
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 March 2012
Sec. Toxicogenomics
Volume 3 - 2012 | https://doi.org/10.3389/fgene.2012.00035
This article is part of the Research Topic Toxicogenomics in non-mammalian species View all 11 articles
 Pablo Cingolani1,2,3 Viral M. Patel1 Melissa Coon2
Pablo Cingolani1,2,3 Viral M. Patel1 Melissa Coon2 Tung Nguyen4
Tung Nguyen4 Susan J. Land2
Susan J. Land2 Douglas M. Ruden1,2*
Douglas M. Ruden1,2* Xiangyi Lu1
Xiangyi Lu1
This paper describes a new program SnpSift for filtering differential DNA sequence variants between two or more experimental genomes after genotoxic chemical exposure. Here, we illustrate how SnpSift can be used to identify candidate phenotype-relevant variants including single nucleotide polymorphisms, multiple nucleotide polymorphisms, insertions, and deletions (InDels) in mutant strains isolated from genome-wide chemical mutagenesis of Drosophila melanogaster. First, the genomes of two independently isolated mutant fly strains that are allelic for a novel recessive male-sterile locus generated by genotoxic chemical exposure were sequenced using the Illumina next-generation DNA sequencer to obtain 20- to 29-fold coverage of the euchromatic sequences. The sequencing reads were processed and variants were called using standard bioinformatic tools. Next, SnpEff was used to annotate all sequence variants and their potential mutational effects on associated genes. Then, SnpSift was used to filter and select differential variants that potentially disrupt a common gene in the two allelic mutant strains. The potential causative DNA lesions were partially validated by capillary sequencing of polymerase chain reaction-amplified DNA in the genetic interval as defined by meiotic mapping and deletions that remove defined regions of the chromosome. Of the five candidate genes located in the genetic interval, the Pka-like gene CG12069 was found to carry a separate pre-mature stop codon mutation in each of the two allelic mutants whereas the other four candidate genes within the interval have wild-type sequences. The Pka-like gene is therefore a strong candidate gene for the male-sterile locus. These results demonstrate that combining SnpEff and SnpSift can expedite the identification of candidate phenotype-causative mutations in chemically mutagenized Drosophila strains. This technique can also be used to characterize the variety of mutations generated by genotoxic chemicals.
There are two types of chemicals that cause developmental abnormities in organisms – genotoxic chemicals and non-genotoxic chemicals. Genotoxic chemicals directly alkylate or oxidize the DNA and cause inappropriate base pairing. This causes permanent genetic mutations after exposing germline cells to genotoxic chemicals. Non-genotoxic chemicals are thought to cause epigenetic changes in the DNA that cause developmental abnormalities. Most non-genotoxic chemicals only affect development or the health of the organism exposed, but some non-genotoxic chemicals such as the estrogenic chemical diethylstilbestrol (DES) can cause developmental abnormalities and increased susceptibility to cancer for several generations (reviewed in Ruden et al., 2005).
Random mutagenesis such as chemical mutagenesis with the genotoxic chemical ethyl methane sulfonate (EMS) is an incredibly powerful tool for generating mutant strains of cells or organisms for purposes of studying all types of biological processes. In mutant bacteria or yeast, identification of the mutated genes is often done by transforming wild-type DNA into the cells and screening for rescue of the mutant phenotype. One could then sequence the DNA that rescues the phenotype to find the gene mutated. In Drosophila melanogaster, a causative DNA lesion for an observable phenotype is traditionally done by meiotic mapping of the mutant locus using a series of visible genetic markers that span the chromosome (Anderson, 1992). Deficiencies that delete defined regions of the chromosome, typically tens to hundreds of kilobases long, can then be used to further refine the boundaries of the mutated gene locus (Parks et al., 2004; Ryder et al., 2007). However, these positional cloning techniques are not only labor-intensive and time consuming, but also without a guarantee of success. This frequently leads to inevitable delays in molecular and functional characterization of the gene involved, even in the post genomic era.
With the development of next-generation DNA sequencing instruments, whole-genome sequencing is becoming feasible to replace labor-intensive positional cloning methods. However, we are limited by the capacity of the current bioinformatic programs to rapidly and reliably process sequence variants including single nucleotide polymorphisms (SNPs), multiple nucleotide polymorphisms (MNPs), insertions, and deletions (InDels) between the wild-type control and the mutant genomes. This is especially the case in dealing with mutant strains isolated from random chemical mutagenesis that typically introduces quite large numbers of background sequence variants and SNPs into the mutant genome, only one of which is likely responsible for the mutant phenotype.
Furthermore, all current next-generation sequencers produce frequent errors, especially when approaching the 3′-ends of each short read. Using current technologies, a short read is typically 70–150 bp long. As the euchromatic genome of D. melanogaster is 117 million base pairs (Mbp), machine-generated errors by themselves are sufficient to produce thousands of false SNPs in whole-genome sequencing data. To expedite the analyses of whole-genome sequencing data and to reduce number of false positives, we have developed the programs SnpEff (Pablo Cingolani and Douglas M. Ruden; Fly, in press; Platts et al., 2009) and SnpSift. These programs can categorize and filter thousands of variants per second, based on their locations in the transcriptional unit and potential mutational effects on transcription or translation. By comparing several sequencing experiments, the number of false positives can be reduced.
Whole-genome sequencing to identify a causative SNP has not been established for D. melanogaster mutants (Hillier et al., 2008; Wang et al., 2010). Here, we describe how SnpEff1 and SnpSift2 can be used together to identify causative gene candidate using just two alleles of a male-sterile Drosophila locus. Both programs have web based interfaces available via the Galaxy project3.
Two allelic male-sterile mutations, X1 and X2, were identified in a F3 genetic screen (Yang et al., 2011). Briefly, males isogenic for the third chromosome were fed the chemical mutagen ethyl methane sulfonate (EMS) for 12 h (10 mM in 1% sucrose solution; Ruden et al., 1997) and then mated with virgin females of the genotype w1118; TM2/TM6,Sb. Approximately 10,000 of the F1 males (w1118; */TM2 or w1118: */TM6, Sb; * represents the mutagenized third chromosome) were then mated individually to w1118; TM2/TM6,Sb virgin females to generate ~6,000 lines, each carrying a mutagenized third chromosome. From the F3 flies, males homozygous for the mutagenized chromosome (*/*) were tested for low fertility by crossing to virgin females from a wild-type stock (y1w1). From this genetic screen, approximately 50 lines were saved that have low male fertility. They were placed into complementation groups by crossing to each other in ~1,275 crosses (i.e., 1,275 = N(N + 1)/2, where N = 50). The characterization of two alleles of the same complementation group that we call X1 and X2 are presented. Details of the other male-sterile mutations isolated in the screen and phenotypic analyses of X1 and X2 will be presented elsewhere.
Males homozygous for X1 and X2 were sequenced (see Materials and Methods), producing over 90 million combined sequencing reads (~76 bp per read), ~10% of which are of insufficient quality and discarded. The remaining sequence reads represent approximately 20- to 29-fold coverage of the euchromatic DNA (Figure 1). These unique sequence reads were aligned to the reference genome (y1; cn1bw1sp1 strain, dm5.30), variant calls were performed, and 204,250 homozygous SNPs were found with a quality score greater than 70 (Figure 2). There were also 97,574 heterozygous SNPs, but they were not analyzed further because the sequenced genomic DNA samples were purified from the X1/X1 and X2/X2 homozygous flies. We found that greater than 99.99% of the homozygous SNPs were identical for X1 and X2 and these have to be common background variants because X1 and X2 were derived from the same parental strain. The remaining SNPs differ between X1 and X2 and they are associated with 141 genes, which were examined further (Figure 3, see below).
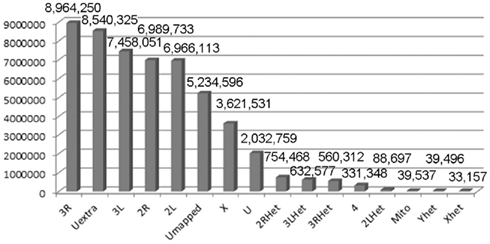
Figure 1. Mapping X1 to the reference genome. The reference genome used was the latest FlyBase version (dm5.30). The quality score was arbitrarily set at 70 and above for this table. The numbers indicate the numbers of reads mapped to the indicated genomic region. U, unmapped regions. Het, heterochromatic regions.
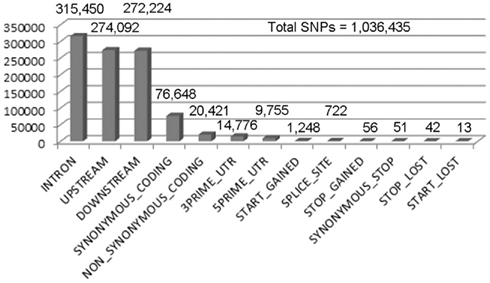
Figure 2. Single nucleotide polymorphism calling for X1 SNPs with a quality score greater than or equal to 70. We performed SNP calling using Samtools, which produced 1,943,047 SNPs with a quality score > 1. Out of these, 1,036,435 are homozygous SNPs. The low quality SNPs were filtered out using an arbitrary threshold of 70 (the peak of the distribution) leaving 204,205 homozygous SNPs. A summary of the remaining homozygous SNPs found in each category is shown in the numbers above the bars.
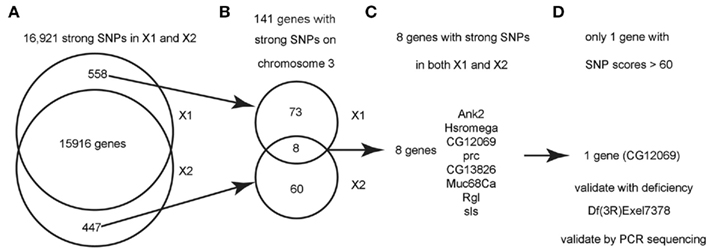
Figure 3. Flowchart for finding the causative SNPs in X1 and X2. (A) SnpEeff identified 16,921 “class 1” SNPs (see text) with a quality score > 1 in both X1 and X2 (zero quality scores are usually resulted from reads mapping to multiple genomic regions). There are 558 SNPs that are only present in X1 and 447 SNPs that are only present in X2. (B) Since we know that X1 and X2 are on chromosome 3, we focused on the 141 strong SNPs on chromosome 3 that are present in X1 or X2 but not both. There are only eight genes that are commonly affected by unique SNPs in both X1 and X2 (note that the eight genes have at least two SNPs at different bases). (C) List of the eight genes with SNPs in both X1 and X2. See Table 1 for more details. (D) Only one gene, CG12069/Pka-like, contained SNPs with scores > 60. These SNPs were validated by capillary sequencing of PCR-amplified DNA from the genetic interval of the male-sterile locus as defined by meiotic and deletion mapping data (see text). ca.
Figure 3 shows a flowchart of how the causative SNPs in X1 and X2 were identified. In order to identify the phenotype-causative candidate SNPs, we first assumed that they change an amino acid, splice site, reading frame, start or stop codon since these types of SNPs potentially alter the activity of the protein produced (we call these class 1 SNPs). Other types of SNPs such as intronic, intergenic, 5′UTR, 3′UTR, upstream, and downstream are less likely to affect gene function and they are considered secondarily only if no candidate genes could be identified from the first category of SNPs (we call these class 2 SNPs). Second, we considered the differential SNPs that are unique to either X1 or X2, but not common for X1 and X2 (Figure 3A). The way that the male-sterile screen was conducted ensured that X1 and X2 carried independently mutagenized chromosomes, so it is very unlikely that they have identical phenotype-causative SNPs (see Materials and Methods). Out of the 16,921 class 1 SNPs in X1 and X2, we found that 558 SNPs are uniquely present in X1 and 447 SNPs are uniquely present in X2 (Figure 3A). For this analysis, thresholds above a certain level, such as 70, were not used because we did not want to eliminate a candidate SNP because it fell below an arbitrary threshold. For Figure 2, for illustrative purposes, we used a threshold score of 70, based on the quality score distribution for this sequencing run (McCarthy, 2010). Quality score, is defined by SAMtools as the probability of error in decibels, that is q = −10 log(p), where p is the error probability and the logarithm is in base 10. Typically range for quality scores is from 1 to 100 with the higher score having a greater probability of being a real SNP and, therefore, not a sequencing artifact (McCarthy, 2010).
Next, we analyzed only the class 1 SNPs on the chromosome 3 since the X1 and X2 mutant strains were generated by using the third chromosome balancer (Figure 3B). As a general exercise, we did not begin our analysis by focusing on the third chromosome alone because this may not be applicable to other experimental settings. Considering just the third chromosome, there are 81 class 1 SNPs associating with 81 genes in X1, and 68 class 1 SNPs in 68 genes in X2. Of most interest are the eight genes that are commonly affected in both X1 and X2; i.e., the SNPs differ, but these SNPs associate with the same eight genes. Since the male-sterile phenotypes of X1 and X2 are presumably caused by two different SNPs affecting the same gene, we focused on these eight genes, which are Ank2, Hsromega, CG12069, prc, CG13826, Muc68Ca, Rgl, and sls (Figure 3C; Table 1). However, CG12069 has SNPs with scores of 102 in X1 and 66 in X2 (Table 1). The score of 66 can be considered significant and it is substantially higher than the scores for the other seven candidate genes which have scores ranging from 1 to 36 with the majority having scores less than 5 (Table 1). CG12069 was named as Pka-like in the Flybase because it encodes a protein with 51% amino acid identity to the adjacent Pka-C2 which encodes a cAMP-dependent protein kinase A catalytic subunit (Figure 4A).

Table 1. Gene candidates for X1 and X2.
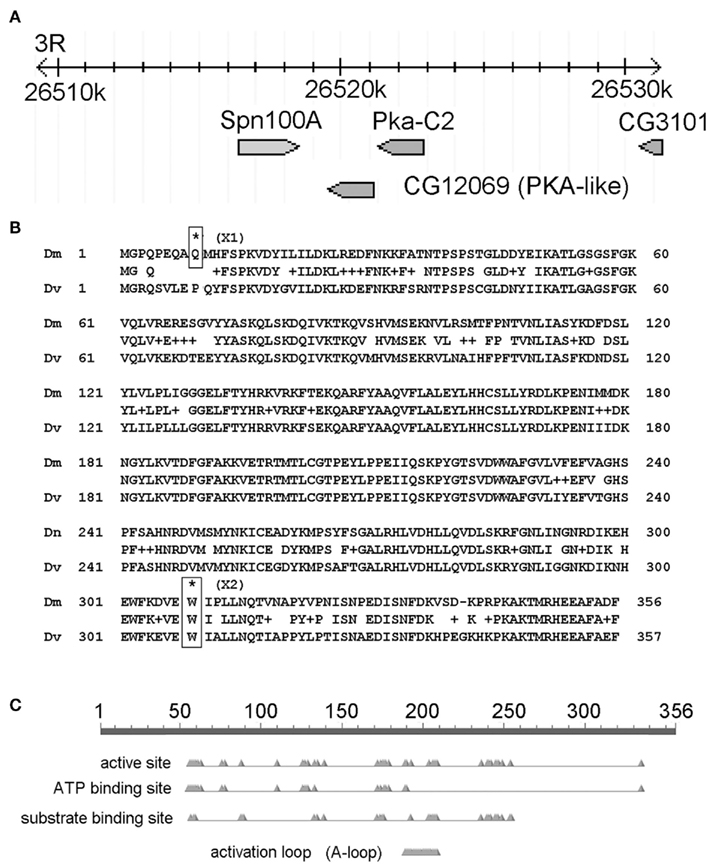
Figure 4. The candidate gene mutated in X1 and X2 is CG12069/Pka-like. (A) Map of the CG12069/Pka-like region on chromosome 3R. The image is adapted from the FlyBase genome browser. The genomic location (26,520 k) is indicated in kilobase pairs. (B) Location of X1 and X2 SNPs. (C) Conserved domains in CG12069/Pka-like.
Further analysis of the two SNPs in CG12069 of X1 and X2 indicated that both of them are nonsense mutations causing pre-mature translational termination at different amino acid residues of the Pka-like protein. X1 contains a TGG/TGA SNP that converts the tryptophan (W) residue 308 to a stop codon whereas X2 contains a CAG/TAG SNP that converts the glutamine (Q) residue 9 to a stop codon (Figure 4B). X1 will make the first 308 out of 356 amino acids of Pka-like. However, the Pka-like function is likely diminished because the conserved region of Pka-like with Drosophila virilis extends beyond amino acid 308. Also, the conserved ATP-binding domain of Pka-like extends beyond amino acid 308 (Figure 4C). X2 will only make the first eight amino acids of Pka-like, but there is another in-frame ATG codon at amino acid 10 that, if it supports translation initiation, would make a functional protein. However, there is a poor match to the Kozak consensus sequence, 5′-ACC-ATG-G-3′, flanking the downstream ATG site, 5′-CAG-ATG-C-3′. Since a good match to the Kozak sequence is generally required for efficient translation, (Kozak, 1987) it is possible that the downstream ATG is not used for translation. We note that the correct translation start sequence, 5′-GCA-ATG-C-3′, has a slightly better match to the Kozak sequence.
Since the male-sterile phenotypes of X1 and X2 homozygotes are nearly as strong as that of the males of the mutation over Df(3R)Exel7378 that deletes CG12069, it is likely that the pre-mature stop codon mutations in CG12069 are the causative loss-of-function mutations. To confirm this, we crossed X1 or X2 with chromosomal deletions that overlap with Df(3R)Exel7378. We found that the male-sterile phenotypes of X1 and X2 failed to complement Df(3R)Exel7378 (3R:26388946;26620677), but complemented Df(3R)BSC504 (3R:26253789;26512985) and Df(3R)Exel8194 (3R:26582117;26713967). These localize the genetic boundary of X1 and X2 to a 69,132-bp of DNA interval from 26,512,985 to 26,582,1174. The ~69 kb of DNA encodes 10 annotated genes, of which five are highly expressed in the testis, including CG12069. No SNPs were found in the remaining four candidate genes expressed in the testes (CG12066, CG31010, CG1340, CG15543), suggesting that CG12069 is a strong candidate gene for the sperm storage defects of X1 and X2.
To further confirm the SNPs identified by SnpEff and SnpSift, genomic DNA samples were isolated from X1 and X2 homozygous mutant males and regions containing exons were amplified by polymerase chain reaction (PCR), cloned into pGEMT (Promega), and sequenced by capillary DNA sequencing (Applied Biosystems, Inc.). Sequencing confirmed the presence of stop codon SNPs in CG12069 in both X1 and X2 at the expected locations. Thus, we conclude that the male-sterile alleles of X1 and X2 probably contain mutations in the CG12069 gene. Complete validation will require a CG12069 rescue transgene that is expressed in the male testes. However, phenotypic rescue of the male-sterile and sperm motility phenotypes of X1 and X2 is beyond the scope of this paper and will be presented elsewhere.
In this paper, we show that SnpEff and SnpSift can be used to identify causative SNPs in EMS-generated alleles of a new male-sterile mutant locus that we isolated from random chemical mutagenesis screens. We performed whole-genome shotgun sequencing of the two non-complementing alleles, X1 and X2, and showed that only a single gene, CG12069/Pka-like, was affected by SNPs at two different places, generating two different truncated proteins. The SNPs were confirmed by PCR amplification and capillary sequencing and further genetic mapping of the mutant locus using overlapping chromosomal deletions. From these, we conclude that a single lane of next-generation sequencing on the GAIIx instrument is probably sufficient for identifying homozygous causative SNP candidates in Drosophila. It should be emphasized that, in this case, we sequenced the DNA from homozygous flies. We were also able to use this technique to identify heterozygous SNPs isolated in a separate genetic screen (data not shown; Ruden et al., 1999). It was lucky that X1 and X2 were both nonsense mutations that designate strong SNPs and these occurred at two different codon positions in the same gene. Nevertheless, SnpEff and SnpSift can also analyze weak SNPs such as those located in the 5′ UTR or promoter regions and it should be possible to use a similar strategy to identify mutations that contain SNPs at regulatory regions of the genes, such as in many examples of population studies.
Recently, the Bellen laboratory developed rapid meiotic mapping techniques to map a recessive-lethal mutation to within a few kilobases to transposons containing easily visualized marker genes such as mini-w+ or y+ (Zhai et al., 2003). Meiotic mapping can be used to further delimit the regions of the genome and facilitate identification of candidate genes by whole-genome sequencing approach. We know of at least one other laboratory that has used next-generation sequencing to identify chemically induced mutations in Drosophila, but this was done with PCR-amplified DNA fragment from the ~1-Mbp region of interest (Wang et al., 2010). Deficiencies, such as in the Exelixis and DrosDel collections that have known breakpoints, (Parks et al., 2004; Ryder et al., 2007) can be used to fine map the mutant locus further, often to a region small enough to PCR amplify and sequence with conventional capillary sequencing techniques.
Although we sequenced homozygous DNA, it is conceivable that larger fold of sequence coverage should overcome complication of data resulting from sequencing heterozygous DNA when the mutation is lethal. Langley et al. (2011) have recently shown that one can “circumvent heterozygosity” by sequencing the genome of a single haploid D. melanogaster embryo. The haploid embryo is gynogenetically produced by mating females with males homozygous for the recessive male-sterile mutation ms(3)K81, which jumps start embryogenesis without incorporating the sperm DNA in the developing embryo (Langley et al., 2011). Another alternative method to circumvent heterozygosity for recessive-lethal mutations is to use “green balancers” that carry, for example, Kr-Gal4 driving GFP expression in the embryo and thus allowing the enrichment of homozygous mutant embryos prior DNA sequencing (Casso et al., 1999, 2000). The Bloomington stock center has green balancer stocks for the X chromosome (FM7), the second chromosome (CyO), and the third chromosome (TM3,Sb5). When a recessive-lethal allele is balanced with a green balancer, one needs only to select for non-GFP expressing embryos to ensure that the flies are homozygous in genotypes (Casso et al., 1999, 2000).
In summary, we describe a new tool, SnpSift that can be used to help identify causative SNPs in mutants derived from random chemical mutagenesis screens. This tool, along with SnpEff, has currently set to analyze and identify SNPs associated with phenotypes of not only Drosophila mutant strains but also other organisms including humans.
Drosophila genomic DNA from the strains X1 and X2 was prepared using an AutoPure LS (Qiagen) Kit. A genomic DNA library was prepared from 5 μg purified Drosophila DNA according to the standard protocol using a Paired-End Sample Prep Kit for the GAIIx (Illumina). The DNA library was then used for cluster generation and sequencing analysis using the Genome Analyzer IIx using Illumina standard protocols. Methods for DNA manipulation, including sample preparation, formation of single-molecule arrays, cluster growth, and sequencing were all done by the standard protocols from Illumina, Inc. All sequencing was performed using two lanes (one for X1 and one for X2) in paired-end sequencing mode on an Illumina Genome Analyzer version 2 (GA2X) that was equipped with a 1-megapixel camera. The Illumina sequencing kits used allowed for 76 base single-end reads. Each lane of DNA sequencing had over 90 million reads.
Image analysis software was provided as part of the Genome Analyzer analysis pipeline and configured for fully automatic parameter selection. Single-end reads were 76 bases in total length. Quality control was performed using FastQC, showing overall low error rates. The reference genome used was the latest FlyBase version at the time (y1; cn1bw1sp1 strain, Dm5.30). The data was aligned using the BWA algorithm (Li and Durbin, 2009). A total of 5,234,506 reads were NOT mapped to the genome (i.e., 10.01%). This is usually due to low quality reads or reads have missing base calling information (i.e., “B” in the quality stream). The rest of the reads for X1 and X2 were mapped as indicated. Gap estimation: according to the mapping software, the gap between pair-end reads is 360 ± 20 bp. The distribution percentiles are 345 (25%), 360 (50%), and 375 (75%). The set of6 and to the NCBI’s map of RefSeq and candidate Drosophila genes7.
Reads were filtered using a minimum mapping quality of 20 (MAPQ). Variant calling was performed using SamTools (Li et al., 2009) and BcfTools. When using individual calls without base alignment quality (BAQ) model, (Li, 2011) a total of 1,036,435 homozygous SNPs were detected. Using multi-sample calling methods and BAQ model, (Li, 2011) the number of homozygous SNPs was reduced to 204,250. Variant annotation and filtering was performed using the software SnpEff (Cingolani et al., Fly, in press) and SnpSift, described below.
Variant filtering was performed using an in-house development tool set called SnpSift8. This tool set works almost exclusively on variant call format (VCF) files according to the specification for versions 4 or 4.1 (Danecek et al., 2011). The two main components used in this work were “SnpSift caseControl” and “SnpSift filter.” Frequently asked questions (FAQs) are addressed on our web site.
This tool counts the number of genotypes present in two user-defined groups (“case” and “control”), and then it calculates a p-value based on Fisher exact test. For each group, either homozygous, heterozygous, or both kinds of variants can be used.
This module performs filtering based on arbitrary expressions. In order to be able to parse arbitrary expressions, we created a top-down recursive grammar [also known as LL(*) grammar] using ANTLR (Parr, 2007). Using the lexer and parser created by ANTLR we are able to parse expressions by creating an abstract syntax tree (AST) for the expression. An AST is a well-known structure, very common in compiler design, that is used to represent the arbitrary input expressions from the user. The AST tree is converted into an interpreter syntax tree (IST), which is a tree composed of objects capable of interpreting conditions, expressions, and functions. This means that the IST is like AST, but it is also capable of performing expression evaluation. The result of the filter expression is the value of the root node in the IST.
There are well-known variables pre-defined according to the VCF format specification. Other additional variables and their respective data types are parsed from VCF meta-information in the file header. As specified in the norm, INFO meta-information lines define the type and the number of values (e.g., an array) in each INFO sub-field. Automatic variable conversion is implemented (e.g., INT is automatically converted to FLOAT whenever required). Genotype fields are similarly parsed by using FORMAT meta-information header lines.
Each VCF entry (i.e., each non-header line in a VCF file) is converted into a set of “variable = value” tuples, which are feed into the interpreter tree. The IST, created using the user expression, interprets the user-defined expression from top to bottom trying to assign a Boolean value to the root node. If the result from evaluating the IST is “true” then the VCF line is either printed to standard output or marked as PASS in the FILTER field; likewise, if it is “false,” the line is filtered out (i.e., not printed) or marked as failed in the FILTER field. Table A1 in Appendix shows a list of allowed operators used in SnpSift and Table A2 in Appendix shows some functions commonly used in SnpSift expressions. Language definition and examples are shown in Appendix.
SnpSift is platform independent and available as an open source as part of the SnpEff project9. A web based interface is available via the Galaxy project (see text foot note 1).
SnpEff and SnpSift Data can be accessed from the data file for X1 and X2 by contacting Douglas M. Ruden.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by a Michigan Core Technology grant from the State of Michigan’s 21st Century Fund Program to the Wayne State University Applied Genomics Technology Center. This work was also supported by the Environmental Health Sciences Center in Molecular and Cellular Toxicology with Human Applications Grant P30 ES06639 at Wayne State University, NIH R01 grants (ES012933) to Douglas M. Ruden, and DK071073 to Xiangyi Lu.
Anderson, K. (1992). The Making of a fly – the genetics of animal design – Lawrence, Pa. Science 256, 1053–1054.
Casso, D., Ramirez-Weber, F., and Kornberg, T. B. (2000). GFP-tagged balancer chromosomes for Drosophila melanogaster. Mech. Dev. 91, 451–454.
Casso, D., Ramirez-Weber, F. A., and Kornberg, T. B. (1999). GFP-tagged balancer chromosomes for Drosophila melanogaster. Mech. Dev. 88, 229–232.
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., Handsaker, R. E., Lunter, G., Marth, G. T., Sherry, S. T., McVean, G., Durbin, R., and 1000 Genomes Project Analysis Group. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158.
Hillier, L. W., Marth, G. T., Quinlan, A. R., Dooling, D., Fewell, G., Barnett, D., Fox, P., Glasscock, J. I., Hickenbotham, M., Huang, W., Magrini, V. J., Richt, R. J., Sander, S. N., Stewart, D. A., Stromberg, M., Tsung, E. F., Wylie, T., Schedl, T., Wilson, R. K., and Mardis, E. R. (2008). Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods 5, 183–188.
Kozak, M. (1987). An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148.
Langley, C. H., Crepeau, M., Cardeno, C., Corbett-Detig, R., and Stevens, K. (2011). Circumventing heterozygosity: sequencing the amplified genome of a single haploid Drosophila melanogaster embryo. Genetics 188, 239–246.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R., and 1000 Genome Project Data Processing Subgroup. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078.
McCarthy, A. (2010). Third generation DNA sequencing: pacific biosciences’ single molecule real time technology. Chem. Biol. 17, 675–676.
Parks, A. L., Cook, K. R., Belvin, M., Dompe, N. A., Fawcett, R., Huppert, K., Tan, L. R., Winter, C. G., Bogart, K. P., Deal, J. E., Deal-Herr, M. E., Grant, D., Marcinko, M., Miyazaki, W. Y., Robertson, S., Shaw, K. J., Tabios, M., Vysotskaia, V., Zhao, L., Andrade, R. S., Edgar, K. A., Howie, E., Killpack, K., Milash, B., Norton, A., Thao, D., Whittaker, K., Winner, M. A., Friedman, L., Margolis, J., Singer, M. A., Kopczynski, C., Curtis, D., Kaufman, T. C., Plowman, G. D., Duyk, G., and Francis-Lang, H. L. (2004). Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292.
Parr, T. (2007). The Definitive ANTLR Reference: Building Domain-Specific Languages. Lewisville: Pragmatic Bookshelf.
Platts, A. E., Land, S. J., Chen, L., Page, G. P., Rasouli, P., Wang, L., Lu, X., and Ruden, D. M. (2009). Massively parallel resequencing of the isogenic Drosophila melanogaster strain w (1118); iso-2; iso-3 identifies hotspots for mutations in sensory perception genes. Fly (Austin) 3, 192–203.
Ruden, D. M., Cui, W., Sollars, V., and Alterman, M. (1997). A Drosophila kinesin-like protein, Klp38B, functions during meiosis, mitosis, and segmentation. Dev. Biol. 191, 284–296.
Ruden, D. M., Wang, X., Cui, W., Mori, D., and Alterman, M. (1999). A novel follicle-cell-dependent dominant female sterile allele, StarKojak, alters receptor tyrosine kinase signaling in Drosophila. Dev. Biol. 207, 393–407.
Ruden, D. M., Xiao, L., Garfinkel, M. D., and Lu, X. (2005). Hsp90 and environmental impacts on epigenetic states: a model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum. Mol. Genet. 14, R149–R155.
Ryder, E., Ashburner, M., Bautista-Llacer, R., Drummond, J., Webster, J., Johnson, G., Morley, T., Chan, Y. S., Blows, F., Coulson, D., Reuter, G., Baisch, H., Apelt, C., Kauk, A., Rudolph, T., Kube, M., Klimm, M., Nickel, C., Szidonya, J., Maróy, P., Pal, M., Rasmuson-Lestander, A., Ekström, K., Stocker, H., Hugentobler, C., Hafen, E., Gubb, D., Pflugfelder, G., Dorner, C., Mechler, B., Schenkel, H., Marhold, J., Serras, F., Corominas, M., Punset, A., Roote, J., and Russell, S. (2007). The DrosDel deletion collection: a Drosophila genomewide chromosomal deficiency resource. Genetics 177, 615–629.
Wang, H., Chattopadhyay, A., Li, Z., Daines, B., Li, Y., Gao, C., Gibbs, R., Zhang, K., and Chen, R. (2010). Rapid identification of heterozygous mutations in Drosophila melanogaster using genomic capture sequencing. Genome Res. 20, 981–988.
Yang, Y., Cochran, D. A., Gargano, M. D., King, I., Samhat, N. K., Burger, B. P., Sabourin, K. R., Hou, Y., Awata, J., Parry, D. A., Marshall, W. F., Witman, G. B., and Lu, X. (2011). Regulation of flagellar motility by the conserved flagellar protein CG34110/Ccdc135/FAP50. Mol. Biol. Cell 22, 976–987.
Zhai, R. G., Hiesinger, P. R., Koh, T. W., Verstreken, P., Schulze, K. L., Cao, Y., Jafar-Nejad, H., Norga, K. K., Pan, H., Bayat, V., Greenbaum, M. P., and Bellen, H. J. (2003). Mapping Drosophila mutations with molecularly defined P element insertions. Proc. Natl. Acad. Sci. U.S.A. 100, 10860–10865.
This section shows the language definition for SnpSift filter. Operators (see Table A1) and functions (see Table A2) can be used to create arbitrary expressions that are evaluated using the information in each VCF line.
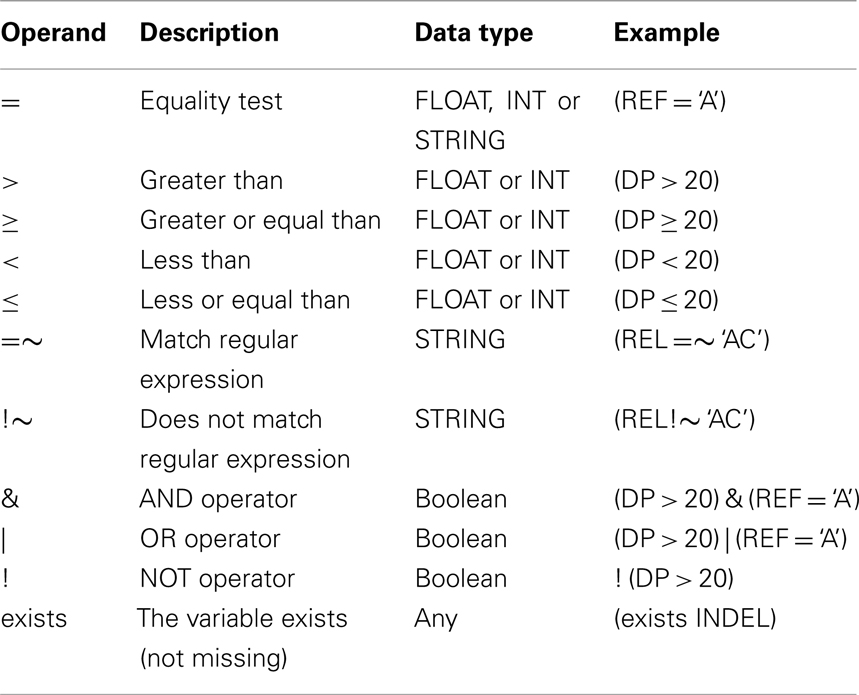
Table A1. Operators allowed in SnpSift filter.
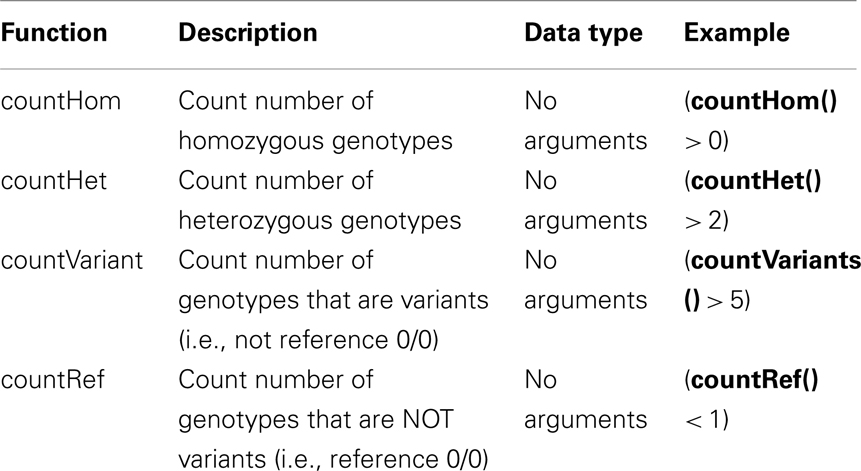
Table A2. Functions implemented in SnpSift filter.
Using the SnpSift filter, arbitrary expressions can be evaluated. Since an arbitrary number of conditions can be combined using Boolean operators, the expressions can be complex, allowing significant flexibility.
Some examples:
1-) Filter out variants with quality less than 30:
cat variants.vcf | java -jar SnpSift.jar ″ ( QUAL >= 30 )″ > filtered.vcf
2-)Filter out variants with quality less than 30 but keep InDels that have quality 20 or more:
cat variants.vcf | java -jar SnpSift.jar ″(( exists INDEL ) & (QUAL > = 20)) | (QUAL > = 30 )″ > filtered.vcf
3-)Same as example 2, but keeping also any homozygous variant present in more than 3 samples:
cat variants.vcf | java -jar SnpSift.jar ″(countHom > 3) | (( exists INDEL ) & (QUAL > = 20)) | (QUAL > = 30 )″ > filtered.vcf
4-)Same as example 3, but keeping also heterozygous variants with coverage 25 or more:
cat variants.vcf | java -jar SnpSift.jar ″((countHet > 0) && (DP > = 25)) | (countHom > 3) | (( exists INDEL ) & (QUAL > = 20)) | (QUAL > = 30 )″ > filtered.vcf
For each VCF entry, the variables are populated and made available in the analyzed expressions. The values used to populate the variables are obtained from different fields of the VCF entry. There are four main groups of variables:
• Fields: these are mandatory valued from the VCF specification and are the first columns in a VCF file (“CHROM, POS, ID, REF, ALT, QUAL, or FILTER”).
• INFO field: each value defined in the info field is made available using the type specified according to the VCF meta-information lines in the header section. Some “well-known” variables are pre-defined and do not need corresponding header entries (see VCF specification for a list of well-known INFO fields).
• Genotype fields: each genotype field is available using the GEN[] array. Subfields of this array include all variables in each genotype field. Types are casted according to the VCF meta-information lines in the header section.
• Effect fields: the “EFF” sub-field from the INFO field (created by SnpEff program) is further parsed and made available. This is parsed as an array since one variant can be annotated with more than one effect.
• Sets: expressions can test if a value belongs to a set. Sets are defined in files having one value per line. This files are parsed when using the “–set” command line option. Values from sets can be used in expressions by using the “in” operator.
Available variable names are: “CHROM, POS, ID, REF, ALT, QUAL, or FILTER.”
Examples:
1-) Any variant in chromosome 1:
″( CHROM = ’chr1’ )″
2-) Variants between two positions:
″( POS > 123456 ) & ( POS < 654321 )″
3-) Variants having an ID and it matches the regular expression “rs”:
″(exists ID) & ( ID = ’rs’ )″
4-) Variants having reference “A”:
″( REF = ’A’ )″
5-) Variants having an alternative “T”:
″( ALT = ’T’ )″
6-) Variants having quality over 30:
″( QUAL > 30 )″
6-) Variants having Filter value is either “PASS” or it is missing:
″( na FILTER ) | (FILTER = ’PASS’)″
Variable names from INFO field. E.g., if the info field has “DP=48;AF1=0;…” e.g.,:
( DP > 10 ) & ( AF1 = 0 )
Info field variables can have multiple values (comma separated). These multiple valued fields are represented as an array. Individual values can be accessed using an index. E.g., If the INFO field has “CI95=0.04167,0.5417,” then the following expression is valid:
″( CI95[0] > 0.1 ) & (CI95[1] < = 0.3)″
An asterisk may be used to represent “ANY” variable index. So the following example is “true” if any of the values in the CI95 field is more than 0.1:
″( CI95[*] > 0.1 )″
Variables from genotype fields are represented as an array. The individual values are accessed using an index (sample number) followed by a variable name. E.g., If the genotypes are “GT:PL:GQ 1/1:255,66,0:63 0/1:245,0,255:99,” then the following expression is “true”:
″( GEN[0].GQ > 60 ) & ( GEN[1].GQ > 90 )″
An asterisk may be used to represent “ANY” variable index
″( GEN[*].GQ > 60 )″
These are represented as arrays, so individual values can be accessed using an index (sample number) followed by a variable name and then another index. E.g., If the genotypes are “GT:PL:GQ 1/1:255,66,0:63 0/1:245,0,255:99,” then the following expression is valid:
″( GEN[0].PL[2] = 0 )″
Also in this case, an asterisk may be used to represent “ANY” variable index, e.g.,:
″( GEN[0].PL[*] = 0 )″
And another asterisk may be used to represent “ANY” genotype index, e.g.,:
″( GEN[*].PL[*] = 0 )″
are defined by the “-s” (or “–set”) command line option. Each file must have one string per line. They are named based on the order used in the command line (e.g., the first one is “SET[0],” the second one is “SET[1],” etc.) An example of the set expression (assuming your command line was “-s set1.txt -s set2.txt -s set3.txt”):
″( ID in SET[2] )″
Effect fields created by SnpEff are accessed using an index (effect number) followed by a sub-field name. Available sub-field are:
• EFFECT: effect (e.g., SYNONYMOUS_CODING, NON_SYNONYMOUS_CODING, FRAME_SHIFT, etc.)
• IMPACT: [ HIGH, MODERATE, LOW, MODIFIER ]
• FUNCLASS: [ NONE, SILENT, MISSENSE, NONSENSE ]
• CODON: codon change (e.g., “ggT/ggG”)
• AA: amino acid change (e.g., “G156”)
• GENE: gene name (e.g., “PSD3”)
• BIOTYPE: gene biotype, as described by the annotations (e.g., “protein_coding”)
• CODING: gene is [ CODING, NON_CODING ]
• TRID: transcript ID
• EXID: exon ID
Examples:
1-) The following expression is true if the first effect is NON_SYNONYMOUS:
″( EFF[0].EFFECT = ’NON_SYNONYMOUS_CODING’ )″
2-) This expression is true if ANY effect is NON_SYNONYMOUS:
″( EFF[*].EFFECT = ’NON_SYNONYMOUS_CODING’ )″
3-) This expression is true if ANY effect is NON_SYNONYMOUS on gene TCF7L2:
″( EFF[*].EFFECT = ’NON_SYNONYMOUS_CODING’ ) & ( EFF[*].GENE = ’TCF7L2’ )″
Keywords: personal genomes, Drosophila melanogaster, whole-genome SNP analysis, next-generation DNA sequencing
Citation: Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM and Lu X (2012) Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Gene. 3:35. doi: 10.3389/fgene.2012.00035
Received: 20 February 2012; Accepted: 24 February 2012;
Published online: 15 March 2012.
Edited by:
Michael Aschner, Vanderbilt University Medical Center, USAReviewed by:
Michael Aschner, Vanderbilt University Medical Center, USACopyright: © 2012 Cingolani, Patel, Coon, Nguyen, Land, Ruden and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Douglas M. Ruden, Department of Obstetrics and Gynecology, Wayne State University School of Medicine, C.S. Mott Center, Detroit, MI 48201, USA. e-mail:ZG91Z2xhc3JAd2F5bmUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.