
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. For. Glob. Change , 12 January 2023
Sec. Forest Soils
Volume 5 - 2022 | https://doi.org/10.3389/ffgc.2022.1114390
This article is part of the Research Topic Forests' Soil-Environment Interactions Under Global Change View all 4 articles
 Minsoo Jeong1
Minsoo Jeong1 Setu Bazie Tagele1
Setu Bazie Tagele1 Min-Ji Kim1
Min-Ji Kim1 Suk-Hyung Ko2Kwon-Su Kim2Jung-Goon Koh2
Suk-Hyung Ko2Kwon-Su Kim2Jung-Goon Koh2 Da-Ryung Jung1
Da-Ryung Jung1 YoungJae Jo1
YoungJae Jo1 YeonGyun Jung1
YeonGyun Jung1 Yeong-Jun Park1Min-Sueng Kim1
Yeong-Jun Park1Min-Sueng Kim1 Kyeongmo Lim1
Kyeongmo Lim1 Jae-Ho Shin1,3,4*
Jae-Ho Shin1,3,4*The population of the Korean fir, Abies koreana, is declining at an accelerating rate, and the average mortality rate in 2019 exceeded 36.43% on Mt. Hallasan in Jeju Island, Republic of Korea. Several prior studies have reported different reasons with various interpretations, indicating that additional data, such as data on microbial communities that promote plant growth and resistance to abiotic stresses, are required to understand the phenomenon further. This is the first investigation that documents the changes in the soil microbial and fungal community and soil physicochemical properties resulting from the death of the Korean fir. In our case, high throughput sequencing data have been provided for the soil microbiome and mycobiome of Korean fir trees, identifying the microbial composition differences before and after the decline in the health of Korean fir trees. The results showed that the soil fungal community was considerably shaped in response to the decline in the health of Korean fir rather than the soil bacterial community. The decline in health or the death of Korean fir trees contributed to the decrease in diversity and dominance of symbiotic fungi such as Russula, Sebacina, and Phenoliferia in the forest ecosystem. It also weakened the complexity and ecological competition of the fungal co-occurrence network. Structural equation modeling showed that the death of Korean fir was strongly associated with the concentrations of soil nutrients such as available phosphorus (P2O5) and potassium ion (K+) concentrations and low moisture content, adversely affecting the symbiotic relationship with ectomycorrhizal fungi. Our findings shed light on the critical taxa of mycobiome of live and dead A. koreana plants and their relationship with ecological edaphic factors, highlighting their potential role as biomarkers for the death of Korean fir.
Abies koreana, the scientific name for Korean fir, is an evergreen tree known as a high-quality Christmas tree in demand for its unique appearance and beautiful fragrance. The Korean fir grows only in sub-alpine southern regions at an altitude of 1,000–1,900 m in the Republic of Korea (Ihm et al., 2000), and Mt. Hallasan is the world’s largest habitat for Korean fir and a preservation site for many endemic species in Korea. However, the Korean fir, growing wild on the Korean Peninsula for tens of thousands of years, has faced severe decline and death for the last 20 years. According to the natural resources survey of the Jeju World Natural Heritage Headquarters reported in 2020 (Ahn et al., 2020), the cumulative mortality rate has consistently increased from 2000 to 2019, and the average mortality rate has been over 36.43% in 2019. Korean fir was classified as Endangered (EN) in the IUCN 3.1 Red List of Threatened Species in 2011 (Kim et al., 2011).
For the last 40 years, the death of the Korean fir forest has been studied in various fields, such as physiological ecology, landscape ecology, and population ecology, to prevent the destruction of the natural balance of the plant ecosystem of Mt. Hallasan due to the threat of the extinction of the species (Koo and Kim, 2020). In previous studies, geographic, climatic, and soil physicochemical characteristics, including tree density, solar radiation, terrain slope (Ahn and Yun, 2020), and wind intensity changes (Seo et al., 2019), have been reported as factors contributing to the decline phenomenon, although findings diverge between studies. For example, Gwon et al. (2013) attributed the decline to the decreased soil moisture content following increased temperature and insufficient winter snow cover. As the conflicting research continues to be reported, more studies that can support previous studies remain to be conducted.
Plant-associated soil microbiome plays a cornerstone of one health (Banerjee and van der Heijden, 2022) and confer fitness advantages to the plant host, including growth promotion, nutrient uptake, stress tolerance, and resistance to pathogens (Dubey et al., 2019). Plant-microbe interactions shape the assembly of plant-associated microbiomes and modulate their beneficial traits, such as nutrient acquisition and plant health, in addition to highlighting knowledge gaps and future directions (Trivedi et al., 2020). The soil microbiome has complex interactions with plant-mediated soil legacies (Hannula et al., 2021) and plays a vital role in plant growth interacting with various biotic and abiotic factors (Kumar et al., 2019). An investigation of the alteration in the microbiome in the soil of fir trees reflecting in the health of Korean fir could be expected to discover potential factors that have correlations with plant death, such as specific taxa and soil physicochemical properties. More importantly, ecological studies of the Korean fir soil microbiome containing bacterial and fungal communities and soil physicochemistry have not yet been documented.
In this study, we investigated the soil microbiome structure of healthy Korean fir (HKF) and dead Korean fir (DKF) trees sampled from the colony of Korean fir trees. The current study used 16S rRNA and ITS2 gene amplicon sequencing to explore the structure and diversity of both bacterial and fungal communities. For a detailed soil ecosystem investigation, we divided the soil into two soil compartments based on their proximity to the host plant: the rhizospheric soil affected by the plant itself and root exudates, and the bulk soil outside the rhizosphere not penetrated by plant roots (Youssef et al., 1989; Smalla et al., 2006; Edwards et al., 2015). We determined the changes in the soil microbial ecosystem and their association with the death of Korean fir and evaluated the relationship between the dominant microorganisms for each sample group and the physicochemical properties of the soil. The co-occurrence network identified ecological relationships of microbial communities on the health status of Korean fir by soil compartment. Investigations were also conducted to determine the relationship between soil physicochemical properties and symbiotic microbes and the potential contribution of the rhizosphere microbiome to the maintenance of the health of Korean fir trees. This study aimed (i) to elucidate soil microbiome and mycobiome changes in rhizosphere and bulk soils between HKF and DKF, and the resulting differences in soil physicochemical properties and (ii) to develop an understanding of the soil microbiome of Korean fir in the current scenario, and (iii) to provide a new biological perspective from the analysis of bacterial and fungal communities.
All samples were obtained with site access and sampling collection permission from the World Heritage Office of Hallasan National Park. This study was conducted in Mt. Hallasan National Park, Jeju-si, Jeju Province, Jeju Island, Republic of Korea (33°21′ N, 126°31′ E). A total of 40 soil samples (20 HKF and 20 DKF each) were obtained from fir forest distribution areas in October 2020 (Figure 1). In order to exclude the influence on the microbiome from vegetation present in the topsoil, samples were taken from soil depths of 15–30 cm and within a distances no greater than 0.5 m from the trunk of each tree. Rhizospheric soil was acquired from the ground firmly attached to the roots of Korean fir trees. Samples for measuring physical properties were collected using a DIK-1,630 soil picker (DAIKI, Saitama, Japan). Soil samples for measuring chemical properties were collected while considering the layer after removing the organic layer at the exact location where the samples were collected for measuring physical properties. Soil physicochemical analysis was performed using routine methods (NIAST, 2010). Soil samples were air-dried and passed through a 2-mm sieve for chemical analysis. The samples were used for measuring soil physicochemical parameters, including pH (H2O, CaCl2, KCl, NaF), electrical conductivity, OM, total nitrogen, available phosphate, phosphate absorption coefficient, exchangeable cations (K+, Ca2+, Mg2+, Na+, Al3+), soil texture (sand, silt, clay), particle density, bulk density, porosity, soil three-phase distribution, and hydraulic conductivity. In the case of soil pH, in addition to the standard method (H2O) for measuring pH, other methods (CaCl2, KCl, NaF) of measuring pH are also used in soil surveys for specific purposes, especially those required for some taxonomic criteria in Soil Taxonomy (Staff and Staff, 1951). For example, the use of 0.01M CaCl2 solution has the advantage of reducing seasonal variations in pH, while the use of 1N KCl solution is to test for the presence of exchangeable Al (Schofield and Taylor, 1955). The role of 1M NaF solution is to infer the presence of short-range order minerals (Staff and Staff, 1951). Sand, silt, clay percentage information, and soil type in the sampling area are listed in Supplementary Table 1. Soil samples for microbial DNA analysis were frozen at −70°C without air-drying and stored in polyethylene sample bottles.
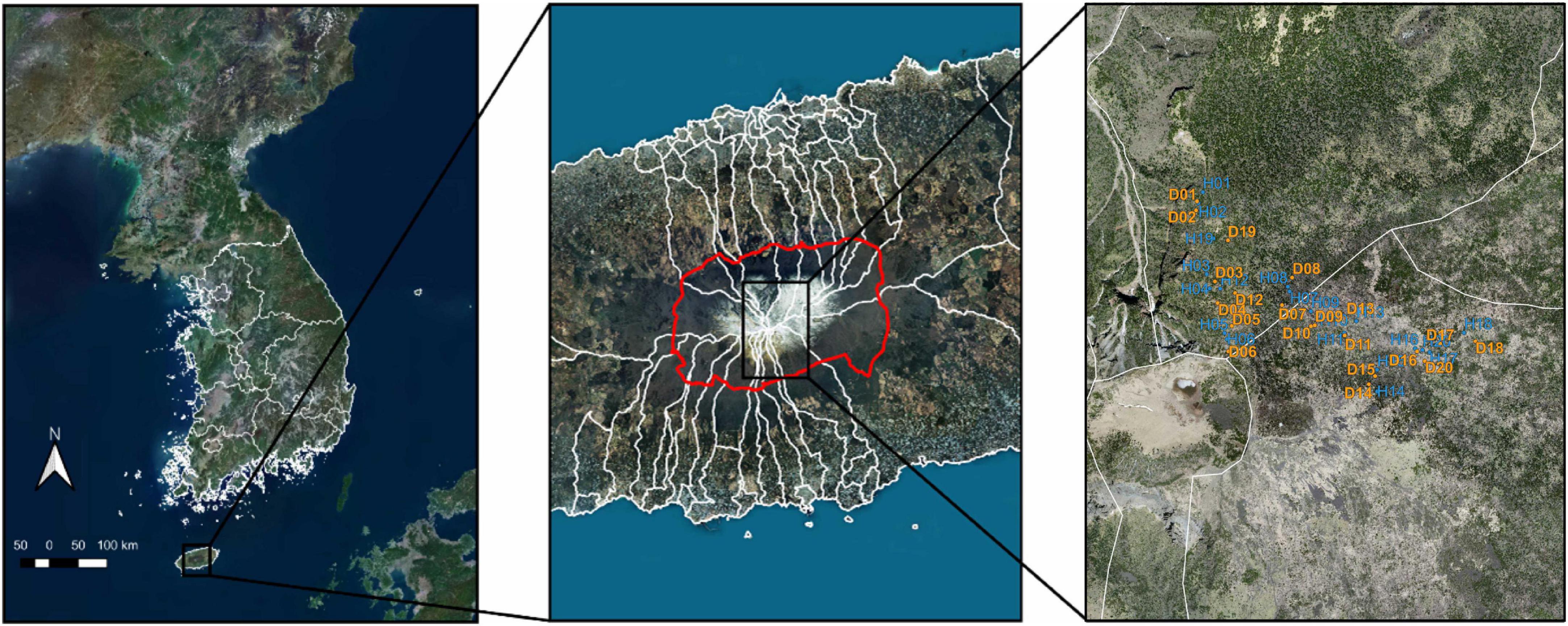
Figure 1. Sampling site of the Korean fir tree. The sampling site is a Korean fir forest (green) near Baengnokdam Crater Lake, the summit of Mt. Hallasan. The boundaries of Mt. Hallasan National Park are marked in red. Sampling points were marked: Healthy Korean Fir (blue) and Dead Korean Fir (yellow).
Soil microbial DNA extraction was performed using the DNeasy PowerSoil Pro Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. The extracted DNA was quantified based on the DNA absorbance ratio at 260/280 nm (∼1.9) and 260/230 nm (∼2.0) using the NanoDrop One UV-Vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The DNA concentration was remeasured for accuracy using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). After DNA extraction, the prokaryotic 16S rRNA V4-V5 hypervariable region was amplified through primer pair 515F/907R (515F, 5′- ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT GTG NCA GCB GCC GCG GTR A –3′ and 907R, 5′- GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC CGY CWA TTY HTT TRA GTT T –3′) (Apprill et al., 2015; Parada et al., 2016), and the eukaryotic ITS2 region was amplified through primer pair ITS86F/ITS4R (ITS86F, 5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT GTG AAT CAT CGA ATC TTT GAA-3′ and ITS4R, 5′-GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TCC TCC GCT TAT TGA TAT GC-3′) (Turenne et al., 1999; Ferrer et al., 2001). For the polymerase chain reaction (PCR) to amplify purified DNA, 25-ml EmeraldAmp MAX HS PCR Master Mix (Takara Bio, Shiga, Japan), 2 ml extracted DNA, 1 ml each primer, and 21-ml sterile distilled water were used in a 50-ml reaction using Eppendorf Mastercycler Nexus PCR Cycler (Eppendorf, Hamburg, Germany). For the prokaryotic 16S rRNA, a denaturation step of 95°C was performed for 3 min, followed by 20 cycles at 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 3 min. For the eukaryotic Internal Transcribed Spacer (ITS) 2 region, a denaturation step of 95°C was performed for 5 min, followed by 30 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 5 min. The second PCR process for attaching the dual multiplexing index and specific adaptor was as follows: for the prokaryotic 16S rRNA, a denaturation step of 95°C was performed for 3 min, followed by 25 cycles at 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 3 min. For the eukaryotic d region, a denaturation step of 95°C was performed for 5 min, followed by 30 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 5 min. The size and quality of the amplicon were verified through 2% (w/v) agarose gel electrophoresis, and amplicon product impurities were eliminated using QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). The amplified and purified DNA amplicon was adjusted to the same DNA concentration and pooled considering the PCR product size of the 16S rRNA and ITS2. Paired-end sequencing was performed using the Illumina MiSeq instrument (Illumina, San Diego, USA) with adjustable read lengths of about 2 × 250 bp using MiSeq Reagent Kit v3 (Illumina, 600 cycles). Fastq files have been deposited into an NCBI Sequence Read Archive (SRA, NCBI1) under the accession numbers PRJNA802974. Sequencing was performed at the next-generation sequencing core facility of Kyungpook National University (Daegu, South Korea).
The demultiplexed raw FASTQ data acquired from the MiSeq instrument were filtered for quality and analyzed using the Quantitative Insights Into Microbial Ecology 2 (QIIME2) (ver 2020.11) software (Bolyen et al., 2019). Since the merged paired sequences are mostly filtered owing to low-quality reverse reads, only forward sequences were used for subsequent analysis. To generate amplicon sequence variants, the single-end sequence was filtered (mean frequency; 26,260 for bacteria, and 37,312 for fungi), trimmed (with lengths of 200 nucleotides), and denoised (Q score threshold > 30) using the DADA2 software (Callahan et al., 2016). The ASVs were assigned taxonomy with a 99% cutoff value by the classify-sklearn program using the SILVA 16S/18S rRNA full-length sequences (Release 138) (Quast et al., 2012) and UNITE database (ver 2019.02) (Abarenkov et al., 2010) as the reference database. The ASVs classified as mitochondria, chloroplast, eukaryote, or unassigned were eliminated from the bacterial ones, and the ASVs classified as mitochondria, prokaryote, or unassigned were removed from the fungal ones. Alignment was conducted using de novo multiple sequence alignment with an align-to-tree-mafft-fasttree plugin of QIIME2. The sequences were rarefied (825,840 raw reads including 356,920 bacterial reads, an average of 8,923 per sample; and 468,920 fungal reads, an average of 11,723 per sample).
Statistical analysis and visualization were conducted using the R software (ver 4.0.3). All QIIME2 data inputs to R software were processed after pre-processing using the “Phyloseq” R package (ver 1.34.0) and the “qiime2R” R package (ver 0.99.4). The normality test was performed using the Shapiro-test function in the “stat” R package (ver 4.0.3), and the test of equal variances was performed with the Levene-test function in the “car” R package (ver 3.0-10) for two independent groups. Regarding the amplicon sequencing results and soil physicochemical properties, the statistical significance of the two independent variables was evaluated using the following statistical analysis: (i) the unpaired two-sample t-test when it follows normality and satisfies equal variance, (ii) Welch’s correction paired t-test when following normality but not equal variance, (iii) Wilcoxon rank-sum test when both normality and equal variance are violated. Shannon, InvSimpson, Chao1, and Evenness indices for alpha diversity analysis were computed using the “vegan” R package. Beta diversity was conducted to evaluate differences between groups using principal coordinates analysis (PCoA) based on the original Bray–Curtis dissimilarity, unweighted UniFrac distance, and canonical analysis of principal coordinates (CAP). Permutational multivariate analysis of variance was conducted using an Adonis test from the “vegan” R package. To investigate the biomarkers characterizing the differences in the microbial communities of tree soil in different health states, linear discriminant analysis effect size (LEfSe) (Segata et al., 2011) was performed using the “microbiomeMarker” R package (ver 0.0.1.9000). Correlation analysis of the matrix between genus biomarkers and soil physicochemical qualities within each group was conducted using Spearman’s rho rank correlation coefficients with the “Hmisc” R package (ver 4.4-2). To identify the functional guild of the fungal communities, the Fungi Functional Guild (FUNGuild2) version 1.0, an open annotation tool for parsing fungal community datasets, Guilds_v1.0 (Nguyen et al., 2016). In brief, the fungal ASVs at the genus level according to sequencing platforms and analysis pipelines are assigned to the guild annotated database, a more manageable ecological unit of taxonomically complex communities.
Inter-kingdom co-occurrence network analysis in HKF and DKF soil samples was performed using the sparse inverse covariance estimation for the Ecological Association and Statistical Inference (SpiecEasi, ver 1.1.03) R package. To reduce complexity and noise in the network, genera with a relative abundance of less than 0.01% were excluded. The quality of the network model was estimated through information criteria, and the sparse graphical lasso was used to adjust the parameters with a penalty method (Layeghifard et al., 2018). For the HKF soil microbial network, 116 bacterial and 85 fungal ASVs were used, whereas 122 bacterial and 85 fungal ASVs were used for the DKF network. The topological role of each genus is determined by the z-values (zi, within-module connectivity) and p-values (pi, among-module connectivity) and is divided into four subcategories: (i) peripheral nodes (Zi ≤ 2.5, pi ≤ 0.62), (ii) connectors (Zi ≤ 2.5, pi > 0.62), (iii) module hubs (Zi > 2.5, pi ≤ 0.62), and (iv) network hubs (Zi > 2.5, pi > 0.62) (Olesen et al., 2006).
Structural equation modeling (SEM) was conducted to quantify the direct and indirect relationships among soil physicochemical properties, fungal functional guild, fungal diversity, and the decline of the health of Korean fir. Fungal diversity was characterized by Shannon, inversed Simpson, and Evenness, whereas fungal composition was characterized with the first axis of PCoA using an unweighted UniFrac distance matrix. Based on a priori and theoretical knowledge, we assumed a conceptual model that the changes in the soil physicochemical properties affect the fungal abundance, diversity, composition, and the decline of the health of Korean fir. The maximum likelihood estimation method was used to compare the SEM with the observation. Model adequacy was determined by χ2-tests, chi-Square/degrees of freedom (CMIN/DF), comparative fit index (CFI), and root square mean errors of approximation (RMSEA), and we revised our conceptual model according to these indexes. Adequate model fits were indicated by a non-significant χ2-test (p > 0.05), high CMIN/DF (<3), high CFI (>0.90), and low RMSEA (<0.05) (Grace, 2006). SEM analysis was performed using the “Lavaan” R package (Rosseel, 2012).
The bacterial and fungal community changes associated with the health status of Korean fir were analyzed using the 16S rRNA and ITS2 gene amplicon sequencing method from the HKF and DKF soil samples (Janda and Abbott, 2007; Ihrmark et al., 2012). After filtering rare taxa with a relative abundance below 0.01%, 1,358 and 1,216 bacterial amplicon sequence variants (ASVs) and 533 and 519 fungal ASVs were identified in the HKF and DKF groups, respectively. For taxonomy-assigned ASVs in the HKF group, thirty bacterial and ten fungal phyla with 200 and 181 genera were obtained, respectively. Meanwhile, in the DKF group, 28 bacterial and seven fungal phyla with 192 and 170 genera were obtained, respectively.
We performed the differential abundance and diversity analysis of the soil microbiome in both the bulk and rhizosphere soil compartments of HKF and DKF soil to determine the association of soil microbiome structure and diversity with the health of Korean fir and their soil compartment niches. We presented the top ten dominant phyla within HKF and DKF for the relative abundance analysis. We did not observe any changes in the relative abundance of various bacterial phyla between the soil compartments of HKF and DKF soil (Figure 2A). On the other hand, the relative abundance of the fungal phyla mainly differed depending on the health of Korean fir rather than their soil compartment niches (Figure 2B). Specifically, in the fungal community, a significant decrease in the relative abundance of the phylum Basidiomycota (p-value = 0.024) and an increase in that of the phylum Ascomycota (p-value = 0.033) were observed following the death of Korean fir (Supplementary Figures 1A, B).
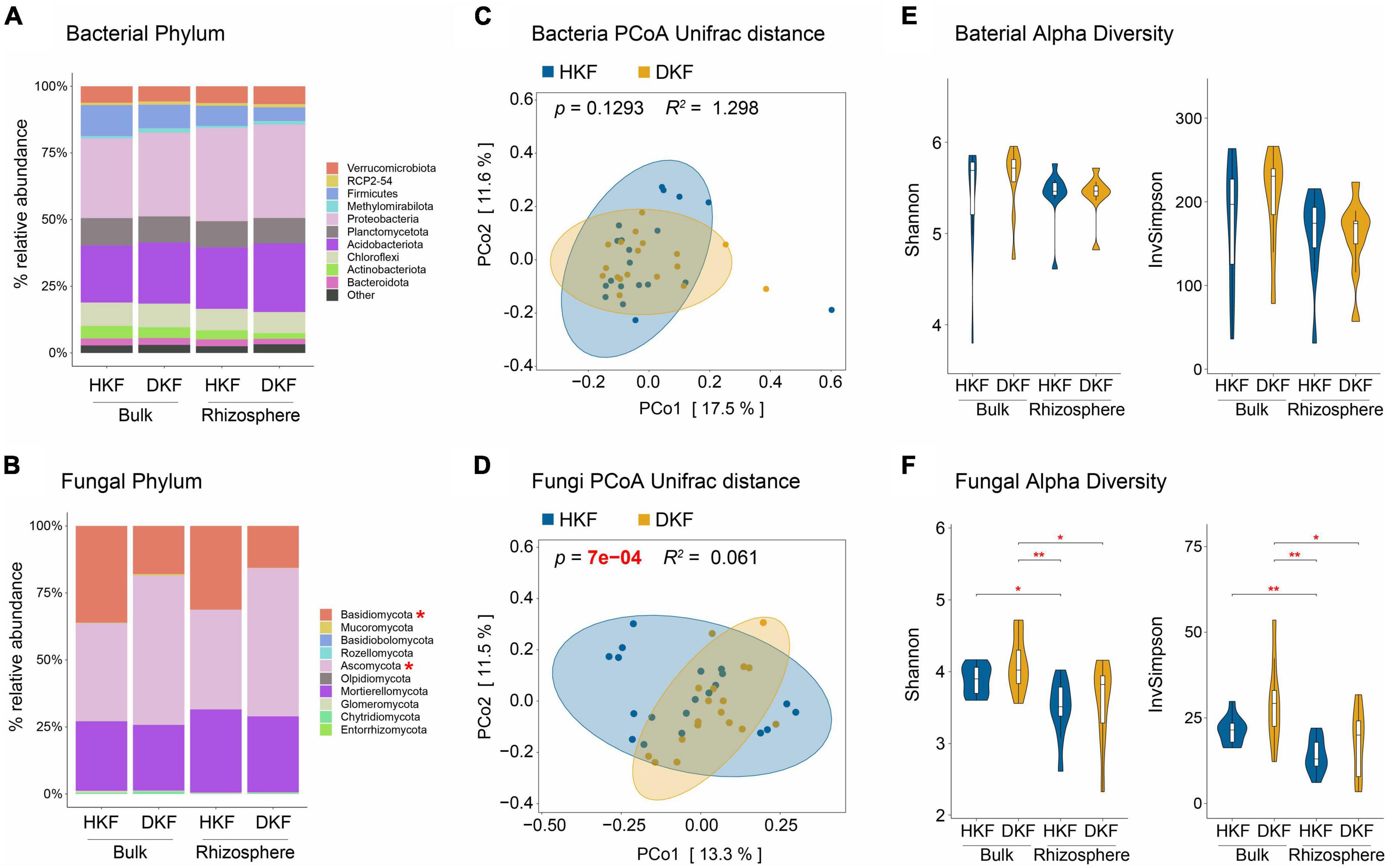
Figure 2. Comparisons of the microbiome community structure and diversity based on the decline of Korean fir health and soil compartment. The upper panel (A,C,E) presents the analysis of the bacterial community, and the lower panel (B,D,F) presents the analysis of the fungal community. (A,B) Relative abundance of top ten dominant phyla of healthy Korean fir (HKF) and dead Korean fir (DKF) in different soil compartments. Other phyla are included as “Others”. (C,D) Principal coordinates analysis (PCoA) based on unweighted UniFrac distance and microbial diversity differences between the HKF (blue) and DKF (yellow) groups. Adonis R2 and p-values are provided for each plot. (E,F) Shannon and inverse Simpson diversity index of the soil microbiota based on heath decline of Korean fir and soil compartment. Significance levels: *p < 0.05, **p < 0.01.
We also investigated the beta diversity to identify the differences in the microbial communities between each group at the ASV level. The microbial community differences according to the comparison groups were conducted via PCoA beta diversity based on the unweighted UniFrac distance (Lozupone et al., 2011). For the alpha diversity analysis, we used the Shannon–Wiener index (Shannon, 1948), which summarizes both richness and evenness, and the inverse Simpson index (Hunter and Gaston, 1988), which reflects only evenness. The soil fungal community was shaped significantly depending on the health status of Korean fir (Figure 2D, Adonis, p-value = 7e–04, R2 = 0.061) rather than the soil bacterial community (Figure 2C, Adonis, p-value = 0.1293, R2 = 1.298), consistent with the results of relative abundance analysis (Figure 2B). On the other hand, the shapes of both the bacterial and fungal communities were not significantly different for the two soil compartments (Supplementary Figures 2A, B). Notably, these mycobiome changes based on the health status of Korean fir were observed in rhizosphere soil rather than bulk soil (Supplementary Figures 2C, D).
Meanwhile, the fungal alpha diversity was mainly different depending on the soil compartment (Figure 2F). Although not significant, the fungal alpha diversity of DKF was slightly higher than that of HKF in both soil compartments, suggesting that increased fungal diversity was still associated with ecosystem changes related to the decline of Korean fir health (Figure 2F). As with beta diversity, bacterial alpha diversity did not differ significantly (Figure 2E). In summary, changes in the structure and diversity of the soil microbiome, specifically of the fungal community, were more dependent on the health status of Korean fir than on the soil compartment niches.
As only the fungal community differed significantly depending on the decline of Korean fir health, we focused on the fungal community in subsequent analyzes while excluding the bacterial community. Figure 3 indicates only genera for which the sum of their relative abundances is above the 0.01% threshold. Circular packing plots display differences in the relative abundance of various fungal genera in each of the two soil compartments (p < 0.05) depending on the health of Korean fir. The circle packing plot indicated that 13 and 14 fungal genera with significantly different abundance were detected in the rhizosphere and bulk soil, respectively (Figures 3A, B). Regardless of the soil compartments, the statistically significant relative abundances of the fungal genera Russula, Sebacina, and Phenoliferia of the phylum Basidiomycota were exclusive to the HKF group (Figures 3A, B), consistent with the increased relative abundance of Basidiomycota observed in the HKF group compared to the DKF group (Supplementary Figure 1A). On the other hand, for the DKF group, Loramyces, Ascocoryne, and Hypochinicium were higher in the rhizosphere soil, and Trichoderma, Paraphoma, Leptodontium, Ascocoryne, and Hypholoma were more related to the bulk soil samples.
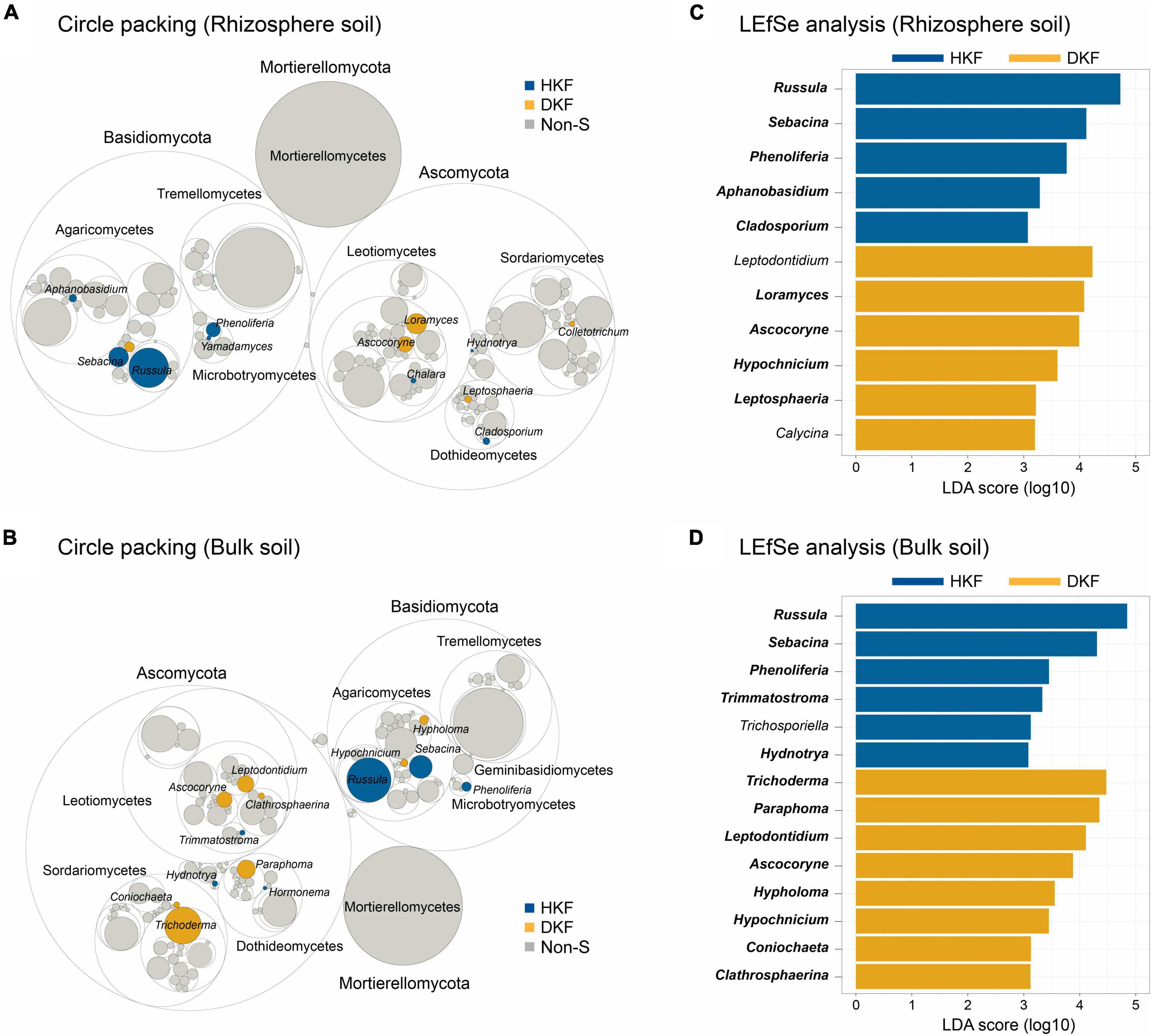
Figure 3. Representative biomarkers at fungal genera level. The upper panel (A,C) is the analysis of the rhizospheric soil, and the lower panel (B,D) is the analysis of the bulk soil. (A,B) Circle packing plots represent the relative abundance of fungal genera observed in the healthy Korean fir (HKF) and dead Korean fir (DKF) groups. The statistically significant abundance of fungal genus for each group is indicated by HKF (blue) and DKF (yellow), and non-S (gray) when a significant difference was not met. (C,D) Linear discriminant analysis effect size analysis based on LDA score to detect biomarkers indicating differences between the groups (with LDA score > 3.0).
Additionally, we investigated functional guild assignments using FUNGuild (Nguyen et al., 2016) to determine the ecological function of fungi. Based on FUNGuild analysis, the genera Russula, Sebacina, and Hydnotrya, dominant in the HKF group, were ectomycorrhizas (ECMs), i.e., symbiotrophs, whereas the genus Phenoliferia was not assigned a functional guild (Supplementary Table 2). In particular, these symbiotic fungi were predominantly abundant and diverse in HKF groups regardless of soil fraction (Supplementary Figure 3). We also confirmed that most of the fungal genus dominant in DKF soil belonged to the Saprotrophs guild. Furthermore, we cross-checked fungal biomarkers with differential abundance between the HKF and DKF groups using LEfSe analysis with a log (LDA score) over 3.0 (Figures 3C, D). Our analysis results indicate that most genera identified in the circular packing analysis were observed as bio-markers in the LEfSe analysis.
We conducted mycobiome network analysis for an overall understanding of the fungal matrix in the soil environment. Co-occurrence networks were analyzed using Sparse InversE Covariance estimation for the Ecological Association and Statistical Inference (Kurtz et al., 2015) to investigate the microbial relationship. The four networks showed different topological properties, and overall, the DKF networks (Figures 4B, D) were less complex and less dense than the HKF networks (Figures 4A, C). Following the decline in the health of Korean fir, the interaction of soil fungi decreased qualitatively and quantitatively. Our results indicated that significant correlations were defined as the total number of nodes and edges decreased (Supplementary Table 3). The edge density and ratio of the negative edge also decreased with tree death. Specifically, the DKF network in rhizosphere soil consisted of the fewest 250 network edges and the lowest edge density compared to other groups (Figure 4B and Supplementary Table 3). In addition, this network was composed of the lowest ratio of negative correlations among the four groups, including the bulk group.
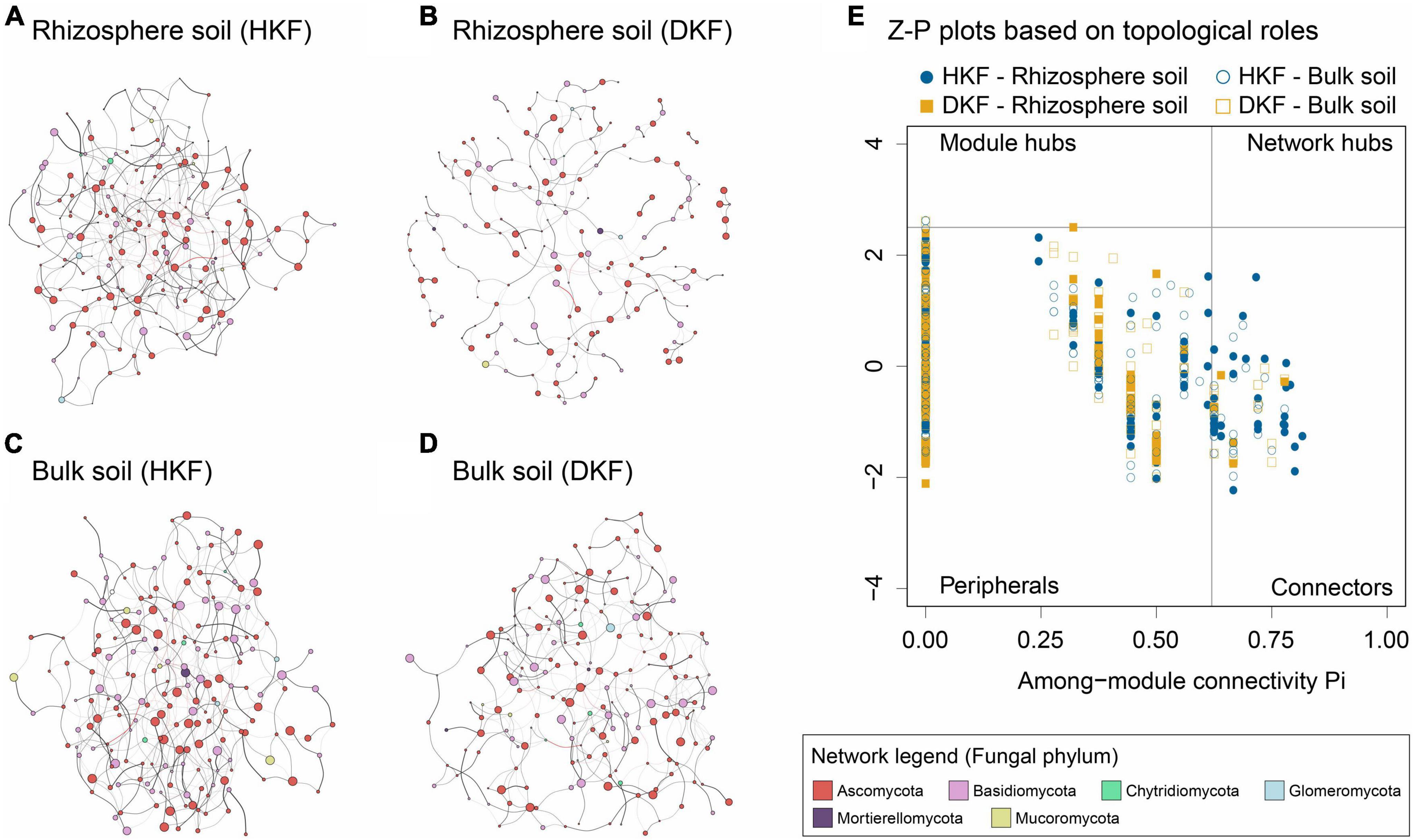
Figure 4. The co-occurrence correlation network analysis of soil mycobiome for the (A,C) healthy Korean fir (HKF) and (B,D) dead Korean fir (DKF) groups. The relative abundance of taxa was represented through the node size and genera through the node color. Positive and negative correlations were indicated as black and red edges, respectively. The edge thickness shows correlation strength. (E) z-plot representing the topological roles of fungal genera based on within-module connectivity (zi) and among-module connectivity (pi). Each point shows a fungal genus in the HKF (blue) and DKF (yellow) soil communities. Filled and blank points indicate rhizospheric and bulk soil, respectively.
The Z-P plot describes the topological roles of the nodes (genus) constituting the microbial network for each group. The two parameters of these Z-P plots are within-module connectivity (zi), representing the connectivity of a node to other nodes within its module, and connectivity among modules (pi), reflecting the connectivity of a node to other modules. In this study, all fungal networks consisted mainly of peripheral nodes having only a few links and almost always to the genus within their modules, followed by connectors indicating close association with multiple modules (Figure 4E). However, there were no network hubs observed in all of the networks. 16.9% of the fungal genus in the HKF group were connectors, whereas, in the DKF group, there was only 5.8%. In the DKF rhizosphere soil network, which showed the weakest network tendency, 96.6% of the total genera were peripheral nodes except for six connectors. The network analysis highlights that reduced complexity and intensity of fungal communities resulting from a decrease in the number of edges, edge density, and connectors were associated with the decline of tree health.
We measured twenty-eight soil physicochemical properties for all collected samples and compared differences between HKF and DKF soil properties (Supplementary Table 4). The concentrations of potassium (K+) and sodium (Na+) ions and available phosphorus (P2O5), sand percentage, and the cation exchange capacity (CEC) of the Korean fir soil differed significantly between the two groups (Supplementary Figure 4). Sand percent (47.03 ± 12.27) belonging to soil physical properties was higher in DKF soil than HKF soil, while P2O5 (14.68 ± 7.71), CEC (38.81 ± 7.42), K+ (0.53 ± 0.43), and Na+ (0.19 ± 0.09) belonging to soil chemical properties were higher in HKF soil. As a result of heatmap correlation analysis, the discriminated soil characteristics except sand percent did not have a significant correlation with individual ECMs, which are major biomarkers (Supplementary Figure 5). However, our Mantel statistics highlight that Sand percent, P2O5, and CEC among the five different physicochemical properties have an overall strong association with soil fungal communities (Supplementary Table 5).
Drivers for changes in mycobiome and environmental variables related to the decline of the health status of Korean fir were evaluated using structural equation modeling (SEM). The decline in the health status of Korean fir and the relative abundance of symbiotrophs in the HKF group had direct and indirect correlations with soil physicochemistry, including the percentages of sand and P2O5 and the CEC of the soil (Figure 5). The decline of the health status of Korean fir had significant direct correlations with the concentrations of K+ ions (λ = −0.550, p < 0.001), P2O5 (λ = −0.516, p < 0.001), and organic matter (OM) (λ = 0.444, p < 0.001) in soil, and the combination of adverse indirect effects of soil chemical properties primarily associated with pH and CEC. The decline in the health status of Korean fir had a direct negative relationship with symbiotrophs (λ = −0.606, p < 0.001), which had a strong negative correlation with saprotrophs (λ = −0.663, p < 0.01).
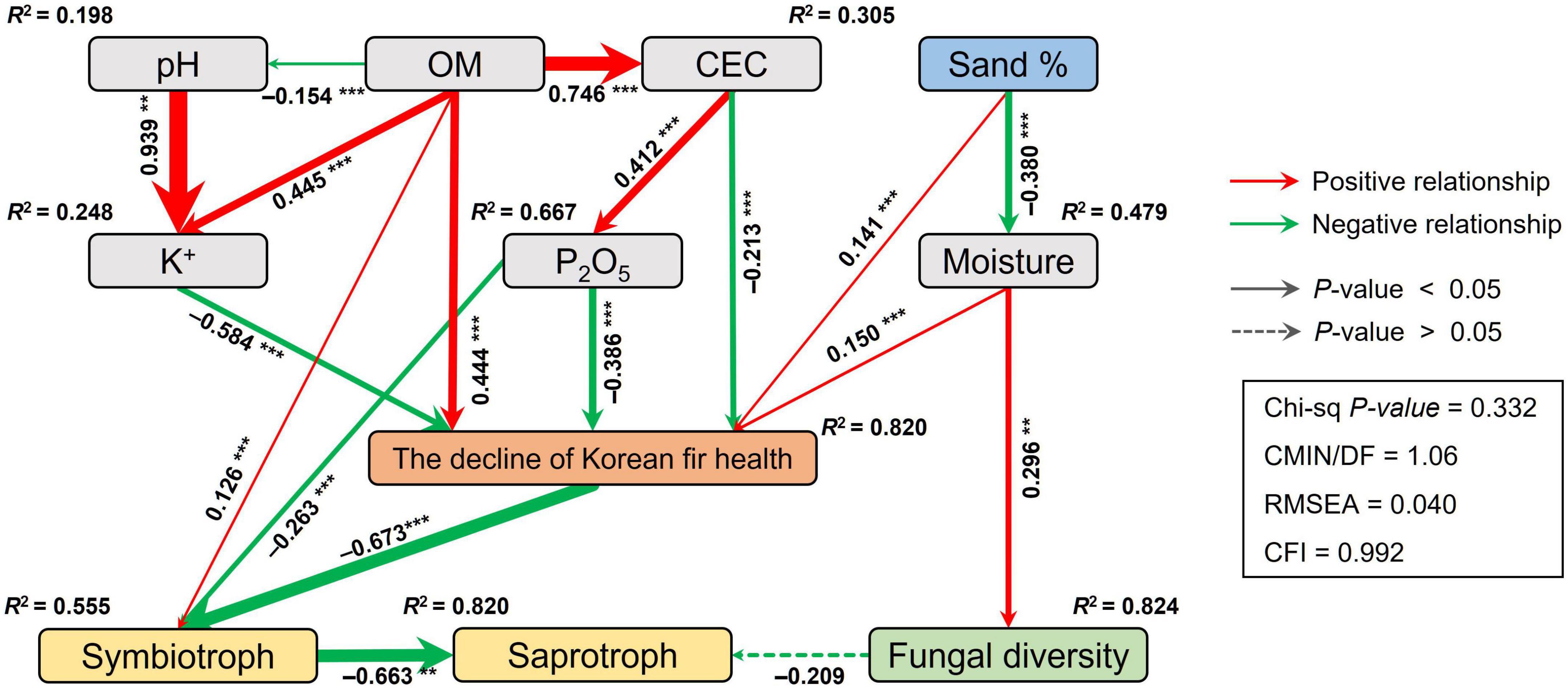
Figure 5. Structural equation model (SEM) illustrating the direct and indirect effects of the soil physico-chemical properties on the decline of Korean fir health and the fungal community. Continuous and dashed arrows represent the significant and non-significant relationships, respectively. The adjacent numbers that are labeled in the same direction as the arrow represent path coefficients, and the width of the arrow is in proportion to the degree of path coefficients. Red and green arrows indicate positive and negative relationships, respectively. R2 values denote the proportion of variance explained by each variable. OM, organic matter; CEC, cation exchange capacity. Significance levels: *p < 0.05, **p < 0.01, ***p < 0.001.
The death of Korean fir is a severe problem for which no clear cause has been ascertained (Ahn and Yun, 2020). Most previous studies have reported associations between tree death and environmental factors, including typhoons (Kim et al., 2017), droughts (Kim et al., 2015), excess moisture (Ahn et al., 2019; Ahn and Yun, 2020) and other climatic changes (Kim et al., 2016; Koo and Kim, 2020). It is necessary to provide appropriate clues for new biological interpretations as conflicts resulting from inconsistent claims of previous studies have created several questions. On the other hand, although the soil microbiota plays an essential role in the forest ecosystem processes (Lladó et al., 2017), the soil microbiome and mycobiome structure of Korean fir trees remain unknown. In addition, plant-associated soil microbial communities are associated strongly with soil health by stimulating plant growth, supplying nutrients, and increasing resistance to biological and abiotic stresses, such as climate change (Berg et al., 2017). Thus, understanding the soil microbiome of the Korean fir and its association with edaphic factors will advance our knowledge about the response of microbial communities to abiotic stresses for such endangered tree species. It will also help design effective alternative strategies for conserving Korean fir by providing new biological perspectives. To the best of our knowledge, this study is the first report about the soil bacterial and fungal structure of Korean fir trees and the association between microbiome and soil physicochemical features.
According to relative abundance and beta diversity analysis, the microbiome changes in Korean fir soils were mainly shaped by the decline of Korean fir health, not by soil compartment niches. In the relative abundance analysis, the Ascomycota phylum increased relatively after tree death, and many ascomycetes are pathogens, both of animals, including humans, and of plants (Berbee, 2001). However, the association of pathogenic fungi belonging to the phylum Ascomycota with the death of fir trees cannot be definitively established.
In particular, the fungal community, the mycobiome, was more affected than the bacterial community. We predict that these fungal community differences are due to differences in fungal composition adapted to the environment before and after tree death. We infer that fungi, which are relatively higher eukaryotes, not only interacted flexibly with Korean fir before tree death but also adapted to the environment better after a large environmental change called death. These observations indicate that the fungal community assembly is comparatively more sensitive than the bacterial community to the stress-induced changes of the fir tree rhizosphere, emphasizing fungi as a good bioindicator of habitat transition (Bai et al., 2018). Bacteria have high plasticity to environmental stresses, which supports the idea that soil bacterial communities differ in their vulnerability to stresses (Classen et al., 2015) and host health status does not affect members of soil microbiota equally (Preiswerk et al., 2018). Moreover, the bacterial community response to abiotic factors observed in our study was similar to that reported in previous studies (Justice et al., 2008).
Our results provided evidence that plant health status can strongly influence fungal community composition in Korean fir soil ecosystems, especially within rhizosphere soils. The rhizosphere mycobiome is closely interconnected to plant health, fitness, and growth, and this result is also supported by previous studies (Anal et al., 2020). Different compartments have different physicochemical properties due to structural differences and exposure to different environmental niches, which can lead to the selective recruitment of microbial communities (Yang et al., 2022). It is consistent with previous studies showing that the microbial diversity of bulk soil was higher than that of the rhizosphere (Xiong et al., 2021).
We expect that these differences are due to fungal diversity, which differs significantly by soil compartment and tree health status. The fungal diversity of the DKF soil sample was slightly higher than that of the HKF sample, which may be attributed to woody plants or crops manufacturing exudates to control an environment suitable for self-growth, which regulates symbiotic soil microorganisms through chemical interaction (Prudence et al., 2021). Fungal diversity appears to be influenced comparatively more by stress-induced rhizosphere changes in the Korean fir soil. However, more research is needed to confirm the evidence of such relationships. These observations indicate that soil serves as a primary reservoir for the plant-associated microbiome, and plants can recruit or filter microbes inhabiting the rhizosphere and endosphere. Alternatively, dead plant roots in DKF soil could create a conducive environment for different fungi groups, especially necrotroph microbes (Prudence et al., 2021).
On the other hand, our study showed that insignificant changes in bacterial diversity between two compartments were observed. According to previous studies, the differences in bacterial communities by compartment were not significant in coniferous soil, unlike other plant species (Ling et al., 2022). Bacterial differences between soil compartments are mainly derived from rhizosphere effects, which depend not only on plant species but also on human agricultural activities such as organic amendments. These agricultural practices, including organic amendments, can reduce rhizosphere effects on chitinolysis, methylotrophy, and methanol oxidation (Ling et al., 2022). These peculiar soil compartment-specific bacterial characteristics of forest ecosystems may be an effect of the absence of practices such as land use, flora, and fertilization regimes. There is not enough research to address this precisely, and it is an issue that must be addressed in future research.
Circle packing and LEfSe analysis at the genus level between HKF and DKF showed that ECMs, including Russula, Sebacina, and Hydnotrya, had the highest abundance in the HKF group. Interestingly, these ECM fungi were present in bulk soil as well as rhizosphere soil. These observations predict that Korean fir roots not only directly interact with the symbiotic fungus in the rhizosphere but also indirectly in a wider area as well. Russula was observed to be one of the most essential and abundant ECM fungi having a symbiotic relationship with diverse higher plants in the mountain rainforest, which conforms to the findings of previous studies (Wang et al., 2015; Ważny and Kowalski, 2017; Bzdyk et al., 2019; Izumi, 2019). Interestingly, Kohout et al. (2018) highlighted that ECMs, including Russula, mostly drove the rapid dynamics in the fungal community composition. In addition to Russula, a previous study has reported Sebacina, as the commonly observed ECM in the forest ecosystem (Long et al., 2016). Hydnotrya has also been reported as one of the most dominant ECM symbiont fungal genera (Leski and Rudawska, 2012), which may be because of rhizodeposits from the live fir tree (Kohout et al., 2018). In contrast, there was a decrease in the abundance of such ECMs in dead plant roots (Wei et al., 2021), which might be attributed to the lack of plant-derived active carbon inputs, including exudates from the dead plant roots (Yu et al., 2020).
The saprotrophs such as Ascocoryne, Loramyces, and Hypochnicium were highly abundant in the DKF group. These finding also supports the fact that saprotrophs are present in extremely low abundance in actively growing plant roots (Kutiel and Shaviv, 1992), and they mainly feed on non-living organic matter, such as dead plant roots (Promputtha et al., 2007). Trichoderma spp., a critical genus specializing in the breakdown of complex compounds, such as lignin and cellulose (Pusz et al., 2021), commonly arising after plant death (Williams and de Vries, 2020), were likewise abundant in DKF soil. These findings indicate a reduction in the abundance of symbiotic fungi in the rhizosphere of the dead tree and an increase in saprotrophic fungi. Nevertheless, pathogenic fungal genera were not as abundant in DKF soil as in HKF soil, consistent with previous findings that the survival of microbes, including ECMs, is related to soil nutrient availability. Therefore, ECMs fungi including Russula, Sebacina, and Phenoliferia have significance as potential biomarkers for fir tree death. Saprotrophs differed at the genus level by soil compartment, and these observations indicate that saprotrophs do not play a role as potential biomarkers for reduced tree health.
Network analyses and Z-P plots were used to understand the relationship patterns of mycobiome both within and between the HKF and the DKF groups in the rhizosphere and bulk soil compartments. The results showed that the DKF networks, which have few keystone taxa of known plant symbionts, are less stable and complex than the HKF networks. In addition, these have fewer topological properties, including transitivity, the number of nodes, total edges, and the ratio of negative edges, than the HKF rhizosphere network, which conforms to previous studies that plants supply high nutrients to microbes, leading to more complex microbial networks (Yang et al., 2021). Additionally, mutual negative interactions indicative of ecological competition can enhance microbiome stability by weakening the destabilizing effects of cooperation (Coyte et al., 2015).
Soil physical properties affect soil chemistry, such as organic nutrients and permeability (Kutiel and Shaviv, 1992), and also cause changes in the soil ecosystem, including host plants and microorganisms (Marschner et al., 2001). Previous studies emphasized that the decline in the health of the Korean fir tree is caused by the interactions of physical damage due to strong winds and typhoons and soil erosion (Seo et al., 2021). Based on the previous studies, changes in the physical characteristics of habitats may have influenced the decline in the health of Korean fir trees and simultaneously caused changes in the soil microbial community, affecting the ECMs in the soil. On the other hand, our study did not show a strong correlation between well-known ECMs and environmental variables (Supplementary Figure 5).
Our SEM analysis revealed that the decline in the health of Korean fir had direct negative correlations with soil nutrient-related characteristics, e.g., K+ ion concentrations, P2O5, and CEC, directly related to OM content in the soil. The K+ ions and P2O5 are well-known essential nutrients for plant growth, and their soil concentrations and availability depend on soil CEC (Brady et al., 2008). Soil moisture, which negatively correlated with the sand percentage in soil, had a positive correlation with the decline in the health of Korean fir, consistent with previous studies suggesting tree death due to excess moisture (Ahn et al., 2019).
Indeed, Mt. Hallasan, the native habitat of Korean fir derived from volcanic ash, has a high ash OM content and phosphoric acid adsorption capacity, preventing the absorption of essential nutrients for plant growth (Hyun, 2011). In addition, Mt. Hallasan soil with low moisture content and easy root extension due to large soil porosity would have influenced the adaptive evolution of Korean fir. We believe that soil properties such as high OM content and increased moisture, caused by various environmental factors mentioned in previous studies, are related to soil nutrients and moisture content and may have had a direct or indirect effect on the decline in the health of Korean fir. Furthermore, we speculate that changes in soil physicochemical properties and the death of Korean fir may have caused changes in soil fungal communities, specifically ECMs.
We acknowledge the following shortcomings in this experiment. First, the study only provides a potential association between the decline of the Korean fir tree and the soil fungal community. Therefore, further research is required on the direct cause between the death phenomenon and the soil microbiome. Second, large sample sizes are needed in future studies to completely address the cause of the decline in tree health and understand the contribution of the Korean fir forest soil microbiome and ECMs in particular. Nevertheless, this study is valuable, being the first to document the decline in the health of the Korean fir tree from an ecological perspective through the association between soil microbiome and soil physicochemical properties. In addition, it has an advantage in that it provides new information for understanding the phenomenon of death in that it is consistent with the results of previous studies.
Our results showed that the soil fungal community was more affected than the bacterial community following the death of Korean fir taking the above information into account. ECMs fungi such as Russula, Sebacina, and Phenoliferia, which dramatically decreased after tree death regardless of soil compartment, are valuable as potential biomarkers. The fungal community of dead Korean fir constituted a weaker soil microbial ecosystem network compared to those of relatively healthy trees and were correlated with changes in soil physicochemical properties known to be associated with the Korean fir decline. Specifically, the decline in the health of Korean fir had strong correlations with organic matter content and soil nutrient-related characteristics such as K+ ion concentrations, P2O5, and CEC. Although this is a baseline study about the soil microbiome and mycobiome of Korean fir trees, our research provides a new microbiological and ecological perspective on the phenomenon of such unknown death of Korean fir.
Fastq files have been deposited into an NCBI Sequence Read Archive (SRA, NCBI, http://www.ncbi.nlm.nih.gov/sra) under the accession numbers PRJNA802974.
MJ, YGJ, S-HK, K-SK, J-GK, and J-HS conceived and designed the experiments. MJ, S-HK, K-SK, J-GK, YGJ, YJJ, Y-JP, M-SK, and KL performed the experiments. MJ, M-JK, D-RJ, and Y-JP analyzed the data. MJ, ST, and J-HS wrote the manuscript. All authors contributed to the study design and conception, wrote, commented, and accepted the final version of the manuscript.
This research was supported by a project to train professional personnel in biological materials of the Ministry of Environment.
We express our special thanks for the support and cooperation of the Leaders in the INdustry-university Cooperation 3.0 project by the Ministry of Education and National Research Foundation of Korea.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffgc.2022.1114390/full#supplementary-material
Abarenkov, K., Nilsson, R.H., Larsson, K.-H., Alexander, I.J., Eberhardt, U., Erland, S., et al. (2010). The UNITE database for molecular identification of fungi–recent updates and future perspectives. New Phytol. 186, 281-285. doi: 10.1111/j.1469-8137.2009.03160.x
Ahn, U. S., Yun, Y. S., and Kim, D. S (2020). A study on the distribution characteristics of korean fir trees (19th Research Report). Jeju: Jeju Island World Natural Heritage.
Ahn, U.S., and Yun, Y.S. (2020). Causes of decline in the Korean fir based on spatial distribution in the Mt. Halla region in Korea: A meta-analysis. Forests 11:391. doi: 10.3390/f11040391.
Ahn, U.S., Kim, D.S., Yun, Y.S., Ko, S.H., Kim, K.S., and Cho, I.S. (2019). The inference about the cause of death of Korean Fir in Mt. Halla through the analysis of spatial dying pattern-Proposing the possibility of excess soil moisture by climate changes. Korean J. Agric. For. Meteorol. 21, 1-28. doi: 10.5532/KJAFM.2019.21.1.1.
Anal, A. K. D., Rai, S., Singh, M., and Solanki, M. K. (2020). “Plant mycobiome: Current research and applications,” in Phytobiomes: Current insights and future vistas, (Berlin: Springer Nature), 81–104.
Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137. doi: 10.3354/ame01753
Bai, Y., Wang, Q., Liao, K., Jian, Z., Zhao, C., and Qu, J. (2018). Fungal community as a bioindicator to reflect anthropogenic activities in a river ecosystem. Front. Microbiol. 9:3152. doi: 10.3389/fmicb.2018.03152
Banerjee, S., and van der Heijden, M. G. (2022). Soil microbiomes and one health. Nat. Rev. Microbiology 21, 1–15.
Berbee, M. L. (2001). The phylogeny of plant and animal pathogens in the Ascomycota. Physiol. Mol. Plant Pathol. 59, 165–187.
Berg, G., Köberl, M., Rybakova, D., Müller, H., Grosch, R., and Smalla, K. (2017). Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 93, fix050. doi: 10.1093/femsec/fix050
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Brady, N. C., Weil, R. R., and Weil, R. R. (2008). The nature and properties of soils. Upper Saddle River, NJ: Prentice Hall.
Bzdyk, R. M., Olchowik, J., Studnicki, M., Nowakowska, J. A., Oszako, T., Urban, A., et al. (2019). Ectomycorrhizal colonisation in declining oak stands on the Krotoszyn Plateau, Poland. Forests 10:30. doi: 10.3390/f10010030
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Classen, A. T., Sundqvist, M. K., Henning, J. A., Newman, G. S., Moore, J. A., Cregger, M. A., et al. (2015). Direct and indirect effects of climate change on soil microbial and soil microbial-plant interactions: What lies ahead? Ecosphere 6, 1–21. doi: 10.1890/ES15-00217.1
Coyte, K. Z., Schluter, J., and Foster, K. R. (2015). The ecology of the microbiome: Networks, competition, and stability. Science 350, 663–666. doi: 10.1126/science.aad2602
Dubey, A., Malla, M. A., Khan, F., Chowdhary, K., Yadav, S., Kumar, A., et al. (2019). Soil microbiome: A key player for conservation of soil health under changing climate. Biodivers. Conserv 28, 2405–2429. doi: 10.1007/s10531-019-01760-5
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. U.S.A. 112, E911–E920.
Ferrer, C., Colom, F., Frasés, S., Mulet, E., Abad, J. L., and Alió, J. L. (2001). Detection and identification of fungal pathogens by PCR and by ITS2 and 5.8 S ribosomal DNA typing in ocular infections. J. Clin. Microbiol. 39, 2873–2879. doi: 10.1128/JCM.39.8.2873-2879.2001
Grace, J. B. (2006). Structural equation modeling and natural systems. Cambridge: Cambridge University Press.
Gwon, J.-H., Sin, M.-K., Kwon, H. J., and Song, H.-K. (2013). A study on the forest vegetation of Jirisan National Park. J. Korean Soc. Environ. Eng. 16, 93–118. doi: 10.13087/kosert.2013.16.5.93
Hannula, S. E., Heinen, R., Huberty, M., Steinauer, K., De Long, J. R., Jongen, R., et al. (2021). Persistence of plant-mediated microbial soil legacy effects in soil and inside roots. Nat. Commun. 12, 1–13. doi: 10.1038/s41467-021-25971-z
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466. doi: 10.1128/jcm.26.11.2465-2466.1988
Hyun, H. N. (2011). “Soil environment, a key to open up Jeju society and culture,” in Proceedings of the korean society of environmental agriculture, Gyeonggi-do.
Ihm, B.-S., Lee, J.-s, Kim, M.-H., and Kim, H.-S. (2000). A comparative study on the habitat of abies koreana wilson between Mt. Jiri and Mt. Halla. Plant Resour. 3, 138–147.
Ihrmark, K., Bödeker, I., Cruz-Martinez, K., Friberg, H., Kubartova, A., Schenck, J., et al. (2012). New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 82, 666–677. doi: 10.1111/j.1574-6941.2012.01437.x
Izumi, H. (2019). Temporal and spacial dynamics of metabolically active bacteria associated with ectomycorrhizal roots of Betula pubescens. Biol. Fertility Soils 55, 777–788. doi: 10.1007/s00374-019-01393-4
Janda, J. M., and Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Justice, S. S., Hunstad, D. A., Cegelski, L., and Hultgren, S. J. (2008). Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 6, 162–168. doi: 10.1038/nrmicro1820
Kim, E.-S., Oh, C. H., Park, H. C., Lee, S.-H., Choi, J., Lee, S.-H., et al. (2016). Disturbed regeneration of saplings of Korean fir (Abies koreana Wilson), an endemic tree species, in Hallasan National Park, a UNESCO Biosphere Reserve, Jeju Island, Korea. J. Mar. Isl. Cult. 5, 68–78. doi: 10.1016/j.imic.2016.02.001
Kim, J.-K., Koh, J.-G., Yim, H.-T., and Kim, D.-S. (2017). Changes of spatial distribution of Korean fir forest in Mt. Hallasan for the past 10 years (2006, 2015). Korean J. Environ. 31, 549–556. doi: 10.13047/KJEE.2017.31.6.549
Kim, N.-S., Han, D., Cha, J.-Y., Park, Y.-S., Cho, H.-J., Kwon, H.-J., et al. (2015). A detection of novel habitats of Abies koreana by using species distribution models (SDMs) and its application for plant conservation. J. Korean Soc. Environ. Eng. 18, 135–149. doi: 10.13087/kosert.2015.18.6.135
Kim, Y.-S., Chang, C.-S., Kim, C.-S., and Gardner, M. (2011). “Abies koreana. The IUCN Red List of Threatened Species 2011,” in The IUCN red list of threatened species, (Incheon: National Institute of Biological Resources.).
Kohout, P., Charvátová, M., Štursová, M., Mašínová, T., Tomšovskı, M., and Baldrian, P. (2018). Clearcutting alters decomposition processes and initiates complex restructuring of fungal communities in soil and tree roots. ISME J. 12, 692–703. doi: 10.1038/s41396-017-0027-3
Koo, K. A., and Kim, D.-B. (2020). Review forty-year studies of Korean fir (Abies koreana Wilson). Korean J. Environ. 34, 358–371. doi: 10.13047/KJEE.2020.34.5.358
Kumar, I., Mondal, M., Gurusamy, R., Balakrishnan, S., and Natarajan, S. (2019). “Plant-Microbiome interaction and the effects of biotic and abiotic components in agroecosystem,” in Microbial Interventions in Agriculture and Environment, (Berlin: Springer), 517–546.
Kurtz, Z. D., Müller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., and Bonneau, R. A. (2015). Sparse and compositionally robust inference of microbial ecological networks. PLoS Comp. Biol. 11:e1004226. doi: 10.1371/journal.pcbi.1004226
Kutiel, P., and Shaviv, A. (1992). Effects of soil type, plant composition and leaching on soil nutrients following a simulated forest fire. For. Ecol. Manag. 53, 329–343. doi: 10.1016/0378-1127(92)90051-A
Layeghifard, M., Hwang, D. M., and Guttman, D. S. (2018). “Constructing and analyzing microbiome networks in R,” in Microbiome analysis, (Berlin: Springer), 243–266. doi: 10.1007/978-1-4939-8728-3_16
Leski, T., and Rudawska, M. (2012). Ectomycorrhizal fungal community of naturally regenerated European larch (Larix decidua) seedlings. Symbiosis 56, 45–53. doi: 10.1007/s13199-012-0164-4
Ling, N., Wang, T., and Kuzyakov, Y. (2022). Rhizosphere bacteriome structure and functions. Nature communications 13, 1–13.
Lladó, S., López-Mondéjar, R., and Baldrian, P. (2017). Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 81, e63–e16. doi: 10.1128/MMBR.00063-16
Long, D., Liu, J., Han, Q., Wang, X., and Huang, J. (2016). Ectomycorrhizal fungal communities associated with Populus simonii and Pinus tabuliformis in the hilly-gully region of the Loess Plateau, China. Sci. Rep. 6, 1–10. doi: 10.1038/srep24336
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2011). UniFrac: An effective distance metric for microbial community comparison. ISME J. 5, 169–172.
Marschner, P., Yang, C.-H., Lieberei, R., and Crowley, D. E. (2001). Soil and plant specific effects on bacterial community composition in the rhizosphere. Soil Biol. Biochem. 33, 1437–1445. doi: 10.1016/S0038-0717(01)00052-9
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fung. Ecol. 20, 241–248.
NIAST (2010). “Method of soil and plant analysis,” in Rural development administration, (Thrissur: NIAST).
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2006). The smallest of all worlds: Pollination networks. J. Theor. Biol. 240, 270–276. doi: 10.1016/j.jtbi.2005.09.014
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Preiswerk, D., Walser, J.-C., and Ebert, D. (2018). Temporal dynamics of microbiota before and after host death. ISME J. 12, 2076–2085. doi: 10.1038/s41396-018-0157-2
Promputtha, I., Lumyong, S., Dhanasekaran, V., McKenzie, E. H. C., Hyde, K. D., and Jeewon, R. (2007). A phylogenetic evaluation of whether endophytes become saprotrophs at host senescence. Microb. Ecol. 53, 579–590. doi: 10.1007/s00248-006-9117-x
Prudence, S. M., Newitt, J. T., Worsley, S. F., Macey, M. C., Murrell, J. C., Lehtovirta-Morley, L. E., et al. (2021). Soil, senescence and exudate utilisation: Characterisation of the Paragon var. spring bread wheat root microbiome. Environ. Microbiome 16, 1–21. doi: 10.1186/s40793-021-00381-2
Pusz, W., Baturo-Cieśniewska, A., Kaczmarek-Pieńczewska, A., Patejuk, K., and Czarnota, P. (2021). Is the survivability of silver fir under condition of strong ungulate pressure related to mycobiota of bark-stripping wounds? Forests 12:976. doi: 10.3390/f12080976
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rosseel, Y. (2012). lavaan: An R package for structural equation modeling. Journal of statistical software 48, 1–36. doi: 10.3389/fpsyg.2014.01521
Schofield, R., and Taylor, A. W. (1955). The measurement of soil pH. Soil Sci. Soc. Am. J. 19, 164–167.
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, 1–18. doi: 10.1186/gb-2011-12-6-r60
Seo, J.-W., Choi, E.-B., Park, J.-H., Kim, Y.-J., and Lim, H.-I. (2021). The Role of Aging and Wind in Inducing Death and/or Growth Reduction in Korean Fir (Abies Koreana Wilson) on Mt. Halla, Korea. Atmosphere 12:1135. doi: 10.3390/atmos12091135
Seo, J.-W., Kim, Y.-J., Choi, E.-B., Park, J.-H., and Kim, J.-H. (2019). Investigation of death years and inter-annual growth reduction of Korean firs (Abies koreana) at Yeongsil in Mt. Halla. J. Korean Soc. Environ. Eng. 22, 1–14. doi: 10.13087/kosert.2019.22.3.1
Smalla, K., Sessitsch, A., and Hartmann, A. (2006). The Rhizosphere:‘soil compartment influenced by the root’. Oxford: Blackwell Publishing Ltd.
Trivedi, P., Leach, J. E., Tringe, S. G., Sa, T., and Singh, B. K. (2020). Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621.
Turenne, C. Y., Sanche, S. E., Hoban, D. J., Karlowsky, J. A., and Kabani, A. M. (1999). Rapid identification of fungi by using the ITS2 genetic region and an automated fluorescent capillary electrophoresis system. J. Clin. Microbiol. 37, 1846–1851. doi: 10.1128/JCM.37.6.1846-1851.1999
Wang, P., Zhang, Y., Mi, F., Tang, X., He, X., Cao, Y., et al. (2015). Recent advances in population genetics of ectomycorrhizal mushrooms Russula spp. Mycology 6, 110–120. doi: 10.1080/21501203.2015.1062810
Ważny, R., and Kowalski, S. (2017). Ectomycorrhizal fungal communities of silver-fir seedlings regenerating in fir stands and larch forecrops. Trees 31, 929–939. doi: 10.1007/s00468-016-1518-y
Wei, F., Feng, H., Zhang, D., Feng, Z., Zhao, L., Zhang, Y., et al. (2021). Composition of rhizosphere microbial communities associated with healthy and Verticillium wilt diseased cotton plants. Front. Microbiol. 12:618169. doi: 10.3389/fmicb.2021.618169
Williams, A., and de Vries, F. T. (2020). Plant root exudation under drought: Implications for ecosystem functioning. New Phytol. 225, 1899–1905. doi: 10.1111/nph.16223
Xiong, C., Zhu, Y. G., Wang, J. T., Singh, B., Han, L. L., Shen, J. P., et al. (2021). Host selection shapes crop microbiome assembly and network complexity. New Phytol. 229, 1091–1104. doi: 10.1111/nph.16890
Yang, H., Yang, Z., Wang, Q.-C., Wang, Y.-L., Hu, H.-W., He, J.-Z., et al. (2022). Compartment and Plant Identity Shape Tree Mycobiome in a Subtropical Forest. Microbiol. Spectr. 10, e1347–e1322. doi: 10.1128/spectrum.01347-22
Yang, Y., Shi, Y., Kerfahi, D., Ogwu, M. C., Wang, J., Dong, K., et al. (2021). Elevation-related climate trends dominate fungal co-occurrence network structure and the abundance of keystone taxa on Mt. Norikura, Japan. Sci. Total Environ. 799:149368. doi: 10.1016/j.scitotenv.2021.149368
Youssef, R., Kanazawa, S., and Chino, M. (1989). Distribution of microbial biomass across the rhizosphere of barley (Hordeum vulgare L.) in soils. Biol. Fertility Soils 7, 341–345.
Keywords: Abies koreana, mycobiome, microbiome, symbiotic fungi, Mt. Hallasan, structural equation modeling, Korean fir death
Citation: Jeong M, Tagele SB, Kim M-J, Ko S-H, Kim K-S, Koh J-G, Jung D-R, Jo YJ, Jung YG, Park Y-J, Kim M-S, Lim K, and Shin J-H (2023) The death of Korean fir (Abies koreana) affects soil symbiotic fungal microbiome: Preliminary findings. Front. For. Glob. Change 5:1114390. doi: 10.3389/ffgc.2022.1114390
Received: 02 December 2022; Accepted: 29 December 2022;
Published: 12 January 2023.
Edited by:
Shri Kant Tripathi, Mizoram University, IndiaReviewed by:
Hang Wang, Southwest Forestry University, ChinaCopyright © 2023 Jeong, Tagele, Kim, Ko, Kim, Koh, Jung, Jo, Jung, Park, Kim, Lim and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jae-Ho Shin,  amhzaGluQGtudS5hYy5rcg==
amhzaGluQGtudS5hYy5rcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.