 Mariana Handelman
Mariana Handelman Nir Osherov
Nir Osherov
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Fungal Biol. , 23 August 2022
Sec. Fungal Pathogenesis
Volume 3 - 2022 | https://doi.org/10.3389/ffunb.2022.957577
This article is part of the Research Topic Experimental evolution and anti-fungal resistance View all 4 articles
The leading fungal pathogens causing systemic infections in humans are Candida spp., Aspergillus fumigatus, and Cryptococcus neoformans. The major class of antifungals used to treat such infections are the triazoles, which target the cytochrome P450 lanosterol 14-α-demethylase, encoded by the ERG11 (yeasts)/cyp51A (molds) genes, catalyzing a key step in the ergosterol biosynthetic pathway. Triazole resistance in clinical fungi is a rising concern worldwide, causing increasing mortality in immunocompromised patients. This review describes the use of serial clinical isolates and in-vitro evolution toward understanding the mechanisms of triazole resistance. We outline, compare, and discuss how these approaches have helped identify the evolutionary pathways taken by pathogenic fungi to acquire triazole resistance. While they all share a core mechanism (mutation and overexpression of ERG11/cyp51A and efflux transporters), their timing and mechanism differs: Candida and Cryptococcus spp. exhibit resistance-conferring aneuploidies and copy number variants not seen in A. fumigatus. Candida spp. have a proclivity to develop resistance by undergoing mutations in transcription factors (TAC1, MRR1, PDR5) that increase the expression of efflux transporters. A. fumigatus is especially prone to accumulate resistance mutations in cyp51A early during the evolution of resistance. Recently, examination of serial clinical isolates and experimental lab-evolved triazole-resistant strains using modern omics and gene editing tools has begun to realize the full potential of these approaches. As a result, triazole-resistance mechanisms can now be analyzed at increasingly finer resolutions. This newfound knowledge will be instrumental in formulating new molecular approaches to fight the rapidly emerging epidemic of antifungal resistant fungi.
Systemic fungal infections are an emerging and serious public health concern, with ~1.5 million deaths occurring every year (Brown et al., 2012). The most common fungal pathogens are Candida spp., Aspergillus fumigatus, and Cryptococcus neoformans.
Currently, there are three leading families of antifungals used to treat systemic fungal infections in the clinic: polyenes, echinocandins and triazoles (Gintjee et al., 2020). Triazoles are a first-line treatment, inhibiting fungal growth by targeting the cytochrome P450 lanosterol 14-α-demethylase, encoded by the ERG11/cyp51A gene, catalyzing a key step in the ergosterol biosynthetic pathway. All genes mentioned in this review are listed in Table 1. This review will focus on the evolution of triazole-resistance in serial fungal isolates from infected patients, and in-vitro experiments in which a fungus is passaged under increasing antifungal concentrations.

Table 1 List of genes mentioned in this review.
Triazole resistance is an increasing problem worldwide (Sanglard, 2016). Candida spp. cause human infection ranging from systemic life-threatening to mucosal diseases (Whaley et al., 2017). Currently known triazole-resistance mechanisms in Candida spp. include mutations in the ERG11 gene, which can be either homozygous or heterozygous, or overexpression via its transcriptional activator, Upc2 (Whaley et al., 2017; Ben-Ami and Kontoyiannis, 2021). Other mechanisms include inactivation of ERG3, encoding sterol C5,6-desaturase, leading to the incorporation of alternative sterols into the cell membrane, overexpression of efflux ATP-binding cassette (ABC) transporters due to mutations in the transcriptional activators TAC1 (activating CDR1 and CDR2) and MRR1 (activating MDR1, formerly known as BENr), and loss of heterozygosity (LOH) and aneuploidy of key genes or chromosomes, such as chromosome 5 on which ERG11 and TAC1 are encoded (Whaley et al., 2017; Ben-Ami and Kontoyiannis, 2021). Amplification or deletion of a chromosomal segment is defined as copy number variation (CNV).
Cryptococcus spp. (mainly C. neoformans and Cryptococcus gattii) are encapsulated yeast that can cause life-threatening infections, primarily in immunocompromised patients (Zafar et al., 2019). While not many ERG11 mutations have been identified in resistant strains, C. neoformans has a known transient resistance and hetero-resistance mechanism mediated by aneuploidy of key chromosomes (primarily chromosome 1 that encodes ERG11 and the efflux transporter AFR1), that can result in upregulation of ERG11 or AFR1 (Zafar et al., 2019; Ben-Ami and Kontoyiannis, 2021).
A. fumigatus is an environmental filamentous fungus and the most common mold pathogen in humans. It can cause a wide range of diseases in humans, including invasive infections with high mortality rates in immunocompromised patients (Latgé and Chamilos, 2019; Nywening et al., 2020). The majority of resistance cases in A. fumigatus are due to mutations or overexpression of ERG11/cyp51A genes (Sanglard, 2016; Nywening et al., 2020). While single point mutations can elevate resistance, a tandem repeat (TR) of the SrbA transcriptional activator binding site, combined with a specific set of mutations in the reading frame, results in overexpression of the cyp51A gene and therefore elevates resistance. Additional triazole-resistance associated mechanisms include, among others, mutations in hmg1 encoding HMG-CoA reductase (one of the first steps in the ergosterol biosynthesis pathway), mutations in hapE encoding a subunit of CCAAT-binding transcription factor complex (CBC) and overexpression of multidrug efflux transporters (Sanglard, 2016; Nywening et al., 2020).
Evolutionary experiments are an important tool to study the processes occurring within a fungus in response to different stressors. There are two categories of evolutionary experiments: collection of serial fungal isolates from infected triazole-treated patients, and in-vitro experiments in which a fungus is passaged under increasing triazole concentrations, either in liquid broth or on agar plates (Figure 1). The advantage of collecting serial clinical isolates is the possibility of monitoring real-life changes within the host during drug treatment. The disadvantage is the lack of identical growth conditions of the isolates and having many non-triazole-related differences between them as they are not always isogenic. The advantage of in-vitro evolution is the ability to control every aspect of the experiment, using all-isogenic strains and monitoring every generation. The clear disadvantage is the lack of an actual physiological environment.
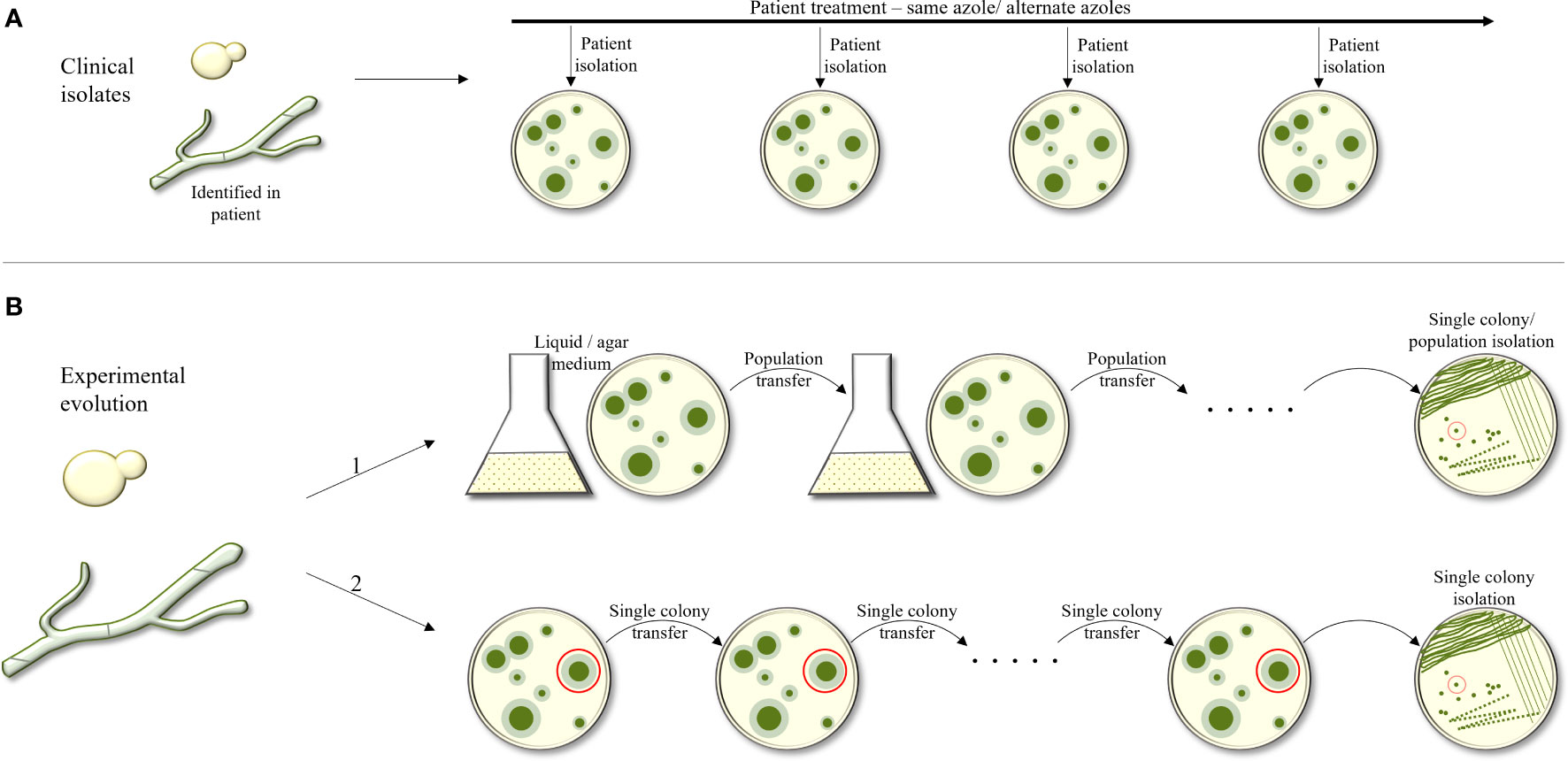
Figure 1 Outline of experimental approaches used to study evolution of triazole resistance. (A) Collection of serial fungal isolates from an infected patient over time. Isogenic strains are identified and compared. (B) Experimental evolution in–vitro. The fungus is passaged under increasing tiazole concentrations, in liquid broth or on agar plates. In B1, a population of the strain under selection is collected and passaged onto the next agar plate or broth tube, containing increasing antifungal concentrations. In B2, during each passage, only one colony or CFU is selected from the agar plate and transferred to the next plate.
In-vitro evolution experiments can again be divided into two categories: in the first, a population of a specific starting strain is collected and passaged to the next agar or broth plate with increasing or steady antifungal concentration. In the second, in each passage, only one colony forming unit (CFU) is picked from one agar plate and transferred to the next. The advantage in the first approach is that it allows competition among the mutating colonies, favoring the most fitness-effective mutations that allow antifungal resistance. In the second, since only one colony is passaged, there is no selection against antifungal-resistance mutations that also have a fitness cost.
Regardless of the approach used to perform in-vitro evolution, a control lineage must be passaged in triazole-free media, in parallel to the evolving triazole-treated lineages. Based on our experience, the evolved and control strains can differ by several hundred SNPs by WGS, and most are probably unrelated to triazole resistance. Therefore, multiple triazole-resistant lineages are evolved, and only SNPs occurring in several of the lineages independently are further analyzed by reintroduction into a susceptible laboratory strain.
This review will describe the in-vitro evolution experiments and serial clinical isolate studies performed in pathogenic fungi during triazole exposure. We will outline the identified resistance mechanisms and compare the findings reported using the various experimental approaches, while focusing more closely on recent publications.
Patients infected with Candida spp. are treated primarily with fluconazole, and fungal resistance can develop over time. The following section will describe a few studies performed with serial clinical isolates, and by in-vitro evolution, and discuss the commonalities and differences between species and experiments. The number of studies using serial clinical isolates of Candida albicans is impressive. Therefore, the citations and examples described here should be considered representative rather than comprehensive.
In this approach, strains were isolated from patients during treatment and tested for drug susceptibility and isogeneity. Several experimental approaches were used to study the isolated strains, including, in earlier studies, sequencing of specific genes for the detection of SNPs, and Southern and Northern blot analysis of specific genes to detect sequence and expression variations. In later studies, omics approaches, including RNAseq, proteomics and whole-genome sequencing (WGS) were used to identify SNPs, expression changes, and gene copy number variations in the entire genome that may be associated with triazole resistance. The subsequent introduction of resistance-associated alleles into susceptible yeasts is a valuable tool for determining a mutation’s importance in the acquisition of triazole resistance. In C. albicans (Table 2), most serial clinical isolate studies support the connection between increased expression of CDR1 and CDR2, encoding efflux ABC transporters, and MDR1, encoding an efflux transporter from the major facilitator superfamily (MFS), to fluconazole resistance (Sanglard et al., 1995; White, 1997; Sanglard et al., 1998; Perea et al., 2001; Rogers and Barker, 2003; Coste et al., 2006; Coste et al., 2007; Dunkel et al., 2008; Ford et al., 2015; Bhattacharya et al., 2016; Song et al., 2022). In some cases, this was linked to hyperactive alleles of their transcriptional activators, TAC1 (Coste et al., 2007; Ford et al., 2015) and MRR1 (Dunkel et al., 2008; Ford et al., 2015), respectively. ERG11 triazole-resistance-associated substitutions, either homozygous or heterozygous, were found in several clinical isolates sets (White, 1997; Sanglard et al., 1998; Perea et al., 2001; Coste et al., 2006; Coste et al., 2007; Ford et al., 2015). Several studies also described elevated expression of ERG11 in resistant isolates (White, 1997; Perea et al., 2001; Coste et al., 2007; Heilmann et al., 2010; Hoot et al., 2011; Bhattacharya et al., 2016; Latgé and Chamilos, 2019), compared to their isogenic susceptible strains, that in some cases can be linked to mutations or overexpression in their transcriptional activator UPC2 (Heilmann et al., 2010; Hoot et al., 2011). Loss of heterozygosity (LOH) can also contribute to triazole resistance by enabling mutated resistance-associated alleles, such as TAC1, ERG11, or areas on chromosome 3 (on which CDR1, CDR2 and MRR1 are encoded), to duplicate and exert a stronger effect (White, 1997; Coste et al., 2007; Dunkel et al., 2008; Selmecki et al., 2008; Ford et al., 2015). Aneuploidy and isochromosome formation increases the copy number of the genes located in the duplication area, such as chromosome 5 (on which ERG11 and TAC1 are encoded, or isochromosome forming i(5L)) (Coste et al., 2007, Selmecki et al., 2008), which results in elevated expression. These main resistance mechanisms are repeatedly evolved in clinical strains isolated from different infection sites. Less common findings are overexpression of several ERG genes, including ERG1, ERG2, ERG3, ERG5, ERG6, ERG9, and ERG10 (Rogers and Barker, 2003; Hoot et al., 2011).
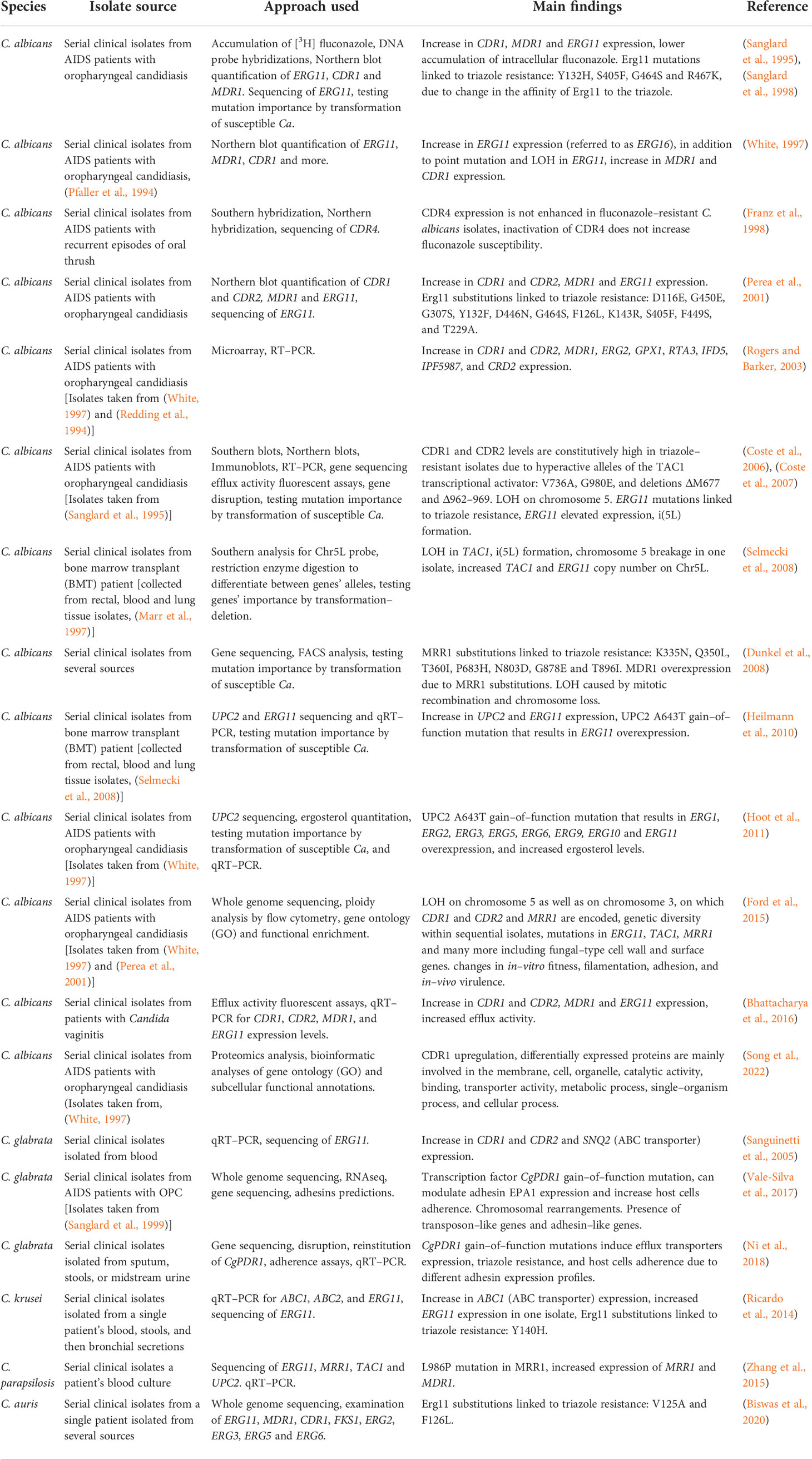
Table 2 Development of triazole resistance in serial clinical isolates of Candida spp.
There seems to be agreement that the first step of triazole-resistance evolution in serial clinical isolate studies in C. albicans involves the acquisition of gain-of-function/hyperactive/resistance-associated mutations or alleles in triazole-resistance associated genes, such as ERG11, TAC1 or MRR1, often accompanied by CDR1, CDR2 and MDR1 overexpression (Sanglard et al., 1995; White, 1997; Sanglard et al., 1998; Perea et al., 2001; Rogers and Barker, 2003; Coste et al., 2006; Dunkel et al., 2008; Ford et al., 2015; Bhattacharya et al., 2016; Latgé and Chamilos, 2019; Song et al., 2022) (Figure 2A). The next steps include LOH of key genes (for example, ERG11) or chromosome areas (for example, chromosome 3 or chromosome 5), aneuploidy of chromosome 5 and i(5L), although there are conflicting results on what event precedes the others. Some researchers hypothesized that aneuploidy of chromosomes such as i(5L) can promote LOH (Ford et al., 2015).
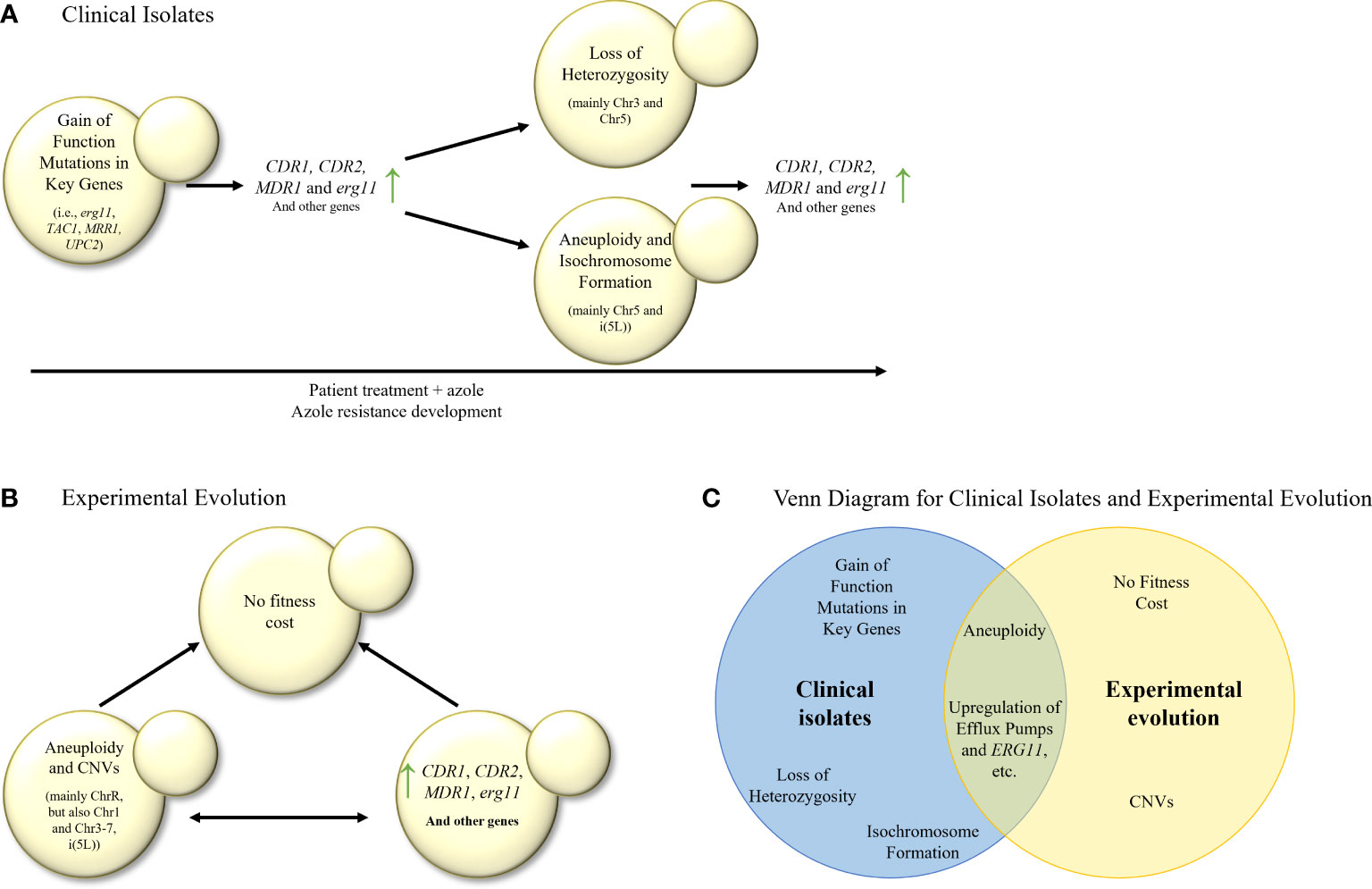
Figure 2 (A) Stepwise evolution of triazole resistance in serial clinical isolates of C. albicans. Gain–of–function mutations are followed by elevation in the expression of efflux transporters and ergosterol biosynthesis (erg) genes, then by LOH, aneuploidy and isochromosome formation, and finally by another increase in expression of efflux transporters and erg genes, leading to triazole resistance. (B) In–vitro evolution of triazole resistance in C. albicans isolates. Elevation in the expression of efflux transporters, erg genes and many other genes is commonly observed, as well as aneuploidies and CNVs in most of the chromosomes. It is unknown whether overexpression of key genes precedes aneuploidies and CNVs, or vice versa. Usually, no fitness cost was found in the evolved isolates. (C) Venn diagram of azole resistance mechanisms in serial clinical isolates and in in–vitro evolution in C. albicans isolates. Aneuploidy and upregulation of efflux pumps and ERG11 are common to both evolutionary tracks.
In Candida glabrata, serial clinical isolate analysis identified a gain-of-function mutation in the transcription factor CgPDR1 (Vale-Silva et al., 2017; Ni et al., 2018), associated with increased expression of efflux transporters CDR1, CDR2, and SNQ2 (Sanguinetti et al., 2005; Ni et al., 2018) (Table 2). Similarly, a gain–of–function mutation in the transcription factor MRR1 was described in Candida parapsilosis and was linked to increased expression of both MRR1 and MDR1 (Zhang et al., 2015). In Candida krusei, ABC1 overexpression was described as well, along with ERG11 point mutation and increased ERG11 expression in one isolate (Ricardo et al., 2014), while in Candida auris only ERG11 point mutations were found (Biswas et al., 2020).
When comparing C. albicans to other Candida species, serial clinical isolate studies suggest that while ERG11 mutation or overexpression is a common triazole–resistance mechanism in C. albicans, C. auris and C. krusei, in other Candida species such as C. glabrata and C. parapsilosis, expression of efflux transporters is mainly seen, resulting from gain–of–function mutations in their respective transcription factors. Although ERG11 mutations have been previously described in clinical isolates of C. glabrata (Silva et al., 2016) and C. parapsilosis (Whaley et al., 2017), none were found in the serial clinical isolates described here. In addition, unlike other Candida species, in C. glabrata, ERG11 alterations are not a major resistance mechanism (Whaley et al., 2017; Ben-Ami and Kontoyiannis, 2021).
In this approach, a small number of strains, often a single strain, are serially passaged in broth containing either constant or increasing triazole concentrations. The last generation is either analyzed as a population or isolated to a single CFU that is studied. All generations are stored and can be analyzed for the timing of individual mutations during the evolutionary process. At the same time, a control group is usually transferred for the same number of generations without triazole. Several experimental approaches are used to study the strains or populations. They include, in earlier studies, checking for ploidy changes, testing of resistance stability as a sign for an underlying genetic mechanism, sequencing of specific genes for the detection of SNPs, and Southern and Northern blot analysis of specific genes. Later studies use WGS and RNAseq to identify SNPs, changes in expression and gene copy number, that may be associated with triazole resistance. The final stage is the verification of mutations by reintroduction, alone and in combination, into a susceptible strain to assess their relative contributions to resistance.
In C. albicans, most in–vitro evolution studies, similarly to clinical isolate studies, describe an increased expression of ERG11, CDR1, CDR2 (Cowen et al., 2000), and MDR1 (Cowen et al., 2000; Dunkel et al., 2008). Increased expression of MDR1 can be partially explained by transcription factor MRR1 gain–of–function mutations and LOH (Dunkel et al., 2008) (Table 3; Figure 2B). Increased expression of CDR1 and CDR2 may be explained by mutations in transcription factor TAC1, or other unknown factors. In addition, changes in expression (either induction or repression of expression) were observed in ERG1, ERG3, and ERG13 (Silva et al., 2016). ChrR (chromosome R, encoding rDNA genes) changes have also been frequently described (Cowen et al., 2000; Huang et al., 2011; Kukurudz et al., 2022), along with Chr3 (containing CDR1 and CDR2), as well as Chr4 and Chr6 aneuploidies, where the resistance mechanism remains unclear (Huang et al., 2011; Todd et al., 2019; Todd and Selmecki, 2020; Kukurudz et al., 2022). Overexpression and CNVs of genes involved in membrane and cell wall integrity (YPL88, YPX98, PDR16, CRZ1, CDR3, NCP1, ECM21, MNN23, RHB1, and KRE6), stress response (HYR1, GRE99, YNL229C, HSP70, CGR1, ERO1, TPK1, ASR1, and PBS2) have also been reported, none of which have been proven to cause triazole resistance (Table 3) (Cowen et al., 2002; Todd and Selmecki, 2020). A high copy number of chromosomal segments, including i(5L) containing ERG11, was described, but not recurrently (Todd and Selmecki, 2020). Despite all this data, it has yet to be shown which processes occur at the beginning of the experimental evolution and support later adaptations (Figure 2B). No ERG11 mutations were described in any of the described studies.
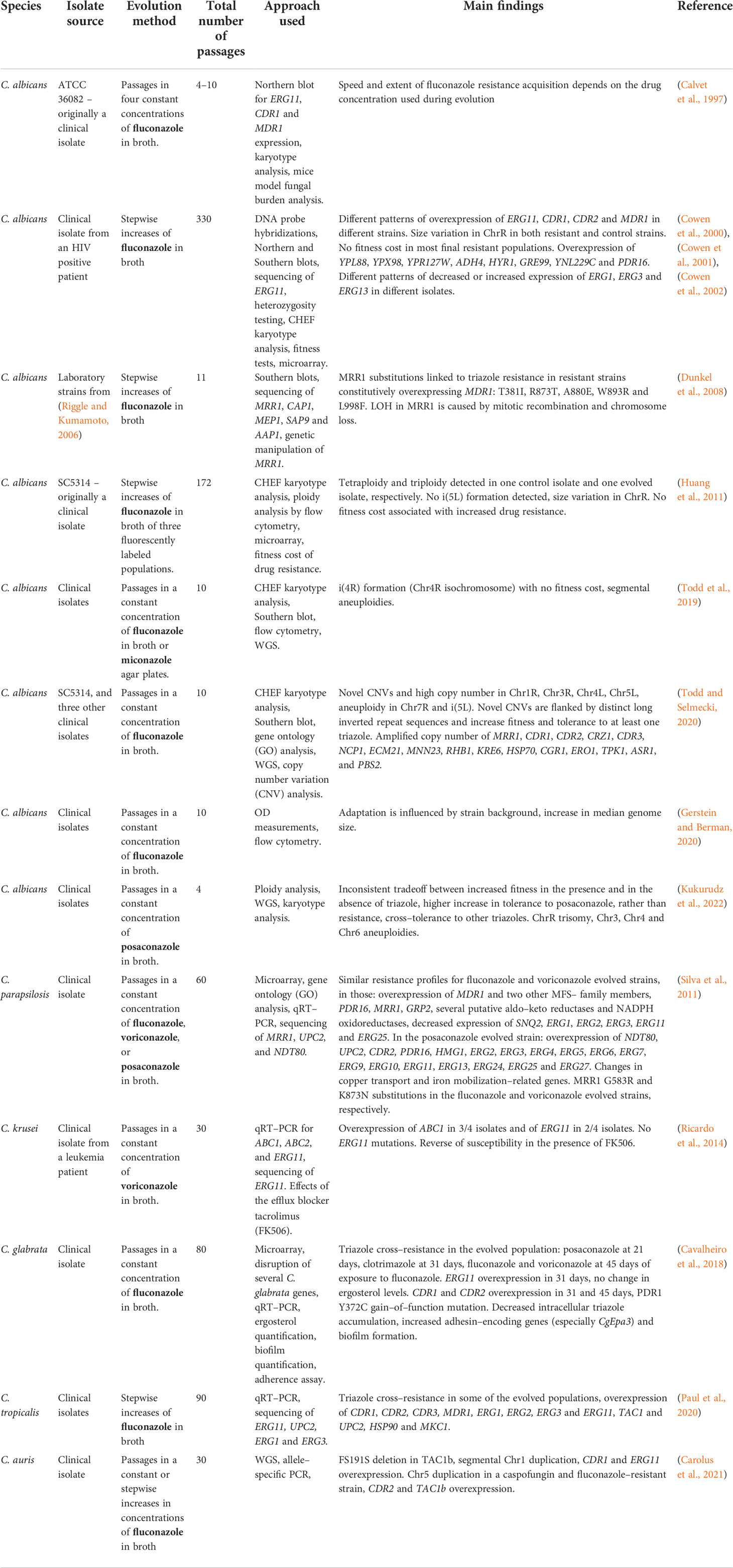
Table 3 Development of triazole resistance in Candida spp. isolates undergoing in–vitro evolution.
Compared with the clinical isolate studies described above, the laboratory evolutionary studies find less gain–of–function mutations in TAC1 and UPC2, with less isochromosome formation and more aneuploidies of chromosomes other than Chr5 (Huang et al., 2011; Todd et al., 2019; Todd and Selmecki, 2020; Kukurudz et al., 2022). In addition, in–vitro evolution studies more frequently describe the overexpression or CNVs of genes outside the ergosterol–biosynthesis pathway (Silva et al., 2016; Todd and Selmecki, 2020). In contrast, both study approaches report the overexpression of ERG11, CDR1, CDR2, and MDR1 (Sanglard et al., 1995, White, 1997; Perea et al., 2001; Rogers and Barker, 2003; Coste et al., 2006; Dunkel et al., 2008, Heilmann et al., 2010; Hoot et al., 2011; Bhattacharya et al., 2016; Song et al., 2022), and gain–of–function mutations in MRR1 (Dunkel et al., 2008; Ford et al., 2015).
Interestingly, clinical isolate studies describe ERG11 mutations while in–vitro evolution studies do not. One hypothesis for this is that in clinical studies, isolates experience a hostile in–host environment, which requires any adaptations to be minimalist and with no fitness cost, such as a single point mutation yielding azole resistance. On the other hand, in–vitro evolution studies are conducted in a clean, constant environment, allowing more room for fitness–costing changes that might yield higher azole resistance.
In general, Candida spp., other than C. albicans, also show high involvement of efflux transporters overexpressed in laboratory–evolved triazole–resistant strains (Table 3). In C. parapsilosis under fluconazole or voriconazole selection, there is strong elevation in expression of efflux transporters and aldo–keto reductases and NADPH oxidoreductases, which may help protect the cells from oxidative stress caused by triazole treatment (Silva et al., 2011). In contrast, in posaconazole–evolved strains, the data suggests that increased production of ergosterol is the primary mechanism of triazole resistance, as many genes of the ergosterol pathway are overexpressed. In C. krusei (Ricardo et al., 2014), overexpression of the ABC1 transporter and ERG11 was observed. The addition of the efflux blocker tacrolimus (FK506) reversed the susceptibility of the evolved strains to voriconazole.
Surprisingly, and for unknown reasons, C. glabrata (Cavalheiro et al., 2018), that was exposed to fluconazole, first acquired posaconazole resistance, followed by resistance to clotrimazole and then fluconazole and voriconazole. A PDR1 transcription factor Y372C gain–of–function mutation was followed by ERG11, CDR1 and CDR2 overexpression. Strains showed decreased intracellular triazole accumulation, increased expression of adhesin–encoding genes (especially CgEpa3) and biofilm formation.
In C. tropicalis (Paul et al., 2020), overexpression of CDR1, CDR2, CDR3, MDR1, ERG1, ERG2, ERG3, ERG11, TAC1, UPC2, HSP90 and MKC1 was observed in the resistant isolates, along with triazole cross–resistance in some. In C. auris (Carolus et al., 2021), one study detected a TAC1b FS191S deletion in the fluconazole–resistant strain, as well as a Chr1 aneuploidy in a segment containing the ERG11 gene, resulting in increased expression. In a C. auris strain that was exposed to both caspofungin and fluconazole, a Chr5 duplication was also observed, correlating with overexpression of TAC1b (which is encoded on Chr5) and CDR2, but not CDR1 (Carolus et al., 2021).
Patients infected with Cryptococcus spp. are often treated with amphotericin B and flucytosine combination therapy followed by fluconazole monotherapy (Ben-Ami and Kontoyiannis, 2021), leading to the development of triazole resistance. Triazole resistance studies based on serial clinical isolates or lab evolution have focused on Cryptococcus neoformans, the major pathogen in this group. Few resistance mutations in ERG11 have been documented in resistant clinical C. neoformans isolates. The main triazole resistance mechanisms found were mediated by aneuploidy of key chromosomes (primarily chromosome 1 that encodes ERG11 and the efflux transporter AFR1), that in turn cause elevated expression of the genes encoded on them (Stone et al., 2019). These unstable genomic reorganizations lead to transient resistance and hetero–resistance, a state in which a resistant subpopulation exists within a largely susceptible population (Stone et al., 2019).
As stated above, in C. neoformans, the main resistance mechanism activated in response to prolonged triazole exposure is through the generation of hetero–resistance and aneuploidies. Indeed, several studies analyzing serial clinical isolates, identified key aneuploidies in Chr1 (encoding AFR1 (an ABC transporter) and ERG11), Chr12 (encoding several oxidative stress related genes, such as dehydrogenases), occurring in that order, and Chr4, Chr5, as well as one deletion in Chr3 (Mondon et al., 1999; Ormerod et al., 2013; Chen et al., 2017; Stone et al., 2019), for which the timeline of occurrence is unknown (Table 4; Figure 3A). In addition, a correlation between hetero–resistance and Chr1 aneuploidy was found, with reversion of said aneuploidy in the absence of the triazole, suggesting that aneuploids bear a fitness cost (Stone et al., 2019). Even though ERG11 mutations are not a significant resistance mechanism in C. neoformans, an Erg11 G484S mutation was found in a series of clinical isolates (Rodero et al., 2003), corresponding with the known G464S gain–of–function mutation resistance mutation in CaErg11. A third, less documented, resistance mechanism includes the deletion mutation in an ARID (AT–rich interaction domain)–containing gene = AVC1, a regulator of virulence traits and carbon assimilation (Ormerod et al., 2013, Chen et al., 2017). However, the resistance mechanism has not been found.

Table 4 Development of triazole resistance in serial clinical isolates of C. neoformans.
Similarly to the results described in serial clinical isolate studies, aneuploidies are also described in the few triazole resistance evolutionary studies performed in C. neoformans (Sionov et al., 2010), with the same loss of aneuploidy in the absence of triazole stress (Table 5; Figure 3B). More specifically, disomy of Chr1 (encoding AFR1 and ERG11), Chr4, Chr10 and Chr14 were identified (Sionov et al., 2010). Overexpression of key genes (ERG11, AFR1 and MDR1 in C. neoformans, and ERG11 in C. gattii) were also described (Bastos et al., 2017), although they were not linked to aneuploidies. As found in the serial clinical isolate studies, one experimental evolution study linked a novel G344S mutation in ERG11 to voriconazole resistance (Kano et al., 2017).
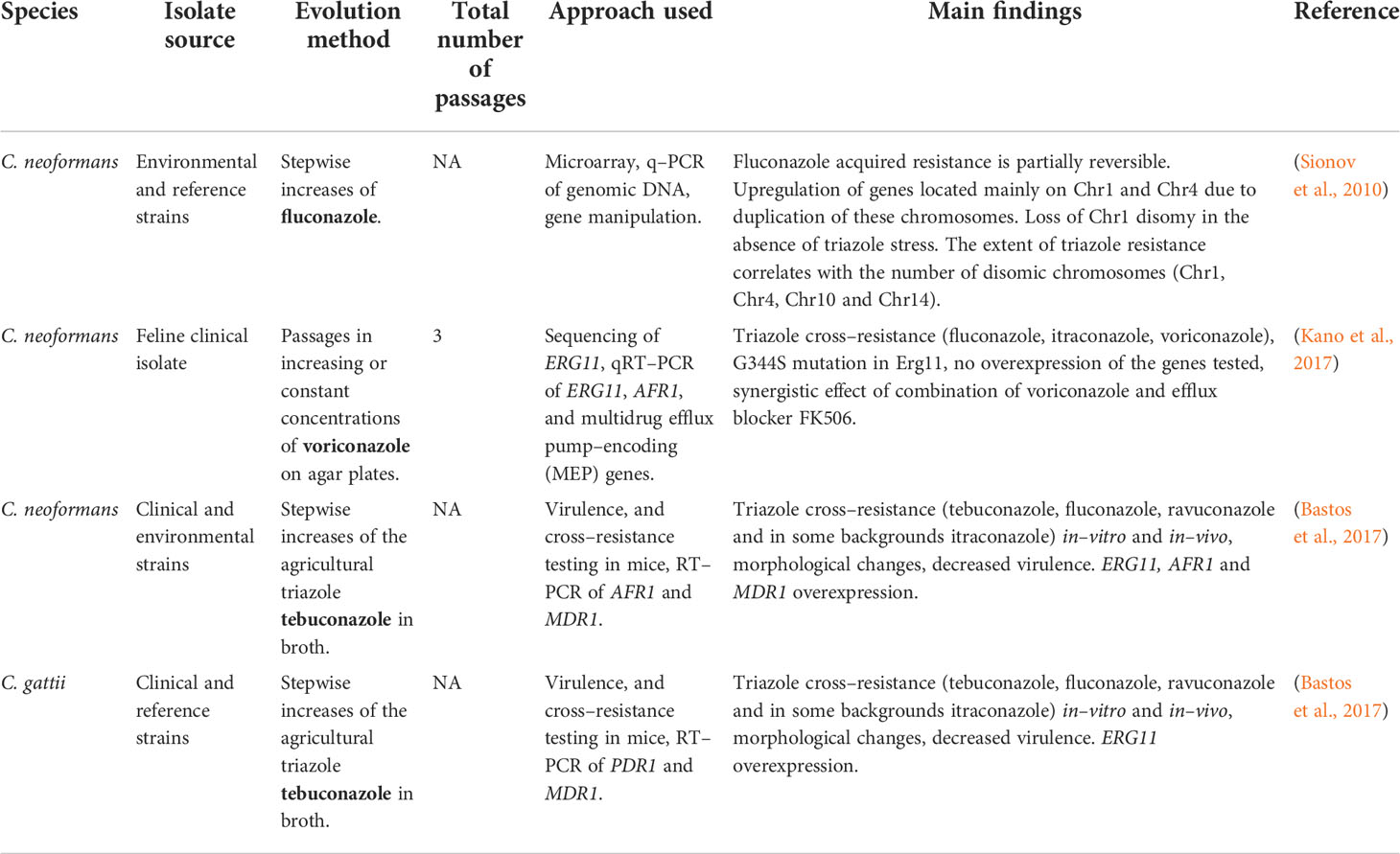
Table 5 Development of triazole resistance in Cryptococcus spp. isolates undergoing in–vitro evolution.
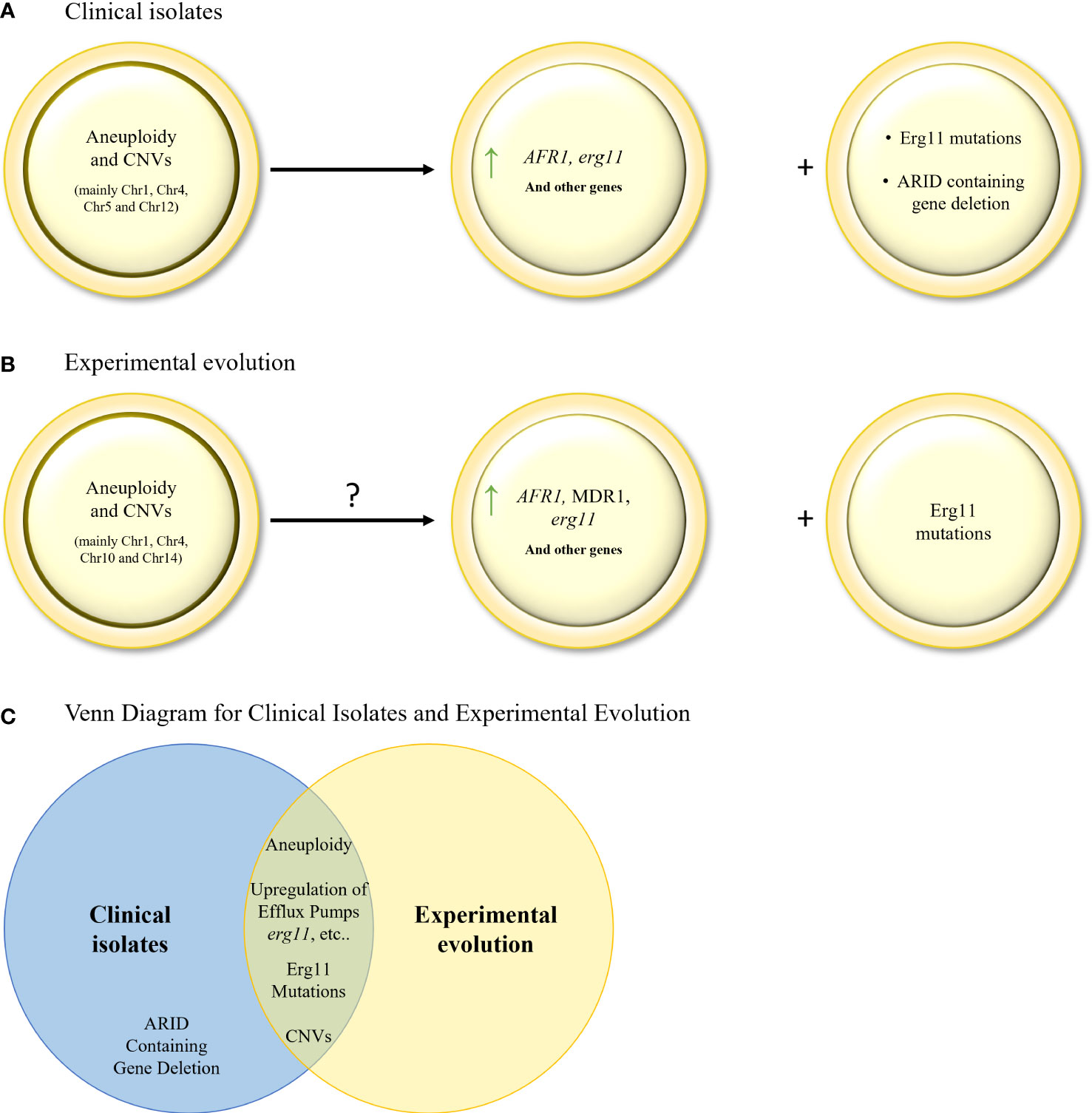
Figure 3 (A). Evolution of triazole resistance in serial clinical isolates of C neoformans. Aneuploidies and CNVs in key chromosomes, leading to gene (AFR1 and ERG11) overexpression, are the main drivers of triazole resistance in C neoformans. ERG11 mutations and ARID containing gene deletion are also found, but to a lesser extent. (B) Mechanisms of triazole resistance identified in in–vitro evolution isolates of C neoformans and C gattii. Aneuploidies and CNVs in key chromosomes, leading to gene overexpression, have been identified. ERG11 mutations were also found but to a lesser extent. (C) Venn diagram of azole resistance mechanisms in serial clinical isolates and in in–vitro evolution in C neoformans and C gattii isolates. Aneuploidy, upregulation of efflux pumps, upregulation and mutations in ERG11, and CNVs are common to both evolutionary tracks.
Aspergillus species known to cause diseases in humans are mainly A. fumigatus, A. flavus, A. niger, and A. terreus (Ben-Ami and Kontoyiannis, 2021). This section will focus on A. fumigatus, the primary pathogen in this group (Latgé and Chamilos, 2019). A. fumigatus is innately resistant to fluconazole (Leonardelli et al., 2016), therefore the triazoles in use in the clinic today are voriconazole, itraconazole, posaconazole, and isavuconazole. We will describe studies involving these clinical triazoles, as well as the agricultural triazole tebuconazole.
Serial clinical isolates are usually collected from patients with prolonged disease, such as chronic pulmonary aspergillosis, where there is sufficient time to develop increasing resistance and collect the evolving strains. The number of studies where serial clinical isolates originated from invasive aspergillosis is relatively low as the disease is rapid and does not allow sufficient time for evolution (Table 6).
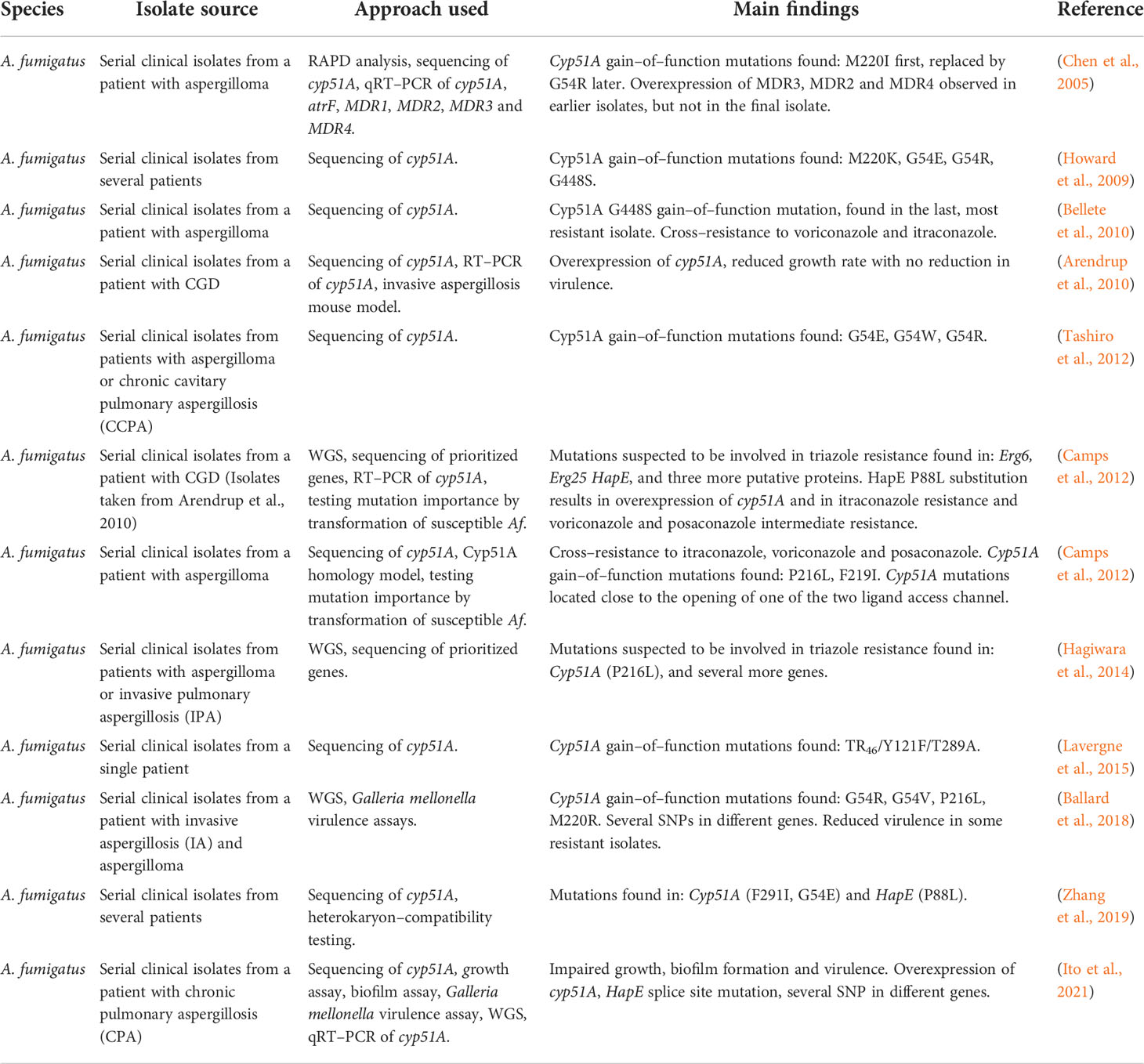
Table 6 Development of triazole resistance in serial clinical isolates of A. fumigatus.
In agreement with what is long known concerning the acquisition of triazole resistance in A. fumigatus, serial clinical isolate studies repeatedly document mutation in cyp51A, the target enzyme of triazole, which includes the known mutations G54E/R/V/W (Chen et al., 2005; Arendrup et al., 2010; Lavergne et al., 2015; Ballard et al., 2018; Rybak et al., 2019), M220I/K/R (Chen et al., 2005; Lavergne et al., 2015; Rybak et al., 2019), P216L (Camps et al., 2012; Camps et al., 2012; Lavergne et al., 2015), F219I (Camps et al., 2012), G448S (Chen et al., 2005; Howard et al., 2009) and TR46/Y121F/T289A (Hagiwara et al., 2014) (Table 6; Figure 4A). Cyp51A mutations P216L and F219I were proven to cause resistance to itraconazole and posaconazole, and were located close to the opening of one of the two ligand access channels in Cyp51A. Overexpression of cyp51A was also observed in several studies (Bellete et al., 2010; Tashiro et al., 2012; Hagiwara et al., 2014), which in some cases can be explained by a mutation in the cyp51A transcriptional repressor, HapE (Tashiro et al., 2012; Hagiwara et al., 2014; Ballard et al., 2018), or from a tandem repeat (TR) mutation in the cyp51A promoter (Hagiwara et al., 2014), which increases binding of the genes transcriptional activator, SrbA (Nywening et al., 2020). From the studies described here, it appears that overexpression of efflux transporters leading to intermediate resistance is the first step towards triazole resistance acquisition (Rybak et al., 2019). Subsequently cyp51A resistance mutations and HapE gain–of–function mutations can occur (Chen et al., 2005; Howard et al., 2009; Arendrup et al., 2010; Camps et al., 2012; Camps et al., 2012; Hagiwara et al., 2014; Lavergne et al., 2015; Ballard et al., 2018; Rybak et al., 2019, Tashiro et al., 2012; Zhang et al., 2019; Ito et al., 2021), which in turn can elevate expression of other key genes such as cyp51A (Figure 4A). These mutations and overexpression can, in many cases, come at a fitness cost, resulting in impaired growth, biofilm formation, and virulence (Bellete et al., 2010; Tashiro et al., 2012; Hagiwara et al., 2014; Lavergne et al., 2015).
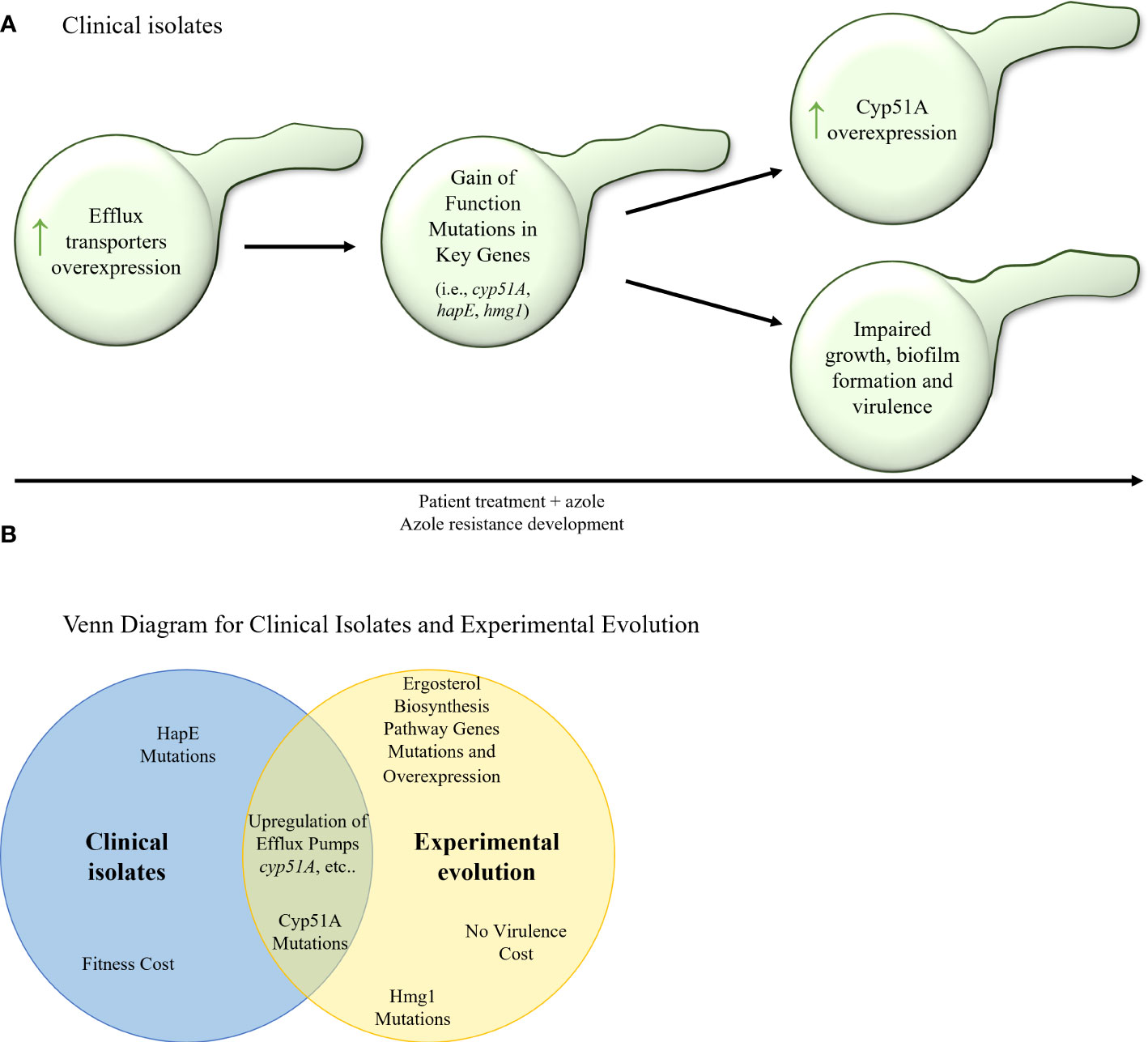
Figure 4 (A) Stepwise evolution of triazole resistance acquisition in serial clinical isolates of A. fumigatus. An increase of efflux transporter activity is followed by gain–of– function mutations in key genes (cyp51A, hapE, hmg1) and overexpression of key genes (such as cyp51A and other erg genes). In many cases, these genetic alterations can carry a fitness cost and cause impaired fungal growth, biofilm formation, and virulence. (B) Venn diagram of azole resistance mechanisms in serial clinical isolates and in in–vitro evolution in isolates of A fumigatus. Upregulation of efflux pumps and upregulation and mutations in Cyp51A are common to both evolutionary tracks.
As expected, most in–vitro evolution studies discovered cyp51A resistance mutations in the evolved strains, including the known mutations G54E/R/W (da Silva et al., 2004; Losada et al., 2015; Chen et al., 2020), M220I/K/R (Zhang et al., 2019; Chen et al., 2020), G138S (Zhang et al., 2017), and the novel mutations N248K/V436A, Y433N (Chen et al., 2020) (Table 7). In addition, hmg1 mutations that cause resistance in non–serial clinical isolates (Rybak et al., 2019), were also described (Losada et al., 2015; Zhang et al., 2017). Interestingly, but not surprisingly, mutations were also found in other genes of the ergosterol biosynthesis pathway (da Silva Ferreira et al., 2004, Losada et al., 2015), as well as overexpression (da Silva Ferreira et al., 2004; Aruanno et al., 2021), but were not proven to confer triazole resistance directly. Another similarity to what is known from clinical isolates is the correlation between triazole resistance and overexpression of efflux transporters observed in several experimental evolution studies (da Silva Ferreira et al., 2004; Aruanno et al., 2021). It is worth noting that in the studies performed with agricultural triazoles, cross resistance with the medical triazoles itraconazole, posaconazole and voriconazole was also seen (Toyotome et al., 2021), suggesting that the use of triazoles in agriculture can give rise to triazole–resistant strains that infect patients. Curiously, while serial clinical isolates often show impaired virulence, isolates from experimental evolution do not (Chen et al., 2020). Overall, it seems that the evolutionary studies repeat the findings of serial clinical isolate studies, although not fully duplicating the timeline suggested above, as cyp51A or hmg1 mutations, for example, can occur early in the experimental evolutionary process at the same time in which enhanced expression of efflux transporters takes place (da Silva Ferreira et al., 2004; Zhang et al., 2017) (Figure 4B).
Table 7 Development of triazole resistance in A. fumigatus isolates undergoing in–vitro evolution.
This article reviewed studies describing the development of fungal triazole resistance. They were performed either by “natural” evolution in infected patients resulting in serial clinical isolates, or by experimental evolution under controlled laboratory conditions. The main resistance mechanisms described in this review are visualized in Figure 5. In general, resistance mechanisms identified by experimental evolution match the results seen in serial clinical isolates. For example, in all three organisms reviewed here, key shared drivers of resistance are the overexpression of ERG11/cyp51A, encoding the target enzyme of triazoles, and of efflux transporters from the ABC (CDR1/2 in Candida spp., AFR1 in Cryptococcus spp., abcB/D in A. fumigatus) and MFS (MDR1 in Candida spp. and Cryptococcus spp., mdrA in A. fumigatus) families. Likewise, gain–of–function mutations, mainly in the ERG11/cyp51A gene, resulting in alteration of the interaction with triazoles, can be found in serial clinical and experimental resistant isolates of both A. fumigatus and Candida spp., and in experimental resistant isolates of Cryptococcus spp. Similarities between Candida spp. and C. neoformans can be found in aneuploidy and CNVs mechanisms, found in both approaches in both yeasts (Mondon et al., 1999; Coste et al., 2007; Selmecki et al., 2008; Sionov et al., 2010; Huang et al., 2011; Ormerod et al., 2013; Chen et al., 2017; Stone et al., 2019; Todd et al., 2019; Todd and Selmecki, 2020; Carolus et al., 2021; Kukurudz et al., 2022). Aneuploidies have not been reported in A. fumigatus. They are unlikely to be an triazole resistance mechanism in this organism, possibly due to its multinuclear filamentous structure, which dilutes the effects of aneuploidy. One major feature differentiating Cryptococcus neoformans from A. fumigatus and Candida spp. is that transient aneuploidy is the main triazole–coping mechanism in Cryptococcus (Sionov et al., 2010; Stone et al., 2019).
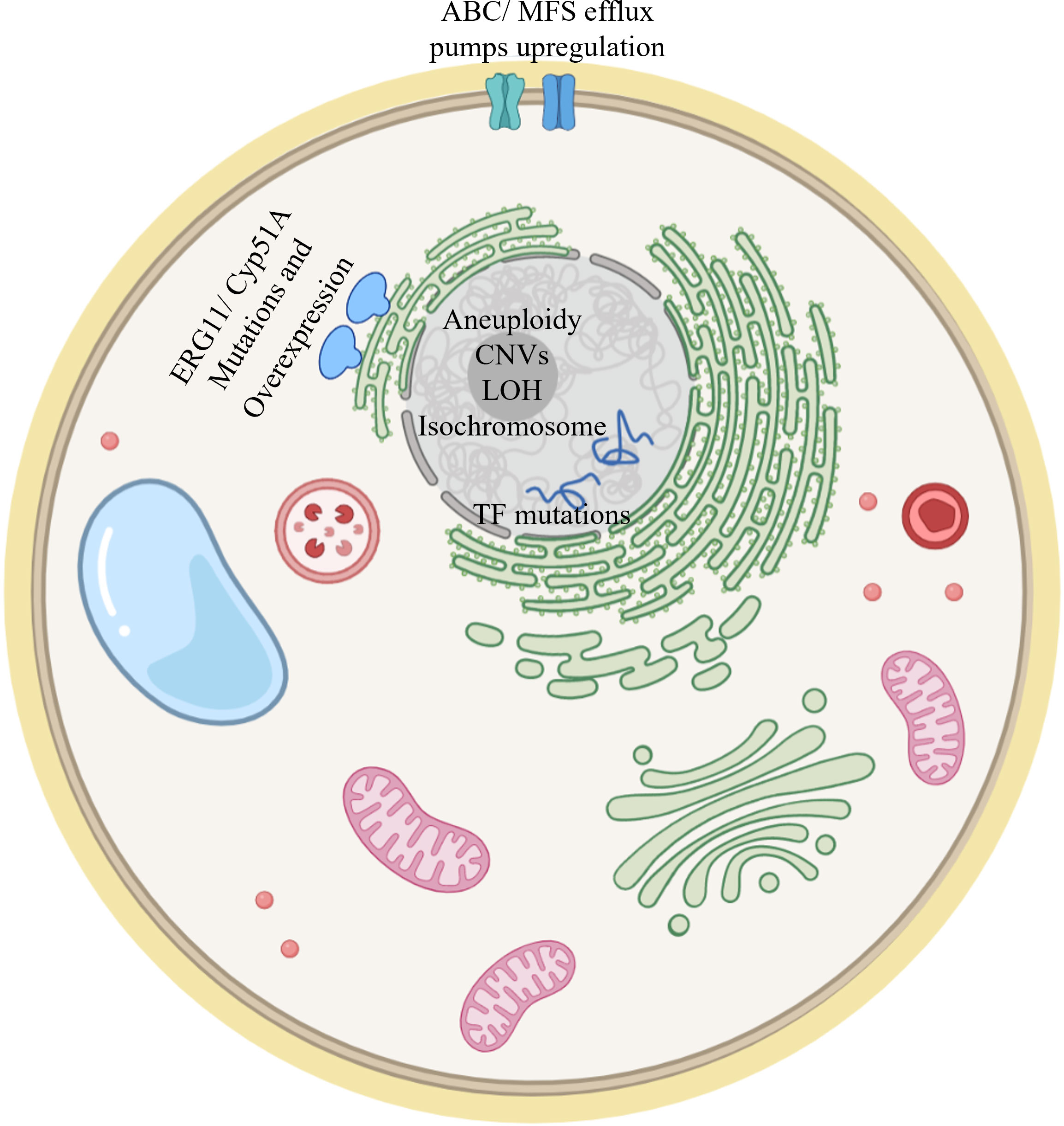
Figure 5 Model of a fungal cell showing the main resistance mechanisms described in this review. ERG11/Cyp51A mutation and overexpression have been documented in Candida spp, Cryptococcus spp and A. fumigatus. Upregulation of efflux pumps has also been described in both clinical and experimental evolution studies for the three fungi addressed in this review. Mutations in transcription factors (TFs) modulating ERG11/cyp51A gene expression have been described. Aneuploidy, CNVs, LOH and isochromosome formation have been documented in Candida spp and Cryptococcus spp.
In serial clinical isolates of Candida spp., mutations in transcription factors Upc2, Tac1 and Mrr1 occur and then lead to increased expression of key genes such as ERG11 and efflux transporters, while in A. fumigatus increased efflux transporter expression precedes gene mutations in serial clinical isolates. A sequence of events was not suggested for experimental evolution isolates of Candida spp. In Cryptococcus spp., serial clinical and experimental evolutionary isolates aneuploidies and CNVs lead to elevated expression of the key genes mentioned above.
The study of serial clinical isolates and experimental evolution studies yielded valuable information, including novel mutations found in C. neoformans (Erg11 G344S, Kano et al., 2017) and A. fumigatus (HapE P88L, Camps et al., 2012), and CNVs in C. albicans (Todd and Selmecki, 2020).
In summary, experimental evolution studies of triazole resistance in pathogenic fungi are valuable tools for determining the mechanisms of resistance acquisition. Despite not replicating the patient environment, they repeat the main findings from serial clinical isolates. They also go beyond them in their ability to monitor the timeline of changes occurring during triazole exposure in proven isogenic strains. Recently, with the advent of omics tools and CRISPR–Cas9 editing technology, the full value of experimental evolution studies has become more evident. This includes the ability to sequence intermediate isolates to precisely deduce the timeline of resistance acquisition, the use of CRISPR–Cas9 editing technology to more rapidly analyze the contribution of mutations in non–coding regions, hypothetical genes and in multiple gene combinations, and the evolution of mutant strains that can highlight alternative pathways of evolution.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This review was supported by the Israel Science Foundation (ISF) grant 2444/18 to NO.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Arendrup M. C., Mavridou E., Mortensen K. L., Snelders E., Frimodt-Møller N., Khan H., et al. (2010). Development of triazole resistance in aspergillus fumigatus during triazole therapy associated with change in virulence. PloS One 5 (4), e10080. doi: 10.1371/journal.pone.0010080
Aruanno M., Gozel S., Mouyna I., Parker J. E., Bachmann D., Flamant P., et al. (2021). Insights in the molecular mechanisms of an triazole stress adapted laboratory-generated aspergillus fumigatus strain. Med. Mycol. 59 (8), 763–772. doi: 10.1093/mmy/myaa118
Ballard E., Melchers W., Zoll J., Brown A., Verweij P. E., Warris A. (2018). In-host microevolution of aspergillus fumigatus: A phenotypic and genotypic analysis. Fungal Genet. Biol. FG B 113, 1–13. doi: 10.1016/j.fgb.2018.02.003
Bastos R. W., Carneiro H., Oliveira L., Rocha K. M., Freitas G., Costa M. C., et al. (2017). Environmental triazole induces cross-resistance to clinical drugs and affects morphophysiology and virulence of cryptococcus gattii and c. neoformans. Antimicrobial Agents Chemotherapy 62 (1), e01179–e01117. doi: 10.1128/AAC.01179-17
Bellete B., Raberin H., Morel J., Flori P., Hafid J., Manhsung R. T. (2010). Acquired resistance to voriconazole and itraconazole in a patient with pulmonary aspergilloma. Med. Mycol. 48 (1), 197–200. doi: 10.3109/13693780902717018
Ben-Ami R., Kontoyiannis D. P. (2021). Resistance to antifungal drugs. Infect. Dis. Clin. North Am. 35 (2), 279–311. doi: 10.1016/j.idc.2021.03.003
Bhattacharya S., Sobel J. D., White T. C. (2016). A combination fluorescence assay demonstrates increased efflux pump activity as a resistance mechanism in triazole-resistant vaginal candida albicans isolates. Antimicrobial Agents Chemotherapy 60 (10), 5858–5866. doi: 10.1128/AAC.01252-16
Biswas C., Wang Q., van Hal S. J., Eyre D. W., Hudson B., Halliday C. L., et al. (2020). Genetic heterogeneity of Australian candida auris isolates: Insights from a nonoutbreak setting using whole-genome sequencing. Open Forum Infect. Dis. 7 (5), ofaa158. doi: 10.1093/ofid/ofaa158
Brown G. D., Denning D. W., Gow N. A., Levitz S. M., Netea M. G., White T. C. (2012). Hidden killers: human fungal infections. Sci. Transl. Med. 4 (165), 165rv13. doi: 10.1126/scitranslmed.3004404
Calvet H. M., Yeaman M. R., Filler S. G. (1997). Reversible fluconazole resistance in candida albicans: a potential in-vitro model. Antimicrobial Agents Chemotherapy 41 (3), 535–539. doi: 10.1128/AAC.41.3.535
Camps S. M., Dutilh B. E., Arendrup M. C., Rijs A. J., Snelders E., Huynen M. A., et al. (2012). Discovery of a HapE mutation that causes triazole resistance in aspergillus fumigatus through whole genome sequencing and sexual crossing. PloS One 7 (11), e50034. doi: 10.1371/journal.pone.0050034
Camps S. M., van der Linden J. W., Li Y., Kuijper E. J., van Dissel J. T., Verweij P. E., et al. (2012). Rapid induction of multiple resistance mechanisms in aspergillus fumigatus during triazole therapy: a case study and review of the literature. Antimicrobial Agents Chemotherapy 56 (1), 10–16. doi: 10.1128/AAC.05088-11
Carolus H., Pierson S., Muñoz J. F., Subotić A., Cruz R. B., Cuomo C. A., et al. (2021). Genome-wide analysis of experimentally evolved candida auris reveals multiple novel mechanisms of multidrug resistance. mBio 12 (2), e03333–e03320. doi: 10.1128/mBio.03333-20
Cavalheiro M., Costa C., Silva-Dias A., Miranda I. M., Wang C., Pais P., et al. (2018). A transcriptomics approach to unveiling the mechanisms of in-vitro evolution towards fluconazole resistance of a candida glabrata clinical isolate. Antimicrobial Agents Chemotherapy 63 (1), e00995–e00918. doi: 10.1128/AAC.00995-18
Chen Y., Farrer R. A., Giamberardino C., Sakthikumar S., Jones A., Yang T., et al. (2017). Microevolution of serial clinical isolates of cryptococcus neoformans var. grubii and c. gattii. mBio 8 (2), e00166–e00117. doi: 10.1128/mBio.00166-17
Chen J., Li H., Li R., Bu D., Wan Z. (2005). Mutations in the cyp51A gene and susceptibility to itraconazole in aspergillus fumigatus serially isolated from a patient with lung aspergilloma. J. Antimicrob. Chemother. 55 (1), 31–37. doi: 10.1093/jac/dkh507
Chen P., Liu M., Zeng Q., Zhang Z., Liu W., Sang H., et al. (2020). Uncovering new mutations conferring triazole resistance in the aspergillus fumigatus cyp51A gene. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.03127
Coste A., Selmecki A., Forche A., Diogo D., Bougnoux M. E., d'Enfert C., et al. (2007). Genotypic evolution of triazole resistance mechanisms in sequential candida albicans isolates. Eukaryotic Cell 6 (10), 1889–1904. doi: 10.1128/EC.00151-07
Coste A., Turner V., Ischer F., Morschhäuser J., Forche A., Selmecki A., et al. (2006). A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in candida albicans. Genetics 172 (4), 2139–2156. doi: 10.1534/genetics.105.054767
Cowen L. E., Kohn L. M., Anderson J. B. (2001). Divergence in fitness and evolution of drug resistance in experimental populations of candida albicans. J. bacteriology 183 (10), 2971–2978. doi: 10.1128/JB.183.10.2971-2978.2001
Cowen L. E., Nantel A., Whiteway M. S., Thomas D. Y., Tessier D. C., Kohn L. M., et al. (2002). Population genomics of drug resistance in candida albicans. Proc. Natl. Acad. Sci. U. S. A. 99 (14), 9284–9289. doi: 10.1073/pnas.102291099
Cowen L. E., Sanglard D., Calabrese D., Sirjusingh C., Anderson J. B., Kohn L. M. (2000). Evolution of drug resistance in experimental populations of candida albicans. J. Bacteriology 182 (6), 1515–1522. doi: 10.1128/JB.182.6.1515-1522.2000
da Silva Ferreira M. E., Capellaro J. L., dos Reis Marques E., Malavazi I., Perlin D., Park S., et al. (2004). In-vitro evolution of itraconazole resistance in aspergillus fumigatus involves multiple mechanisms of resistance. Antimicrobial Agents Chemotherapy 48 (11), 4405–4413. doi: 10.1128/AAC.48.11.4405-4413.2004
Dunkel N., Blass J., Rogers P. D., Morschhäuser J. (2008). Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant candida albicans strains. Mol. Microbiol. 69 (4), 827–840. doi: 10.1111/j.1365-2958.2008.06309.x
Ford C. B., Funt J. M., Abbey D., Issi L., Guiducci C., Martinez D. A., et al. (2015). The evolution of drug resistance in clinical isolates of candida albicans. eLife 4, e00662. doi: 10.7554/eLife.00662
Franz R., Michel S., Morschhäuser J. (1998). A fourth gene from the candida albicans CDR family of ABC transporters. Gene 220 (1-2), 91–98. doi: 10.1016/S0378-1119(98)00412-0
Gerstein A. C., Berman J. (2020). Candida albicans genetic background influence mean and heterogeneity of drug response and genome stability during evolution in fluconazole. mSphere 5, e00480–e00420. doi: 10.1128/mSphere.00480-20
Gintjee T. J., Donnelley M. A., Thompson G. R. 3rd (2020). Aspiring antifungals: Review of current antifungal pipeline developments. J. fungi 6 (1), 28. doi: 10.3390/jof6010028
Hagiwara D., Takahashi H., Watanabe A., Takahashi-Nakaguchi A., Kawamoto S., Kamei K., et al. (2014). Whole-genome comparison of aspergillus fumigatus strains serially isolated from patients with aspergillosis. J. Clin. Microbiol. 52 (12), 4202–4209. doi: 10.1128/JCM.01105-14
Heilmann C. J., Schneider S., Barker K. S., Rogers P. D., Morschhäuser J. (2010). An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in candida albicans. Antimicrobial Agents Chemotherapy 54 (1), 353–359. doi: 10.1128/AAC.01102-09
Hoot S. J., Smith A. R., Brown R. P., White T. C. (2011). An A643V amino acid substitution in Upc2p contributes to triazole resistance in well-characterized clinical isolates of candida albicans. Antimicrobial Agents Chemotherapy 55 (2), 940–942. doi: 10.1128/AAC.00995-10
Howard S. J., Cerar D., Anderson M. J., Albarrag A., Fisher M. C., Pasqualotto A. C., et al. (2009). Frequency and evolution of triazole resistance in aspergillus fumigatus associated with treatment failure. Emerging Infect. Dis. 15 (7), 1068–1076. doi: 10.3201/eid1507.090043
Huang M., McClellan M., Berman J., Kao K. C. (2011). Evolutionary dynamics of candida albicans during in-vitro evolution. Eukaryotic Cell 10 (11), 1413–1421. doi: 10.1128/EC.05168-11
Ito Y., Takazono T., Koga S., Nakano Y., Ashizawa N., Hirayama T., et al. (2021). Clinical and experimental phenotype of triazole-resistant aspergillus fumigatus with a HapE splice site mutation: a case report. BMC Infect. Dis. 21 (1), 573. doi: 10.1186/s12879-021-06279-1
Kano R., Okubo M., Hasegawa A., Kamata H. (2017). Multi-triazole-resistant strains of cryptococcus neoformans var. grubii isolated from a FLZ-resistant strain by culturing in medium containing voriconazole. Med. Mycol. 55 (8), 877–882. doi: 10.1093/mmy/myw101
Kukurudz R. J., Chapel M., Wonitowy Q., Adamu Bukari A. R., Sidney B., Sierhuis R., et al. (2022). Acquisition of cross-triazole tolerance and aneuploidy in candida albicans strains evolved to posaconazole. bioRxiv. doi: 10.1101/2022.01.06.475277
Latgé J. P., Chamilos G. (2019). Aspergillus fumigatus and aspergillosis in 2019. Clin. Microbiol. Rev. 33 (1), e00140–e00118. doi: 10.1128/CMR.00140-18
Lavergne R. A., Morio F., Favennec L., Dominique S., Meis J. F., Gargala G., et al. (2015). First description of triazole-resistant aspergillus fumigatus due to TR46/Y121F/T289A mutation in France. Antimicrobial Agents Chemotherapy 59 (7), 4331–4335. doi: 10.1128/AAC.00127-15
Leonardelli F., Macedo D., Dudiuk C., Cabeza M. S., Gamarra S., Garcia-Effron G. (2016). Aspergillus fumigatus intrinsic fluconazole resistance is due to the naturally occurring T301I substitution in Cyp51Ap. Antimicrobial Agents Chemotherapy 60 (9), 5420–5426. doi: 10.1128/AAC.00905-16
Losada L., Sugui J. A., Eckhaus M. A., Chang Y. C., Mounaud S., Figat A., et al. (2015). Genetic analysis using an isogenic mating pair of aspergillus fumigatus identifies triazole resistance genes and lack of MAT locus's role in virulence. PloS Pathog. 11 (4), e1004834. doi: 10.1371/journal.ppat.1004834
Marr K. A., White T. C., van Burik J. A., Bowden R. A. (1997). Development of fluconazole resistance in candida albicans causing disseminated infection in a patient undergoing marrow transplantation. Clin. Infect. Dis. 25 (4), 908–910. doi: 10.1086/515553
Mondon P., Petter R., Amalfitano G., Luzzati R., Concia E., Polacheck I., et al. (1999). Heteroresistance to fluconazole and voriconazole in cryptococcus neoformans. Antimicrobial Agents Chemotherapy 43 (8), 1856–1861. doi: 10.1128/AAC.43.8.1856
Ni Q., Wang C., Tian Y., Dong D., Jiang C., Mao E., et al. (2018). CgPDR1 gain-of-function mutations lead to triazole-resistance and increased adhesion in clinical candida glabrata strains. Mycoses 61 (7), 430–440. doi: 10.1111/myc.12756
Nywening A. V., Rybak J. M., Rogers P. D., Fortwendel J. R. (2020). Mechanisms of triazole resistance in aspergillus fumigatus. Environ. Microbiol. 22 (12), 4934–4952. doi: 10.1111/1462-2920.15274
Ormerod K. L., Morrow C. A., Chow E. W., Lee I. R., Arras S. D., Schirra H. J., et al. (2013). Comparative genomics of serial isolates of cryptococcus neoformans reveals gene associated with carbon utilization and virulence. G3 3 (4), 675–686. doi: 10.1534/g3.113.005660
Paul S., Singh S., Sharma D., Chakrabarti A., Rudramurthy S. M., Ghosh A. K. (2020). Dynamics of in-vitro development of triazole resistance in candida tropicalis. J. Glob Antimicrob. Resist. 22, 553–561. doi: 10.1016/j.jgar.2020.04.018
Perea S., López-Ribot J. L., Kirkpatrick W. R., McAtee R. K., Santillán R. A., Martínez M., et al. (2001). Prevalence of molecular mechanisms of resistance to triazole antifungal agents in candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrobial Agents Chemotherapy 45 (10), 2676–2684. doi: 10.1128/AAC.45.10.2676-2684.2001
Pfaller M. A., Rhine-Chalberg J., Redding S. W., Smith J., Farinacci G., Fothergill A. W., et al. (1994). Variations in fluconazole susceptibility and electrophoretic karyotype among oral isolates of candida albicans from patients with AIDS and oral candidiasis. J. Clin. Microbiol. 32 (1), 59–64. doi: 10.1128/jcm.32.1.59-64.1994
Redding S., Smith J., Farinacci G., Rinaldi M., Fothergill A., Rhine-Chalberg J., et al. (1994). Resistance of candida albicans to fluconazole during treatment of oropharyngeal candidiasis in a patient with AIDS: documentation by in-vitro susceptibility testing and DNA subtype analysis. Clin. Infect. Dis. 18 (2), 240–242. doi: 10.1093/clinids/18.2.240
Ricardo E., Miranda I. M., Faria-Ramos I., Silva R. M., Rodrigues A. G., Pina-Vaz C. (2014). In-vivo and in-vitro acquisition of resistance to voriconazole by candida krusei. Antimicrobial Agents Chemotherapy 58 (8), 4604–4611. doi: 10.1128/AAC.02603-14
Riggle P. J., Kumamoto C. A. (2006). Transcriptional regulation of MDR1, encoding a drug efflux determinant, in fluconazole-resistant candida albicans strains through an Mcm1p binding site. Eukaryotic Cell 5 (12), 1957–1968. doi: 10.1128/EC.00243-06
Rodero L., Mellado E., Rodriguez A. C., Salve A., Guelfand L., Cahn P., et al. (2003). G484S amino acid substitution in lanosterol 14-alpha demethylase (ERG11) is related to fluconazole resistance in a recurrent cryptococcus neoformans clinical isolate. Antimicrobial Agents Chemotherapy 47 (11), 3653–3656. doi: 10.1128/AAC.47.11.3653-3656.2003
Rogers P. D., Barker K. S. (2003). Genome-wide expression profile analysis reveals coordinately regulated genes associated with stepwise acquisition of triazole resistance in candida albicans clinical isolates. Antimicrobial Agents Chemotherapy 47 (4), 1220–1227. doi: 10.1128/AAC.47.4.1220-1227.2003
Rossi S. A., Trevijano-Contador N., Scorzoni L., Mesa-Arango A. C., de Oliveira H. C., Werther K., et al. (2016). Impact of resistance to fluconazole on virulence and morphological aspects of cryptococcus neoformans and cryptococcus gattii isolates. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00153
Rybak J. M., Ge W., Wiederhold N. P., Parker J. E., Kelly S. L., Rogers P. D., et al. (2019). Mutations in hmg1, challenging the paradigm of clinical triazole resistance in aspergillus fumigatus. mBio 10 (2), e00437–e00419. doi: 10.1128/mBio.00437-19
Sanglard D. (2016). Emerging threats in antifungal-resistant fungal pathogens. Front. Med. 3. doi: 10.3389/fmed.2016.00011
Sanglard D., Ischer F., Calabrese D., Majcherczyk P. A., Bille J. (1999). The ATP binding cassette transporter gene CgCDR1 from candida glabrata is involved in the resistance of clinical isolates to triazole antifungal agents. Antimicrobial Agents Chemotherapy 43 (11), 2753–2765. doi: 10.1128/AAC.43.11.2753
Sanglard D., Ischer F., Koymans L., Bille J. (1998). Amino acid substitutions in the cytochrome p-450 lanosterol 14alpha-demethylase (CYP51A1) from triazole-resistant candida albicans clinical isolates contribute to resistance to triazole antifungal agents. Antimicrobial Agents Chemotherapy 42 (2), 241–253. doi: 10.1128/AAC.42.2.241
Sanglard D., Kuchler K., Ischer F., Pagani J. L., Monod M., Bille J. (1995). Mechanisms of resistance to triazole antifungal agents in candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrobial Agents Chemotherapy 39 (11), 2378–2386. doi: 10.1128/AAC.39.11.2378
Sanguinetti M., Posteraro B., Fiori B., Ranno S., Torelli R., Fadda G. (2005). Mechanisms of triazole resistance in clinical isolates of candida glabrata collected during a hospital survey of antifungal resistance. Antimicrobial Agents Chemotherapy 49 (2), 668–679. doi: 10.1128/AAC.49.2.668-679.2005
Selmecki A., Gerami-Nejad M., Paulson C., Forche A., Berman J. (2008). An isochromosome confers drug resistance in-vivo by amplification of two genes, ERG11 and TAC1. Mol. Microbiol. 68 (3), 624–641. doi: 10.1111/j.1365-2958.2008.06176.x
Silva A. P., Miranda I. M., Guida A., Synnott J., Rocha R., Silva R., et al. (2011). Transcriptional profiling of triazole-resistant candida parapsilosis strains. Antimicrobial Agents Chemotherapy 55 (7), 3546–3556. doi: 10.1128/AAC.01127-10
Silva D. B., Rodrigues L. M., Almeida A. A., Oliveira K. M., Grisolia A. B. (2016). Novel point mutations in the ERG11 gene in clinical isolates of triazole-resistant candida species. Memorias do Instituto Oswaldo Cruz 111 (3), 192–199. doi: 10.1590/0074-02760150400
Sionov E., Lee H., Chang Y. C., Kwon-Chung K. J. (2010). Cryptococcus neoformans overcomes stress of triazole drugs by formation of disomy in specific multiple chromosomes. PloS Pathog. 6 (4), e1000848. doi: 10.1371/journal.ppat.1000848
Song N., Zhou X., Li D., Li X., Liu W. (2022). A proteomic landscape of candida albicans in the stepwise evolution to fluconazole resistance. Antimicrob. Agents Chemother. 66 (4), e0210521. doi: 10.1128/aac.02105-21
Stone N. R., Rhodes J., Fisher M. C., Mfinanga S., Kivuyo S., Rugemalila J., et al. (2019). Dynamic ploidy changes drive fluconazole resistance in human cryptococcal meningitis. J. Clin. Invest. 129 (3), 999–1014. doi: 10.1172/JCI124516
Sullivan D., Haynes K., Moran G., Shanley D., Coleman D. (1996). Persistence, replacement, and microevolution of cryptococcus neoformans strains in recurrent meningitis in AIDS patients. J. Clin. Microbiol. 34 (7), 1739–1744. doi: 10.1128/JCM.34.7.1739-1744.1996
Tashiro M., Izumikawa K., Hirano K., Ide S., Mihara T., Hosogaya N., et al. (2012). Correlation between triazole treatment history and susceptibility in clinically isolated aspergillus fumigatus. Antimicrobial Agents Chemotherapy 56 (9), 4870–4875. doi: 10.1128/AAC.00514-12
Todd R. T., Selmecki A. (2020). Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. eLife 9, e58349. doi: 10.7554/eLife.58349
Todd R. T., Wikoff T. D., Forche A., Selmecki A. (2019). Genome plasticity in candida albicans is driven by long repeat sequences. eLife 8, e45954. doi: 10.7554/eLife.45954
Toyotome T., Onishi K., Sato M., Kusuya Y., Hagiwara D., Watanabe A., et al. (2021). Identification of novel mutations contributing to triazole tolerance of aspergillus fumigatus through in-vitro exposure to tebuconazole. Antimicrobial Agents Chemotherapy 65 (9), e0265720. doi: 10.1128/AAC.02657-20
Vale-Silva L., Beaudoing E., Tran V., Sanglard D. (2017). Comparative genomics of two sequential candida glabrata clinical isolates. G3 7 (8), 2413–2426. doi: 10.1534/g3.117.042887
Van Wyk M., Govender N. P., Mitchell T. G., Litvintseva A. P. (2014). Multilocus sequence typing of serially collected isolates of cryptococcus from HIV-infected patients in south Africa. J. Clin. Microbiol. 52 (6), 1921–1931. doi: 10.1128/JCM.03177-13
Whaley S. G., Berkow E. L., Rybak J. M., Nishimoto A. T., Barker K. S., Rogers P. D. (2017). Triazole antifungal resistance in candida albicans and emerging non-albicans candida species. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.02173
White T. C. (1997). Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in triazole resistance in candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrobial Agents Chemotherapy 41 (7), 1482–1487. doi: 10.1128/AAC.41.7.1482
Zafar H., Altamirano S., Ballou E. R., Nielsen K. (2019). A titanic drug resistance threat in cryptococcus neoformans. Curr. Opin. Microbiol. 52, 158–164. doi: 10.1016/j.mib.2019.11.001
Zhang J., Debets A. J. M., Verweij P. E., Melchers W. J. G., Zwaan B. J., Schoustra S. E. (2015). Asexual sporulation facilitates adaptation: The emergence of triazole resistance in aspergillus fumigatus. Evolution 69, 2573–2586. doi: 10.1111/evo.12763
Zhang J., Snelders E. E., Zwaan B. J., Schoustra S. E., Kuijper E. J., Arendrup M. C., et al. (2019). Relevance of heterokaryosis for adaptation and triazole-resistance development in aspergillus fumigatus. Proc. Biol. Sci. 286 (1896), 20182886. doi: 10.1098/rspb.2018.2886
Zhang J., van den Heuvel J., Debets A., Verweij P. E., Melchers W., Zwaan B. J., et al. (2017). Evolution of cross-resistance to medical triazoles in aspergillus fumigatus through selection pressure of environmental fungicides. Proc. Biol. Sci. 284 (1863), 20170635. doi: 10.1098/rspb.2017.0635
Keywords: Aspergillus fumigatus, antifungal resistance, triazole antifungals, evolution of triazole resistance, serial clinical isolates
Citation: Handelman M and Osherov N (2022) Experimental and in-host evolution of triazole resistance in human pathogenic fungi. Front. Fungal Biol. 3:957577. doi: 10.3389/ffunb.2022.957577
Received: 31 May 2022; Accepted: 19 July 2022;
Published: 23 August 2022.
Edited by:
Chaoyang Xue, Rutgers University, United StatesReviewed by:
Michelle Momany, University of Georgia, United StatesCopyright © 2022 Handelman and Osherov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nir Osherov, bm9zaGVyb3ZAcG9zdC50YXUuYWMuaWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.