
 Miguel Cacho Teixeira1,2
Miguel Cacho Teixeira1,2


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 19 April 2012
Sec. Toxicogenomics
Volume 3 - 2012 | https://doi.org/10.3389/fgene.2012.00063
This article is part of the Research Topic Toxicogenomics in non-mammalian species View all 11 articles
The emerging transdisciplinary field of Toxicogenomics aims to study the cell response to a given toxicant at the genome, transcriptome, proteome, and metabolome levels. This approach is expected to provide earlier and more sensitive biomarkers of toxicological responses and help in the delineation of regulatory risk assessment. The use of model organisms to gather such genomic information, through the exploitation of Omics and Bioinformatics approaches and tools, together with more focused molecular and cellular biology studies are rapidly increasing our understanding and providing an integrative view on how cells interact with their environment. The use of the model eukaryote Saccharomyces cerevisiae in the field of Toxicogenomics is discussed in this review. Despite the limitations intrinsic to the use of such a simple single cell experimental model, S. cerevisiae appears to be very useful as a first screening tool, limiting the use of animal models. Moreover, it is also one of the most interesting systems to obtain a truly global understanding of the toxicological response and resistance mechanisms, being in the frontline of systems biology research and developments. The impact of the knowledge gathered in the yeast model, through the use of Toxicogenomics approaches, is highlighted here by its use in prediction of toxicological outcomes of exposure to pesticides and pharmaceutical drugs, but also by its impact in biotechnology, namely in the development of more robust crops and in the improvement of yeast strains as cell factories.
The transdisciplinary field of Toxicogenomics is defined as the merging of Omics approaches with toxicology to elucidate the response at the genome level to environmental stressors, drugs, and other toxicants (Hamadeh et al., 2002; Gomase and Tagore, 2008; North and Vulpe, 2010). The coupling of such approaches with classical toxicology studies combined with bioinformatics has the potential to provide a more comprehensive knowledge of the molecular and cellular effects of chemicals in biological systems than more traditional approaches. Understanding these complex responses is of paramount importance in fields ranging from Environmental Health to Pharmacology and drug development and to Biotechnology in general (Hamadeh et al., 2002; Guerreiro et al., 2003; Teixeira et al., 2007). Toxicity testing using animal models has a number of limitations that make it difficult to deal with the increasingly large number of potentially toxic compounds found in the environment and in the pharmaceutical industry for which toxicity data available is scarce (North and Vulpe, 2010). Mechanism-centered analysis represents an alternative approach to animal testing and, in this context, the yeast Saccharomyces cerevisiae can be an invaluable asset.
S. cerevisiae is a thoroughly established and widely used eukaryotic model for molecular and cellular biology studies. Yeast also plays a significant role in biotechnology, where it is used as a cell factory with diverse applications (Botstein and Fink, 2011). There are several inherent features that make yeast such a proficient model system: (1) it is a unicellular non-pathogenic microorganism with rapid and inexpensive growth, (2) it is amenable to genetic manipulation, (3) genome-wide analyses are easily implemented, with a vast array of experimental tools and biological material readily available, (4) it possesses a strikingly high-level of functional conservation within the human genome and other higher eukaryotes, and (5) it has the unique advantage of possessing functional information available for nearly every gene. The paradigm of research using S. cerevisiae changed with the publication of its genome sequence more than 15 years ago (Goffeau et al., 1996). A wealth of biological information has been gathered over several years of post-genomic research facilitated by easy access to public databases (Saccharomyces Genome Database, SGD1; and YEASTRACT2, among others). Research on S. cerevisiae pioneered the development of several post-genomic experimental approaches and computational tools, changing the field of yeast research with the application of innovative methodologies in functional genomics and proteomics (Mager and Winderickx, 2005; Smith et al., 2010; Botstein and Fink, 2011). In summary, yeast is a robust and inexpensive experimental platform where molecular studies difficult to carry out in more complex and less accessible eukaryotes are deeply facilitated (Wuster and Madan Babu, 2008; Smith et al., 2010; Botstein and Fink, 2011). Moreover, although many cytotoxic compounds act on their target organisms via physiological mechanisms that do not exist in yeast, many of the basic mechanisms underlying toxicity, adaptation, and resistance to chemical and environmental stresses are apparently conserved between yeast and phylogenetically distant organisms (Foury, 1997; Hohmann and Mager, 1997; Parsons et al., 2003; Mager and Winderickx, 2005).
In this review we will focus on the field of yeast toxicogenomics and how it can be exploited to obtain mechanistic insights into the action of drugs or toxicants with relevance in environmental health and agriculture (pesticides and environmental pollutants), medicinal and biomedical research (drugs used in the treatment of cancer, malaria, bacterial infections, etc.), and biotechnology (wine, beer, and other alcoholic fermentation processes, including the production of bio-ethanol, etc.; Figure 1), with a particular emphasis on the contribution of our research group to the field.
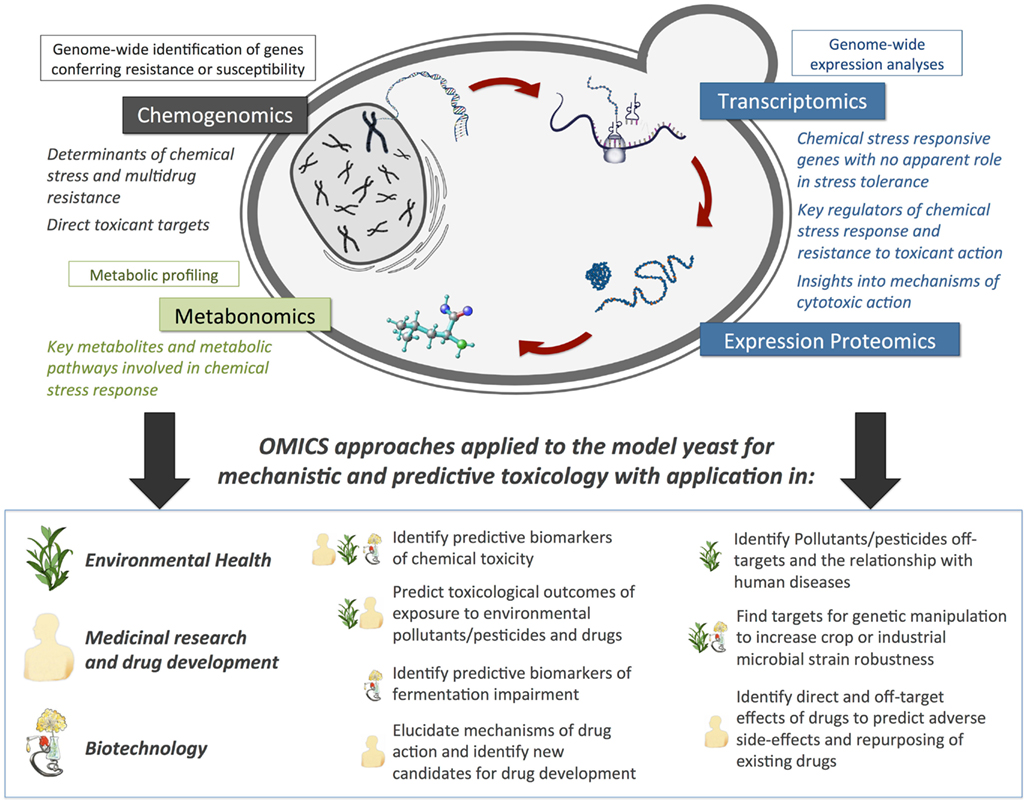
Figure 1. Predicted contribution of Omics approaches applied in the yeast Saccharomyces cerevisiae to obtain toxicological mechanistic insights with application in environmental health, agriculture, drug development, and biotechnology.
Variations in gene and protein expression or metabolite levels following exposure to a toxicant can contribute to identify the cellular components and pathways that are most relevant to a toxicological response (Hamadeh et al., 2002; North and Vulpe, 2010). S. cerevisiae provides a privileged experimental system for obtaining an integrated assessment and genome-wide perspective of toxicity mechanisms (Figure 1), through the combination of transcriptomics and quantitative proteomics, for the evaluation of genome-wide expression changes occurring as part of the yeast response to environmental toxicants, metabonomics, for the study of the cell’s small-molecule metabolite profile as the ultimate response to the toxicant, and chemogenomics, for the identification of cellular toxicity molecular targets (Figure 1). It is noteworthy that, as stand-alone, these analyses are not expected to provide decisive insights into the role of genes or proteins in a toxicological response, and should instead be integrated and coupled with suitable bioinformatics tools. Future research in the field will require the development of computational tools aiming the integration of high-throughput data and the collaborative activity of multidisciplinary teams with expertise in biological sciences, functional and comparative genomics, and bioinformatics.
A considerable amount of toxicity assessment data is available from yeast DNA microarrays. Upon toxicant exposure, yeast cells reprogram mRNA expression in order to adapt to the new environmental conditions, which produces a gene expression pattern (or “signature”) characteristic of the compound (Simmons and Portier, 2002; Schwartz et al., 2004). These transcriptional signatures are shared by compounds with similar modes of action and can be used to infer mechanisms of action and predict toxicological outcomes of uncharacterized toxicants (Marton et al., 1998; Lamb et al., 2006; Teixeira et al., 2007). In fact, alterations of global gene expression can occur almost immediately after exposure, and the assessment of these changes could potentially provide an earlier and more sensitive biomarker of a toxic response than traditional toxicological methods (Hamadeh et al., 2002; Simmons and Portier, 2002). However, gene expression alone is not adequate to fully understand a toxicant’s action and the resulting outcome (Hamadeh et al., 2002), since abnormalities in protein production and/or function are also expected to occur. As such, proteomics approaches are employed to identify the protein alterations associated with toxicant exposure (Teixeira et al., 2005, 2009b; Santos et al., 2009; Sa-Correia and Teixeira, 2010). Finally, genomic and proteomics methods do not address how the cell’s dynamic metabolic status is affected by exposure to a toxicant. Metabonomics is an approach that allows the study of metabolic profiles based on the premise that toxicant-induced alterations will provide information on chemical toxicity (Nicholson et al., 2002). Data in yeast are still scarce, however it has already contributed to increase the current understanding of weak acid toxicity (Hasunuma et al., 2011; Lourenço et al., 2011).
Functional toxicogenomics is defined as the global study of the biological function of genes regarding the toxic effect of a compound or environmental stress, providing a direct link between gene and toxicant (Hamadeh et al., 2002; North and Vulpe, 2010). In this context, a major breakthrough with respect to studies on the mechanism of action and one of the main advantages of using yeast bioassays for toxicity assessment was the generation of heterozygous/homozygous diploid and haploid gene deletion collections (Figure 2; Scherens and Goffeau, 2004). Fitness is the primary phenotypic descriptor for yeast studies, and these collections enable comprehensive and systematic genetic screens that provide direct links between a specific gene and the requirement for that gene product function in the cellular response to a particular condition (Auerbach et al., 2005; Hoon et al., 2008b; Wuster and Madan Babu, 2008). The different types of fitness-based assays that can be used to identify toxicant-induced phenotypes include homozygous (knock-out deletion, gene dosage = 0%), haploinsufficiency (heterozygous deletion strains, gene dosage = 50%) and multicopy- and overexpression (gene dosage > 100%) screens (Figure 2; Hoon et al., 2008b). In homozygous profiling, each non-essential gene is knocked-out leading to complete loss-of-function. These screens are often used to identify genes that are important for conferring stress resistance, for example by genetic interaction with the toxicant’s target. It is also applied to identify biological functions that are affected by a given stress, and infer from those the mechanisms of toxicological action. Finally, it is possible to screen for deletions that confer resistance to a compound in conditions that are deleterious to the wild-type strain. The deleted genes can be direct targets, or be involved in modifications or pathways that enable the compound’s cytotoxic action. Naturally, one obvious downside of homozygous or haploid gene deletion strains is that only non-essential genes can be deleted, but the mechanism of action of many toxicants might target essential gene products. On the other hand, haploinsufficiency screens are based on the premise that lowering the gene dosage of the molecular target increases susceptibility, and thus enables direct identification of a toxicant’s cellular target (see Hoon et al., 2008b; Wuster and Madan Babu, 2008). Libraries of double mutants have also been generated to uncover interactions between genes through synthetic lethality, which can be integrated with functional screens data to elucidate toxicity mechanisms and modes of action (Costanzo et al., 2010). Many bioinformatics tools are available to facilitate interpretation of the results. For example, Gene Ontology (GO) annotation and GO-based resources such as GOToolBox3 (Martin et al., 2004) allow the identification of biological functions that are enriched within datasets, thus creating a “functional fingerprint” comparable to transcriptional signatures by expression profiling. These can be used to identify responses and pathways that are common to different classes of toxicants.
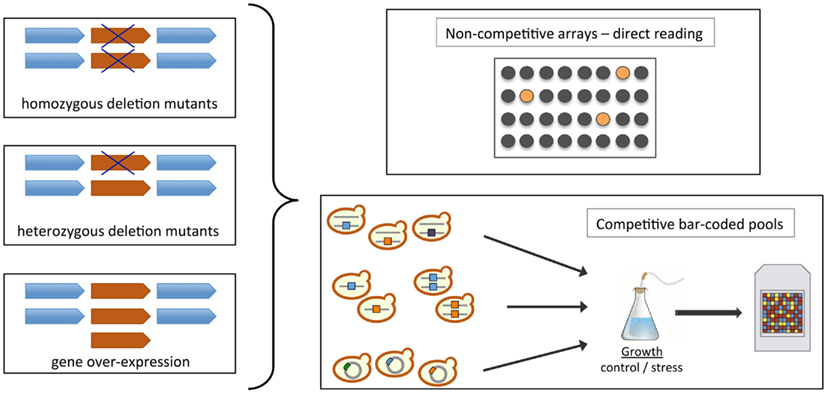
Figure 2. Construction and screening of yeast collections. Schematic representation of methodologies and cell libraries available for chemogenomics testing in S. cerevisiae (homozygous or haploid deletion – gene dosage 0%, heterozygous deletion – gene dosage 50%, and overexpression – gene dosage > 100%; see Section “Functional Toxicogenomics using Yeast Gene Deletion Collections”; Auerbach et al., 2005; Hoon et al., 2008b; Wuster and Madan Babu, 2008; North and Vulpe, 2010; Smith et al., 2010). The fitness of strains upon chemical treatment is usually assessed in non-competitive arrays or in competitive bar-coded pools. In the first case, the toxicant can be added to a well plate and each mutant occupies a separate well; the effects are observed directly by comparison with wild-type strain fitness. In the second case, the screen is executed in a pooled format where uniquely tagged (“bar-coded”) strains are grown together in the presence of a toxicant. Fitness is assessed by determining the abundance of the different mutant strains using microarrays coupled with a PCR strategy that amplifies the molecular bar-codes associated with each mutant. Strain depletion in the toxicant-treated pool indicates chemical hypersensitivity.
Our current literature survey found at least 80 publications that have used yeast deletion collections in the context of toxicity testing, in which hundreds of different stresses were explored and phenotypes were attributed to over 90% of all yeast genes (see Table 1 for an overview). Remarkably, although many of these conditions had been previously scrutinized using classical methodologies or even DNA microarrays, many of the genes identified using deletion collections had not been known to be involved in the toxicological pathways investigated. One major contribution arising from the use of homozygous and heterozygous yeast deletion collections was the so-called “chemical genomic portrait of yeast” (Hillenmeyer et al., 2008). The authors carried out over 1100 assays in the presence of chemical or environmental stress conditions, ranging from approved therapeutic drugs to compounds with uncertain activity. Functional enrichment analysis led to the identification of several biological functions that were required for growth in at least 20% of the assays performed, in particular endosome transport, vacuolar degradation, and transcription, a coordinated system that is conserved from yeast to humans (Hillenmeyer et al., 2008). The remarkable amount of data generated can be applied, for example, to predict toxicological modes of action for the compounds tested and other structurally related, or to predict the effects of synergies between some of these stresses. Other important contributions using yeast deletion collections will be described throughout this review.

Table 1. Selected publications in yeast toxicogenomics studies using deletion mutant collections.
To achieve a more complete understanding of the mechanisms of action and toxicological response, it is necessary to intersect and integrate the genome-wide data coming from the different approaches (Figure 1). Interestingly, several studies have now shown that the previous belief that genes that are up-regulated under a given stress are also required for growth under those conditions is often false (Giaever et al., 2002; Mettetal et al., 2008; Batova et al., 2010; Landstetter et al., 2010; dos Santos and Sá-Correia, 2011). Although this seems to suggest that deletion collections provide a better assessment of genes required for the response to a toxicant treatment, an integrated approach can lead to other important insights, for example the identification of key regulators of stress response. In yeast, the identification of the transcription factors predicted to underlie the transcriptomic response to stress is facilitated by freely accessible databases and computational tools such as YEASTRACT, a database focused on the delineation of yeast transcription regulatory networks, at a genomic scale (Teixeira et al., 2006b; Abdulrehman et al., 2011). Comprising more than 48,000 documented regulatory associations between yeast transcription factors and target genes (Abdulrehman et al., 2011), YEASTRACT offers one of the best platforms for the study and understanding of genomic regulation in an integrative perspective. The growing use of mathematical descriptions of the dynamical behavior of such networks (Teixeira et al., 2010a) is now allowing the prediction of the possible behaviors of biological systems under the action of pollutants, pesticides, drugs, and other chemical stresses, emphasizing the outstanding position of the yeast model in the field of toxicogenomics. Moreover, yeast toxicogenomics data can be integrated with studies from other sources, such as toxicological results obtained in the toxicant’s target organism when appropriate, to obtain a view of a compound’s toxicity at the systems level. This raises the potential of yeast toxicogenomics very high but poses additional challenges to the tasks of data integration and usage.
The toxicological outcome of sudden or chronic exposure to environmental pollutants (e.g., metal ions or organic solvents, including benzene, or phenol derived compounds), is scarcely understood at the molecular and cellular levels. However the genome-wide yeast response to toxic concentrations of metal ions, such as nickel, cadmium, copper, chromium, arsenic, cobalt, manganese, and zinc has been studied by exploring chemogenomics and transcriptomics approaches. These studies led to the identification of several functional groups that are important in the yeast response to all or to part of the metal ions tested, mostly involved in sulfur amino acid and iron metabolism, oxidative stress response, vacuolar function, protein modification, transport and degradation, enzyme inactivation, cation and transition metal transport, mRNA decay, and DNA metabolism (Momose and Iwahashi, 2001; Jin et al., 2008; Ruotolo et al., 2008; Serero et al., 2008; Yasokawa et al., 2008; Takumi et al., 2010; Bleackley et al., 2011). The toxicological outcome of the exposure to agrochemicals, including herbicides and agricultural fungicides, is difficult to predict, since many times it takes years to develop. Genome-wide analyses in yeast have been successfully used to identify the genes responsible for response and resistance to stresses induced by pesticides of agricultural interest (Cabrito et al., 2009, 2011). These pesticides include the herbicide sulfometuron methyl (Jia et al., 2000), the dithiocarbamate fungicides mancozeb (Santos et al., 2009; Dias et al., 2010), thiuram, zineb and maneb (Kitagawa et al., 2003), the benzimidazole fungicide benomyl (Lucau-Danila et al., 2005), the pesticide lindane (Parveen et al., 2003), the herbicide 2,4-D (Teixeira et al., 2005, 2006a, 2007), and the herbicides paraquat and cyperquat (1-methyl-4-phenylpyridinium – MPP+; Doostzadeh et al., 2007). Toxicogenomics approaches have been applied to define and predict new toxicological outcomes of exposure to pesticides, including the agricultural fungicide mancozeb (Santos et al., 2009; Dias et al., 2010) and the herbicide 2,4-D (Teixeira et al., 2005, 2006a). A review of the main results of these two case studies follows.
Mancozeb, a mixture of manganese- and zinc-ethylene-bis-dithiocarbamate (Mn:Zn, 9:1), is an agricultural fungicide with a broad spectrum of action and multiple cell targets, widely used against phytopathogenic fungi in several crops and vineyards (Maroni et al., 2000; Ballantyne, 2004). This compound displays low acute toxicity, however, in recent years, mounting evidence suggests that chronic exposure to this fungicide increases the probability of developing Parkinson’s disease and certain forms of cancer (Belpoggi et al., 2002; Zhou et al., 2004; Calviello et al., 2006).
The early global response to mancozeb and the genome-wide resistance mechanisms established by S. cerevisiae were analyzed by expression proteomics (Santos et al., 2009) and chemogenomics (Dias et al., 2010; see Figure 3 for a schematic representation of the main findings obtained in these studies with mancozeb). Interestingly, 70% of the proteins differently expressed in cells exposed to mancozeb (Santos et al., 2009) and 53% of the determinants of yeast resistance to the fungicide (Dias et al., 2010) have human orthologs. This is the case for proteins involved in V-ATPase function (Vma4 and Vma13), protein synthesis, folding (e.g., Kar2), protein degradation/proteasome sub-units (e.g., Pre3, Pre7, Pre8, Pre9, Nas2, Sem1, and Ubp6), and in the oxidative stress and anti-oxidant response (e.g., Tsa1, Tsa2, Glr1, Gsh1, Gsh2, Sod1, Sod2, and Yap1). Interestingly, V-ATPases are overexpressed in several metastatic cancers (Sennoune et al., 2004) and the overexpression of proteasome sub-units leads to increased survival rate of human cell lines following oxidative stress, due to a higher proteasome degradation of oxidized modified protein (Chondrogianni et al., 2005). Other human orthologs related with the oxidative stress response (PRDX2, PRDX3, GSR, GCLC, GSS, SOD1, and SOD2), have been involved in the onset and progression of neurodegenerative diseases, namely Parkinson’s disease, by protecting the cell, acting as anti-oxidant agents and neuroprotectors (Chang et al., 2004; Ihara et al., 2005; Cumming et al., 2007). Besides, PAK1, MAP2K1, and LCP1, three other genes described as being involved in tumor development and invasiveness (Wang et al., 2006a,b), are orthologs of yeast determinants of mancozeb resistance CLA4, PBS2, and SAC6 (Dias et al., 2010).
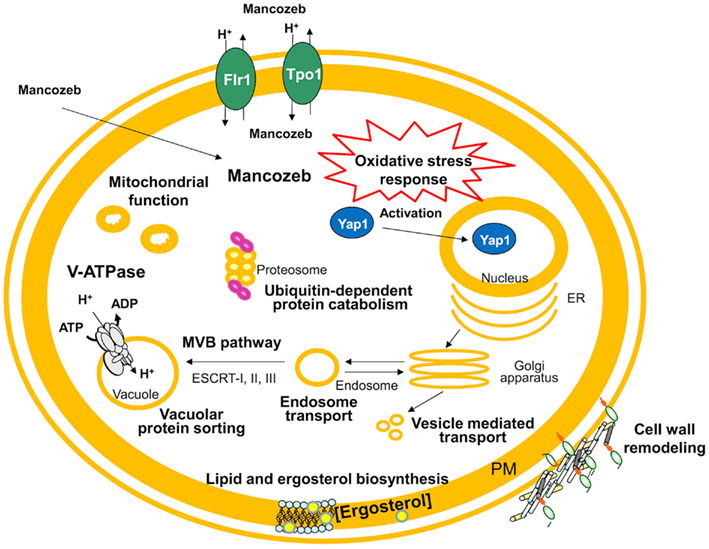
Figure 3. Proposed model for the action of mancozeb in S. cerevisiae cells. This model results from the integration of yeast chemogenomics (Dias et al., 2010) and proteomics (Santos et al., 2009) approaches. The complex mancozeb-induced expression changes and mancozeb determinants of yeast resistance, were found to be related to oxidative stress, V-ATPase function, protein translation initiation and protein folding, disassembling of protein aggregates and degradation of damaged proteins, lipid and ergosterol biosynthesis, mitochondrial function, cell wall remodeling, and multidrug resistance transporters.
Using the YEASTRACT database, more than 90% of the genes that encode proteins up-regulated under mancozeb imposed stress were found to be known targets of Yap1 (Santos et al., 2009), the major oxidative stress regulator in yeast. Yap1 was also found, based on a chemogenomics study (Dias et al., 2010), to be a determinant of yeast resistance to the fungicide, and also to control the regulatory network underlying the up-regulation of the multidrug transporter encoding gene FLR1 (Teixeira et al., 2010a; Monteiro et al., 2011). Remarkably, the human orthologs of Yap1, Jun, and Jdp2, are activated during acute and chronic phases of several neurodegenerative diseases (Shaulian and Karin, 2002), establishing a possible link between the predicted response to mancozeb toxicity and neurodegenerative disease progression through the Yap1/Jun/Jdp2 regulators. Although mancozeb was reported to induce reactive oxygen species (ROS) production as a consequence of mitochondrial dysfunction in mesencephalic cells (Domico et al., 2007), Dias et al. (2010) registered no increase in ROS production in yeast in response to the fungicide, probably due to the fact that in glucose fermenting yeast the level of mitochondrial respiration is reduced. In the absence of mitochondrial electron leakage, which masks other aspects of mancozeb toxicity, a direct role of mancozeb in protein damage, as a thiol-reactive compound, was thus identified in the yeast model, with a possible parallel in human cells (Dias et al., 2010).
The herbicide 2,4-D is the most commonly used member of the auxin-like herbicide family. Although being considered relatively safe, exposure to 2,4-D has been linked to the development of non-Hodgkin lymphoma and sarcoma (Ibrahim et al., 1991). Furthermore, several cases of 2,4-D resistant weeds have been described4, raising the need to increase herbicidal application rates, with the risk of reaching environmental toxic levels.
S. cerevisiae genome-wide approaches, including transcriptomics (Teixeira et al., 2006a) and expression proteomics (Teixeira et al., 2005), were used to gain insights into the mechanisms of response and resistance to 2,4-D. The results obtained in yeast have been used to guide studies on the molecular mechanisms underlying 2,4-D toxicity and response in plants and other higher eukaryotes. Interestingly, the quantification of the relative toxicity of 2,4-D compared to other herbicides measuring electrophysiological parameters and vitality of an animal nervous system (the frog’s sciatic nerve) was comparable to the results obtained when yeast growth inhibition due the same herbicides was tested (Papaefthimiou et al., 2004). The early transcriptional response of yeast to 2,4-D includes the up-regulation of several genes involved in oxidative stress and anti-oxidant response (Teixeira et al., 2006a), which correlates with the increase in hydroxyl radicals and lipid peroxidation levels registered as a consequence of acute 2,4-D stress in yeast (Teixeira et al., 2004). Significantly, 2,4-D was found to induce fatty acid β-oxidation and also electron leakage from the mitochondrial respiration and catalase activity in rat cells (Bradberry et al., 2000, 2004). Given the fact that oxidative stress is associated with neurological diseases, aging, and cancer, these results can give clues on the effect of massive or repeated human exposure to the herbicide. In parallel to the implications of these studies in environmental health, results of herbicide resistance obtained in the yeast model have also proven useful to study herbicide toxicity mechanisms in plants, with expected impact in agriculture and plant biotechnology (Cabrito et al., 2009; Cabrito et al., unpublished results).
In acidified growth medium, yeast cells challenged with the herbicide 2,4-D suffer a strong reduction in their cytosolic and vacuolar pH (Fernandes et al., 2003; Simões et al., 2003), which is counteracted by the activation of the plasma and vacuolar membrane H+-ATPases (Fernandes et al., 2003; Teixeira et al., 2005). In fact, auxins, similar to 2,4-D, were also shown to induce the activity of the model plant Arabidopsis plasma membrane H+-ATPase, contributing to maintain the intracellular pH in plant roots (Shen et al., 2006).
Both the microarray and the proteomic analyses carried out indicated the up-regulation of a large number of genes involved in carbon and nutrient source metabolism and amino acid homeostasis in response to 2,4-D (Teixeira et al., 2005, 2006a), which can be a response to the damaging effects of the herbicide on the plasma membrane lipid and sterol organization and permeability, with consequences in terms of nutrient uptake. Teixeira et al. (2007) suggested that the auxin-like herbicide 2,4-D exerts a repressing effect over the TOR (target of rapamycin) pathway, recognized as a central controller of cell growth in all eukaryotes, which control the balance between protein synthesis and degradation in response to nutrient availability and quality (Crespo and Hall, 2002). Interestingly, in plants, namely Arabidopsis thaliana, the existence of a functional TOR kinase pathway has been reported and linked to cell proliferation and growth regulation, auxin being suggested as a possible signaling molecule in the pathway (Berkowitz et al., 2008).
Additionally, several other mechanisms of yeast resistance to the herbicide are similar to processes described in plants, namely those involved in cytosolic detoxification (Smart and Fleming, 1996; Teixeira and Sá-Correia, 2002; Ito and Gray, 2006; Teixeira et al., 2006a; Cabrito et al., 2009). Exposure to the herbicide leads to the up-regulation of several genes encoding MDR transporters in yeast (Teixeira et al., 2006a). The MDR transporters Tpo1, Pdr5, and Pdr18 were confirmed as determinants of yeast resistance to 2,4-D (Teixeira et al., 2004; Cabrito et al., 2011). 2,4-D induces the expression of PDR5 orthologs, SpTUR2, in the aquaphyte Spirodela polyrrhiza, and AtPDR9, in the model plant A. thaliana, the latter catalyzing 2,4-D extrusion from root cells (Smart and Fleming, 1996; Ito and Gray, 2006). Recently, Tpo1 homologs encoding putative plasma membrane MFS transporters from A. thaliana were analyzed by Cabrito et al. (2009) for a possible role in 2,4-D resistance. At5g13750/ZIFL1 transcript levels were found to increase in 2,4-D stressed plants. The functional heterologous expression of AtZIFL1 in yeast was found to confer increased resistance to the herbicide in wild-type and Δtpo1 cells, through the reduction of the intracellular concentration of 2,4-D counter-ion (Cabrito et al., 2009). Interestingly, Zifl1 was the first eukaryotic transporter of the MFS identified as a multidrug resistance determinant, opening an entirely new field of research with promising repercussions in medicine, biotechnology, and agriculture.
These case studies highlight the similarities of toxicological effects of these pesticides from yeast to higher eukaryotes, such as humans and plants. Hence, the use of the yeast model system is expected to continue to contribute to the understanding of the molecular mechanisms underlying pesticide toxicity in more complex and less easily accessible eukaryotes.
The use of yeast as a eukaryotic model is particularly important in the field of medicinal research and drug discovery (Simon and Bedalov, 2004; Mager and Winderickx, 2005; Menacho-Marquez and Murguia, 2007; Hoon et al., 2008b). Approximately 17% of all yeast genes are members of orthologous gene families associated with human disease, and for the majority of these genes their mammalian homolog is functional in yeast and complements the yeast deletion mutant (Foury, 1997; Heinicke et al., 2007). Modern medicine faces the challenge of developing safer and more effective therapies to treat human diseases; toxicogenomics represents a new paradigm in drug development and risk assessment, particularly in mechanistic and predictive toxicology as well as in biomarker discovery (Guerreiro et al., 2003; Gomase and Tagore, 2008). To better evaluate drug-associated adverse effects, the drug’s specific mode of action needs to be elucidated first. However, the successful identification of drug targets and mechanisms of action requires a prior understanding of the high-level functional interaction between the key components of cells and systems (Guerreiro et al., 2003; North and Vulpe, 2010).
The primary advantage of yeast in drug discovery is the contribution to identify the mechanisms of action of compounds when they are unknown. Moreover, yeast is currently the only system where it is possible to assess all targets in the cell simultaneously and in vivo (Smith et al., 2010). These strategies are important not only to identify new drugs for further development, but also to find new uses for already approved drugs. The contribution of yeast toxicogenomics to this field takes on a pharmacogenomics perspective. This emerging post-genomic discipline is characterized by having a genome-wide perspective on the action of drugs, making use of global approaches to identify candidate drug targets and off-target effects (Swen et al., 2007; Wang and Weinshilboum, 2008; Ruderfer et al., 2009). Off-target effects are more difficult to detect and are often the cause of deleterious side effects. Such effects can arise, for example, when the direct binding interaction between one protein and a target results in an interaction with a second protein (Parsons et al., 2006; Ericson et al., 2008; West et al., 2010). In recent years, yeast fitness screens and other Omics approaches have been employed to search for new targets and elucidate the mode of action of different compounds, including anticancer drugs, antimalarials, antimicrobials, and other bioactive compounds (see Table 1). Yeast methodologies have also been used to study human disease genes and model human disorders (Steinmetz et al., 2002; Outeiro and Lindquist, 2003; Willingham et al., 2003; Gammie et al., 2007; Yuen et al., 2007), screen for new drugs to treat cancer, obesity, prion disease, etc. (Hammonds et al., 1998; Bach et al., 2003; Tribouillard et al., 2006; Marjanovic et al., 2010), and predict drug responses in relation to indicators such as genotype and expression levels (Perlstein et al., 2007; Ruderfer et al., 2009; Chen et al., 2010).
In an illustrative example, Ericson et al. (2008) identified 81 psychoactive drugs that affected yeast fitness at the level of evolutionarily conserved cellular processes such as secretion, protein folding, RNA processing, and chromatin structure. These processes might constitute secondary drug targets and point to additional, previously uncharacterized mechanisms of action for these drugs in humans. Information of this nature can be used to guide the rational design of new compound derivatives with fewer side effects and for tailoring drug treatment to individual patient genotypes, in a personalized medicine perspective. In another interesting study, yeast was applied to identify secondary targets of nitrogen-containing bisphosphonates, drugs commonly used to treat bone-related disorders including cancer (Bivi et al., 2009). The only known target of these compounds was farnesyl pyrophosphate synthase, but the authors showed that the action of this drug in yeast involves additional mechanisms, predominantly at the level of DNA damage, and cytoskeleton dynamics. The dataset obtained from the yeast screen was validated in a mammalian system, and confirmed the involvement of new biological processes and specific genes that represent potential new targets for compounds with antitumor activity (Bivi et al., 2009). Genome-wide expression patterns were applied for target validation and identification of secondary drug targets of FK506, an immunosuppressant drug known to inhibit the protein phosphatase calcineurin (Marton et al., 1998). The authors identified a transcriptional signature of FK506 and found that it closely resembled that of the calcineurin null mutant. However, an increase of the drug dosage resulted in a different expression profile, suggesting that targets other than calcineurin mediated FK506 effects. Using a similar approach, Hughes et al. (2000) compared the transcriptomes of 13 compounds with those of 286 deletion mutants representing a variety of functional classes, identifying novel targets and off-targets for several drugs.
Yeast deletion mutants have also been used to identify targets for 78 compounds with diverse chemical structures and therapeutic relevance (Lum et al., 2004), resulting in identification of a lanosterol synthase in the sterol biosynthetic pathway as a target of the antianginal drug molsidomine, while the rRNA processing exosome was singled-out as a potential target of the cell growth inhibitor 5-fluorouracil. A similar study screened 10 different compounds (including anticancer and antifungal agents) against a deletion collection in 80 competitive pool assays (Giaever et al., 2004) and identified a chemical core structure that is shared by three compounds that inhibit the ERG24 deletion strain, suggesting that cells respond similarly to compounds of related structure. Several compounds with in vivo activity against yeast and mammalian prions were identified in a yeast-based screen (Tribouillard et al., 2006), establishing this method as an economic and efficient high-throughput approach to identify novel prion inhibitors or to carry out comprehensive structure-activity studies for already isolated anti-mammalian prion drugs. The results also seem to highlight the extensive conservation of biochemical pathways controlling prion formation and/or maintenance from yeast to human.
A comprehensive example of yeast toxicogenomics is its application to the study of the antimalarial quinine. It was the first effective treatment against malaria and is still the drug of choice for chloroquine-resistant or severe malaria (WHO, 2006; Alkadi, 2007), but the molecular mechanism of action and toxicity of quinine in the Plasmodium parasite are still a topic of debate (Fitch, 2004). In a clear demonstration of the merging of toxicogenomics with pharmacogenomics, Khozoie et al. (2009) screened a yeast deletion collection to address the mode of action of quinine and its adverse side effects. The authors observed an enrichment of genes involved in tryptophan biosynthesis, and additional assays seemed to demonstrate that quinine causes tryptophan starvation and that dietary tryptophan supplements could help to avert the toxic effects of quinine (Khozoie et al., 2009). Another chemogenomics-based analysis identified for the first time several genes encoding ribosome protein sub-units whose deletion leads to increased quinine resistance (dos Santos and Sá-Correia, 2011). The particular involvement of phosphate signaling and transport in quinine tolerance was also studied, with indications that phosphate-starvation responsive genes are activated in response to quinine. P. falciparum homology searches identified several relevant functional homologs in the parasite, suggesting that the quinine targets identified in the yeast model are good candidates to be transposed and explored in a parasitic context (Figure 4A). The transcriptomic analysis of the yeast early response to quinine unveiled glucose de-repression reprogramming (Figure 4A; dos Santos et al., 2009). Moreover, quinine was shown to inhibit the uptake of glucose into yeast cells following a competitive inhibition kinetic model. These findings have an important parallel in the malaria parasite, where glucose uptake is vital and mediated by PfHT1, a single-copy transporter homologous to yeast’s hexose HXT transporters (Woodrow et al., 1999; Saliba et al., 2004). The mechanism by which quinine enters and accumulates in the parasitic cell is not clear, but it is believed that a carrier-mediated import system is involved (Sanchez et al., 2008). The yeast results suggested PfHT1 as a quinine target, possibly involved in quinine uptake into the cell and depriving the parasite of glucose (dos Santos et al., 2009).
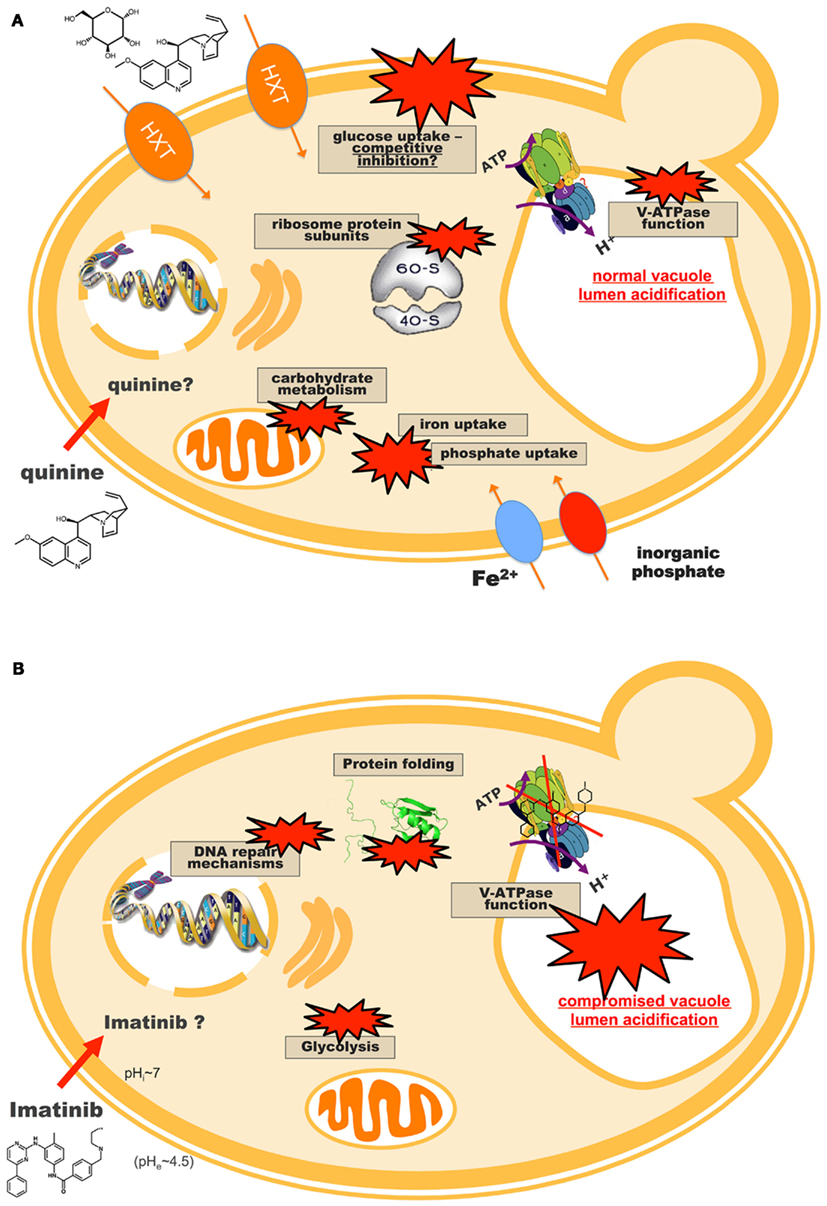
Figure 4. Proposed model for the action of (A) quinine and (B) imatinib in S. cerevisiae cells. These models result from the integration of chemogenomics, transcriptomics and proteomics approaches (dos Santos and Sá-Correia, 2009; dos Santos et al., 2009; dos Santos and Sá-Correia, 2011; dos Santos and Sá-Correia, unpublished results), suggesting new targets and modes of action for quinine and imatinib that possess extensive functional conservation in the organisms of interest, Plasmodium falciparum, and human cells, respectively. The most important results are the identification of PfHT1 as a potential target of quinine, as well as the vacuolar H+-ATPase (V-ATPase) as a target of imatinib (see Genome-wide Responses and Determinants of Resistance to Antimalarial Drugs and Genome-wide Responses and Determinants of Resistance to Anticancer Drugs).
Yeast has also been used as model for mechanistic studies with other antimalarial drugs, namely mefloquine (Delling et al., 1998), chloroquine (Emerson et al., 2002), artemisinins (Li et al., 2005; Alenquer et al., 2006), and quinidine (Delling et al., 1998; Nunes et al., 2001; Felder et al., 2002; Tenreiro et al., 2002; Vargas et al., 2004).
Anticancer drugs have been the subject of several yeast pharmacogenomics studies. For example, at least three genome-wide screens for cisplatin susceptibility have been performed using yeast deletion collections (Wu et al., 2004; Huang et al., 2005; Liao et al., 2007). The vacuolar H+-ATPase (V-ATPase) and its action in cytoplasmic pH maintenance was shown to have an important role in sensitivity to this drug (Liao et al., 2007), an important result given how cisplatin cytotoxicity is potentiated by synergistic treatment with a V-ATPase inhibitor in human cell lines (Murakami et al., 2001). In a demonstration of how results obtained in the yeast model can be extended to human cells, Schenk et al. (2001, 2002) identified SKY1 as a cisplatin sensitivity gene whose disruption conferred cisplatin resistance in yeast; later work in human ovarian carcinoma cell lines showed that inactivation of its human homolog, SRPK1, induces cisplatin resistance as well. The paradigmatic anticancer drug imatinib mesylate (Glivec, Novartis) was also studied in yeast. Imatinib is a selective tyrosine kinase inhibitor used in chronic myeloid leukemia with outstanding results, but drug resistance is an arising problem (Quintás-Cardama et al., 2009; Volpe et al., 2009). Fifty-one genes emerged as determinants of resistance to imatinib from the screening of a yeast deletion mutant collection, including 83% human homologs (dos Santos and Sá-Correia, 2009). Imatinib was also shown to act as a potent inhibitor of the highly conserved yeast V-ATPase, both in vivo and in vitro (dos Santos and Sá-Correia, 2009; dos Santos and Sá-Correia, unpublished results), suggesting that vacuolar function is a novel imatinib target. In fact, V-ATPase activity has been shown to be necessary to limit the deleterious effects of several drugs in yeast, namely the genotoxic tirapazamine and cisplatin, weak acids, mancozeb, toxins, and others (Parsons et al., 2004; Hellauer et al., 2005; Liao et al., 2007; Mira et al., 2009; Dias et al., 2010). However, in none of these studies vacuolar acidification was affected directly, which is in striking contrast with imatinib, where a clear loss of vacuolar acidification in cells treated with this drug was reported (dos Santos and Sá-Correia, 2009). Quantitative- and phospho-proteomic analyses identified 18 proteins altered at the content level or displaying imatinib-repressed phosphorylation (dos Santos and Sá-Correia, unpublished results). All these proteins have human homologs and are mainly involved in glycolytic pathways, translation, and protein folding. A role for HSP70 proteins in the response to imatinib in yeast, as well as decreased glycolysis as a metabolic marker of imatinib action were suggested, consistent with findings from studies in human cell lines (Nowicki et al., 2003; Ferrari et al., 2007; Pocaly et al., 2008; Kominsky et al., 2009). The previously proposed effect of imatinib as an inhibitor of V-ATPase function was supported by the identification of an under-expressed subunit of this complex in imatinib-stressed yeast cells (Figure 4B).
S. cerevisiae has been used for millennia in fermentation processes behind wine, beer, and spirits production. Its remarkable capability of carrying out alcoholic fermentation very efficiently, leading to the production of very high ethanol titers, has also launched S. cerevisiae as a preferable host for the production of bio-ethanol, as a renewable biofuel. Furthermore, genetic engineering and synthetic biology methods have allowed the development of S. cerevisiae strains to be used as cell factories for the production of a number of interesting biomolecules, of biotechnological and pharmaceutical interest. In all these industrial processes, yeast cells have to cope with stressful environmental conditions, including chemical stress coming from the raw material composition, and from the accumulation of ethanol, weak acids, and other toxic byproducts of the yeast metabolism (Teixeira et al., 2011).
Toxicogenomics tools have been used with success to characterize the toxicological outcome of yeast exposure to fermentation-related chemical stress inducers. Such an approach has the potential to elucidate the mechanisms of yeast tolerance to fermentation stressors, thus providing clues on how to improve process conditions and to engineer yeast strains to increase fermentation yield. A particularly good example of the use of such an approach can be found in the efforts to improve bio-ethanol production process whose efficiency is compromised by several stress factors throughout fermentation. First generation bio-ethanol production relies on the use of very high gravity (VHG) media, highly enriched in fermentable carbon sources, which induce osmotic stress in the beginning of the fermentation process. In recent years, the interest in the production of bio-ethanol from agricultural lignocellulosic residues, the so-called second-generation bio-ethanol, has gained strength. These residues appear to be preferable for a sustainable large-scale production of bio-ethanol since they are largely available and do not compete with food resources (van Maris et al., 2006). However, in lignocellulosic hydrolysate fermentations, the first phase of the process is hindered by the presence of toxic concentrations of inhibitory side-products of the raw material hydrolysis process, including acetic acid, furfural, and vanillin. During the later stages of alcoholic fermentation for first or second-generation bio-ethanol production, the accumulation of toxic concentrations of ethanol and weak organic acids are responsible for lower fermentation productivity and, eventually, for fermentation arrest, limiting the final ethanol concentration achieved. Having this in mind, transcriptomics, expression proteomics, and metabolomics approaches have been used to study the expression and metabolic profile of yeast cells exposed to sudden stress induced by ethanol (Alexandre et al., 2001; Hirasawa et al., 2007; Stanley et al., 2010), weak acids (Mira et al., 2009, 2010a; Hasunuma et al., 2011), high sugar concentrations (Erasmus et al., 2003; Pham et al., 2006), but also throughout industrial or industrial-like fermentation processes (Devantier et al., 2005; Marks et al., 2008; Ding et al., 2009; Li et al., 2010). Using such toxicogenomics tools, the involvement of three signaling pathways mediated by the transcription factors War1 (Schüller et al., 2004), Haa1 (Mira et al., 2010a), and Rim101 (Mira et al., 2009) in the yeast response to weak acids was recently characterized. Weak acid toxicity mechanisms are additionally interesting in this context, given that they are widely used as food-preservatives against spoilage yeasts and molds and because S. cerevisiae is arising as an interesting alternative host for the industrial production of carboxylic acids, being more tolerant to their toxicity than currently used bacterial systems (Abbott et al., 2009). In this context, the use of transcriptomic and chemogenomic (Mollapour et al., 2004; Schüller et al., 2004) screenings focused on the food preservative sorbate has further highlighted the importance of vacuolar acidification and redox homeostasis for weak acid stress resistance. Clues on the mechanisms of weak acid toxicity have also come from metabolomics approaches (Hasunuma et al., 2011; Lourenço et al., 2011). For example, in a S. cerevisiae strain, modified through metabolic engineering tools to be able to ferment xylose, metabolomics data revealed that metabolites involved in the pentose phosphate pathway (PPP) were significantly accumulated by the addition of acetate during xylose fermentation, suggesting that acetic acid slows down the flux of the pathway (Hasunuma et al., 2011). Based on this result, a gene encoding a PPP-related enzyme was overexpressed in the xylose-fermenting yeast, conferring increased ethanol productivity in the presence of acetic acid (Hasunuma et al., 2011).
A particularly successful approach, in this context, has proven to be the use of the yeast deletion mutant collections to identify the determinants of yeast resistance to all these stresses, individually, or in combination. This chemogenomics strategy was used to unveil the global mechanisms and determinants of yeast resistance to stresses occurring during alcoholic fermentation, in particular to high ethanol (Fujita et al., 2006; van Voorst et al., 2006; Teixeira et al., 2009a; Yoshikawa et al., 2009), high glucose (Teixeira et al., 2010b), and acetic acid (Mira et al., 2010b) concentrations. Based on these results, the aquaglyceroporin Fps1 was proposed as a major determinant of yeast resistance to ethanol and shown to play a role in reducing intracellular ethanol accumulation. The manipulation of FPS1 expression levels was found to result in an increase of the final concentration of ethanol produced under conditions close to high gravity industrial fermentation (Teixeira et al., 2009a). The chemogenomics analysis of acetic acid stress resistance has further highlighted, among other things, the importance the potassium concentration in this process, suggesting that the control of potassium levels in the fermentation broth may be crucial to increase fermentation performance (Mira et al., 2010b).
Since it is the combination of all fermentation stresses, and not their individual effect, that affects yeast fermentative capacity, a recent study (Pereira et al., 2011) focused on the integration of several chemogenomics studies to identify the few genes that are able to increase yeast tolerance to: (1) ethanol (Fujita et al., 2006; van Voorst et al., 2006; Teixeira et al., 2009a; Yoshikawa et al., 2009), acetate (Mira et al., 2010b), and high glucose (Teixeira et al., 2010b) concentrations, (2) ethanol (Fujita et al., 2006; van Voorst et al., 2006; Teixeira et al., 2009a; Yoshikawa et al., 2009), acetate (Mira et al., 2010b), and vanillin (Endo et al., 2008), and (3) ethanol (Fujita et al., 2006; van Voorst et al., 2006; Teixeira et al., 2009a; Yoshikawa et al., 2009), acetate (Mira et al., 2010b), and furfural (Gorsich et al., 2006). The effect of the deletion of these genes in VHG or biomass fermentation performance, respectively, was evaluated. The identified genes, including BUD31, HPR1, PHO85, VRP1, and YGL024w, found to contribute to improved performances in VHG, and ERG2, PRS3, RAV1, RPB4, and VMA8, required for improved performance in wheat straw hydrolysate fermentations, stand as preferential targets for genetic engineering in order to generate more robust industrial yeast strains, better suited for industrial bio-ethanol production (Pereira et al., 2011).
Functional genomics approaches applied to the simple single cell model S. cerevisiae have deeply modified the understanding of drug/chemical stress resistance and response mechanisms. The gathered data reinforces the idea that observations made in yeast are very likely to have a parallel in more complex eukaryotes, validating the use of this model organism in the context of toxicogenomics. The extrapolation of these results to higher eukaryotes, which has been so far carried out with relative success, is obviously of paramount importance.
This article emphasizes how yeast toxicogenomics enables a rapid and reproducible assessment of the mechanisms of toxicity of, and resistance to, many chemicals, requiring small amounts of growth medium and compound under testing, leading to reduced costs and reduced toxic wastes, effectively contributing to reduce, refine, and replace (3R) the use of animals in toxicological testing of pesticides (Gad, 1990). However, and in spite of these numerous advantages, yeast is not without its drawbacks. The simplicity of yeast is a disadvantage, since unicellularity is not conducive to study complex phenomena and cannot provide data on organ or tissue-specific toxicity. Moreover, studies in yeast do not provide accurate indicators for determination of toxic doses of a compound, since S. cerevisiae is usually much more tolerant to high doses of toxicants than higher eukaryotic cells. This is likely due to the barrier presented by the cell wall as well as the expression of numerous active efflux pumps and detoxification mechanisms that are highly abundant in yeast cells, who together make it virtually impossible to know the real concentration that is acting on the toxicant targets (Sá-Correia et al., 2009; Smith et al., 2010). Another important limitation is the possible absence of adequate molecular targets in yeast, since many cytotoxic compounds act in their target organisms via physiological mechanisms that do not exist in yeast (Foury, 1997; Hohmann and Mager, 1997; Parsons et al., 2003; Mager and Winderickx, 2005). Furthermore, the finding of homology between yeast and human genes does not necessarily imply that they are orthologs (Foury, 1997; Heinicke et al., 2007), demanding experimental verification of which (if any) is the true homolog of interest.
Nonetheless, the exploitation of high-throughput technologies and the global molecular analyses of the effects of drugs and other chemicals using the yeast model are revealing previously unsuspected on unknown molecular targets or adverse effects. It has proven a valuable first platform for the screening and prediction of the toxicological outcome of new or still unstudied drugs/chemicals and for the study of toxicity mechanisms. These first analyses are very much facilitated by the exploitation of the yeast model, given that signaling and regulatory pathways are highly conserved and may uncover the interactions of a chemical with its expected and unexpected gene/protein/metabolite targets. The use of yeast, together with other model organisms, and cross-species comparison of important genes/proteins in the toxicological response will facilitate the understanding of the response of an organism to toxic insults at a systems level. This strategy is expected to allow the description of all toxicological interactions occurring in a living system under chemical stress and the prediction of action of similar compounds in other species.
In conclusion, and based on the results reviewed in this paper, we believe that yeast stands out as an unavoidable and preferential system for toxicogenomics studies, through a combination of large-scale experimental approaches and expertise with biological and computational tools.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research carried out at IBB-IST and overviewed in this paper was financially supported by the EU (HERBICBIOREM project QLK3-CT-1999-00041) and by FEDER and “Fundação para a Ciência e a Tecnologia” (FCT, Portugal) – contracts POCTI/AGG/38110/2001, POCTI/AGR/45347/2002, POCTI/BME/46526/2002, POCTI/BIO/56838/2004, PTDC/SAU-FCF/71760/2006, PTDC/BIO/72063/2006, PTDC/AGR-AAM/67858/2006, PTDC/AGR-AAM/102967/2008, PTDC/AGR-ALI/102608/2008, and ERA-IB/0002/2010, and postdoctoral fellowships to Sandra C. dos Santos (SFRH/BPD/75483/2010) and Tânia R. Cabrito (SFRH/BPD/81221/2011).
Abbott, D. A., Zelle, R. M., Pronk, J. T., and Van Maris, A. J. (2009). Metabolic engineering of Saccharomyces cerevisiae for production of carboxylic acids: current status and challenges. FEMS Yeast Res. 9, 1123–1136.
Abdulrehman, D., Monteiro, P. T., Teixeira, M. C., Mira, N. P., Lourenco, A. B., dos Santos, S. C., Cabrito, T. R., Francisco, A. P., Madeira, S. C., Aires, R. S., Oliveira, A. L., Sa-Correia, I., and Freitas, A. T. (2011). YEASTRACT: providing a programmatic access to curated transcriptional regulatory associations in Saccharomyces cerevisiae through a web services interface. Nucleic Acids Res. 39, D136–D140.
Alenquer, M., Tenreiro, S., and Sá-Correia, I. (2006). Adaptive response to the antimalarial drug artesunate in yeast involves Pdr1p/Pdr3p-mediated transcriptional activation of the resistance determinants TPO1 and PDR5. FEMS Yeast Res. 6, 1130–1139.
Alexandre, H., Ansanay-Galeote, V., Dequin, S., and Blondin, B. (2001). Global gene expression during short-term ethanol stress in Saccharomyces cerevisiae. FEBS Lett. 498, 98–103.
Aouida, M., Pagé, N., Leduc, A., Peter, M., and Ramotar, D. (2004). A genome-wide screen in Saccharomyces cerevisiae reveals altered transport as a mechanism of resistance to the anticancer drug bleomycin. Cancer Res. 64, 1102–1109.
Auerbach, D., Arnoldo, A., Bogdan, B., and Fetchko, M. (2005). Drug discovery using yeast as a model system: a functional genomic and proteomic view. Curr. Proteomics 2, 1–13.
Bach, S., Talarek, N., Andrieu, T., Vierfond, J. M., Mettey, Y., Galons, H., Dormont, D., Meijer, L., Cullin, C., and Blondel, M. (2003). Isolation of drugs active against mammalian prions using a yeast-based screening assay. Nat. Biotechnol. 21, 1075–1081.
Ballantyne, B. (2004). “Toxicology in fungicides,” in Pesticide Toxicology and International Regulation, eds T. C. Marrs and B. Ballantyne (Chichester: John Wiley and Sons), 191–303.
Batova, M., Klobucnikova, V., Oblasova, Z., Gregan, J., Zahradnik, P., Hapala, I., Subik, J., and Schuller, C. (2010). Chemogenomic and transcriptome analysis identifies mode of action of the chemosensitizing agent CTBT (7-chlorotetrazolo[5,1-c]benzo[1,2,4]triazine). BMC Genomics 11, 153. doi:10.1186/1471-2164-11-153
Belpoggi, F., Soffritti, M., Guarino, M., Lambertini, L., Cevolani, D., and Maltoni, C. (2002). Results of long-term experimental studies on the carcinogenicity of ethylene-bis-dithiocarbamate (Mancozeb) in rats. Ann. N. Y. Acad. Sci. 982, 123–136.
Berkowitz, O., Jost, R., Pollmann, S., and Masle, J. (2008). Characterization of TCTP, the translationally controlled tumor protein, from Arabidopsis thaliana. Plant Cell 20, 3430–3447.
Bivi, N., Romanello, M., Harrison, R., Clarke, I., Hoyle, D. C., Moro, L., Ortolani, F., Bonetti, A., Quadrifoglio, F., Tell, G., and Delneri, D. (2009). Identification of secondary targets of N-containing bisphosphonates in mammalian cells via parallel competition analysis of the barcoded yeast deletion collection. Genome Biol. 10, R93.
Blackburn, A. S., and Avery, S. V. (2003). Genome-wide screening of Saccharomyces cerevisiae to identify genes required for antibiotic insusceptibility of eukaryotes. Antimicrob. Agents Chemother. 47, 676–681.
Bleackley, M. R., Young, B. P., Loewen, C. J., and Macgillivray, R. T. (2011). High density array screening to identify the genetic requirements for transition metal tolerance in Saccharomyces cerevisiae. Metallomics 3, 195–205.
Botstein, D., and Fink, G. R. (2011). Yeast: an experimental organism for 21st century biology. Genetics 189, 695–704.
Bradberry, S. M., Proudfoot, A. T., and Vale, J. A. (2004). Poisoning due to chlorophenoxy herbicides. Toxicol. Rev. 23, 65–73.
Bradberry, S. M., Watt, B. E., Proudfoot, A. T., and Vale, J. A. (2000). Mechanisms of toxicity, clinical features, and management of acute chlorophenoxy herbicide poisoning: a review. J. Toxicol. Clin. Toxicol. 38, 111–122.
Cabrito, T. R., Teixeira, M. C., Duarte, A. A., Duque, P., and Sa-Correia, I. (2009). Heterologous expression of a Tpo1 homolog from Arabidopsis thaliana confers resistance to the herbicide 2,4-D and other chemical stresses in yeast. Appl. Microbiol. Biotechnol. 84, 927–936.
Cabrito, T. R., Teixeira, M. C., Singh, A., Prasad, R., and Sa-Correia, I. (2011). The yeast ABC transporter Pdr18 (ORF YNR070w) controls plasma membrane sterol composition, playing a role in multidrug resistance. Biochem. J. 440, 195–202.
Calviello, G., Piccioni, E., Boninsegna, A., Tedesco, B., Maggiano, N., Serini, S., Wolf, F. I., and Palozza, P. (2006). DNA damage and apoptosis induction by the pesticide Mancozeb in rat cells: involvement of the oxidative mechanism. Toxicol. Appl. Pharmacol. 211, 87–96.
Chang, D. J., Lee, S. H., Lim, C. S., Jang, D. H., Lee, C. H., Lee, Y. D., and Kaang, B. K. (2004). Thiol oxidation-mediated cell death in Aplysia cultured sensory neurons. Brain Res. 1007, 71–77.
Chen, B.-J., Causton, H. C., Mancenido, D., Goddard, N. L., Perlstein, E. O., and Pe’er, D. (2010). Harnessing gene expression to identify the genetic basis of drug resistance. Mol. Syst. Biol. 5, 1–14.
Chen, Y., Feldman, D. E., Deng, C., Brown, J. A., De Giacomo, A. F., Gaw, A. F., Shi, G., Le, Q. T., Brown, J. M., and Koong, A. C. (2005). Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol. Cancer Res. 3, 669–677.
Chondrogianni, N., Tzavelas, C., Pemberton, A. J., Nezis, I. P., Rivett, A. J., and Gonos, E. S. (2005). Overexpression of proteasome beta5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem. 280, 11840–11850.
Costanzo, M., Baryshnikova, A., Bellay, J., Kim, Y., Spear, E. D., Sevier, C. S., Ding, H., Koh, J. L., Toufighi, K., Mostafavi, S., Prinz, J., St Onge, R. P., Vandersluis, B., Makhnevych, T., Vizeacoumar, F. J., Alizadeh, S., Bahr, S., Brost, R. L., Chen, Y., Cokol, M., Deshpande, R., Li, Z., Lin, Z. Y., Liang, W., Marback, M., Paw, J., San Luis, B. J., Shuteriqi, E., Tong, A. H., Van Dyk, N., Wallace, I. M., Whitney, J. A., Weirauch, M. T., Zhong, G., Zhu, H., Houry, W. A., Brudno, M., Ragibizadeh, S., Papp, B., Pal, C., Roth, F. P., Giaever, G., Nislow, C., Troyanskaya, O. G., Bussey, H., Bader, G. D., Gingras, A. C., Morris, Q. D., Kim, P. M., Kaiser, C. A., Myers, C. L., Andrews, B. J., and Boone, C. (2010). The genetic landscape of a cell. Science 327, 425–431.
Crespo, J. L., and Hall, M. N. (2002). Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 66, 579–591.
Cumming, R. C., Dargusch, R., Fischer, W. H., and Schubert, D. (2007). Increase in expression levels and resistance to sulfhydryl oxidation of peroxiredoxin isoforms in amyloid beta-resistant nerve cells. J. Biol. Chem. 282, 30523–30534.
Delling, U., Raymond, M., and Schurr, E. (1998). Identification of Saccharomyces cerevisiae genes conferring resistance to quinoline ring-containing antimalarial drugs. Antimicrob. Agents Chemother. 42, 1034–1041.
Devantier, R., Scheithauer, B., Villas-Boas, S. G., Pedersen, S., and Olsson, L. (2005). Metabolite profiling for analysis of yeast stress response during very high gravity ethanol fermentations. Biotechnol. Bioeng. 90, 703–714.
Dias, P. J., Teixeira, M. C., Telo, J. P., and Sá-Correia, I. (2010). Insights into the mechanisms of toxicity and tolerance to the agricultural fungicide mancozeb in yeast, as suggested by a chemogenomic approach. OMICS 14, 211–227.
Ding, M. Z., Cheng, J. S., Xiao, W. H., Qiao, B., and Yuan, Y. J. (2009). Comparative metabolomic analysis on industrial continuous and batch ethanol fermentation processes by GC-TOF-MS. Metabolomics 5, 229–238.
Domico, L. M., Cooper, K. R., Bernard, L. P., and Zeevalk, G. D. (2007). Reactive oxygen species generation by the ethylene-bis-dithiocarbamate (EBDC) fungicide mancozeb and its contribution to neuronal toxicity in mesencephalic cells. Neurotoxicology 28, 1079–1091.
Doostzadeh, J., Davis, R. W., Giaever, G. N., Nislow, C., and Langston, J. W. (2007). Chemical genomic profiling for identifying intracellular targets of toxicants producing Parkinson’s disease. Toxicol. Sci. 95, 182–187.
dos Santos, S. C., and Sá-Correia, I. (2009). Genome-wide identification of genes required for yeast growth under imatinib stress: vacuolar H+-ATPase function is an important target of this anticancer drug. OMICS 13, 185–198.
dos Santos, S. C., and Sá-Correia, I. (2011). A genome-wide screen identifies yeast genes required for protection against or enhanced cytotoxicity of the antimalarial drug quinine. Mol. Genet. Genomics 286, 333–346.
dos Santos, S. C., Tenreiro, S., Palma, M., Becker, J., and Sa-Correia, I. (2009). Transcriptomic profiling of the Saccharomyces cerevisiae response to quinine reveals a glucose limitation response attributable to drug-induced inhibition of glucose uptake. Antimicrob. Agents Chemother. 53, 5213–5223.
Emerson, L. R., Nau, M. E., Martin, R. K., Kyle, D. E., Vahey, M., and Wirth, D. F. (2002). Relationship between chloroquine toxicity and iron acquisition in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 46, 787–796.
Endo, A., Nakamura, T., Ando, A., Tokuyasu, K., and Shima, J. (2008). Genome-wide screening of the genes required for tolerance to vanillin, which is a potential inhibitor of bioethanol fermentation, in Saccharomyces cerevisiae. Biotechnol. Biofuels 1, 3.
Erasmus, D. J., Van Der Merwe, G. K., and Van Vuuren, H. J. (2003). Genome-wide expression analyses: metabolic adaptation of Saccharomyces cerevisiae to high sugar stress. FEMS Yeast Res. 3, 375–399.
Ericson, E., Gebbia, M., Heisler, L. E., Wildenhain, J., Tyers, M., Giaever, G., and Nislow, C. (2008). Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet. 4, e1000151. doi:10.1371/journal.pgen.1000151
Felder, T., Bogengruber, E., Tenreiro, S., Ellinger, A., Sá-Correia, I., and Briza, P. (2002). Dtrlp, a multidrug resistance transporter of the major facilitator superfamily, plays an essential role in spore wall maturation in Saccharomyces cerevisiae. Eukaryotic Cell 1, 799–810.
Fernandes, A. R., Durão, P. J., Santos, P. M., and Sá-Correia, I. (2003). Activation and significance of vacuolar H+-ATPase in Saccharomyces cerevisiae adaptation and resistance to the herbicide 2,4-dichlorophenoxyacetic acid. Biochem. Biophys. Res. Commun. 312, 1317–1324.
Ferrari, G., Pastorelli, R., Buchi, F., Spinelli, E., Gozzini, A., Bosi, A., and Santini, V. (2007). Comparative proteomic analysis of chronic myelogenous leukemia cells: inside the mechanism of imatinib resistance. J. Proteome Res. 6, 367–375.
Fitch, C. D. (2004). Ferriprotoporphyrin IX, phospholipids, and the antimalarial actions of quinoline drugs. Life Sci. 74, 1957–1972.
Fujita, K., Matsuyama, A., Kobayashi, Y., and Iwahashi, H. (2006). The genome-wide screening of yeast deletion mutants to identify the genes required for tolerance to ethanol and other alcohols. FEMS Yeast Res. 6, 744–750.
Gad, S. C. (1990). Recent developments in replacing, reducing, and refining animal use in toxicologic research and testing. Fundam. Appl. Toxicol. 15, 8–16.
Gammie, A. E., Erdeniz, N., Beaver, J., Devlin, B., Nanji, A., and Rose, M. D. (2007). Functional characterization of pathogenic human MSH2 missense mutations in Saccharomyces cerevisiae. Genetics 177, 707–721.
Giaever, G., Chu, A. M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., Arkin, A. P., Astromoff, A., El-Bakkoury, M., Bangham, R., Benito, R., Brachat, S., Campanaro, S., Curtiss, M., Davis, K., Deutschbauer, A., Entian, K. D., Flaherty, P., Foury, F., Garfinkel, D. J., Gerstein, M., Gotte, D., Guldener, U., Hegemann, J. H., Hempel, S., Herman, Z., Jaramillo, D. F., Kelly, D. E., Kelly, S. L., Kotter, P., Labonte, D., Lamb, D. C., Lan, N., Liang, H., Liao, H., Liu, L., Luo, C., Lussier, M., Mao, R., Menard, P., Ooi, S. L., Revuelta, J. L., Roberts, C. J., Rose, M., Ross-Macdonald, P., Scherens, B., Schimmack, G., Shafer, B., Shoemaker, D. D., Sookhai-Mahadeo, S., Storms, R. K., Strathern, J. N., Valle, G., Voet, M., Volckaert, G., Wang, C. Y., Ward, T. R., Wilhelmy, J., Winzeler, E. A., Yang, Y., Yen, G., Youngman, E., Yu, K., Bussey, H., Boeke, J. D., Snyder, M., Philippsen, P., Davis, R. W., and Johnston, M. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391.
Giaever, G., Flaherty, P., Kumm, J., Proctor, M., Nislow, C., Jaramillo, D. F., Chu, A. M., Jordan, M. I., Arkin, A. P., and Davis, R. W. (2004). Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. U.S.A. 101, 793–798.
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J. D., Jacq, C., Johnston, M., Louis, E. J., Mewes, H. W., Murakami, Y., Philippsen, P., Tettelin, H., and Oliver, S. G. (1996). Life with 6000 genes. Science 274, 546, 563–547.
Gorsich, S. W., Dien, B. S., Nichols, N. N., Slininger, P. J., Liu, Z. L., and Skory, C. D. (2006). Tolerance to furfural-induced stress is associated with pentose phosphate pathway genes ZWF1, GND1, RPE1, and TKL1 in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 71, 339–349.
Guerreiro, N., Staedtler, F., Grenet, O., Kehren, J., and Chibout, S. D. (2003). Toxicogenomics in drug development. Toxicol. Pathol. 31, 471–479.
Hamadeh, H. K., Amin, R. P., Paules, R. S., and Afshari, C. A. (2002). An overview of toxicogenomics. Curr. Issues Mol. Biol. 4, 45–56.
Hammonds, T. R., Maxwell, A., and Jenkins, J. R. (1998). Use of a rapid throughput in vivo screen to investigate inhibitors of eukaryotic topoisomerase II enzymes. Antimicrob. Agents Chemother. 42, 889–894.
Hasunuma, T., Sanda, T., Yamada, R., Yoshimura, K., Ishii, J., and Kondo, A. (2011). Metabolic pathway engineering based on metabolomics confers acetic and formic acid tolerance to a recombinant xylose-fermenting strain of Saccharomyces cerevisiae. Microb. Cell Fact. 10, 2.
Heinicke, S., Livstone, M. S., Lu, C., Oughtred, R., Kang, F., Angiuoli, S. V., White, O., Botstein, D., and Dolinski, K. (2007). The Princeton protein orthology database (P-POD): a comparative genomics analysis tool for biologists. PLoS ONE 2, e766. doi:10.1371/journal.pone.0000766
Hellauer, K., Lesage, G., Sdicu, A.-M., and Turcotte, B. (2005). Large-scale analysis of genes that alter sensitivity to the anticancer drug tirapazamine in Saccharomyces cerevisiae. Mol. Pharmacol. 68, 1365–1375.
Hillenmeyer, M. E., Fung, E., Wildenhain, J., Pierce, S. E., Hoon, S., Lee, W., Proctor, M., St Onge, R. P., Tyers, M., Koller, D., Altman, R. B., Davis, R. W., Nislow, C., and Giaever, G. (2008). The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365.
Hirasawa, T., Yoshikawa, K., Nakakura, Y., Nagahisa, K., Furusawa, C., Katakura, Y., Shimizu, H., and Shioya, S. (2007). Identification of target genes conferring ethanol stress tolerance to Saccharomyces cerevisiae based on DNA microarray data analysis. J. Biotechnol. 131, 34–44.
Hohmann, S., and Mager, W. H. (1997). “Stress response mechanisms in the yeast Saccharomyces cerevisiae,” in Yeast Stress Responses, eds S. Hohmann and W. H. Mager (Austin, TX: Molecular Biology Intelligence Unit, R.G. Landes Company), 1–5.
Holland, S., Lodwig, E., Sideri, T., Reader, T., Clarke, I., Gkargkas, K., Hoyle, D. C., Delneri, D., Oliver, S. G., and Avery, S. V. (2007). Application of the comprehensive set of heterozygous yeast deletion mutants to elucidate the molecular basis of cellular chromium toxicity. Genome Biol. 8, R268.
Hoon, S., Smith, A. M., Wallace, I. M., Suresh, S., Miranda, M., Fung, E., Proctor, M., Shokat, K. M., Zhang, C., Davis, R. W., Giaever, G., St Onge, R. P., and Nislow, C. (2008a). An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol. 4, 498–506.
Hoon, S., St Onge, R. P., Giaever, G., and Nislow, C. (2008b). Yeast chemical genomics and drug discovery: an update. Trends Pharmacol. Sci. 29, 499–504.
Huang, R.-Y., Eddy, M., Vujcic, M., and Kowalski, D. (2005). Genome-wide screen identifies genes whose inactivation confer resistance to cisplatin in Saccharomyces cerevisiae. Cancer Res. 65, 5890–5897.
Hughes, T. R., Marton, M. J., Jones, A. R., Roberts, C. J., Stoughton, R., Armour, C. D., Bennett, H. A., Coffey, E., Dai, H., He, Y. D., Kidd, M. J., King, A. M., Meyer, M. R., Slade, D., Lum, P. Y., Stepaniants, S. B., Shoemaker, D. D., Gachotte, D., Chakraburtty, K., Simon, J., Bard, M., and Friend, S. H. (2000). Functional discovery via a compendium of expression profiles. Cell 102, 109–126.
Ibrahim, M. A., Bond, G. G., Burke, T. A., Cole, P., Dost, F. N., Enterline, P. E., Gough, M., Greenberg, R. S., Halperin, W. E., Mcconnell, E., Munro, I. C., Swenberg, J. A., Zahm, S. H., and Graham, J. D. (1991). Weight of the evidence on the human carcinogenicity of 2,4-D. Environ. Health Perspect. 96, 213–222.
Ihara, Y., Nobukuni, K., Takata, H., and Hayabara, T. (2005). Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurol. Res. 27, 105–108.
Ito, H., and Gray, W. M. (2006). A gain-of-function mutation in the Arabidopsis pleiotropic drug resistance transporter PDR9 confers resistance to auxinic herbicides. Plant Physiol. 142, 63–74.
Jia, M. H., Larossa, R. A., Lee, J. M., Rafalski, A., Derose, E., Gonye, G., and Xue, Z. (2000). Global expression profiling of yeast treated with an inhibitor of amino acid biosynthesis, sulfometuron methyl. Physiol. Genomics 3, 83–92.
Jin, Y. H., Dunlap, P. E., Mcbride, S. J., Al-Refai, H., Bushel, P. R., and Freedman, J. H. (2008). Global transcriptome and deletome profiles of yeast exposed to transition metals. PLoS Genet. 4, e1000053. doi:10.1371/journal.pgen.1000053
Khozoie, C., Pleass, R. J., and Avery, S. V. (2009). The antimalarial drug quinine disrupts Tat2p-mediated tryptophan transport and causes tryptophan starvation. J. Biol. Chem. 284, 17968–17974.
Kitagawa, E., Momose, Y., and Iwahashi, H. (2003). Correlation of the structures of agricultural fungicides to gene expression in Saccharomyces cerevisiae upon exposure to toxic doses. Environ. Sci. Technol. 37, 2788–2793.
Kominsky, D. J., Klawitter, J., Brown, J. L., Boros, L. G., Melo, J. V., Eckhardt, S. G., and Serkova, N. J. (2009). Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin. Cancer Res. 15, 3442–3450.
Lamb, J., Crawford, E. D., Peck, D., Modell, J. W., Blat, I. C., Wrobel, M. J., Lerner, J., Brunet, J. P., Subramanian, A., Ross, K. N., Reich, M., Hieronymus, H., Wei, G., Armstrong, S. A., Haggarty, S. J., Clemons, P. A., Wei, R., Carr, S. A., Lander, E. S., and Golub, T. R. (2006). The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935.
Landstetter, N., Glaser, W., Gregori, C., Seipelt, J., and Kuchler, K. (2010). Functional genomics of drug-induced ion homeostasis identifies a novel regulatory crosstalk of iron and zinc regulons in yeast. OMICS 14, 651–663.
Li, B. Z., Cheng, J. S., Qiao, B., and Yuan, Y. J. (2010). Genome-wide transcriptional analysis of Saccharomyces cerevisiae during industrial bioethanol fermentation. J. Ind. Microbiol. Biotechnol. 37, 43–55.
Li, W., Mo, W., Shen, D., Sun, L., Wang, J., Lu, S., Gitschier, J. M., and Zhou, B. (2005). Yeast model uncovers dual roles of mitochondria in action of artemisinin. PLoS Genet. 1, e36. doi:10.1371/journal.pgen.0010036
Liao, C., Hu, B., Arno, M. J., and Panaretou, B. (2007). Genomic screening in vivo reveals the role played by vacuolar H+- ATPase and cytosolic acidification in sensitivity to DNA-damaging agents such as cisplatin. Mol. Pharmacol. 71, 416–425.
Lourenço, A. B., Ascenso, J. R., and Sá-Correia, I. (2011). Metabolic insights into the yeast response to propionic acid based on high resolution 1H NMR spectroscopy. Metabolomics 7, 457–468.
Lucau-Danila, A., Lelandais, G., Kozovska, Z., Tanty, V., Delaveau, T., Devaux, F., and Jacq, C. (2005). Early expression of yeast genes affected by chemical stress. Mol. Cell. Biol. 25, 1860–1868.
Lum, P. Y., Armour, C. D., Stepaniants, S. B., Cavet, G., Wolf, M. K., Butler, J. S., Hinshaw, J. C., Garnier, P., Prestwich, G. D., Leonardson, A., Garrett-Engele, P., Rush, C. M., Bard, M., Schimmack, G., Phillips, J. W., Roberts, C. J., and Shoemaker, D. D. (2004). Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 116, 121–137.
Mager, W. H., and Winderickx, J. (2005). Yeast as a model for medical and medicinal research. Trends Pharmacol. Sci. 26, 265–273.
Marjanovic, J., Chalupska, D., Patenode, C., Coster, A., Arnold, E., Ye, A., Anesi, G., Lu, Y., Okun, I., Tkachenko, S., Haselkorn, R., and Gornicki, P. (2010). Recombinant yeast screen for new inhibitors of human acetyl-CoA carboxylase 2 identifies potential drugs to treat obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 9093–9098.
Markovich, S., Yekutiel, A., Shalit, I., Shadkchan, Y., and Osherov, N. (2004). Genomic approach to identification of mutations affecting caspofungin susceptibility in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 48, 3871–3876.
Marks, V. D., Ho Sui, S. J., Erasmus, D., Van Der Merwe, G. K., Brumm, J., Wasserman, W. W., Bryan, J., and Van Vuuren, H. J. (2008). Dynamics of the yeast transcriptome during wine fermentation reveals a novel fermentation stress response. FEMS Yeast Res. 8, 35–52.
Maroni, M., Colosio, C., Ferioli, A., and Fait, A. (2000). Biological monitoring of pesticide exposure: a review. Toxicology 143, 1–118.
Martin, D., Brun, C., Remy, E., Mouren, P., Thieffry, D., and Jacq, B. (2004). GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol. 5, R101.
Marton, M. J., Derisi, J. L., Bennett, H. A., Iyer, V. R., Meyer, M. R., Roberts, C. J., Stoughton, R., Burchard, J., Slade, D., Dai, H., Bassett, D. E. Jr., Hartwell, L. H., Brown, P. O., and Friend, S. H. (1998). Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat. Med. 4, 1293–1301.
Menacho-Marquez, M., and Murguia, J. R. (2007). Yeast on drugs: Saccharomyces cerevisiae as a tool for anticancer drug research. Clin. Transl. Oncol. 9, 221–228.
Mettetal, J. T., Muzzey, D., Gomez-Uribe, C., and Van Oudenaarden, A. (2008). The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science 319, 482–484.
Mira, N. P., Becker, J. D., and Sa-Correia, I. (2010a). Genomic expression program involving the Haa1p-regulon in Saccharomyces cerevisiae response to acetic acid. OMICS 14, 587–601.
Mira, N. P., Palma, M., Guerreiro, J. F., and Sa-Correia, I. (2010b). Genome-wide identification of Saccharomyces cerevisiae genes required for tolerance to acetic acid. Microb. Cell Fact. 9, 79.
Mira, N. P., Lourenço, A. B., Fernandes, A. R., Becker, J. D., and Sá-Correia, I. (2009). The RIM101 pathway has a role in Saccharomyces cerevisiae adaptive response and resistance to propionic acid and other weak acids. FEMS Yeast Res. 9, 202–216.
Miyamoto, M., Furuichi, Y., and Komiyama, T. (2011). Genome-wide screen of Saccharomyces cerevisiae for killer toxin HM-1 resistance. Yeast 28, 27–41.
Mollapour, M., Fong, D., Balakrishnan, K., Harris, N., Thompson, S., Schuller, C., Kuchler, K., and Piper, P. W. (2004). Screening the yeast deletant mutant collection for hypersensitivity and hyper-resistance to sorbate, a weak organic acid food preservative. Yeast 21, 927–946.
Momose, Y., and Iwahashi, H. (2001). Bioassay of cadmium using a DNA microarray: genome-wide expression patterns of Saccharomyces cerevisiae response to cadmium. Environ. Toxicol. Chem. 20, 2353–2360.
Monteiro, P. T., Dias, P. J., Ropers, D., Oliveira, A. L., Sá-Correia, I., Teixeira, M. C., and Freitas, A. T. (2011). Qualitative modeling and formal verification of the FLR1 gen mancozeb response in Saccharomyces cerevisiae. IET Syst. Biol. 5, 308–316.
Morton, C. O., dos Santos, S. C., and Coote, P. (2007). An amphibian-derived, cationic, alpha-helical antimicrobial peptide kills yeast by caspase-independent but AIF-dependent programmed cell death. Mol. Microbiol. 65, 494–507.
Murakami, T., Shibuya, I., Ise, T., Chen, Z. S., Akiyama, S., Nakagawa, M., Izumi, H., Nakamura, T., Matsuo, K., Yamada, Y., and Kohno, K. (2001). Elevated expression of vacuolar proton pump genes and cellular PH in cisplatin resistance. Int. J. Cancer 93, 869–874.
Nicholson, J. K., Connelly, J., Lindon, J. C., and Holmes, E. (2002). Metabonomics: a platform for studying drug toxicity and gene function. Nat. Rev. Drug Discov. 1, 153–161.
North, M., and Vulpe, C. D. (2010). Functional toxicogenomics: mechanism-centered toxicology. Int. J. Mol. Sci. 11, 4796–4813.
Nowicki, M. O., Pawlowski, P., Fischer, T., Hess, G., Pawlowski, T., and Skorski, T. (2003). Chronic myelogenous leukemia molecular signature. Oncogene 22, 3952–3963.
Nunes, P. A., Tenreiro, S., and Sá-Correia, I. (2001). Resistance and adaptation to quinidine in Saccharomyces cerevisiae: role of QDR1 (YIL120w), encoding a plasma membrane transporter of the major facilitator superfamily required for multidrug resistance. Antimicrob. Agents Chemother. 45, 1528–1534.
Outeiro, T. F., and Lindquist, S. (2003). Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775.
Papaefthimiou, C., Cabral Mde, G., Mixailidou, C., Viegas, C. A., Sá-Correia, I., and Theophilidis, G. (2004). Comparison of two screening bioassays, based on the frog sciatic nerve and yeast cells, for the assessment of herbicide toxicity. Environ. Toxicol. Chem. 23, 1211–1218.
Parsons, A. B., Brost, R. L., Ding, H., Li, Z., Zhang, C., Sheikh, B., Brown, G. W., Kane, P. M., Hughes, T. R., and Boone, C. (2004). Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotechnol. 22, 62–69.
Parsons, A. B., Geyer, R., Hughes, T. R., and Boone, C. (2003). Yeast genomics and proteomics in drug discovery and target validation. Prog. Cell Cycle Res. 5, 159–166.
Parsons, A. B., Lopez, A., Givoni, I. E., Williams, D. E., Gray, C. A., Porter, J., Chua, G., Sopko, R., Brost, R. L., Ho, C. H., Wang, J., Ketela, T., Brenner, C., Brill, J. A., Fernandez, G. E., Lorenz, T. C., Payne, G. S., Ishihara, S., Ohya, Y., Andrews, B., Hughes, T. R., Frey, B. J., Graham, T. R., Andersen, R. J., and Boone, C. (2006). Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126, 611–625.
Parveen, M., Momose, Y., Kitagawa, E., Kurita, S., Kodama, O., and Iwahashi, H. (2003). Bioassay of pesticide Lindane using yeast-DNA microarrays technology. Chem-Bio Informatics J. 3, 12–29.
Pereira, F. B., Guimaraes, P. M., Gomes, D. G., Mira, N. P., Teixeira, M. C., Sa-Correia, I., and Domingues, L. (2011). Identification of candidate genes for yeast engineering to improve bioethanol production in very-high-gravity and lignocellulosic biomass industrial fermentations. Biotechnol. Biofuels 4, 57.
Perlstein, E. O., Ruderfer, D. M., Roberts, D. C., Schreiber, S. L., and Kruglyak, L. (2007). Genetic basis of individual differences in the response to small-molecule drugs in yeast. Nat. Genet. 39, 496–502.
Pham, T. K., Chong, P. K., Gan, C. S., and Wright, P. C. (2006). Proteomic analysis of Saccharomyces cerevisiae under high gravity fermentation conditions. J. Proteome Res. 5, 3411–3419.
Pocaly, M., Lagarde, V., Etienne, G., Dupouy, M., Lapaillerie, D., Claverol, S., Vilain, S., Bonneu, M., Turcq, B., Mahon, F.-X., and Pasquet, J.-M. (2008). Proteomic analysis of an imatinib-resistant K562 cell line highlights opposing roles of heat shock cognate 70 and heat shock 70 proteins in resistance. Proteomics 8, 2394–2406.
Quintás-Cardama, A., Kantarjian, H. M., and Cortes, J. E. (2009). Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control 16, 122–131.
Ruderfer, D. M., Roberts, D. C., Schreiber, S. L., Perlstein, E. O., and Kruglyak, L. (2009). Using expression and genotype to predict drug response in yeast. PLoS ONE 4, e6907. doi:10.1371/journal.pone.0006907
Ruotolo, R., Marchini, G., and Ottonello, S. (2008). Membrane transporters and protein traffic networks differentially affecting metal tolerance: a genomic phenotyping study in yeast. Genome Biol. 9, R67.
Sá-Correia, I., dos Santos, S. C., Teixeira, M. C., Cabrito, T. R., and Mira, N. P. (2009). Drug:H+ antiporters in chemical stress response in yeast. Trends Microbiol. 17, 22–31.
Sa-Correia, I., and Teixeira, M. C. (2010). 2D electrophoresis-based expression proteomics: a microbiologist’s perspective. Expert Rev. Proteomics 7, 943–953.
Saliba, K. J., Krishna, S., and Kirk, K. (2004). Inhibition of hexose transport and abrogation of pH homeostasis in the intraerythrocytic malaria parasite by an O-3-hexose derivative. FEBS Lett. 570, 93–96.
Sanchez, C. P., Stein, W. D., and Lanzer, M. (2008). Dissecting the components of quinine accumulation in Plasmodium falciparum. Mol. Microbiol. 67, 1081–1093.
Santos, P. M., Simões, T., and Sá-Correia, I. (2009). Insights into yeast adaptive response to the agricultural fungicide mancozeb: a toxicoproteomics approach. Proteomics 9, 657–670.
Schenk, P. W., Boersma, A. W., Brandsma, J. A., Den Dulk, H., Burger, H., Stoter, G., Brouwer, J., and Nooter, K. (2001). SKY1 is involved in cisplatin-induced cell kill in Saccharomyces cerevisiae, and inactivation of its human homologue, SRPK1, induces cisplatin resistance in a human ovarian carcinoma cell line. Cancer Res. 61, 6982–6986.
Schenk, P. W., Boersma, A. W. M., Brok, M., Burger, H., Stoter, G., and Nooter, K. (2002). Inactivation of the Saccharomyces cerevisiae SKY1 gene induces a specific modification of the yeast anticancer drug sensitivity profile accompanied by a mutator phenotype. Mol. Pharmacol. 61, 659–666.
Scherens, B., and Goffeau, A. (2004). The uses of genome-wide yeast mutant collections. Genome Biol. 5, 229.
Schüller, C., Mamnun, Y. M., Mollapour, M., Krapf, G., Schuster, M., Bauer, B. E., Piper, P. W., and Kuchler, K. (2004). Global phenotypic analysis and transcriptional profiling defines the weak acid stress response regulon in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 706–720.
Schwartz, D. A., Freedman, J. H., and Linney, E. A. (2004). Environmental genomics: a key to understanding biology, pathophysiology and disease. Hum. Mol. Genet. 13, R217–R224.
Sennoune, S. R., Bakunts, K., Martínez, G. M., Chua-Tuan, J. L., Kebir, Y., Attaya, M. N., and Martínez-Zaguilán, R. (2004). Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am. J. Physiol. Cell Physiol. 286, C1443–C1452.
Serero, A., Lopes, J., Nicolas, A., and Boiteux, S. (2008). Yeast genes involved in cadmium tolerance: identification of DNA replication as a target of cadmium toxicity. DNA Repair (Amst.) 7, 1262–1275.
Shaulian, E., and Karin, M. (2002). AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136.
Shen, H., Chen, J., Wang, Z., Yang, C., Sasaki, T., Yamamoto, Y., Matsumoto, H., and Yan, X. (2006). Root plasma membrane H+-ATPase is involved in the adaptation of soybean to phosphorus starvation. J. Exp. Bot. 57, 1353–1362.
Simmons, P. T., and Portier, C. J. (2002). Toxicogenomics: the new frontier in risk analysis. Carcinogenesis 23, 903–905.
Simões, T., Teixeira, M. C., Fernandes, A. R., and Sá-Correia, I. (2003). Adaptation of Saccharomyces cerevisiae to the herbicide 2,4-dichlorophenoxyacetic acid, mediated by Msn2p- and Msn4p-regulated genes: important role of SPI1. Appl. Environ. Microbiol. 69, 4019–4028.
Simon, J. A., and Bedalov, A. (2004). Yeast as a model system for anticancer drug discovery. Nat. Rev. Cancer 4, 481–492.
Sletta, H., Klinkenberg, G., Winnberg, A., Kvitvang, H. F., Nilsen, M. B., Krokan, H. E., Otterlei, M., and Bruheim, P. (2011). A new high resolution screening method for study of phenotype stress responses of Saccharomyces cerevisiae mutants. J. Microbiol. Methods 87, 363–367.
Smart, C. C., and Fleming, A. J. (1996). Hormonal and environmental regulation of a plant PDR5-like ABC transporter. J. Biol. Chem. 271, 19351–19357.
Smith, A. M., Ammar, R., Nislow, C., and Giaever, G. (2010). A survey of yeast genomic assays for drug and target discovery. Pharmacol. Ther. 127, 156–164.
Stanley, D., Chambers, P. J., Stanley, G. A., Borneman, A., and Fraser, S. (2010). Transcriptional changes associated with ethanol tolerance in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 88, 231–239.
Steinmetz, L. M., Scharfe, C., Deutschbauer, A. M., Mokranjac, D., Herman, Z. S., Jones, T., Chu, A. M., Giaever, G., Prokisch, H., Oefner, P. J., and Davis, R. W. (2002). Systematic screen for human disease genes in yeast. Nat. Genet. 31, 400–404.
Swen, J. J., Huizinga, T. W., Gelderblom, H., De Vries, E. G. E., Assendelft, W. J. J., Kirchheiner, J., and Guchelaar, H.-J. (2007). Translating pharmacogenomics: challenges on the road to the clinic. PLoS Med. 4, e209. doi:10.1371/journal.pmed.0040209
Takumi, S., Kimura, H., Matsusaki, H., Kawazoe, S., Tominaga, N., and Arizono, K. (2010). DNA microarray analysis of genomic responses of yeast Saccharomyces cerevisiae to nickel chloride. J. Toxicol. Sci. 35, 125–129.
Teixeira, M. C., Dias, P. J., Monteiro, P. T., Sala, A., Oliveira, A. L., Freitas, A. T., and Sá-Correia, I. (2010a). Refining current knowledge on the yeast FLR1 regulatory network by combined experimental and computational approaches. Mol. Biosyst. 6, 2471–2481.
Teixeira, M. C., Raposo, L. R., Palma, M., and Sa-Correia, I. (2010b). Identification of genes required for maximal tolerance to high-glucose concentrations, as those present in industrial alcoholic fermentation media, through a chemogenomics approach. OMICS 14, 201–210.
Teixeira, M. C., Duque, P., and Sá-Correia, I. (2007). Environmental genomics: mechanistic insights into toxicity of and resistance to the herbicide 2,4-D. Trends Biotechnol. 25, 363–370.
Teixeira, M. C., Fernandes, A. R., Mira, N. P., Becker, J. D., and Sá-Correia, I. (2006a). Early transcriptional response of Saccharomyces cerevisiae to stress imposed by the herbicide 2,4-dichlorophenoxyacetic acid. FEMS Yeast Res. 6, 230–248.
Teixeira, M. C., Monteiro, P., Jain, P., Tenreiro, S., Fernandes, A. R., Mira, N. P., Alenquer, M., Freitas, A. T., Oliveira, A. L., and Sá-Correia, I. (2006b). The YEASTRACT database: a tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 34, D446–D451.
Teixeira, M. C., Mira, N. P., and Sa-Correia, I. (2011). A genome-wide perspective on the response and tolerance to food-relevant stresses in Saccharomyces cerevisiae. Curr. Opin. Biotechnol. 22, 150–156.
Teixeira, M. C., Raposo, L. R., Mira, N. P., Lourenço, A. B., and Sá-Correia, I. (2009a). Genome-wide identification of Saccharomyces cerevisiae genes required for maximal tolerance to ethanol. Appl. Environ. Microbiol. 75, 5761–5772.
Teixeira, M. C., Santos, P. M., Rodrigues, C., and Sa-Correia, I. (2009b). Teaching expression proteomics: from the wet-lab to the laptop. Biochem. Mol. Biol. Educ. 37, 279–286.
Teixeira, M. C., and Sá-Correia, I. (2002). Saccharomyces cerevisiae resistance to chlorinated phenoxyacetic acid herbicides involves Pdr1p-mediated transcriptional activation of TPO1 and PDR5 genes. Biochem. Biophys. Res. Commun. 292, 530–537.
Teixeira, M. C., Santos, P. M., Fernandes, A. R., and Sá-Correia, I. (2005). A proteome analysis of the yeast response to the herbicide 2,4-dichlorophenoxyacetic acid. Proteomics 5, 1889–1901.
Teixeira, M. C., Telo, J. P., Duarte, N. F., and Sá-Correia, I. (2004). The herbicide 2,4-dichlorophenoxyacetic acid induces the generation of free-radicals and associated oxidative stress responses in yeast. Biochem. Biophys. Res. Commun. 324, 1101–1107.
Tenreiro, S., Nunes, P. A., Viegas, C. A., Neves, M. S., Teixeira, M. C., Cabral, M. G., and Sá-Correia, I. (2002). AQR1 gene (ORF YNL065w) encodes a plasma membrane transporter of the major facilitator superfamily that confers resistance to short-chain monocarboxylic acids and quinidine in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 292, 741–748.
Thorpe, G. W., Fong, C. S., Alic, N., Higgins, V. J., and Dawes, I. W. (2004). Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc. Natl. Acad. Sci. U.S.A. 101, 6564–6569.
Tribouillard, D., Bach, S., Gug, F., Desban, N., Beringue, V., Andrieu, T., Dormont, D., Galons, H., Laude, H., Vilette, D., and Blondel, M. (2006). Using budding yeast to screen for anti-prion drugs. Biotechnol. J. 1, 58–67.
Tucker, C. L., and Fields, S. (2004). Quantitative genome-wide analysis of yeast deletion strain sensitivities to oxidative and chemical stress. Comp. Funct. Genomics 5, 216–224.
van Maris, A. J., Abbott, D. A., Bellissimi, E., Van Den Brink, J., Kuyper, M., Luttik, M. A., Wisselink, H. W., Scheffers, W. A., Van Dijken, J. P., and Pronk, J. T. (2006). Alcoholic fermentation of carbon sources in biomass hydrolysates by Saccharomyces cerevisiae: current status. Antonie Van Leeuwenhoek 90, 391–418.
van Voorst, F., Houghton-Larsen, J., Jonson, L., Kielland-Brandt, M. C., and Brandt, A. (2006). Genome-wide identification of genes required for growth of Saccharomyces cerevisiae under ethanol stress. Yeast 23, 351–359.
Vargas, R. C., Tenreiro, S., Teixeira, M. C., Fernandes, A. R., and Sá-Correia, I. (2004). Saccharomyces cerevisiae multidrug transporter Qdr2p (Yil121wp): localization and function as a quinidine resistance determinant. Antimicrob. Agents Chemother. 48, 2531–2537.
Volpe, G., Panuzzo, C., Ulisciani, S., and Cilloni, D. (2009). Imatinib resistance in CML. Cancer Lett. 274, 1–9.
Wang, H. P., Long, X. H., Sun, Z. Z., Rigaud, O., Xu, Q. Z., Huang, Y. C., Sui, J. L., Bai, B., and Zhou, P. K. (2006a). Identification of differentially transcribed genes in human lymphoblastoid cells irradiated with 0.5 Gy of gamma-ray and the involvement of low dose radiation inducible CHD6 gene in cell proliferation and radiosensitivity. Int. J. Radiat. Biol. 82, 181–190.
Wang, R. A., Zhang, H., Balasenthil, S., Medina, D., and Kumar, R. (2006b). PAK1 hyperactivation is sufficient for mammary gland tumor formation. Oncogene 25, 2931–2936.
Wang, L., and Weinshilboum, R. M. (2008). Pharmacogenomics: candidate gene identification, functional validation and mechanisms. Hum. Mol. Genet. 17, R174–R179.
West, G. M., Tucker, C. L., Xu, T., Park, S. K., Han, X., Yates, J. R. III, and Fitzgerald, M. C. (2010). Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci. U.S.A. 107, 9078–9082.
Willingham, S., Outeiro, T. F., Devit, M. J., Lindquist, S. L., and Muchowski, P. J. (2003). Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 302, 1769–1772.
Woodrow, C. J., Penny, J. I., and Krishna, S. (1999). Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J. Biol. Chem. 274, 7272–7277.
Wu, H. I., Brown, J. A., Dorie, M. J., Lazzeroni, L., and Brown, J. M. (2004). Genome-wide identification of genes conferring resistance to the anticancer agents cisplatin, oxaliplatin, and mitomycin C. Cancer Res. 64, 3940–3948.
Wuster, A., and Madan Babu, M. (2008). Chemogenomics and biotechnology. Trends Biotechnol. 26, 252–258.
Yasokawa, D., Murata, S., Kitagawa, E., Iwahashi, Y., Nakagawa, R., Hashido, T., and Iwahashi, H. (2008). Mechanisms of copper toxicity in Saccharomyces cerevisiae determined by microarray analysis. Environ. Toxicol. 23, 599–606.
Yoshikawa, K., Tanaka, T., Furusawa, C., Nagahisa, K., Hirasawa, T., and Shimizu, H. (2009). Comprehensive phenotypic analysis for identification of genes affecting growth under ethanol stress in Saccharomyces cerevisiae. FEMS Yeast Res. 9, 32–44.
Yuen, K. W. Y., Warren, C. D., Chen, O., Kwok, T., Hieter, P., and Spencer, F. A. (2007). Systematic genome instability screens in yeast and their potential relevance to cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 3925–3930.
Zhang, L., Mchale, C. M., Rothman, N., Li, G., Ji, Z., Vermeulen, R., Hubbard, A. E., Ren, X., Shen, M., Rappaport, S. M., North, M., Skibola, C. F., Yin, S., Vulpe, C., Chanock, S. J., Smith, M. T., and Lan, Q. (2010). Systems biology of human benzene exposure. Chem. Biol. Interact. 184, 86–93.
Keywords: yeast model, toxicogenomics, molecular systems biology, genome-wide approaches, response to stress, toxicity mechanisms, predictive toxicology
Citation: dos Santos SC, Teixeira MC, Cabrito TR and Sá-Correia I (2012) Yeast toxicogenomics: genome-wide responses to chemical stresses with impact in environmental health, pharmacology, and biotechnology. Front. Gene. 3:63. doi: 10.3389/fgene.2012.00063
Received: 16 February 2012; Paper pending published: 09 March 2012;
Accepted: 03 April 2012; Published online: 19 April 2012.
Edited by:
Stephen Sturzenbaum, King’s College London, UKReviewed by:
Ralph Menzel, Humboldt-Universität zu Berlin, GermanyCopyright: © 2012 dos Santos, Teixeira, Cabrito and Sá-Correia. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Isabel Sá-Correia, Institute for Biotechnology and Bioengineering, Centre for Biological and Chemical Engineering, Department of Bioengineering, Instituto Superior Técnico, Technical University of Lisbon, Av. Rovisco Pais, 1049-001 Lisbon, Portugal. e-mail:aXNhY29ycmVpYUBpc3QudXRsLnB0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.