

- Metabolic Research Laboratories, Institute of Metabolic Science, Addenbrooke’s Hospital, University of Cambridge, Cambridge, UK
Metabolic disorders have seen an increased prevalence in recent years in developed as well as developing countries. While it is clear lifestyle choices and habits have contributed to this epidemic, mounting evidence suggests the nutritional milieu during critical stages of development in early life can “program” individuals to develop the metabolic syndrome later in life. Extensive epidemiological data presents an association between maternal obesity and nutrition during pregnancy and offspring obesity, and a number of animal models have been established in order to uncover the underlying mechanisms contributing to the programming of physiological systems. It is hard to distinguish the causal factors due to the complex nature of the maternal–fetal relationship; however, in order to develop adequate prevention strategies it is vital to identify which maternal factor(s) – be it the diet, diet-induced obesity or weight gain – and at which time during early development instigate the programmed phenotype. Curtailing the onset of obesity at this early stage in life presents a promising avenue through which to stem the growing epidemic of obesity.
Introduction
Recent decades have seen a rapid rise in metabolic disorders throughout the world. Given the health risks associated with obesity (often grouped as the metabolic syndrome), with 68% of the US adult population reported overweight and obese in 2008 (Flegal et al., 2010) and 60% of the UK population predicted to be obese by 2050 (Musingarimi, 2009), it is presenting itself as a significant health challenge. The rise in obesity, diabetes, and related disorders has been primarily attributed to changes in lifestyle and diet; with increased food intake and reduced energy expenditure leading to the storage of excess energy in the form of fat. Genome wide association studies (GWAS) have highlighted the importance of an individual’s genetic make up and their predisposition to metabolic disorders (Frayling, 2007). However, the rapid rise in obesity suggests it is being driven by environmental factors. Although much focus has been directed toward the role of current diet, recent insights have also highlighted the importance of nutrition during early life in the development of metabolic disorders. It is now well established that the phenotype of an individual can be driven by in utero and early postnatal environmental conditions, such as the nutritional state of the mother. This has given rise to the notion of “developmental programming” or the “developmental origins of health and disease” (DOHaD) that proposes that the conditions presented during a critical window of development can lead to permanent programmed alterations in physiological systems.
Early Life Programming
Maternal Undernutrition and the “Thrifty Phenotype” Hypothesis
A wealth of evidence from epidemiological studies has revealed a relationship between birth weight and increased risk of metabolic disease (reviewed in Jones and Ozanne, 2009). Early studies focused on the effect of maternal undernutrition and low offspring birth weight. Forsdahl hypothesized that poverty in childhood or adolescence programs the organism for low energy intake and not for today’s high-energy food that is readily available. He therefore suggested that changes in food availability in recent times contributed to the increased incidence of cardiovascular disease (Forsdahl, 1977). Further studies of a Hertfordshire cohort, of 64-year-old men in the UK, demonstrated that individuals with low birth weight were at increased risk of heart disease (Barker and Osmond, 1986), glucose intolerance/type 2 diabetes (Hales et al., 1991) and metabolic syndrome. This was interpreted as demonstrating that the fetal environment played an important role in the risk of metabolic disease (Barker et al., 1993). These observations formed the basis of the “thrifty phenotype” hypothesis by Hales and Barker (1992). They proposed that poor fetal nutrition drives metabolic adaptations to maximize the chances of survival in conditions of on-going nutritional deprivation. These adaptations would be beneficial in continued poor conditions post-birth, but are not suited to a postnatal environment providing plentiful nutrition; and therefore increasing risk of developing type 2 diabetes and other features of the metabolic syndrome.
Maternal Obesity
Given the surge in obesity, maternal obesity, and consequently gestational diabetes, recent studies have focused on the potential detrimental effects of fetal overnutrition. These have revealed a relationship between maternal obesity and the onset of the metabolic syndrome in the offspring. Many epidemiological studies have shown an association between maternal BMI [a proxy measure of human body fat based on an individual’s weight/(height)2] and gestational diabetes with offspring adiposity (Lake et al., 1997; Laitinen et al., 2001; Boney et al., 2005). A possible explanation for the higher incidence of overweight and obesity in offspring exposed to maternal diabetes and obesity is that these women transmit a greater number of susceptibility genes to their offspring compared to normal weight women. However, sibpair studies in Pima Indians discordant for maternal diabetes have shown a greater prevalence for type 2 diabetes and higher BMI in siblings born after the mother had been diagnosed with type 2 diabetes (Dabelea et al., 2000). Maternal weight gain and increased BMI between pregnancies has also been found to increase the risk of overweight in offspring compared with their siblings (Villamor and Cnattingius, 2006). Furthermore, interventional strategies to reduce the weight of obese women via bariatric surgery reduce the risk of obesity and other adverse effects in subsequent offspring compared to those born before maternal surgery (Kral et al., 2006; Smith et al., 2009). These data show a disproportionate risk in offspring from the same mother under different in utero conditions and reinforces the concept that environmental conditions, both in addition to and independent of genetic susceptibilities, are capable of physiologically programming offspring during development.
These findings are relevant since the rates of overweight and obesity in women of childbearing age continue to increase worldwide. In the USA the prevalence of obese women aged 20–39 years old tripled between 1960 and 2000 (Flegal et al., 2002). Similarly in the UK, between 1990 and 2004 the BMI of pregnant women at their first prenatal booking was increased by an average of 1.37 kg/m2 and the prevalence of obesity tripled (Kanagalingam et al., 2005). This presents a worrying picture of a cyclical transgenerational transmission of obesity. Obesity is also associated with a whole host of metabolic abnormalities including hypertension, hepatic steatosis, and cardiovascular disease, making it a pertinent issue.
Early Postnatal Growth and Overnutrition
The early postnatal period also appears to be a key time point in which offspring development can be influenced. The postnatal period is a period of continual growth and development, and rapid growth, as well as factors associated with it increase obesity risk. Previous studies have implicated rapid postnatal growth following maternal undernutrition leads to adult onset obesity, with extensive research attributing this to the developmental effects of undernutrition on predisposing offspring to obesity owing to discordant nutrition post-birth (reviewed in Chen et al., 2010). However, rapid postnatal growth in itself is associated with increased risk of obesity and metabolic traits in adulthood (Baird et al., 2005; Tzoulaki et al., 2010). Weight gain during infancy has been associated with adiposity, blood pressure, fasting triglyceride levels, and other metabolic risk factors independent of birth weight (Ekelund et al., 2007).
The above findings suggest factors before or during infancy that are related to infant growth influence the risk of later obesity. Dietary composition during this period has been shown to affect growth patterns, such as the use of formula milk and its composition. Nutrient enriched formula feed is commonly used as an alternative or supplement to breastfeeding and has been implicated in neonatal and infant growth acceleration and subsequent obesity risk (Stettler et al., 2005; Singhal et al., 2010). Formula milk feeding results in increased dietary intake at 4 months of age and increased risk of childhood obesity (Ong et al., 2006) as well as risk of overweight (Harder et al., 2005) and high cholesterol in adulthood (Singhal et al., 2004) compared to breast feeding. Rapid growth has been attributed to differences in intake of metabolisable substrates, namely protein. Children fed protein enriched formula feed had higher body weight gain in the first 2 years of life compared to low protein and breast-fed children (Koletzko et al., 2009).
Therefore, developmental programming is not limited to the in utero environment and as physiological systems continue to develop after birth, overnutrition may have a considerable impact. The associations between early growth and obesity risk have been made through epidemiological studies, and insights into the contributory factors and mechanisms are starting to emerge.
Insights from Animal Models
Although investigations of human cohorts are of great value in establishing associations between maternal nutritional status and offspring outcome, they are confounded by the influence of uncontrollable variables of genetic and environmental origin. Here, the use of animal models have provided an invaluable insight into the mechanisms of developmental programming, as the conditions to which the fetuses are exposed can be controlled and tissue sampling from the mother, fetus, or offspring can be performed during various stages of the exposure. A great deal of research is now being undertaken in order to understand the mechanisms underlying developmental programming in response to maternal obesity. A variety of animal models have been established for the purpose of dissecting the mechanisms leading to the onset of increasing adiposity. By far the most common models used are of rodents, however larger animals have also been used, both providing important insight into underlying mechanisms.
Non-human primate models have the advantage of their close relation to the human species and greater similarities in terms of development patterns during gestation. However, long gestation periods and increased costs for the maintenance of larger animals render them less favorable. An early report in baboons showed that overfeeding during early infancy led to elevated triglyceride levels and increased adiposity through hypertrophy of adipocytes in adulthood (Lewis et al., 1986). Others showed changes in placental morphology and function of diet-induced obese mothers, similar to those described for large for gestational age human fetuses, which may result in increased nutrient delivery to the fetus (Farley et al., 2009). Rhesus macaques are also a well-characterized model used to study maternal overnutrition. One study found fetal offspring of chronically high fat diet (HFD) fed mothers developed features of non-alcoholic fatty liver disease including triglyceride accumulation, which persisted into the postnatal period and included a two-fold increase in percent body fat. The effects were prevented in offspring from subsequent pregnancies after switching the mother to the control diet (McCurdy et al., 2009) reinforcing it as a developmental effect in response to the environment.
Sheep models have also been widely studied. Similar to the primate models, maternal diet-induced obesity (DIO) has been shown to cause increased placental fatty acid transporter expression resulting in higher triglyceride levels in fetuses (Zhu et al., 2010a) as well as upregulated inflammatory signaling (Zhu et al., 2010b). Other studies demonstrate upregulated lipogenic genes (Muhlhausler et al., 2007a), increased fat deposition in the offspring and defects in skeletal muscle development (Zhu et al., 2008; Tong et al., 2009; Yan et al., 2010). Since the skeletal muscle is a major depot of glucose stores, reduced development would be expected to result in insulin resistance.
Rodents are predominantly the species of choice in establishing models of maternal overnutrition. Advantages include their short gestation period and lifespan, cost effectiveness, and broad similarities to humans in terms of diet, albeit differences in gestational development. However, drawbacks to these models have been established. The term “overnutrition” can be interpreted in a number of different ways, and is reflected in the variety of animal models published. Early studies of dietary manipulation involved feeding dams HFDs. Offspring of high fat-fed dams demonstrate increased adiposity, insulin resistance (Buckley et al., 2005), and hypertension (Khan et al., 2005). However, modern day dietary intake comprises high sugar as well as fat and so models reflecting this would be of greater relevance in unveiling the subsequent phenotype and underlying mechanisms. More recently, models where maternal obesity is induced by providing dams with a highly palatable high fat, high sugar obesogenic diet have been developed. Offspring of such dams are hyperphagic, insulin resistant, hypertensive, and glucose intolerant in adulthood (Samuelsson et al., 2008; Nivoit et al., 2009). Offspring of dams fed a “junk food” diet (consisting of highly palatable high street snacks such as doughnuts high in carbohydrates, sugar, and salt) demonstrate hyperphagia, a preference for “junk food,” increased weight gain after weaning onto the junk food diet (Bayol et al., 2007) and reduced skeletal muscle mass (Bayol et al., 2005). Other forms of dietary manipulation include providing a high protein diet (Daenzer et al., 2002) all of which result in the onset of components of the metabolic syndrome. Dietary manipulation in itself provides complications for interpretation, as increasing one dietary component will require reduction of another. In most cases increasing fat content is compensated for with reduced carbohydrate (Samuelsson et al., 2008; Nivoit et al., 2009) or protein content (Bayol et al., 2005). Although it is assumed that increased fat content is the causal factor of the resulting phenotype, we cannot rule out low protein or carbohydrate as contributing to the effect in such studies.
Interestingly, non-genetic paternal transmission of metabolic disorders to female offspring has been demonstrated in a rat model of chronic high fat feeding of males which results in low birth weight, adiposity, and impaired glucose tolerance (Ng et al., 2010). This suggests that paternal obesity influences offspring risk of childhood and adult obesity. Mother–child associations for obesity are significantly greater than father–child associations (Whitaker et al., 2010). Although this could reflect a role for maternally imprinted genes in conferring obesity risk to offspring it is more commonly interpreted as reflecting a direct effect of maternal obesity on the fetal development.
Rodent models have also investigated the effects of early postnatal overnutrition. These have primarily used litter size manipulation post-birth to adjust the nutritional exposure. Plagemann et al. (1992) found that reducing litter sizes post-birth results in offspring hyperphagia and hyperinsulinemia while others report rearing DIO rats in large litters protects against obesity by enhancing leptin sensitivity (Patterson et al., 2010). Cross fostering experiments have also been used to demonstrate the potential effects of postnatal overnutrition in contributing to the obese phenotype later in life. Models show control rat offspring suckled by fat-fed dams develop hypertension and endothelial cell dysfunction similar to those exposed to high fat during pregnancy (Khan et al., 2005). Further, maintaining offspring on nutritionally rich diets at weaning after exposure to maternal overnutrition exacerbates the onset of metabolic syndrome features (Bayol et al., 2007; Mitra et al., 2009; Page et al., 2009).
The various animal models appear to converge on a common offspring phenotype of hyperphagia, insulin resistance, increased adiposity, and hypertension (Figure 1). Although offspring obesity could compound the effects of maternal obesity on offspring insulin resistance and hypertension, these both arise before the development of increased adiposity, suggesting independent programming of these systems occurs in response to maternal overnutrition. Whilst the programming of obesity is undoubtedly a multifactoral process, the diversity of models with similar phenotypic outcomes suggests some common mechanistic pathways.
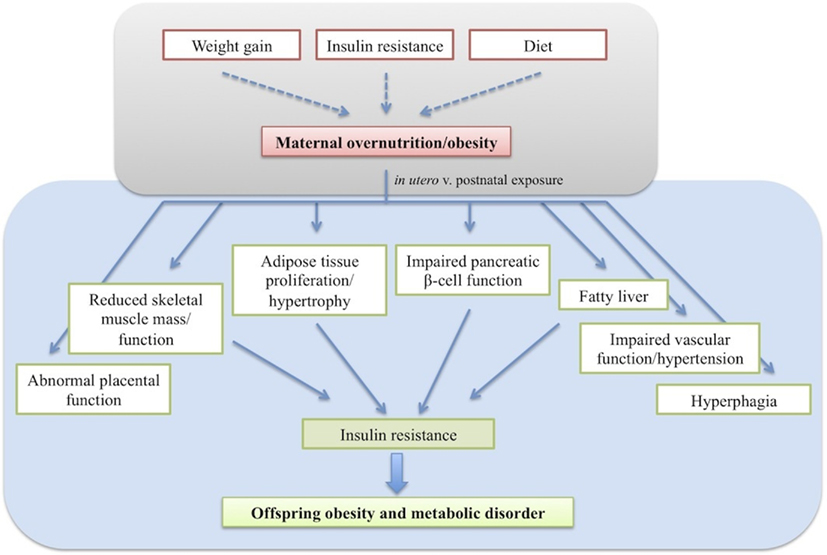
Figure 1. The effects of developmental programming on offspring phenotype resulting in insulin resistance and obesity.
Contributing Factors to Offspring Developmental Programming
The diversity of animal models present a challenge in determining which component(s) and at which time point is key in causing adverse effects in offspring leading to increased risk of metabolic disorders. Whether exposure to the insult at conception, in utero or postnatally primarily results in developmental programming and the nature of the insult that leads to programming in the developing fetus remains to be determined.
Relative Contributions of in Utero and Postnatal Exposure to Maternal Obesity
Standard animal models in which dams are fed an obesogenic/HFD throughout pregnancy and lactation demonstrate that exposure of the offspring throughout this period results in increased adiposity in the offspring (Samuelsson et al., 2008; Nivoit et al., 2009). However this does not distinguish between gestational and lactational exposure to maternal overnutrition. Models in which the timing of exposure to maternal overnutrition has been limited to pregnancy or lactation are extremely useful in deciphering the contribution of each element to programming offspring physiology.
Studies in which offspring exposure to maternal obesity were limited to the gestational period, by cross fostering offspring to lean rat dams after birth, show no changes in offspring body weight or adiposity at weaning determined by CT analysis, but increased serum insulin and leptin levels and upregulated lipid synthesis. However, offspring exposed to maternal obesity during gestation and lactation maintained on a HFD after weaning showed accelerated weight gain and percent fat mass (Shankar et al., 2008, 2010). This suggests the risk of obesity in adulthood is subject to programming from the onset of pregnancy, with an important role of postnatal diet in modifying adiposity.
The Lactation Period as a Critical Time Point
Postnatal factors have been shown to be able, to a certain degree, to modulate both genetic predisposition and prenatal factors. Cross fostering offspring of diet resistant (DR) dams to DIO dams during lactation resulted in obesity and insulin resistance suggesting excess nutrition during lactation increases the risk of obesity even in obesity resistant animals, while DIO offspring fostered by DR dams had improved insulin sensitivity, again pointing to the importance of the suckling period (Gorski et al., 2006). Raising pups in small litters post-birth receiving increased nutritional uptake from mothers’ milk, results in obesity and hyperphagia (Plagemann et al., 1999). Dietary manipulation after birth provides further evidence demonstrating the potential programming in the neonatal period. Providing pups with a high carbohydrate formula milk immediately after birth resulted in adult onset obesity in female offspring, which was transmitted to the next generation offspring even after females received a standard chow diet after weaning (Srinivasan et al., 2008). This is particularly interesting in suggesting that early life insults in mothers alone can influence offspring metabolic programming. Studies by Bayol et al. (2007) also identified the lactation period as a critical time window. Offspring exposed to maternal obesity during gestation and lactation and weaned onto junk food exhibited greater hyperphagia compared to those offspring of control dams. However, offspring exposed to the junk diet during gestation alone and weaned onto chow diet did not exhibit hyperphagia (Bayol et al., 2007). This demonstrates the importance of the postnatal period in predisposing offspring to metabolic disorders.
Maternal Factors Influencing Offspring Developmental Programming
It is unknown whether maternal diet itself or the maternal metabolic parameters associated with obesity imprints the metabolic phenotype onto the fetus, and it is hard to dissect the two in the human context. The molecular parameters driving the relationship between maternal obesity and offspring risk of obesity are therefore unclear. Obese women are often consuming a calorie dense diet and it is unknown whether a leaner woman eating a calorically dense diet can equally pass on the risk to her offspring. Recent studies have therefore attempted to distinguish between the independent contributions of maternal obesity per se and an obesogenic maternal diet.
Maternal weight gain
Epidemiological evidence suggests maternal weight gain in itself significantly increases the risk of large for gestational age babies and childhood obesity independently of maternal BMI or glucose tolerance (Oken et al., 2007).Women exceeding the Institute of Medicine recommended gestational weight gain (GWG) are more likely to have children with greater adiposity and associated metabolic traits (Fraser et al., 2010). It is, however, hard to isolate the effects of maternal GWG and maternal diet in animal studies. Due to satiety induced self-limiting of food intake, the majority of models available induce maternal weight gain and obesity by feeding dams an obesogenic diet. Therefore, any observable outcomes in offspring may be a result of the diet, maternal obesity, or an interaction between the two. Further study is essential into this avenue as it has major implications in potential preventative measures. If there is strong evidence to confirm epidemiological data demonstrating that excess GWG increases offspring obesity risk, simply monitoring GWG and advising prospective mothers accordingly could potentially control the obesity epidemic.
Maternal diet per se
Others show that infant birth weight and macrosomia have a stronger association with maternal triglycerides than maternal glucose tolerance (Di Cianni et al., 2005), suggesting dietary components may influence fetal development during pregnancy. Howie et al. (2009) demonstrated increased body weight and adiposity in offspring of HFD rat dams (45% fat) regardless of whether the dam had been fed the HFD since her own weaning (and was therefore obese) or restricted to pregnancy and lactation (no significant difference in body weight and GWG compared to control dams). This suggests diet has the potential to impart the effects on offspring phenotype independently of obesity and diet during pregnancy and lactation in particular can program the offspring (Howie et al., 2009). The importance of high fat in the diet has also been established in non-human primates. Maternal HFD consumption (35% fat) during pregnancy led to significant increases in FFA levels in the fetus (McCurdy et al., 2009), which could lead to insulin resistance. Carmody et al. (2010) also highlighted the importance of dietary fat, over other maternal factors such as insulin resistance. They suggested that HFD plays a role per se in influencing the offspring phenotype independently of insulin resistance through influencing offspring body weight and liver triglyceride content. However, maternal insulin resistance and HFD combined did have additional effects on adiposity in early life (Carmody et al., 2010).
Maternal Obesity
Although current insights into weight gain during gestation in animal models are limited, many studies have explored the effect of maternal obesity onset in the periconceptional and lactation periods. Nonetheless, in order to dissect the independent contributions of diet and obesity it is necessary to induce obesity in dams without altering the diet, and few studies have attempted this. Maternal obesity has been reported to have an independent effect to HFD in programming offspring adiposity, hyperphagia, and insulin resistance. Only obese rat dams fed a HFD (60% fat) for 4 weeks as opposed to non-obese dams fed a LFD (10% fat) or a HFD but pair fed for calorific intake transmitted the risk to the offspring (White et al., 2009). Further, Shankar et al. (2008, 2010) modeled maternal obesity in dams through intragastric feeding of liquid diets, thus varying the caloric intake alone. Their findings demonstrate an independent effect of maternal obesity, limited to the gestational period, in programming offspring adiposity, and insulin resistance when weaned on a HFD (Shankar et al., 2008, 2010). Maternal obesity and maternal diet are reported to have differential effects on fetal growth during gestation. Akyol et al. (2009) varied dam exposure to cafeteria diet to either before gestation, resulting in obese dams, or during gestation. They reported that the in utero environment associated with obesity resulted in reduced fetal weight, whereas a cafeteria diet during gestation caused abnormal placental growth (Akyol et al., 2009).
These, together with the epidemiological evidence of reduced offspring obesity risk in mothers post-weight-reducing surgery, have implications for future interventional strategies. A deeper understanding behind the causal factors associated with maternal obesity, such as hyperglycemia, hyperinsulinemia, and hyperleptinemia, require further investigation.
Mechanisms of the Programming of Obesity
Numerous factors have been implicated in the onset of offspring obesity and type 2 diabetes in response to maternal DIO (Figure 2). The use of animal models is critical in distinguishing between cause and effect. Longitudinal studies, cataloging the progression of offspring metabolic dysfunction at specific time points enable determination of the root causes of developmental programming, which may in turn provide insights into prevention or intervention.
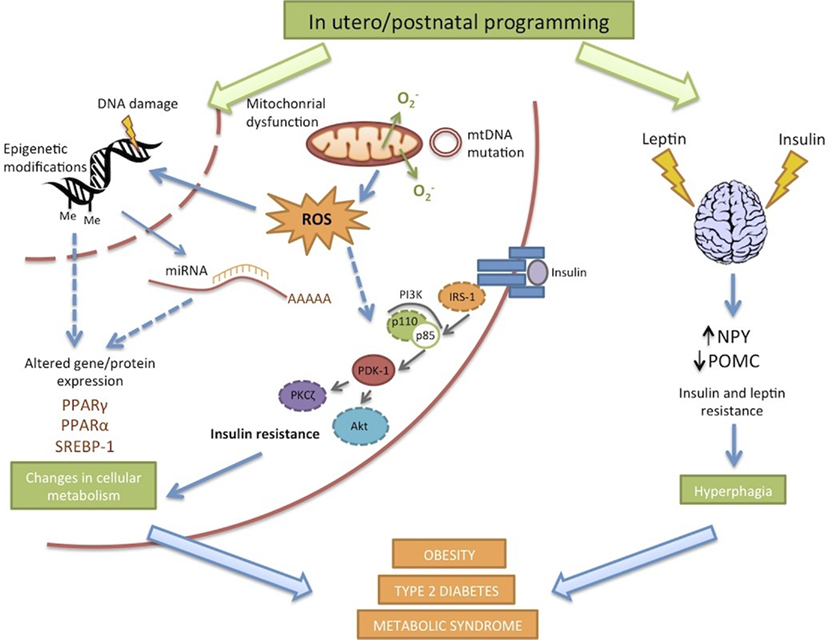
Figure 2. Potential mechanisms of developmental programming in response to maternal overnutrition.
Altered Regulation of Food Intake
The development of hyperphagia in offspring of obese dams soon after weaning suggest persistent alteration in regulation of feeding/appetite-controlling pathways may play a causative role in the development of increased adiposity. The hypothalamus is the central brain region mediating regulation of short-term and long-term dietary intake via the synthesis of various orexigenic and anorectic neuropeptides, integrating them with peripheral signals of nutritional status (Arora and Anubhuti, 2006). The arcuate nucleus (ARC) acts as a feeding control center containing neuronal populations expressing orexigenic neuropeptide Y (NPY) and agouti-related peptide (AgRP) and the anorectic pro-opiomelanocortin (POMC). The ARC lies immediately above the median eminence, the interface being specially modified to allow entry of peripheral peptides and proteins, thus making it the ideal site to integrate hormonal signals for energy homeostasis.
Dysregulated expression of hypothalamic neuropeptides is associated with obesity. NPY synthesis and secretion have been found to be increased compared to controls in various models of obesity (Beck et al., 1990; Wilding et al., 1993) as well as reduced expression of POMC and its cleaved product α-MSH (Kim et al., 2000). These changes may be due to disturbances in the peptides that regulate their production and release, namely leptin and insulin. Inappropriate concentrations or a shift in these hormones during a critical window of neuronal development and feeding pathway differentiation may have permanent structural consequences.
Neurotrophic actions of leptin
Long-term feeding regulation is provided by the main circulating hormones leptin and insulin. The roles of leptin and insulin in regulating feeding behavior in adults are well established. The functional leptin receptor ObRb is largely expressed in brain regions known to control energy balance and following leptin administration in postnatal mice, the hormone has been shown to act on NPY and POMC neurons (Caron et al., 2010). Leptin treatment in Lepob/ob mice reduces body weight and food intake (Campfield et al., 1995) via stimulating the expression of POMC and down regulating NPY and AgRP expression (Jéquier, 2002).
Recent evidence is emerging to support the role of leptin in neuronal differentiation during the neonatal period. In both rats and mice there is a surge in circulating leptin concentration during the early postnatal period, independent of fat mass; yet studies revealed that neonatal rodents do not lose weight in response to leptin treatment the same way adults do suggesting the immaturity of hypothalamic circuits (Bouret et al., 2004). Leptin treatment increases the density and length of axons from the ARC in vitro and could reverse the abnormality in ARC connectivity in ob/ob mice in neonates but not adults, suggesting its neurodevelopmental effects are restricted to the neonatal period. Leptin signaling in ependymal cells of the third ventricle are suggested to play a role in leptin action over this restricted period as the leptin surge correlated with the expression of leptin receptors in this region during this period (Cottrell et al., 2009). These data suggest that inappropriate levels of leptin or leptin action during this critical period of neuronal development may cause permanent abnormalities in the circuitry. Exogenous leptin treatment during the early postnatal period causes abnormal expression of NPY, AgRP, and POMC expression in the ARC (Proulx et al., 2002).However, there is also evidence suggesting a beneficial role of leptin during lactation in preventing obesity later in life. Studies have reported the oral intake of physiological doses of leptin over lactation reduced food intake and body weight gain and protected against HFD-induced leptin resistance (Picó et al., 2007), insulin resistance and food preferences in adult rats (Sánchez et al., 2008).
Various alterations in hypothalamic structures, neuropeptide levels, neuronal activity, and hormonal responsiveness have been reported following manipulation of litter size post-birth. Small litter sizes were associated with increased orexigenic neuropeptide expression (NPY and galanin) with reduced responsiveness to leptin within the ARC and PVN (Davidowa et al., 2002, 2003; Glavas et al., 2010) and an inhibitory as opposed to stimulatory effect of leptin on VMH neurons, which reduce food intake when stimulated (Davidowa and Plagemann, 2000). Offspring of rat dams fed an obesogenic diet resulted in an amplified and prolonged neonatal leptin surge. At postnatal day 30, before the onset of hyperphagia, leptin induced appetite suppression, and leptin signaling in the ARC were attenuated (Kirk et al., 2009). This suggests hyperleptinemia during the immediate postnatal period in which neuronal development occurs results in the developmental programming of leptin resistance and thus obesity in these rats. This may be true of altricial species, such as rodents, where significant development occurs post-birth; however recent evidence questions whether the same occurs in precocial species. A sheep model, in which newborn lambs are born at an equivalent level of maturity to humans, observed an absence of the normal leptin peak and a trend toward a premature one in offspring of obese ewes accompanied by increased appetite in the lambs (Long et al., 2011). Thus, neonatal leptin treatment may potentially reverse the programming effects of maternal obesity, as it does in offspring of undernourished dams in which the leptin surge is reduced (Vickers et al., 2005, 2008).
Neurotrophic actions of insulin
Like leptin, the expression level of the insulin receptor throughout various brain regions is developmentally regulated, being higher in the neonate and lower in the adult. Insulin administration increases POMC mRNA expression as well as reducing NPY expression and protecting against diet-induced obesity in the adult (Air et al., 2002). A neurotrophic role of insulin has also emerged in addition to its role in regulating food intake. Various studies have shown its involvement in synapse formation, dendritic morphogenesis and plasticity (reviewed in Chiu and Cline, 2010). Suboptimal fetal and neonatal hyperinsulinemia may therefore act as a programming cue, conferring malformation of hypothalamic structures. This is supported by evidence showing that neonatal insulin treatment induces morphological alterations in hypothalamic structures with reduced neuronal density and volume, and results in obesity and adult hyperinsulinemia (Harder et al., 1998). Further, the negative feedback effect of insulin on median ARC neurons (predominantly consisting of NPY and AgRP neurons) is reportedly reduced with insulin having a stimulatory as opposed to inhibitory action in small litter offspring exposed to hyperinsulinemia (Davidowa and Plagemann, 2007). Plagemann et al. (2010) reported hypermethylation of the hypothalamic insulin receptor promoter in small litter rats exposed to increased neonatal feeding, suggesting this as a mechanism of hypothalamic insulin resistance and resultant hyperphagia and obesity. Hypermethylation of the POMC gene promoter has also been reported in the same rats within sequences that are essential for the mediation of leptin and insulin effects in POMC expression (Plagemann et al., 2009).
Altered Regulation of Reward Pathways
The regulation of food intake is controlled on two levels: first, factors regulating appetite and satiety; second, motivation to eat – appetite stimulation through reward pathways. Cortico-limbic reward pathways running through multiple brain areas including the nucleus accumbens, ventral tegmental area, hippocampus, and amygdala process reward from food. Palatable foods activate the dopaminergic and opioid signaling pathways within the mesolimbic system and disruption of the systems have been shown to alter feeding behavior (reviewed in Fulton, 2010). As above, it is suffice to presume that any alterations in the development of these reward pathways would alter motivation to eat, and may affect food preferences to stimulate one to choose food with high fat content. Hyperphagia is commonly seen in models of maternal obesity and there is evidence to suggest that dysregulation of reward pathways contribute to this phenotype. Maternal DIO in models has been found to alter food preferences in offspring toward highly palatable foods, along with altered expression of components of mesolimbic dopamine and opioid reward pathways (Bayol et al., 2007; Vucetic et al., 2010; Ong and Muhlhausler, 2011).
These findings demonstrate the importance of the developmental window during neonatal life in influencing neuronal function and responsiveness. However, the critical developmental periods that are susceptible to programming are likely to vary in humans compared to rodent models due to differences in timing of the development of neuronal circuitry. Although the same neuropeptides and hormonal signals are important in hypothalamic circuits in rodents and primates, the timing of development of these circuits differ between them. ARC projections develop during the postnatal period in rodents but during the third trimester in primates. Administration of regulatory hormones during the first few weeks in rodents does not appear to have a significant effect in regulating food intake, but alters the development of circuits. It remains to be determined whether similar patterns are seen in primates (reviewed in Grove et al., 2005).
Glucose Homeostasis
Glucose homeostasis requires two components: adequate insulin production by pancreatic beta-cells and responsiveness to insulin in peripheral tissues. Therefore, defects in one or the other makes one vulnerable to the effects of postnatal overfeeding or to the natural effects of aging. The development of insulin resistance is commonly seen in patients prior to the progression of type 2 diabetes, in which higher concentrations of insulin are required to maintain glucose homeostasis. Progressive impairment of glucose homeostasis is also noted in many models of maternal DIO. Recent studies suggest insulin resistance may result from persistent alterations in insulin-signaling molecules in insulin responsive tissues, discussed below. It is likely that insulin resistance precedes beta-cell failure as studies report increased insulin concentrations and pancreatic content in young offspring followed by an age-related decline in beta-cell function in models of maternal obesity due to persistent overstimulation (Samuelsson et al., 2008; Srinivasan et al., 2008).
Skeletal Muscle Dysfunction
Skeletal muscle is a major insulin responsive tissue, acting as a glucose storage depot. Abnormal skeletal muscle development has been reported in a number of models. Offspring of dams fed a “junk food” diet show reduced muscle mass proliferation and insulin receptor mRNA levels as well as higher intramuscular lipid content which would be expected to disrupt insulin action (Bayol et al., 2005). Disruption in the insulin-signaling pathway has been associated with insulin resistance in response to maternal overnutrition. Reduced serine 473 protein kinase B/Akt phosphorylation as well as insulin receptor mRNA expression is reported in offspring of ewes fed an obesogenic diet; the main downstream signaling pathway in insulin signaling (Yan et al., 2010). This has also been reported in rodent models along with reduced expression of P110β, a catalytic subunit of phosphoinositide 3-kinase (PI3K; Shelley et al., 2009).
Altered Adipose Physiology
Exposure to maternal obesity may be associated with programmed alterations in genes important for adipocyte differentiation and function. Altered body composition and excess fat deposition is commonly seen in response to maternal overnutrition. At 3 months of age, offspring of obese mothers demonstrated adipocyte hypertrophy with increased PPARγ as well as reduction in β2- and β3-adrenoreceptors mRNA expression, indicative of upregulated lipogenesis and downregulated lipolysis respectively (Samuelsson et al., 2008). Adipocyte hypertrophy independent of hyperplasia has similarly been reported in offspring of cafeteria diet fed dams during gestation and lactation. In larger animal models also, periconceptional or gestational overnutrition programs offspring fat mass through upregulation of genes involved in adipogenesis and lipogenesis (Muhlhausler et al., 2007a,2007b; Rattanatray et al., 2010). Increased fat mass may be a compensatory response to ensure excess lipids are stored in adipocytes rather than other metabolic organs leading to their dysfunction. However, increased adiposity in itself is a causal factor in insulin resistance from the release of various adipokines and inflammatory cytokines. Therefore, the programming of adipose gene expression may be a mechanism through which the exposure to overnutrition during development leads to adiposity in adult life. The increased secretion of leptin as a result of adiposity may also be a key event in the resulting leptin resistance within hypothalamic neurons.
Non-Alcoholic Fatty Liver Disease
The liver plays a central role in global metabolism, regulating a range of biochemical reactions. NAFLD occurs commonly with the metabolic syndrome, linked to excessive triglyceride accumulation, which may be the result of insulin resistance. Elevated triglyceride concentrations and fatty liver have been associated with maternal overnutrition in a number of models (Buckley et al., 2005; Samuelsson et al., 2008; McCurdy et al., 2009; Oben et al., 2010a). Altered expression of genes involved in lipogenesis and lipolysis would be expected to alter the balance of hepatic lipid accumulation. Exposure to maternal obesity though increased calorific uptake or HF feeding results in increased liver weight and lipid accumulation. This is attributed to upregulated lipogenesis and down regulated lipolysis. Critical nodes of regulation in the respective pathways have been reported with upregulated SREBP-1 and 20 downstream genes involved in hepatic lipid biosynthesis and reduced expression of PPARα and related genes involved in fatty acid oxidation (Shankar et al., 2010). Increased gluconeogenesis is reported in fetuses of HFD mothers, therefore the programming effects of maternal diet appear to occur during the development of metabolic systems. Offspring weaned onto a HFD after fetal exposure are reported to have a more progressive form of NAFLD with increased fibrosis and inflammation (Bruce et al., 2009). Disruption in insulin signaling has been consistently reported alongside these changes suggesting insulin resistance as a causal factor in this metabolic phenotype. Reduced expression of insulin receptor β, insulin receptor substrate-1 and p85α have been demonstrated with increased protein kinase Cζ at 3 months of age (Buckley et al., 2005; Martin-Gronert et al., 2010).
Altered Pancreatic Function
The decline in glucose homeostasis develops with age in models of developmental programming. Offspring of obese dams were hyperinsulinemic at 3 months of age progressing to diabetes by 6 months of age (Samuelsson et al., 2008) which was associated with increased pancreatic insulin content early in life that diminished in adulthood (Han et al., 2005; Taylor et al., 2005; Srinivasan et al., 2006). Programming of pancreatic fibrosis and non-alcoholic fatty pancreas disease is also observed in mice following exposure to maternal overnutrition both during gestation or lactation alone (Oben et al., 2010b). In sheep, maternal obesity is associated with increased fetal pancreatic weight and increased number of insulin-positive cells per unit area due to proliferation (Ford et al., 2009). In late gestation however (day 135) and at lambing, offspring beta-cell numbers were reduced with increased beta-cell apoptotic rate, predisposing to glucose intolerance in adulthood (Zhang et al., 2011). Interestingly, a mouse model of paternal HFD overfeeding impairs beta-cell function in the female progeny in early life; presenting reduced relative islet area and insulin secretion following a glucose tolerance test. This is accompanied by altered gene expression of insulin and glucose metabolic pathway components indicating the programming of impaired insulin-granule exocytosis (Ng et al., 2010).
Oxidative Stress
Reactive oxygen species (ROS) are well recognized as double-edged swords. At low/moderate concentrations they may be beneficial in defense against infectious agents, in the function of a number of cellular signaling pathways and the induction of a mitogenic response. However, overproduction results in oxidative stress; a deleterious process that can lead to damage of cell structures, including lipids and membranes, proteins, and DNA (Valko et al., 2007).
Studies have shown extensive association of markers of oxidative stress with obesity, insulin resistance, and diabetes through interference of insulin signaling (Urakawa et al., 2003; Furukawa et al., 2004; Nishikawa et al., 2007). Upregulation of pathways for ROS production and oxidative stress in insulin responsive tissues (liver and adipose tissue) have been observed before the onset of insulin resistance (Matsuzawa-Nagata et al., 2008), which could be suppressed by dietary restriction or antioxidant supplementation (Hotamisligil et al., 1995; Dandona et al., 2001; Campión et al., 2006).
Recent studies suggest ROS production may be an initial key event preceding the onset of insulin resistance and obesity in response to maternal nutrition. The expression of inflammation-related genes and those linked to oxidative stress are elevated in the placenta of obese women (Radaelli et al., 2003) as well as in fetal skeletal muscle (Yan et al., 2010). Therefore, maternal obesity contributes to an abnormal environment in which the embryo and fetus develop; suggesting that oxidative stress may be a triggering mechanism in offspring, behind changes in gene expression, and insulin resistance, which subsequently leads to the metabolic phenotype observed.
Altered insulin signaling in offspring of obese and HFD mothers has been reported with increased inflammation and oxidative stress in numerous models. Increased oxidative damage markers 8-hydroxy-2-deoxy Guanosine and 4-hydroxy-2-nonenal were reported in liver of fetal offspring of HFD fed macaques (McCurdy et al., 2009) and increased mRNA expression of enzymes involved in oxidative stress in 15-week-old rodent offspring (Bruce et al., 2009). These were correlated with upregulation of lipogenic genes, suggesting association of ROS and fatty acid accumulation. Further support to this hypothesis would be a demonstration that reducing levels of ROS in models of maternal obesity, as has been shown in models of obesity, results in reversal or reduction in the metabolic syndrome phenotype. Vitamin C antioxidant supplementation to dams fed a western HFD was able to ameliorate adiposity, reduce oxidative stress, and inflammation in embryos (Sen and Simmons, 2010).
Mitochondrial Mechanisms
Mitochondria play an important role in metabolism, providing the energy source as well as being the site of fatty acid oxidation. They are also a source of ROS; overstimulation of the oxidative phosphorylation/electron transport chain gives rise to the production of free radicals. Mitochondrial dysfunction is also implicated in a range of disorders including insulin resistance and type 2 diabetes. Mitochondrial DNA mutations have also been suggested to contribute to diabetes development (Wang et al., 2010). Mitochondrial dysfunction has been reported as early as embryogenesis in obese mothers. Mouse oocytes and embryos of obese dams are reported to have increased mitochondrial membrane potential, higher levels of oxidative phosphorylation and increased ROS production compared to those of lean mothers. mtDNA copy number was also increased, which usually occurs in response to oxidative damage (Igosheva et al., 2010). At 3 months however mtDNA was reduced in liver and kidney in the same model, which the authors suggest, if indicative of global reduction in all tissues, would impair the glucose sensing ability by beta-cells and subsequent insulin release (Taylor et al., 2005). Disrupted ETC activity has also been found in skeletal muscle (Shelley et al., 2009) and liver (Bruce et al., 2009) of offspring from over-nourished mothers, although direct evidence of oxidative damage as a result was not demonstrated.
Epigenetic Programming
Recent years have seen increased investigation into epigenetic mechanisms in human disease. These are changes in gene expression not involving changes in DNA sequence, such as DNA methylation, histone modification, and chromatin packing. Epigenetic dysregulation has been reported to mediate the effects of early nutrition on adult disease susceptibility. DNA methylation is the most common mechanism studied. As the source of methyl donors and cofactors involved in methyl metabolism is dietary and methylation patterns are established during development, it is hypothesized that early donor malnutrition could alter epigenetic patterns, increasing susceptibility to chronic disease later in life (Waterland, 2004).
Early evidence showing changes in early nutrition affecting epigenetic modification came from Waterland and Garza (2002). They reported epigenetic alterations in pancreatic genes of rat offspring reared in small litter sizes including that of insulin 2 (indicating a 65% reduction in expression), which was associated with reduced glucose-stimulated insulin release (Waterland and Garza, 2002). In a similar model, Plagemann et al. (2009) demonstrated hypermethylation of the hypothalamic POMC and insulin receptor (Plagemann et al., 2010) promoter regions, which correlated with blood glucose concentrations. Hypomethylation of pancreatic il13ra2, a gene involved in Jak-Stat signaling is reported alongside beta-cell dysfunction in female offspring of in a model of paternal obesity (Ng et al., 2010). microRNAs (miRNA) have also been implicated in developmental programming. Analysis of hepatic miRNA levels showed differential expression of several miRNAs whose predicted targets were proteins important for epigenetic regulation in offspring of HFD fed dams such as methylCpG binding domain proteins (Zhang et al., 2009). Although epigenetic changes have been implicated in animal models, there is little direct evidence relating this to humans. One study has, however, correlated epigenetic RXRA (retinoid X receptor alpha – induces transcription of PPARs) promoter methylation with increased adiposity in children of mothers with lower carbohydrate intake in two independent cohorts (Godfrey et al., 2011). Although this fails to confirm a causal relationship, it may provide an objective marker in identifying children at risk of obesity.
Data is also emerging on the transgenerational effects of obesity and diet with studies reporting maternal HFD influencing body length and insulin sensitivity in second (Dunn and Bale, 2009) and third generation mice (Dunn and Bale, 2011). The observed transmission to the third generation mice solely via the paternal lineage supports the role of a germ-line based model of epigenetic programming, possibly involving editing of imprinted genes. A model of transgenerational programming in response to paternal low-protein diet has reported differences in sperm gene expression, including those of transcription factors and chromatin regulators. This shows that the sperm epigenome is regulated by dietary conditions, which may result in altered offspring physiology (Carone et al., 2010). Taken together, these suggest maternal nutrition is able to influence future generations through stable programming of germ cells as a means of ensuring offspring receive important adaptive information. It would also help explain the population wide increase in obesity prevalence through generations, however the mechanisms behind this remain to be explored.
Future Directions and Possible Interventions
Once obesity has onset, the higher body weight is defended as the new “set point,” resistant to permanent reduction via therapy in many individuals (Levin, 2006). Although bariatric surgery is able to ameliorate the situation, it is an extreme form of intervention and is not possible to sustain for all obese patients population wide. Therefore, it is vital that we gain better insights into the mechanisms promoting the development of obesity, in order to develop prevention strategies.
The perinatal environment is evidently an important determinant in programming an individuals metabolic system and the use of animal models have been extremely useful for eliciting the possible contributory factors and underlying mechanisms. A variety of perturbations in both the pre and postnatal environment alter feeding behavior and metabolism in the offspring. Teasing out the relative contributions of the fetal and postnatal periods will be important in considering interventional strategies, as postnatal intervention is likely to prove an easier avenue due to direct access to the offspring. The entwined relationship between the factors makes this difficult but it can be done through developing standardized experimental models in which a clear separation can be made between: the individual effects of gestation, lactation, and weaning; the relative effects of maternal or neonatal dietary components and mechanistic triggers. From the available models, it seems the gestational period is sufficient to program changes during fetal development, and that continual insult post-birth exacerbates the phenotype.
Promising possible interventional strategies are emerging from animal studies. Dietary restriction prior to pregnancy is able to ameliorate the effects of maternal obesity on offspring programming (Rattanatray et al., 2010; Zambrano et al., 2010). Weight loss in obese women prior to pregnancy has also demonstrated improved offspring metabolic phenotype (Kral et al., 2006; Smith et al., 2009). Further, during pregnancy, mild gestational food restriction or switching to control diet of dams on HFD is able to reverse the programming effects and even protect against HFD post-weaning (Gallou-Kabani et al., 2007; Giraudo et al., 2010).Therefore, weight-loss strategies or simple dietary changes to improve the metabolic milieu in mothers prior to pregnancy are likely to positively impact offspring phenotype. Thus, increasing public awareness of the importance of a balanced diet before and during pregnancy and a reduction in the incidence of obesity during pregnancy must be a long-term goal in the reducing risks for future generations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Maria Z. Alfaradhi holds a Wellcome Trust Studentship. Susan E. Ozanne is a British Heart Foundation Senior Fellow.
References
Air, E. L., Strowski, M. Z., Benoit, S. C., Conarello, S. L., Salituro, G. M., Guan, X.-M., Liu, K., Woods, S. C., and Zhang, B. B. (2002). Small molecule insulin mimetics reduce food intake and body weight and prevent development of obesity. Nat. Med. 8, 179–183.
Akyol, A., Langley-Evans, S. C., and McMullen, S. (2009). Obesity induced by cafeteria feeding and pregnancy outcome in the rat. Br. J. Nutr. 102, 1601–1610.
Arora, S., and Anubhuti. (2006). Role of neuropeptides in appetite regulation and obesity – a review. Neuropeptides 40, 375–401.
Baird, J., Fisher, D., Lucas, P., Kleijnen, J., Roberts, H., and Law, C. (2005). Being big or growing fast: systematic review of size and growth in infancy and later obesity. BMJ 331, 929.
Barker, D. J., Hales, C. N., Fall, C. H., Osmond, C., Phipps, K., and Clark, P. M. (1993). Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36, 62–67.
Barker, D. J., and Osmond, C. (1986). Diet and coronary heart disease in England and Wales during and after the second world war. J. Epidemiol. Community Health 40, 37–44.
Bayol, S. A., Farrington, S. J., and Stickland, N. C. (2007). A maternal ‘junk food’ diet in pregnancy and lactation promotes an exacerbated taste for ‘junk food’ and a greater propensity for obesity in rat offspring. Br. J. Nutr. 98, 843–851.
Bayol, S. A., Simbi, B. H., and Stickland, N. C. (2005). A maternal cafeteria diet during gestation and lactation promotes adiposity and impairs skeletal muscle development and metabolism in rat offspring at weaning. J. Physiol. 567(Pt 3), 951–961.
Beck, B., Burlet, A., Nicolas, J. P., and Burlet, C. (1990). Hyperphagia in obesity is associated with a central peptidergic dysregulation in rats. J. Nutr. 120, 806–811.
Boney, C. M., Verma, A., Tucker, R., and Vohr, B. R. (2005). Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 115, e290–e296.
Bouret, S. G., Draper, S. J., and Simerly, R. B. (2004). Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J. Neurosci. 24, 2797–2805.
Bruce, K. D., Cagampang, F. R., Argenton, M., Zhang, J., Ethirajan, P. L., Burdge, G. C., Bateman, A. C., Clough, G. F., Poston, L., Hanson, M. A., McConnell, J. M., and Byrne, C. D. (2009). Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 50, 1796–1808.
Buckley, A. J., Keserü, B., Briody, J., Thompson, M., Ozanne, S. E., and Thompson, C. H. (2005). Altered body composition and metabolism in the male offspring of high fat-fed rats. Metab. Clin. Exp. 54, 500–507.
Campfield, L. A., Smith, F. J., Guisez, Y., Devos, R., and Burn, P. (1995). Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546–549.
Campión, J., Milagro, F. I., Fernández, D., and Martínez, J. A. (2006). Differential gene expression and adiposity reduction induced by ascorbic acid supplementation in a cafeteria model of obesity. J. Physiol. Biochem. 62, 71–80.
Carmody, J. S., Wan, P., Accili, D., Zeltser, L. M., and Leibel, R. L. (2010). Respective contributions of maternal insulin resistance and diet to metabolic and hypothalamic phenotypes of progeny. Obesity (Silver Spring) 19, 492–499.
Caron, E., Sachot, C., Prevot, V., and Bouret, S. G. (2010). Distribution of leptin-sensitive cells in the postnatal and adult mouse brain. J. Comp. Neurol. 518, 459–476.
Carone, B. R., Fauquier, L., Habib, N., Shea, J. M., Hart, C. E., Li, R., Bock, C., Li, Chengjian, Gu, H., Zamore, P. D., Meissner, A., Weng, Z., Hofmann, H. A., Friedman, N., and Rando, O. J. (2010). Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143, 1084–1096.
Chen, J.-H., Cottrell, E. C., and Ozanne, S. E. (2010). Early growth and ageing. Nestlé Nutr. Workshop Ser. Paediatr. Program. 65, 41–50; discussion 50–54.
Chiu, S.-L., and Cline, H. T. (2010). Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 5, 7.
Cottrell, E. C., Cripps, R. L., Duncan, J. S., Barrett, P., Mercer, J. G., Herwig, A., and Ozanne, S. E. (2009). Developmental changes in hypothalamic leptin receptor: relationship with the postnatal leptin surge and energy balance neuropeptides in the postnatal rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R631–R639.
Dabelea, D., Hanson, R. L., Lindsay, R. S., Pettitt, D. J., Imperatore, G., Gabir, M. M., Roumain, J., Bennett, P. H., and Knowler, W. C. (2000). Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 49, 2208–2211.
Daenzer, M., Ortmann, S., Klaus, S., and Metges, C. C. (2002). Prenatal high protein exposure decreases energy expenditure and increases adiposity in young rats. J. Nutr. 132, 142–144.
Dandona, P., Mohanty, P., Ghanim, H., Aljada, A., Browne, R., Hamouda, W., Prabhala, A., Afzal, A., and Garg, R. (2001). The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J. Clin. Endocrinol. Metab. 86, 355–362.
Davidowa, H., Li, Y., and Plagemann, A. (2002). Hypothalamic ventromedial and arcuate neurons of normal and postnatally overnourished rats differ in their responses to melanin-concentrating hormone. Regul. Pept. 108, 103–111.
Davidowa, H., Li, Y., and Plagemann, A. (2003). Altered responses to orexigenic (AGRP, MCH) and anorexigenic (alpha-MSH, CART) neuropeptides of paraventricular hypothalamic neurons in early postnatally overfed rats. Eur. J. Neurosci. 18, 613–621.
Davidowa, H., and Plagemann, A. (2000). Different responses of ventromedial hypothalamic neurons to leptin in normal and early postnatally overfed rats. Neurosci. Lett. 293, 21–24.
Davidowa, H., and Plagemann, A. (2007). Insulin resistance of hypothalamic arcuate neurons in neonatally overfed rats. Neuroreport 18, 521–524.
Di Cianni, G., Miccoli, R., Volpe, L., Lencioni, C., Ghio, A., Giovannitti, M. G., Cuccuru, I., Pellegrini, G., Chatzianagnostou, K., Boldrini, A., and Del Prato, S. (2005). Maternal triglyceride levels and newborn weight in pregnant women with normal glucose tolerance. Diabet. Med. 22, 21–25.
Dunn, G. A., and Bale, T. L. (2009). Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology 150, 4999–5009.
Dunn, G. A., and Bale, T. L. (2011). Maternal high-fat diet effects on third-generation female body size via the paternal lineage. Endocrinology 152, 2228–2236.
Ekelund, U., Ong, K. K., Linné, Y., Neovius, M., Brage, S., Dunger, D. B., Wareham, N. J., and Rössner, S. (2007). Association of weight gain in infancy and early childhood with metabolic risk in young adults. J. Clin. Endocrinol. Metab. 92, 98–103.
Farley, D., Tejero, M. E., Comuzzie, A. G., Higgins, P. B., Cox, L., Werner, S. L., Jenkins, S. L., Li, C., Choi, J., Dick, E. J., Hubbard, G. B., Frost, P., Dudley, D. J., Ballesteros, B., Wu, G., Nathanielsz, P. W., and Schlabritz-Loutsevitch, N. E. (2009). Feto-placental adaptations to maternal obesity in the baboon. Placenta 30, 752–760.
Flegal, K. M., Carroll, M. D., Ogden, C. L., and Curtin, L. R. (2010). Prevalence and trends in obesity among US adults, 1999–2008. JAMA 303, 235–241.
Flegal, K. M., Carroll, M. D., Ogden, C. L., and Johnson, C. L. (2002). Prevalence and trends in obesity among US adults, 1999–2000. JAMA 288, 1723–1727.
Ford, S. P., Zhang, L., Zhu, M., Miller, M. M., Smith, D. T., Hess, B. W., Moss, G. E., Nathanielsz, P. W., and Nijland, M. J. (2009). Maternal obesity accelerates fetal pancreatic beta-cell but not alpha-cell development in sheep: prenatal consequences. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R835–R843.
Forsdahl, A. (1977). Are poor living conditions in childhood and adolescence an important risk factor for arteriosclerotic heart disease? Br. J. Prev. Soc. Med. 31, 91–95.
Fraser, A., Tilling, K., Macdonald-Wallis, C., Sattar, N., Brion, M.-J., Benfield, L., Ness, A., Deanfield, J., Hingorani, A., Nelson, S. M., Smith, G. D., and Lawlor, D. A. (2010). Association of maternal weight gain in pregnancy with offspring obesity and metabolic and vascular traits in childhood. Circulation 121, 2557–2564.
Frayling, T. M. (2007). Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat. Rev. Genet. 8, 657–662.
Furukawa, S., Fujita, T., Shimabukuro, M., Iwaki, M., Yamada, Y., Nakajima, Y., Nakayama, O., Makishima, M., Matsuda, M., and Shimomura, I. (2004). Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114, 1752–1761.
Gallou-Kabani, C., Vigé, A., Gross, M.-S., Boileau, C., Rabes, J.-P., Fruchart-Najib, J., Jais, J.-P., and Junien, C. (2007). Resistance to high-fat diet in the female progeny of obese mice fed a control diet during the periconceptual, gestation, and lactation periods. Am. J. Physiol. Endocrinol. Metab. 292, E1095–E1100.
Giraudo, S. Q., Della-Fera, M. A., Proctor, L., Wickwire, K., Ambati, S., and Baile, C. A. (2010). Maternal high fat feeding and gestational dietary restriction: effects on offspring body weight, food intake and hypothalamic gene expression over three generations in mice. Pharmacol. Biochem. Behav. 97, 121–129.
Glavas, M. M., Kirigiti, M. A., Xiao, X. Q., Enriori, P. J., Fisher, S. K., Evans, A. E., Grayson, B. E., Cowley, M. A., Smith, M. S., and Grove, K. L. (2010). Early overnutrition results in early-onset arcuate leptin resistance and increased sensitivity to high-fat diet. Endocrinology 151, 1598–1610.
Godfrey, K. M., Sheppard, A., Gluckman, P. D., Lillycrop, K. A., Burdge, G. C., McLean, C., Rodford, J., Slater-Jefferies, J. L., Garratt, E., Crozier, S. R., Emerald, B. S., Gale, C. R., Inskip, H. M., Cooper, C., and Hanson, M. A. (2011). Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 60, 1528–1534.
Gorski, J. N., Dunn-Meynell, A. A., Hartman, T. G., and Levin, B. E. (2006). Postnatal environment overrides genetic and prenatal factors influencing offspring obesity and insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R768–R778.
Grove, K. L., Grayson, B. E., Glavas, M. M., Xiao, X. Q., and Smith, M. S. (2005). Development of metabolic systems. Physiol. Behav. 86, 646–660.
Hales, C. N., and Barker, D. J. (1992). Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35, 595–601.
Hales, C. N., Barker, D. J., Clark, P. M., Cox, L. J., Fall, C., Osmond, C., and Winter, P. D. (1991). Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303, 1019–1022.
Han, J., Xu, J., Epstein, P. N., and Liu, Y. Q. (2005). Long-term effect of maternal obesity on pancreatic beta-cells of offspring: reduced beta cell adaptation to high glucose and high-fat diet challenges in adult female mouse offspring. Diabetologia 48, 1810–1818.
Harder, T., Bergmann, R., Kallischnigg, G., and Plagemann, A. (2005). Duration of breastfeeding and risk of overweight: a meta-analysis. Am. J. Epidemiol. 162, 397–403.
Harder, T., Plagemann, A., Rohde, W., and Dörner, G. (1998). Syndrome X-like alterations in adult female rats due to neonatal insulin treatment. Metab. Clin. Exp. 47, 855–862.
Hotamisligil, G. S., Arner, P., Caro, J. F., Atkinson, R. L., and Spiegelman, B. M. (1995). Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Invest. 95, 2409–2415.
Howie, G. J., Sloboda, D. M., Kamal, T., and Vickers, M. H. (2009). Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J. Physiol. 587(Pt 4), 905–915.
Igosheva, N., Abramov, A. Y., Poston, L., Eckert, J. J., Fleming, T. P., Duchen, M. R., and McConnell, J. (2010). Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS ONE 5, e10074. doi:10.1371/journal.pone.0010074
Jéquier, E. (2002). Leptin signaling, adiposity, and energy balance. Ann. N. Y. Acad. Sci. 967, 379–388.
Jones, R. H., and Ozanne, S. E. (2009). Fetal programming of glucose-insulin metabolism. Mol. Cell. Endocrinol. 297, 4–9.
Kanagalingam, M. G., Forouhi, N. G., Greer, I. A., and Sattar, N. (2005). Changes in booking body mass index over a decade: retrospective analysis from a Glasgow Maternity Hospital. BJOG 112, 1431–1433.
Khan, I. Y., Dekou, V., Douglas, G., Jensen, R., Hanson, M. A., Poston, L., and Taylor, P. D. (2005). A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R127–R133.
Kim, E. M., O’Hare, E., Grace, M. K., Welch, C. C., Billington, C. J., and Levine, A. S. (2000). ARC POMC mRNA and PVN alpha-MSH are lower in obese relative to lean zucker rats. Brain Res. 862, 11–16.
Kirk, S. L., Samuelsson, A.-M., Argenton, M., Dhonye, H., Kalamatianos, T., Poston, L., Taylor, P. D., and Coen, C. W. (2009). Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS ONE 4, e5870. doi:10.1371/journal.pone.0005870
Koletzko, B., von Kries, R., Closa, R., Escribano, J., Scaglioni, S., Giovannini, M., Beyer, J., Demmelmair, H., Gruszfeld, D., Dobrzanska, A., Sengier, A., Langhendries, J.-P., Rolland Cachera, M.-F., and Grote, V. (2009). Lower protein in infant formula is associated with lower weight up to age 2 y: a randomized clinical trial. Am. J. Clin. Nutr. 89, 1836–1845.
Kral, J. G., Biron, S., Simard, S., Hould, F.-S., Lebel, S., Marceau, S., and Marceau, P. (2006). Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics 118, e1644–e1649.
Laitinen, J., Power, C., and Järvelin, M. R. (2001). Family social class, maternal body mass index, childhood body mass index, and age at menarche as predictors of adult obesity. Am. J. Clin. Nutr. 74, 287–294.
Lake, J. K., Power, C., and Cole, T. J. (1997). Child to adult body mass index in the 1958 British birth cohort: associations with parental obesity. Arch. Dis. Child. 77, 376–381.
Levin, B. E. (2006). Metabolic imprinting: critical impact of the perinatal environment on the regulation of energy homeostasis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1107–1121.
Lewis, D. S., Bertrand, H. A., McMahan, C. A., McGill, H. C., Carey, K. D., and Masoro, E. J. (1986). Preweaning food intake influences the adiposity of young adult baboons. J. Clin. Invest. 78, 899–905.
Long, N. M., Ford, S. P., and Nathanielsz, P. W. (2011). Maternal obesity eliminates the neonatal lamb plasma leptin peak. J. Physiol. 589(Pt 6), 1455–1462.
Martin-Gronert, M. S., Fernandez-Twinn, D. S., Poston, L., and Ozanne, S. E. (2010). Altered hepatic insulin signalling in male offspring of obese mice. J. Dev. Orig. Health Dis. 1, 184–191.
Matsuzawa-Nagata, N., Takamura, T., Ando, H., Nakamura, S., Kurita, S., Misu, H., Ota, T., Yokoyama, M., Honda, M., Miyamoto, K., and Kaneko, S. (2008). Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metab. Clin. Exp. 57, 1071–1077.
McCurdy, C. E., Bishop, J. M., Williams, S. M., Grayson, B. E., Smith, M. S., Friedman, J. E., and Grove, K. L. (2009). Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 119, 323–335.
Mitra, A., Alvers, K. M., Crump, E. M., and Rowland, N. E. (2009). Effect of high-fat diet during gestation, lactation, or postweaning on physiological and behavioral indexes in borderline hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R20–R28.
Muhlhausler, B. S., Duffield, J. A., and McMillen, I. C. (2007a). Increased maternal nutrition stimulates peroxisome proliferator activated receptor-gamma, adiponectin, and leptin messenger ribonucleic acid expression in adipose tissue before birth. Endocrinology 148, 878–885.
Muhlhausler, B. S., Duffield, J. A., and McMillen, I. C. (2007b). Increased maternal nutrition increases leptin expression in perirenal and subcutaneous adipose tissue in the postnatal lamb. Endocrinology 148, 6157–6163.
Musingarimi, P. (2009). Obesity in the UK: a review and comparative analysis of policies within the devolved administrations. Health Policy 91, 10–16.
Ng, S.-F., Lin, R. C. Y., Laybutt, D. R., Barres, R., Owens, J. A., and Morris, M. J. (2010). Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467, 963–966.
Nishikawa, T., Kukidome, D., Sonoda, K., Fujisawa, K., Matsuhisa, T., Motoshima, H., Matsumura, T., and Araki, E. (2007). Impact of mitochondrial ROS production in the pathogenesis of insulin resistance. Diabetes Res. Clin. Pract. 77(Suppl. 1), S161–S164.
Nivoit, P., Morens, C., Van Assche, F. A., Jansen, E., Poston, L., Remacle, C., and Reusens, B. (2009). Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 52, 1133–1142.
Oben, J. A., Mouralidarane, A., Samuelsson, A.-M., Matthews, P. J., Morgan, M. L., McKee, C., Soeda, J., Fernandez-Twinn, D. S., Martin-Gronert, M. S., Ozanne, S. E., Sigala, B., Novelli, M., Poston, L., and Taylor, P. D. (2010a). Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J. Hepatol. 52, 913–920.
Oben, J. A., Patel, T., Mouralidarane, A., Samuelsson, A. M., Matthews, P., Pombo, J., Morgan, M., McKee, C., Soeda, J., Novelli, M., Poston, L., and Taylor, P. (2010b). Maternal obesity programmes offspring development of non-alcoholic fatty pancreas disease. Biochem. Biophys. Res. Commun. 394, 24–28.
Oken, E., Taveras, E. M., Kleinman, K. P., Rich-Edwards, J. W., and Gillman, M. W. (2007). Gestational weight gain and child adiposity at age 3 years. Am. J. Obstet. Gynecol. 196, 322.e1–e8.
Ong, K. K., Emmett, P. M., Noble, S., Ness, A., and Dunger, D. B. (2006). Dietary energy intake at the age of 4 months predicts postnatal weight gain and childhood body mass index. Pediatrics 117, e503–e508.
Ong, Z. Y., and Muhlhausler, B. S. (2011). Maternal ‘junk-food’ feeding of rat dams alters food choices and development of the mesolimbic reward pathway in the offspring. FASEB J. doi: 10.1096/fj.10-178392. [Epub ahead of print].
Page, K. C., Malik, R. E., Ripple, J. A., and Anday, E. K. (2009). Maternal and postweaning diet interaction alters hypothalamic gene expression and modulates response to a high-fat diet in male offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1049–R1057.
Patterson, C. M., Bouret, S. G., Park, S., Irani, B. G., Dunn-Meynell, A. A., and Levin, B. E. (2010). Large litter rearing enhances leptin sensitivity and protects selectively bred diet-induced obese rats from becoming obese. Endocrinology 151, 4270–4279.
Picó, C., Oliver, P., Sánchez, J., Miralles, O., Caimari, A., Priego, T., and Palou, A. (2007). The intake of physiological doses of leptin during lactation in rats prevents obesity in later life. Int. J. Obes. (Lond), 31, 1199–1209.
Plagemann, A., Harder, T., Brunn, M., Harder, A., Roepke, K., Wittrock-Staar, M., Ziska, T., Schellong, K., Rodekamp, E., Melchior, K., and Dudenhausen, J. W. (2009). Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J. Physiol. 587(Pt 20), 4963–4976.
Plagemann, A., Harder, T., Rake, A., Voits, M., Fink, H., Rohde, W., and Dörner, G. (1999). Perinatal elevation of hypothalamic insulin, acquired malformation of hypothalamic galaninergic neurons, and syndrome x-like alterations in adulthood of neonatally overfed rats. Brain Res. 836, 146–155.
Plagemann, A., Heidrich, I., Götz, F., Rohde, W., and Dörner, G. (1992). Obesity and enhanced diabetes and cardiovascular risk in adult rats due to early postnatal overfeeding. Exp. Clin. Endocrinol. 99, 154–158.
Plagemann, A., Roepke, K., Harder, T., Brunn, M., Harder, A., Wittrock-Staar, M., Ziska, T., Schellong, K., Rodekamp, E., Melchior, K., and Dudenhausen, J. W. (2010). Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. J. Perinat. Med. 38, 393–400.
Proulx, K., Richard, D., and Walker, C.-D. (2002). Leptin regulates appetite-related neuropeptides in the hypothalamus of developing rats without affecting food intake. Endocrinology 143, 4683–4692.
Radaelli, T., Varastehpour, A., Catalano, P., and Hauguel-de Mouzon, S. (2003). Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes 52, 2951–2958.
Rattanatray, L., MacLaughlin, S. M., Kleemann, D. O., Walker, S. K., Muhlhausler, B. S., and McMillen, I. C. (2010). Impact of maternal periconceptional overnutrition on fat mass and expression of adipogenic and lipogenic genes in visceral and subcutaneous fat depots in the postnatal lamb. Endocrinology 151, 5195–5205.
Samuelsson, A.-M., Matthews, P. A., Argenton, M., Christie, M. R., McConnell, J. M., Jansen, E. H. J. M., Piersma, A. H., Ozanne, S. E., Twinn, D. F., Remacle, Claude, Rowlerson, A., Poston, L., and Taylor, P. D. (2008). Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51, 383–392.
Sánchez, J., Priego, T., Palou, M., Tobaruela, A., Palou, A., and Picó, C. (2008). Oral supplementation with physiological doses of leptin during lactation in rats improves insulin sensitivity and affects food preferences later in life. Endocrinology 149, 733–740.
Sen, S., and Simmons, R. A. (2010). Maternal antioxidant supplementation prevents adiposity in the offspring of Western diet-fed rats. Diabetes 59, 3058–3065.
Shankar, K., Harrell, A., Liu, X., Gilchrist, J. M., Ronis, M. J. J., and Badger, T. M. (2008). Maternal obesity at conception programs obesity in the offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R528–R538.
Shankar, K., Kang, P., Harrell, A., Zhong, Y., Marecki, J. C., Ronis, M. J. J., and Badger, T. M. (2010). Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 151, 2577–2589.
Shelley, P., Martin-Gronert, M. S., Rowlerson, A., Poston, L., Heales, S. J. R., Hargreaves, I. P., McConnell, J. M., Ozanne, S. E., and Fernandez-Twinn, D. S. (2009). Altered skeletal muscle insulin signaling and mitochondrial complex II-III linked activity in adult offspring of obese mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R675–R681.
Singhal, A., Cole, T. J., Fewtrell, M., and Lucas, A. (2004). Breastmilk feeding and lipoprotein profile in adolescents born preterm: follow-up of a prospective randomised study. Lancet 363, 1571–1578.
Singhal, A., Kennedy, K., Lanigan, J., Fewtrell, M., Cole, T. J., Stephenson, T., Elias-Jones, A., Weaver, L. T., Ibhanesebhor, S., MacDonald, P. D., Bindels, J., and Lucas, A. (2010). Nutrition in infancy and long-term risk of obesity: evidence from 2 randomized controlled trials. Am. J. Clin. Nutr. 92, 1133–1144.
Smith, J., Cianflone, K., Biron, S., Hould, F. S., Lebel, S., Marceau, S., Lescelleur, O., Biertho, L., Simard, S., Kral, J. G., and Marceau, P. (2009). Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J. Clin. Endocrinol. Metab. 94, 4275–4283.
Srinivasan, M., Katewa, S. D., Palaniyappan, A., Pandya, J. D., and Patel, M. S. (2006). Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 291, E792–E799.
Srinivasan, M., Mitrani, P., Sadhanandan, G., Dodds, C., Shbeir-ElDika, S., Thamotharan, S., Ghanim, H., Dandona, P., Devaskar, S. U., and Patel, M. S. (2008). A high-carbohydrate diet in the immediate postnatal life of rats induces adaptations predisposing to adult-onset obesity. J. Endocrinol. 197, 565–574.
Stettler, N., Stallings, V. A., Troxel, A. B., Zhao, J., Schinnar, R., Nelson, S. E., Ziegler, E. E., and Strom, B. L. (2005). Weight gain in the first week of life and overweight in adulthood: a cohort study of European American subjects fed infant formula. Circulation 111, 1897–1903.
Taylor, P. D., McConnell, J., Khan, I. Y., Holemans, K., Lawrence, K. M., Asare-Anane, H., Persaud, S. J., Jones, P. M., Petrie, L., Hanson, M. A., and Poston, L. (2005). Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R134–R139.
Tong, J. F., Yan, X., Zhu, M. J., Ford, S. P., Nathanielsz, P. W., and Du, M. (2009). Maternal obesity downregulates myogenesis and beta-catenin signaling in fetal skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 296, E917–E924.
Tzoulaki, I., Sovio, U., Pillas, D., Hartikainen, A.-L., Pouta, A., Laitinen, Jaana, Tammelin, T. H., Jarvelin, M.-R., and Elliott, P. (2010). Relation of immediate postnatal growth with obesity and related metabolic risk factors in adulthood: the northern Finland birth cohort 1966 study. Am. J. Epidemiol. 171, 989–998.
Urakawa, H., Katsuki, A., Sumida, Y., Gabazza, E. C., Murashima, S., Morioka, K., Maruyama, N., Kitagawa, N., Tanaka, T., Hori, Y., Nakatani, K., Yano, Y., and Adachi, Y. (2003). Oxidative stress is associated with adiposity and insulin resistance in men. J. Clin. Endocrinol. Metab. 88, 4673–4676.
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T. D., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84.
Vickers, M. H., Gluckman, P. D., Coveny, A. H., Hofman, P. L., Cutfield, W. S., Gertler, A., Breier, B. H., and Harris, M. (2005). Neonatal leptin treatment reverses developmental programming. Endocrinology 146, 4211–4216.
Vickers, M. H., Gluckman, P. D., Coveny, A. H., Hofman, P. L., Cutfield, W. S., Gertler, A., Breier, B. H., and Harris, M. (2008). The effect of neonatal leptin treatment on postnatal weight gain in male rats is dependent on maternal nutritional status during pregnancy. Endocrinology 149, 1906–1913.
Villamor, E., and Cnattingius, S. (2006). Interpregnancy weight change and risk of adverse pregnancy outcomes: a population-based study. Lancet 368, 1164–1170.
Vucetic, Z., Kimmel, J., Totoki, K., Hollenbeck, E., and Reyes, T. M. (2010). Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 151, 4756–4764.
Wang, C.-H., Wang, C.-C., and Wei, Y.-H. (2010). Mitochondrial dysfunction in insulin insensitivity: implication of mitochondrial role in type 2 diabetes. Ann. N. Y. Acad. Sci. 1201, 157–165.
Waterland, R. (2004). Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition 20, 63–68.
Waterland, R. A., and Garza, C. (2002). Early postnatal nutrition determines adult pancreatic glucose-responsive insulin secretion and islet gene expression in rats. J. Nutr. 132, 357–364.
Whitaker, K. L., Jarvis, M. J., Beeken, R. J., Boniface, D., and Wardle, J. (2010). Comparing maternal and paternal intergenerational transmission of obesity risk in a large population-based sample. Am. J. Clin. Nutr. 91, 1560–1567.
White, C. L., Purpera, M. N., and Morrison, C. D. (2009). Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R1464–R1472.
Wilding, J. P., Gilbey, S. G., Bailey, C. J., Batt, R. A., Williams, G., Ghatei, M. A., and Bloom, S. R. (1993). Increased neuropeptide-Y messenger ribonucleic acid (mRNA) and decreased neurotensin mRNA in the hypothalamus of the obese (ob/ob) mouse. Endocrinology 132, 1939–1944.
Yan, X., Zhu, M. J., Xu, W., Tong, J. F., Ford, S. P., Nathanielsz, P. W., and Du, M. (2010). Up-regulation of Toll-like receptor 4/nuclear factor-kappaB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology 151, 380–387.
Zambrano, E., Martínez-Samayoa, P. M., Rodríguez-González, G. L., and Nathanielsz, P. W. (2010). Dietary intervention prior to pregnancy reverses metabolic programming in male offspring of obese rats. J. Physiol. 588(Pt 10), 1791–1799.
Zhang, J., Zhang, F., Didelot, X., Bruce, K. D., Cagampang, F. R., Vatish, M., Hanson, M., Lehnert, H., Ceriello, A., and Byrne, C. D. (2009). Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics 10, 478. doi: 10.1186/1471-2164-10-478
Zhang, L., Long, N. M., Hein, S. M., Ma, Y., Nathanielsz, P. W., and Ford, S. P. (2011). Maternal obesity in ewes results in reduced fetal pancreatic β-cell numbers in late gestation and decreased circulating insulin concentration at term. Domest. Anim. Endocrinol. 40, 30–39.
Zhu, M. J., Du, M., Nathanielsz, P. W., and Ford, S. P. (2010a). Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 31, 387–391.
Zhu, M. J., Ma, Y., Long, N. M., Du, M., and Ford, S. P. (2010b). Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1224–R1231.
Zhu, M. J., Han, B., Tong, J., Ma, C., Kimzey, J. M., Underwood, K. R., Xiao, Y., Hess, B. W., Ford, S. P., Nathanielsz, P. W., and Du, M. (2008). AMP-activated protein kinase signalling pathways are down regulated and skeletal muscle development impaired in fetuses of obese, over-nourished sheep. J. Physiol. (Lond.) 586, 2651–2664.
Keywords: developmental programming, obesity, maternal overnutrition, epigenetics
Citation: Alfaradhi MZ and Ozanne SE (2011) Developmental programming in response to maternal overnutrition. Front. Gene. 2:27. doi: 10.3389/fgene.2011.00027
Received: 03 March 2011;
Accepted: 23 May 2011;
Published online: 03 June 2011.
Edited by:
Jeff Schwartz, Griffith University, AustraliaReviewed by:
Claire Joanne Stocker, University of Buckingham, UKAndy Levy, University of Bristol, UK
Sarah McMullen, University of Nottingham, UK
Copyright: © 2011 Alfaradhi and Ozanne. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Susan E. Ozanne, Metabolic Research Laboratories, Institute of Metabolic Science, Addenbrooke’s Hospital, University of Cambridge, Cambridge CB2 0QQ, UK. e-mail:c2VvMTBAbW9sZS5iaW8uY2FtLmFjLnVr