 Ashley L. Cooney
Ashley L. Cooney Jennifer A. Wambach3
Jennifer A. Wambach3 Paul B. McCray
Paul B. McCray 
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genome Ed., 14 January 2022
Sec. Genome Editing in Blood Disorders
Volume 3 - 2021 | https://doi.org/10.3389/fgeed.2021.785829
This article is part of the Research TopicGenome Editing to Treat Cystic Fibrosis and Other Pulmonary DiseasesView all 5 articles
Pulmonary surfactant is critically important to prevent atelectasis by lowering the surface tension of the alveolar lining liquid. While respiratory distress syndrome (RDS) is common in premature infants, severe RDS in term and late preterm infants suggests an underlying genetic etiology. Pathogenic variants in the genes encoding key components of pulmonary surfactant including surfactant protein B (SP-B, SFTPB gene), surfactant protein C (SP-C, SFTPC gene), and the ATP-Binding Cassette transporter A3 (ABCA3, ABCA3 gene) result in severe neonatal RDS or childhood interstitial lung disease (chILD). These proteins play essential roles in pulmonary surfactant biogenesis and are expressed in alveolar epithelial type II cells (AEC2), the progenitor cell of the alveolar epithelium. SP-B deficiency most commonly presents in the neonatal period with severe RDS and requires lung transplantation for survival. SFTPC mutations act in an autosomal dominant fashion and more commonly presents with chILD or idiopathic pulmonary fibrosis than neonatal RDS. ABCA3 deficiency often presents as neonatal RDS or chILD. Gene therapy is a promising option to treat monogenic lung diseases. Successes and challenges in developing gene therapies for genetic disorders of surfactant dysfunction include viral vector design and tropism for target cell types. In this review, we explore adeno-associated virus (AAV), lentiviral, and adenoviral (Ad)-based vectors as delivery vehicles. Both gene addition and gene editing strategies are compared to best design treatments for lung diseases resulting from pathogenic variants in the SFTPB, SFTPC, and ABCA3 genes.
Pulmonary surfactant is a complex mixture of phospholipids and proteins that is secreted into the alveolar space, reduces surface tension, prevents end expiratory alveolar collapse, and is required for gas exchange. Surfactant is synthesized in lamellar bodies, specialized intracellular organelles derived from lysosomes in alveolar epithelial type II cells (AEC2, aka ATII or AT2 cells). Phospholipids are transported into the lamellar bodies by ABCA3 and assemble with surfactant proteins B and C to form surfactant. The lamellar bodies are released into the alveolar lumen via exocytosis. Pathogenic variants in genes that encode key components of pulmonary surfactant include surfactant protein B (SP-B, SFTPB gene), surfactant protein C (SP-C, SFTPC gene), and the ATP-binding cassette transporter A3 (ABCA3, ABCA3 gene) and are leading inherited causes of neonatal respiratory distress syndrome (RDS) and childhood interstitial lung disease (chILD) (Garmany et al., 2008). Treatments for these monogenic pulmonary diseases are limited and non-specific, and for many patients, lung transplant may be the only option for survival beyond the first months of life (Wambach et al., 2014). Recent advances offer renewed opportunities to develop gene therapies for genetic disorders of surfactant dysfunction resulting from pathogenic variants in SFTPB, SFTPC, and ABCA3. There is no “one size fits all” approach for developing gene therapy vectors for SP-B, SP-C, and ABCA3 deficiencies. Non-viral vectors (i.e., plasmid DNA, nanoparticles, and or in vitro transcribed mRNA) and Epstein-Barr based plasmid DNA each have important qualities and have advanced the lung gene transfer field (reviewed in (Stribling et al., 1992; Tu et al., 2000)). This review will focus on adeno-associated virus (AAV), lentiviral, or adenoviral (Ad)-based delivery vehicles for a gene addition approach or delivery of gene editing tools to complement or repair disorders of surfactant dysfunction, as well as appropriate models to assess gene transfer efficacy. Our goal for this review is to introduce potential viral vector-mediated gene therapy options for surfactant diseases and provide enough details about each deficiency to highlight why gene therapy may be particularly challenging for this disease class.
Pulmonary surfactant production is a highly regulated process of synthesis, secretion, degradation, and recycling. Surfactant is composed of 90% phospholipids, specifically phosphatidylcholine and phosphatidylglycerol, and cholesterol, and 10% surfactant proteins A, B, C, and D (Agassandian and Mallampalli, 2013). These components are synthesized in the endoplasmic reticulum in AEC2s and assembled and stored in lamellar bodies (Figure 1). Surfactant proteins B and C play a role in reducing surface tension while surfactant proteins A and D are collectins and play important roles in innate immunity (Pastva et al., 2007). Surfactant is released from lamellar bodies via exocytosis into the alveolar lumen. Upon release, the lamellar bodies unwind to form tubular myelin, an ordered structure which forms a film at the air-liquid interface (Cañadas et al., 2020).
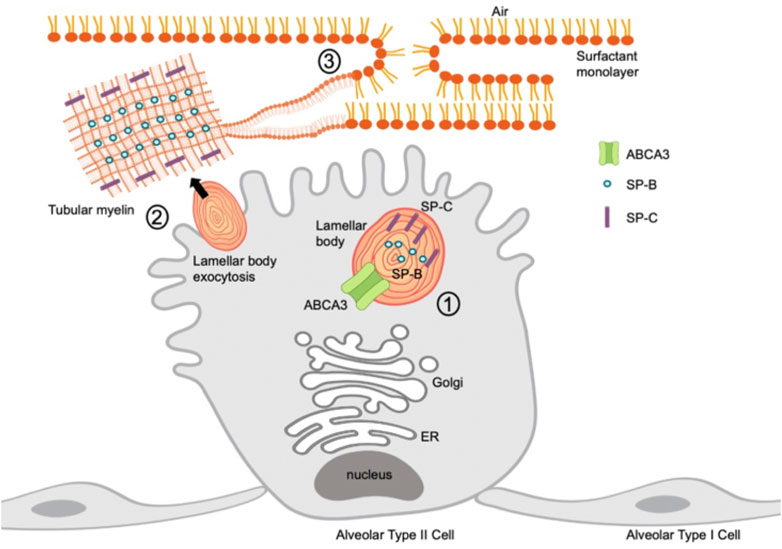
FIGURE 1. Schematic of SP-B, SP-C, and ABCA3 protein localization in an alveolar type II cell. 1) ABCA3 transports phospholipids into lamellar bodies; SP-B and SP-C provide support during surfactant assembly. 2) Lamellar bodies undergo exocytosis from alveolar type II cell and unravel into tubular myelin. 3) Tubular myelin disassembles into a surfactant monolayer through adsorption into a film at an air-liquid interface.
SP-B, SP-C, and ABCA3 each play distinct roles in surfactant production and homeostasis. SP-B is a hydrophobic protein required for surfactant formation and is a key structural support, conferring surface tension lowering properties with phospholipid molecules to enhance surfactant spreading (Cochrane and Revak, 1991). SP-C enhances adsorption at the air-liquid interface to reduce surface tension. SP-C also plays an immunomodulatory role in clearing pulmonary infections, as SP-C deficient mice exhibit inflammation upon loss of SP-C (Glasser et al., 2009). ABCA3 is involved in lamellar body formation and phospholipid transport. Loss of ABCA3 results in surfactant lacking phosphatidylcholine and increased surface tension (Garmany et al., 2006).
Newborns with SP-B deficiency typically present with RDS shortly after birth and die of progressive respiratory failure in the first few months of life without a lung transplant. A few children with biallelic SFTPB variants and chronic respiratory insufficiency have been reported (Dunbar et al., 2000). SP-B deficiency is an autosomal recessive disease and the most common pathogenic variant p. Pro133Glnfs*95 (previously known as ‘121ins2’) results in a frameshift and nonsense-mediated decay of the mRNA transcript. Treatment with exogenous surfactant enriched in SP-B protein is ineffective (Thompson, 2001) and lung transplant remains infrequent due to inavailability of suitable neonatal donor lungs (Eldridge et al., 2017). Gene addition (Barnett et al., 2017; Leibel et al., 2019; Kang et al., 2020) and gene editing approaches (Mahiny et al., 2015; Jacob et al., 2017) have demonstrated that complementing SP-B expression in AEC2s restores the phenotypic defect in vitro and in vivo. Although gene editing approaches may be successful, the diversity of pathogenic variants suggests that a gene addition approach may be a more near term goal for SP-B deficiency.
SP-B plays a major role in the assembly and function of pulmonary surfactant and is required for proper lamellar body biogenesis in AEC2s (Figure 1). It commonly exists as a homodimer and is a small, 79 amino acid hydrophobic protein that permeabilizes, cross-links, mixes, and fuses cell membranes. Mature SP-B results from proteolytic modifications involving the 381 amino acid preproSP-B peptide followed by additional glycosylation and proteolytic events of proSP-B (Guttentag, 2008) (Figure 2A). Pathogenic SFTPB variants that disrupt glycosylation or proteolytic cleavage sites have been identified (Lin et al., 2000). While both viral and nonviral delivery strategies offer promise, here we will focus on viral vectors.
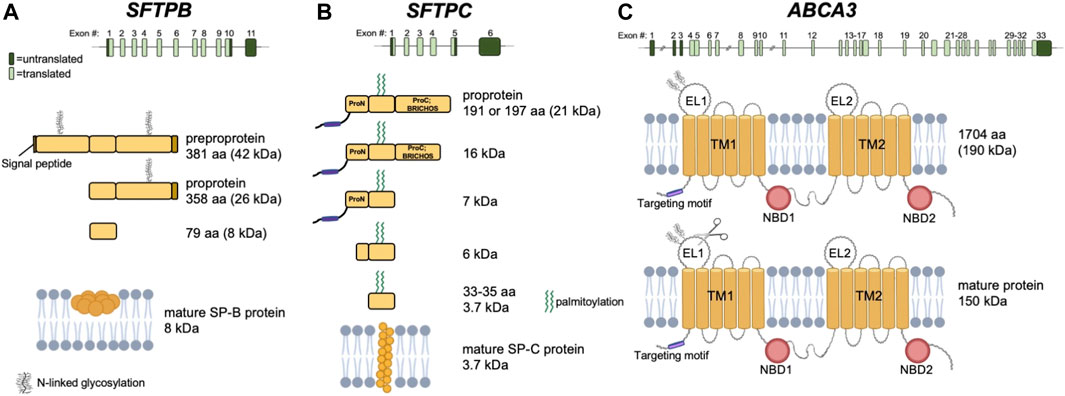
FIGURE 2. (A) Top panel: Exon representation of SFTPB on chromosome 2. Middle panels: progression of protein processing. Glycosylation at residues 129-131 and 311-313 occurs in ER. Bottom panel: yellow circles indicate oligomer of at least 6 homo-dimers inserted into a lipid bilayer. (B) Top panel: Exon representation of SFTPC on chromosome 8. Middle panels: progression of protein processing. Palmitoylation occurs in the Golgi apparatus. Cleavage events resulting in shortened protein forms are the result of Cathepsin H and Pepsinogen C enzymatic activities. Bottom panel: yellow circles indicate mature membrane bound protein. (C) Top panel: Exon representation of ABCA3 on chromosome 16. Middle panel: N-linked glycosylation at positions N124 and N140 indicated by N-linked glycosylation symbol. Bottom panel: N-terminus is proteolytically cleaved by cathepsins L and B, as indicated by scissors icon in EL1. TM, transmembrane domain; EL, extracellular loop; NBD, nucleotide binding domain; aa, amino acid.
AAV vectors have gained interest with increasing clinical applications following the FDA approval of Luxturna (Maguire et al., 2008), Zolgensma (Mendell et al., 2017), and promising studies for Duchenne muscular dystrophy (Duan, 2018). AAV vectors are episomal, have excellent safety profiles, and persist long-term in mitotically quiescent cells. Additionally, AAV can be produced to high titers and has a scalability to produce clinical grade vector (Selvaraj et al., 2021). However, the ∼4.7 kb packaging capacity limit is challenging for large transgenes. The relatively small coding sequence of SP-B (∼1.2 kb) makes AAV a feasible vector candidate. Indeed, Kang et al. used AAV to deliver SFTPB and restored surfactant production and improved survival in the conditional lethal SP-B knockout mouse model (Kang et al., 2020). AAV6 carrying a proSFTPB cDNA was delivered to conditional SP-B null mice, which typically die within 2 days of birth due to respiratory failure. In this study, survival increased to ∼200 days. Additionally, lamellar body formation was restored and lung structure and function were improved. Other approaches such as delivering truncated versions of SP-B demonstrated increased survival in SP-B deficient mice. These studies also revealed that the C-terminal portion of the protein is important for surfactant formation (Akinbi et al., 1997).
When designing a gene therapy vector, an important goal is the efficient delivery to and the persistent expression of the transgene in a target cell type. Endpoints to be quantified include the transduction efficiency, transgene expression (mRNA and protein), and functional correction. To achieve this with AAV, capsid selection is necessary for transducing AEC2s. AAV2, AAV6, and AAV6 variant capsids transduce AEC2 organoid models (Meyer-Berg et al., 2020) as well as both airway and alveolar epithelial cells (Kang et al., 2020). The route of administration is an important consideration for successfully transducing target cells of interest. AAV9 administered intravenously transduces heart, skeletal muscle, and pancreas with better efficacy compared to an AAV8 capsid (Inagaki et al., 2006). Whether topical lung delivery or systemic delivery is superior for AEC2 transduction remains to be determined.
Following efficient transduction of AEC2s, achieving persistent expression in the lung likely will require integration in a progenitor cell population. AAV is generally considered to be non-integrating, but its persistence in AEC2s is unknown. Incorporating an integrating system such as the piggyBac transposon into AAV is a potential strategy to achieve persistent expression in the alveoli (Cooney et al., 2015; Brommel et al., 2020). AEC2s are an important progenitor cell in the alveolus (Olajuyin et al., 2019) and while they retain secretory functions for surfactant production, they are also critical for alveolar homeostasis, through self-renewal or differentiation to alveolar type I cells (Mason and Williams, 1977; Fehrenbach, 2001). Lastly, promoter and polyadenylation (pA) tail choice is important to consider. Promoter size is another key factor in decision making, specifically a short promoter with sufficient activity. Synthetic promoters such as F5Tg83 and a short polyA have been created to maximize space within AAV vectors (Yan et al., 2015). AEC2-specific promoters for cell specific expression of SP-B and ABCA3 have yet to be evaluated. The SP-C promoter is a candidate for directing AEC2 specific gene expression (Degiulio et al., 2010).
Lentiviral vectors demonstrated therapeutic efficacy in ex vivo gene transfer to hematopoietic stem cells for as ADA-SCID (Kohn et al., 2021), β-Thalassemia (Thompson et al., 2018), and Wiskott-Aldrich syndrome (Sereni et al., 2019). Insertional mutagenesis is a potential risk with any integrating vector. The functional consequences of lentiviral vector integration has received considerable attention (reviewed in (Schlimgen et al., 2016; Milone and O’Doherty, 2018)). However, using the current generation of self-inactivating vectors, the clonal expansion of corrected cells has not been observed following ex vivo or systemic delivery of lentiviral vectors. The risk versus benefit must be taken into consideration when evaluating the use of an integrating vector. Lentiviral vectors are recognized for their ability to transduce both dividing and non-dividing cells, integrate for long-term expression, and allow readministration without blocking immune responses (Sinn et al., 2008). Lentiviral vectors encoding SFTPB delivered to alveolar organoids conferred SP-B expression and reversed the surfactant deficiency phenotype in vitro (Leibel et al., 2019; Munis et al., 2021). A key consideration in designing a therapeutic lentiviral vector for SP-B deficiency is envelope selection to transduce the appropriate cell type. Vesicular stomatitis virus glycoprotein (VSV-G) is the most common envelope used to pseudotype retroviruses and is reported to transduce AEC2s (Borok et al., 2001). Other envelopes such as baculovirus GP64 (Sinn et al., 2012) and Sendai virus F/HN (Griesenbach et al., 2012) also transduce alveolar epithelia. Options for promoter choice include cell type specific or constitutive elements. Production of clinical grade vector for in vivo somatic cell targeting represents a challenge, but advances in lentivirus manufacturing (Valkama et al., 2018; Martínez-Molina et al., 2020; Soldi et al., 2020) make this a promising therapeutic for SP-B deficiency.
SP-C is a 35-amino acid hydrophobic protein that organizes with SP-B to lower the surface tension of surfactant (Figure 1). The SFTPC locus spans ∼3,500 bp and is expressed from six exons on chromosome 8. A 197-amino acid protein is first produced with an N-terminal propeptide domain and C-terminal BRICHOS domain (pro-SPC). As the mature protein is processed, the N-terminal domain is cleaved from the proSPC form to SP-C. The mature protein has a shortened N-terminal domain, linker, and C-terminal BRICHOS domain (Figure 2B).
Pathogenic variants in SFTPC can arise de novo (∼50% of cases) or are inherited (∼50% of cases) (Nogee et al., 2002). A pathogenic splicing variant in SFTPC within the first base of intron 4 (c.460+1G > A) was first reported in 2001 in an infant and mother with interstitial pneumonitis (Nogee et al., 2001). Subsequently, additional pathogenic SFTPC variants have been identified, the most frequent of which is p. I73T (Cameron et al., 2005). Individuals with pathogenic SFTPC variants most commonly present with sporadic or familial interstitial lung disease as infants, children, or adults, and less frequently with neonatal RDS (Nogee et al., 2002). The disease course is highly variable with some infants presenting with severe respiratory failure requiring lung transplant, others exhibiting chronic disease managed with long-term mechanical ventilation (Liptzin et al., 2015), and others remaining relatively asymptomatic (Thomas et al., 2002). Genotype alone is not predictive of disease presentation, severity, and or course. Pathogenic SFTPC variants located in the C-terminal BRICHOS domain, which acts as a chaperone to promote protein stability, result in increased endoplasmic reticulum stress, inflammation, and spontaneous pulmonary fibrosis in a murine model (Katzen et al., 2019). Non-BRICHOS variants, which include p. I73T, result in defective AEC2 macroautophagy, inflammation, remodeling, and fibrosis (Nureki et al., 2018).
SP-C associated interstitial lung disease is a gain-of-toxic-function disease inherited in an autosomal dominant pattern with variable penetrance (Thomas et al., 2002). Expression of a mutant SFTPC allele is responsible for the surfactant dysfunction phenotype. The resultant gain-of-toxic-function requires silencing of the mutant allele. Therefore, a gene addition approach is not an option. An allele specific gene knockout or knockdown approach can be used to disrupt and inactivate or reduce the abundance of the mutant SFTPC product. CRISPR/Cas9 nuclease-mediated gene knockout has been validated in mice in utero by targeting the p.173T allele, resulting in improved lung morphology, and increased survival of offspring (Alapati et al., 2019). RNAi and antisense RNA strategies can also be envisioned.
Common gene editing approaches, such as CRISPR/Cas9, may be employed using either homologous recombination or non-homologous end joining approaches to repair or inactivate the mutant allele. Alternatively, the advent of cytosine and adenine base editors and prime editing may allow single base changes without causing indels (Levy et al., 2020). AAV is commonly used to deliver gene editing tools, including incorporating a split-intein system to allow for co-delivery of cassettes larger than the AAV packaging capacity. Alternatively, the smaller Staphylococcus aureus Cas9 can be delivered with a sgRNA using a single AAV vector (Lau and Suh, 2017). As with developing a gene editing treatment for any disease, an individualized approach may be required for the array of pathogenic SFTPC variants that cause interstitial lung disease.
ABCA3 deficiency, an autosomal recessive disorder, and is caused by pathogenic variants in ABCA3. Biallelic variants in ABCA3 cause severe neonatal RDS, chILD, and adult pulmonary fibrosis (Shulenin et al., 2004; Klay et al., 2020; Tomer et al., 2021). Infants with biallelic ABCA3 frameshift or nonsense variants present with neonatal RDS at birth and die within the first year of life without lung transplant (Wambach et al., 2014). The respiratory phenotypes and disease courses for individuals with ABCA3 missense variants are variable and include neonatal RDS and interstitial lung disease.
ABCA3 is a phospholipid transporter located at the lysosomal-derived lamellar body limiting membrane and plays a critical role in surfactant assembly and lamellar body formation (Figure 1). The ABCA3 cDNA encodes a 1,704 amino acid polypeptide. Mature ABCA3 protein is folded in the endoplasmic reticulum and undergoes glycosylation in the Golgi (Beers and Mulugeta, 2017). A second post-translational modification involves the N-terminal proteolytic cleavage of the 190 kD protein, shortening the protein to 150 kD (Engelbrecht et al., 2010) (Figure 2C). The lamellar bodies from infants and children with ABCA3 deficiency are small with dense bodies and are described as having a “fried-egg appearance” (Wert et al., 2009). Two mechanistic classes of ABCA3 missense variants have been identified: disruption of intracellular trafficking and impaired phospholipid transport (Matsumura et al., 2006; Denman et al., 2018; Wambach et al., 2020). The most common pathogenic variant is p. E292V, a missense variant that impairs phospholipid transport (Wambach et al., 2016). This variant is commonly associated with chILD (Wambach et al., 2014) and is present in 0.4% of individuals in Genome Aggregation database (gnomad.broadinstitute.org, accessed September 2021).
The overall goal for gene therapy to treat loss-of-function diseases is to efficiently and persistently (long-term) express ABCA3 at physiological levels in AEC2s. A gene addition strategy involves complementing the loss of function with a full length ABCA3 cDNA. Because most ABCA3 variants are rare and private, this poses a challenge for developing variant-specific gene editing or targeted drug approaches. The size constraints of some viral vectors present challenges for a transgene as large as ABCA3.
Gene addition of ABCA3 cDNA using lentiviral vectors is a promising therapeutic approach. Lentiviral vectors have a packaging capacity of at least 7.5 kb and could readily accommodate the 5.1 kb ABCA3 transgene and promoter. Additionally, lentiviral vectors are amenable to pseudotyping with various envelopes to modify tropism. As discussed in the lentiviral section of SP-B deficiency, envelope, promoter, and polyadenylation signal are important considerations in designing a gene therapy vector for ABCA3 deficiency. The integrating properties of lentiviral vectors make this vector class an attractive option for ABCA3 deficiency.
Given that the ABCA3 5.1 kb cDNA size surpasses the 4.7 kb packaging limit of AAV vectors, an AAV approach to deliver the full length ABCA3 is challenging with current technologies. Approaches using AAV to restore ABCA3 deficiency can be modeled from other diseases, including insertion of a partial super exon cDNA as described for another ABC transporter protein, and cystic fibrosis transmembrane conductance regulator (CFTR also know as ABCC7) (Bednarski et al., 2016). A dual vector approach using split inteins has been employed to deliver gene editing tools, specifically splitting the Cas9 transgene between two AAV vectors which are joined by a C-terminal and N-terminal inteins (Tornabene et al., 2019); however, this approach may not be feasible in proteins with multiple transmembrane domains. Further studies are required to investigate whether ABCA3 could be delivered using such dual vector platforms.
Adenoviral (Ad)-based vectors provide robust expression and a relatively large carrying capacity (∼10 kb). Additional features of Ad vectors are: 1) they transduce both dividing and non-dividing cells, 2) they have broad tissue tropism, and 3) they are scalable for clinical platforms. Still, their greatest hurdle is the innate and adaptive immune responses which can clear transduced cells. In efforts to overcome limitations due to immune responses, helper-dependent Ad (HDAd) vectors were created by deleting all viral genes, leaving only a packaging signal. The required viral components are provided by a helper virus in trans during virus production. A second limitation is that Ad is not an integrating vector. However, Ad-based delivery of gene editing tools has shown long-term phenotypic correction of Hemophilia B in a mouse model (Stephens et al., 2019). Furthermore, numerous studies have incorporated transposon systems such as Sleeping Beauty and piggyBac to create hybrid integrating adenoviral vectors (Cooney et al., 2015; Boehme et al., 2016). Ad-based vectors remain a candidate platform for gene therapy and vaccine developments. An advantage for the production of Ad-based vectors is their ability to be grown to high titers and stringently purified (Danthinne and Imperiale, 2000; Nadeau and Kamen, 2003).
Given the 5.1 kb ABCA3 transgene, Ad-based vectors have been extensively used to study ABCA3 complementation and the impact of various variants. For example, Ad vectors carrying ABCA3 variants including p. L101P (mistrafficking mutant) and p. E292V (impaired phospholipid transport mutant) were used to functionally characterize mutant effects in a pulmonary epithelial cell line (A549 cells) (Wambach et al., 2016; Hu et al., 2020; Wambach et al., 2020). These studies helped establish how the proteins from each mutant mechanistic class traffic through the cell, affect lamellar body formation, and transport phosphatidylcholine. Identification of variant-specific mechansims may inform disease course or therapeutic approach.
Immortalized pulmonary epithelial cell lines with characteristics of AEC2s such as A549 cells are a first line model system because they are easy to passage and maintain, form lamellar body-like structures which can be visualized by electron microscopy, and are readily transduced by viral vectors. A549 ABCA3−/− cells are established and lack well-developed lamellar body-like structures (Wambach et al., 2020). However, a major limitation for tumor-derived cell lines such as A549 is that they do not produce surfactant. Alveolar cells need to differentiate in order to produce surfactant and express SP-C, a common marker used to identify AEC2s.
Alveolar epithelial cell organoids derived from human embryonic or pluripotent stem cells generate alveolospheres in culture which display functional properties of AEC2s. These cells can proliferate, differentiate, and be modified to encompass surfactant dysfunction phenotypes (Jacob et al., 2017; Jacob et al., 2019). iPSC organoids derived from a patient with SP-B deficiency mirrored the disease phenotype including decreased surfactant production. This defect was restored when transduced with a lentiviral vector carrying SFTPB, including proper lamellar body formation (Leibel et al., 2019). In total, AEC2 organoids are a useful model to assess strategies to restore function of surfactant components.
Measuring restoration of surfactant function is challenging. Surfactant proteins B and C are typically quantified to confirm the presence of surfactant proteins in cell lysates or secretions. The measurement of phospholipid transport using dipalmitylphosphatidylcholine (DPPC) release is one method to assess surfactant production (Jacob et al., 2017). Instruments such as the pulsating bubble surfactometer can be used to measure the surface tension lowering properties of cell secretions.
Mouse models of each of these surfactant-based genetic diseases have been generated. SP-B knockout mice develop severe respiratory distress and die within hours of birth and exhibit dense, abnormal lamellar body-like organelles (Clark et al., 1995; Stahlman et al., 2000). Restoring SP-B using an AAV vector improved survival in conditional SP-B knockout mice (Kang et al., 2020). Alternatively, SP-C knockout mice are viable and can survive to adulthood. They produce lamellar bodies and surfactant proteins A, B, and D. Phenotypic responses include decreased stability of surfactant at low volumes, pneumonitis and emphysema (Glasser et al., 2001; Whitsett and Weaver, 2002). ABCA3 knockout mice have a similar outcome as SP-B knockout mice and do not survive (Ban et al., 2007; Fitzgerald et al., 2007). Conditionally null ABCA3 mice have also been generated (Besnard et al., 2010).
Gene based therapies for disorders of surfactant dysfunction resulting from pathogenic variants in SFTPB, SFTPC, and ABCA3 are now realizable. Generating a delivery vector for either a gene addition or gene editing approach requires the careful consideration of vector design, transgene expression cassette, preclinical models used, and assays to assess the correction of surfactant production and function.
AC drafted and edited article; JW, PS, and PM critically reviewed and edited article.
This work was supported by the National Institutes of Health (UG3 HL147366, P01 HL51670, P01 HL091842, P01 HL152960, and R01 HL133089, the Cystic Fibrosis Foundation, the University of Iowa Center for Gene Therapy (DK54759), and the Roy J. Carver Chair in Pulmonary Research (PM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. PM is on the SAB consults and performs sponsored research for Spirovant Sciences.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Anthony Fischer, Christian Brommel, Camilla Hippee, and Lorellin Durnell for their critical review of this article.
Agassandian, M., and Mallampalli, R. K. (2013). Surfactant Phospholipid Metabolism. Biochim. Biophys. Acta (Bba) - Mol. Cel Biol. Lipids 1831, 612–625. doi:10.1016/j.bbalip.2012.09.010
Akinbi, H. T., Breslin, J. S., Ikegami, M., Iwamoto, H. S., Clark, J. C., Whitsett, J. A., et al. (1997). Rescue of SP-B Knockout Mice with a Truncated SP-B Proprotein. J. Biol. Chem. 272, 9640–9647. doi:10.1074/jbc.272.15.9640
Alapati, D., Zacharias, W. J., Hartman, H. A., Rossidis, A. C., Stratigis, J. D., Ahn, N. J., et al. (2019). In Utero gene Editing for Monogenic Lung Disease. Sci. Transl. Med. 11, eaav8375. doi:10.1126/scitranslmed.aav8375
Ban, N., Matsumura, Y., Sakai, H., Takanezawa, Y., Sasaki, M., Arai, H., et al. (2007). Abca3 as a Lipid Transporter in Pulmonary Surfactant Biogenesis. J. Biol. Chem. 282, 9628–9634. doi:10.1074/jbc.M611767200
Barnett, R. C., Lin, X., Barravecchia, M., Norman, R. A., de Mesy Bentley, K. L., Fazal, F., et al. (2017). Featured Article: Electroporation-Mediated Gene Delivery of Surfactant Protein B (Sp-b) Restores Expression and Improves Survival in Mouse Model of Sp-B Deficiency. Exp. Biol. Med. (Maywood) 242, 1345–1354. doi:10.1177/1535370217713000
Bednarski, C., Tomczak, K., vom Hövel, B., Weber, W.-M., and Cathomen, T. (2016). Targeted Integration of a Super-Exon into the Cftr Locus Leads to Functional Correction of a Cystic Fibrosis Cell Line Model. PLoS One 11, e0161072. doi:10.1371/journal.pone.0161072
Beers, M. F., and Mulugeta, S. (2017). The Biology of the Abca3 Lipid Transporter in Lung Health and Disease. Cell Tissue Res 367, 481–493. doi:10.1007/s00441-016-2554-z
Besnard, V., Matsuzaki, Y., Clark, J., Xu, Y., Wert, S. E., Ikegami, M., et al. (2010). Conditional Deletion ofAbca3in Alveolar Type II Cells Alters Surfactant Homeostasis in Newborn and Adult Mice. Am. J. Physiology-Lung Cell Mol. Physiol. 298, L646–L659. doi:10.1152/ajplung.00409.2009
Boehme, P., Zhang, W., Solanki, M., Ehrke-Schulz, E., and Ehrhardt, A. (2016). A High-Capacity Adenoviral Hybrid Vector System Utilizing the Hyperactive Sleeping beauty Transposase Sb100x for Enhanced Integration. Mol. Ther. - Nucleic Acids 5, e337. doi:10.1038/mtna.2016.44
Borok, Z., Harboe-Schmidt, J. E., Brody, S. L., You, Y., Zhou, B., Li, X., et al. (2001). Vesicular Stomatitis Virus G-Pseudotyped Lentivirus Vectors Mediate Efficient Apical Transduction of Polarized Quiescent Primary Alveolar Epithelial Cells. J. Virol. 75, 11747–11754. doi:10.1128/JVI.75.23.11747-11754.2001
Brommel, C. M., Cooney, A. L., and Sinn, P. L. (2020). Adeno-Associated Virus-Based Gene Therapy for Lifelong Correction of Genetic Disease. Hum. Gene Ther. 31, 985–995. doi:10.1089/hum.2020.138
Cameron, H. S., Somaschini, M., Carrera, P., Hamvas, A., Whitsett, J. A., Wert, S. E., et al. (2005). A Common Mutation in the Surfactant Protein C Gene Associated with Lung Disease. J. Pediatr. 146, 370–375. doi:10.1016/j.jpeds.2004.10.028
Cañadas, O., Olmeda, B., Alonso, A., and Pérez-Gil, J. (2020). Lipid-Protein and Protein-Protein Interactions in the Pulmonary Surfactant System and Their Role in Lung Homeostasis. Int. J. Mol. Sci. 21, 3708. doi:10.3390/ijms21103708
Clark, J. C., Wert, S. E., Bachurski, C. J., Stahlman, M. T., Stripp, B. R., Weaver, T. E., et al. (1995). Targeted Disruption of the Surfactant Protein B Gene Disrupts Surfactant Homeostasis, Causing Respiratory Failure in Newborn Mice. Proc. Natl. Acad. Sci. 92, 7794–7798. doi:10.1073/pnas.92.17.7794
Cochrane, C. G., and Revak, S. D. (1991). Pulmonary Surfactant Protein B (Sp-b): Structure-Function Relationships. Science 254, 566–568. doi:10.1126/science.1948032
Cooney, A. L., Singh, B. K., and Sinn, P. L. (2015). Hybrid Nonviral/Viral Vector Systems for Improved Piggybac DNA Transposon In Vivo Delivery. Mol. Ther. 23, 667–674. doi:10.1038/mt.2014.254
Danthinne, X., and Imperiale, M. J. (2000). Production of First Generation Adenovirus Vectors: A Review. Gene Ther. 7, 1707–1714. doi:10.1038/sj.gt.3301301
Degiulio, J. V., Kaufman, C. D., and Dean, D. A. (2010). The Sp-C Promoter Facilitates Alveolar Type Ii Epithelial Cell-Specific Plasmid Nuclear Import and Gene Expression. Gene Ther. 17, 541–549. doi:10.1038/gt.2009.166
Denman, L., Yonker, L. M., and Kinane, T. B. (2018). The Classification of Atp-Binding Cassette Subfamily a Member 3 Mutations Using the Cystic Fibrosis Transmembrane Conductance Regulator Classification System. Pediatr. Invest. 2, 17–24. doi:10.1002/ped4.12020
Duan, D. (2018). Micro-Dystrophin Gene Therapy Goes Systemic in Duchenne Muscular Dystrophy Patients. Hum. Gene Ther. 29, 733–736. doi:10.1089/hum.2018.012
Dunbar, A. E., Ikegami, S. E. M., Whitsett, J. A., Hamvas, A., White, F. V., Piedboeuf, B., et al. (2000). Prolonged Survival in Hereditary Surfactant Protein B (Sp-b) Deficiency Associated with a Novel Splicing Mutation. Pediatr. Res. 48, 275–282. doi:10.1203/00006450-200009000-00003
Eldridge, W. B., Zhang, Q., Faro, A., Sweet, S. C., Eghtesady, P., Hamvas, A., et al. (2017). Outcomes of Lung Transplantation for Infants and Children with Genetic Disorders of Surfactant Metabolism. J. Pediatr. 184, 157–164. e152. doi:10.1016/j.jpeds.2017.01.017
Engelbrecht, S., Kaltenborn, E., Griese, M., and Kern, S. (2010). The Surfactant Lipid Transporter Abca3 Is N-Terminally Cleaved inside Lamp3-Positive Vesicles. FEBS Lett. 584, 4306–4312. doi:10.1016/j.febslet.2010.09.026
Fehrenbach, H. (2001). Alveolar Epithelial Type Ii Cell: Defender of the Alveolus Revisited. Respir. Res. 2, 33–46. doi:10.1186/rr36
Fitzgerald, M. L., Xavier, R., Haley, K. J., Welti, R., Goss, J. L., Brown, C. E., et al. (2007). Abca3 Inactivation in Mice Causes Respiratory Failure, Loss of Pulmonary Surfactant, and Depletion of Lung Phosphatidylglycerol. J. Lipid Res. 48, 621–632. doi:10.1194/jlr.M600449-JLR200
Garmany, T. H., Moxley, M. A., White, F. V., Dean, M., Hull, W. M., Whitsett, J. A., et al. (2006). Surfactant Composition and Function in Patients with Abca3 Mutations. Pediatr. Res. 59, 801–805. doi:10.1203/01.pdr.0000219311.14291.df
Garmany, T. H., Wambach, J. A., Heins, H. B., Watkins-Torry, J. M., Wegner, D. J., Bennet, K., et al. (2008). Population and Disease-Based Prevalence of the Common Mutations Associated with Surfactant Deficiency. Pediatr. Res. 63, 645–649. doi:10.1203/PDR.0b013e31816fdbeb
Glasser, S. W., Burhans, M. S., Korfhagen, T. R., Na, C.-L., Sly, P. D., Ross, G. F., et al. (2001). Altered Stability of Pulmonary Surfactant in Sp-C-Deficient Mice. Proc. Natl. Acad. Sci. 98, 6366–6371. doi:10.1073/pnas.101500298
Glasser, S. W., Witt, T. L., Senft, A. P., Baatz, J. E., Folger, D., Maxfield, M. D., et al. (2009). Surfactant Protein C-Deficient Mice Are Susceptible to Respiratory Syncytial Virus Infection. Am. J. Physiology-Lung Cell Mol. Physiol. 297, L64–L72. doi:10.1152/ajplung.90640.2008
Griesenbach, U., Inoue, M., Meng, C., Farley, R., Chan, M., Newman, N. K., et al. (2012). Assessment of F/hn-Pseudotyped Lentivirus as a Clinically Relevant Vector for Lung Gene Therapy. Am. J. Respir. Crit. Care Med. 186, 846–856. doi:10.1164/rccm.201206-1056OC
Guttentag, S. (2008). Posttranslational Regulation of Surfactant Protein B Expression. Semin. Perinatology 32, 367–370. doi:10.1053/j.semperi.2008.08.003
Hu, J. Y., Yang, P., Wegner, D. J., Heins, H. B., Luke, C. J., Li, F., et al. (2020). Functional Characterization of Four ATP‐Binding Cassette Transporter A3 Gene ( ABCA3 ) Variants. Hum. Mutat. 41, 1298–1307. doi:10.1002/humu.24014
Inagaki, K., Fuess, S., Storm, T. A., Gibson, G. A., Mctiernan, C. F., Kay, M. A., et al. (2006). Robust Systemic Transduction with Aav9 Vectors in Mice: Efficient Global Cardiac Gene Transfer superior to that of Aav8. Mol. Ther. 14, 45–53. doi:10.1016/j.ymthe.2006.03.014
Jacob, A., Morley, M., Hawkins, F., McCauley, K. B., Jean, J. C., Heins, H., et al. (2017). Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 21, 472–488. e410. doi:10.1016/j.stem.2017.08.014
Jacob, A., Vedaie, M., Roberts, D. A., Thomas, D. C., Villacorta-Martin, C., Alysandratos, K.-D., et al. (2019). Derivation of Self-Renewing Lung Alveolar Epithelial Type Ii Cells from Human Pluripotent Stem Cells. Nat. Protoc. 14, 3303–3332. doi:10.1038/s41596-019-0220-0
Kang, M. H., van Lieshout, L. P., Xu, L., Domm, J. M., Vadivel, A., Renesme, L., et al. (2020). A Lung Tropic Aav Vector Improves Survival in a Mouse Model of Surfactant B Deficiency. Nat. Commun. 11, 3929. doi:10.1038/s41467-020-17577-8
Katzen, J., Wagner, B. D., Venosa, A., Kopp, M., Tomer, Y., Russo, S. J., et al. (2019). A Sftpc Brichos Mutant Links Epithelial ER Stress and Spontaneous Lung Fibrosis. JCI Insight 4, e126125. doi:10.1172/jci.insight.126125
Klay, D., Platenburg, M. G. J. P., van Rijswijk, R. H. N. A. J., Grutters, J. C., and van Moorsel, C. H. M. (2020). Abca3 Mutations in Adult Pulmonary Fibrosis Patients: A Case Series and Review of Literature. Curr. Opin. Pulm. Med. 26, 293–301. doi:10.1097/MCP.0000000000000680
Kohn, D. B., Booth, C., Shaw, K. L., Xu-Bayford, J., Garabedian, E., Trevisan, V., et al. (2021). Autologous Ex Vivo Lentiviral Gene Therapy for Adenosine Deaminase Deficiency. N. Engl. J. Med. 384, 2002–2013. doi:10.1056/NEJMoa2027675
Lau, C.-H., and Suh, Y. (2017). In Vivo Genome Editing in Animals Using Aav-Crispr System: Applications to Translational Research of Human Disease. F1000Res 6, 2153. doi:10.12688/f1000research.11243.1
Leibel, S. L., Winquist, A., Tseu, I., Wang, J., Luo, D., Shojaie, S., et al. (2019). Reversal of Surfactant Protein B Deficiency in Patient Specific Human Induced Pluripotent Stem Cell Derived Lung Organoids by Gene Therapy. Sci. Rep. 9, 13450. doi:10.1038/s41598-019-49696-8
Levy, J. M., Yeh, W.-H., Pendse, N., Davis, J. R., Hennessey, E., Butcher, R., et al. (2020). Cytosine and Adenine Base Editing of the Brain, Liver, Retina, Heart and Skeletal Muscle of Mice via Adeno-Associated Viruses. Nat. Biomed. Eng. 4, 97–110. doi:10.1038/s41551-019-0501-5
Lin, Z., Pearson, C., Chinchilli, V., Pietschmann, S., Luo, J., Pison, U., et al. (2000). Polymorphisms of humanSP-A,SP-B, andSP-Dgenes: Association ofSP-BThr131Ile with ARDS. Clin. Genet. 58, 181–191. doi:10.1034/j.1399-0004.2000.580305.x
Liptzin, D. R., Patel, T., and Deterding, R. R. (2015). Chronic Ventilation in Infants with Surfactant Protein C Mutations: An Alternative to Lung Transplantation. Am. J. Respir. Crit. Care Med. 191, 1338–1340. doi:10.1164/rccm.201411-1955LE
Maguire, A. M., Simonelli, F., Pierce, E. A., Pugh, E. N., Mingozzi, F., Bennicelli, J., et al. (2008). Safety and Efficacy of Gene Transfer for Leber's Congenital Amaurosis. N. Engl. J. Med. 358, 2240–2248. doi:10.1056/NEJMoa0802315
Mahiny, A. J., Dewerth, A., Mays, L. E., Alkhaled, M., Mothes, B., Malaeksefat, E., et al. (2015). In Vivo Genome Editing Using Nuclease-Encoding mRNA Corrects SP-B Deficiency. Nat. Biotechnol. 33, 584–586. doi:10.1038/nbt.3241
Martínez-Molina, E., Chocarro-Wrona, C., Martínez-Moreno, D., Marchal, J. A., and Boulaiz, H. (2020). Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges. Pharmaceutics 12, 1051. doi:10.3390/pharmaceutics12111051
Mason, R. J., and Williams, M. C. (1977). Type Ii Alveolar Cell. Defender of the Alveolus. Am. Rev. Respir. Dis. 115, 81–91. doi:10.1164/arrd.1977.115.S.81
Matsumura, Y., Ban, N., Ueda, K., and Inagaki, N. (2006). Characterization and Classification of Atp-Binding Cassette Transporter Abca3 Mutants in Fatal Surfactant Deficiency. J. Biol. Chem. 281, 34503–34514. doi:10.1074/jbc.M600071200
Mendell, J. R., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., et al. (2017). Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 377, 1713–1722. doi:10.1056/NEJMoa1706198
Meyer-Berg, H., Zhou Yang, L., Pilar de Lucas, M., Zambrano, A., Hyde, S. C., and Gill, D. R. (2020). Identification of Aav Serotypes for Lung Gene Therapy in Human Embryonic Stem Cell-Derived Lung Organoids. Stem Cel Res Ther 11, 448. doi:10.1186/s13287-020-01950-x
Milone, M. C., and O’Doherty, U. (2018). Clinical Use of Lentiviral Vectors. Leukemia 32, 1529–1541. doi:10.1038/s41375-018-0106-0
Munis, A. M., Hyde, S. C., and Gill, D. R. (2021). A Human Surfactant B Deficiency Air-Liquid Interface Cell Culture Model Suitable for Gene Therapy Applications. Mol. Ther. - Methods Clin. Develop. 20, 237–246. doi:10.1016/j.omtm.2020.11.013
Nadeau, I., and Kamen, A. (2003). Production of Adenovirus Vector for Gene Therapy. Biotechnol. Adv. 20, 475–489. doi:10.1016/s0734-9750(02)00030-7
Nogee, L. M., Dunbar, A. E., Wert, S., Askin, F., Hamvas, A., and Whitsett, J. A. (2002). Mutations in the Surfactant Protein C Gene Associated with Interstitial Lung Disease. Chest 121, 20S–21S. doi:10.1378/chest.121.3_suppl.20s
Nogee, L. M., Dunbar, A. E., Wert, S. E., Askin, F., Hamvas, A., and Whitsett, J. A. (2001). A Mutation in the Surfactant Protein C Gene Associated with Familial Interstitial Lung Disease. N. Engl. J. Med. 344, 573–579. doi:10.1056/NEJM200102223440805
Nureki, S.-I., Tomer, Y., Venosa, A., Katzen, J., Russo, S. J., Jamil, S., et al. (2018). Expression of Mutant Sftpc in Murine Alveolar Epithelia Drives Spontaneous Lung Fibrosis. J. Clin. Invest. 128, 4008–4024. doi:10.1172/JCI99287
Olajuyin, A. M., Zhang, X., and Ji, H.-L. (2019). Alveolar Type 2 Progenitor Cells for Lung Injury Repair. Cel Death Discov. 5, 63. doi:10.1038/s41420-019-0147-9
Pastva, A. M., Wright, J. R., and Williams, K. L. (2007). Immunomodulatory Roles of Surfactant Proteins a and D: Implications in Lung Disease. Proc. Am. Thorac. Soc. 4, 252–257. doi:10.1513/pats.200701-018AW
Schlimgen, R., Howard, J., Wooley, D., Thompson, M., Baden, L. R., Yang, O. O., et al. (2016). Risks Associated with Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 58, 1159–1166. doi:10.1097/JOM.0000000000000879
Selvaraj, N., Wang, C.-K., Bowser, B., Broadt, T., Shaban, S., Burns, J., et al. (2021). Detailed Protocol for the Novel and Scalable Viral Vector Upstream Process for Aav Gene Therapy Manufacturing. Hum. Gene Ther. 32, 850–861. doi:10.1089/hum.2020.054
Sereni, L., Castiello, M. C., Di Silvestre, D., Della Valle, P., Brombin, C., Ferrua, F., et al. (2019). Lentiviral Gene Therapy Corrects Platelet Phenotype and Function in Patients with Wiskott-Aldrich Syndrome. J. Allergy Clin. Immunol. 144, 825–838. doi:10.1016/j.jaci.2019.03.012
Shulenin, S., Nogee, L. M., Annilo, T., Wert, S. E., Whitsett, J. A., and Dean, M. (2004). ABCA3Gene Mutations in Newborns with Fatal Surfactant Deficiency. N. Engl. J. Med. 350, 1296–1303. doi:10.1056/NEJMoa032178
Sinn, P. L., Arias, A. C., Brogden, K. A., and McCray, P. B. (2008). Lentivirus Vector Can Be Readministered to Nasal Epithelia without Blocking Immune Responses. J. Virol. 82, 10684–10692. doi:10.1128/JVI.00227-08
Sinn, P. L., Cooney, A. L., Oakland, M., Dylla, D. E., Wallen, T. J., Pezzulo, A. A., et al. (2012). Lentiviral Vector Gene Transfer to Porcine Airways. Mol. Ther. - Nucleic Acids 1, e56. doi:10.1038/mtna.2012.47
Soldi, M., Sergi Sergi, L., Unali, G., Kerzel, T., Cuccovillo, I., Capasso, P., et al. (2020). Laboratory-Scale Lentiviral Vector Production and Purification for Enhanced Ex Vivo and In Vivo Genetic Engineering. Mol. Ther. - Methods Clin. Develop. 19, 411–425. doi:10.1016/j.omtm.2020.10.009
Stahlman, M. T., Gray, M. P., Falconieri, M. W., Whitsett, J. A., and Weaver, T. E. (2000). Lamellar Body Formation in Normal and Surfactant Protein B-Deficient Fetal Mice. Lab. Invest. 80, 395–403. doi:10.1038/labinvest.3780044
Stephens, C. J., Lauron, E. J., Kashentseva, E., Lu, Z. H., Yokoyama, W. M., and Curiel, D. T. (2019). Long-Term Correction of Hemophilia B Using Adenoviral Delivery of Crispr/cas9. J. Controlled Release 298, 128–141. doi:10.1016/j.jconrel.2019.02.009
Stribling, R., Brunette, E., Liggitt, D., Gaensler, K., and Debs, R. (1992). Aerosol Gene Delivery In Vivo. Proc. Natl. Acad. Sci. 89, 11277–11281. doi:10.1073/pnas.89.23.11277
Thomas, A. Q., Lane, K., Phillips, J., Prince, M., Markin, C., Speer, M., et al. (2002). Heterozygosity for a Surfactant Protein C Gene Mutation Associated with Usual Interstitial Pneumonitis and Cellular Nonspecific Interstitial Pneumonitis in One Kindred. Am. J. Respir. Crit. Care Med. 165, 1322–1328. doi:10.1164/rccm.200112-123OC
Thompson, A. A., Walters, M. C., Kwiatkowski, J., Rasko, J. E. J., Ribeil, J. A., Hongeng, S., et al. (2018). Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 378, 1479–1493. doi:10.1056/NEJMoa1705342
Thompson, M. W. (2001). Surfactant Protein B Deficiency: Insights into Surfactant Function through Clinical Surfactant Protein Deficiency. Am. J. Med. Sci. 321, 26–32. doi:10.1097/00000441-200101000-00005
Tomer, Y., Wambach, J., Knudsen, L., Zhao, M., Rodriguez, L. R., Murthy, A., et al. (2021). The Common ABCA3E292V Variant Disrupts AT2 Cell Quality Control and Increases Susceptibility to Lung Injury and Aberrant Remodeling. Am. J. Physiology-Lung Cell Mol. Physiol. 321, L291–L307. doi:10.1152/ajplung.00400.2020
Tornabene, P., Trapani, I., Minopoli, R., Centrulo, M., Lupo, M., de Simone, S., et al. (2019). Intein-Mediated Protein Trans-splicing Expands Adeno-Associated Virus Transfer Capacity in the Retina. Sci. Transl. Med. 11, eaav4523. doi:10.1126/scitranslmed.aav4523
Tu, G., Kirchmaier, A. L., Liggitt, D., Liu, Y., Liu, S., Yu, W. H., et al. (2000). Non-Replicating Epstein-Barr Virus-Based Plasmids Extend Gene Expression and Can Improve Gene Therapy In Vivo. J. Biol. Chem. 275, 30408–30416. doi:10.1074/jbc.M004782200
Valkama, A. J., Leinonen, H. M., Lipponen, E. M., Turkki, V., Malinen, J., Heikura, T., et al. (2018). Optimization of Lentiviral Vector Production for Scale-Up in Fixed-Bed Bioreactor. Gene Ther. 25, 39–46. doi:10.1038/gt.2017.91
Wambach, J. A., Casey, A. M., Fishman, M. P., Wegner, D. J., Wert, S. E., Cole, F. S., et al. (2014). Genotype-phenotype Correlations for Infants and Children with Abca3 Deficiency. Am. J. Respir. Crit. Care Med. 189, 1538–1543. doi:10.1164/rccm.201402-0342OC
Wambach, J. A., Yang, P., Wegner, D. J., Heins, H. B., Kaliberova, L. N., Kaliberov, S. A., et al. (2016). Functional Characterization of Atp-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome. Am. J. Respir. Cel Mol Biol 55, 716–721. doi:10.1165/rcmb.2016-0008OC
Wambach, J. A., Yang, P., Wegner, D. J., Heins, H. B., Luke, C., Li, F., et al. (2020). Functional Genomics of Abca3 Variants. Am. J. Respir. Cel Mol Biol 63, 436–443. doi:10.1165/rcmb.2020-0034MA
Wert, S. E., Whitsett, J. A., and Nogee, L. M. (2009). Genetic Disorders of Surfactant Dysfunction. Pediatr. Dev. Pathol. 12, 253–274. doi:10.2350/09-01-0586.1
Whitsett, J. A., and Weaver, T. E. (2002). Hydrophobic Surfactant Proteins in Lung Function and Disease. N. Engl. J. Med. 347, 2141–2148. doi:10.1056/NEJMra022387
Keywords: surfactant deficiency, viral vectors, alveoli, pulmonary disease, AEC2, ATII, AT2
Citation: Cooney AL, Wambach JA, Sinn PL and McCray PB (2022) Gene Therapy Potential for Genetic Disorders of Surfactant Dysfunction. Front. Genome Ed. 3:785829. doi: 10.3389/fgeed.2021.785829
Received: 29 September 2021; Accepted: 15 December 2021;
Published: 14 January 2022.
Edited by:
Sriram Vaidyanathan, Stanford University, United StatesReviewed by:
Nejat Duzgunes, University of the Pacific, United StatesCopyright © 2022 Cooney, Wambach, Sinn and McCray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ashley L. Cooney, YXNobGV5LXBldGVyc29uQHVpb3dhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.