 Stefanie Klaver-Flores1
Stefanie Klaver-Flores1 Hidde A. Zittersteijn2
Hidde A. Zittersteijn2 Kirsten Canté-Barrett1
Kirsten Canté-Barrett1 Arjan Lankester3
Arjan Lankester3 Rob C. Hoeben2
Rob C. Hoeben2 Manuel A. F. V. Gonçalves2
Manuel A. F. V. Gonçalves2 Karin Pike-Overzet1
Karin Pike-Overzet1 Frank J. T. Staal1*
Frank J. T. Staal1*- 1Department of Immunology, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Cell and Chemical Biology, Leiden University Medical Center, Leiden, Netherlands
- 3Department of Pediatrics, Willem-Alexander Children's Hospital, Leiden University Medical Center, Leiden, Netherlands
Many gene editing techniques are developed and tested, yet, most of these are optimized for transformed cell lines, which differ from their primary cell counterparts in terms of transfectability, cell death propensity, differentiation capability, and chromatin accessibility to gene editing tools. Researchers are working to overcome the challenges associated with gene editing of primary cells, namely, at the level of improving the gene editing tool components, e.g., the use of modified single guide RNAs, more efficient delivery of Cas9 and RNA in the ribonucleoprotein of these cells. Despite these efforts, the low efficiency of proper gene editing in true primary cells is an obstacle that needs to be overcome in order to generate sufficiently high numbers of corrected cells for therapeutic use. In addition, many of the therapeutic candidate genes for gene editing are expressed in more mature blood cell lineages but not in the hematopoietic stem cells (HSCs), where they are tightly packed in heterochromatin, making them less accessible to gene editing enzymes. Bringing HSCs in proliferation is sometimes seen as a solution to overcome lack of chromatin access, but the induction of proliferation in HSCs often is associated with loss of stemness. The documented occurrences of off-target effects and, importantly, on-target side effects also raise important safety issues. In conclusion, many obstacles still remain to be overcome before gene editing in HSCs for gene correction purposes can be applied clinically. In this review, in a perspective way, we will discuss the challenges of researching and developing a novel genetic engineering therapy for monogenic blood and immune system disorders.
Introduction
During the last decade, a wide range of scientific advances have emerged in the field of genomic engineering. Those advances vary from γ-retroviruses to self-inactivating lentiviruses, and from designed meganucleases to the more versatile, hence more powerful, CRISPR/Cas-based systems. What makes gene editing technologies interesting for researchers and clinicians, but also for the general public is their potential for therapeutic application in a range of genetic and acquired diseases, such as inborn errors of immunity (IEI) (Gatti et al., 1968), hemoglobinopathies including sickle cell disease (SCD) (Johnson et al., 1984; Lucarelli et al., 1984), cystic fibrosis, certain types of cancers, and viral diseases such as AIDS (White and Khalili, 2016; Shim et al., 2017; Porteus, 2019; Shahryari et al., 2019). However, these promising state-of-the-art technologies face a number of obstacles that prompt questions regarding their safety and efficiency especially when considering clinical applications. Preeminent amongst these obstacles are the generation of off-target effects with associated potential tumorigenicity, and immune responses triggered by the delivery vehicles and/or the gene editing reagents themselves (Doudna and Charpentier, 2014; Shim et al., 2017). In this perspective, we provide a brief overview of hematopoietic stem cell (HSC) biology and ex vivo expansion protocols, followed by a critical discussion about the scientific basis for the development of novel HSC gene editing therapies for blood and immune disorders.
Hematopoietic Stem Cells
Stem cells are cells of embryonic, fetal or adult origin, capable of dividing indefinitely (Staal et al., 2011). All stem cells, regardless of their origin, have three characteristics that distinguish them from other cell types: (i) they are undifferentiated and non-specialized cells; (ii) are able to divide and renew themselves indefinitely; and (iii) are able to differentiate into specialized cells when subjected to certain physiological or experimental conditions. Those cells can be classified, according to their origin or their differentiation capacity, into embryonic and non-embryonic stem cells that can be pluripotent or multipotent, respectively.
Hematopoietic stem cells (HSC) comprise a heterogeneous and relatively small group of cells that have the ability to self-renew and differentiate into specialized cells of the blood tissue and the immune system. Those cells are characterized by being the most immature in the differentiation hierarchy for blood cells.
In the classic model of hematopoiesis, the most primitive HSC progenitor cells (phenotypically defined as CD34+ CD38− CD90+ CD45RA− and CD49f−), differentiate into progenitors that further give rise to other blood cells (Notta et al., 2011). The recently identified Junction adhesion molecule-2 (Jam2) is highly expressed in HSCs and can generate T cells, have been suggested as novel surface markers in HSCs (Radulovic et al., 2019). Also, recently other two molecules have been identified as a relatively robust surface marker in human HSCs. The Endothelial protein C receptor (EPCR) is highly conserved in LT-HSCs (Fares et al., 2017), and the Endothelial cell-selective adhesion molecule (ESAM) is highly expressed in HSCs and MPPs, in a long-term lifetime. Thus, the ESAM seems to have a big influence in HSC differentiation path in different studies (Ooi et al., 2009; Yokota et al., 2009; Ishibashi et al., 2016; Roch et al., 2017).
For clinical applications, the interest of using HSCs has been increasing over the years. Among the difficulties faced by the researchers are the number of cells extracted from the patient, and also the fact that those cells undergo symmetrical and asymmetrical cell divisions when cultured. In an ex vivo expansion approach, the symmetrical cell division leads to an increase in the number of cells (Morrison and Kimble, 2006), achieved by the use of different combinations of growth factors and cytokines, such as SCF, TPO, Flt3-L, IL-3, and IL-6 (Sauvageau et al., 2004; Buza-Vidas et al., 2006; Hofmeister et al., 2007; Metcalf, 2008). Aside from that, other compounds are screened and tested for their potential for in vitro HSC expansion, including Stemregenin1 (SR1) and UM171 molecules (Boitano et al., 2010; Fares et al., 2014). The SR1 molecule was the first identified with the property of supporting the expansion of human and murine HSCs in vitro (Boitano et al., 2010), and has clinical benefit when cultured with the aforementioned cytokines cocktail (Wagner et al., 2016). The UM171 has been shown to be a good and promising candidate for ex vivo expansion of human cord blood HSCs (Fares et al., 2014). A recent clinical trial is using the UM171 with the purpose of ex vivo expansion of HSCs for allogeneic transplantation and gene therapy (NCT02668315), which suggests the potential use in ex vivo gene therapy. An interesting recently identified compound is CPI203, which acts at the epigenetic level to expand human CD34+ cells in NSG mouse models and may support ex vivo expansion of human HSCs (Hua et al., 2020; Staal and Fibbe, 2020).
State-of-the-Art Genome Engineering of HSCs
Allogeneic-hematopoietic stem cell transplants (allo-HSCT) have been used since the late 1960's to offer a potential lifetime cure for a variety of monogenic hematological diseases (Thomas et al., 1975). The main benefit of successful allo-HSCT is that the patient is cured for life, highlighting the concept that transplantation of healthy donor-derived HSCs containing the correct gene variant can reconstitute a functional hematopoietic system. While allo-HSCT can cure multiple blood and immune system disorders, clinical problems remain due to the challenge of finding a suitable HLA-matched bone marrow donor together with need for strong conditioning regimens for HSC engraftment, potentially resulting in subsequent complications such as graft-vs.-host disease (GvHD) or incomplete reconstitution of blood cell lineages. Moreover, chemotherapeutic conditioning regimens may result in infertility or development of lymphomas later in life. In patient genotype-specific cases when a suitable HLA-matched donor is not available, mismatched related donors are often used, however at the cost of increased morbidity and incomplete immune recovery leading to lower quality of life. To overcome these limitations of allo-HSCT, researchers initially have developed retroviral vectors that carry a recombinant version of the correct gene for permanent transfer into autologous CD34+ cell-enriched HSCs that. The ex vivo, genetically modified CD34+ cells that include HSCs are infused back into the patient and the genetically modified cells engraft and subsequently produce hematopoietic cells expressing the therapeutic gene (Figure 1). This ex vivo gene therapy principle has been shown to be efficacious in diseases such as severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID) (Aiuti et al., 2009), X-linked severe combined immunodeficiency (Hacein-Bey-Abina et al., 2002, 2010; Pavel-Dinu et al., 2019) and more recently for hemoglobinopathies including SCD, conditions that require high levels of therapeutic gene expression to attain phenotypic rescue (Woods et al., 2006; Badat and Davies, 2017). Currently, departing from “classic” gene therapy, gene editing technology based on programmable nucleases is offering the perspective for changing the genome of HSCs with unprecedented specificity and accuracy. Together with increased knowledge of the mechanisms that regulate human hematopoiesis, this has created the possibility to further developing cell and gene therapies for inherited diseases of the blood cell compartment. Backed by many years of fundamental research and, at times serendipity, the discovery of restriction enzymes was followed by that of other classes of DNA-modifying tools, including site-specific recombinases and programmable nucleases, such as meganucleases (MGN), zinc-finger nucleases (ZNFs), transcription activator-like effector (TALE) nucleases (TALENs), and more recently, powerful RNA-guided nucleases based on clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated endonuclease (Cas) systems (Chandrasegaran and Carroll, 2016; Chen and Goncalves, 2018). In this context, the non-integrating adeno-associated vector (AVV) has become a widely exploited vehicle of donor DNA template that is required for homology directed repair (HDR) in HSCs (Bak et al., 2018). Single-strand and double-strand oligodeoxynucleotides (ODN) are also emerging as effective means to deliver donor template for HDR in many clinical relevant settings (Chen et al., 2015). The engineering of meganucleases with new DNA-binding specificities has been challenging in large part due to the fact that the DNA recognition and cleavage sites are located in the same domain. In contrast to the meganucleases, the DNA binding domains of ZFNs and TALENs are distinct from that of their FokI cleavage domains whose (catalytic) activation depends on target DNA binding of a working ZFN or TALEN pair resulting in local dimerization (Urnov et al., 2010). The ZFNs and TALENs DNA-binding domain consist of zing-finger motif and TALE repeat arrays, respectively, with each zinc-finger motif binding to specific nucleotide triplets and each TALE repeat recognizing individual single nucleotides. The changes of the zinc-finger motifs can be done by the nucleotides that are surrounding its triple target. As a consequence of this sequence context dependency, generating robust and highly specific ZFNs often requires complex protein engineering methods involving reiterative optimization cycles and/or screening of large zinc-finger libraries (Cathomen and Keith Joung, 2008). The straightforward TALE repeat-to-nucleotide one-to-one recognition code together with the fact that binding of a TALE repeat to its target nucleotide is not substantially altered by neighboring nucleotides (Mussolino and Cathomen, 2012), makes the assembly of functional and highly specific TALENs easier and more flexible than that of ZFNs (Jinek et al., 2013). While each programmable nuclease platform is at different stages of clinical development, RNA-guided CRISPR/Cas-based systems are becoming the tools of choice for pursuing genetic therapies based on genome editing principles and technologies. This principally stems from their high efficiency and increasingly improving specificity, as well as from their versatile RNA-dependent programmability and easy-to-use versatile design.
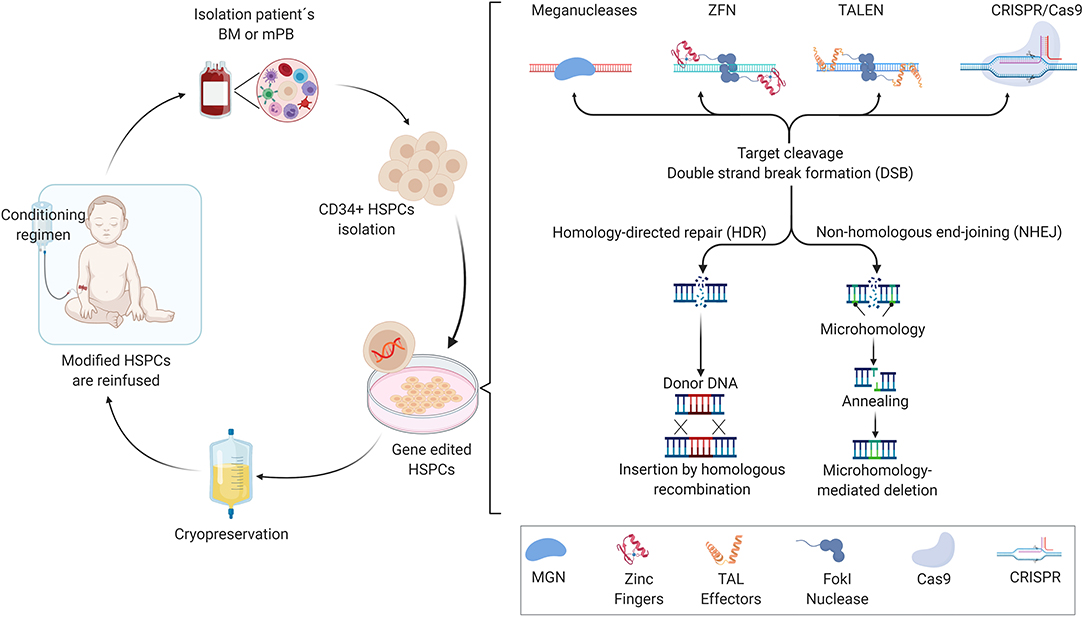
Figure 1. Representation of ex vivo HSCs gene editing, showing the crucial steps of the process. After harvesting the hematopoietic stem and progenitor cells (HSPCs) from mobilized peripheral blood or bone marrow, the CD34+ cells are enriched and cultured ex vivo in the presence of growth factors, which allows the maintenance and expansion of self-renewing stem cells, and are then subjected to gene editing tool transfer (e.g., meganucleases, ZFNs, TALENs, or CRISPR/Cas-derived nucleases). When the nuclease induces a standard DNA double-strand break (DSB) at the desired genomic loci, the homology-directed repair (HDR) machinery are recruited in order to repair the DNA, where a template donor DNA is supplied for the homologous recombination between the template and chromosomal DNA, or by non-homologous end-joining (NHEJ) without a homologous template DNA, resulting in small indels generation (insertions and deletions) if there is only one cut, or triggers large DNA deletions if two cuts. After the treatment, the patient receives a specific conditioning regimen that depletes endogenous HSPCs from the bone marrow and makes space for the ex vivo engineered cells to engraft. The gene-corrected cells are then reinfused intravenously and engraft in the bone marrow.
CRISPR sequences together with CRISPR-associated (Cas) protein genes form CRISPR/Cas loci as part of the adaptive immune systems in prokaryotes organisms, evolved as a strategy to fend off infectious agents, e.g., bacteriophages and foreign plasmids (Horvath and Barrangou, 2010; Wiedenheft et al., 2012; Rath et al., 2015). Scientists have been investigating the properties of these exquisite defense mechanisms encoded in various CRISPR loci for over 20 years. Crucially, in 2012, the real potential of CRISPR/Cas systems for genomic engineering purposes was uncovered in seminal studies by Gasiunas et al. (2012) and Jinek et al. (2012). In particular, through these eminent in vitro biochemical studies, these teams found that Cas9 proteins from Streptococcus thermophilus and Streptococcus pyogenes, respectively, are RNA-programmable site-specific endonucleases. Later, the CRISPR system was readily adapted by independent research groups that had the aim of turning the technique into a powerful genome editing platform for genome editing purposes in mammalian cells (Cho et al., 2013; Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013).
Key adaptations involved codon-optimization of Cas9 reading frames encoding nuclease localization motifs and fusion of native trans-activating CRISPR RNA and CRISPR RNA moieties to form a so-called single-guide gRNA (sgRNA). The latter component binds to the Cas9 protein and address it to a target sequence consisting of a protospacer adjacent motif (NGG; in the case of S. pyogenes Cas9) and a typically 20 nucleotide-long sequence complementary to the 5′ end of the sgRNA (spacer). Upon target site binding and sgRNA-DNA hybridization, the HNH and RuvC-like nuclease domains of Cas9 become active resulting in site-specific DNA cleavage, of inducing double-stranded DNA (dsDNA) breaks at a specific genomic target region, homologous to the crRNA spacer sequence.
Two major DNA repair pathways exist in humans. The endogenous non-homologous end-joining (NHEJ) and homology-directed repair (HDR) pathways are responsible for repair of the double-stranded chromosomal breaks made by programmable nucleases allowing for the removal or insertion of new genetic information at specific genomic loci (Jinek et al., 2012). Typically, NHEJ processes are exploited for knocking-out preexisting genetic information after the exclusive transfer of programmable nucleases, whilst the HDR mechanism is mostly used for knocking-in new genetic information after the delivery of programmable nucleases together with exogenous (donor) DNA templates.
The prokaryotic-CRISPR/Cas9 genome editing tool has changed our ability to change and manipulate specific sequences of DNA and RNA in living cells from diverse species, including mammalian cells. The CRISPR/Cas9 system for genetic engineering is an exciting advancement for HSC gene therapy, although it potentially comes with safety risks, such as suboptimal specificity correlated with off-target effects and on-target but unwanted mutations, immunogenicity, and unfavorable bio-distribution.
The Challenges of Genome Engineering in HSCs
Genome-editing tools in the form of the aforementioned programmable nucleases and their derivatives can, in principle, be projected for correcting or disrupting any disease-causing gene typically via knocking-in and knocking-out exogenous and endogenous DNA sequences, respectively, or via the introduction of specific point mutations (Byrne et al., 2014). HSCs are optimal target cells for therapeutic genome editing technologies owing to their self-renewal and differentiation capabilities (Hoke et al., 2012; Liu et al., 2012; Lee et al., 2019). However, these genome editing tools and strategies are initially mostly developed and tested in transformed cell lines that differ from their primary cell counterparts in key aspects, such as transfectability, cell death propensity, loss of differentiation capabilities, ploidy, and chromatin accessibility (Figure 2). Primary cells, unlike immortalized or full-fledged transformed cells, for the most part maintain their biological identity in proper culture systems, yet, they can only be propagated for a few generations in vitro before reaching senescence and, in the case of true HSCs, they are difficult to expand in vitro.
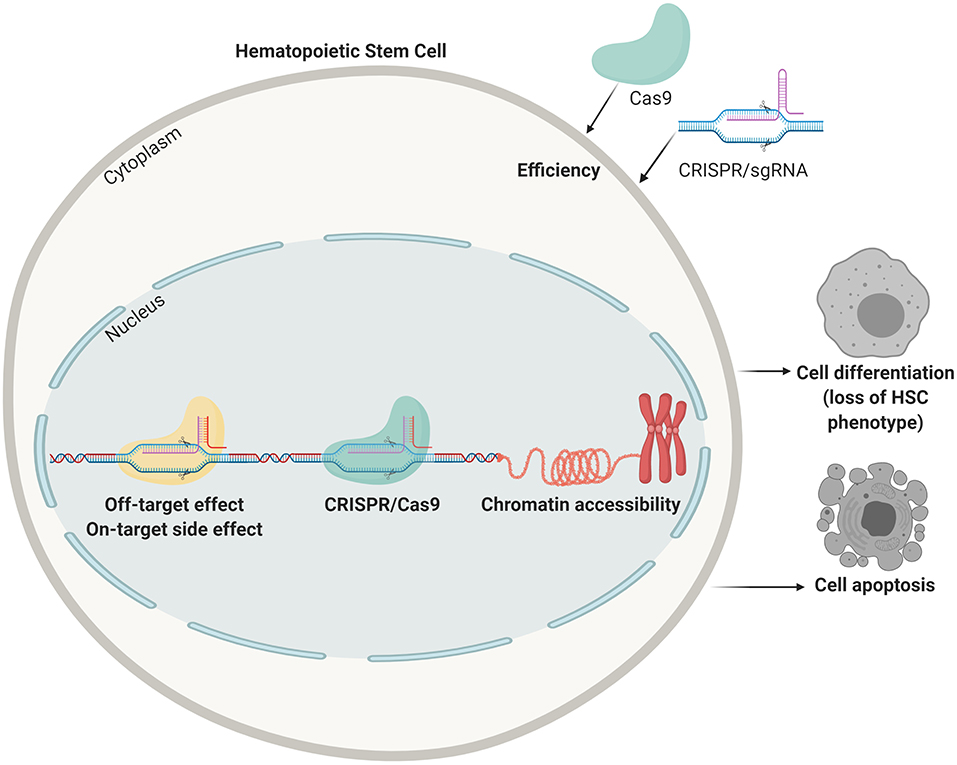
Figure 2. Points of improvement in CRISPR mediated gene correction in HSCs. Efficiency of delivery, off-target effects (targeting the wrong locus), side effects on the target sites (unwanted indels, translocations, and mutations), lack of accessible chromatin, apoptosis due to the harsh procedures, and loss of stemness are all problems that need to be tackled before obtaining clinically relevant HSC numbers that can be transplanted in patients.
Hence, when thinking about applying these genome editing tools and strategies to primary cells, and in particular HSCs, one faces numerous challenges associated with the aforementioned intrinsic characteristics of these target cells and the sub-optimal performance of gene editing procedures, such as on-target and off-target side effects, as well as insertional mutagenesis risks and unregulated transgene expression resulting from random chromosomal integration of exogenous (donor) DNA templates (Crisostomo et al., 2006). In order to meet the safety requirements and other important criteria such as a high efficacy, high quality and good reproducibility, it is crucial that the genome-editing tool is proper developed and tested in appropriate cell types.
One of the most challenging issues of ex vivo genome-editing of HSCs, besides the low viability and the decreased differentiation potential of these cells upon prolonged culture, is the difficulty in achieving high gene delivery efficiencies. Part of the difficulty is the absence of methods that permit the in vitro identification and, thus, selection of bona fide HSCs (gene-modified or otherwise) from cultured hematopoietic cells. Another component of the difficulty concerns the gene transfer into HSCs. Because the existing protocols do not employ drug selection, the gene transduction methods need to yield enough functionally reconstituted cells for a good therapeutic response. Another limitation when applying genome-editing in HSCs is the low transplanted cell engraftment capacity due to their poor viability after gene-editing, especially when high percentages of non-edited cells are present after the ex vivo modification (Naldini, 2019). For those cases, the enrichment of the CD34+ fraction using a combination of additional hematopoietic surface markers may be important for the improvement of cell engraftment and repopulation, although these additional cell manipulations might lead to loss of stemness and cell death. Along these lines, it is necessary to identify which specific gene-editing tools and strategies are the most appropriate for each disease, and consider whether, on the basis of the disease phenotype, the modified cells present a selective advantage that might reduce conditioning regimens and increase the cell engraftment capacity.
Genome Editing Techniques: Transition to the Clinic
The introduction of gene-editing tools in the form of engineered nucleases has provided strong support to the idea that targeted genetic therapies for treating monogenetic diseases of the hematopoietic system is achievable. Yet, there are multi-tier bottlenecks on the path to transitioning from applying HSC-directed gene-editing laboratory technologies to the clinic. To overcome these bottlenecks it will be crucial to develop and combine delivery agents and gene-editing reagents that allow for efficient and precise gene-editing at the HSCs level. Further, these integrated gene editing procedures need to be scalable under good manufacturing practice conditions, and, clearly, neither cytotoxic, or genotoxic. Moreover, there are other points that should have important improvements, such as the delivery of the homologous donor templates and the nuclease of choice.
Regardless of their class, programmable nucleases are capable of achieving high specificity, especially once individual reagents are identified and optimized for cleaving target sequences and not off-target sites (Akcakaya et al., 2018), but it is important to know that none of them are perfect. However, even when using the highly specific nucleases, when making changes at the desired target site, unintentional changes can be induced elsewhere in the genome due to, for instance, differences in nuclease amounts and chromosomal accessibility in different cell types. Indeed, these parameters might influence DNA cleavage and NHEJ-mediated repair (profiles) at secondary sites (White et al., 2017). These unwanted genome-modifying events present a modest hazard in experimental systems, where conclusions can be validated by (i) comparing independent gene-edited cells and organisms, (ii) “cleaning-up” the genetic background by out-breeding/cross-breeding and (ii) complementing gene knockouts via introducing wild-type gene sequences. However, for therapeutic applications off-target effects are more problematic. Methods have been developed for detecting, locating and quantifying those off-target effects (Koo et al., 2015). When applied in human therapy, we need to be assured that the adverse effects of the treatment are as minimal as possible while the one originally addressed gene is repaired.
Besides off-target effects, adverse effects caused by cleavage at the desired side of modification have also been reported (Kosicki et al., 2018; Chen et al., 2020). These unwarranted on-target effects can affect not only the genotype but also the phenotype of gene-edited cells (Chen et al., 2020) and are more difficult to assess, but clearly are undesired. In addition, as aforementioned, the efficiency of gene modification can be reduced due to the limited accessibly of target sequences tightly packed in heterochromatic regions (Chen et al., 2016; Daer et al., 2017), resulting in a lack of efficient delivery of the Cas enzyme or the DNA template needed for repair via homologous recombination. The limited access of gene editing reagents to the DNA can perhaps be overcome at some target loci by forcing the HSCs to enter into the S and M phases. However, ex vivo proliferation of HSCs without losing their stemness properties is still a daunting task (Tajer et al., 2019).
Despite these problems, researchers have reported significant advances in gene editing of HSCs for SCID. For instance, Genovese et al. have shown that gene editing for X-SCID is in principle possible (Genovese et al., 2014). In this report, ZFNs were used and the efficacy was relatively low, but some correction was obtained in human long-term repopulating HSCs transplanted in immune-deficient mice. The next improvement consisted of using RGN nucleofection for introducing an IL2RG transgene delivered via an adeno-associated viral vector pseudo-type (i.e., AAV6) into the first exon of the IL2RG gene that is deficient in X-SCID (Pavel-Dinu et al., 2019). The reported gene correction efficiencies were much higher but the phenotypic differences between corrected and uncorrected HSCs transplanted were only minor, with modest increases in T and NK cells, the two lineages affected in this type of SCID (Pavel-Dinu et al., 2019). This indicated that even for a relatively easy target such as an X-linked gene which only requires correction in one allele, efficacies need to be significantly improved for clinical application. Gene editing is particularly attractive for diseases where the expression of affected gene normally is strictly regulated. While gene addition approaches work well for X-linked SCID (Hacein-Bey-Abina et al., 2002; Pavel-Dinu et al., 2019), ADA-SCID (Aiuti et al., 2009), Wiskott–Aldrich syndrome (WAS) (Braun et al., 2014), RAG1-SCID (Garcia-Perez et al., 2020), and b-globin disease (Dong and Rivella, 2017), for IL7Rα-SCID and for the Hyper IgM syndrome (caused by mutations in the CD40L gene), gene addition with constitutively expressing vectors will cause severe side effects (Kuo et al., 2018). However, also for diseases caused by defects in such genes significant progress is being made. Indeed, Kohn et al. reported specific insertion of a recombinant CD40L sequence downstream of the endogenous CD40L promoter using RGNs and an AAV-delivered donor template (Kuo et al., 2018). Relevant levels of gene modification were achieved in primary HSCs and in patient-derived T cells. Therefore, significant progress is made to clinical implementation of these techniques. Nevertheless, clinical trials using CRISPR and HSCs have been confined to gene deletion strategies rather than editing of mutant genes. Examples include a gene disruption approach to delete the CCR5 HIV coreceptor and the disruption of erythroid lineage-specific enhancer of the BCL11A suppressor protein in the g-globin gene to induce re-expression of fetal g-globin in thalassemia patients (NCT03745287) (Psatha et al., 2018). Indeed, for bona fide gene editing in Hyper IgM syndrome due to CD40L mutations, T cells rather than HSCs are being proposed as target cells in clinical trials.
Regulatory Authorities
In the last three decades, the ex vivo gene therapy in HSCs has been progressing substantially from the pre-clinical stage to clinical trials (Thrasher and Williams, 2017; Staal et al., 2019). With the FDA-approved first clinical trial gene-editing of HSCs for the treatment of HIV using the ZFNs CCR5 (Tebas et al., 2014), a new paradigm treatment in cell and gene therapy had been started. Before wide-spread clinical approval, however, there are several regulatory hurdles. Regulation may be complex and vary across countries and continents because gene-editing medicine entails the unprecedented introduction of designed alterations in the genetic make-up of some of the patient's cells. As a minimum, regulators will focus on whether the gene disruption/restoration is based on robust preclinical evidence, as illustrated by the US FDA approval of multiple clinical trials.
Although there is a great promise for gene-editing in the future of medicine, the regulatory approval by the competent authorities will not be granted in the short term. One of the reasons is because the authorities strictly guard safety and well-being of patients (White et al., 2017). One of the major obstacles is that there is no clear consensus regarding the occurrence of on-target and off-target alterations by the gene-editing tools, and also it is not clear when and how these effects should be monitored in the clinical applications (Joung, 2015). Regulatory authorities and the pharmaceutical industry of Europe, Japan, and the USA have developed some consideration documents regarding gene therapy (Coppens et al., 2018; de Wilde et al., 2018), indicating that more regulatory harmonization is indispensable in order to realize the therapeutic benefits of genome editing worldwide.
The versatility and robustness of gene-editing approaches are expected to positively contribute to the development of novel somatic disease treatments. However, the technology could also lead to some unfavorable social phenomena due to high prices and public misconceptions. The general public should understand that so far only a few gene therapy products have been approved by health regulators worldwide. Moreover, scientists have the obligation to provide the public with accurate and realistic information regarding the prospects, as well as the problems associated with the use of somatic gene-editing therapy. In addition, good communication between researchers and the regulatory authorities are key to fulfill the promises and to achieve the medical benefits of genome editing. Communication and cooperation should foster an increase in worldwide regulatory harmonization. This should eventually lead to clinical benefit for those affected with inborn diseases.
Final Remarks
Tremendous progress has been made in the field of gene editing over the last few years. However, no clinical trials using this technology have been used so far to treat immune deficiencies via gene editing for reasons of efficiency and safety. To have HSC gene editing working safely at the scale needed for clinical application remains challenging and will require carefully designed protocols using the correct target cells, assays to detect potential side effects, and comparisons with more conventional allo-HSCT and gene addition therapy methods. Such efforts will hopefully lead to the clinical application of gene editing techniques to cure monogenetic diseases of the hematopoietic system.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Work in our laboratories was supported in part by a ZonMW E-RARE grant (40-419000-98-020) and EU H2020 grant RECOMB (755170-2) and has received funding from the European Union Horizon 2020 research and innovation program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aiuti, A., Cattaneo, F., Galimberti, S., Benninghoff, U., Cassani, B., Callegaro, L., et al. (2009). Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 360, 447–458. doi: 10.1056/NEJMoa0805817
Akcakaya, P., Bobbin, M. L., Guo, J. A., Malagon-Lopez, J., Clement, K., Garcia, S. P., et al. (2018). In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561, 416–419. doi: 10.1038/s41586-018-0500-9
Badat, M., and Davies, J. (2017). Gene therapy in a patient with sickle cell disease. N. Engl. J. Med. 376, 2093–2094. doi: 10.1056/NEJMc1704009
Bak, R. O., Dever, D. P., and Porteus, M. H. (2018). CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 13, 358–376. doi: 10.1038/nprot.2017.143
Boitano, A. E., Wang, J., Romeo, R., Bouchez, L. C., Parker, A. E., Sutton, S. E., et al. (2010). Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329, 1345–1348. doi: 10.1126/science.1191536
Braun, C. J., Boztug, K., Paruzynski, A., Witzel, M., Schwarzer, A., Rothe, M., et al. (2014). Gene therapy for wiskott-aldrich syndrome–long-term efficacy and genotoxicity. Sci. Transl. Med. 6:227ra33. doi: 10.1126/scitranslmed.3007280
Buza-Vidas, N., Antonchuk, J., Qian, H., Mansson, R., Luc, S., Zandi, S., et al. (2006). Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev. 20, 2018–2023. doi: 10.1101/gad.385606
Byrne, S. M., Mali, P., and Church, G. M. (2014). Genome editing in human stem cells. Meth. Enzymol. 546, 119–138. doi: 10.1016/B978-0-12-801185-0.00006-4
Cathomen, T., and Keith Joung, J. (2008). Zinc-finger nucleases: the next generation emerges. Mol. Ther. 16, 1200–1207. doi: 10.1038/mt.2008.114
Chandrasegaran, S., and Carroll, D. (2016). Origins of programmable nucleases for genome engineering. J. Mol. Biol. 428(5 Pt B), 963–89. doi: 10.1016/j.jmb.2015.10.014
Chen, F., Pruett-Miller, S. M., and Davis, G. D. (2015). Gene editing using ssODNs with engineered endonucleases. Methods Mol. Biol. 1239, 251–265. doi: 10.1007/978-1-4939-1862-1_14
Chen, X., and Goncalves, M. (2018). DNA, RNA, and protein tools for editing the genetic information in human cells. iScience 6, 247–263. doi: 10.1016/j.isci.2018.08.001
Chen, X., Rinsma, M., Janssen, J. M., Liu, J., Maggio, I., and Goncalves, M. A. (2016). Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 44, 6482–6492. doi: 10.1093/nar/gkw524
Chen, X., Tasca, F., Wang, Q., Liu, J., Janssen, J. M., Brescia, M. D., et al. (2020). Expanding the editable genome and CRISPR-Cas9 versatility using DNA cutting-free gene targeting based on in trans paired nicking. Nucleic Acids Res. 48, 974–995. doi: 10.1093/nar/gkz1121
Cho, S. W., Kim, S., Kim, J. M., and Kim, J. S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Coppens, D. G. M., de Wilde, S., Guchelaar, H. J., De Bruin, M. L., Leufkens, H. G. M., Meij, P., et al. (2018). A decade of marketing approval of gene and cell-based therapies in the united states, European union and Japan: an evaluation of regulatory decision-making. Cytotherapy 20, 769–778. doi: 10.1016/j.jcyt.2018.03.038
Crisostomo, P. R., Wang, M., Wairiuko, G. M., Morrell, E. D., Terrell, A. M., Seshadri, P., et al. (2006). High passage number of stem cells adversely affects stem cell activation and myocardial protection. Shock 26, 575–580. doi: 10.1097/01.shk.0000235087.45798.93
Daer, R. M., Cutts, J. P., Brafman, D. A., and Haynes, K. A. (2017). The impact of chromatin dynamics on cas9-mediated genome editing in human cells. ACS Synth. Biol. 6, 428–438. doi: 10.1021/acssynbio.5b00299
de Wilde, S., Coppens, D. G. M., Hoekman, J., de Bruin, M. L., Leufkens, H. G. M., Guchelaar, H. J., et al. (2018). EU decision-making for marketing authorization of advanced therapy medicinal products: a case study. Drug Discov. Today 23, 1328–1333. doi: 10.1016/j.drudis.2018.03.008
Dong, A. C., and Rivella, S. (2017). Gene addition strategies for beta-thalassemia and sickle cell anemia. Adv. Exp. Med. Biol. 1013, 155–176. doi: 10.1007/978-1-4939-7299-9_6
Doudna, J. A., and Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. doi: 10.1126/science.1258096
Fares, I., Chagraoui, J., Gareau, Y., Gingras, S., Ruel, R., Mayotte, N., et al. (2014). Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science 345, 1509–1512. doi: 10.1126/science.1256337
Fares, I., Chagraoui, J., Lehnertz, B., MacRae, T., Mayotte, N., Tomellini, E., et al. (2017). EPCR expression marks UM171-expanded CD34(+) cord blood stem cells. Blood 129, 3344–3351. doi: 10.1182/blood-2016-11-750729
Garcia-Perez, L., van Eggermond, M., van Roon, L., Vloemans, S. A., Cordes, M., Schambach, A., et al. (2020). Successful preclinical development of gene therapy for recombinase-activating gene-1-Deficient SCID. Mol. Ther. Methods Clin. Dev. 17, 666–682. doi: 10.1016/j.omtm.2020.03.016
Gasiunas, G., Barrangou, R., Horvath, P., and Siksnys, V. (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 109, E2579–E2586. doi: 10.1073/pnas.1208507109
Gatti, R. A., Meuwissen, H. J., Allen, H. D., Hong, R., and Good, R. A. (1968). Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet 2, 1366–1369. doi: 10.1016/S0140-6736(68)92673-1
Genovese, P., Schiroli, G., Escobar, G., Tomaso, T. D., Firrito, C., Calabria, A., et al. (2014). Targeted genome editing in human repopulating haematopoietic stem cells. Nature 510, 235–240. doi: 10.1038/nature13420
Hacein-Bey-Abina, S., Hauer, J., Lim, A., Picard, C., Wang, G. P., Berry, C. C., et al. (2010). Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 363, 355–364. doi: 10.1056/NEJMoa1000164
Hacein-Bey-Abina, S., Le Deist, F., Carlier, F., Bouneaud, C., Hue, C., De Villartay, J. P., et al. (2002). Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N. Engl. J. Med. 346, 1185–1193. doi: 10.1056/NEJMoa012616
Hofmeister, C. C., Zhang, J., Knight, K. L., Le P, and Stiff, P. J. (2007). Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone Marrow Transplant. 39, 11–23. doi: 10.1038/sj.bmt.1705538
Hoke, N. N., Salloum, F. N., Kass, D. A., Das, A., and Kukreja, R. C. (2012). Preconditioning by phosphodiesterase-5 inhibition improves therapeutic efficacy of adipose-derived stem cells following myocardial infarction in mice. Stem Cells 30, 326–335. doi: 10.1002/stem.789
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Hua, P., Hester, J., Adigbli, G., Li, R., Psaila, B., Roy, A., et al. (2020). The BET inhibitor CPI203 promotes ex vivo expansion of cord blood long-term repopulating HSCs and megakaryocytes. Blood 136, 2410–2415. doi: 10.1182/blood.2020005357
Ishibashi, T., Yokota, T., Tanaka, H., Ichii, M., Sudo, T., Satoh, Y., et al. (2016). ESAM is a novel human hematopoietic stem cell marker associated with a subset of human leukemias. Exp. Hematol. 44, 269.e1–81.e1. doi: 10.1016/j.exphem.2015.12.010
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jinek, M., East, A., Cheng, A., Lin, S., Ma, E., and Doudna, J. (2013). RNA-programmed genome editing in human cells. Elife 2:e00471. doi: 10.7554/eLife.00471
Johnson, F. L., Look, A. T., Gockerman, J., Ruggiero, M. R., Dalla-Pozza, L., and Billings, F. T. III. (1984). Bone-marrow transplantation in a patient with sickle-cell anemia. N. Engl. J. Med. 311, 780–783. doi: 10.1056/NEJM198409203111207
Joung, J. K. (2015). Unwanted mutations: standards needed for gene-editing errors. Nature 523:158. doi: 10.1038/523158a
Koo, T., Lee, J., and Kim, J. S. (2015). Measuring and reducing off-target activities of programmable nucleases including CRISPR-Cas9. Mol. Cells 38, 475–481. doi: 10.14348/molcells.2015.0103
Kosicki, M., Tomberg, K., and Bradley, A. (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771. doi: 10.1038/nbt.4192
Kuo, C. Y., Long, J. D., Campo-Fernandez, B., de Oliveira, S., Cooper, A. R., Romero, Z., et al. (2018). Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 23, 2606–2616. doi: 10.1016/j.celrep.2018.04.103
Lee, J., Bayarsaikhan, D., Arivazhagan, R., Park, H., Lim, B., Gwak, P., et al. (2019). CRISPR/Cas9 edited sRAGE-MSCs protect neuronal death in parkinsons disease model. Int. J. Stem Cells 12, 114–124. doi: 10.15283/ijsc18110
Liu, X., Wu, Y., Li, Z., Yang, J., Xue, J., Hu, Y., et al. (2012). Targeting of the human coagulation factor IX gene at rDNA locus of human embryonic stem cells. PLoS ONE 7:e37071. doi: 10.1371/journal.pone.0037071
Lucarelli, G., Polchi, P., Izzi, T., Manna, M., Agostinelli, F., Delfini, C., et al. (1984). Allogeneic marrow transplantation for thalassemia. Exp. Hematol. 12, 676–681.
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Morrison, S. J., and Kimble, J. (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068–1074. doi: 10.1038/nature04956
Mussolino, C., and Cathomen, T. (2012). TALE nucleases: tailored genome engineering made easy. Curr. Opin. Biotechnol. 23, 644–650. doi: 10.1016/j.copbio.2012.01.013
Naldini, L. (2019). Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 11:e9958. doi: 10.15252/emmm.201809958
Notta, F., Doulatov, S., Laurenti, E., Poeppl, A., Jurisica, I., and Dick, J. E. (2011). Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 333, 218–221. doi: 10.1126/science.1201219
Ooi, A. G., Karsunky, H., Majeti, R., Butz, S., Vestweber, D., Ishida, T., et al. (2009). The adhesion molecule esam1 is a novel hematopoietic stem cell marker. Stem Cells 27, 653–661. doi: 10.1634/stemcells.2008-0824
Pavel-Dinu, M., Wiebking, V., Dejene, B. T., Srifa, W., Mantri, S., Nicolas, C. E., et al. (2019). Author correction: gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 10:5624. doi: 10.1038/s41467-019-13620-5
Porteus, M. H. (2019). A new class of medicines through DNA editing. N. Engl. J. Med. 380, 947–959. doi: 10.1056/NEJMra1800729
Psatha, N., Reik, A., Phelps, S., Zhou, Y., Dalas, D., Yannaki, E., et al. (2018). Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with beta-thalassemia major. Mol. Ther. Methods Clin. Dev. 10, 313–326. doi: 10.1016/j.omtm.2018.08.003
Radulovic, V., van der Garde, M., Koide, S., Sigurdsson, V., Lang, S., Kaneko, S., et al. (2019). Junctional adhesion molecule 2 represents a subset of hematopoietic stem cells with enhanced potential for T lymphopoiesis. Cell Rep. 27, 2826–36.e5. doi: 10.1016/j.celrep.2019.05.028
Rath, D., Amlinger, L., Rath, A., and Lundgren, M. (2015). The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie 117, 119–128. doi: 10.1016/j.biochi.2015.03.025
Roch, A., Giger, S., Girotra, M., Campos, V., Vannini, N., Naveiras, O., et al. (2017). Single-cell analyses identify bioengineered niches for enhanced maintenance of hematopoietic stem cells. Nat. Commun. 8:221. doi: 10.1038/s41467-017-00291-3
Sauvageau, G., Iscove, N. N., and Humphries, R. K. (2004). In vitro and in vivo expansion of hematopoietic stem cells. Oncogene 23, 7223–7232. doi: 10.1038/sj.onc.1207942
Shahryari, A., Saghaeian Jazi, M., Mohammadi, S., Razavi Nikoo, H., Nazari, Z., Hosseini, E. S., et al. (2019). Development and clinical translation of approved gene therapy products for genetic disorders. Front. Genet. 10:868. doi: 10.3389/fgene.2019.00868
Shim, G., Kim, D., Park, G. T., Jin, H., Suh, S. K., and Oh, Y. K. (2017). Therapeutic gene editing: delivery and regulatory perspectives. Acta Pharmacol. Sin. 38, 738–753. doi: 10.1038/aps.2017.2
Staal, F. J., Baum, C., Cowan, C., Dzierzak, E., Hacein-Bey-Abina, S., Karlsson, S., et al. (2011). Stem cell self-renewal: lessons from bone marrow, gut and iPS toward clinical applications. Leukemia 25, 1095–1102. doi: 10.1038/leu.2011.52
Staal, F. J. T., Aiuti, A., and Cavazzana, M. (2019). Autologous stem-cell-based gene therapy for inherited disorders: state of the art and perspectives. Front. Pediatr. 7:443. doi: 10.3389/fped.2019.00443
Staal, F. J. T., and Fibbe, W. E. (2020). BETting on stem cell expansion. Blood 136, 2364–2365. doi: 10.1182/blood.2020007759
Tajer, P., Pike-Overzet, K., Arias, S., Havenga, M., and Staal, F. J. T. (2019). Ex vivo expansion of hematopoietic stem cells for therapeutic purposes: lessons from development and the niche. Cells 8:169. doi: 10.3390/cells8020169
Tebas, P., Stein, D., Tang, W. W., Frank, I., Wang, S. Q., Lee, G., et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910. doi: 10.1056/NEJMoa1300662
Thomas, E. D., Storb, R., Clift, R. A., Fefer, A., Johnson, L., Neiman, P. E., et al. (1975). Bone-marrow transplantation (second of two parts). N. Engl. J. Med. 292, 895–902. doi: 10.1056/NEJM197504242921706
Thrasher, A. J., and Williams, D. A. (2017). Evolving gene therapy in primary immunodeficiency. Mol. Ther. 25, 1132–1141. doi: 10.1016/j.ymthe.2017.03.018
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., and Gregory, P. D. (2010). Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646. doi: 10.1038/nrg2842
Wagner, J. E. Jr., Brunstein, C. G., Boitano, A. E., DeFor, T. E., McKenna, D., Sumstad, D., et al. (2016). Phase I/II trial of StemRegenin-1 expanded umbilical cord blood hematopoietic stem cells supports testing as a stand-alone graft. Cell Stem Cell 18, 144–155. doi: 10.1016/j.stem.2015.10.004
White, M., Whittaker, R., Gandara, C., and Stoll, E. A. (2017). A guide to approaching regulatory considerations for lentiviral-mediated gene therapies. Hum. Gene Ther. Methods 28, 163–176. doi: 10.1089/hgtb.2017.096
White, M. K., and Khalili, K. (2016). CRISPR/Cas9 and cancer targets: future possibilities and present challenges. Oncotarget 7, 12305–12317. doi: 10.18632/oncotarget.7104
Wiedenheft, B., Sternberg, S. H., and Doudna, J. A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338. doi: 10.1038/nature10886
Woods, N. B., Bottero, V., Schmidt, M., von Kalle, C., and Verma, I. M. (2006). Gene therapy: therapeutic gene causing lymphoma. Nature 440:1123. doi: 10.1038/4401123a
Keywords: CRISPR-Cas9, gene editing, hematopoietic stem cells, stem cell biology, genomic engineering, therapeutic, clinic
Citation: Klaver-Flores S, Zittersteijn HA, Canté-Barrett K, Lankester A, Hoeben RC, Gonçalves MAFV, Pike-Overzet K and Staal FJT (2021) Genomic Engineering in Human Hematopoietic Stem Cells: Hype or Hope? Front. Genome Ed. 2:615619. doi: 10.3389/fgeed.2020.615619
Received: 09 October 2020; Accepted: 22 December 2020;
Published: 22 January 2021.
Edited by:
Annarita Miccio, INSERM U1163 Institut Imagine, FranceReviewed by:
Daniel E. Bauer, Boston Children's Hospital and Harvard Medical School, United StatesAndreas Reinisch, Medizinische Universität Graz, Austria
Copyright © 2021 Klaver-Flores, Zittersteijn, Canté-Barrett, Lankester, Hoeben, Gonçalves, Pike-Overzet and Staal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank J. T. Staal, Zi5qLnQuc3RhYWxAbHVtYy5ubA==