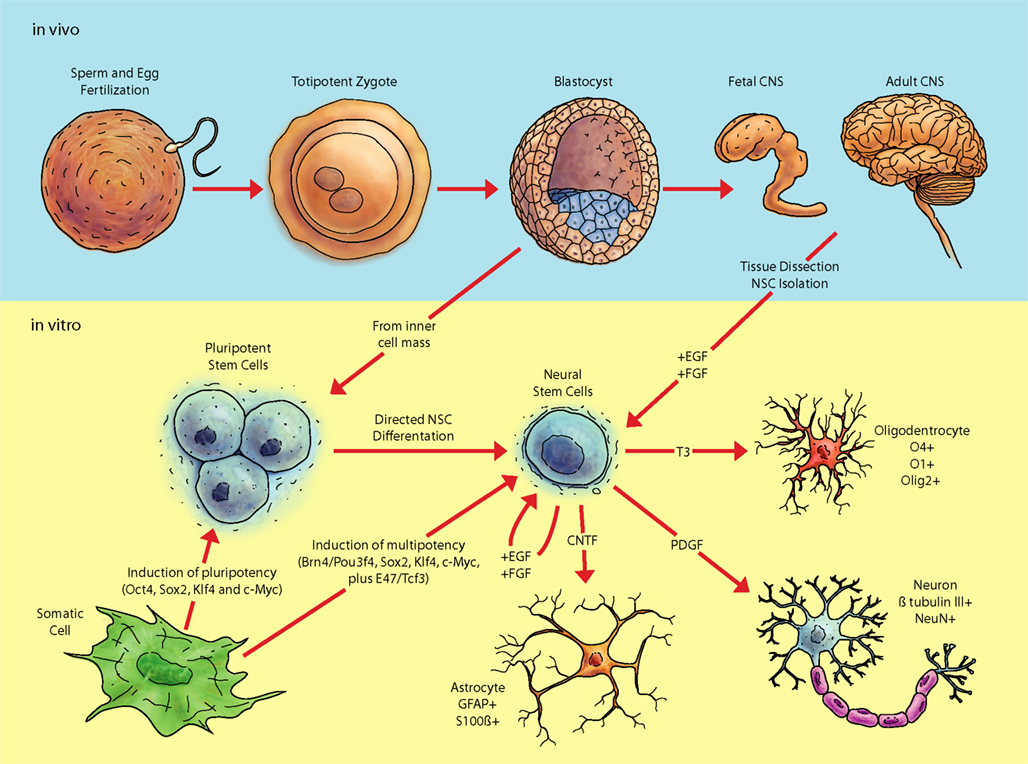
Figure 1. Pathway of pluripotent stem cells to neural cell populations. Fertilization and subsequent cellular divisions create the embryonic blastocyst, where pluripotent ESC are derived (from the inner cell mass; ICM). Additionally, pluripotent and multipotent-like cells can be created via transduction of various factors into differentiated tissue, such as fibroblasts. In vitro analyses of pluripotent and multipotent neural stem cells are integral for understanding aspects of neural differentiation. The in vivo niche of stem cells contains a considerable diversity of biomolecules whose roles still need be deciphered. Exposure of ESC in vitro to various growth factors in serum free media such as fibroblast growth factor 2 (FGF2) and epithelial growth factor (EGF) allows selection of cell lines possessing a neural fate. Neural stem cells can also be acquired from adult tissue and expanded in vitro.
In the developing embryo, NSC sense and respond to signals that influence anterior–posterior (AP) and dorso-ventral (DV) axis of the CNS (Rubenstein and Beachy, 1998; Hoch et al., 2009). During development, NSC give rise to a wide range of cell types and neurons within the CNS along the body axis. It is fascinating how NSC can generate particular neurons in specific locations with appropriate extensions projecting toward target tissues and properly reaching other neurons. A major part of these instructions are dictated by the expression of a large family of genes with a common DNA sequence called the “homeobox (HB),” a 180-bp DNA sequence, encoding for a DNA-binding domain called the “homeodomain” (HD). The homeodomain was discovered in the fruit fly “Drosophila Melanogaster” about three decades ago as a highly conserved DNA-binding domain found within the developmentally important genes (McGinnis et al., 1984). These genes were referred to as the “homeotic genes,” currently known as the “Hox genes” (Barber and Rastegar, 2010). Since then, many HD-containing proteins have been characterized in mammalians sharing a similar 60 amino acid-long DNA-binding domain. Eight such Hox genes exist in fruit fly, and 39 Hox genes are identified in mammals. Hox genes are key regulators of the hindbrain segmentation into rhombomeres (r) and the “Hox code” expression within each rhombomere instructs the cellular composition of each individual hindbrain segment. The hindbrain, that is also named the “rhombencephalon,” is the most posterior compartment of the brain in a developing embryo, and anterior to that we find the midbrain followed by the forebrain (Tvrdik and Capecchi, 2006; Narita and Rijli, 2009). Hox genes are master regulators of development and cellular differentiation that determine the identity of individual cells within the AP and dorso-ventral axes of the hindbrain including segmental regulation within the CNS. Hox genes will further dictate and direct the cellular migration, axonal direction and the process of somatogenesis during embryogenesis and development (Studer et al., 1996; Gavalas et al., 1997; Gaufo et al., 2004; Oury et al., 2006; Oury and Rijli, 2007).
The action of homeobox transcription factors is mediated via regulation of their downstream target genes. As a result, HOX proteins will determine the precise time and space of any given cell type to develop and differentiate along the AP axis of a developing embryo during embryogenesis. Hox genes will further control the proper function of each developed organ within the body. Such a precise action of Hox genes requires their tight control of expression at the exact location during development and in adulthood. The mis-regulation of Hox genes leads to human disease and cancer (Barber and Rastegar, 2010). HOX proteins also regulate the differentiation of the AP and transverse planes of the spinal cord through a collinear expression in a temporal and spatial activation order with the genes located 3′ of each cluster expressed earlier and more anteriorly compared to the more 5′ genes within the same cluster. Hox gene expression in the hindbrain is controlled by different factors and stimuli including retinoic acid (RA), a “vitamin A”-derivative, which functions in the embryonic anterior-posterior patterning. RA treatment has been used to study the transcriptional expression of Hox genes in vitro (Rastegar et al., 2004; Kobrossy et al., 2006; Nolte et al., 2006). The response to RA is mediated through retinoic acid response elements (RARE), which have been identified and characterized in the regulatory elements of many Hox genes including Hoxa1, Hoxb1, Hoxa4, Hoxb4, and Hoxd4 (Langston and Gudas, 1992; Marshall et al., 1994; Studer et al., 1994; Dupe et al., 1997; Gould et al., 1998; Packer et al., 1998; Zhang et al., 2000; Huang et al., 2002; Rastegar et al., 2004; Nolte et al., 2006). There are auto- and cross-regulatory loops that control the transcriptional expression of Hox genes, examples of which are characterized in the expression of the most 3′ gene within the B cluster (Hoxb1) within the rhomobomere 4. Hoxb1 expression in r4 is controlled by a network of signaling molecules, involving Hoxb1, Hoxb2, Hoxa1, and Hoxa2 (Tumpel et al., 2007; Barber and Rastegar, 2010).
HOXB1 regulates the development of the VII facial nerve of the peripheral nervous system, which derives from r4 and has a transient expression prior to the formation of r5, r6, and r7 (Trainor and Krumlauf, 2001; Tumpel et al., 2009). Hoxb1 loss-of-function in mice indicates a non-autonomous role for Hoxb1 in facial nerve development, delineating an essential function in controlling facial branchiomotor neuron development at the level of r4 in the hindbrain (Arenkiel et al., 2003). The cranial neural crest cells are highly dynamic and generate complex derivatives such as cartilage, bone, muscle, glands, neurons, and glia and represent a population of stem cells with restricted cellular commitments (Cornell and Eisen, 2005; Trainor, 2005; Schlosser, 2008). Hoxb1 expressing neural crest cells preferentially generate glial progenitors associated with the development of motor component of VII facial nerve complex and are destined to become peripheral glial cells (Arenkiel et al., 2003). In addition to their role in AP embryonic patterning and neurogenesis, Hox genes are also involved in different types of human cancer such as leukemia (Slany, 2009), breast cancer (Duriseti et al., 2006), and brain tumors (Abdel-Fattah et al., 2006). HOX proteins regulate the expression of their target genes through DNA-binding with different partners, influencing their binding activity and specificity (Huang et al., 2005; Barber and Rastegar, 2010). The best-characterized HOX cofactors are the proto-oncogene Pre-B cell leukemia transcription factor 1 (PBX1) and murine ectopic integration site (MEIS); both affecting HOX DNA-binding activity/specificity (Huang et al., 2005; Barber and Rastegar, 2010). HOX proteins have been the subject of intensive research studies during embryonic development (Rastegar et al., 2004; Stern et al., 2006), ESC neurogenesis (Morey et al., 2007), and embryonic carcinoma cell (ECC) differentiation (Rastegar et al., 2004; Huang et al., 2005; Kobrossy et al., 2006; Nolte et al., 2006). However, due to the redundancies, which exist between the members of each paralog groups, our knowledge about their downstream target genes, besides Hox genes themselves, is currently very limited.
Pluripotent Embryonic Stem Cells
Following the unionization of an egg and a sperm, a single fertilized totipotent zygote will be formed that is capable of developing into all embryonic and extra-embryonic tissues. The resulted zygote will subsequently divide many times toward the generation of a sphere-shaped conglomerate, known as the “blastocyst” (Rossant and Tam, 2009). This early embryonic structure has an outer layer called the trophoblast, which will form the placenta, and inside resides the ICM (Rossant et al., 2003), where ESCs can be isolated (Figure 1). ICM-derived ESCs are pluripotent with the potential of indefinite self-renewal and ability to differentiate into any cell type and tissue within the body (Reubinoff et al., 2000; Odorico et al., 2001; Rastegar et al., 2011). Herein lays ethical controversy in stem cell research, as harvesting cells of the ICM will resultantly destroy the embryo; one of the reasons why alternative methods for acquiring pluripotent cells are eagerly being sought recently (Takahashi and Yamanaka, 2006; Robinton and Daley, 2012). Perhaps one of the most achievements in the field, is the in vitro generation of pluripotent stem cells termed “induced pluripotent stem (iPS) cells” from fibroblast cells in both mice and humans. Shinya Yamanaka was the first to demonstrate that retroviral transduction of cDNA comprising the pluripotent factors, Oct4, Sox2, Klf4, and c-Myc, can transform mature, lineage restricted somatic cells into de-differentiated cells with ESC-like properties (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). Since then, many labs have successfully generated iPS cells from different cell types and with different inducing techniques (Wernig et al., 2007; Aoi et al., 2008; Okita et al., 2008; Park et al., 2008; Hotta et al., 2009), such as the use of piggy back vectors (Woltjen et al., 2009), recombinant proteins (Zhou et al., 2009), nanotechnology (Ruan et al., 2011), and small chemical compounds (Huangfu et al., 2008; Shi et al., 2008). Recently, the transcription factors, Brn4/Pou3f4, Sox2, Klf4, c-Myc, plus E47/Tcf3, have been used to directly induce neural stem-like multipotent stem cells from murine fibroblasts (Han et al., 2012). In general, the processes of re-programming the nuclear transcriptome would synchronize with transformation of the epigenome (Sasaki and Matsui, 2008; Fussner et al., 2011b; Mattout et al., 2011). Overall, the generation of iPS cells has brought great promise and excitement into the translational research, regenerative medicine and modern innovative therapeutic approaches with regards to addressing the current shortage of organs and tissues for transplantation applications. Currently, there is an intensive effort in our scientific community in search of perfecting the iPS cell generation and to better develop technologies for in vivo application of iPS cells. Regardless, this technology has already given us an opportunity to model human disease such as neurological disorders, with no accessible neurons to study the mechanism of disease prior to the development of the iPS technology. Although iPS cell research is still at its infancy period, it has already given us hope by the success of curing limited diseases in mouse models, such as sickle cell anemia in mice (Hanna et al., 2007).
To maintain pluripotency, ESCs continuously express key transcription factors such as OCT4 and NANOG along with the SOX2 protein during their self-renewal. These proteins contribute and share at least in part in the regulation of downstream developmentally important genes. Additionally, OCT4 controls the expression H3K9me2/3 demethylases and as a consequence contributes in maintaining the epigenetic landscape during ESC self-renewal (Loh et al., 2007; Calvanese and Fraga, 2012). Moreover, through the simultaneous existence of both active and inactive histone marks (referred to as “bivalent marks,” which will be discussed later) on the specific developmentally regulated genes, they would remain at the poised state of transcription during ESC self-renewal. Subsequently, during differentiation, these genes may lose one type of marks and become active or silenced depending on the direction of differentiation. In general, chromatin structure in ESCs is highly dynamic (Meshorer et al., 2006) and during differentiation, histone modifications of open chromatin such as histone H3 and H4 acetylation are reduced globally, with a general increase in the repressive marks such as H3K9me3. This leads to more condensed chromatin at specific genomic loci (and corresponding genes) for proper differentiation (Lee et al., 2004; Meshorer et al., 2006). Chromatin condensation, in part, is controlled by linker histone H1 with a high ratio close to 1/nucleosome in differentiated cells and 0.5 in ESCs. Depletion of three histone H1 isoforms is lethal in mice, but such ESCs (Triple histone H1 Knock Out) with 0.25 histone H1/nucleosome, have generated perfect tools and opportunity to study the role of chromatin compaction in controlling specific genomic loci (Fan et al., 2005).
Multipotent Neural Stem Cells
The human brain is a central part of our nervous system, with a constitution of over one-hundred billion neurons, with an average of 7000 connections for each neuron (Drachman, 2005). This large number of neurons seems to be able to compensate for the loss of activity in part of the cells in the brain. However, depending on location, neuronal activity loss may have severe consequences leading to neurological disorders. In this respect, NSC have attracted much attention because of their potential to replace neurons and glia which are lost or damaged in trauma, stroke, spinal cord injury, amyotrophic lateral sclerosis (ALS), Parkinson’s, or Alzheimer’s disease. Altman (1962) observed and reported the incorporation of tritiated thymidine in neural cell types within traumatized regions of rat brains. However, it took many years, with key experimental evidence to prove to the scientific community that the adult nervous system can produce new neurons. There is now a wider acknowledgment that neurons are being produced from NSC not only during embryonic development, but also in the postnatal brain. In the developing embryo, NSC sense, and respond to signals that influence the AP and dorso-ventral axes of the CNS (Rubenstein and Beachy, 1998; Hoch et al., 2009). NSC are primary progenitors capable of self-renewal and differentiation into the three main cell types of the mammalian nervous system; neurons, astrocytes, and oligodendrocytes (Namihira et al., 2008). During the progression of nervous system development, neural stem cell will behave differently while dividing, with respect to the particular fate commitment of their daughter cells (Hirabayashi and Gotoh, 2005). During early gestation, NSC exist as bipolar neuroepithelial cells across the neural tube (Figure 2). At this stage NSC self-renew and symmetrically divide into two identical daughter cells, similar to the parental cell to increase the number of NSC. Later in development and during mid-gestation, NSC will divide asymmetrically, producing a new NSC and a neuron, or a neural stem cell and a neural progenitor cell. Neural progenitor (or precursor) cells are intermediary between the stem cell and more differentiated cells and have more limited self-renewal capability. However, the distinction between neural precursor cells and NSC are not always clearly defined in experimental analyses, and the terms are often used interchangeably. In later stages of embryonic development, NSC are capable of both self-renewal and differentiation and may give rise to neurons, astrocytes, and oligodendrocytes (Figure 2; Qian et al., 2000).
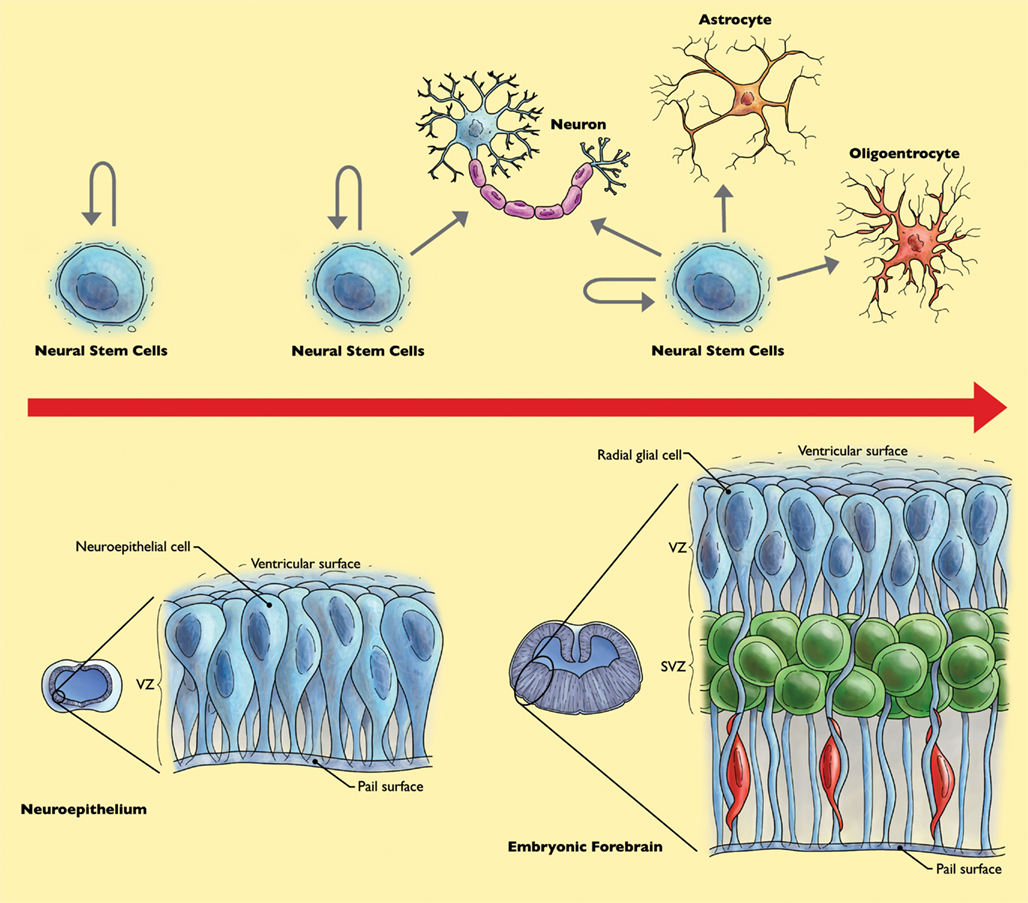
Figure 2. Behavior of neural stem cells (NSC) throughout forebrain development. Early during development, NSC increase their population of stem cells through symmetrical cell divisions. At mid-gestation, at the “neurogenic phase,” NSC divide asymmetrically, yielding a NSC and a neuron or neural progenitor cell and neuron. Neural progenitor cells can further divide symmetrically to produce two neurons. During the “gliogenic phase” in late gestation, populations of not only neurons, but astrocytes and oligodendrocytes are created (Merkle and Alvarez-Buylla, 2006).
As mentioned earlier, functional neurogenesis is not limited to embryonic development and will continue throughout life (Ma et al., 2010) even in the adulthood (Altman, 1962; Goldman and Nottebohm, 1983; Eriksson et al., 1998). In the adult mammalian brain, NSC reside in the sub granular zone of the dentate gyrus in the hippocampus, and in the sub ventricular zone of the lateral ventricles (Aimone et al., 2010; Gage, 2010; Bonfanti and Peretto, 2011; Lee et al., 2012). Adult neurogenesis has been shown to be modulated by a wide range of influences from exercise (van Praag et al., 2005; Brandt et al., 2010) and learning (Sultan et al., 2010; Anderson et al., 2011), to pharmacological treatment with anti-depressants (Duman et al., 2001; Anacker et al., 2011). As a result of these observations, there is promise for discovering new therapeutic modes of neural induction, for those who have lost or damaged neural tissue. Since epigenetic activities are critical for cellular differentiation (Schofer and Weipoltshammer, 2008; Delcuve et al., 2009; Meissner, 2010), we will briefly discuss the involved molecular mechanisms with development, concerning the transition of embryonic to NSC and mature neural cells, including the recognition of key epigenetic activities.
Embryonic Stem Cells Embark on the Neural Road
We have discussed about the major epigenetic mechanisms, neural development, ESCs, and the role of NSC in development. We will now focus on the metamorphoses of the epigenome that occurs during neural commitment of ESCs; and briefly address the corresponding extrinsic factors, genetic transcriptional networks, and epigenetic events (Darr and Benvenisty, 2006; Ng and Surani, 2011; Xue et al., 2011).
Extrinsic Factors Elicit the Actions of Transcriptional Equipment
Embryonic stem cells can be directly differentiated into specially differentiated cell types, yet their default differentiation pathway is into NSC (Smukler et al., 2006). In vivo, NSC are isolated from different parts of the CNS both during embryonic development and from the postnatal brain. Extrinsic factors that affect stem cell differentiation include a variety of signaling molecules and growth factors that are critical for maintenance of ESC pluripotency and self-renewal, or triggering differentiation (Pera and Tam, 2010). In case of NSC, the extrinsic factors mainly originate from the cellular niche, referring to the microenvironment where these cells reside within the CNS. In NSC, the extrinsic factors tightly control the maintenance of multipotency of these cells. Important extrinsic factors include the NOTCH Signaling family, Transforming Growth Factor-β (TGF- β) superfamily members, Fibroblast Growth Factors (FGF), Neuregulins (NRG), and Platelet Derived Growth Factors (PDGF), to name a few (Mizutani et al., 2007; Pera and Tam, 2010). EGF and FGF2 are known to regulate neural programming (Reynolds and Weiss, 1996; Kuhn et al., 1997; Palmer et al., 1999), and are used for in vitro isolation and expansion of NSC (Gage, 2000; Reubinoff et al., 2001; Abranches et al., 2009; Figure 1). One particular member of the EGF family, Betacellulin, has been recently shown to promote cell proliferation and drive NSC neurogenesis (Gomez-Gaviro et al., 2012). Interestingly, insulin like growth factors (IGF1 and IGF2) predispose ESCs toward a mesodermal fate (Piecewicz et al., 2012), yet IGF2 regulates adult hippocampal neurogenesis (Bracko et al., 2012).
Transcription Factors Bridge the State of Stem Cells
During ESC self-renewal, key transcription factors such as OCT4, NANOG, as well as SOX2 control ESC pluripotency via regulating the expression of specific downstream target genes (Heng and Ng, 2010). Recently, other proteins are also shown to have similar roles including TAF3, E-cadherin, KLF4, and PBX1 (Chan et al., 2009; Liu et al., 2011a; Redmer et al., 2011). To point out another level of control to these factors, the microRNA, mir-145, can target the 3′ untranslated region of Oct4, Sox2 (and Klf4) transcripts; and up-regulation of mir-145 is evident during ESC differentiation (Xu et al., 2009). The zinc finger nuclear protein, ZFP521, has been recently shown to be a key regulator for driving neural differentiation of ESCs; activating early neural genes in association with the co-activator p300 (Kamiya et al., 2011). In NSC, the SOXB1 and GLI families, CBF-1, HMGA2, HES1/5, BMI-1, HESR1/2, TLX, MASH1, PAX6, and REST have been associated with self-renewal, growth, and differentiation (Ahmed et al., 2009). There is ultimately a cascade of epigenomic re-organization from the extrinsic cues to the genomic machinery for modulating the stem cell state.
General Changes in Genomic Accessibility during Lineage Commitment
The chromatin structure in ESCs is relatively flexible and dynamically open with particular epigenetic signature, having a “closure” in many regions as ESCs commit to a particular lineage (Delcuve et al., 2009; Rastegar et al., 2011). This is valid for genes associated with differentiation, being “silenced” by DNA methylation and inactive histone marks in pluripotent cells compared to multipotent stem cells (Aranda et al., 2009; Barber and Rastegar, 2010; Rastegar et al., 2011). For example, during the transition of ESCs into the three germ layers, there is an increase in number of common DNA promoter methylation regions (CpG sites; Delcuve et al., 2009; Isagawa et al., 2011; Rastegar et al., 2011). Both histones (H1, H2B, H3) and non-histone chromatin proteins, such as the heterochromatin protein 1 (HP1) are shown to be loosely affiliated with chromatin structure in ESC, leading to a plastic nature of pluripotent stem cells compared to more lineage committed cells (Meshorer et al., 2006). Further, higher global levels of histone acetylation are detected in pluripotent stem cells in contrast to multipotent, and tissue specific stem cells as well as somatic cell types (Efroni et al., 2008). These findings compliment the observation that histone de-acetylase activity is required for ESC differentiation (Lee et al., 2004). Pluripotent ESC also possesses high levels of transcriptional activity, yet a large proportion of gene transcripts are only expressed at low levels. Based on a variety of global transcript analyses, it is hypothesized that as ESCs commit toward a specific lineage, downregulation of many different gene transcripts increases the effectiveness of other genes whose transcripts are not downregulated (Bibikova et al., 2008). As a unique epigenetic characteristic of ESCs; possessing a “poised” transcriptional state or bivalent chromatin marks are detected at many developmentally important genomic loci such as the Hox clusters (Bernstein et al., 2006; Delcuve et al., 2009; Barber and Rastegar, 2010).
The Role of Bivalent Histone Marks during Pluripotency and Differentiation
Transcriptionally actively genes in ESCs, such as Oct4 or Nanog are characterized by histone acetylation and histone H3-Lysine 4 tri-methylation. In the other hand, most key developmental genes remain inactive during ESC self-renewal carrying both “active” and “inactive” histone marks. This has led to the simultaneous detection of histone H3-lysine 27 tri-methylation and lysine 4 methylation, referring to as the category of “bivalent chromatin” marks. Among genes with bivalent chromatin marks are the master regulators of embryonic development, namely the members of all four Hox clusters (Barber and Rastegar, 2010). As described previously, HOX homeoproteins are highly conserved transcription factors that instruct the AP axes of the developing embryo (Rastegar et al., 2004; Huang et al., 2005; Kobrossy et al., 2006; Nolte et al., 2006; Barber and Rastegar, 2010). The 39 mammalian Hox genes are organized in four clusters A–D, in four different chromosomes with unique collinear gene activation across each chromosome (Rastegar et al., 2004; Barber and Rastegar, 2010) along the AP axis. It has been suggested that the underlying mechanism is a unidirectional chromatin opening and de-condensation along each chromosome, resulting in precise expression pattern that is corresponding to Hox physical position within each cluster, the phenomenon which is called “colinearity” (Rastegar et al., 2004; Barber and Rastegar, 2010). We have shown such unidirectional chromatin opening at the Hoxd4 locus during embryonic development and in differentiated P19 ECCs (Rastegar et al., 2004; Barber and Rastegar, 2010). HOX proteins play key roles in ESCs differentiation and their transcriptional expression is tightly regulated by histone marks, chromatin condensation, other transcription factors (Chambeyron and Bickmore, 2004; Chambeyron et al., 2005; Huang et al., 2005; Kobrossy et al., 2006; Nolte et al., 2006), and non-coding microRNAs (Barber and Rastegar, 2010).
During differentiation of pluripotent ESCs, the “poised” state essentially readies the stem cells for multiple descendancies; as losing one of the bivalent marks can lock a gene at a “off” or “on” state of transcription. Perhaps the most well characterized examples of developmentally important genes with these bivalent chromatin marks are the Hox genes. The Polycomb Repressive Complexes (PRC) play a role in regulating the state of ESCs (Boyer et al., 2006) as PRC1 can compact chromatin and catalyze ubiquitination of histone H2A, whereas PRC2 can aid in compaction as well as tri-methylation of H3K27 (Delcuve et al., 2009; Margueron and Reinberg, 2011; Rastegar et al., 2011). PRC2 is associated at genes necessary to preserve a pluripotent state (Lee et al., 2006), but it has been now challenged whether the member PRC2 is actually necessary to maintain ESC pluripotency (Chamberlain et al., 2008). More recently, a component of PRC2, Polycomb-like 3 (Pcl3), has been shown to be instrumental for ESC self-renewal by mediating PRC2 binding, promoting H3K27me3 formation (Hunkapiller et al., 2012). Also, the Polycomb family repressor, Bmi-1, is important for neural stem cell self-renewal (Molofsky et al., 2003). Overexpression of Bmi-1 in in vitro cultures of NSC correlates with the increased proliferation and neurogenesis; however, this was not reflected with the in vivo overexpression in a transgenic murine model (He et al., 2009). The Trithorax Group (TrxG) counteracts the Polycomb Group’s repressive effect on histone bivalency (Schuettengruber et al., 2011) with tri-methylation of H3K4 accomplished by mixed lineage leukemia (MLL) of the Trx group (Wang et al., 2009).
Bivalent marks may become univalent during neural differentiation; securing the epigenetic identity of a particular genomic region (Delcuve et al., 2009). The MLL member, MLL1, has been shown to be integral for resolving bivalent loci in NSC and during neurogenesis (Lim et al., 2009). A specific demethylase of H3K27me3, JMJD3, acts upon the promoter for Nestin, a neurofilament gene important in the process of neural differentiation. This results is the activation of the Nestin gene, and a step toward the differentiation of ESCs into NSC (Burgold et al., 2008). Furthermore, the H3K4me2/3 demethylase JARID1B, is essential for ESC commitment toward a neural lineage (Schmitz et al., 2011). Examples of other neural genes that resolve the repressive mark of bivalency as cells commit toward a neural lineage fate commitment include: neurogenins (Ngns), paired box gene 6 (Pax6), SRY-Box 1 (Sox1), Sox3, Mash1 (achaete–scute complex homolog 1, or Ascl1), and NK2 transcription factor related locus 2 (Nkx2.2; Mikkelsen et al., 2007; Hirabayashi and Gotoh, 2010). Interestingly, the DNA mark of 5hmC, is prevalent at the promoters of “poised” regions in self-renewing ESCs, and at the genes that are upregulated during differentiation. Therefore, it seems that 5hmC may function by priming regions for transcription as ESCs differentiate (Pastor et al., 2011).
Biomolecules that can be found in niche microenvironments of the developing embryo, or neurogenic regions of the adult CNS, trigger signal transduction pathways leading to intrinsic responses necessary for neural differentiation. Overall, as ESCs ascribe to the neural lineage, non-neural and pluripotency-related genes become silenced and inaccessible for transcription, whereas the promoters and enhancers of neural related genes will become accessible (Hirabayashi and Gotoh, 2010). The NSC, descendants of ESCs, do not possess the plasticity of their parents. In the other words they have a more constricted directional potential, and the cellular identity of their generated daughter cells is in part impacted by the context of the environment in which they reside. Many cells in their niche such as astrocytes, neurons, endothelial cells, ependymal cells, and immature NSC are thought to be crucial components for NSC maintenance and development (Duan et al., 2008). Finally, at the last stages of the developmental journey, NSC will potentially become specialized nerve cells; as neurons are produced during neurogenesis and glia by the process of gliogenesis.
Nearing the End of the Road: Neural Differentiation
We have discussed a collection of factors that impact ESC and neural stem cell fate decisions during self-perpetuation and toward lineage differentiation. We will now briefly touch-base on a selection of genomic machinery and miRNA that are implicated in the processes of gliogenesis and neurogenesis; weighted more toward players in the formation of neurons.
REST, a Factor for Preventing Neurogenesis
First discovered in 2005 (Chong et al., 1995; Schoenherr and Anderson, 1995), the repressor element 1 (RE1) silencing transcription factor (REST), also known as neuron restrictive silencing factor (NRSF), is responsible for silencing neuronal genes in non-neuronal tissues, neural precursor cells, ESCs, and NSC (Chen et al., 1998; Ballas and Mandel, 2005). REST contributes to the regulation of ESC pluripotency, and it is also involved in neural differentiation (Soldati et al., 2012), and controlling ncRNA (Qureshi and Mehler, 2009). The RE1 is another designation for the neuron restrictive silencer element (NRSE), which is found within many neuronal genes (Ooi and Wood, 2007). When cells commit to a neuronal lineage, the REST complex may degrade; resulting in de-repression of its target genes. REST binds to the 21-bp, RE1, and recruits HDACs as well as other chromatin modifiers to silence genes. Furthermore, REST can recruit co-repressor protein complexes such as HDAC1/2, CoREST, and mSin3 (Dewald et al., 2011). Additionally, REST has been observed to mediate the recruitment of PRC1 and PRC2 in certain circumstances (Dietrich et al., 2012).
Micro RNAs Target Neural Regulatory Transcripts
After the discovery of their modulatory role in neural regulation within the last several years (Stark et al., 2005; Krichevsky et al., 2006), miRNAs have been under extensive investigation. One miRNA that is prevalent within the nervous system, and particularly in neuronal cells, is mir-124a (Lagos-Quintana et al., 2002). It has shown to be critical in the process of neural differentiation (Makeyev et al., 2007) specifically in neurogenesis, by targeting Sox9, an important player in neural stem cell self-renewal maintenance (Cheng et al., 2009). Recently, in post-ischemic injured rats, mir-124a has been shown to affect Jag1 levels, as part of the NOTCH signaling pathway. Subsequently, the downregulation of mir-124a induces Jag1 and proliferation of neural precursor cells, that has been hypothesized to result from the increase of the homeoprotein DLX2, whose corresponding transcripts are targeted by mir-124a (Cheng et al., 2009; Liu et al., 2011b). The nuclear receptor TLX, also plays key roles in neural stem cell self-renewal, and is targeted by mir-9, resulting in increased neuronal production through a negative regulatory feedback loop (Zhao et al., 2009). Expression of mir-125b was similar to mir-124a in that they both increase percentage of differentiated neuroblastoma cells (SH-SY5Y) with neurite outgrowth. Also and some gene targets of the mir-125b have been connected with the negative control of neuronal related genes (Le et al., 2009).
The Link between miRNA and Rest
Multiple microRNAs including, mir-9, mir-124a, and mir-132 appear to be controlled by REST (Conaco et al., 2006). For example, mir-9 targets the REST sequence forming a negative feedback loop (Packer et al., 2008). It has been demonstrated that mir-124a directly targets the SCP1-3’ untranslated region of small C-terminal domain phosphatase 1 (SCP1; Visvanathan et al., 2007). The phosphatase, is found in non-neuronal tissues and recruited to RE1 by REST, and has anti-neuronal function (Yeo et al., 2005). More recently, mir-26b has been shown to represses the REST cofactor, CTDSP2 to regulate neurogenesis (Dill et al., 2012). Additionally, MeCP2 may interact with REST to suppress particular sites, and MeCP2 itself, is targeted by mir-132 (Klein et al., 2007).
Other Nuclear Factors Implicated in Neuronal Regulation
In the adult murine hippocampus, GADD45B, is responsible for demethylating the promoter region of genes which are critical for neurogenesis, such as BDNF and FGF (Ma et al., 2009). Nuclear factor kappa B (NFκB) signaling has been observed to have novel functionality by initiating early neurogenesis in NSC (Zhang et al., 2012). Recently, the Querkopf (QKF), a histone acetyltransferase, has been shown to be a useful marker of self-renewal and multipotency in NSC (Sheikh et al., 2012), and it is also essential for normal adult neurogenesis (Merson et al., 2006).
The function of STAT–p300/CBP–SMAD in Astrocyte Formation
Astrocytic differentiation of NSC can be induced through the activation of the Janus kinase (JAK)–signal transducer and activator of the transcription (STAT) pathway. Cytokines of the interleukin-6 (IL-6) family trigger JAK–STAT, and include the following members; leukemia inhibitory factor (LIF), cardiotrophin-1 (CT-1), and ciliary neurotrophic factor (CNTF; Bonni et al., 1997). The bone morphogenic protein (BMP) family cytokine members of the TGF-β superfamily activate SMAD transcription factors. Afterward, a complex can be formed between STAT and SMAD proteins by the transcription co-activators, p300/CBP, which possesses histone acetylating functionality (Nakashima et al., 1999). The STAT–p300/CBP–SMAD complex can promote transcriptional activation of associated genomic regions.
Moreover, the Glial fibrillary acidic protein (GFAP) is an astrocytic marker whose promoter region is methylated during early gestation, while it will later become demethylated. Within the promoter of Gfap, there is a highly methylated CpG dinucleotide at a STAT binding element when cells are in a neuronal phase. This methylated CpG prevents the STAT–p300/CBP–SMAD complex from facilitating transcriptional activation at the site (Takizawa et al., 2001). In another astrocytic marker, S100β it is shown that it has a methylated cytosine in its promoter region, and similarly becomes demethylated later in gestation (Namihira et al., 2004) much like the Gfap promoter, allowing the STAT–P300/CBP–SMAD complex to bind and transcription to commence. MeCP2 can also bind 5mC in the exon 1 of Gfap and the CpG at the transcriptional start sites of S100β, suppressing these astrocytic markers and subsequent differentiation (Setoguchi et al., 2006).
Applying Knowledge of the Neural Epigenome in Regenerative Medicine
There is much excitement surrounding the application of pluripotent and multipotent stem cells in the hope of replenishing damaged cells and tissues in various medical circumstances, such as in neurodegenerative disease. Clinical trials are currently underway that involve neural stem cell transplantation in patients with ischemic injury and ALS, with preclinical studies being implemented for future cell-based treatments of brain cancer, ischemic spastic paraplegia, chronic spinal cord injury, and chronic stroke. In order to attain pure populations of differentiated cell types in vitro that can be designated for safe therapeutic use, such as differentiated neurons or glia, a thorough comprehension of the epigenetic mechanisms is needed. This is imperative with induced cell types (i.e., iPS cells or induced NSC) where the understanding of re-programmed cells is a bit further than its infancy, as well as other in vitro cultured cell types. Nonetheless, patient-specific stem cells hold great promise in regenerative medicine by supplying patients with genetically identical matches for treatment, although heed must be taken to ensure that any molecular variances (with epigenome, proteome, etc.), if any, do not perturb their potential benefit. Therefore, we believe that a comprehensive mapping of the epigenome would be invaluable for this cause; with further investigation of novel marks such as 5hmC, histone variants, and factors contributing to their presence.
Conclusion
A selection of key epigenetic events studied in neural stem cell differentiation have been discussed, including novel findings to date as well as more established results in the field. Overall, ESCs can be described as having a dynamic increase of “closed” chromatin marks, and decrease of “open” chromatin marks during lineage commitment, with unique modification patterns attributed to lineage orientated genes. To travel further down the neural path, a resolution of bivalent marks is required for stem cells. The differentiation of NSC into neurons or glial cells requires regulation of multiple miRNAs including mir-124a, mir-125b, mir-26b, and epigenetic modifying transcriptional machinery, such as Gadd45b, QKF, REST, or STAT–p300/CBP–SMAD complexes. These examples are only a few of many that demonstrate the elaborate organization of the epigenome in ESCs as they take the journey to become neural cell types.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Jeff Dixon for the art work and all members of the Rastegar lab for helpful discussions. Mojgan Rastegar is supported by funds from the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant 372405-2009), Manitoba Institute of Child Health (MICH), Manitoba Health Research Council (MHRC; Establishment and Operating Funds), Scottish Rites Charitable Foundation of Canada (SRCFC, Grant 10110), Health Sciences Centre Foundation (HSCF), Manitoba Medical Service Foundation (MMSF), Canada Foundation for Innovation-Leaders Opportunity Fund (CFI-LOF), and CFI Infrastructure Operating Fund (CFI-IOF). The research of Brendan M. Olynik is supported by Natural Sciences and Engineering Research Council of Canada and MHRC Operating Funds (grants to Mojgan Rastegar).
References
Abdel-Fattah, R., Xiao, A., Bomgardner, D., Pease, C. S., Lopes, M. B., and Hussaini, I. M. (2006). Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J. Pathol. 209, 15–24.
Abranches, E., Silva, M., Pradier, L., Schulz, H., Hummel, O., Henrique, D., and Bekman, E. (2009). Neural differentiation of embryonic stem cells in vitro: a road map to neurogenesis in the embryo. PLoS ONE 4, e6286. doi:10.1371/journal.pone.0006286
Ahmed, S., Gan, H. T., Lam, C. S., Poonepalli, A., Ramasamy, S., Tay, Y., Tham, M., and Yu, Y. H. (2009). Transcription factors and neural stem cell self-renewal, growth and differentiation. Cell Adh. Migr. 3, 412–424.
Aimone, J. B., Deng, W., and Gage, F. H. (2010). Adult neurogenesis: integrating theories and separating functions. Trends Cogn. Sci. (Regul. Ed.) 14, 325–337.
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Anacker, C., Zunszain, P. A., Cattaneo, A., Carvalho, L. A., Garabedian, M. J., Thuret, S., Price, J., and Pariante, C. M. (2011). Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol. Psychiatry 16, 738–750.
Anderson, M. L., Sisti, H. M., Curlik, D. M. II, and Shors, T. J. (2011). Associative learning increases adult neurogenesis during a critical period. Eur. J. Neurosci. 33, 175–181.
Aoi, T., Yae, K., Nakagawa, M., Ichisaka, T., Okita, K., Takahashi, K., Chiba, T., and Yamanaka, S. (2008). Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 321, 699–702.
Aranda, P., Agirre, X., Ballestar, E., Andreu, E. J., Roman-Gomez, J., Prieto, I., Martin-Subero, J. I., Cigudosa, J. C., Siebert, R., Esteller, M., and Prosper, F. (2009). Epigenetic signatures associated with different levels of differentiation potential in human stem cells. PLoS ONE 4, e7809. doi:10.1371/journal.pone.0007809
Arenkiel, B. R., Gaufo, G. O., and Capecchi, M. R. (2003). Hoxb1 neural crest preferentially form glia of the PNS. Dev. Dyn. 227, 379–386.
Ballas, N., Grunseich, C., Lu, D. D., Speh, J. C., and Mandel, G. (2005). REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121, 645–657.
Ballas, N., and Mandel, G. (2005). The many faces of REST oversee epigenetic programming of neuronal genes. Curr. Opin. Neurobiol. 15, 500–506.
Barber, B. A., and Rastegar, M. (2010). Epigenetic control of Hox genes during neurogenesis, development, and disease. Ann. Anat. 192, 261–274.
Barros, S. P., and Offenbacher, S. (2009). Epigenetics: connecting environment and genotype to phenotype and disease. J. Dent. Res. 88, 400–408.
Bassett, A., Cooper, S., Wu, C., and Travers, A. (2009). The folding and unfolding of eukaryotic chromatin. Curr. Opin. Genet. Dev. 19, 159–165.
Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., Fry, B., Meissner, A., Wernig, M., Plath, K., Jaenisch, R., Wagschal, A., Feil, R., Schreiber, S. L., and Lander, E. S. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326.
Bernstein, E., Kim, S. Y., Carmell, M. A., Murchison, E. P., Alcorn, H., Li, M. Z., Mills, A. A., Elledge, S. J., Anderson, K. V., and Hannon, G. J. (2003). Dicer is essential for mouse development. Nat. Genet. 35, 215–217.
Bibikova, M., Laurent, L. C., Ren, B., Loring, J. F., and Fan, J. B. (2008). Unraveling epigenetic regulation in embryonic stem cells. Cell Stem Cell 2, 123–134.
Bonfanti, L., and Peretto, P. (2011). Adult neurogenesis in mammals – a theme with many variations. Eur. J. Neurosci. 34, 930–950.
Bonni, A., Sun, Y., Nadal-Vicens, M., Bhatt, A., Frank, D. A., Rozovsky, I., Stahl, N., Yancopoulos, G. D., and Greenberg, M. E. (1997). Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278, 477–483.
Boulard, M., Bouvet, P., Kundu, T. K., and Dimitrov, S. (2007). Histone variant nucleosomes: structure, function and implication in disease. Subcell. Biochem. 41, 71–89.
Boyer, L. A., Plath, K., Zeitlinger, J., Brambrink, T., Medeiros, L. A., Lee, T. I., Levine, S. S., Wernig, M., Tajonar, A., Ray, M. K., Bell, G. W., Otte, A. P., Vidal, M., Gifford, D. K., Young, R. A., and Jaenisch, R. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353.
Bracko, O., Singer, T., Aigner, S., Knobloch, M., Winner, B., Ray, J., Clemenson, G. D. Jr., Suh, H., Couillard-Despres, S., Aigner, L., Gage, F. H., and Jessberger, S. (2012). Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J. Neurosci. 32, 3376–3387.
Brandl, A., Heinzel, T., and Kramer, O. H. (2009). Histone deacetylases: salesmen and customers in the post-translational modification market. Biol. Cell 101, 193–205.
Brandt, M. D., Maass, A., Kempermann, G., and Storch, A. (2010). Physical exercise increases Notch activity, proliferation and cell cycle exit of type-3 progenitor cells in adult hippocampal neurogenesis. Eur. J. Neurosci. 32, 1256–1264.
Burgold, T., Spreafico, F., De Santa, F., Totaro, M. G., Prosperini, E., Natoli, G., and Testa, G. (2008). The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS ONE 3, e3034. doi:10.1371/journal.pone.0003034
Calvanese, V., and Fraga, M. F. (2012). Epigenetics of embryonic stem cells. Adv. Exp. Med. Biol. 741, 231–253.
Cedar, H., and Bergman, Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10, 295–304.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., and Zoghbi, H. Y. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229.
Chamberlain, S. J., Yee, D., and Magnuson, T. (2008). Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 26, 1496–1505.
Chambeyron, S., and Bickmore, W. A. (2004). Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 18, 1119–1130.
Chambeyron, S., Da Silva, N. R., Lawson, K. A., and Bickmore, W. A. (2005). Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development 132, 2215–2223.
Chan, K. K., Zhang, J., Chia, N. Y., Chan, Y. S., Sim, H. S., Tan, K. S., Oh, S. K., Ng, H. H., and Choo, A. B. (2009). KLF4 and PBX1 directly regulate NANOG expression in human embryonic stem cells. Stem Cells 27, 2114–2125.
Chen, Z. F., Paquette, A. J., and Anderson, D. J. (1998). NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 20, 136–142.
Cheng, L. C., Pastrana, E., Tavazoie, M., and Doetsch, F. (2009). miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 12, 399–408.
Chong, J. A., Tapia-Ramirez, J., Kim, S., Toledo-Aral, J. J., Zheng, Y., Boutros, M. C., Altshuller, Y. M., Frohman, M. A., Kraner, S. D., and Mandel, G. (1995). REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80, 949–957.
Conaco, C., Otto, S., Han, J. J., and Mandel, G. (2006). Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl. Acad. Sci. U.S.A. 103, 2422–2427.
Cornell, R. A., and Eisen, J. S. (2005). Notch in the pathway: the roles of Notch signaling in neural crest development. Semin. Cell Dev. Biol. 16, 663–672.
Darr, H., and Benvenisty, N. (2006). Factors involved in self-renewal and pluripotency of embryonic stem cells. Handb. Exp. Pharmacol. 174, 1–19.
Dawson, M. A., Bannister, A. J., Gottgens, B., Foster, S. D., Bartke, T., Green, A. R., and Kouzarides, T. (2009). JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461, 819–822.
de Ruijter, A. J., van Gennip, A. H., Caron, H. N., Kemp, S., and van Kuilenburg, A. B. (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749.
Delcuve, G. P., Rastegar, M., and Davie, J. R. (2009). Epigenetic control. J. Cell. Physiol. 219, 243–250.
Dewald, L. E., Rodriguez, J. P., and Levine, J. M. (2011). The RE1 binding protein REST regulates oligodendrocyte differentiation. J. Neurosci. 31, 3470–3483.
Dietrich, N., Lerdrup, M., Landt, E., Agrawal-Singh, S., Bak, M., Tommerup, N., Rappsilber, J., Sodersten, E., and Hansen, K. (2012). REST-mediated recruitment of Polycomb repressor complexes in mammalian cells. PLoS Genet. 8, e1002494. doi:10.1371/journal.pgen.1002494
Dill, H., Linder, B., Fehr, A., and Fischer, U. (2012). Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev. 26, 25–30.
Dobosy, J. R., and Selker, E. U. (2001). Emerging connections between DNA methylation and histone acetylation. Cell. Mol. Life Sci. 58, 721–727.
Duan, X., Kang, E., Liu, C. Y., Ming, G. L., and Song, H. (2008). Development of neural stem cell in the adult brain. Curr. Opin. Neurobiol. 18, 108–115.
Dubois-Dauphin, M., Theler, J. M., Zaganidis, N., Dominik, W., Tribollet, E., Pevet, P., Charpak, G., and Dreifuss, J. J. (1991). Expression of vasopressin receptors in hamster hypothalamus is sexually dimorphic and dependent upon photoperiod. Proc. Natl. Acad. Sci. U.S.A. 88, 11163–11167.
Duman, R. S., Nakagawa, S., and Malberg, J. (2001). Regulation of adult neurogenesis by antidepressant treatment. Neuropsychopharmacology 25, 836–844.
Dupe, V., Davenne, M., Brocard, J., Dolle, P., Mark, M., Dierich, A., Chambon, P., and Rijli, F. M. (1997). In vivo functional analysis of the Hoxa-1 3′ retinoic acid response element (3′RARE). Development 124, 399–410.
Duriseti, S., Winnard, P. T. Jr., Mironchik, Y., Vesuna, F., Raman, A., and Raman, V. (2006). HOXA5 regulates hMLH1 expression in breast cancer cells. Neoplasia 8, 250–258.
Efroni, S., Duttagupta, R., Cheng, J., Dehghani, H., Hoeppner, D. J., Dash, C., Bazett-Jones, D. P., Le Grice, S., McKay, R. D., Buetow, K. H., Gingeras, T. R., Misteli, T., and Meshorer, E. (2008). Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2, 437–447.
Ellis, J., Hotta, A., and Rastegar, M. (2007). Retrovirus silencing by an epigenetic TRIM. Cell 131, 13–14.
Elsaesser, S. J., Goldberg, A. D., and Allis, C. D. (2010). New functions for an old variant: no substitute for histone H3.3. Curr. Opin. Genet. Dev. 20, 110–117.
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A., and Gage, F. H. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317.
Fan, Y., Nikitina, T., Zhao, J., Fleury, T. J., Bhattacharyya, R., Bouhassira, E. E., Stein, A., Woodcock, C. L., and Skoultchi, A. I. (2005). Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212.
Faust, T., Frankel, A., and D’Orso, I. (2012). Transcription control by long non-coding RNAs. Transcription 3, 78–86.
Fuks, F. (2005). DNA methylation and histone modifications: teaming up to silence genes. Curr. Opin. Genet. Dev. 15, 490–495.
Fussner, E., Ahmed, K., Dehghani, H., Strauss, M., and Bazett-Jones, D. P. (2011a). Changes in chromatin fiber density as a marker for pluripotency. Cold Spring Harb. Symp. Quant. Biol. 75, 245–249.
Fussner, E., Djuric, U., Strauss, M., Hotta, A., Perez-Iratxeta, C., Lanner, F., Dilworth, F. J., Ellis, J., and Bazett-Jones, D. P. (2011b). Constitutive heterochromatin reorganization during somatic cell reprogramming. EMBO J. 30, 1778–1789.
Gage, F. H. (2010). Molecular and cellular mechanisms contributing to the regulation, proliferation and differentiation of neural stem cells in the adult dentate gyrus. Keio J. Med. 59, 79–83.
Gaufo, G. O., Wu, S., and Capecchi, M. R. (2004). Contribution of Hox genes to the diversity of the hindbrain sensory system. Development 131, 1259–1266.
Gavalas, A., Davenne, M., Lumsden, A., Chambon, P., and Rijli, F. M. (1997). Role of Hoxa-2 in axon pathfinding and rostral hindbrain patterning. Development 124, 3693–3702.
Goldberg, A. D., Banaszynski, L. A., Noh, K. M., Lewis, P. W., Elsaesser, S. J., Stadler, S., Dewell, S., Law, M., Guo, X., Li, X., Wen, D., Chapgier, A., DeKelver, R. C., Miller, J. C., Lee, Y. L., Boydston, E. A., Holmes, M. C., Gregory, P. D., Greally, J. M., Rafii, S., Yang, C., Scambler, P. J., Garrick, D., Gibbons, R. J., Higgs, D. R., Cristea, I. M., Urnov, F. D., Zheng, D., and Allis, C. D. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678–691.
Goldman, S. A., and Nottebohm, F. (1983). Neuronal production, migration, and differentiation in a vocal control nucleus of the adult female canary brain. Proc. Natl. Acad. Sci. U.S.A. 80, 2390–2394.
Gomez-Gaviro, M. V., Scott, C. E., Sesay, A. K., Matheu, A., Booth, S., Galichet, C., and Lovell-Badge, R. (2012). Betacellulin promotes cell proliferation in the neural stem cell niche and stimulates neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 1317–1322.
Gould, A., Itasaki, N., and Krumlauf, R. (1998). Initiation of rhombomeric Hoxb4 expression requires induction by somites and a retinoid pathway. Neuron 21, 39–51.
Han, D. W., Tapia, N., Hermann, A., Hemmer, K., Hoing, S., Arauzo-Bravo, M. J., Zaehres, H., Wu, G., Frank, S., Moritz, S., Greber, B., Yang, J. H., Lee, H. T., Schwamborn, J. C., Storch, A., and Scholer, H. R. (2012). Direct reprogramming of fibroblasts into neural stem cells by defined factors. Cell Stem Cell 10, 465–472.
Hanna, J., Wernig, M., Markoulaki, S., Sun, C. W., Meissner, A., Cassady, J. P., Beard, C., Brambrink, T., Wu, L. C., Townes, T. M., and Jaenisch, R. (2007). Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318, 1920–1923.
He, S., Iwashita, T., Buchstaller, J., Molofsky, A. V., Thomas, D., and Morrison, S. J. (2009). Bmi-1 over-expression in neural stem/progenitor cells increases proliferation and neurogenesis in culture but has little effect on these functions in vivo. Dev. Biol. 328, 257–272.
Hendrich, B., and Bird, A. (1998). Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18, 6538–6547.
Heng, J. C., and Ng, H. H. (2010). Transcriptional regulation in embryonic stem cells. Adv. Exp. Med. Biol. 695, 76–91.
Hirabayashi, Y., and Gotoh, Y. (2010). Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci. 11, 377–388.
Hirabayashi, Y., and Gotoh, Y. (2005). Stage-dependent fate determination of neural precursor cells in mouse forebrain. Neurosci. Res. 51, 331–336.
Hoch, R. V., Rubenstein, J. L., and Pleasure, S. (2009). Genes and signaling events that establish regional patterning of the mammalian forebrain. Semin. Cell Dev. Biol. 20, 378–386.
Horvath, J. E., Bailey, J. A., Locke, D. P., and Eichler, E. E. (2001). Lessons from the human genome: transitions between euchromatin and heterochromatin. Hum. Mol. Genet. 10, 2215–2223.
Hotta, A., Cheung, A. Y., Farra, N., Vijayaragavan, K., Seguin, C. A., Draper, J. S., Pasceri, P., Maksakova, I. A., Mager, D. L., Rossant, J., Bhatia, M., and Ellis, J. (2009). Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods 6, 370–376.
Huang, D., Chen, S. W., and Gudas, L. J. (2002). Analysis of two distinct retinoic acid response elements in the homeobox gene Hoxb1 in transgenic mice. Dev. Dyn. 223, 353–370.
Huang, H., Rastegar, M., Bodner, C., Goh, S. L., Rambaldi, I., and Featherstone, M. (2005). MEIS C termini harbor transcriptional activation domains that respond to cell signaling. J. Biol. Chem. 280, 10119–10127.
Huang, Y., Pastor, W. A., Shen, Y., Tahiliani, M., Liu, D. R., and Rao, A. (2010). The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 5, e8888. doi:10.1371/journal.pone.0008888
Huangfu, D., Maehr, R., Guo, W., Eijkelenboom, A., Snitow, M., Chen, A. E., and Melton, D. A. (2008). Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 26, 795–797.
Hunkapiller, J., Shen, Y., Diaz, A., Cagney, G., McCleary, D., Ramalho-Santos, M., Krogan, N., Ren, B., Song, J. S., and Reiter, J. F. (2012). Polycomb-like 3 promotes Polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self-renewal. PLoS Genet. 8, e1002576. doi:10.1371/journal.pgen.1002576
Isagawa, T., Nagae, G., Shiraki, N., Fujita, T., Sato, N., Ishikawa, S., Kume, S., and Aburatani, H. (2011). DNA methylation profiling of embryonic stem cell differentiation into the three germ layers. PLoS ONE 6, e26052. doi:10.1371/journal.pone.0026052
Ito, S., D’Alessio, A. C., Taranova, O. V., Hong, K., Sowers, L. C., and Zhang, Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133.
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., He, C., and Zhang, Y. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303.
Jin, B., Tao, Q., Peng, J., Soo, H. M., Wu, W., Ying, J., Fields, C. R., Delmas, A. L., Liu, X., Qiu, J., and Robertson, K. D. (2008). DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 17, 690–709.
Jones, P. L., Veenstra, G. J., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N., Strouboulis, J., and Wolffe, A. P. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191.
Kamiya, D., Banno, S., Sasai, N., Ohgushi, M., Inomata, H., Watanabe, K., Kawada, M., Yakura, R., Kiyonari, H., Nakao, K., Jakt, L. M., Nishikawa, S., and Sasai, Y. (2011). Intrinsic transition of embryonic stem-cell differentiation into neural progenitors. Nature 470, 503–509.
Klein, M. E., Lioy, D. T., Ma, L., Impey, S., Mandel, G., and Goodman, R. H. (2007). Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 10, 1513–1514.
Kobrossy, L., Rastegar, M., and Featherstone, M. (2006). Interplay between chromatin and trans-acting factors regulating the Hoxd4 promoter during neural differentiation. J. Biol. Chem. 281, 25926–25939.
Kornberg, R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871.
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930.
Krichevsky, A. M., Sonntag, K. C., Isacson, O., and Kosik, K. S. (2006). Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 24, 857–864.
Kuhn, H. G., Winkler, J., Kempermann, G., Thal, L. J., and Gage, F. H. (1997). Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J. Neurosci. 17, 5820–5829.
Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739.
Langston, A. W., and Gudas, L. J. (1992). Identification of a retinoic acid responsive enhancer 3′ of the murine homeobox gene Hox-1.6. Mech. Dev. 38, 217–227.
Le, M. T., Xie, H., Zhou, B., Chia, P. H., Rizk, P., Um, M., Udolph, G., Yang, H., Lim, B., and Lodish, H. F. (2009). MicroRNA-125b promotes neuronal differentiation in human cells by repressing multiple targets. Mol. Cell. Biol. 29, 5290–5305.
Lee, J. H., Hart, S. R., and Skalnik, D. G. (2004). Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis 38, 32–38.
Lee, S. W., Clemenson, G. D., and Gage, F. H. (2012). New neurons in an aged brain. Behav. Brain Res. 227, 497–507.
Lee, T. I., Jenner, R. G., Boyer, L. A., Guenther, M. G., Levine, S. S., Kumar, R. M., Chevalier, B., Johnstone, S. E., Cole, M. F., Isono, K., Koseki, H., Fuchikami, T., Abe, K., Murray, H. L., Zucker, J. P., Yuan, B., Bell, G. W., Herbolsheimer, E., Hannett, N. M., Sun, K., Odom, D. T., Otte, A. P., Volkert, T. L., Bartel, D. P., Melton, D. A., Gifford, D. K., Jaenisch, R., and Young, R. A. (2006). Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313.
Li, E., Beard, C., and Jaenisch, R. (1993). Role for DNA methylation in genomic imprinting. Nature 366, 362–365.
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., Amit, I., Lajoie, B. R., Sabo, P. J., Dorschner, M. O., Sandstrom, R., Bernstein, B., Bender, M. A., Groudine, M., Gnirke, A., Stamatoyannopoulos, J., Mirny, L. A., Lander, E. S., and Dekker, J. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293.
Lim, D. A., Huang, Y. C., Swigut, T., Mirick, A. L., Garcia-Verdugo, J. M., Wysocka, J., Ernst, P., and Alvarez-Buylla, A. (2009). Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458, 529–533.
Liu, Z., Scannell, D. R., Eisen, M. B., and Tjian, R. (2011a). Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell 146, 720–731.
Liu, X. S., Chopp, M., Zhang, R. L., Tao, T., Wang, X. L., Kassis, H., Hozeska-Solgot, A., Zhang, L., Chen, C., and Zhang, Z. G. (2011b). MicroRNA profiling in subventricular zone after stroke: MiR-124a regulates proliferation of neural progenitor cells through Notch signaling pathway. PLoS ONE 6, e23461. doi:10.1371/journal.pone.0023461
Loh, Y. H., Zhang, W., Chen, X., George, J., and Ng, H. H. (2007). Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 21, 2545–2557.
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., Flavell, R. A., Lu, B., Ming, G. L., and Song, H. (2009). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077.
Ma, D. K., Marchetto, M. C., Guo, J. U., Ming, G. L., Gage, F. H., and Song, H. (2010). Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat. Neurosci. 13, 1338–1344.
Makeyev, E. V., Zhang, J., Carrasco, M. A., and Maniatis, T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448.
Malecova, B., and Morris, K. V. (2010). Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr. Opin. Mol. Ther. 12, 214–222.
Margueron, R., and Reinberg, D. (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349.
Marshall, H., Studer, M., Popperl, H., Aparicio, S., Kuroiwa, A., Brenner, S., and Krumlauf, R. (1994). A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb-1. Nature 370, 567–571.
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U.S.A. 78, 7634–7638.
Mattout, A., Biran, A., and Meshorer, E. (2011). Global epigenetic changes during somatic cell reprogramming to iPS cells. J. Mol. Cell Biol. 3, 341–350.
McGinnis, W., Levine, M. S., Hafen, E., Kuroiwa, A., and Gehring, W. J. (1984). A conserved DNA sequence in homoeotic genes of the Drosophila Antennapedia and bithorax complexes. Nature 308, 428–433.
Meehan, R. R., Lewis, J. D., and Bird, A. P. (1992). Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 20, 5085–5092.
Meissner, A. (2010). Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol. 28, 1079–1088.
Merkle, F. T., and Alvarez-Buylla, A. (2006). Neural stem cells in mammalian development. Curr. Opin. Cell Biol. 18, 704–709.
Merson, T. D., Dixon, M. P., Collin, C., Rietze, R. L., Bartlett, P. F., Thomas, T., and Voss, A. K. (2006). The transcriptional coactivator Querkopf controls adult neurogenesis. J. Neurosci. 26, 11359–11370.
Meshorer, E., Yellajoshula, D., George, E., Scambler, P. J., Brown, D. T., and Misteli, T. (2006). Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell 10, 105–116.
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., Alvarez, P., Brockman, W., Kim, T. K., Koche, R. P., Lee, W., Mendenhall, E., O’Donovan, A., Presser, A., Russ, C., Xie, X., Meissner, A., Wernig, M., Jaenisch, R., Nusbaum, C., Lander, E. S., and Bernstein, B. E. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560.
Mizuguchi, G., Shen, X., Landry, J., Wu, W. H., Sen, S., and Wu, C. (2004). ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303, 343–348.
Mizutani, K., Yoon, K., Dang, L., Tokunaga, A., and Gaiano, N. (2007). Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449, 351–355.
Mohandas, T., Sparkes, R. S., and Shapiro, L. J. (1981). Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science 211, 393–396.
Molofsky, A. V., Pardal, R., Iwashita, T., Park, I. K., Clarke, M. F., and Morrison, S. J. (2003). Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425, 962–967.
Morey, C., Da Silva, N. R., Perry, P., and Bickmore, W. A. (2007). Nuclear reorganisation and chromatin decondensation are conserved, but distinct, mechanisms linked to Hox gene activation. Development 134, 909–919.
Morris, K. V. (2009). Non-coding RNAs, epigenetic memory and the passage of information to progeny. RNA Biol. 6, 242–247.
Munzel, M., Globisch, D., and Carell, T. (2011). 5-Hydroxymethylcytosine, the sixth base of the genome. Angew. Chem. Int. Ed. Engl. 50, 6460–6468.
Nakashima, K., Yanagisawa, M., Arakawa, H., Kimura, N., Hisatsune, T., Kawabata, M., Miyazono, K., and Taga, T. (1999). Synergistic signaling in fetal brain by STAT3-Smad1 complex bridged by p300. Science 284, 479–482.
Namihira, M., Kohyama, J., Abematsu, M., and Nakashima, K. (2008). Epigenetic mechanisms regulating fate specification of neural stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 2099–2109.
Namihira, M., Nakashima, K., and Taga, T. (2004). Developmental stage dependent regulation of DNA methylation and chromatin modification in a immature astrocyte specific gene promoter. FEBS Lett. 572, 184–188.
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., and Bird, A. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389.
Narita, Y., and Rijli, F. M. (2009). Hox genes in neural patterning and circuit formation in the mouse hindbrain. Curr. Top. Dev. Biol. 88, 139–167.
Nestor, C. E., Ottaviano, R., Reddington, J., Sproul, D., Reinhardt, D., Dunican, D., Katz, E., Dixon, J. M., Harrison, D. J., and Meehan, R. R. (2012). Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 22, 467–477.
Ng, H. H., and Surani, M. A. (2011). The transcriptional and signalling networks of pluripotency. Nat. Cell Biol. 13, 490–496.
Nolte, C., Rastegar, M., Amores, A., Bouchard, M., Grote, D., Maas, R., Kovacs, E. N., Postlethwait, J., Rambaldi, I., Rowan, S., Yan, Y. L., Zhang, F., and Featherstone, M. (2006). Stereospecificity and PAX6 function direct Hoxd4 neural enhancer activity along the antero-posterior axis. Dev. Biol. 299, 582–593.
Odorico, J. S., Kaufman, D. S., and Thomson, J. A. (2001). Multilineage differentiation from human embryonic stem cell lines. Stem Cells 19, 193–204.
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257.
Okita, K., Nakagawa, M., Hyenjong, H., Ichisaka, T., and Yamanaka, S. (2008). Generation of mouse induced pluripotent stem cells without viral vectors. Science 322, 949–953.
Ooi, L., and Wood, I. C. (2007). Chromatin crosstalk in development and disease: lessons from REST. Nat. Rev. Genet. 8, 544–554.
Oury, F., Murakami, Y., Renaud, J. S., Pasqualetti, M., Charnay, P., Ren, S. Y., and Rijli, F. M. (2006). Hoxa2- and rhombomere-dependent development of the mouse facial somatosensory map. Science 313, 1408–1413.
Oury, F., and Rijli, F. M. (2007). [Hoxa2: a key gene for the facial somatosensory map]. Med. Sci. (Paris) 23, 247–249.
Packer, A. I., Crotty, D. A., Elwell, V. A., and Wolgemuth, D. J. (1998). Expression of the murine Hoxa4 gene requires both autoregulation and a conserved retinoic acid response element. Development 125, 1991–1998.
Packer, A. N., Xing, Y., Harper, S. Q., Jones, L., and Davidson, B. L. (2008). The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 28, 14341–14346.
Palmer, T. D., Markakis, E. A., Willhoite, A. R., Safar, F., and Gage, F. H. (1999). Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J. Neurosci. 19, 8487–8497.
Pan, G., and Thomson, J. A. (2007). Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 17, 42–49.
Park, I. H., Lerou, P. H., Zhao, R., Huo, H., and Daley, G. Q. (2008). Generation of human-induced pluripotent stem cells. Nat. Protoc. 3, 1180–1186.
Pastor, W. A., Pape, U. J., Huang, Y., Henderson, H. R., Lister, R., Ko, M., McLoughlin, E. M., Brudno, Y., Mahapatra, S., Kapranov, P., Tahiliani, M., Daley, G. Q., Liu, X. S., Ecker, J. R., Milos, P. M., Agarwal, S., and Rao, A. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397.
Pera, M. F., and Tam, P. P. (2010). Extrinsic regulation of pluripotent stem cells. Nature 465, 713–720.
Pfaffeneder, T., Hackner, B., Truss, M., Munzel, M., Muller, M., Deiml, C. A., Hagemeier, C., and Carell, T. (2011). The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. Engl. 50, 7008–7012.
Piecewicz, S. M., Pandey, A., Roy, B., Hua Xiang, S., Zetter, B. R., and Sengupta, S. (2012). Insulin-like growth factors promote vasculogenesis in embryonic stem cells. PLoS ONE 7, e32191. doi:10.1371/journal.pone.0032191
Qian, X., Shen, Q., Goderie, S. K., He, W., Capela, A., Davis, A. A., and Temple, S. (2000). Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 28, 69–80.
Qureshi, I. A., and Mehler, M. F. (2009). Regulation of non-coding RNA networks in the nervous system – what’s the REST of the story? Neurosci. Lett. 466, 73–80.
Rastegar, M., Delcuve, G. P., and Davie, J. R. (2011). “Epigenetic analysis of pluripotent cells,” in Human Stem Cell Technology and Biology: A Research Guide and Laboratory Manual, eds G. S. Stein, M. Borowski, M. X. Luong, M.-J. Shi, K. P. Smith, and P. Vazquez (Hoboken: John Wiley and Sons, Inc.), 273–288.
Rastegar, M., Hotta, A., Pasceri, P., Makarem, M., Cheung, A. Y., Elliott, S., Park, K. J., Adachi, M., Jones, F. S., Clarke, I. D., Dirks, P., and Ellis, J. (2009). MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS ONE 4, e6810. doi:10.1371/journal.pone.0006810
Rastegar, M., Kobrossy, L., Kovacs, E. N., Rambaldi, I., and Featherstone, M. (2004). Sequential histone modifications at Hoxd4 regulatory regions distinguish anterior from posterior embryonic compartments. Mol. Cell. Biol. 24, 8090–8103.
Redmer, T., Diecke, S., Grigoryan, T., Quiroga-Negreira, A., Birchmeier, W., and Besser, D. (2011). E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep. 12, 720–726.
Reik, W., Dean, W., and Walter, J. (2001). Epigenetic reprogramming in mammalian development. Science 293, 1089–1093.
Reubinoff, B. E., Itsykson, P., Turetsky, T., Pera, M. F., Reinhartz, E., Itzik, A., and Ben-Hur, T. (2001). Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 19, 1134–1140.
Reubinoff, B. E., Pera, M. F., Fong, C. Y., Trounson, A., and Bongso, A. (2000). Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat. Biotechnol. 18, 399–404.
Reynolds, B. A., and Weiss, S. (1996). Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 175, 1–13.
Robinton, D. A., and Daley, G. Q. (2012). The promise of induced pluripotent stem cells in research and therapy. Nature 481, 295–305.
Rossant, J., Chazaud, C., and Yamanaka, Y. (2003). Lineage allocation and asymmetries in the early mouse embryo. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 1341–1318; discussion 1349.
Rossant, J., and Tam, P. P. (2009). Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development 136, 701–713.
Roth, S. Y., Denu, J. M., and Allis, C. D. (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120.
Ruan, J., Shen, J., Wang, Z., Ji, J., Song, H., Wang, K., Liu, B., Li, J., and Cui, D. (2011). Efficient preparation and labeling of human induced pluripotent stem cells by nanotechnology. Int. J. Nanomedicine 6, 425–435.
Rubenstein, J. L., and Beachy, P. A. (1998). Patterning of the embryonic forebrain. Curr. Opin. Neurobiol. 8, 18–26.
Ruzov, A., Tsenkina, Y., Serio, A., Dudnakova, T., Fletcher, J., Bai, Y., Chebotareva, T., Pells, S., Hannoun, Z., Sullivan, G., Chandran, S., Hay, D. C., Bradley, M., Wilmut, I., and De Sousa, P. (2011). Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res. 21, 1332–1342.
Santos, F., Hendrich, B., Reik, W., and Dean, W. (2002). Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev. Biol. 241, 172–182.
Sasaki, H., and Matsui, Y. (2008). Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat. Rev. Genet. 9, 129–140.
Schlosser, G. (2008). Do vertebrate neural crest and cranial placodes have a common evolutionary origin? Bioessays 30, 659–672.
Schmitz, S. U., Albert, M., Malatesta, M., Morey, L., Johansen, J. V., Bak, M., Tommerup, N., Abarrategui, I., and Helin, K. (2011). Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 30, 4586–4600.
Schoenherr, C. J., and Anderson, D. J. (1995). The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 267, 1360–1363.
Schofer, C., and Weipoltshammer, K. (2008). Gene dynamics and nuclear architecture during differentiation. Differentiation 76, 41–56.
Schuettengruber, B., Martinez, A. M., Iovino, N., and Cavalli, G. (2011). Trithorax group proteins: switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 12, 799–814.
Setoguchi, H., Namihira, M., Kohyama, J., Asano, H., Sanosaka, T., and Nakashima, K. (2006). Methyl-CpG binding proteins are involved in restricting differentiation plasticity in neurons. J. Neurosci. Res. 84, 969–979.
Sheikh, B. N., Dixon, M. P., Thomas, T., and Voss, A. K. (2012). Querkopf is a key marker of self-renewal and multipotency of adult neural stem cells. J. Cell. Sci. 125, 295–309.
Shi, Y., Desponts, C., Do, J. T., Hahm, H. S., Scholer, H. R., and Ding, S. (2008). Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 3, 568–574.
Sinkkonen, L., Hugenschmidt, T., Berninger, P., Gaidatzis, D., Mohn, F., Artus-Revel, C. G., Zavolan, M., Svoboda, P., and Filipowicz, W. (2008). MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 15, 259–267.
Skene, P. J., Illingworth, R. S., Webb, S., Kerr, A. R., James, K. D., Turner, D. J., Andrews, R., and Bird, A. P. (2010). Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 37, 457–468.
Smukler, S. R., Runciman, S. B., Xu, S., and van der Kooy, D. (2006). Embryonic stem cells assume a primitive neural stem cell fate in the absence of extrinsic influences. J. Cell Biol. 172, 79–90.
Soldati, C., Bithell, A., Johnston, C., Wong, K. Y., Teng, S. W., Beglopoulos, V., Stanton, L. W., and Buckley, N. J. (2012). Repressor element 1 silencing transcription factor couples loss of pluripotency with neural induction and neural differentiation. Stem Cells 30, 425–434.
Squillaro, T., Alessio, N., Cipollaro, M., Melone, M. A., Hayek, G., Renieri, A., Giordano, A., and Galderisi, U. (2012). Reduced expression of MECP2 affects cell commitment and maintenance in neurons by triggering senescence, new perspective for Rett syndrome. Mol. Biol. Cell 23, 1435–1445.
Stancheva, I. (2005). Caught in conspiracy: cooperation between DNA methylation and histone H3K9 methylation in the establishment and maintenance of heterochromatin. Biochem. Cell Biol. 83, 385–395.
Stark, A., Brennecke, J., Bushati, N., Russell, R. B., and Cohen, S. M. (2005). Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123, 1133–1146.
Stern, C. D., Charite, J., Deschamps, J., Duboule, D., Durston, A. J., Kmita, M., Nicolas, J. F., Palmeirim, I., Smith, J. C., and Wolpert, L. (2006). Head-tail patterning of the vertebrate embryo: one, two or many unresolved problems? Int. J. Dev. Biol. 50, 3–15.
Studer, M., Lumsden, A., Ariza-McNaughton, L., Bradley, A., and Krumlauf, R. (1996). Altered segmental identity and abnormal migration of motor neurons in mice lacking Hoxb-1. Nature 384, 630–634.
Studer, M., Popperl, H., Marshall, H., Kuroiwa, A., and Krumlauf, R. (1994). Role of a conserved retinoic acid response element in rhombomere restriction of Hoxb-1. Science 265, 1728–1732.
Sultan, S., Mandairon, N., Kermen, F., Garcia, S., Sacquet, J., and Didier, A. (2010). Learning-dependent neurogenesis in the olfactory bulb determines long-term olfactory memory. FASEB J. 24, 2355–2363.
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476.
Szenker, E., Ray-Gallet, D., and Almouzni, G. (2011). The double face of the histone variant H3.3. Cell Res. 21, 421–434.
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., Agarwal, S., Iyer, L. M., Liu, D. R., Aravind, L., and Rao, A. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., and Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872.
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676.
Takizawa, T., Nakashima, K., Namihira, M., Ochiai, W., Uemura, A., Yanagisawa, M., Fujita, N., Nakao, M., and Taga, T. (2001). DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell 1, 749–758.
Talbert, P. B., and Henikoff, S. (2010). Histone variants – ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 11, 264–275.
Tate, P. H., and Bird, A. P. (1993). Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 3, 226–231.
Temple, S. (1989). Division and differentiation of isolated CNS blast cells in microculture. Nature 340, 471–473.
Thambirajah, A. A., Ng, M. K., Frehlick, L. J., Li, A., Serpa, J. J., Petrotchenko, E. V., Silva-Moreno, B., Missiaen, K. K., Borchers, C. H., Adam Hall, J., Mackie, R., Lutz, F., Gowen, B. E., Hendzel, M., Georgel, P. T., and Ausio, J. (2011). MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 40, 2884–2897.
Tjeertes, J. V., Miller, K. M., and Jackson, S. P. (2009). Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 28, 1878–1889.
Trainor, P. A. (2005). Specification of neural crest cell formation and migration in mouse embryos. Semin. Cell Dev. Biol. 16, 683–693.
Trainor, P. A., and Krumlauf, R. (2001). Hox genes, neural crest cells and branchial arch patterning. Curr. Opin. Cell Biol. 13, 698–705.
Tumpel, S., Cambronero, F., Ferretti, E., Blasi, F., Wiedemann, L. M., and Krumlauf, R. (2007). Expression of Hoxa2 in rhombomere 4 is regulated by a conserved cross-regulatory mechanism dependent upon Hoxb1. Dev. Biol. 302, 646–660.
Tumpel, S., Wiedemann, L. M., and Krumlauf, R. (2009). Hox genes and segmentation of the vertebrate hindbrain. Curr. Top. Dev. Biol. 88, 103–137.
Tvrdik, P., and Capecchi, M. R. (2006). Reversal of Hox1 gene subfunctionalization in the mouse. Dev. Cell 11, 239–250.
van Praag, H., Shubert, T., Zhao, C., and Gage, F. H. (2005). Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 25, 8680–8685.
Visvanathan, J., Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2007). The microRNA miR-124 antagonizes the anti-neural REST/SCP1 pathway during embryonic CNS development. Genes Dev. 21, 744–749.
Waddington, C. H. (1942). Canalization of development and the inheritance of acquired characters. Nature 150, 563–565.
Wade, P. A. (2001). Methyl CpG-binding proteins and transcriptional repression. Bioessays 23, 1131–1137.
Wang, P., Lin, C., Smith, E. R., Guo, H., Sanderson, B. W., Wu, M., Gogol, M., Alexander, T., Seidel, C., Wiedemann, L. M., Ge, K., Krumlauf, R., and Shilatifard, A. (2009). Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 29, 6074–6085.
Watt, F., and Molloy, P. L. (1988). Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 2, 1136–1143.
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., Bernstein, B. E., and Jaenisch, R. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324.
Woltjen, K., Michael, I. P., Mohseni, P., Desai, R., Mileikovsky, M., Hamalainen, R., Cowling, R., Wang, W., Liu, P., Gertsenstein, M., Kaji, K., Sung, H. K., and Nagy, A. (2009). piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770.
Wyatt, G. R., and Cohen, S. S. (1953). The bases of the nucleic acids of some bacterial and animal viruses: the occurrence of 5-hydroxymethylcytosine. Biochem. J. 55, 774–782.
Xu, N., Papagiannakopoulos, T., Pan, G., Thomson, J. A., and Kosik, K. S. (2009). MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 137, 647–658.
Xue, K., Ng, J. H., and Ng, H. H. (2011). Mapping the networks for pluripotency. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 2238–2246.
Yeo, M., Lee, S. K., Lee, B., Ruiz, E. C., Pfaff, S. L., and Gill, G. N. (2005). Small CTD phosphatases function in silencing neuronal gene expression. Science 307, 596–600.
Zachariah, R. M., and Rastegar, M. (2012). Linking epigenetics to human disease and Rett syndrome: the emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012, 1–10.
Zhang, F., Nagy Kovacs, E., and Featherstone, M. S. (2000). Murine hoxd4 expression in the CNS requires multiple elements including a retinoic acid response element. Mech. Dev. 96, 79–89.
Zhang, K., Williams, K. E., Huang, L., Yau, P., Siino, J. S., Bradbury, E. M., Jones, P. R., Minch, M. J., and Burlingame, A. L. (2002). Histone acetylation and deacetylation: identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Mol. Cell Proteomics 1, 500–508.
Zhang, Y., Liu, J., Yao, S., Li, F., Xin, L., Lai, M., Bracchi-Ricard, V., Xu, H., Yen, W., Meng, W., Liu, S., Yang, L., Karmally, S., Zhu, H., Gordon, J., Khalili, K., Srinivasan, S., Bethea, J. R., Mo, X., and Hu, W. (2012). Nuclear factor kappa B signaling initiates early differentiation of neural stem cells. Stem Cells 30, 510–524.
Zhao, C., Sun, G., Li, S., and Shi, Y. (2009). A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat. Struct. Mol. Biol. 16, 365–371.
Keywords: epigenetics, embryonic stem cells, neural stem cells, neurogenesis, gliogenesis, homeobox transcription factors, regenerative medicine
Citation: Olynik BM and Rastegar M (2012) The genetic and epigenetic journey of embryonic stem cells into mature neural cells. Front. Gene. 3:81. doi: 10.3389/fgene.2012.00081
Received: 27 January 2012; Accepted: 25 April 2012;
Published online: 18 May 2012.
Edited by:
Halyna R. Shcherbata, Max Planck Society, GermanyReviewed by:
Chris Anthony Murgatroyd, Manchester Metropolitan University, UKLuigi Bouchard, Université de Sherbrooke, Canada
Copyright: © 2012 Olynik and Rastegar. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mojgan Rastegar, Regenerative Medicine Program, Faculty of Medicine, Department of Biochemistry and Medical Genetics, University of Manitoba, 745 Bannatyne Avenue, Room 627, Basic Medical Sciences Building, Winnipeg, MB, Canada R3E 0J9. e-mail:cmFzdGVnYXJAY2MudW1hbml0b2JhLmNh