 Alec Brown
Alec Brown Matthew E. Mead
Matthew E. Mead Jacob L. Steenwyk
Jacob L. Steenwyk Gustavo H. Goldman
Gustavo H. Goldman Antonis Rokas
Antonis Rokas
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Fungal Biol. , 07 July 2022
Sec. Fungal Genomics and Evolution
Volume 3 - 2022 | https://doi.org/10.3389/ffunb.2022.802494
This article is part of the Research Topic Genomic approaches for understanding the genetics of traits in filamentous fungi View all 4 articles
Invasive aspergillosis is a deadly fungal disease; more than 400,000 patients are infected worldwide each year and the mortality rate can be as high as 50-95%. Of the ~450 species in the genus Aspergillus only a few are known to be clinically relevant, with the major pathogen Aspergillus fumigatus being responsible for ~50% of all invasive mold infections. Genomic comparisons between A. fumigatus and other Aspergillus species have historically focused on protein-coding regions. However, most A. fumigatus genes, including those that modulate its virulence, are also present in other pathogenic and non-pathogenic closely related species. Our hypothesis is that differential gene regulation – mediated through the non-coding regions upstream of genes’ first codon – contributes to A. fumigatus pathogenicity. To begin testing this, we compared non-coding regions upstream of the first codon of single-copy orthologous genes from the two A. fumigatus reference strains Af293 and A1163 and eight closely related Aspergillus section Fumigati species. We found that these non-coding regions showed extensive sequence variation and lack of homology across species. By examining the evolutionary rates of both protein-coding and non-coding regions in a subset of orthologous genes with highly conserved non-coding regions across the phylogeny, we identified 418 genes, including 25 genes known to modulate A. fumigatus virulence, whose non-coding regions exhibit a different rate of evolution in A. fumigatus. Examination of sequence alignments of these non-coding regions revealed numerous instances of insertions, deletions, and other types of mutations of at least a few nucleotides in A. fumigatus compared to its close relatives. These results show that closely related Aspergillus species that vary greatly in their pathogenicity exhibit extensive non-coding sequence variation and identify numerous changes in non-coding regions of A. fumigatus genes known to contribute to virulence.
Invasive aspergillosis (IA), a human disease caused by members of the fungal genus Aspergillus, is responsible for >400,000 cases worldwide per year with a mortality rate between 50-95% (Bongomin et al., 2017). More than 90% of IA cases are caused by Aspergillus fumigatus, with about a dozen other species such as Aspergillus lentulus, Aspergillus thermomutatus, and Aspergillus udagawae accounting for the rest (Steinbach et al., 2012; Rokas et al., 2020). Studies in both environmental (Flores et al., 2014) and hospital settings (Wirmann et al., 2018) show that asexual spores (conidia) of A. fumigatus and many other Aspergillus species are present in the air, yet A. fumigatus causes IA more frequently than its close relatives.
IA begins with inhalation of Aspergillus asexual spores and subsequent interaction between the asexual spores and the epithelium of the lung (Chotirmall et al., 2013). Several defense mechanisms including physical removal of asexual spores (Croft et al., 2016), secretion of antimicrobial peptides (Wiesner and Klein, 2017), and recruitment of specialized immune cells are employed by the human host to prevent spore germination (Bertuzzi et al., 2018). To cause infection, A. fumigatus must overcome these challenges and adapt to the host environment. The dynamics and intricacies of the interaction between A. fumigatus and host responses have yet to be fully elucidated. Decades of work have identified at least 206 genetic determinants of A. fumigatus virulence, that is genes whose deletion is known to modulate the virulence of A. fumigatus (for a detailed list, see: Steenwyk et al., 2021a). These genetic determinants of virulence are involved in a wide range of activities including gene regulation, RNA processing, protein modification, production of secondary metabolites, amino acid biosynthesis, cell cycle regulation, morphological regulation, and others (Steenwyk et al., 2021b).
The phylogeny of the genus Aspergillus reveals that pathogenic species are often more closely related to nonpathogenic species than to other pathogenic ones (Steenwyk et al., 2009; Houbraken et al., 2014; de Vries et al., 2017; Rokas et al., 2020; Mead et al., 2021). For example, A. fischeri is a close relative of A. fumigatus (the two share >90% average nucleotide sequence similarity and >95% average amino acid sequence similarity between orthologs), yet A. fischeri is less virulent and is not considered clinically relevant (Mead et al., 2019; Steenwyk et al., 2020a). Given the large disparity of IA cases caused by A. fumigatus and closely related species, early studies looked to species-specific genes in A. fumigatus as a potential contributor (Fedorova et al., 2008). However, a recent examination found that 206 known genetic determinants of virulence in A. fumigatus are shared between A. fumigatus and at least one other closely related species (Mead et al., 2021).
Variation in non-coding regions can also contribute to phenotypic diversity (Carroll, 2008; Li and Johnson, 2010) and disease (Ropero et al., 2017; Jang et al., 2018; Caron et al., 2019). In fungi, non-coding regions found immediately upstream of genes’ protein-coding regions are bound by transcription factors (TFs), impact transcriptional activity (Kim et al., 2019), and play roles in vital biological processes such as zinc homeostasis (Eide, 2020) and thermotolerance (Yamamoto et al., 2008). Differences in gene expression have become an important focus in understanding A. fumigatus virulence (Chung et al., 2014; de Castro et al., 2014; Furukawa et al., 2020; Ries et al., 2020; Takahashi et al., 2021; Colabardini et al., 2022). However, the role non-coding regions play in differential gene regulation between A. fumigatus and close relatives remains largely unknown.
Here, we perform genome-wide comparisons of intergenic, non-coding regions upstream of the first codon of single-copy orthologous genes of the reference strains A. fumigatus Af293 and A1163 against those of eight closely related species. We identified 5,215 single-copy orthologous genes across the 10 taxa of interest. Of the 5,215 genes, the non-coding regions of 4,483 genes either lacked homology across the ten taxa or showed extensive sequence variation, such that multiple sequence alignment was not possible. For the remaining 732 genes, each non-coding sequence was ≥500 bp long in all ten taxa and the sequence similarity of the sequence alignment between the A. fumigatus Af293 sequence and those of all other nine strains/species was ≥75%, enabling us to construct accurate multiple sequence alignments. Examination of the evolutionary rates of the non-coding and protein-coding regions of these 732 genes identified 418 upstream non-coding and 100 protein-coding regions whose evolutionary rate was different in A. fumigatus compared to close relatives. These 418 non-coding regions include 25 known genetic determinants of A. fumigatus virulence, such as pkaR (a regulatory subunit essential for protein kinase A pathway), gliG (glutathione S-transferase required for gliotoxin production), and metR (transcription factor required for sulfur assimilation).
All Aspergillus genomes are publicly available and were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/). These strains include A. fumigatus Af293 (Nierman et al., 2005), A. fumigatus A1163 (Fedorova et al., 2008), A. oerlinghausenensis CBS139183 (Steenwyk et al., 2020b), A. fischeri NRRL1881 (Fedorova et al., 2008), A. lentulus IFM54703 (Kusuya et al., 2016), A. novofumigatus IBT 16806 (GenBank accession: MSZS00000000.1) A. fumigatiaffinis 5878 (dos Santos et al., 2020), A. udagawae IFM 46973 (Kusuya et al., 2016), A. turcosus HMR AF 1038 (Parent-Michaud et al., 2019), and A. thermomutatus HMR AF 39 (Parent-Michaud et al., 2019).
To infer single-copy orthologous genes among all protein-coding sequences for all ten taxa, we used OrthoFinder, version 2.4.0 (Emms and Kelly, 2015). OrthoFinder clustered genes into orthogroups from gene-gene sequence similarity information obtained using the program DIAMOND version 2.0.9 (Buchfink et al., 2015) with the proteomes of the ten Aspergillus species as input. The key parameters used in DIAMOND were e-value = 1 x 10-3 with a percent identity cutoff of 30% and percent match cutoff of 70%. This approach identified 5,215 single copy orthologous genes (Table S1).
To identify highly conserved non-coding regions, we first retrieved intergenic sequences directly upstream of the first codon of all 5,215 single-copy orthologous genes for each of the ten Aspergillus species/strains using a custom script (https://github.com/alecbrown24/General_Bio_Scripts; this script was based on a previously available script: https://github.com/shenwei356/bio_scripts). We retrieved the first 500 bp of intergenic sequence directly upstream of each gene’s first codon and used these sequences to generate FASTA files of non-coding regions, as well as FASTA files of single-copy orthologous protein-coding sequences using Python version 3.8.2 (https://www.python.org/). For some of the non-coding regions, there were <500 bp of non-coding sequence between the first codon of the gene of interest and an upstream gene; in these instances, only the intergenic region was used for subsequent analyses.
All multiple sequence alignments were constructed using MAFFT, version 7.453, with default parameter settings (Rozewicki et al., 2019). Analyses were conducted using custom Python scripts that used BioPython, version 1.78 (Cock et al., 2009), and NumPy, version 1.20.3 (Harris et al., 2020), modules. Sequence similarity in protein-coding and non-coding regions was calculated from their corresponding multiple sequence alignment files. The percent sequence similarity for each position in the alignment was calculated by determining if the nucleotide/amino acid at each position was the same as the nucleotide/amino acid for A. fumigatus Af293 and then dividing by 10. The percent similarity for each nucleotide/amino acid in each of the 5,215 non-coding and protein-coding regions of genes was averaged and reported. We discovered that the non-coding regions of only 732 of the 5,215 genes contained ≥500 bp of sequence directly upstream of the first codon in all ten taxa and exhibited sequence similarity ≥75% between the A. fumigatus Af293 sequence and each of the other nine strains/species, enabling us to construct accurate multiple sequence alignments. Thus, we focused our analyses on these 732 genes.
To construct a phylogenomic data matrix, codon-based alignments for all 5,215 single-copy protein-coding orthologs were individually trimmed using ClipKIT, version 1.1.5 (Steenwyk et al., 2020a), with the ‘gappy’ mode and the gaps parameter set to 0.7. The resulting trimmed codon-based alignments were then concatenated into a single matrix with 9,248,205 sites using the ‘create_concat’ function from PhyKIT, version 1.2.1 (Steenwyk et al., 2021a). Next, the evolutionary history of the ten Aspergillus genomes was inferred using IQ-TREE, version 2.0.6 (Minh et al., 2020), and the “GTR+F+I+G4” model of sequence evolution, which was the best fitting one according to the Bayesian Information Criterion (Waddell and Steel, 1997; Vinet and Zhedanov, 2011). Bipartition support was assessed using ultrafast bootstrap approximations (Hoang et al., 2018). All bipartitions received full support. The inferred topology is congruent with known relationships inferred from analyses of single or a few loci as well as from genome-scale analyses (Steenwyk et al., 2009; Hubka et al., 2018; dos Santos et al., 2020).
To identify gene trees whose phylogeny was statistically different from the species phylogeny, we used the approximately unbiased test (Shimodaira, 2002). Protein-coding region and non-coding region trees were inferred using IQ-TREE, version 2.0.6 (Minh et al., 2020), with “GTR+I+G+F” as it was the best fitting substitution model (Waddell and Steel, 1997; Vinet and Zhedanov, 2011). The distributions of branch lengths for protein-coding region and non-coding region trees were determined using the “total_tree_length” function from PhyKIT version 1.2.1 (Steenwyk et al., 2021b) (Table S2).
To determine the rate of sequence evolution in protein-coding region alignments between A. fumigatus and close relatives, we examined variation in the ratio of the rate of nonsynonymous (dN) to the rate of synonymous (dS) substitutions (dN/dS or ω) across the phylogeny. We first obtained codon-based alignments from their corresponding protein sequence alignments using pal2nal, version 14 (Suyama et al., 2006). We next used the codon-based alignments to calculate ω values under two different hypotheses using the codeml module in paml, version 4.9 (Yang, 2007). For each gene tested, the null hypothesis (H0) was that all branches of the phylogeny exhibit the same estimated ω value. We compared H0 to an alternative hypothesis (HA) which allows for the branch leading to A. fumigatus to have a distinct estimated ω value from the rest of the branches. To determine whether HA was significantly different from H0 for each of the codon-based alignments, we used the likelihood ratio test with a statistical significance threshold of α = 0.01.
To determine the rate of sequence evolution in non-coding region alignments between A. fumigatus and close relatives, we examined variation in the ratio of the rate of substitutions in each non-coding region (dNC) to the rate of synonymous (dS) substitutions in its corresponding protein-coding region (dNC/dS or ζ) across the phylogeny. Like the analysis of the protein-coding regions, the null hypothesis (H0) was that all branches of the phylogeny exhibit the same estimated ζ value. We compared H0 to an alternative hypothesis (HA) which allows for the branch leading to A. fumigatus to have a distinct estimated ζ value from the rest of the branches. ζ values were calculated under the different hypotheses using HyPhy version 2.2.2 (Pond et al., 2004) with the “nonCodingSelection.bf” batch file as established by Oliver Fedrigo (Haygood et al., 2007; Fedrigo et al., 2011). To determine whether HA was significantly different from H0 for each of the non-coding region alignments, we used the likelihood ratio test with a statistical significance threshold of α = 0.01.
To determine whether genes with signatures of different evolutionary rates in either their protein-coding or non-coding regions are enriched for particular functional categories, we implemented the Gene Ontology (GO) Term Finder webtool on AspGD (Cerqueira et al., 2013) using default settings. We conducted two separate analyses. The first examined those A. fumigatus genes that exhibited a different evolutionary rate in their non-coding regions, whereas the second examined those A. fumigatus genes with a different evolutionary rate in their protein-coding regions. These gene sets were compared to a general background set that includes all the features/gene names in the database with at least one GO annotation for A. fumigatus. Both analyses used a p-value cutoff of 0.05.
To identify interesting examples of sequence variation between A. fumigatus and the other species for non-coding regions of genes of interest, we visualized and compared multiple sequence alignments using the MView function in EMBL-EBI (Madeira et al., 2019). Workflow of methods can be seen in Figure S1.
To analyze the sequence diversity of non-coding regions in section Fumigati, we first identified 5,215 single-copy orthologous genes amongst ten strains/species in the section (Figure 1A). We then computed the percent similarity between the non-coding and protein-coding regions of each A. fumigatus Af293 ortholog and their homologous non-coding and protein-coding regions in the other nine taxa (Tables S3, S4). Those individual percent similarities were then averaged to get the final percent similarity for the non-coding and protein-coding regions of that ortholog. Averaging the non-coding region percent similarities for the 5,215 single-copy orthologous genes revealed an average similarity of ~72%; 648 alignments exhibited <50% similarity, 3,665 exhibited sequence similarity between ≥50% and <75%, and 902 exhibited ≥75% similarity. Interestingly, three genes exhibited > 90% similarity. These genes were cnaB, whose protein product is a calcineurin regulatory subunit and whose transcript is induced by exposure to human airway epithelial cells (Juvvadi et al., 2011; Oosthuizen et al., 2011), AFUA_6G07800, which is predicted to be a transcription factor with unknown function (Cerqueira et al., 2013), and AFUA_6G04530, which is predicted to have a role in histone acetylation (Cerqueira et al., 2013).
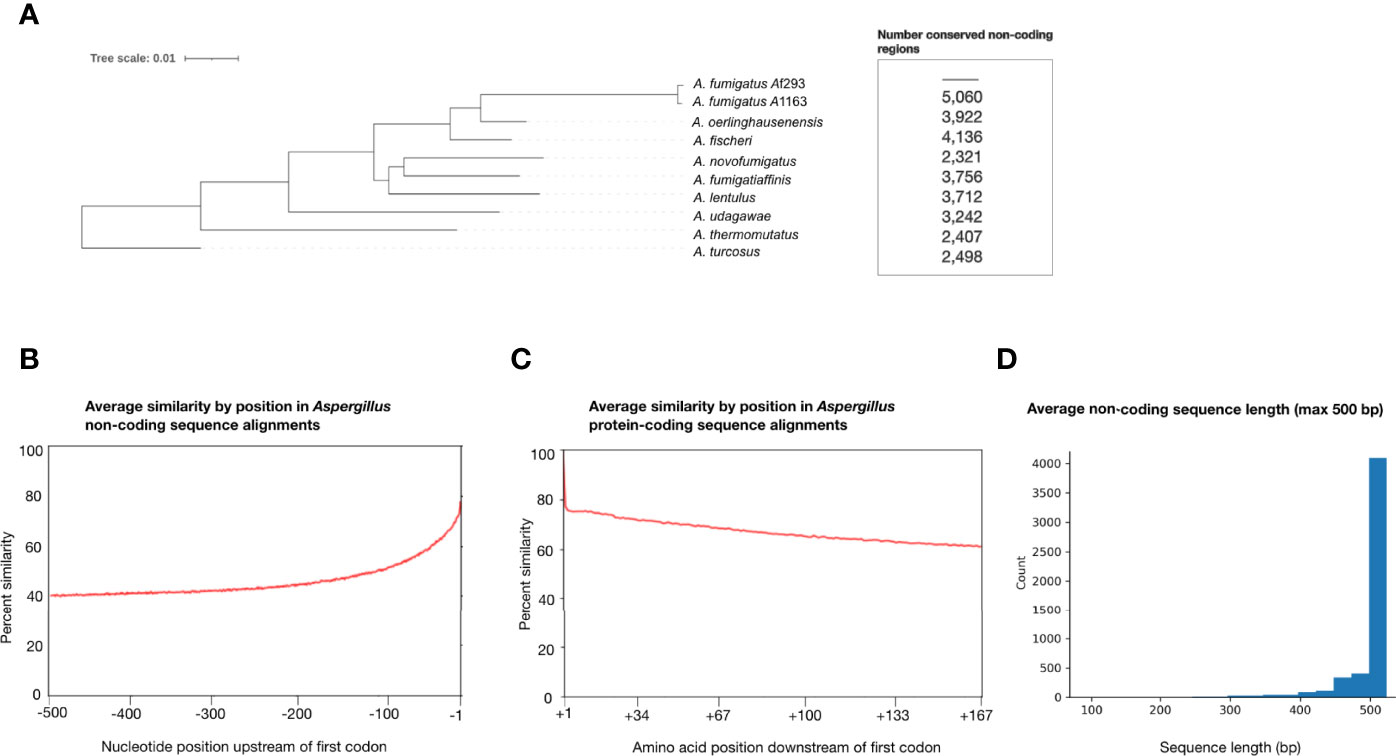
Figure 1 Aspergillus section Fumigati species exhibit sequence variation in non-coding regions that are 500 base pairs upstream of genes’ first codon. (A) Species phylogeny of two A fumigatus reference strains (Af293 and A1163) and closely related Aspergillus section Fumigati species constructed from concatenation analysis of a 5,215-gene data matrix. Branch lengths correspond to nucleotide substitutions/site. Note the long branch leading to A fumigatus, indicative of a greater number of nucleotide substitutions per site in this species. The number of genes whose non-coding regions were conserved (≥ 75% sequence similarity between each species and ≥ 500 bp in length) between A. fumigatus Af293 and each species are shown next to the corresponding taxa. (B) Average percent sequence similarity of non-coding regions of 5,215 genes by position, relative to the gene’s first codon. Sequence alignments of non-coding regions were compared by position and the average percent similarities for each site are reported with -1 indicating the site directly upstream of the first codon and -500 indicating the site 500 bp upstream of the first codon. (C) Average percent sequence similarity by position of the first 167 amino acid sites in the alignments of 5,215 genes, relative to the gene’s first codon. The average percent similarity for each site is reported, with +1 indicating the first amino acid. (D) Average sequence alignment lengths of the 5,215 non-coding regions examined in this study. 4,079 of the 5,215 non-coding regions have ≥ 500 bp in all 10 strains/species used in this study.
Average percent similarity by position in Aspergillus non-coding region alignments (Figure 1B) revealed that the percent similarity directly upstream of the first codon (-1 bp upstream) is higher than 60% and decreases as the distance from the first codon increases, approaching 40% similarity. This result suggests that potentially conserved promoter and cis-regulatory elements occur in these non-coding regions and is consistent with transcription factor binding location in A. fumigatus (Chung et al., 2014; de Castro et al., 2014). For comparison, we also calculated the average percent similarity by position in the protein-coding region alignments of all 5,215 genes (Figure 1C). We found that the percent similarity of the first amino acid (+1) was ~100%, indicative of the first methionine; similarity was high throughout the first 167 sites of amino acid alignment but decreased as the distance from the first amino acid increased, approaching 60% amino acid sequence similarity.
Examination of the 4,567 genes whose non-coding region alignments exhibited ≥50% similarity (i.e., the 3,665 genes whose similarity was ≥50% and <75%, and the 902 that had ≥75% similarity) revealed several instances in which one or more sequences were poorly aligned for stretches of 100 bp or more. This was especially true when sequences in these alignments exhibited large variation in their lengths. Further, we found a low level of synteny as genes immediately upstream of a non-coding region of interest generally differed between species.
We also determined the number of conserved non-coding regions with ≥75% sequence similarity and that were ≥500 bp in length between A. fumigatus Af293 and each of the other nine taxa, separately (Figure 1). We found that A. novofumigatus shares the fewest number of conserved non-coding regions (2,321) despite being more closely related to A. fumigatus than other species included in our phylogeny (Steenwyk et al., 2009; Houbraken et al., 2014; Rokas et al., 2020); this suggests that the quality of annotations may differ across the ten genomes examined and that improvements in the gene annotation of these genomes could increase the number of conserved non-coding regions shared by these taxa. A. fumigatus A1163 shared the greatest number of conserved non-coding regions with A. fumigatus Af293 (5,020). With the exceptions of A. novofumigatus, A. oerlinghausenensis, and A. thermomutatus, the closer a relative is to A. fumigatus Af293, the greater the number of conserved non-coding regions that are shared.
To help determine if differences existed between non-coding and protein-coding regions across our species, we first compared total branch lengths in phylogenetic trees constructed from both non-coding and protein-coding regions from all 5,215 genes (Figure S2). Comparisons of the overall distributions between these two groups revealed a statistically significant difference between the overall branch lengths of protein-coding and non-coding regions (Wilcoxon signed-ranked test; p-value = 0.004), suggesting that non-coding regions of single-copy orthologs evolve faster than protein-coding regions.
Given the uncertainty regarding the homology of some sequences in these 5,215 non-coding region alignments and our finding that most sequence conservation was found near the first codon position, we focused our evolutionary rate analyses only on the 732 non-coding region alignments whose sequences were all ≥500 bp long and exhibited ≥75% sequence similarity between A. fumigatus Af293 and each other strain/species in the phylogeny. Briefly, to determine the rate of sequence evolution in protein-coding and non-coding region alignments between A. fumigatus and close relatives, we examined variation in the ratio of the rate of nonsynonymous (dN) to the rate of synonymous (dS) substitutions (ω value) for protein-coding regions and the variation in the ratio of the rate of non-coding (dNC) to the rate of synonymous (dS) substitutions (dNC/dS or ζ value) for non-coding regions across the phylogeny. To test whether the molecular evolutionary rates of protein-coding and non-coding regions differed between the major pathogen A. fumigatus and its relatives, we statistically examined whether protein-coding and non-coding A. fumigatus sequences evolved at a similar (H0) or different (HA) rate as those of other taxa (Figure 2).
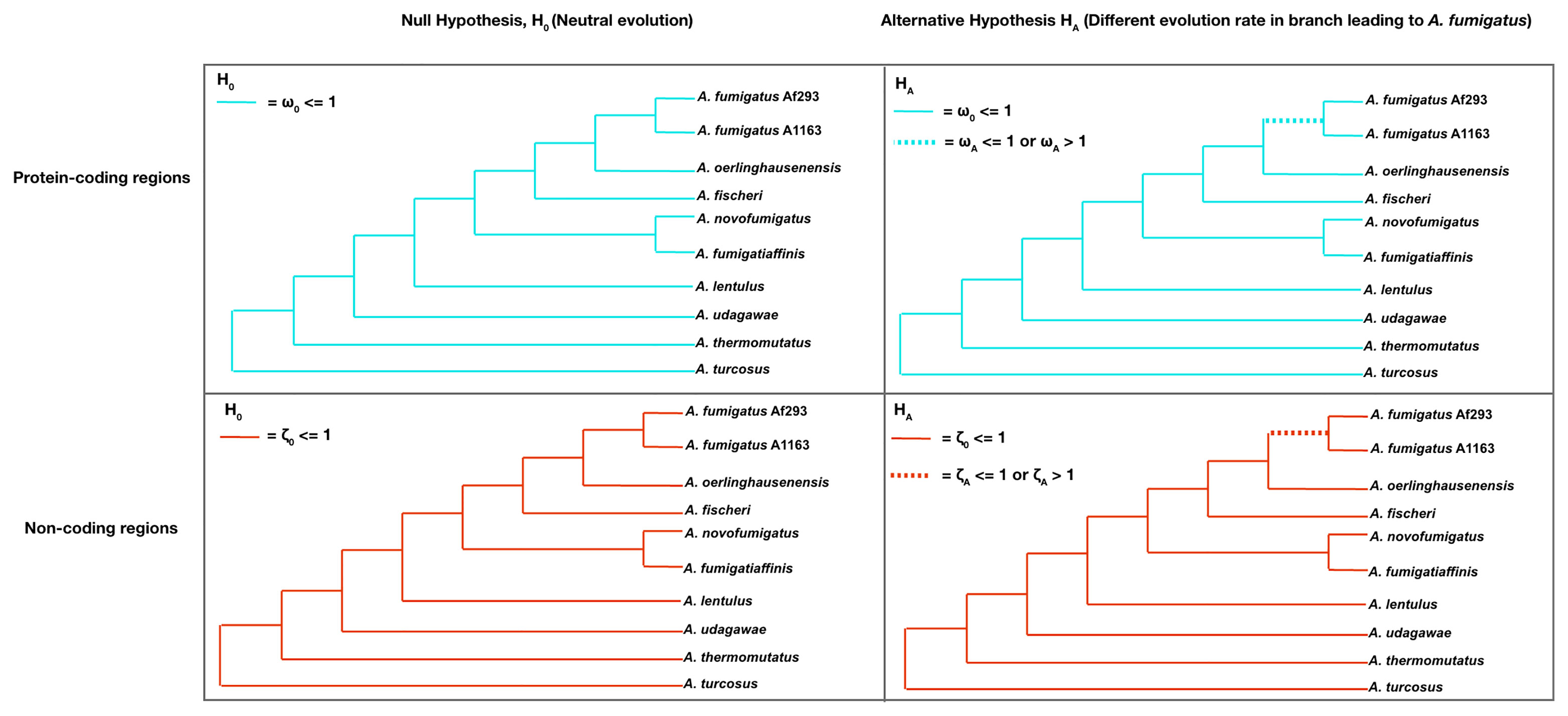
Figure 2 Examining whether the non-coding and protein-coding regions of 732 genes have different evolutionary rates in the major pathogen A. fumigatus. The top two panels present the null and alternative hypotheses for evolutionary rate difference in the protein-coding regions of A. fumigatus genes relative to the other species. The null hypothesis (H0, upper left) constrains all ω (dN/dS) values across all branches to be less than or equal to 1, the neutral evolutionary rate. The alternative hypothesis (HA, upper right) allows the branch leading to A. fumigatus (dashed branch) to have an ω value lower than, equal to, or greater than 1 (indicative of evolutionary rate difference) compared to the background branches. The bottom two panels present the null and alternative hypotheses for evolutionary rate difference in the non-coding regions of A. fumigatus genes relative to other species. Similarly, the null hypothesis (H0, bottom left) constrains all ζ (dNC/dS) values across all branches to less than or equal to 1. The alternative hypothesis (HA, bottom right) allows for the branch leading to A. fumigatus (dashed branch) to have a ζ value lower than, equal to, or greater than 1 (evolutionary rate difference) compared to the background branches. For each protein-coding and non-coding region, a likelihood ratio test was used to determine which hypothesis best fits the data.
Examination of protein-coding regions identified 100/732 genes (Table S5) (13.7% of examined genes) that significantly rejected H0 (under which all branches exhibited the same ω value) (Figure 3A) over HA (which postulates that the ω value of the branch leading to A. fumigatus was distinct from the background ω value of all other branches) (Figure 3B). Examination of non-coding regions identified 418/732 genes (Table S6) (57.1% of examined genes) that significantly rejected H0 (under which all branches exhibited the same ζ value) over HA (which postulates that the ζ value of the branch leading to A. fumigatus was distinct from the background ζ value of all other branches) (Figure 3C). Taken together, these results suggest a much higher amount of variation in non-coding regions than in protein-coding regions between A. fumigatus and relatives. The p-value distribution of protein-coding regions is uniform, while the p-value distribution of non-coding regions is bimodal with nearly all p-values being either under 0.05 or 1.0. This result suggests that the 418 non-coding regions exhibited major differences in their relative fit for the two hypotheses, whereas protein-coding regions exhibited much smaller differences.
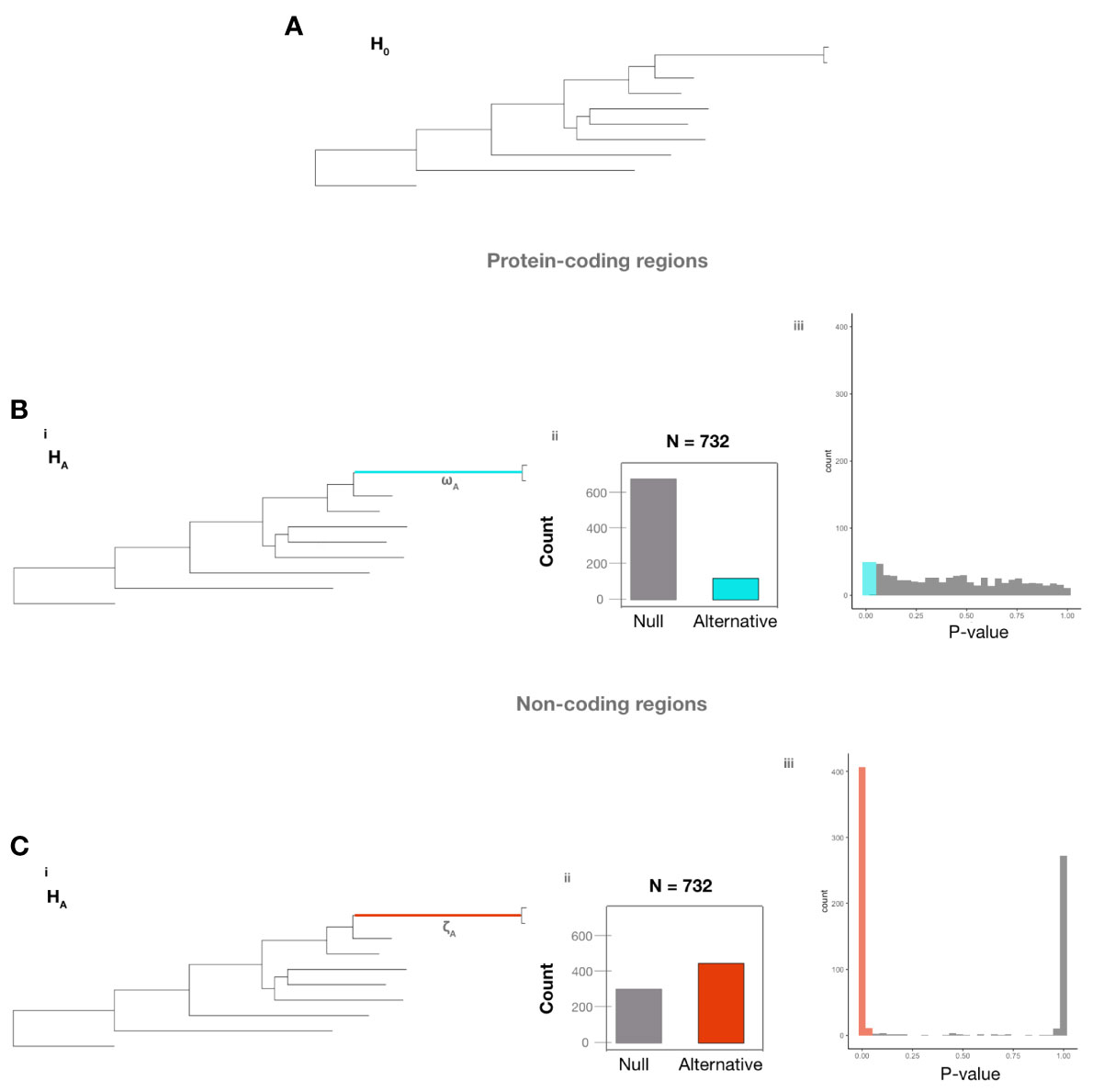
Figure 3 Non-coding regions of A. fumigatus genes exhibit many more signatures of evolutionary rate difference than their corresponding protein-coding regions. (A) The null hypothesis (H0) that all branches have the same evolutionary rate. (B, C) The alternative hypotheses assume that the ω value (Bi) or the ζ value (Ci) in the branch leading to A. fumigatus differs from the value in the rest of the branches of the phylogeny. Bii. 632 of 732 protein-coding regions (84.34%) did not reject H0 (gray) and 100 of 732 (16.66%) rejected H0 (blue). Biii. The distribution of p-values for protein-coding regions that did not (gray) and did (blue) reject H0. Cii. 314 of 732 non-coding regions (42.90%) did not reject H0 (gray) and 418 of 732 non-coding regions (57.10%) rejected H0 (red), which suggests a greater amount of variation in non-coding regions than in protein-coding regions between A. fumigatus and relatives. Ciii The distribution of p-values for non-coding regions that did not (gray) and did (red) reject H0.
To identify functions that were over-represented in the list of genes that rejected H0 for either their coding or non-coding regions, we conducted gene ontology (GO) enrichment analyses. Examination of significantly over-represented GO terms for the 418 genes (Table S7) with signatures of different evolutionary rates in non-coding regions revealed numerous biological processes related to regulation, metabolism, and development (e.g., “cellular component organization or biogenesis”, p = 0.00016; “regulation of protein metabolic process”, p = 0.00101; “hyphal growth”, p = 0.00352; “regulation of cell cycle”; p = 0.0076; “developmental process”, p = 0.01233; “reproduction”. p = 0.00361). Of the 418 genes, 71 lacked any functional GO annotation. In comparison, for the 314 genes that did not exhibit a different evolutionary rate in their non-coding regions (Table S8), the only term that was enriched was “nucleotide binding” (p = 0.06129) and was found associated with 48 genes. For the 100 genes with signatures of different evolutionary rate in their protein-coding regions, only one function was found enriched (“regulation of cellular process”, p = 0.02647). Of note, 74 of the protein-coding genes lacked any functional GO annotation (Table S9).
To identify if any of the A. fumigatus genes with different evolutionary rates in their respective non-coding regions also contain known TF binding sites, we compared the list of 418 non-coding regions to binding sites of two TFs known to be genetic determinants of virulence, CrzA and SrbA (Cramer et al., 2008; Willger et al., 2008). ChIP-seq analysis of CrzA (de Castro et al., 2014) uncovered 110 genes that are directly bound by the TF in A. fumigatus strain Af293. Of these, 28 were reported to exhibit CrzA binding within 500 bp of the first codon, and two genes, AFUA_8G05090 (a putative MFS transporter) and AFUA_3G09960 (Aureobasidin resistance protein), exhibited a different evolutionary rate in the non-coding regions of A. fumigatus strains in our analysis. ChIP-seq analysis of SrbA (Chung et al., 2014) revealed 112 genes directly bound by the TF in A. fumigatus strain A1163. Of these, 57 were reported to exhibit SrbA binding within 500 bp of the first codon, and two genes, AFUB_074100 (a gene of unknown function which appears to interact with sldA, a checkpoint protein kinase) and AFUB_012300 (a gene predicted to be involved in nitrate assimilation), exhibited a different evolutionary rate in the non-coding regions of A. fumigatus strains in our analysis. Importantly, we found that for all four genes (AFUA_8G05090, AFUA_3G09960, AFUB_074100, and AFUB_012300) there was at least a 2 bp sequence difference between A. fumigatus and at least one close relative in their sequences at the TF binding site location (Figure 4). Together, our results suggest that intergenic non-coding regions that bind known TFs can exhibit substantial differences in their evolutionary rates between A. fumigatus and close relatives, which raises the hypothesis that these differences may lead to differences in gene expression.
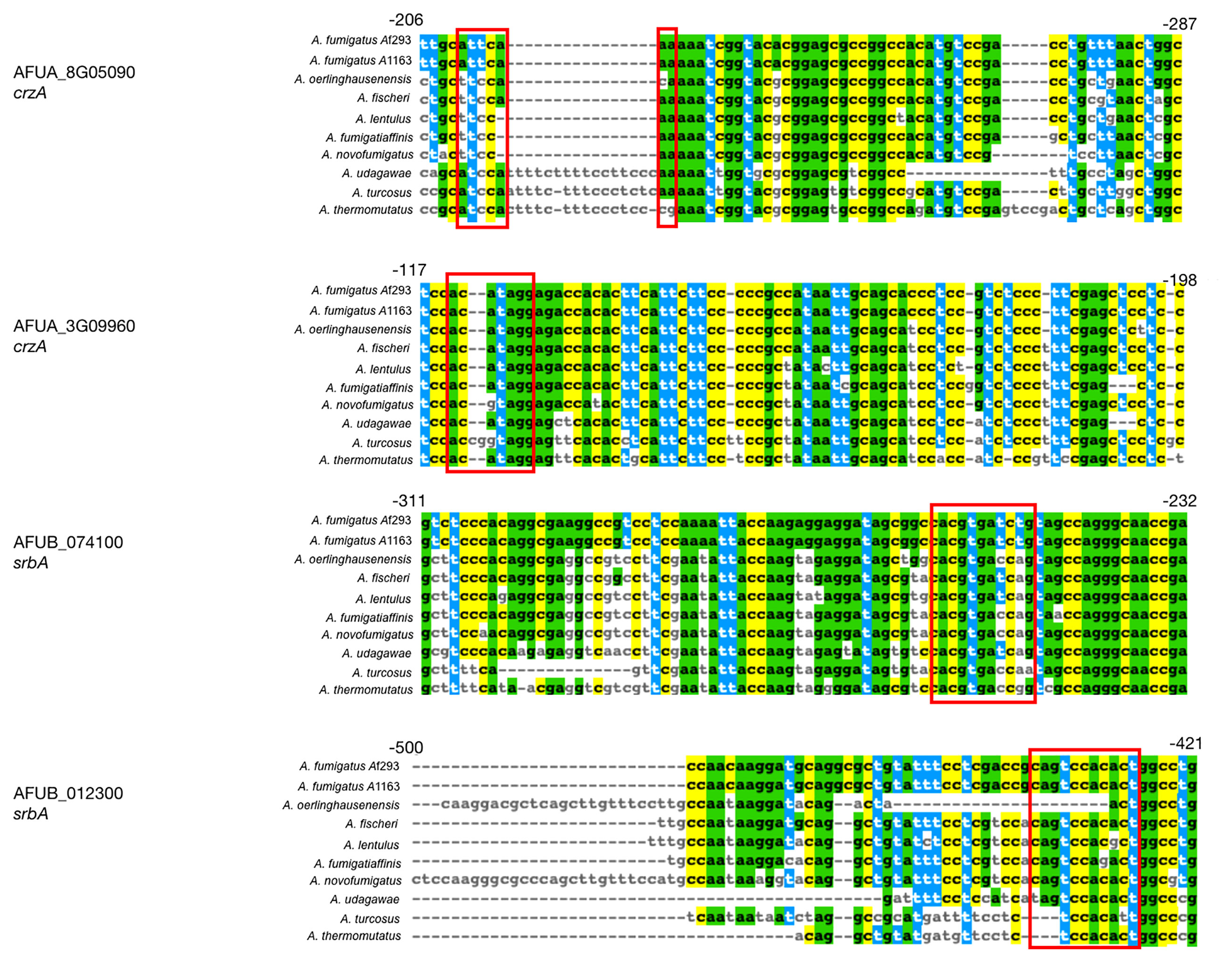
Figure 4 CrzA and SrbA binding locations in 4 genes that exhibit a different evolutionary rate in their non-coding regions. Two A. fumigatus genes (AFUA_8G05090 and AFUA_3G09960) known to bind CrzA in their non-coding regions and two A. fumigatus genes (AFUB_074100 and AFUB_012300) known to bind SrbA in their non-coding regions have at least a 2 bp difference in the TF binding site between A. fumigatus and one or more relatives. Red boxes represent the binding site locations found in previous ChIP-seq experiments.
We identified 25 genetic determinants of virulence whose non-coding regions exhibited a different rate of evolution in A. fumigatus (Table 1). Three genes (metR, his3, and met16) are involved in amino acid biosynthesis, eight genes (chsF, calA, gel2, nrps1, gfa1, csmB, rlmA, and rodA) are involved in cell wall biosynthesis, nine genes (noc3, spe2, gus1, pri1, AFUA_2G10600, mak5, pkaR, ramA, and somA) are involved in cellular metabolism, two genes (aspB and tom40) are involved in hyphal growth, and three genes (gliG, gliI, and gliJ) are involved in gliotoxin biosynthesis. Of the 25, 14 genes (metR, chsF, calA, gel2, nrps1, gfa1, csmB, rlmA, rodA, AFUA_2G10600, pkaR, ramA, somA, and aspB) have been shown to modulate virulence in an animal model of infectious disease. Three genes (gliG, gliI, and gliJ) are required for the biosynthesis of gliotoxin, a secondary metabolite involved in A. fumigatus virulence (Brakhage and Langfelder, 2002; Dagenais and Keller, 2009), and deletions of the eight remaining genes (his3, met16, noc3, spe2, gus1, pri1, mak5, and tom40) have been previously shown to be important for viability and therefore, likely virulence (for a detailed list, see: Steenwyk et al., 2021a). Interestingly, only two of these 25 genes (csmB and rodA) exhibit a signature of different evolutionary rate in their protein-coding region as well, a finding consistent with our result that there are more changes in non-coding regions than in protein-coding regions of Aspergillus genes.
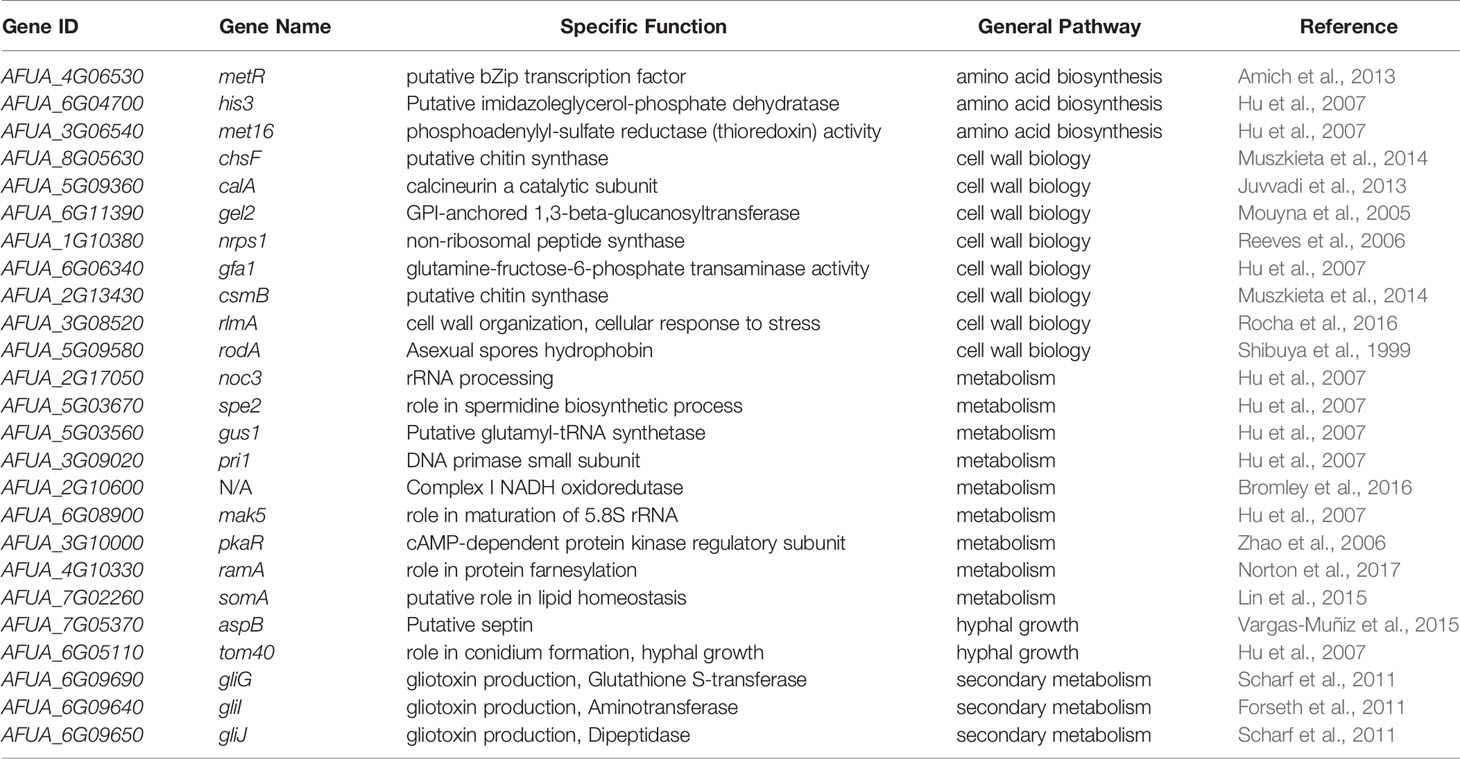
Table 1 Twenty-five genetic determinants of A. fumigatus virulence have a different evolutionary rate in their non-coding regions.
Examination of sequence alignments of non-coding regions of these 25 genes (Table 1) revealed several interesting patterns (Figure 5). For example, the non-coding region of pkaR exhibits a 10 bp stretch from -434 bp to -424 bp upstream of the first codon, which is deleted exclusively in A. fumigatus and present and largely conserved in all other species (Figure 5A). The non-coding region of gfa1 also has a stretch of 5 bp exclusively deleted in A. fumigatus and present in all other species. Sequence alignment of the gliG non-coding region revealed an 11 bp G-rich insertion that is unique to the two A. fumigatus strains (Figure 5B). In addition to A. fumigatus-specific indels, we also observed that the non-coding regions of several genes known to be involved in A. fumigatus virulence exhibited indel variation across the other Aspergillus species examined as well. For example, the non-coding region of metR exhibits a 7 bp pyrimidine rich insertion that is found only in A. fumigatus, A. oerlinghausenensis, and A. lentulus (Figure 5C), while the non-coding regions of calA and pri1 both have small sequences (12 bp and 5 bp, respectively) that are exclusively absent in A. fumigatus strains Af293 andA1163, and in A. oerlinghausenensis.

Figure 5 Notable examples of sequence differences between A fumigatus and its close relatives in non-coding regions located upstream of three known genetic determinants of A fumigatus virulence that exhibited signatures of a different evolutionary rate. (A) pkaR encodes a regulatory subunit involved in the regulation of the cyclic AMP – dependent protein kinase pathway; deletion of pkaR in A fumigatus has been shown to attenuate virulence in a neutropenic mouse model (Zhao et al., 2006). The sequence alignment of the non-coding region of the pkaR gene exhibits a 10 bp region (red box) that is uniquely deleted in A fumigatus but present in all other close relatives. (B) gliG encodes for a glutathione S-transferase (GST) that is part of the gliotoxin biosynthetic gene cluster and is required for gliotoxin production (Scharf et al., 2011). Gliotoxin contributes to the virulence of A fumigatus, inactivating host vital proteins via conjugation (Scharf et al., 2011). The sequence alignment of the non-coding region upstream of the gliG gene exhibits a 15 bp G-rich region that has been inserted in A fumigatus and is absent from all other species. (C) metR encodes for a TF involved in sulfur assimilation and is a known A fumigatus virulence factor (Amich et al., 2013). The sequence alignment of the non-coding region of the metR gene contains a 7 bp region that is absent only in A fumigatus, A oerlinghausenensis, and A lentulus. Colored nucleotides represent sites that are present in A fumigatus Af293 and shared between other species, highlighting differences between the reference strain and close relatives.
We identified a set of 732 genes whose non-coding regions were conserved between the genomes of reference strain A. fumigatus Af293, the more virulent A. fumigatus reference strain A1163, and eight closely related species. In these 732 genes, we also tested whether the branch leading to A. fumigatus exhibited a difference in the evolutionary rate in either its protein-coding or its non-coding regions compared to the other species. We found that the non-coding regions of 418 of these genes exhibit signatures of a different evolutionary rate in A. fumigatus. These 418 genes include 25 that are known genetic determinants of A. fumigatus virulence (Steenwyk et al., 2021b) (Table 1). Given the differences in reported invasive aspergillosis cases caused by A. fumigatus compared to other Aspergillus species (Steinbach et al., 2012), genetic differences in the non-coding regions of these 418 genes, and especially of these 25 genes previously connected to virulence, may play a role in varying pathogenic potentials of Aspergillus section Fumigati species.
Gene Ontology (GO) analysis of the 418 genes which exhibit signatures of a different evolutionary rate in non-coding regions revealed an enrichment for genes involved in regulation of metabolism and development. This is consistent with previous studies of the evolution of non-coding regions in humans (Haygood et al., 2007), which experienced positive selection in non-coding regions for genes involved in metabolism regulation (particularly glucose metabolism) and regulation of development (particularly the nervous system) compared to close relatives. These results raise the possibility that non-coding regions associated with particular functions in diverse taxa are more likely to experience changes in their evolutionary rates.
We compared our list of genes with signatures of different evolutionary rates with previous ChIP-seq studies of the transcription factors CrzA and SrbA, both of which are well studied genetic determinants of virulence for A. fumigatus (Cramer et al., 2008; Willger et al., 2008; Chung et al., 2014; de Castro et al., 2014; Colabardini et al., 2022). We found that the non-coding regions of two genes bound by CrzA (AFUA_8G05090 and AFUA_3G09960) and two genes bound by SrbA (AFUB_074100 and AFUB_012300) also exhibited different evolutionary rates in A. fumigatus. We found several nucleotide differences in these non-coding regions that likely contributed to the observed differences in evolutionary rate. Interestingly, when we examined the sequence alignments of the non-coding regions of these genes, we found differences at the TF binding sites (TFBS) between A. fumigatus and relatives. We hypothesize that the different evolutionary rate we observed in these A. fumigatus genes are due, in part, to changes in the associated TFBS, which may influence the regulation of these genes.
Comparisons between Saccharomyces cerevisiae and Saccharomyces paradoxus non-coding regions that were similar to the ones we report here, revealed that TFBS tend be more conserved in the proximal promoter region (within 200bp of the transcription start site) than the distal region, yet some differences in TFBS were reported between the two species in their respective proximal promoter regions (Schaefke et al., 2015). Evolutionary differences near the transcription start site have also been reported in Drosophila species, with certain species (such as Drosophila pseudoobscura) exhibiting an increased mutation rate upstream of the transcription start site when compared to Drosophila melanogaster (Main et al., 2013). Combined with the results presented here, it is likely that the evolution of non-coding regions is not uniform across closely related species and that these differences may play a functional role in downstream gene expression.
We compared our list of 418 genes with signatures of a different evolutionary rate in the non-coding regions of A. fumigatus to a previously curated set of 206 genetic determinants of A. fumigatus virulence (Steenwyk et al., 2021a) and found that 25 of the 206 exhibited a different evolutionary rate their non-coding regions between A. fumigatus and close relatives (Table S6). We found that the most represented general function amongst these 25 genes was “metabolism”, which raises the question of their impact on virulence, given the role that metabolism has been shown to play in A. fumigatus virulence (Willger et al., 2009). In particular, pkaR is essential for proper protein kinase A signaling (Griffioen and Thevelein, 2002) and plays a key role in the germination and growth of A. fumigatus asexual spores (Zhao et al., 2006). Moreover, pkaR has been shown to be required for A. fumigatus virulence (Lin et al., 2015) in an immunocompromised murine model of invasive aspergillosis (Fuller et al., 2011). We found that the protein-coding region of pkaR does not exhibit a different evolutionary rate in A. fumigatus, suggesting that it is conserved. However, analysis of the sequence alignment of the non-coding region revealed a 11 bp region (CAACTTCTTT) absent in A. fumigatus but present in all other species (Figure 5). Interestingly, this binding site is similar to the predicted TFBS for the S. cerevisiae TF Ste12 (Badis et al., 2008), a homolog to SteA in A. fumigatus. While it has yet to be elucidated if this region is involved in SteA binding, it may be that its absence changed the regulation of pkaR and thus somehow contributed to the evolution of A. fumigatus virulence.
The role of gliotoxin in A. fumigatus-mediated disease has been of increasing interest, due to its ability to inhibit the host immune response (Raffa and Keller, 2019). However, the gliotoxin biosynthetic gene cluster is found in both A. fumigatus and its non-pathogenic close relatives A. oerlinghausenensis and A. fischeri, and all three species are known to produce gliotoxin (Knowles et al., 2020; Steenwyk et al., 2020b). Here, we identify that three genes in the gliotoxin biosynthetic gene cluster (gliG, gliJ, gliI) exhibit a different evolutionary rate in their non-coding regions in A. fumigatus. Gliotoxin genes have been shown to require certain TFs (GliZ and RglT for example) for gliotoxin biosynthesis and/or self-protection (Schrettl et al., 2010; Ries et al., 2020; de Castro et al., 2022). Interestingly, analysis of the sequence alignment of the non-coding region of gliG revealed a G-rich region unique to A. fumigatus (Figure 5). G-rich regions have been previously reported to be found in biologically active sites and to play important roles in regulating cellular processes such as gene expression (Maizels and Gray, 2013; Maity et al., 2020). This A. fumigatus-specific G-rich region may contribute to some unknown gliotoxin expression pattern that contributes to A. fumigatus virulence or the lack of disease caused by other closely related Aspergillus species.
metR encodes a bZIP DNA binding protein required for sulfur metabolism in A. fumigatus and whose gene expression is regulated by LaeA, a major regulator of secondary metabolism (Jain et al., 2018. Pertaining to A. fumigatus virulence, sulfur assimilation plays key roles in oxidative stress response and gliotoxin biosynthesis (Traynor et al., 2019). Recent efforts have identified differences in the transcriptional profiles of A. fumigatus and relatives in response to exogenous gliotoxin, highlighting the pathways relating sulfur assimilation and gliotoxin production (de Castro et al., 2022). The non-coding region of metR contains a 7 bp region (TCACCT) in A. fischeri and five other species; in contrast, the two strains of A. fumigatus, A. oerlinghausenensis and A. lentulus all lack this 7 bp motif (Figure 5). While it remains unclear if this 7 bp motif has a functional role in the expression of metR, this result nicely illustrates the complex patterns of sequence evolution of non-coding regions in this clade of pathogens and non-pathogens.
A major outstanding question emanating from our work is whether this extensive non-coding sequence variation of closely related Aspergillus species that vary in their pathogenicity functionally contributes to differences in gene expression between strains and species. Currently, there are no datasets available that report genome-wide differential expression data for A. fumigatus and close relatives; to our knowledge, the only published differential expression study of A. fumigatus and close relatives focused on expression differences only for genes involved in secondary metabolism (Takahashi et al., 2021). Designing and performing differential gene expression experiments in diverse Aspergillus species will be a future aim. Additional future work will include functionally test if the non-coding region differences we report here play a role in A. fumigatus expression and virulence. Further, testing if non-coding regions in a larger set of A. fumigatus strains exhibit differences in evolutionary rates would help to elucidate more recent evolutionary changes in A. fumigatus and the pathogenic differences observed in these strains as well.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
AB, MM, and AR designed the study and its experiments. AB performed the experiments. JS generated the species phylogeny. AB and AR wrote the manuscript. All authors commented on the manuscript and contributed to its revision.
AB was funded by the Biological Sciences graduate program at Vanderbilt University. Research in AR’s lab is supported by grants from the National Science Foundation (DEB-1442113 and DEB-2110404), the National Institutes of Health/National Institute of Allergy and Infectious Diseases (R56AI146096 and R01AI153356), and the Burroughs Wellcome Fund. JS and AR were supported by the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program.
Author AR is a scientific consultant for LifeMine Therapeutics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Rokas lab for helpful discussion and feedback.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffunb.2022.802494/full#supplementary-material
Amich J., Schafferer L., Haas H., Krappmann S. (2013). Regulation of Sulphur Assimilation Is Essential for Virulence and Affects Iron Homeostasis of the Human-Pathogenic Mould Aspergillus Fumigatus. PloS Pathog. 9, e1003573. doi: 10.1371/journal.ppat.1003573
Badis G., Chan E. T., van Bakel H., Pena-Castillo L., Tillo D., Tsui K., et al. (2008). A Library of Yeast Transcription Factor Motifs Reveals a Widespread Function for Rsc3 in Targeting Nucleosome Exclusion at Promoters. Mol. Cell 32, 878–887. doi: 10.1016/j.molcel.2008.11.020
Bertuzzi M., Hayes G. E., Icheoku U. J., van Rhijn N., Denning D. W., Osherov N., et al. (2018). Anti-Aspergillus Activities of the Respiratory Epithelium in Health and Disease. J. Fungi. (Basel). 4, E8. doi: 10.3390/jof4010008
Bongomin F., Gago S., Oladele R. O., Denning D. W. (2017). Global and Multi-National Prevalence of Fungal Diseases—Estimate Precision. J. Fungi. (Basel). 3, 57. doi: 10.3390/jof3040057
Brakhage A. A., Langfelder K. (2002). Menacing Mold: The Molecular Biology of Aspergillus Fumigatus. Annu. Rev. Microbiol. 56, 433–455. doi: 10.1146/annurev.micro.56.012302.160625
Bromley M., Johns A., Davies E., Fraczek M., Gilsenan J. M., Kurbatova N., et al. (2016). Mitochondrial Complex I Is a Global Regulator of Secondary Metabolism, Virulence and Azole Sensitivity in Fungi. PloS One 11, e0158724. doi: 10.1371/journal.pone.0158724
Buchfink B., Xie C., Huson D. H. (2015). Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Caron B., Luo Y., Rausell A. (2019). NCBoost Classifies Pathogenic non-Coding Variants in Mendelian Diseases Through Supervised Learning on Purifying Selection Signals in Humans. Genome Biol. 20, 32. doi: 10.1186/s13059-019-1634-2
Carroll S. B. (2008). Evo-Devo and an Expanding Evolutionary Synthesis: A Genetic Theory of Morphological Evolution. Cell 134, 25–36. doi: 10.1016/j.cell.2008.06.030
Cerqueira G. C., Arnaud M. B., Inglis D. O., Skrzypek M. S., Binkley G., Simison M., et al. (2013). The Aspergillus Genome Database: Multispecies Curation and Incorporation of RNA-Seq Data to Improve Structural Gene Annotations. Nucleic Acids Res. 42, D705–D710. doi: 10.1093/nar/gkt1029
Chotirmall S. H., Al-Alawi M., Mirkovic B., Lavelle G., Logan P. M., Greene C. M., et al. (2013). Aspergillus-Associated Airway Disease, Inflammation, and the Innate Immune Response. BioMed. Res. Int. 2013, 723129. doi: 10.1155/2013/723129
Chung D., Barker B. M., Carey C. C., Merriman B., Werner E. R., Lechner B. E., et al. (2014). ChIP-Seq and In Vivo Transcriptome Analyses of the Aspergillus Fumigatus SREBP SrbA Reveals a New Regulator of the Fungal Hypoxia Response and Virulence. PloS Pathog. 10, e1004487. doi: 10.1371/journal.ppat.1004487
Cock P. J. A., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., et al. (2009). Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Colabardini A. C., Wang F., Dong Z., Pardeshi L., Rocha M. C., Costa J. H., et al. (2022). Heterogeneity in the Transcriptional Response of the Human Pathogen Aspergillus Fumigatus to the Antifungal Agent Caspofungin. Genetics 220, iyab183. doi: 10.1093/genetics/iyab183
Cramer R. A., Perfect B. Z., Pinchai N., Park S., Perlin D. S., Asfaw Y. G., et al. (2008). Calcineurin Target CrzA Regulates Conidial Germination, Hyphal Growth, and Pathogenesis of Aspergillus Fumigatus. Eukaryot. Cell 7, 1085–1097. doi: 10.1128/EC.00086-08
Croft C. A., Culibrk L., Moore M. M., Tebbutt S. J. (2016). Interactions of Aspergillus Fumigatus Conidia With Airway Epithelial Cells: A Critical Review. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00472
Dagenais T. R. T., Keller N. P. (2009). Pathogenesis of Aspergillus Fumigatus in Invasive Aspergillosis. Clin. Microbiol. Rev. 22, 447–465. doi: 10.1128/CMR.00055-08
de Castro P. A., Chen C., de Almeida R. S. C., Freitas F. Z., Bertolini M. C., Morais E. R., et al. (2014). ChIP-Seq Reveals a Role for CrzA in the Aspergillus Fumigatus High-Osmolarity Glycerol Response (HOG) Signalling Pathway. Mol. Microbiol. 94, 655–674. doi: 10.1111/mmi.12785
de Castro P. A., Moraes M., Colabardini A. C., Horta M. A.C., Knowles S. L., Raja H. A., et al. (2022). Regulation of Gliotoxin Biosynthesis and Protection in Aspergillus Species. PLOS Genetics 18, e1009965. doi: 10.1371/journal.pgen.1009965
de Vries R. P., Riley R., Wiebenga A., Aguilar-Osorio G., Amillis S., Uchima C. A., et al. (2017). Comparative Genomics Reveals High Biological Diversity and Specific Adaptations in the Industrially and Medically Important Fungal Genus Aspergillus. Genome Biol. 18, 28. doi: 10.1186/s13059-017-1151-0
dos Santos R. A. C., Steenwyk J. L., Rivero-Menendez O., Mead M. E., Silva L. P., Bastos R. W., et al. (2020). Genomic and Phenotypic Heterogeneity of Clinical Isolates of the Human Pathogens Aspergillus fumigatus, Aspergillus lentulus and Aspergillus fumigatiaffinis. Front. Genet. 11, 459. doi: 10.3389/fgene.2020.00459
Eide D. J. (2020). Transcription Factors and Transporters in Zinc Homeostasis: Lessons Learned From Fungi. Crit. Rev. Biochem. Mol. Biol. 55, 88–110. doi: 10.1080/10409238.2020.1742092
Emms D. M., Kelly S. (2015). OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Fedorova N. D., Khaldi N., Joardar V. S., Maiti R., Amedeo P., Anderson M. J., et al. (2008). Genomic Islands in the Pathogenic Filamentous Fungus Aspergillus Fumigatus. PloS Genet. 4, e1000046. doi: 10.1371/journal.pgen.1000046
Fedrigo O., Pfefferle A. D., Babbitt C. C., Haygood R., Wall C. E., Wray G. A. (2011). A Potential Role for Glucose Transporters in the Evolution of Human Brain Size. Brain Behav. Evol. 78, 315–326. doi: 10.1159/000329852
Flores M. E. B., Medina P. G., Camacho S. P. D., de Jesús Uribe Beltrán M., de la Cruz Otero M., Ramírez I. O., et al. (2014). Fungal Spore Concentrations in Indoor and Outdoor Air in University Libraries, and Their Variations in Response to Changes in Meteorological Variables. Int. J. Environ. Health Res. 24, 320–340. doi: 10.1080/09603123.2013.835029
Forseth R. R., Fox E. M., Chung D., Howlett B. J., Keller N. P., Schroeder F. C. (2011). Identification of Cryptic Products of the Gliotoxin Gene Cluster Using NMR-Based Comparative Metabolomics and a Model for Gliotoxin Biosynthesis. J. Am. Chem. Soc. 133, 9678–9681. doi: 10.1021/ja2029987
Fuller K. K., Richie D. L., Feng X., Krishnan K., Stephens T. J., Wikenheiser-Brokamp K. A., et al. (2011). Divergent Protein Kinase A Isoforms Co-Ordinately Regulate Conidial Germination, Carbohydrate Metabolism and Virulence in Aspergillus Fumigatus. Mol. Microbiol. 79, 1045–1062. doi: 10.1111/j.1365-2958.2010.07509.x
Furukawa T., van Rhijn N., Fraczek M., Gsaller F., Davies E., Carr P., et al. (2020). The Negative Cofactor 2 Complex is a Key Regulator of Drug Resistance in Aspergillus Fumigatus. Nat. Commun. 11, 427. doi: 10.1038/s41467-019-14191-1
Griffioen G., Thevelein J. M. (2002). Molecular Mechanisms Controlling the Localisation of Protein Kinase A. Curr. Genet. 41, 199–207. doi: 10.1007/s00294-002-0308-9
Harris C. R., Millman K. J., van der Walt S. J., Gommers R., Virtanen P., Cournapeau D., et al. (2020). Array Programming With NumPy. Nature 585, 357–362. doi: 10.1038/s41586-020-2649-2
Haygood R., Fedrigo O., Hanson B., Yokoyama K.-D., Wray G. A. (2007). Promoter Regions of Many Neural- and Nutrition-Related Genes Have Experienced Positive Selection During Human Evolution. Nat. Genet. 39, 1140–1144. doi: 10.1038/ng2104
Hoang D. T., Chernomor O., von Haeseler A., Minh B. Q., Vinh L. S. (2018). UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Houbraken J., de Vries R. P., Samson R. A. (2014). Chapter Four - Modern Taxonomy of Biotechnologically Important Aspergillus and Penicillium Species Advances in Applied Microbiology. 86, 199–249. doi: 10.1016/B978-0-12-800262-9.00004-4
Hubka V., Barrs V., Dudová Z., Sklenář F., Kubátová A., Matsuzawa T., et al. (2018). Unravelling Species Boundaries in the Aspergillus Viridinutans Complex (Section Fumigati): Opportunistic Human and Animal Pathogens Capable of Interspecific Hybridization. Persoonia 41, 142–174. doi: 10.3767/persoonia.2018.41.08
Hu W., Sillaots S., Lemieux S., Davison J., Kauffman S., Breton A., et al. (2007). Essential Gene Identification and Drug Target Prioritization in Aspergillus Fumigatus. PloS Pathog. 3, e24. doi: 10.1371/journal.ppat.0030024
Jain S., Sekonyela R., Knox B. P., Palmer J. M., Huttenlocher A., Kabbage M., et al. (2018). Selenate Sensitivity of a laeA Mutant is Restored by Overexpression of the bZIP Protein MetR in Aspergillus Fumigatus. Fungal Genet. Biol. 117, 1–10. doi: 10.1016/j.fgb.2018.05.001
Jang Y. J., LaBella A. L., Feeney T. P., Braverman N., Tuchman M., Morizono H., et al. (2018). Disease-Causing Mutations in the Promoter and Enhancer of the Ornithine Transcarbamylase Gene. Hum. Mutat. 39, 527–536. doi: 10.1002/humu.23394
Juvvadi P. R., Fortwendel J. R., Rogg L. E., Burns K. A., Randell S. H., Steinbach W. J. (2011). Localization and Activity of the Calcineurin Catalytic and Regulatory Subunit Complex at the Septum is Essential for Hyphal Elongation and Proper Septation in Aspergillus Fumigatus. Mol. Microbiol. 82, 1235–1259. doi: 10.1111/j.1365-2958.2011.07886.x
Juvvadi P. R., Gehrke C., Fortwendel J. R., Lamoth F., Soderblom E. J., Cook E. C., et al. (2013). Phosphorylation of Calcineurin at a Novel Serine-Proline Rich Region Orchestrates Hyphal Growth and Virulence in Aspergillus Fumigatus. PloS Pathog. 9, e1003564. doi: 10.1371/journal.ppat.1003564
Kim M. S., Cho K. H., Park K. H., Jang J., Hahn J.-S. (2019). Activation of Haa1 and War1 Transcription Factors by Differential Binding of Weak Acid Anions in Saccharomyces Cerevisiae. Nucleic Acids Res. 47, 1211–1224. doi: 10.1093/nar/gky1188
Knowles S. L., Mead M. E., Silva L. P., Raja H. A., Steenwyk J. L., Goldman G. H., et al. (2020). Gliotoxin, a Known Virulence Factor in the Major Human Pathogen Aspergillus Fumigatus, Is Also Biosynthesized by Its Nonpathogenic Relative Aspergillus Fischeri. mBio 11, e03361–e03319. doi: 10.1128/mBio.03361-19
Kusuya Y., Sakai K., Kamei K., Takahashi H., Yaguchi T. (2016). Draft Genome Sequence of the Pathogenic Filamentous Fungus Aspergillus Lentulus IFM 54703t. Genome Announc. 4, e01568–e01515. doi: 10.1128/genomeA.01568-15
Li H., Johnson A. D. (2010). Evolution of Transcription Networks — Lessons From Yeasts. Curr. Biol. 20, R746–R753. doi: 10.1016/j.cub.2010.06.056
Lin C.-J., Sasse C., Gerke J., Valerius O., Irmer H., Frauendorf H., et al. (2015). Transcription Factor SomA Is Required for Adhesion, Development and Virulence of the Human Pathogen Aspergillus Fumigatus. PloS Pathog. 11, e1005205. doi: 10.1371/journal.ppat.1005205
Madeira F., Park Y., Lee J., Buso N., Gur T., Madhusoodanan N., et al. (2019). The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Main B. J., Smith A. D., Jang H., Nuzhdin S. V. (2013). Transcription Start Site Evolution in Drosophila. Mol. Biol. Evol. 30, 1966–1974. doi: 10.1093/molbev/mst085
Maity A., Winnerdy F. R., Chang W. D., Chen G., Phan A. T. (2020). Intra-Locked G-Quadruplex Structures Formed by Irregular DNA G-Rich Motifs. Nucleic Acids Res. 48, 3315–3327. doi: 10.1093/nar/gkaa008
Maizels N., Gray L. T. (2013). The G4 Genome. PloS Genet. 9, e1003468. doi: 10.1371/journal.pgen.1003468
Mead M. E., Knowles S. L., Raja H. A., Beattie S. R., Kowalski C. H., Steenwyk J. L., et al. (2019). Characterizing the Pathogenic, Genomic, and Chemical Traits of Aspergillus fischeri, a Close Relative of the Major Human Fungal Pathogen Aspergillus Fumigatus. mSphere 4, e00018-19. doi: 10.1128/mSphere.00018-19
Mead M. E., Steenwyk J. L., Silva L. P., de Castro P. A., Saeed N., Hillmann F., et al. (2021). An Evolutionary Genomic Approach Reveals Both Conserved and Species-Specific Genetic Elements Related to Human Disease in Closely Related Aspergillus Fungi. Genetics 218, iyab066. doi: 10.1093/genetics/iyab066
Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., von Haeseler A., et al. (2020). IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mouyna I., Morelle W., Vai M., Monod M., Léchenne B., Fontaine T., et al. (2005). Deletion of GEL2 Encoding for a Beta(1-3)Glucanosyltransferase Affects Morphogenesis and Virulence in Aspergillus Fumigatus. Mol. Microbiol. 56, 1675–1688. doi: 10.1111/j.1365-2958.2005.04654.x
Muszkieta L., Aimanianda V., Mellado E., Gribaldo S., Alcàzar-Fuoli L., Szewczyk E., et al. (2014). Deciphering the Role of the Chitin Synthase Families 1 and 2 in the In Vivo and In Vitro Growth of Aspergillus Fumigatus by Multiple Gene Targeting Deletion. Cell Microbiol. 16, 1784–1805. doi: 10.1111/cmi.12326
Nierman W. C., Pain A., Anderson M. J., Wortman J. R., Kim H. S., Arroyo J., et al. (2005). Genomic Sequence of the Pathogenic and Allergenic Filamentous Fungus Aspergillus Fumigatus. Nature 438, 1151–1156. doi: 10.1038/nature04332
Norton T. S., Al Abdallah Q., Hill A. M., Lovingood R. V., Fortwendel J. R. (2017). The Aspergillus Fumigatus Farnesyltransferase β-Subunit, RamA, Mediates Growth, Virulence, and Antifungal Susceptibility. Virulence 8, 1401–1416. doi: 10.1080/21505594.2017.1328343
Oosthuizen J. L., Gomez P., Ruan J., Hackett T. L., Moore M. M., Knight D. A., et al. (2011). Dual Organism Transcriptomics of Airway Epithelial Cells Interacting With Conidia of Aspergillus Fumigatus. PloS One 6, e20527. doi: 10.1371/journal.pone.0020527
Parent-Michaud M., Dufresne P. J., Fournier É., Martineau C., Moreira S., Perkins V., et al (2019). Draft Genome Sequences of Azole-Resistant and Azole-Susceptible Aspergillus Turcosus Clinical Isolates Recovered From Bronchoalveolar Lavage Fluid Samples. Microbiol. Resource. Announcements. 8, e01446–e01418. doi: 10.1128/MRA.01446-18
Pond S. L. K., Frost S. D. W., Muse S. V. (2004). HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 21, 676–679. doi: 10.1093/bioinformatics/bti079
Raffa N., Keller N. P. (2019). A Call to Arms: Mustering Secondary Metabolites for Success and Survival of an Opportunistic Pathogen. PloS Pathog. 15, e1007606. doi: 10.1371/journal.ppat.1007606
Reeves E. P., Reiber K., Neville C., Scheibner O., Kavanagh K., Doyle S. (2006). A Nonribosomal Peptide Synthetase (Pes1) Confers Protection Against Oxidative Stress in Aspergillus Fumigatus. FEBS J. 273, 3038–3053. doi: 10.1111/j.1742-4658.2006.05315.x
Ries L. N. A., Pardeshi L., Dong Z., Tan K., Steenwyk J. L., Colabardini A. C., et al. (2020). The Aspergillus Fumigatus Transcription Factor RglT is Important for Gliotoxin Biosynthesis and Self-Protection, and Virulence. PloS Pathog. 16, e1008645. doi: 10.1371/journal.ppat.1008645
Rocha M. C., Fabri J. H. T. M., Franco de Godoy K., Alves de Castro P., Hori J. I., Ferreira da Cunha A., et al. (2016). Aspergillus Fumigatus MADS-Box Transcription Factor rlmA Is Required for Regulation of the Cell Wall Integrity and Virulence. G3 (Bethesda). 6, 2983–3002. doi: 10.1534/g3.116.031112
Rokas A., Mead M. E., Steenwyk J. L., Oberlies N. H., Goldman G. H. (2020). Evolving Moldy Murderers: Aspergillus Section Fumigati as a Model for Studying the Repeated Evolution of Fungal Pathogenicity. PloS Pathog. 16, e1008315. doi: 10.1371/journal.ppat.1008315
Ropero P., Erquiaga S., Arrizabalaga B., Pérez G., de la Iglesia S., Torrejón M. J., et al. (2017). Phenotype of Mutations in the Promoter Region of the β-Globin Gene. J. Clin. Pathol. 70, 874–878. doi: 10.1136/jclinpath-2017-204378
Rozewicki J., Li S., Amada K. M., Standley D. M., Katoh K. (2019). MAFFT-DASH: Integrated Protein Sequence and Structural Alignment. Nucleic Acids Res. 47, W5–W10. doi: 10.1093/nar/gkz342
Schaefke B., Wang T.-Y., Wang C.-Y., Li W.-H. (2015). Gains and Losses of Transcription Factor Binding Sites in Saccharomyces Cerevisiae and Saccharomyces Paradoxus. Genome Biol. Evol. 7, 2245–2257. doi: 10.1093/gbe/evv138
Scharf D. H., Remme N., Habel A., Chankhamjon P., Scherlach K., Heinekamp T., et al. (2011). A Dedicated Glutathione S -Transferase Mediates Carbon–Sulfur Bond Formation in Gliotoxin Biosynthesis. J. Am. Chem. Soc 133, 12322–12325. doi: 10.1021/ja201311d
Schrettl M., Carberry S., Kavanagh K., Haas H., Jones G. W., O’Brien J., et al. (2010). Self-Protection Against Gliotoxin—A Component of the Gliotoxin Biosynthetic Cluster, GliT, Completely Protects Aspergillus Fumigatus Against Exogenous Gliotoxin. PloS Pathog. 6, e1000952. doi: 10.1371/journal.ppat.1000952
Shibuya K., Takaoka M., Uchida K., Wakayama M., Yamaguchi H., Takahashi K., et al. (1999). Histopathology of Experimental Invasive Pulmonary Aspergillosis in Rats: Pathological Comparison of Pulmonary Lesions Induced by Specific Virulent Factor Deficient Mutants. Microb. Pathog. 27, 123–131. doi: 10.1006/mpat.1999.0288
Shimodaira H. (2002). An Approximately Unbiased Test of Phylogenetic Tree Selection. Systematic. Biol. 51, 492–508. doi: 10.1080/10635150290069913
Steenwyk J. L., Buida T. J., Labella A. L., Li Y., Shen X.-X., Rokas A. (2021a). PhyKIT: A Broadly Applicable UNIX Shell Toolkit for Processing and Analyzing Phylogenomic Data. Bioinformatics. 37 (16), 2325–2331. doi: 10.1093/bioinformatics/btab096
Steenwyk J. L., Iii T. J. B., Li Y., Shen X.-X., Rokas A. (2020a). ClipKIT: A Multiple Sequence Alignment Trimming Software for Accurate Phylogenomic Inference. PloS Biol. 18, e3001007. doi: 10.1371/journal.pbio.3001007
Steenwyk J. L., Mead M. E., de Castro P. A., Valero C., Damasio A., dos Santos R. A. C., et al. (2021b). Genomic and Phenotypic Analysis of COVID-19-Associated Pulmonary Aspergillosis Isolates of Aspergillus Fumigatus. Microbiol. Spectr. 9, e00010-21. doi: 10.1128/Spectrum.00010-21
Steenwyk J. L., Mead M. E., Knowles S. L., Raja H. A., Roberts C. D., Bader O., et al. (2020b). Variation Among Biosynthetic Gene Clusters, Secondary Metabolite Profiles, and Cards of Virulence Across Aspergillus Species. Genetics 216, 481–497. doi: 10.1534/genetics.120.303549
Steenwyk J. L., Shen X.-X., Lind A. L., Goldman G. H., Rokas A. (2009). A Robust Phylogenomic Time Tree for Biotechnologically and Medically Important Fungi in the Genera Aspergillus and Penicillium. mBio 10, e00925–e00919. doi: 10.1128/mBio.00925-19
Steinbach W. J., Marr K. A., Anaissie E. J., Azie N., Quan S.-P., Meier-Kriesche H.-U., et al. (2012). Clinical Epidemiology of 960 Patients With Invasive Aspergillosis From the PATH Alliance Registry. J. Infection. 65, 453–464. doi: 10.1016/j.jinf.2012.08.003
Suyama M., Torrents D., Bork P. (2006). PAL2NAL: Robust Conversion of Protein Sequence Alignments Into the Corresponding Codon Alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Takahashi H., Umemura M., Ninomiya A., Kusuya Y., Shimizu M., Urayama S., et al. (2021). Interspecies Genomic Variation and Transcriptional Activeness of Secondary Metabolism-Related Genes in Aspergillus Section Fumigati. Front. Fungal Biol. 2. doi: 10.3389/ffunb.2021.656751
Traynor A. M., Sheridan K. J., Jones G. W., Calera J. A., Doyle S. (2019). Involvement of Sulfur in the Biosynthesis of Essential Metabolites in Pathogenic Fungi of Animals, Particularly Aspergillus Spp.: Molecular and Therapeutic Implications. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.02859
Vargas-Muñiz J. M., Renshaw H., Richards A. D., Lamoth F., Soderblom E. J., Moseley M. A., et al. (2015). The Aspergillus Fumigatus Septins Play Pleiotropic Roles in Septation, Conidiation, and Cell Wall Stress, But are Dispensable for Virulence. Fungal Genet. Biol. 81, 41–51. doi: 10.1016/j.fgb.2015.05.014
Vinet L., Zhedanov A. (2011). A `Missing’ Family of Classical Orthogonal Polynomials. J. Phys. A.: Math. Theor. 44, 85201. doi: 10.1088/1751-8113/44/8/085201
Waddell P. J., Steel M. A. (1997). General Time-Reversible Distances With Unequal Rates Across Sites: Mixing Gamma and Inverse Gaussian Distributions With Invariant Sites. Mol. Phylogenet. Evol. 8, 398–414. doi: 10.1006/mpev.1997.0452
Wiesner D. L., Klein B. S. (2017). Lung Epithelium: Barrier Immunity to Inhaled Fungi and Driver of Fungal-Associated Allergic Asthma. Curr. Opin. Microbiol. 40, 8–13. doi: 10.1016/j.mib.2017.10.007
Willger S. D., Grahl N., Cramer R. A. (2009). Aspergillus Fumigatus Metabolism: Clues to Mechanisms of In Vivo Fungal Growth and Virulence. Med. Mycol. 47 Suppl 1, S72–S79. doi: 10.1080/13693780802455313
Willger S. D., Puttikamonkul S., Kim K.-H., Burritt J. B., Grahl N., Metzler L. J., et al. (2008). A Sterol-Regulatory Element Binding Protein Is Required for Cell Polarity, Hypoxia Adaptation, Azole Drug Resistance, and Virulence in Aspergillus Fumigatus. PloS Pathog. 4, e1000200. doi: 10.1371/journal.ppat.1000200
Wirmann L., Ross B., Reimann O., Steinmann J., Rath P.-M. (2018). Airborne Aspergillus Fumigatus Spore Concentration During Demolition of a Building on a Hospital Site, and Patient Risk Determination for Invasive Aspergillosis Including Azole Resistance. J. Hosp. Infect. 100, e91–e97. doi: 10.1016/j.jhin.2018.07.030
Yamamoto N., Maeda Y., Ikeda A., Sakurai H. (2008). Regulation of Thermotolerance by Stress-Induced Transcription Factors in Saccharomyces Cerevisiae. Eukaryot. Cell 7, 783–790. doi: 10.1128/EC.00029-08
Yang Z. (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Keywords: fungal genomics, evolution, Aspergillus fumigatus, non-coding region, evolutionary rate, virulence factor
Citation: Brown A, Mead ME, Steenwyk JL, Goldman GH and Rokas A (2022) Extensive Non-Coding Sequence Divergence Between the Major Human Pathogen Aspergillus fumigatus and its Relatives. Front. Fungal Biol. 3:802494. doi: 10.3389/ffunb.2022.802494
Received: 26 October 2021; Accepted: 09 June 2022;
Published: 07 July 2022.
Edited by:
John G. Gibbons, University of Massachusetts Amherst, United StatesReviewed by:
Vishukumar Aimanianda, Institut Pasteur, FranceCopyright © 2022 Brown, Mead, Steenwyk, Goldman and Rokas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonis Rokas, YW50b25pcy5yb2thc0B2YW5kZXJiaWx0LmVkdQ==
†ORCID: Alec Brown, orcid.org/0000-0002-1073-7562
Matthew E. Mead, orcid.org/0000-0001-9195-7585
Jacob L. Steenwyk, orcid.org/0000-0002-8436-595X
Gustavo H. Goldman, orcid.org/0000-0002-2986-350X
Antonis Rokas, orcid.org/0000-0002-7248-6551
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.