 Alvine C. Mehinto1
Alvine C. Mehinto1


95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 25 April 2012
Sec. Toxicogenomics
Volume 3 - 2012 | https://doi.org/10.3389/fgene.2012.00062
This article is part of the Research Topic Toxicogenomics in non-mammalian species View all 11 articles
The new technologies for next-generation sequencing (NGS) and global gene expression analyses that are widely used in molecular medicine are increasingly applied to the field of fish biology. This has facilitated new directions to address research areas that could not be previously considered due to the lack of molecular information for ecologically relevant species. Over the past decade, the cost of NGS has decreased significantly, making it possible to use non-model fish species to investigate emerging environmental issues. NGS technologies have permitted researchers to obtain large amounts of raw data in short periods of time. There have also been significant improvements in bioinformatics to assemble the sequences and annotate the genes, thus facilitating the management of these large datasets.The combination of DNA sequencing and bioinformatics has improved our abilities to design custom microarrays and study the genome and transcriptome of a wide variety of organisms. Despite the promising results obtained using these techniques in fish studies, NGS technologies are currently underused in ecotoxicogenomics and few studies have employed these methods. These issues should be addressed in order to exploit the full potential of NGS in ecotoxicological studies and expand our understanding of the biology of non-model organisms.
Research in fish physiology, genetics, evolution, immunology, and endocrinology using non-model species has seen a marked increase in the utilization of genomic information over the last decade. Traditionally, obtaining genomic information was achieved through Sanger sequencing methods which utilizes fluorescent dye-labeled dideoxynucleotide triphosphates as DNA chain terminators. However, Sanger sequencing is limiting because of the high cost and labor intensity. The development of next-generation sequencing (NGS) technologies has facilitated the collection of large amounts of nucleotide information in sequence read-length from 30 to 1,500 nucleotides (nt) for hundreds of thousands to millions of DNA molecules simultaneously. In parallel, the bioinformatics tools required to analyze these large datasets and identify unique gene sequences have also significantly improved. The different steps involved in NGS studies are illustrated in Figure 1. NGS technologies are already considered revolutionary tools in the fields of eukaryotic microorganism (Nowrousian, 2010), plant (Bräutigam and Gowik, 2010), animal, and human genomics (Pareek et al., 2011) and their application has demonstrated great potential to study genome evolution (Holt et al., 2008), gene expression profiling (Wang et al., 2008), and gene regulation (e.g., DNA methylation; Pomraning et al., 2009). With regard to fish studies, the number of publications using NGS technologies has increased approximately 10-fold in the last 3 years (Figure 2).
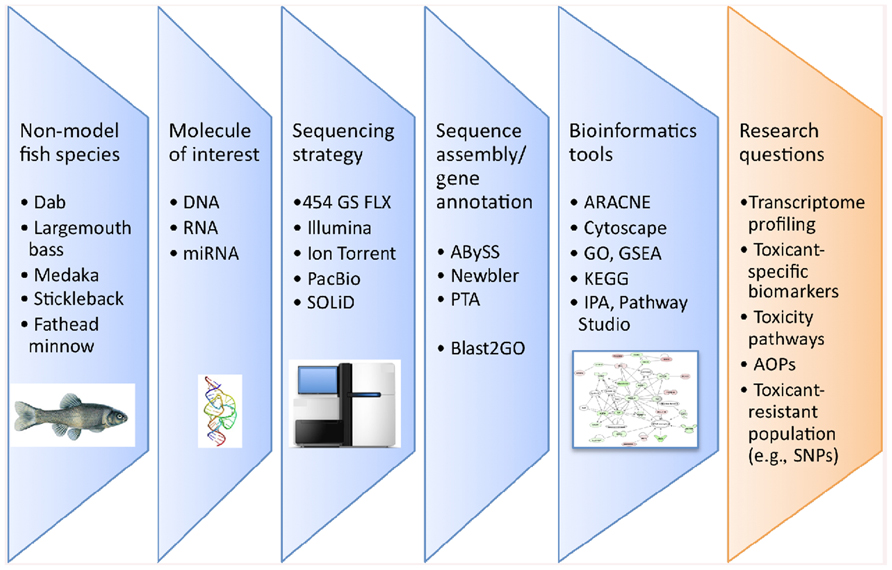
FIGURE 1. Flowchart of the different steps involved in NGS based studies in fish ecotoxicology. AOPs, adverse outcome pathways; GO, gene ontology; GSEA, gene set enrichment analysis; IPA, ingenuity pathway analysis; PTA, paracel transcript assembler; SNPs, single nucleotide polymorphism.
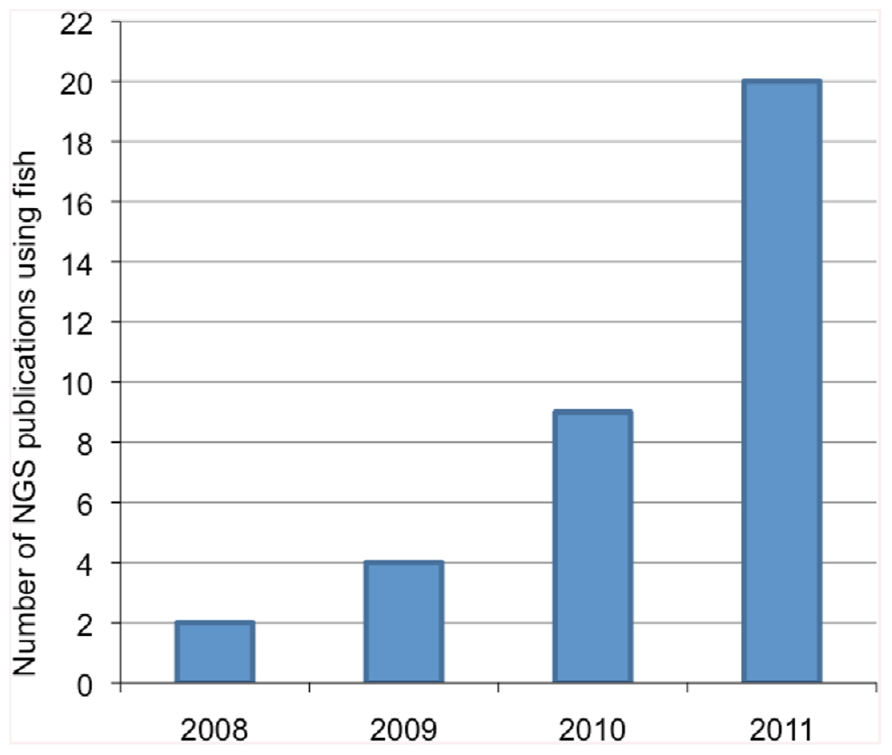
FIGURE 2. Number of publications using next-generation sequencing (NGS) with non-model fish species in the last 4 years. Keywords used for PubMed query include: fish, next-gen sequencing, high throughput sequencing, and toxicology.
Researchers in fish biology stand to gain a great deal of insight using NGS to learn more about genome-wide and transcriptome-wide control of biological processes, discover novel biomarkers for ecotoxicological applications, characterize toxicity pathways, and investigate evolutionary questions to a greater degree of resolution than previously provided by using more traditional population genetic markers such as DNA microsatellites. In ecotoxicogenomics, gene expression profiling using techniques such as microarrays plays a key role for biomarkers characterization and discovery of toxicity pathways (Denslow et al., 2007; Ju et al., 2007). But research in this field often requires the analysis of complex genomic events using extensive time course and dose response studies in multiple tissues of teleost fish, which can be difficult due to logistics and cost. Fortunately the cost of sequencing is now decreasing, permitting the analysis of many biological replicates (i.e., multiple individual genomes) in a single study. The application of NGS technologies will permit to better link knowledge of individual genotype to phenotype and transcriptomic responses under varying environmental conditions and experimental paradigms.
This review describes the latest NGS platforms available and bioinformatics tools that can be employed to examine the transcriptome of non-model fish species. Specifically, we aim to discuss the possible factors involved in platform selection for researchers working with non-model fish species. Studies that have utilized NGS technologies using fish species are also reviewed with the conclusion that NGS data can contribute significantly to our understanding of the detrimental effects of aquatic pollution.
There are five leading instruments that can be classified as part of the NGS technologies: the 454 GS FLX, the Ion Torrent, the SOLiD, the Illumina, and the more recently released PacBio instrument. These can be distinguished from each other based on the chemistry employed for sequencing, the amount of sequence information produced, the length of each sequence read, and the overall price per nt. While next-generation sequencers are reviewed for general purposes elsewhere (e.g., Mardis, 2008), we provide a brief description of the various technologies followed by a discussion of the relative advantages of each platform for fish toxicogenomics research.
The 454 pyrosequencer, manufactured by Roche1, is the NGS instrument most utilized in fish genomics research (Table 1). This platform operates on a principle referred to as “pyrosequencing,” a method of detecting single nucleotide addition by capturing the emission of light produced from the release of the by-product pyrophosphate during the polymerization of the DNA molecule (Droege and Hill, 2008; Rothberg and Leamon, 2008). During 454 sequencing, DNA is fragmented and ligated to sepharose beads with one DNA fragment per bead, optimally. This DNA library is then amplified using a process called emulsion PCR (emPCR), producing many copies of a unique single-stranded template on each bead. Following amplification, a single DNA bead and enzyme beads (sulfurylase, luciferase) are deposited in each well of a picotiter plate where as many as one million sequencing reactions – one per bead – occur in parallel. For each nucleotide added during the polymerization reaction, inorganic pyrophosphate and proton by-products are released, which interact with the luciferase to produce a pulse of light that is read by a high-density camera.
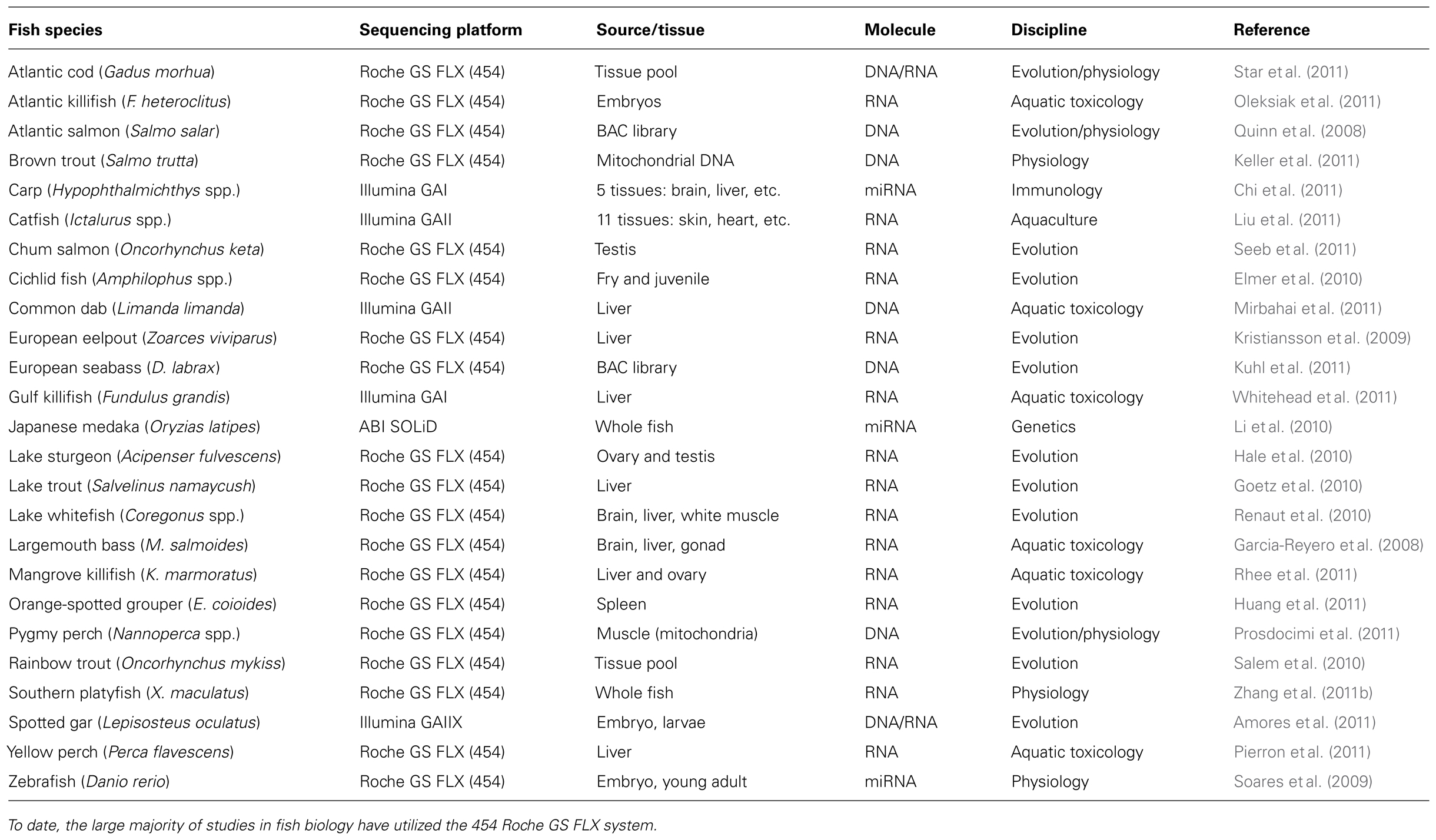
Table 1. Some examples of next-generation sequencing studies in fish.
The Ion Torrent is a modified version of the 454 pyrosequencing approach and operates based on the same sequencing chemistry, except that it makes use of the H+ that is released with every nucleotide incorporated, instead of the pyrophosphate (Rothberg et al., 2011). To detect the H+ released, the picotiter plate sits on top of a massively parallel semiconductor-sensing device or ion chip. The integrated circuits take advantage of metal-oxide semiconductor technology, which significantly reduces the cost of sequencing since luciferase and other costly enzymes and scanners are not needed. To date, this instrument can sequence about 100 nt but it should soon be able to read sequence lengths up to 200 nt (Ion Torrent System, Inc.2). Modifications of this technology to increase the length of the sequences produced will likely increase the use of the Ion Torrent in fish ecotoxicology.
The SOLiD genome sequencer from Applied Biosystems uses an emPCR process similar to 454, but parallel DNA sequencing is achieved by repeatedly ligating two-nucleotide probes instead of a sequencing reaction catalyzed by DNA polymerase (Morozova and Marra, 2008). The two-nucleotide probes are used to query adjacent bases on the DNA fragment, therefore each nucleotide is actually probed twice. This system is designed to make sequence calls on two signals per base, rather than one, resulting in a lower error rate (for more information on this process, see Morozova and Marra, 2008; Rusk and Kiermer, 2008). Originally, SOLiD technology could only read approximately 35 nucleotides (Morozova and Marra, 2008), but current versions of the instrument have increased the read-length to about 50 nucleotides (Applied Biosystems3).
The Illumina/Solexa technology is the second most utilized in fish genomics research (Table 1). This sequencing platform differs from 454 and SOLiD in terms of its amplification strategy. Rather than amplifying DNA-covered beads by emPCR, the Illumina technology amplifies clusters of DNA fragments that are affixed to a glass slide using a strategy called bridge amplification. The parallel sequencing process uses dye-labeled nucleotides (one fluorophore per base) that are added simultaneously, rather than sequentially as in the 454 process. The DNA clusters are then subjected to laser excitation that cleaves the dye and permits the addition of the next nucleotide. In 2008, Illumina sequencer projects reported reads of 25–50 nt. Base-calling algorithms have been improving to increase read-length and base-calling confidence (Rougemont et al., 2008; Smith et al., 2008). Currently, the Illumina sequencer can produce longer reads of 100 nt (Illumina, Inc.4).
The PacBio is a single-molecule sequencing approach that has been developed to further reduce the cost and time required to obtain the sequence of a genome or transcriptome. It is thought of as a “third generation” sequencing platform. This instrument has recently become commercially available and only a few institutions have used it. The PacBio works based on a nanophotonic tool called zero-mode waveguide (ZMW; Levene et al., 2003). ZMW technology allows for a DNA polymerase to work in real time using fluorescently labeled nucleotides and tracks synthesis of a single molecule per DNA fragment (Eid et al., 2009). Like the 454 and Illumina instruments, the PacBio sequences by measuring the burst of light produced when the pyrophosphate and fluorescent label are released during the polymerization reaction. This instrument is able to sequence single molecules up to 1500 nt long, but the error rate (around 15%) is still relatively high (Pacific Biosciences5). However, pairing this instrument with other more robust sequencers can be a real advantage for non-model species, as one can get a relatively long intact scaffold against which to build and assemble genomes or transcriptomes for species of interest.
The instruments described above use different technologies and each approach has its advantages and disadvantages. Currently, Illumina sequencing produces short reads of about 100 nt in length but has the ability to do this from each end of the DNA molecule when paired ends are used. The SOLiD likewise produces reads of approximately 35–60 nt in length. The short sequences yielded by Illumina and SOLiD platforms have proven useful for the detection of miRNA (small RNA molecules of about 22 nt; Chi et al., 2011; Johansen et al., 2011) and comparative genome analysis of different fish populations (Chi et al., 2011). They could also be useful to design microarrays probes for a variety of non-model fish species. However the use of short sequence reads can be challenging for de novo sequencing, sequence assembly and accurate annotation of the genes. It must be noted that the Illumina and SOLiD are working toward increasing the number of base pairs reads and this will improve in the future.
The DNA sequencing techniques employed in Illumina and SOLiD technologies are effective to assess genetic variations in fish at individual (i.e., single nucleotide polymorphisms, SNPs) and population level (Liu et al., 2011). Indeed, while the 454 pyrosequencer determines the length of homopolymers in one step based on the intensity of the light signal (Morozova and Marra, 2008), the Illumina reads all nucleotides individually. In addition, the SOLiD sequencing system can more reliably distinguish between true sequence polymorphisms and sequencing errors. In SOLiD sequencing, each base is probed twice in two independent ligation reactions, rather than one synthesis reaction. If one of the two ligation reactions gives an unexpected nucleotide, this is recognized as an error. If a consistent result is found for both ligation reactions, it is recognized as a polymorphism (Morozova and Marra, 2008). This distinction is paramount for fish genetics studies due to the increased polymorphic loci resulting from genome duplication events. It should also be mentioned that the newer SOLiD instrument is organized in such a way that individual lanes can be run, without having to fill an entire plate which may improve accessibility for smaller projects in non-model fish.
The new Ion Torrent instrument is relatively inexpensive and will allow individual researchers to have one in their laboratories, much like they do for qPCR. However this instrument also produces relatively short reads. The 454 pyrosequencer and the PacBio are superior in term of read-length and are capable of producing up to 700 and 1,500 nt per read respectively, making them ideal techniques for de novo sequencing of fish species as a scaffold is required. The 454 pyrosequencing technology has already shown great potential for whole transcriptome analysis using non-model fish (Garcia-Reyero et al., 2008; Jeukens et al., 2010). We should point out that the PacBio is still in its infancy and the platform still requires a lot of care. With improvements, this instrument will surely become a mainstay for de novo sequencing of non-model fish species.
Researchers should consider carefully each sequencing platform based on the aims of the project (i.e., assessment of genetic variation, de novo sequencing or transcriptome sequencing). In toxicogenomics studies with non-model fish species, it may be more beneficial to use a hybrid sequencing strategy. For example, combining the short pair-ends reads of the Illumina with the longer single-end reads of the 454 will likely enhance sequence assembly and gene annotation. This was demonstrated recently by Jiang et al. (2011). The authors used Illumina and 454 sequencing to investigate the genome of the channel catfish (Ictalurus punctatus), and demonstrated that sequencing data from two NGS platforms improved the sequencing depth and increased the number of contigs assembled.
Next-generation sequencing technologies produce massive amounts of data that need to be processed, annotated, and aligned to the genome before expression analysis (Garber et al., 2011). This is a significant obstacle for ecotoxicogenomics because many researchers are using non-model fish species to study the impacts of aquatic pollutants. Therefore, the advances in DNA sequencing technologies require corresponding improvements in bioinformatics approaches to better manage and interpret genomic and transcriptomics data. There are new algorithms, such as GENE-counter (Cumbie et al., 2011) that can assist with processing and managing the data but these methods have not been tested with non-model fish species. The process to align reads in NGS will not be covered here and there are a number of pipelines for obtaining meaningful sequencing data in order to quantitate transcriptome data (Goncalves et al., 2011). Although some algorithms incorporate splicing events of transcripts into the analysis, the detection of splice variants could be more challenging in teleosts because there are multiple copies of genes. For example, in some teleost species there are four gene variants of the estrogen receptor that show differences in ontogeny and sex expression (Boyce-Derricott et al., 2010). The ER isoforms show high conservation in the DNA and ligand binding domains and are more variable in other regions. Sequencing a gene with multiples isoforms in the conserved region by chance could make interpretation and quantitation difficult, especially when counting differentially expressed tags in RNA-seq studies.
In recent years, there has been a movement away from single gene characterization and toward the integration and quantification of high-throughput sequencing data in ecotoxicology. To supplement and enhance biologically relevant observations made from gene expression analysis using NGS, bioinformatics algorithms have been developed to consider all affected genes, many of which appear functionally unrelated, and to identify cellular processes and molecular functions perturbed by toxicants. This approach circumvents concerns with multiple hypotheses testing of both microarray and RNA-Seq data which severely restrict expression data because genes are grouped into larger categories resulting in fewer comparisons. Bioinformatics approaches implemented for fish ecotoxicogenomics experiments include functional enrichment, gene set enrichment, pathway analysis, and reverse engineering. We provide some brief examples of their use in ecotoxicology.
Gene ontology (GO) is a manually curated database of genes using a standardized vocabulary that includes biological process, molecular function, and cellular component. Using NCBI PubMed for a literature search, more than 40 scientific publications investigating the impact of aquatic pollutants in fish have characterized differentially expressed transcripts using GO to identify functionally enriched biological processes. As an example, there has been valuable insight obtained into the effects of endocrine disrupting chemicals that mimic estrogens. Many studies with different experimental paradigms reported common biological processes and molecular functions affected by environmental estrogens, despite the variety of genes that were differentially regulated. These include electron transport, amino acid synthesis, primary metabolism, cell communication and signaling, steroid binding, and steroid metabolism (Martyniuk et al., 2007; Benninghoff and Williams, 2008; Hoffmann et al., 2008; Garcia-Reyero et al., 2009).
In contrast to functional enrichment that utilizes a user defined gene list based on predetermined criteria (i.e., fold change or p-value cutoff), Gene set enrichment analysis (GSEA) considers the entire list of genes in the analysis (Subramanian et al., 2005) and can be used for microarray and RNA-seq data. GSEA is a computational method that determines whether an a priori defined set of genes shows statistical differences in rank order in a list based on differential gene expression. The advantage of GSEA is that it identifies pathways and cell processes more robustly by reducing the signal-to-noise ratio in a dataset, and there is higher resolution and ability to identify regulated gene groups. GSEA has been utilized in toxicogenomics, for example to study the neurotoxic effects of aquatic pollutants such as fluoxetine, venlafaxine, and carbamazepine (Thomas et al., 2012). In this study, GSEA identified central nervous system development, axonogenesis, brain development, and neurogenesis as the main biological pathways altered in fathead minnows exposed to these three neuroactive contaminants. Until now, GSEA has had limited use in fish transcriptomics studies but it promises to be an important bioinformatics methodology to characterize adverse outcome pathways (AOPs). Another enrichment analysis method called sub-network enrichment analysis (SNEA) can be used in fish transcriptomics studies (Trudeau et al., 2012), but it is not yet widely utilized in fish ecotoxicology This approach identifies gene regulatory pathways underlying chemical perturbation and one can construct informative gene networks in a method similar to pathway analysis (outline below) but the networks are constructed in a post hoc fashion. GSEA and SNEA have shown high potential to characterize biological pathways affected by contaminants but their application remains limited for examining NGS data in fish toxicology as they require high quality gene annotation.
Biochemical pathways are important for characterizing AOPs in toxicogenomics. A number of bioinformatics tools are available to link transcriptomics data to pathway categories such as disease progression, drug effects, and biochemical processes among others. These tools include the Database for Annotation, Visualization and Integrated Discovery (DAVID6), Connectivity Map7, and the Kyoto Encyclopedia of Genes and Genomes (KEGG8). Of interest to ecotoxicogenomics, KEGG MAPPER and Babelomics can be used to integrate metagenomic and transcriptomics with chemical and pathway information (Kawashima et al., 2008; Medina et al., 2010; Kanehisa et al., 2012). Other programs used for pathway analysis in fish ecotoxicology studies include Ingenuity Pathways Analysis (Ingenuity® Systems) and Pathway Studio® (Nikitin et al., 2003; Ariadne Genomics). Networks are built based upon relationships extracted from primary literature and algorithm searches for entity connections based on regulation, interaction, and binding between proteins or cell processes. In ecotoxicology studies, pathway analysis has been used with success to explore relationships among genes that are impacted by aquatic pollutants. Gene interaction pathways have been constructed after exposure to pollutants of concern such as ethinylestradiol, 17β-trenbolone, and fipronil in the hypothalamic–pituitary–gonadal axis of zebrafish (Wang et al., 2010), the pesticide methoxychlor in largemouth bass liver (Martyniuk et al., 2011) and environmental estrogens in fathead minnow ovary (Garcia-Reyero et al., 2009). NGS approaches in ecotoxicogenomics will benefit from these bioinformatics tools to integrate both DNA and transcriptomics data and better predict the adverse effects in non-target aquatic organisms.
The successes of building meaningful interaction pathways in fish toxicology reported in the literature are impressive as genomics information is limited for non-model fish species. Researchers using fish model in genomics studies have to consider the fact that many gene–gene interaction pathways are based on mammalian literature. Therefore, to extract significant functional gene information for pathway analysis, mammalian homologs for fish genes must be retrieved. Fish specific databases for model fishes such as the zebrafish, are currently under development and will include gene information not found in mammals.
Reverse engineering offers a new way of characterizing AOPs in fish toxicology (Perkins et al., 2011). The theory behind reverse engineering and the potential applications in ecotoxicology are well described by Garcia-Reyero and Perkins (2011). Generally, the process of reverse engineering, borrowed from computing sciences and engineering, is to identify the working parts of a system in order to better understand how it functions. This methodology increases the potential to study this system in a different context. In toxicogenomics, multiple Omics datasets can be statistically evaluated to identify key nodes (genes or proteins) that regulate gene networks. A framework for reverse engineering of AOPs in ecotoxicology has been introduced by Perkins et al. (2011). This framework consists of building and integrating gene networks, interrogating the networks with chemical perturbations, defining the AOPs, and predicting phenotypic consequences to the perturbation. The authors provide an example using an impressive 868 microarray datasets from female fathead minnow ovary to investigate environmental contaminants able to disrupt the hypothalamic–pituitary–gonadal axis. The analysis permitted to identify gene networks affected by the anti-androgen flutamide, which were composed of several signaling and receptor genes (both estrogen and androgen responsive) and associated with cell regeneration, development, and antioxidant response. Some of the network nodes included activin A receptor (type 1), aryl hydrocarbon receptor (AHR) interacting protein, and Wnt1 inducible signaling pathway protein 1. This approach offers unique biological perspective on the regulatory pathways affected by flutamide.
Some challenges for reverse engineering, and other methods such as SNEA, have been addressed over a decade ago in the early stages of transcriptomics and network analysis (Szallasi, 1999). These include the stochastic nature of the transcriptome (or variation in the time sequences of gene activation/inhibition), the effective size of the network (i.e., how many interacting entities comprise a “network”), the compartmentalization of genetic networks (e.g., a highly compartmentalized gene network will have few regulators and may be more “buffered” from environmental perturbations), and information content of gene expression matrices (i.e., what information is present on a temporal scale about variation in gene–gene or gene–protein relationships). Despite these challenges, there have been great strides in adopting reverse engineering into aquatic toxicology. It should also be noted that many of the algorithms described above depend upon, by definition, the annotations and curated gene descriptions available. Nevertheless, fish ecotoxicogenomics studies using high-throughput transcript sequencing have benefited tremendously from these bioinformatics approaches and they have been extremely useful for characterizing genes and pathways altered by aquatic pollutants.
Over the last few years, NGS has been used to examine DNA and RNA from over 20 fish species including Atlantic salmon (Salmo salar), bighead carp (Hypophthalmichthys nobilis), European seabass (Dicentrarchus labrax), lake sturgeon (Acipenser fulvescens), mangrove killifish (Kryptolebias marmoratus), pygmy perch (Nannoperca spp.), and spotted gar (Lepisosteus oculatus; Table 1). Most fish sequencing projects have employed the 454 pyrosequencer and have been successful using multipletissuetypes (e.g., liver, gonad, kidney, brain) as well as different life stages (e.g., adult versus embryo). This demonstrates that NGS platforms are versatile and can be used to address a range of biological questions in fish. To date, only a few studies have used NGS technologies to research the impact of environmental contaminants in aquatic organisms. Below, we discuss the application and advantages of these methodologies in fish toxicogenomics.
Next-generation sequencing has already started to have a positive impact in the field of fish transcriptomics. Microarrays are frequently used in the field of fish ecotoxicology (Douglas, 2006; Falciani et al., 2008; Garcia-Reyero et al., 2009; Villeneuve et al., 2010; Gust et al., 2011; Sellin Jeffries et al., 2012). Typically microarray probes were designed based on cDNA contigs produced by suppressive subtractive hybridization (SHH) and cDNA libraries (Blum et al., 2004; Williams et al., 2006; Larkin et al., 2007; Cairns et al., 2008; Katsiadaki et al., 2010). However, a few studies have applied NGS technology to produce oligonucleotide microarrays. Because NGS platforms can generate high numbers of reads, the resulting sequences are often extended which increases the chance to find matching reads and correctly annotate them. Garcia-Reyero et al. (2008) used 454 pyrosequencing technologies to build a 44,000-oligonucleotide microarray for largemouth bass (Micropterus salmoides). This approach resulted in obtaining 31,391 unique sequences, which were compiled with sequences from SHH to produce nearly 16,000 gene sequences (half of them were annotated). The custom-designed microarray was then tested by assessing the impact of 17β-estradiol exposure on endocrine disruption and hormone signaling in adult largemouth bass. The combination of NGS and microarray analyses permitted characterizing several pathways perturbed by the estrogenic compound including gonad development, sex differentiation, signal transduction, and cell communication. In another study, Mirbahai et al. (2011) used NGS technology in combination with methylated DNA immunoprecipitation to design a 14,919-oligonucleotide microarray. This permitted to examine hepatic DNA methylation changes in common Dab (Limanda limanda) living inpolluted environments and tocorrelate methylation levels with gene expression levels.
Recently, RNA-seq analyses are increasingly used (Xiang et al., 2010; Fraser et al., 2011) and the results suggest that this method could replace array-based technology in toxicogenomics research. Indeed, RNA-seq presents the advantage to quantify directly the expression level of mRNAs across the transcriptome from the number of reads for a particular cDNA contig in a sequencing run, allowing for the quantification of low-expressed transcripts. While currently cost-prohibitive, RNA-seq analysis can potentially provide a greater degree of resolution than microarrays and help to identify splice variants. Oleksiak et al. (2011) utilized this technique in supplement to a microarray experiment to determine the genomic differences between a polychlorinated biphenyl (PCB) sensitive and a PCB resistant population of Atlantic killifish (Fundulus heteroclitus). Using 454 pyrosequencing technology, they demonstrated that NGS data can be used to extend the length of array probes, which helped to find new matching sequences and to annotate previously unannotated probes. The RNA-seq study corroborated most of the microarray results and suggested that AHR regulatory pathway may be responsible for the PCB resistance of one of the killifish population. In another study, Whitehead et al. (2011) applied similar techniques to examine the effects of the Deepwater Horizon oil spill on Gulf killifish. RNA sequencing data was acquired using the Illumina platform and over 6000 unique EST sequences were obtained. Both microarray and RNA-seq analyses identified zona pellucida, choriogenin, and vitellogenin as PCB-responsive genes. These early studies provide strong evidence that RNA-seq methods are suitable to investigate the adverse effects of pollutants present in the aquatic environment.
The usefulness of this approach was further demonstrated by Pierron et al. (2011) who conducted RNA-seq to examine the effects of chronic metal exposure in four wild populations of yellow perch. NGS data generated from the yellow perch yielded over 9,000 gene sequences among which 6,000 were annotated. As mentioned previously, annotating EST sequences from non-model fish species is one of the main challenges for fish biologists and NGS technologies could facilitate this task. Pierron et al. (2011) were able to establish relationships between the hepatic expression levels of specific transcripts and the concentrations of copper and cadmium measured in the fish as well as to identify potential adverse effects. In general, these studies have successfully shown that NGS is a powerful technique to study the ecotoxicological responses of non-model fish species living in polluted environments.
Interestingly, most of the research published on the application of NGS in fish has focused on different aspects of fish evolution such as genome evolution (Hale et al., 2010; Amores et al., 2011), phenotypic evolution (Elmer et al., 2010; Goetz et al., 2010; Jeukens et al., 2010), and evolution of immune system (Star et al., 2011; Zhang et al., 2011a). This subject area is outside of the scope of this article, but a few of these studies are reviewed below.
Next-generation sequencing technologies have had a significant impact in the field of ecological divergence and have contributed in elucidating the links between genetic and environmental factors leading to species evolution (Elmer et al., 2010). Most of the research in this field used the 454 GS FLX pyrosequencer for RNA-seq analyses to uncover the molecular basis for the phenotypic and ecological divergences between endemic species. For example, Jeukens et al. (2010) employed this methodology to investigate the genomic differences behind the phenotypic divergence of two populations of lake whitefish (Coreons clupeaformis spp.). The authors discovered that dwarf fish had an over-representation of genes linked to immunity, DNA replication and repair while normal fish over-expressed genes linked to protein synthesis. Elmer et al. (2010) used the same approach to correlate the genomic and phenotypic differences between crater lake cichlids: the benthic species Amphilophus astorquii and the limnetic species Amphilophus zaliosus. Their study revealed that a number of transcripts associated with development, biosynthesis, and metabolic processes were differentially expressed between the two species. Other studies have employed NGS technologies to characterize fish immune system and its evolution. All these studies have concluded that NGS technologies provide a greater scope of understanding of the genetic events that preceded natural selection and fish species evolution. The significant advancement made in these disciplines may provide valuable genetic insights to facilitate ecotoxicogenomic analyses. For example, studies on the Atlantic killifish (Oleksiak et al., 2011) combined principles in ecotoxicology and evolution to better understand adaptation of fish in polluted environments. Combining data on both genetic variation (SNPs) in fish genomes and transcriptomic responses will lead to the characterization of expression quantitative trait loci (eQTL) and genetic architecture that underlies adaptation.
There is great promise for toxicogenomics in non-model fish species. Fish offer unique challenges compared to mammals due to genome duplication events and the presence of multiple isoforms for many genes. Nevertheless, teleost fish are important model organisms for assessing the impact of anthropogenic pollutants in the environment as well as studying certain human diseases (Albertson et al., 2009; Zhang et al., 2010). As the costs for DNA and RNA sequencing decrease, the combination of several NGS platforms should facilitate whole genome sequencing projects and expand our knowledge of ecologically relevant species. Understanding the relationships between environmental chemical exposure and gene expression will provide valuable data for environmental risk assessments (ERA). In 2011, Piña and Barata reviewed the potential for ecotoxicogenomics studies to improve the tests necessary for ERA by discovering biological assays and biomarkers relevant to environmental conditions (Piña and Barata, 2011). Thus, the development of ecotoxicogenomics and bioinformatics tools will greatly benefit the assessment of the impacts of environmental pollutants. In the future, it will be necessary to integrate the extensive genomic data gathered from transcriptomics, gene regulation, and evolutionary biology into a working framework in order to propose new hypotheses in fish research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This review was supported by grant R01 ES015449 from the Superfund Basic Research Program from the National Institute of Environmental Health Sciences (Nancy D. Denslow) and by a Canada Research Chair and NSERC Discovery Grant to Christopher J. Martyniuk. Publication of this article was funded in part by the University of Florida Open-Access Publishing Fund.
Albertson, R. C., Cresko, W., Detrich, H. W. III, and Postlethwait, J. H. (2009). Evolutionary mutant models for human disease. Trends Genet. 25, 74–81.
Amores, A., Catchen, J., Ferrara, A., Fontenot, Q., and Postlethwait, J. H. (2011). Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics 188, 799–808.
Benninghoff, A. D., and Williams, D. E. (2008). Identification of a transcriptional fingerprint of estrogen exposure in rainbow trout liver. Toxicol. Sci. 101, 65–80.
Blum, J. L., Knoebl, I., Larkin, P., Kroll, K. J., and Denslow, N. D. (2004). Use of suppressive subtractive hybridization and cDNA arrays to discover patterns of altered gene expression in the liver of dihydrotestosterone and 11-ketotestosterone exposed adult male largemouth bass (Micropterus salmoides). Mar. Environ. Res. 58, 565–569.
Boyce-Derricott, J., Nagler, J. J., and Cloud, J. G. (2010). The ontogeny of nuclear estrogen receptor isoform expression and the effect of 17beta-estradiol in embryonic rainbow trout (Oncorhynchus mykiss). Mol. Cell. Endocrinol. 315, 277–281.
Bräutigam, A., and Gowik, U. (2010). What can next generation sequencing do for you? Next generation sequencing as a valuable tool in plant research. Plant Biol. 12, 831–841.
Cairns, M. T., Johnson, M. C., Talbot, A. T., Pemmasani, J. K., McNeill, R. E., Houeix, B., Sangrador-Vegas, A., and Pottinger, T. G. (2008). A cDNA microarray assessment of gene expression in the liver of rainbow trout (Oncorhynchus mykiss) in response to a handling and confinement stressor. Comp. Biochem. Physiol. D Genomics Proteomics 3, 51–66.
Chi, W., Tong, C., Gan, X., and He, S. (2011). Characterization and comparative profiling of miRNA transcriptomes in bighead carp and silver carp. PLoS ONE 6, e23549. doi: 10.1371/journal.pone.0023549
Cumbie, J. S., Kimbrel, J. A., Di, Y., Schafer, D. W., Wilhelm, L. J., Fox, S. E., Sullivan, C. M., Curzon, A. D., Carrington, J. C., Mockler, T. C., and Chang, J. H. (2011). GENE-counter: a computational pipeline for the analysis of RNA-seq data for gene expression differences. PLoS ONE 6, e25279. doi: 10.1371/ journal.pone.0025279
Denslow, N. D., Garcia-Reyero, N., and Barber, D. S. (2007). Fish ’n’ chips: the use of microarrays for aquatic toxicology. Mol. Biosyst. 3, 172–177.
Droege, M., and Hill, B. (2008). The Genome Sequencer FLX™ System – longer reads, more applications, straight forward bioinformatics and more complete data sets. J. Biotechnol. 136, 3–10.
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., Peluso, P., Rank, D., Baybayan, P., Bettman, B., Bibillo, A., Bjornson, K., Chaudhuri, B., Christians, F., Cicero, R., Clark, S., Dalal, R., Dewinter, A., Dixon, J., Foquet, M., Gaertner, A., Hardenbol, P., Heiner, C., Hester, K., Holden, D., Kearns, G., Kong, X., Kuse, R., Lacroix, Y., Lin, S., Lundquist, P., Ma, C., Marks, P., Maxham, M., Murphy, D., Park, I., Pham, T., Phillips, M., Roy, J., Sebra, R., Shen, G., Sorenson, J., Tomaney, A., Travers, K., Trulson, M., Vieceli, J., Wegener, J., Wu, D., Yang, A., Zaccarin. D., Zhao, P., Zhong, F., Korlach, J., and Turner, S. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138.
Elmer, K. R., Fan, S., Gunter, H. M., Jones, J. C., Boekhoff, S., Kuraku, S., and Meyer, A. (2010). Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlid fishes. Mol. Ecol. 19, 197–211.
Falciani, F., Diab, A. M., Sabine, V., Williams, T. D., Ortega, F., George, S. G., and Chipman, J. K. (2008). Hepatic transcriptomics profiles of European flounder (Platichthys flesus) from field sites and computational approaches to predict site from stress gene responses following exposure to model toxicants. Aquat. Toxicol . 90, 92–101.
Fraser, B. A., Weadick, C. J., Janowitz, I., Rodd, H., and Hughes, K. A. (2011). Sequencing and characterization of the guppy (Poecilia reticulata) transcriptome. BMC Genomics 12, 202. doi: 10.1186/1471-2164-12-202
Garber, M., Grabherr, M. G., Guttman, M., and Trapnell, C. (2011). Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 8, 469–477.
Garcia-Reyero, N., Griffitt, R. J., Liu, L., Kroll, K. J., Farmerie, W. G., Barber, D. S., and Denslow, N. D. (2008). Construction of a robust microarray from a non-model species (largemouth bass) using pyrosequencing technology. J. Fish Biol. 72, 2354–2376.
Garcia-Reyero, N., and Perkins, E. J. (2011). Systems biology: leading the revolution in ecotoxicology. Environ. Toxicol. Chem. 30, 265–273.
Garcia-Reyero, N., Villeneuve, D. L., Kroll, K. J., Liu, L., Orlando, E. F., Watanabe, K. H., Sepúlveda, M. S., Ankley, G. T., and Denslow, N. D. (2009). Gene expression responses in male fathead minnows exposed to binary mixtures of an estrogen and antiestrogen. BMC Genomics 10, 308. doi: 10.1186/1471-2164-10-308
Goetz, F., Rosauer, D., Sitar, S., Goetz, G., Simchick, C., Roberts, S., Johnson, R., Murphy, C., Bronte, C. R., and Mackenzie, S. (2010). A genetic basis for the phenotypic differentiation between siscowet and lean lake trout (Salvelinus namaycush). Mol. Ecol. 19, 176–196.
Goncalves, A., Tikhonov, A., Brazma, A., and Kapushesky, M. (2011). A pipeline for RNA-seq data processing and quality assessment. Bioinformatics 27, 867–869.
Gust, K. A., Wilbanks, M. S., Guan, X., Pirooznia, M., Habib, T., Yoo, L., Wintz, H., Vulpe, C. D., and Perkins, E. J. (2011). Investigations of transcript expression in fathead minnow (Pimephales promelas) brain tissue reveal toxicological impacts of RDX exposure. Aquat. Toxicol. 101, 135–145.
Hale, M. C., Jackson, J. R., and Dewoody, J. A. (2010). Discovery and evaluation of candidate sex-determining genes and xenobiotics in the gonads of lake sturgeon (Acipenser fulvescens). Genetica 138, 745–756.
Hoffmann, J. L., Thomason, R. G., Lee, D. M., Brill, J. L., Price, B. B., Carr, G. J., and Versteeg, D. J. (2008). Hepatic gene expression profiling using GeneChips in zebrafish exposed to 17alpha-methyldihydrotestosterone. Aquat. To x i c o l . 87, 69–80.
Holt, K. E., Parkhill, J., Mazzoni, C. J., Roumagnac, P., Weill, F. X., Goodhead, I., Rance, R., Baker, S., Maskell, D. J., Wain, J., Dolecek, C., Achtman, M., and Dougan, G. (2008). High-throughput sequencing provides insights into genome variation and evolution in Salmonella typhi. Nat. Genet. 40, 987–993.
Huang, Y., Huang, X., Yan, Y., Cai, J., Ouyang, Z., Cui, H., Wang, P., and Qin, Q. (2011). Transcriptome analysis of orange-spotted grouper (Epinephelus coioides) spleen in response to Singapore grouper iridovirus. BMC Genomics 12, 556. doi: 10.1186/1471-2164-12-556
Jeukens, J., Renaut, S., St-Cyr, J., Nolte, A. W., and Bernatchez, L. (2010). The transcriptomics of sympatric dwarf and normal lake whitefish (Coregonus clupeaformis spp., Salmonidae) divergence as revealed by next-generation sequencing. Mol. Ecol. 19, 5389–5403.
Jiang, Y., Lu, J., Peatman, E., Kucuktas, H., Liu, S., Wang, S., Sun, F., and Liu, Z. (2011). A pilot study for channel catfish whole genome sequencingand de novo assembly. BMC Genomics 12, 629. doi: 10.1186/1471-2164-12-629
Johansen, S. D., Karlsen, B. O., Furmanek, T., Andreassen, M., Jørgensen, T. E., Bizuayehu, T. T., Breines, R., Emblem, A., Kettunen, P., Luukko, K., Edvardsen, R. B., Nordeide, J. T., Coucheron, D. H., and Moum, T. (2011). RNA deep sequencing of the Atlantic cod transcriptome. Comp. Biochem. Physiol. D Genomics Proteomics 6, 18–22.
Ju, Z., Wells, M. C., and Walter, R. B. (2007). DNA microarray technology in toxicogenomics of aquatic models: methods and applications. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 145, 5–14.
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114.
Katsiadaki, I., Williams, T. D., Ball, J. S., Bean, T. P., Sanders, M. B., Wu, H., Santos, E. M., Brown, M. M., Baker, P., Ortega, F., Falciani, F., Craft, J. A., Tyler, C. R., Viant, M. R., and Chipman, J. K. (2010). Hepatic transcriptomic and metabolomic responses in the stickleback (Gasterosteus aculeatus) exposed to ethinyl-estradiol. Aquat. Toxicol. 97, 174–187.
Kawashima, S., Okuda, S., Tokimatsu, T., and Yamanishi, Y. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484.
Keller, I., Taverna, A., and Seehausen, O. (2011). A pyrosequencing assay for the rapid discrimination of mitochondrial lineages in the Salmo trutta species complex. Mol. Ecol. Resour. 11, 196–199.
Kristiansson, E., Asker, N., Förlin, L., and Larsson, D. G. (2009). Characterization of the Zoarces viviparus liver transcriptome using massively parallel pyrosequencing. BMC Genomics 10, 345. doi: 10.1186/1471-2164-10-345
Kuhl, H., Tine, M., Beck, A., Timmermann, B., Kodira, C., and Reinhardt, R. (2011). Directed sequencing and annotation of three Dicentrarchus labrax L. chromosomes by applying Sanger- and pyrosequencing technologies on pooled DNA of comparatively mapped BAC clones. Genomics 98, 202–212.
Larkin, P., Villeneuve, D. L., Knoebl, I., Miracle, A. L., Carter, B. J., Liu, L., Denslow, N. D., and Ankley, G. T. (2007). Development and validation of a 2,000-gene microarray for the fathead minnow (Pimephales promelas). Environ. Toxicol. Chem. 26, 1497–1506.
Levene, M. J., Korlach, J., Turner, S. W., Foquet, M., Craighead, H. G., and Webb, W. W. (2003). Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299, 682–686.
Li, S. C., Chan, W. C., Ho, M. R., Tsai, K. W., Hu, L. Y., Hsu, C. N., Hwang, P. P., and Lin, W. C. (2010). Discovery and characterization of medaka miRNA genes by next generation sequencing platform. BMC Genomics 11(Suppl. 4), S8. doi: 10.1186/1471-2164-11-S4-S8
Liu, S., Zhou, Z., Lu, J., Sun, F., Wang, S., Liu, H., Jiang, Y., Kucuktas, H., Kaltenboeck, L., Peatman, E., and Liu, Z. (2011). Generation of genome-scale gene-associated SNPs in catfish for the construction of a high-density SNP array. BMC Genomics 12, 53. doi: 10.1186/1471-2164-12-53
Mardis, E. R. (2008). Next generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402.
Martyniuk, C. J., Gerrie, E. R., Popesku, J. T., Ekker, M., and Trudeau, V. L. (2007). Microarray analysis in the zebrafish (Danio rerio) liver and telencephalon after exposure to low concentration of 17alpha-ethinylestradiol. Aquat. Toxicol. 84, 38–49.
Martyniuk, C. J., Spade, D. J., Blum, J. L., Kroll, K. J., and Denslow, N. D. (2011). Methoxychlor affects multiple hormone signaling pathways in the largemouth bass (Micropterus salmoides) liver. Aquat. Toxicol. 101, 483–492.
Medina, I., Carbonell, J., Pulido, L., Madeira, S. C., Conesa, A., Tárraga, J., Pascual-Montano, A., Nogales-Cadenas, R., Santoyo, J., García, F., Marbà, M., Montaner, D., and Dopazo, J. (2010). Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomics data with advanced functional profiling. Nucleic Acids Res. 38, W210–W213.
Mirbahai, L., Yin, G., Bignell, J. P., Li, N., Williams, T. D., and Chipman, J. K. (2011). DNA methylation in the liver tumorigenesis in fish from the environment. Epigenetics 6, 1319–1333.
Morozova, O., and Marra, M. A. (2008). Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264.
Nikitin, A., Egorov, S., Daraselia, N., and Mazo, I. (2003). Pathway studio – the analysis and navigation of molecular networks. Bioinformatics 19, 2155–2157.
Nowrousian, M. (2010). Next-generation sequencing techniques for eukaryotic microorganisms: sequencing-based solutions to biological problems. Eukaryot. Cell 9, 1300–1310.
Oleksiak, M. F., Karchner, S. I., Jenny, M. J., Franks, D. G., Welch, D. B., and Hahn, M. E. (2011). Transcriptomic assessment of resistance to effects of an aryl hydrocarbon receptor (AHR) agonist in embryos of Atlantic killifish (Fundulus heteroclitus) from a marine Superfund site. BMC Genomics 12, 263. doi: 10.1186/1471-2164-12-263
Pareek, C. S., Smoczynski, R., and Tretyn, A. (2011). Sequencing technologies and genome sequencing. J. Appl. Genet. 52, 413–435.
Perkins, E. J., Chipman, J. K., Edwards, S., Habib, T., Falciani, F., Taylor, R., Van Aggelen, G., Vulpe, C., Antczak, P., and Loguinov, A. (2011). Reverse engineering adverse outcome pathways. Environ. Toxicol. Chem. 30, 22–38.
Pierron, F., Normandeau, E., Defo, M. A., Campbell, P. G., Bernatchez, L., and Couture, P. (2011). Effects of chronic metal exposure on wild fish populations revealed by high-throughput cDNA sequencing. Ecotoxicology 20, 1388–1399.
Piña, B., and Barata, C. (2011). A genomic and ecotoxicological perspective of DNA array studies in aquatic environmental risk assessment. Aquat. Toxicol. 105S, 40–49.
Pomraning, K. R., Smith, K. M., and Freitag, M. (2009). Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods 47, 142–150.
Prosdocimi, F., de Carvalho, D. C., de Almeida, R. N., and Beheregaray, L. B. (2011). The complete mitochondrial genome of two recently derived species of the fish genus Nannoperca (Perciformes, Percichthyidae). Mol. Biol. Rep. 39, 2767–2772.
Quinn, N. L., Levenkova, N., Chow, W., Bouffard, P., Boroevich, K. A., Knight, J. R., Jarvie, T. P., Lubieniecki, K. P., Desany, B. A., Koop, B. F., Harkins, T. T., and Davidson, W. S. (2008). Assessing the feasibility of GS FLX pyrosequencing for sequencing the Atlantic salmon genome. BMC Genomics 9, 404. doi: 10.1186/ 1471-2164-9-404
Renaut, S., Nolte, A. W., and Bernatchez, L. (2010). Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs (Coregonus spp. Salmonidae). Mol. Ecol. 19, 115–131.
Rhee, J. S., Kim, B. M., Kim, R. O., Choi, B. S., Choi, I. Y., Lee, Y. M., and Lee, J. S. (2011). Analysis of expressed sequence tags from the liver and ovary of the euryhaline hermaphroditic fish, Kryptolebias marmoratus. Comp. Biochem. Physiol. D Genomics Proteomics 6, 244–255.
Rothberg, J. M., Hinz, W., Rearick, T. M., Schultz, J., Mileski, W., Davey, M., Leamon, J. H., Johnson, K., Milgrew, M. J., Edwards, M., Hoon, J., Simons, J. F., Marran, D., Myers, J. W., Davidson, J. F., Branting, A., Nobile, J. R., Puc, B. P., Light, D., Clark, T. A., Huber, M., Branciforte, J. T., Stoner, I. B, Cawley, S. E., Lyons, M., Fu, Y., Homer, N., Sedova, M., Miao, X., Reed, B., Sabina, J., Feierstein, E., Schorn, M., Alanjary, M., Dimalanta, E., Dressman, D., Kasinskas, R., Sokolsky, T., Fidanza, J. A., Namsaraev, E., McKernan, K. J., Williams, A., Roth, G. T., and Bustillo, J. (2011). An integrated semiconductor device enabling non-optical genome sequencing. Nature 475, 348–352.
Rothberg, J. M., and Leamon, J. H. (2008). The development and impact of 454 sequencing. Nat. Biotechnol. 26, 1117–1124.
Rougemont, J., Amzallag, A., Iseli, C., Farinelli, L., Xenarios, I., and Naef, F. (2008). Probabilistic base calling of Solexa sequencing data. BMC Bioinformatics 9, 431. doi: 10.1186/ 1471-2105-9-431
Salem, M., Rexroad, C. E. III, Wang, J., Thorgaard, G. H., and Yao, J. (2010). Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genomics 11, 564. doi: 10.1186/ 1471-2164-11-564
Seeb, J. E., Pascal, C. E, Grau, E. D., Seeb, L. W., Templin, W. D., Harkins, T., and Roberts, S. B. (2011). Transcriptome sequencing and high-resolution. melt analysis advance single nucleotide polymorphism discovery in duplicated salmonids. Mol. Ecol. Resour. 11, 335–348.
Sellin Jeffries, M. K., Mehinto, A. C., Carter, B., Denslow, N. D., and Kolok, A. S. (2012). Taking microarrays to the field: differential hepatic gene expression of caged fathead minnows from Nebraska watersheds. Environ. Sci. Technol. 46, 1877–1885.
Smith, A. D., Xuan, Z., and Zhang, M. Q. (2008). Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinformatics 9, 128. doi: 10.1186/1471-2105-9-128
Soares, A. R., Pereira, P. M., Santos, B., Egas, C., Gomes, A. C., Arrais, J., Oliveira, J. L., Moura, G. R., and Santos, M. A. (2009). Parallel DNA pyrosequencing unveils new zebrafish microRNAs. BMC Genomics 10, 195. doi: 10.1186/1471-2164-10-195
Star, B., Nederbragt, A. J., Jentoft, S., Grimholt, U., Malmstrøm, M., Gregers, T. F., Rounge, T. B., Paulsen, J., Solbakken, M. H., Sharma, A., Wetten, O. F., Lanzén, A., Winer, R., Knight, J., Vogel, J. H., Aken, B., Andersen, O., Lagesen, K., Tooming-Klunderud, A., Edvardsen, R. B., Tina, K. G., Espelund, M., Nepal, C., Previti, C., Karlsen, B. O., Moum, T., Skage, M., Berg, P. R., Gjøen, T., Kuhl, H., Thorsen, J., Malde, K., Reinhardt, R., Du, L., Johansen, S. D., Searle, S., Lien, S., Nilsen, F., Jonassen, I., Omholt, S. W., Stenseth, N. C., and Jakobsen, K. S. (2011). The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210.
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., and Mesirov, J. P. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–1550.
Szallasi, Z. (1999). Genetic network analysis in light of massively parallel biological data acquisition. Pac. Symp. Biocomput. 4, 5–16.
Thomas, M. A., Joshi, P. P., and Klaper, R. D. (2012). Gene-class analysis of expression patterns induced by psychoactive pharmaceutical exposure in fathead minnow (Pimephales promelas) indicates induction of neuronal systems. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 155, 109–120.
Trudeau, V. L., Martyniuk, C. J., Zhao, E., Hu, H., Volkoff, H., Decatur, W. A., and Basak, A. (2012). Is secretoneurin a new hormone? Gen. Comp. Endocrinol. 175, 10–18.
Villeneuve, D. L., Garcia-Reyero, N., Martinović, D., Cavallin, J. E., Mueller, N. D., Wehmas, L. C., Kahl, M. D., Linnum, A. L., Perkins, E. J., and Ankley, G. T. (2010). Influence of ovarian stage on transcript profiles in fathead minnow (Pimephales promelas) ovary tissue. Aquat. Toxicol. 98, 354–366.
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., and Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Wang, R. L., Bencic, D., Villeneuve, D. L., Ankley, G. T., Lazorchak, J., and Edwards, S. (2010). A transcriptomics-based biological framework for studying mechanisms of endocrine disruption in small fish species. Aquat. Toxicol. 98, 230–244.
Whitehead, A., Dubansky, B., Bodinier, C., Garcia, T. I., Miles, S., Pilley, C., Raghunathan, V., Roach, J. L., Walker, N., Walter, R. B., Rice, C. D., and Galvez, F. (2011). Genomic and physiological footprint of the Deepwater Horizon oil spill on resident marsh fishes. Proc. Natl. Acad. Sci. U.S.A. doi: 10.1073/pnas.1109545108 [Epub ahead of print].
Williams, T. D., Diab, A. M., George, S. G., Godfrey, R. E., Sabine, V., Conesa, A., Minchin, S. D., Watts, P.C., and Chipman, J. K. (2006). Development of the GENIPOL European flounder (Platichthys flesus) microarray and determination of temporal transcriptional responses to cadmium at low dose. Environ. Sci. Technol. 40, 6479– 6488.
Xiang, L. X., He, D., Dong, W. R., Zhang, Y. W., and Shab, J. Z. (2010). Deep sequencing-based transcriptome profiling analysis of bacteria-challenged Lateolabrax japonicus reveals insight into the immune-relevant genes in marine fish. BMC Genomics 11, 472. doi: 10.1186/1471-2164-11-472
Zhang, G., Hoersch, S., Amsterdam, A., Whittaker, C. A., and Lees, J. A. (2010). Highly aneuploid zebrafish malignant peripheral nerve sheath tumors have genetic alterations similar to human cancers. Proc. Natl. Acad. Sci. U.S.A. 107, 16940–16945.
Zhang, Y., Stupka, E., Henkel, C. V., Jansen, H. J., Spaink, H. P., and Verbeek, F. J. (2011a). Identification of common carp innate immune genes with whole-genome sequencing and RNA-seq data. J. Integr. Bioinform. 8, 169.
Keywords: toxicogenomics, non-model fish, next-generation sequencing, bioinformatics, pathway analysis
Citation: Mehinto AC, Martyniuk CJ, Spade DJ and Denslow ND (2012) Applications of next-generation sequencing in fish ecotoxicogenomics. Front. Gene. 3:62. doi: 10.3389/fgene.2012.00062
Received: 30 January 2012; Paper pending published: 24 February 2012;
Accepted: 02 April 2012; Published online: 25 April 2012.
Edited by:
Jonathan Freedman, National Institute of Health, USAReviewed by:
Jing-Woei Li, The Chinese University of Hong Kong, Hong KongCopyright: © 2012 Mehinto, Martyniuk, Spade and Denslow. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nancy D. Denslow, Center for Environmental and Human Toxicology, Department of Physiological Sciences, University of Florida, 2187 Mowry Road, Building 471, PO Box 110885, Gainesville, FL 32611, USA. e-mail:bmRlbnNsb3dAdWZsLmVkdQ==
†Current address: Daniel J. Spade, Department of Pathology and Laboratory Medicine, Brown University, Providence, RI, USA
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.