
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 30 May 2024
Sec. Evolutionary and Population Genetics
Volume 12 - 2024 | https://doi.org/10.3389/fevo.2024.1360714
This article is part of the Research TopicChallenges and Prospects for Conservation Genetics at XXI CenturyView all 8 articles
 Natalia Mannise1,2
Natalia Mannise1,2 Mariana Cosse1*
Mariana Cosse1* Gonzalo Greif3
Gonzalo Greif3 Nadia Bou1
Nadia Bou1 Carlos Robello3,4Susana González1
Carlos Robello3,4Susana González1 Andres Iriarte2*
Andres Iriarte2*Pampas and crab-eating foxes are medium-sized canids living in sympatry in the middle east of South America. Studies on the diet composition of these species provide a deep understanding of their ecological roles in the ecosystem structure and regulation. Using the metabarcoding technique, we analyzed the diet of both fox species in order to identify the vertebrate taxa included as food items. A fragment of the 12S ribosomal gene of the mtDNA was amplified using DNA extracted from 27 scat samples collected in south-central Uruguay during cold (June 2015) and warm (January – April 2016) seasons. A fox DNA blocking primer was designed to minimize the host amplicon products, and pooled samples were sequenced through paired-end reads (100 bp library) on a MiSeq Illumina Platform. The generated sequences were compared to a reference database built with sequences available in GenBank. In concordance with previous studies using traditional methods, we found that the most common food taxon were rodents. Qualitative differences in diet composition between both fox species were identified. Armadillo species were only found in pampas fox diet, while a greater variety of amphibians and birds were detected in crab-eating fox feces. Additionally, an innovative approach to differentiate between real and artifact sequences was employed. This method was based on comparing mutations at conserved and non-conserved positions within the secondary structure of the 12S rRNA, combined with network sequence reconstruction. Our results demonstrate the efficacy of the methodology in detecting the food species present in both fox diets, enabling the evaluation of intraspecific diversity among these species and facilitating the discarding of sequencing errors. This makes the methodology applicable to a wide range of studies.
The comprehension of trophic interactions is necessary for understanding ecosystem structure and functionality, it is important in many areas of ecology such as conservation biology, agroecology, and bioenergetics (Pompanon et al., 2012; Shehzad et al., 2012). The suitable identification of the taxa consumed is difficult to obtain from micro and macroscopic evaluations of feces. Soft tissues, e.g., hair or fur, are often degraded, and hard parts are frequently fragmented, contributing to difficult identifications. It depends on expert skills for taxon determination from semi-digested food items (Pompanon et al., 2012). Fecal DNA-based dietary assessment techniques are demonstrably better for detailed taxonomic identification in comparison with traditional methods (Deagle and Tollit, 2007; Pompanon et al., 2012; Deagle et al., 2013; Quemere et al., 2013; De Barba et al., 2014; Buglione et al., 2018). Additionally, diet comparisons among species from the same genus were developed using metabarcoding approaches (Lopes et al., 2015, 2020). Throughout the advent of high-throughput sequencing (HTS) technologies and the increasing number of sequences that could be obtained, the term metabarcoding has recently emerged (Shendure and Ji, 2008; Taberlet et al., 2012). Originally introduced for identifying multiple taxa using DNA isolated from environmental samples, such as soil, water, or feces (Taberlet et al., 2012), DNA metabarcoding has been increasingly utilized for non-invasive biodiversity assessment (Deiner et al., 2017). Despite its high potential, certain traits need to be considered: 1) DNA is typically degraded in environmental samples, and 2) several taxa must be amplified during the same PCR reaction (Taberlet et al., 2012). For taxonomic identification, the sequences obtained are compared to a suitable and trustable reference sequence database (Taberlet et al., 2012). Primers designed to amplify a short DNA fragment in various organisms are highly recommended; thus, they will allow the amplification of degraded fecal DNA from multiple species in the same PCR (Taberlet et al., 2012; Staats et al., 2016). In fecal samples, host DNA is present in higher amounts, and food target concentration differences need to be considered in the data analysis steps throughout this technique (Pompanon et al., 2012). Therefore, it is recommended to employ strategies that minimize the recovery of host DNA, thereby increasing the likelihood of obtaining sequences from prey items present in low abundance. Blocking oligonucleotides have been successfully used in other DNA metabarcoding feeding studies and their use reduced the host DNA (Vestheim and Jarman, 2008; De Barba et al., 2014).
DNA metabarcoding is a non-invasive manner to approach trophic studies at a population scale in a relatively short time, allowing researchers to analyze tens or hundreds of samples simultaneously with a precise identification of food species (Monterroso et al., 2019). Furthermore, it could also be possible to analyze the intraspecific genetic variability of food items identified (Elbrecht et al., 2018). Methods based on sequence abundance have previously been used to explore the populations’ genetic diversity and communities from environmental samples (Elbrecht et al., 2018; Adams et al., 2019; Zizka et al., 2020). These bioinformatic strategies, capable of distinguish between true variants, which are more abundant, and sequencing noise, have been developed to extract haplotype information from metabarcoding studies (Zizka et al., 2020). To identify genuine genetic variations, it is advisable to incorporate positive controls, and utilize multiple replicates (Adams et al., 2019). Subsequently, the reduction of erroneous sequences can be achieved through data denoising and the application of rigorous filtering thresholds (Tsuji et al., 2020; Zizka et al., 2020).
The development and continuous improvement of non-invasive strategies have become increasingly significant in addressing the biodiversity crisis. The ability to detect species present in different areas, new species, cryptic species, and intraspecific diversity by the sequence analysis of prey DNA in generalist predators’ feces, has led to the idea of feces as ‘biodiversity capsules’ (Boyer et al., 2015). Consequently, evaluating the effectiveness of genetic markers used in dietary analyses is crucial. Within fecal samples, this evaluation is particularly important for accurately identifying taxonomic identities and distinguishing authentic genetic variability from low-quality, artifacts, and errors encountered during high-throughput sequencing (HTS).
It is essential to understand the diet of carnivores in order to evaluate their role in resource use and competition (Klare et al., 2011). Medium-sized carnivore species, such as Canidae, often play a crucial role in maintaining the environment’s function, structure, and dynamics, such as controlling the size of prey populations (Prugh et al., 2009; Roemer et al., 2009). In particular, canids can adapt to resource changes over time and space (Farias and Kittlein, 2008). The crab-eating fox, Cerdocyon thous, and the pampas fox, Lycalopex gymnocercus, live in sympatry in southern Brazil, northern Argentina, Paraguay, and Uruguay (Di Bitetti et al., 2009; Bossi et al., 2019). They are mesopredator species with omnivorous generalist diets with similar behavior and are considered conflictive species throughout they distribution range (Rodríguez-Mazzini and Molina-Espinosa, 2000; Di Bitetti et al., 2009; Bossi et al., 2019). Previously published feeding studies on both fox species show similar diet compositions and temporal partitioning of resource utilization (Vieira and Port, 2007; Faria-Correa et al., 2008; Di Bitetti et al., 2009; Bossi et al., 2019). However, comparing them is challenging due to differences in taxonomic resolution among the studies (Hernández Rodriguez, 2007; Faria-Correa et al., 2008; Cravino, 2014).
In this work, we aim to develop an approach for the precise detection of vertebrate taxa present in the diet of crab-eating and pampas foxes. We utilized the partial sequence of the mitochondrial 12S ribosomal RNA gene (12S rRNA) and a DNA metabarcoding technique to achieve deep and comparable taxonomic levels. Additionally, we employed two complementary techniques to discriminate between real and artifact sequence variants. Firstly, a network sequence reconstruction technique was used to evaluate the intraspecific diversity of food species and to discard sequencing errors. Secondly, we developed a methodology to validate the sequence changes by comparing of observed mutations in conserved and non-conserved positions within the secondary structure of the 12S rRNA (Springer and Douzery, 1996). These two approaches may serve as useful tools not only for feeding ecology assessments but also for biodiversity surveys and the evaluation of intraspecific genetic variation in environmental samples.
The study was conducted in the Coronilla Hill within the “Reserva Natural Salus” (34° 23’37.8168” S; 55° 19’43.05” W) of south-central Uruguay (Figure 1). This is a 13 km2 hilly area characterized by a mosaic of woodlands, shrublands, and grasslands. Fecal samples were collected opportunistically from various locations in the study area during 2015-2016, in both cold (June) and warm (January-April) seasons. The field surveys were conducted over three days, and five field assistants were involved in feces collection. Only fresh carnivore feces, identified by their low-humidity signs and morphogenic characteristics, were sampled. These samples were stored in individual plastic tubes in the freezer until DNA extraction to avoid contamination from other environmental sources (Pompanon et al., 2012; De Barba et al., 2014). The three canid species inhabiting the study area are crab-eating fox (C. thous), pampas fox (L. gymnocercus) and domestic dog (Canis lupus familiaris).
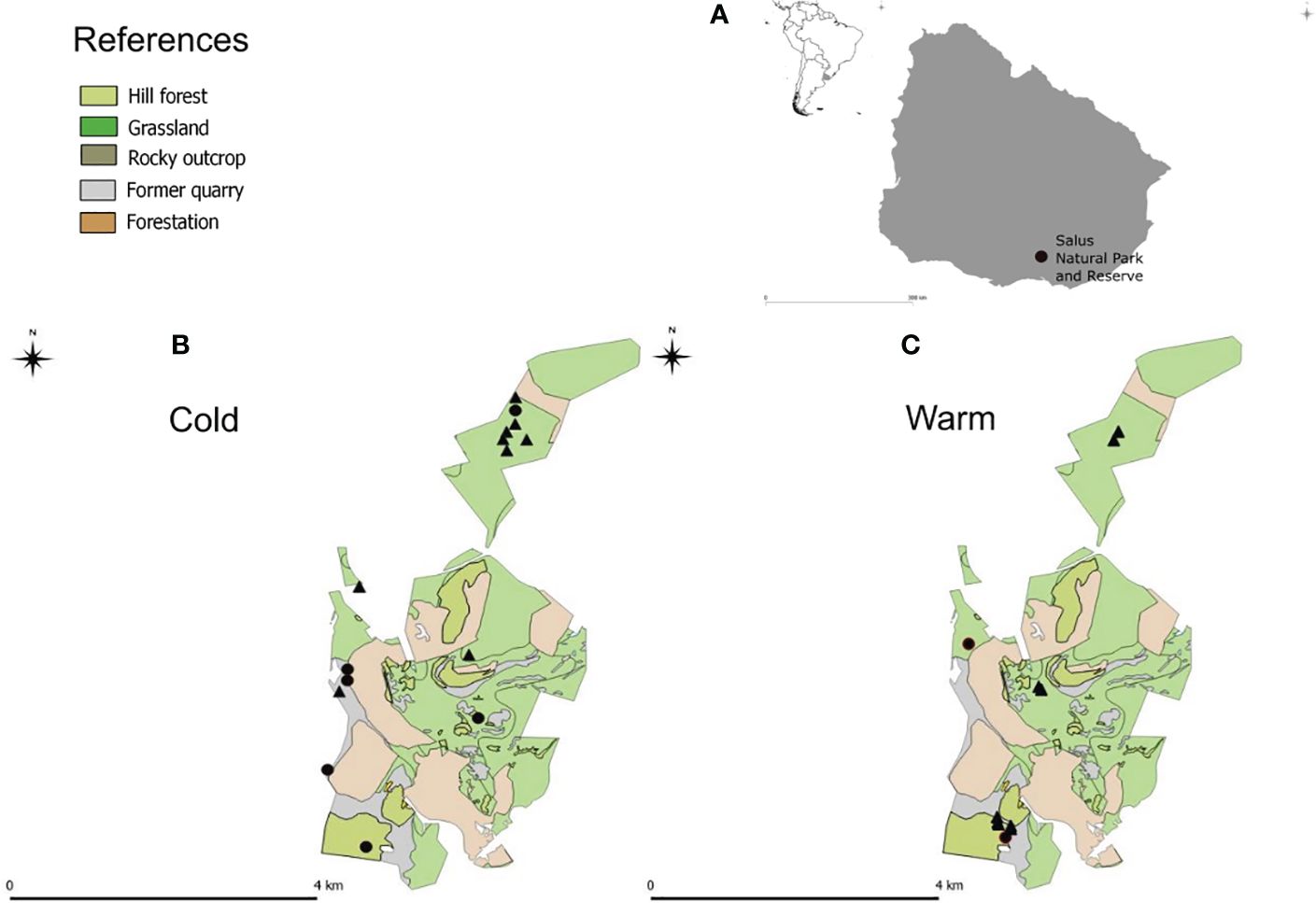
Figure 1 Geographic distribution of collected samples. (A) shows the localization of the sampling area. Pampas fox (triangle) and crab-eating fox (circle) sample locations are indicated for cold (B) and warm (C) seasons in a land cover map.
We extracted the DNA as soon as the samples arrived at the laboratory (3 to 5 days after collection) using the DNA Stool™ Minikit (Qiagen, Hilden, Germany), following manufacturer’s instructions. The feces were disintegrated using sterile tweezers and scissors, thus taking parts of the interior and exterior of the feces in order to homogenize the material for DNA extractions (De Barba et al., 2014).
Donor species were identified using a three TaqManTM probe real-time PCR assay (Cosse et al., 2017), with species-specific dyes included for crab-eating fox C. thous, pampas fox L. gymnocercus, and domestic dog C. lupus familiaris detection. PCR amplifications were conducted on Rotor-Gene ™6000 (Corbett) with SensiFastprobe™ PCR master mix (Bioline, London, UK) following Cosse et al. (Cosse et al., 2017) procedures.
A fox-blocking oligonucleotide was developed following De Barba et al. (2014) and Vestheim and Jarman (15) to avoid host DNA amplification from feces. The fox-blocking oligonucleotide developed with a phosphate (PHO) 3’ extreme modification (5´-CCACTATGCTTAGCCTTAAACGTAAATGATTYTAT PHO- 3’) was included in the 12S fragment PCR from fecal DNA (Section 2.2), using the primer set 12S-V5, which amplifies a fragment with an average length of 98 bp (Riaz et al., 2011). The PCR consisted of 10µl final volume with 0.04U of Taq polymerase (Invitrogen, USA), 1X Taq buffer, 1.5mM of MgCl, 0.2mg/mL of BSA (Bovine Serum Albumin), 0.05mM of dNTPs, 0.1 µM of forward and reverse primers, 1.6µM of fox blocking primer and a 4-8ng/µl final concentration of DNA. The thermal profile started with a denaturalization step of 94°C for 3 minutes, followed by 35 cycles of 94°C for 30 seconds and 58°C for 90 seconds. We used 40 to 45 cycles in degraded fecal DNA. Positive and negative controls were included in each PCR reaction. A second round of indexing PCR amplifications was conducted to add the Nextera indexes for sequencing on a MiSeq Illumina™ platform. The sequencing library preparation followed the 16S sample preparation guide for the Illumina MiSeq system. Each PCR from the same fox species and taken in the same season shared the same index.
Amplification success was confirmed by visualizing PCR products in 2% agarose electrophoresis using Goodview™ (SBS Genetech, Beijing) nucleic acid stain. PCR products were then purified using 0.8U/µl of Exonuclease I and 0.2U/µl of Thermosensitive Alkaline Phosphatase (FastAP) enzymes (ThermoFisher Scientific™, USA); the thermal profile was 37°C for 1 hour and 30 minutes followed by 5 minutes at 75°C. PCR-purified products were quantified using a Nanodrop TMND-1000 UV-vis Spectrophotometer (Nano-Drop Technologies, Inc., Wilmington, DE, USA) and then pooled in equimolar concentrations. Thus, four pools were generated that included libraries prepared from crab-eating and pampas foxes’ feces collected in warm and cold seasons.
Paired-end (2X 100) sequencing was performed using the Illumina MiSeq platform (Illumina, Inc., San Diego, CA, USA), available at the sequencing facility of the Institut Pasteur, Montevideo. The raw reads obtained were trimmed based on quality (Phred score ≥ 30) using SICKLE software (Joshi and Fass, 2011). PCR primers were removed, applying a locally developed script. Paired-end reads were merged using the PEAR program (Zhang et al., 2014), and the consensus sequences were determined.
The reference database was built using the available sequences of the 12S rRNA mitochondrial gene in GenBank (www.ncbi.nlm.nih.gov) in December 2020, including complete vertebrate mitochondrial genomes and partial sequences. The final dataset comprised 306,362 sequences from 28,270 different vertebrate species. Complete taxonomy information for each sequence included in the reference database was retrieved from Genbank. The reference database used is available at Zenodo (zenodo.org/records/10731091), and the raw reads are accessible from the SRA database under BioProject accession PRJNA1082626.
The pipeline described was completed for each experiment and was implemented through locally developed scripts. A detailed description of the bioinformatics workflow was included as supplementary data (Supplementary Figure S1, Supplementary Data 1). First, non-redundant consensus sequences (consensus from paired-end reads) were used for taxonomic identification based on Blast against the local reference database. Thus, each non-redundant consensus sequence was used as query and, the most similar reference sequences (best hits) were defined using the highest blast score. Second, taxonomic information for each best hit was retrieved and further considered. Finally, the list of Molecular Operational Taxonomic Units (MOTUs) comprises species derived from the best hits described in the studied region with a minimum of ten supporting reads. Additionally, higher taxonomic levels such as Genera, Family, or Order were assigned when matched species in the database do not occur in the study area, but there is a species of the same taxon in it. Following Turon et al. (2020), we used networks to analyze observed patterns of variations in generated reads. The reads obtained from the same MOTU in each pool were aligned using MUSCLE (Edgar, 2004). Then, networks were built using the minimum spanning method (Bandelt et al., 1999) in PopArt (Leigh and Bryant, 2015), as described in Supplementary Data 2. Variants with low abundance and only one-point mutations from highly abundant central variants are classified as non-biological (Moreno et al., 2016; Turon et al., 2020). These variants, which arise due to PCR bias, sequencing errors, or degradation (Turon et al., 2020; Zizka et al., 2020), occur punctually in different reads and consequently are highly dispersed in the network and have low relative frequencies. Conversely, highly abundant sequences in central network positions (centroids) are considered genuine biological sequences.
To further support our distinction between erroneous sequences and real biological intra-MOTU variants, we analyzed the pattern of observed mutations in conserved and non-conserved positions. Highly conserved positions within the 12S rRNA sequence were identified by referring to available rRNA secondary structures on the Rfam website (rfam.xfam.org) (Kalvari et al., 2020) (Supplementary Data 3). We reasoned that these specific sequence positions remain stable due to the constraints imposed by purifying selection on the structural integrity. Therefore, variations observed within conserved regions were deemed more likely to be errors, and were utilized to estimate the frequency of noisy variants per site. We estimated the overall mean p-distance for conserved and non-conserved positions using MEGA X (Tamura et al., 2011). Finally, we compared the network reconstructions (see above) and mean p-distance for conserved and non-conserved positions for the most frequent food species identified in each pool.
A total of 80 samples were collected, forty-four were collected during the cold season and thirty-six during the warm season. Six crab-eating and ten pampas fox feces were assigned through molecular methods from samples collected in the cold season; meanwhile, two crab-eating fox and nine pampas fox feces were identified from samples collected in the warm season (Figure 1). The remaining samples either corresponded to another carnivore species, or the DNA was too degraded to be amplified. The sequences of pampas and crab-eating foxes for the 12S rRNA gene fragment were deposited in the GenBank database (crab-eating fox: accession numbers MT272186 and MT272187; pampas fox: accession numbers MT272188 and MT272189).
We assessed that the fox-blocking oligonucleotide reduced the amplification of donor sequences to 4.7% and 24.0% of the generated reads in crab-eating foxes, and to 6.5% and 16.4% in pampas foxes during the cold and warm seasons, respectively. Therefore, for the food item identification analyses, after filtering and reads processing, we retained a total of 1,395 non-ambiguous consensus sequences in pool I (crab-eating fox in cold sampling), 1,242 in pool II (crab-eating fox in warm sampling), 2,125 in pool III (pampas fox in cold sampling), and 999 in pool IV (pampas fox in warm sampling).
The Blast search results show a distribution of identity values of best hits ranging from 93 to 100% (Supplementary Figure S2). Mammals showed higher read and variant counts across all cases. During the cold season, crab-eating fox feces revealed the presence of cavy (Cavia aperea) and cow (Bos taurus). A wide variety of toads taxa were detected in the crab-eating fox feces. Specifically, during the warm sampling period, the genus Rhinella was detected, while Pseudis, Leptodactylus, and Physalaemus were identified only in the cold season samples (Table 1). Several bird food items were also detected in crab-eating fox feces. The food items with the highest number of reads during cold and warm seasons were Cavia aperea and Lepus europaeus, respectively (Table 1). On the other hand, in pampas fox feces, the food items with the highest number of reads were Cavia aperea during the cold season and Mus sp. during the warm seasons sampling (Table 1). Other items identified in pampas fox feces include three armadillo species, all of which are present in the study area. Additionally, only one amphibian species and one Passeriform bird MOTU were identified in pampas fox feces during the cold season (Table 1).
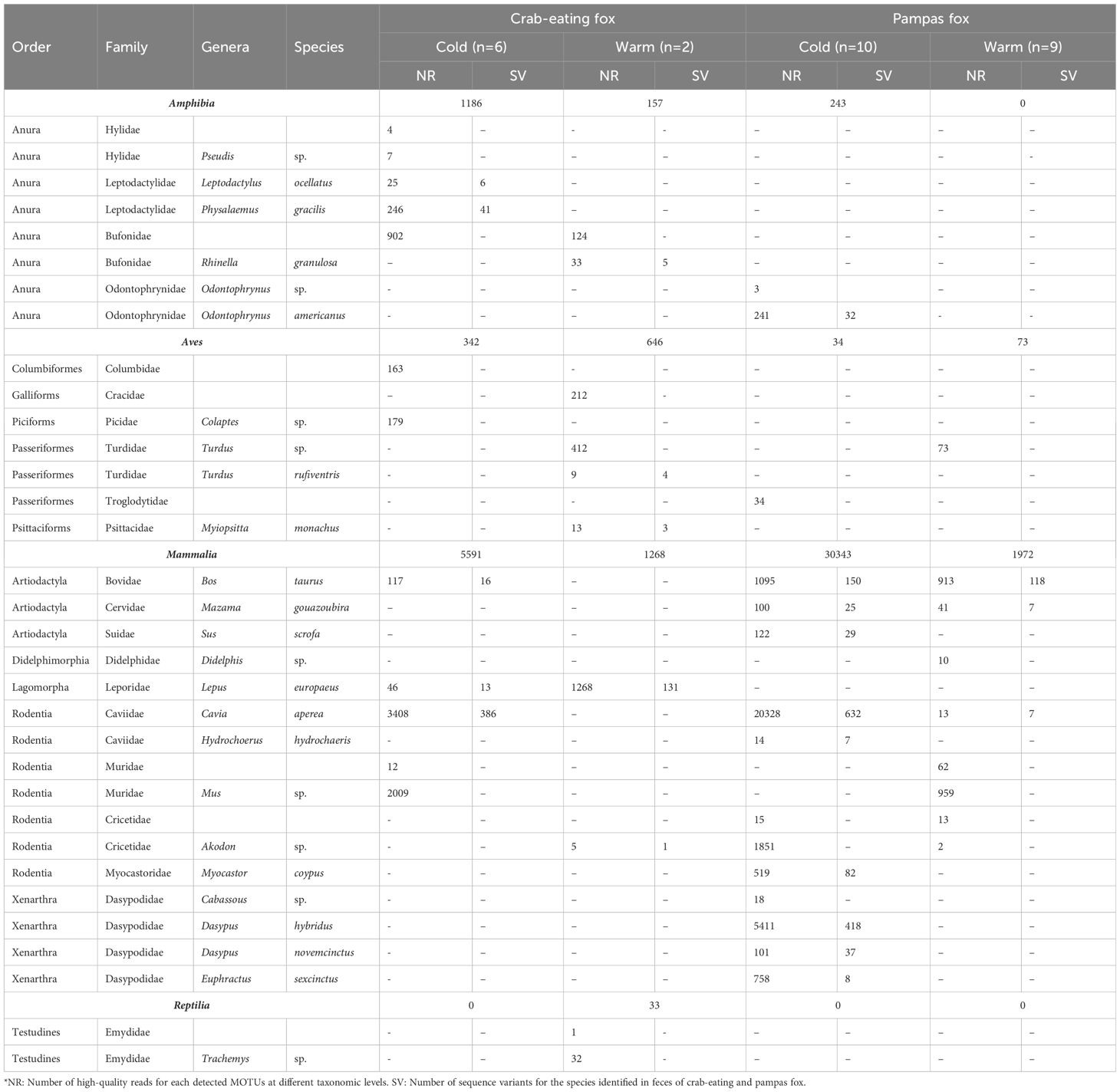
Table 1 Summary of the taxonomic identification from the sequences generated in this study.* .
Most food items consumed by crab-eating foxes were mammals, both in cold and warm seasons (78.5 and 60.3% of reads, respectively). Also, in this species, amphibians represented 16.6% of the reads in the cold season, and birds 30.7% in the warm season (Figure 2). In pampas fox, mammals were the substantial majority of food items consumed, 99.1% of reads in the cold season and 96.4% in the warm season (Figure 2). For both fox species, rodents constituted the most prevalent order among mammals. During the cold season, 67.0% of reads belonged to the Caviidae family, while in the warm season, 51.8% were attributed to the Muridae family (Figure 2).
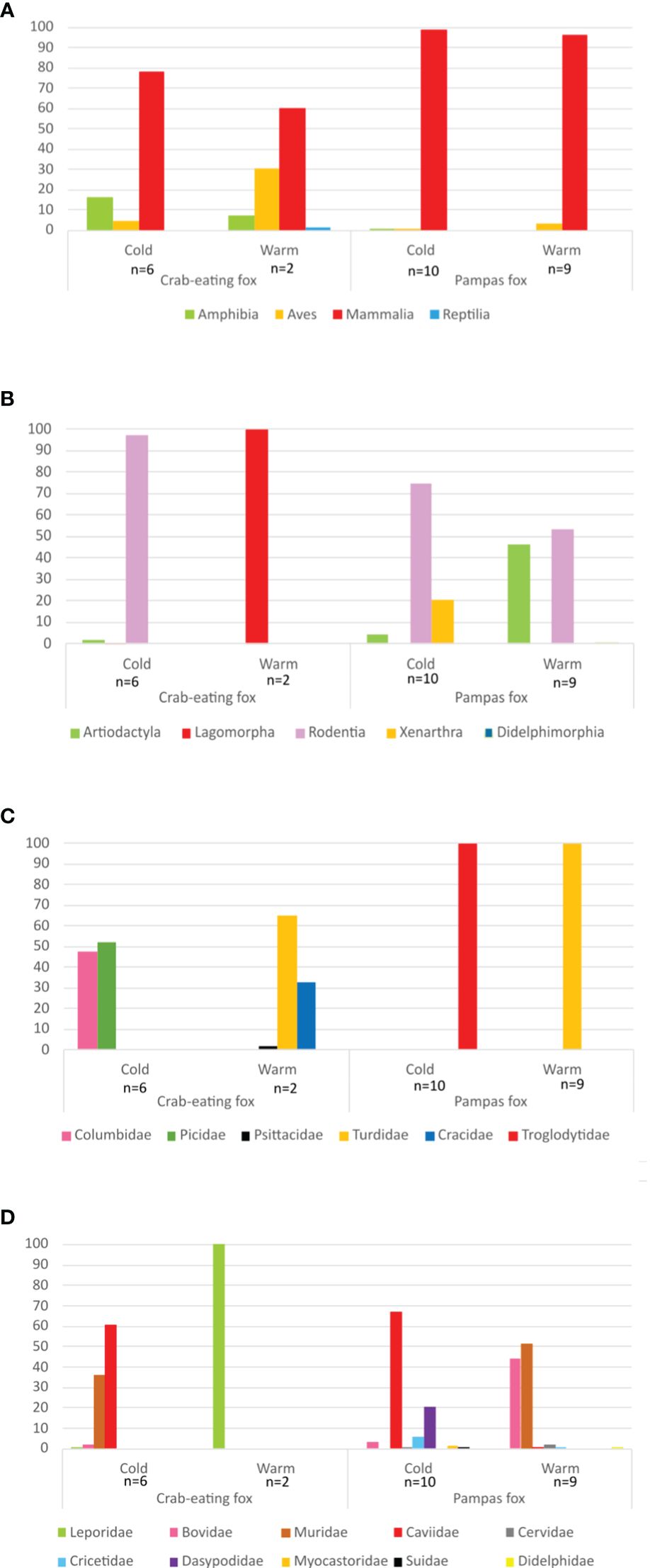
Figure 2 Distribution of vertebrate MOTUs detected in crab-eating and pampas fox diets during cold and warm sampling periods. MOTUs are distributed into vertebrate classes (A), mammalian orders (B), avian families (C) and mammalian families (D).
The sequence networks obtained in almost all cases showed a typical star shape, with a unique most frequent central sequence variant and less frequent sequence variant located in the periphery separated by few changes from the central one (Figure 3, Supplementary Data 2). Our analyses reveal no difference in the estimated variability (measure as overall mean p-distance) between the structural conserved and the non-conserved sequence regions. Moreover, no main differences can be observed between the network generated using both conserved and non-conserved sequences (Figure 3). This result suggests that intraspecific variation observed in food items comes from erroneous reads. Taken together, we conclude that there is only one real biological variant identified in C. aperea in the cold season on both fox species. Also, in the warm season, only one natural variant could be considered in L. europaeus on crab-eating fox and only one in B. taurus on pampas fox (Figure 3).
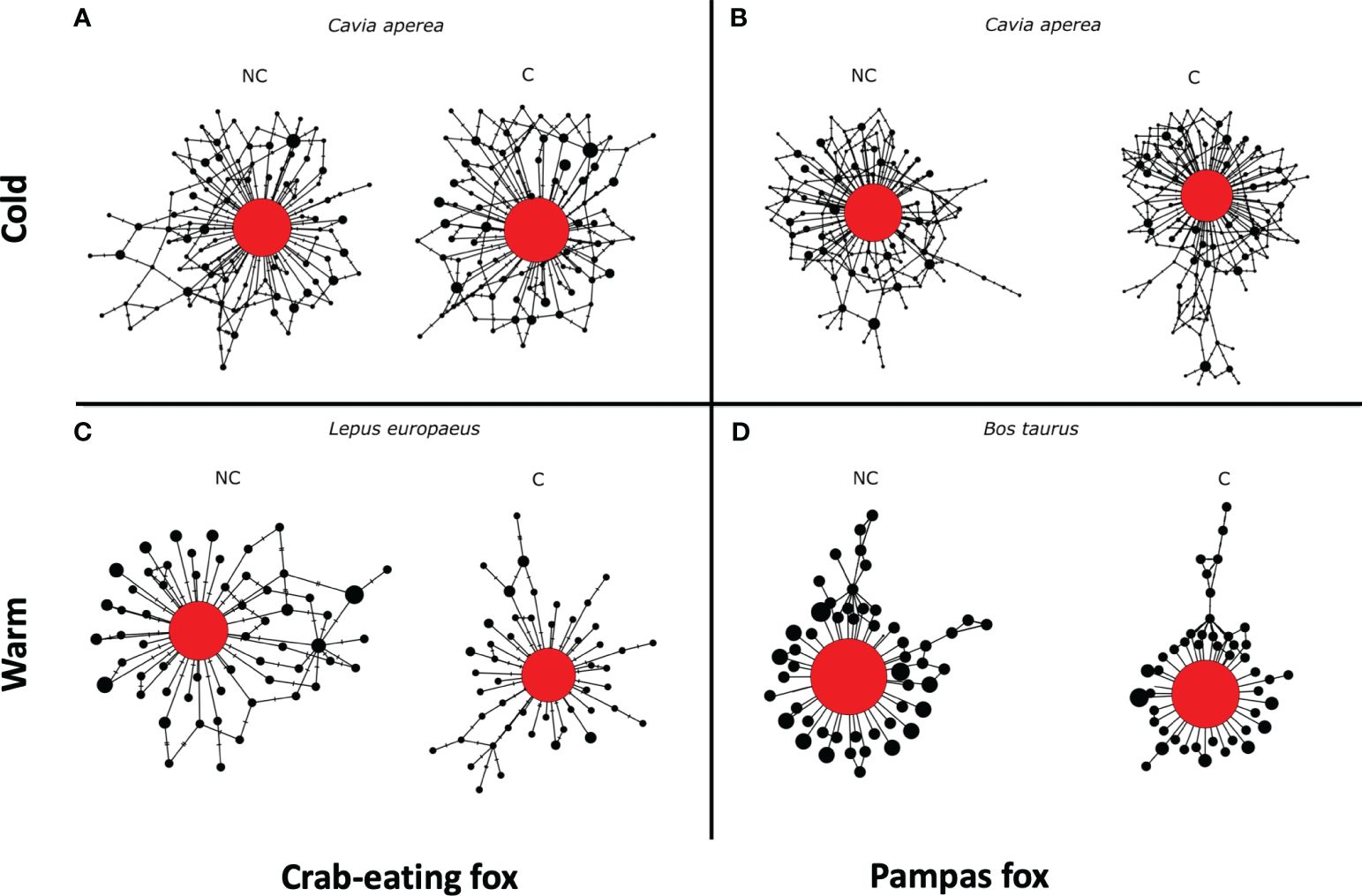
Figure 3 Network reconstruction based on the structurally conserved (C) and non-conserved (NC) sequences variants of the main food items identified in our study in cold (A, B) and warm (C, D) seasons in Crab-eating and Pampas foxes, respectively. Putative biological sequence variants are red-colored, while noisy sequence variants are black-colored.
The experimental design used in this study for carnivore species detection and diet assessment proved to be sensitive enough to allow qualitative trophic evaluations (Buglione et al., 2018). We also present a novel and strong blocking oligonucleotide for pampas and crab-eating fox for the 12S rRNA gene fragment. The use of this kind of oligonucleotides is an issue that needs to be addressed since the predator DNA is much more abundant than the consumed taxon DNA. Despite the advantages of the speed, accuracy, and potential of the HTS approach for dietary studies, some considerations should be taken into account (Pompanon et al., 2012). The taxonomic resolution of the DNA fragment chosen should have broad taxonomic coverage, suitable discrimination power, and high amplification efficiency. The latter is usually linked to short amplicon length (100-250bp) (Pompanon et al., 2012). In this sense, the DNA fragment chosen here fulfills all these conditions and has been previously successfully used in several carnivores and omnivorous metabarcoding feeding studies in which they evaluated its usefulness and efficiency (Ficetola et al., 2009; Roemer et al., 2009; Shehzad et al., 2012; De Barba et al., 2014). Taxonomic bias or underrepresentation of local species in the reference database may affect the analysis, as can be observed in the distribution of MOTUs among the identified food items (Figure 2). Thus, it is mandatory to improve and increase the quality and taxonomy coverage of the sequence database, including local species data. Meanwhile, the definition of MOTUs at higher taxonomic levels may help explore diet composition based on available data on public databases (Pompanon et al., 2012). Several DNA sequences for the 12S rRNA gene from vertebrate species inhabiting the study area were not found in the GenBank database. However, in some cases, the identification of taxon at the Genera level was possible (e.g., Pseudis and Colaptes). A similar approach was used when two or more species in the database displayed equal similarity scores due to the insufficient resolution of 12S rRNA for species assignment. In some cases, the identification at the Genera level was enough since only one species of the group is present in the study area (e.g., Cavia aperea).
Both false-positive and false-negative results are frequent in diet metabarcoding approaches (Taberlet et al., 2012; De Barba et al., 2014; Corse et al., 2019). To prevent these methodological shortcomings, we used universal PCR primers, blocking oligonucleotide primers fox species and included negative controls, and as a result, relatively low-frequency taxa were detected (De Barba et al., 2014; Corse et al., 2019).
In line with published diet studies that rely on morphological characteristics of taxon remains from the feces of both fox species, this study also identifies mammals (rodents and hares) as the most common vertebrate food items. Cavies (Cavia aperea) were detected in almost all seasons and consumed by both fox species. The only exception was seen during the warm season in crab-eating fox (Table 1), where minor consumption of meat animal proteins may occur because large amounts of fruits are accessible and there are less energetic requirements (Di Bitetti et al., 2009). Also, in accordance with our results, published trophic analyses concluded that rodents, particularly Cavies, were the main taxon consumed by these foxes (Farias and Kittlein, 2008; Di Bitetti et al., 2009; Bossi et al., 2019). The brown hare, Lepus europaeus, is also one of the most abundant food items identified, and species from this Genera have been previously described as important prey for both fox species (García and Kittlein, 2005; Farias and Kittlein, 2008; Bossi et al., 2019).
Interestingly, some dietary composition differences were detected between fox species (Table 1). As reported by Bossi et al. (2019), several amphibian taxa remains were present, especially in crab-eating fox feces, while different armadillo species are specific prey items for pampas fox. Birds were found as food items in both fox species, but only the Passeriform Order was identified in pampas fox. Moreover, during the warm season, Mus sp. was found in pampas fox feces in the study area near houses; this is not surprising, given that this rodent species frequently inhabits areas near human settlements.
Despite the smaller number of crab-eating fox feces sampled, many more taxa were detected in their diet, even those previously reported in other areas of its geographic distribution (Di Bitetti et al., 2009; Bossi et al., 2019). Although the limitation of the pooling strategy used here and the consequences of the relatively low number of samples included, the metabarcoding approach represents an improvement from the macroscopic trophic analysis. In the latter, the comparison of the results from different studies is challenging, mainly due to differences in taxonomic level prey resolution (Di Bitetti et al., 2009; Pompanon et al., 2012).
Cow DNA was identified in three of the four pools analyzed. The only exception was crab-eating fox during the warm season, the pool with a reduced number of collected feces (Table 1). This observation is not surprising since the study area is a Reserve with a popular picnic zone with barbecues. Thus, foxes could be consuming leftovers because predation attacks on cows are improbable (Farias and Kittlein, 2008). Moreover, livestock consumption was earlier related to their scavenger habits (Farias and Kittlein, 2008; Bossi et al., 2019). The presence of livestock in fox’s diet needs to be further analyzed and clarified because canids are persecuted by predation of domestic animals (mainly sheep) and are recurrent targets of poisoning and hunting by ranch owners (García and Kittlein, 2005; Bossi et al., 2019). Note, however, that no sheep DNA was found among food items in our study. This result is highly significant for conservation biology because these fox species are considered livestock predators in the countries where they occur. Unfortunately, additional studies in other areas dedicated to sheep breeding, including methodological approaches that distinguish carrion consumption from predation, are necessary. For this reason, future feeding metabarcoding DNA studies should include new sampling and molecular methods to elucidate trophic interactions such as secondary predation, scavenging, coprophagy, or parasitism.
The study of the genetic variability of food species was achieved through network reconstruction (Figure 3). This methodology utilizes the frequency and network position of detected variants to discriminate between biological variability and variability generated from erroneous reads (see above). The development of new methods to distinguish between both sources of observed variation is much needed to improve the accuracy of metabarcoding based on HTS. This is particularly important in environmental samples, where DNA is typically of low quality and degraded. Therefore, PCR cycles or sequencing depth need to be increased to achieve amplification success (Adams et al., 2019). To identify genuine genetic variations, it is advisable to incorporate positive controls, utilize multiple replicates, and employ high-quality filtering (Adams et al., 2019). Additionally, the reduction of erroneous sequences can be achieved through data denoising by in-silico methods (Tsuji et al., 2020; Zizka et al., 2020). Unfortunately, low abundant real biological variants could also be lost (Elbrecht et al., 2018). In this work, variations observed in sequence sites that are part of the conserved secondary structure of rRNA helices among vertebrate species, are presumed to be primarily errors. The null or marginal differences found in the estimated p-distances between structurally conserved and non-conserved sequences suggest that most of the variation observed is mainly random and comes from erroneous reads (Supplementary Data 3). Thus, we can state that only one variant was detected in all cases; the most frequent and central sequence in each network reconstruction should be considered the only real biological variant (Figure 3). There are non-mutually exclusive explanations for this lack of variability in the consumed taxon identified. First, our study was focused on a small area, and the number of samples per pool was low. Therefore, the low genetic variability observed may characterize the populations analyzed in the study area. Second, the DNA fragment used here, which allows species and taxon discrimination, may be too conserved for intraspecific genetic analysis (Riaz et al., 2011). This is a primary approach, further analyses, including non-coding intraspecific variable DNA fragments, should be done to confirm the efficiency of this strategy. In addition, expanding the database of sequences of neotropical species for the 12S rRNA enables us to better understand natural variability through traditional population genetic analysis methods.
This work is the first vertebrate diet composition analysis on Neotropical canids conducted using a metabarcoding approach. A sensitive and accurate methodology was applied for crab-eating fox and pampas fox diet characterization to achieve this goal. Our results agree with previous trophic analyses in these fox species based on macroscopic methods, validating the methodological strategy. Moreover, a first approach for detecting consumed species sequence variants was presented based on network reconstruction using non-coding DNA fragments. The extensive use of these techniques will allow going further in the understanding of South American wild canids feeding ecology and in the knowledge about their role in ecosystem regulation. Further analyses are needed to validate the approach for estimating intraspecific diversity through sequence analysis of prey DNA in generalist predators’ feces. In this regard, it is necessary to compare these results with more traditional population genetic methods.
The datasets presented in this study can be found in online repositories. The data presented in the study are deposited in the SRA database repository, accession number PRJNA1082626.
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because fresh feces of pampas fox and crab-eating fox were collected for this study.
NM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. MC: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – review & editing. GG: Methodology, Writing – review & editing. NB: Conceptualization, Investigation, Writing – review & editing. CR: Funding acquisition, Project administration, Resources, Writing – review & editing. SG: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Writing – review & editing. AI: Conceptualization, Investigation, Methodology, Resources, Software, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We obtained financial support from Programa de Desarrollo de Ciencias Básicas (PEDECIBA- Uruguay), Agencia Nacional de Investigación e Innovación Uruguay (ANII FSSA_2015_1_1005292), NGO Vida Silvestre Uruguay and Salus Company. Salus Company was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
We thank Antonella Bruno, Yanina Leone, Daniel Hernández, Claudia Elizondo, Hernán Juan, Eduardo Méndez, Alejandro Rodríguez, Andrés de Mello, Natalia Zaldua, for their assistance on field and laboratory work; Andres Ligrone for data shape file; Felipe Montenegro for his comments during data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1360714/full#supplementary-material
Adams C. I. M., Knapp M., Gemmell N. J., Jeunen G. J., Bunce M., Lamare M. D., et al. (2019). Beyond biodiversity: Can environmental DNA (eDNA) cut it as a population genetics tool? Genes 10, 1–24. doi: 10.3390/genes10030192
Bandelt H. J., Forster P., Röhl A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Bossi M. A. S., Migliorini R. P., Santos T. G., Kasper C. B. (2019). Comparative trophic ecology of two sympatric canids in the Brazilian Pampa. J. Zool. 307, 215–222. doi: 10.1111/jzo.12636
Boyer S., Cruickshank R. H., Wratten S. D. (2015). Faeces of generalist predators as ‘biodiversity capsules’: A new tool for biodiversity assessment in remote and inaccessible habitats. Food Webs. 3, 1–6. doi: 10.1016/j.fooweb.2015.02.001
Buglione M., Maselli V., Rippa D., de Filippo G., Trapanese M., Fulgione D. (2018). A pilot study on the application of DNA metabarcoding for non-invasive diet analysis in the Italian hare. Mamm. Biol. 88, 31–42. doi: 10.1016/j.mambio.2017.10.010
Corse E., Tougard C., Archambaud G., Duneau D., Zinger L., Chappaz R., et al. (2019). One-locus-several-primers: A strategy to improve the taxonomic and haplotypic coverage in diet metabarcoding studies. Ecol. Evo. 9, 4603–4620. doi: 10.1002/ece3.5063
Cosse M., Grattarola F., Mannise N. (2017). A novel real-time TaqManTM PCR assay for simultaneous detection of Neotropical fox species using noninvasive samples based on cytochrome c oxidase subunit II. Mamm. Res. 62, 405–411. doi: 10.1007/s13364-017-0328-y
Cravino A. (2014). El ensamble de carnívoros (Orden Carnívora) del área protegida Parque Nacional San Miguel (Rocha, Uruguay): uso de hábitat, dieta y valor indicador [dissertation/degree thesis] (Montevideo, Uruguay: Facultad de Ciencias, Universidad de la República Oriental del Uruguay).
Deagle B. E., Thomas A. C., Shaffer A. K., Trites A. W. (2013). Quantifying sequence proportions in a DNA-based diet study using Ion Torrent amplicon sequencing: which counts count? Mol. Ecol. Resour. 13, 620–633. doi: 10.1111/1755-0998.12103
Deagle B. E., Tollit D. J. (2007). Quantitative analysis of prey DNA in pinniped faeces: potential to estimate diet composition? Conserv. Genet. 8, 743–747. doi: 10.1007/s10592-006-9197-7
De Barba M., Miquel C., Boyer F., Mercier C., Rioux D., Coissac E., et al. (2014). DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet. Mol. Ecol. Resour. 14, 306–323. doi: 10.1111/1755-0998.12188
Deiner K., Bik H. M., Mächler E., Seymour M., Lacoursière-Roussel A., Altermatt F., et al. (2017). Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 26, 5872–5895. doi: 10.1111/mec.14350
Di Bitetti M. S., Di Blanco Y. E., Pereira J. A., Paviolo A., Pérez I. J. (2009). Time Partitioning Favors the Coexistence of Sympatric Crab-Eating Foxes (Cerdocyon thous) and Pampas Foxes (Lycalopex gymnocercus). J. Mammal. 90, 479–490. doi: 10.1644/08-MAMM-A-113.1
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Elbrecht V., Vamos E. E., Steinke D., Leese F. (2018). Estimating intraspecific genetic diversity from community DNA metabarcoding data. PeerJ 6, 1–13. doi: 10.7717/peerj.4644
Faria-Correa M., Balbueno R. A., Vieira E. M., de Freitas. T. R. O. (2008). Activity, habitat use, density, and reproductive biology of the crab-eating fox (Cerdocyon thous) and comparison with the pampas fox (Lycalopex gymnocercus) in a Restinga area in the southern Brazilian Atlantic Forest. Mamm. Biol. 74, 220–229. doi: 10.1016/j.mambio.2008.12.005
Farias A. A., Kittlein M. J. (2008). Small-scale spatial variability in the diet of pampas foxes (Pseudalopex gymnocercus) and human-induced changes in prey base. Ecol. Res. 23, 543–550. doi: 10.1007/s11284-007-0407-7
Ficetola G. F., Coissac E., Zundel S., Riaz T., Shehzad W., Bessiere J., et al. (2009). An In silico approach for the evaluation of DNA barcodes. BMC Genomics 11, 434. doi: 10.1186/1471-2164-11-434
García V. B., Kittlein M. J. (2005). Diet, habitat use, and relative abundance of pampas fox (Pseudalopex gymnocercus) in northern Patagonia, Argentina. Mamm. Biol. 70, 218–226. doi: 10.1016/j.mambio.2004.11.019
Hernández Rodriguez Y. (2007). Importancia del zorro de monte Cerdocyon thous entrerianus (Carnívora: Canidae) para la conservación del monte nativo en el Parque Nacional San Miguel. [dissertation/degree thesis] (Montevideo, Uruguay: Facultad de Ciencias, Universidad de la República Oriental del Uruguay).
Joshi N. A., Fass J. N. (2011). Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available at: https://github.com/najoshi/sickle.
Kalvari I., Nawrocki E. P., Ontiveros-Palacios N., Argasinska J., Lamkiewicz K., Marz M., et al. (2020). Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, 1. doi: 10.1093/nar/gkaa1047
Klare U., Kamler J. F., MacDonald D. W. (2011). A comparison and critique of different scat-analysis methods for determining carnivore diet. Mamm. Rev. 41, 294–312. doi: 10.1111/j.1365-2907.2011.00183.x
Leigh J. W., Bryant D. (2015). Popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Lopes C., De Barba M., Boyer F., Mercier C., da Silva Filho P. J. S., Heidtmann L. M., et al. (2015). DNA metabarcoding diet analysis for species with parapatric vs sympatric distribution: a case study on subterranean rodents. Heredity 114, 525–536. doi: 10.1111/mec.15549
Lopes C. M., De Barba M., Boyer F., Mercier C., Galiano D., Busnello Kubiak B., et al. (2020). Ecological specialization and niche overlap of subterranean rodents inferred from DNA metabarcoding diet analysis. Mol. Ecol. 29, 3143–3153. doi: 10.1111/mec.15549
Monterroso P., Godinho R., Oliveira T., Ferreras P., Kelly M. J., Morin D. J., et al. (2019). Feeding ecological knowledge: the underutilised power of faecal DNA approaches for carnivore diet analysis. Mamm. Rev. 49, 97–112. doi: 10.1111/mam.12144
Moreno F., Figueiro G., Mannise N., Iriarte A., González S., Duarte J. M. B., et al. (2016). Use of next-generation molecular tools in archaeological neotropical deer sample analysis. J. Archaeol. Sci. Rep. 10, 403–410. doi: 10.1016/j.jasrep.2016.11.006
Pompanon F., Deagle B. E., Symondson W. O. C., Brown D. S., Jarman S. N., Taberlet P. (2012). Who is eating what: diet assessment using next generation sequencing. Mol. Ecol. 21, 1931–1950. doi: 10.1111/j.1365-294X.2011.05403.x
Prugh L. R., Stoner C. J., Epps C. W., Bean W. T., Ripple W. J., Laliberte A. S., et al. (2009). The rise of the mesopredator. BioScience 59, 779–791. doi: 10.1111/j.1365-294X.2011.05403.x
Quemere E., Hibert F., Miquel C., Lhuillier E., Rasolondraibe E., Champeau J., et al. (2013). A DNA metabarcoding study of a primate dietary diversity and plasticity across its entire fragmented range. PloS One 8, e58971. doi: 10.1371/journal.pone.0058971
Riaz T., Shehzad W., Viari A., Pompanon F., Taberlet P., Coissac E. (2011). EcoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res. 39, e145. doi: 10.1093/nar/gkr732
Rodríguez-Mazzini R., Molina-Espinosa B. (2000) El zorro de monte (Cerdocyon thous) como agente dispersor de semillas de palma: estudios realizados en la Estación Biológica Potrerillo de Santa Teresa PROBIDES (No. 599.742. 1 ROD). Available at: https://www.probides.org.uy/imagenes/ckfinder/files/files/Documentos%20de%20Trabajo/DT30.pdf.
Roemer G. W., Gompper M. E., Van Valkenburgh B. (2009). The ecological role of the mammalian mesocarnivore. Bioscience 59, 165–173. doi: 10.1525/bio.2009.59.2.9
Shehzad W., Riaz T., Nawaz M. A., Miquel C., Poillot C., Shah S. A., et al. (2012). Carnivore diet analysis based on next- generation sequencing: application to the leopard cat (Prionailurus bengalensis) in Pakistan. Mol. Ecol. 21, 1951–1965. doi: 10.1111/j.1365-294X.2011.05424.x
Shendure J., Ji H. (2008). Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145. doi: 10.1038/nbt1486
Springer M. S., Douzery E. (1996). Secondary structure and patterns of evolution among mammalian mitochondrial 12S rRNA molecules. J. Mol. Evol. 43, 357–373. doi: 10.1007/BF02339010
Staats M., Arulandhu A. J., Gravendeel B., Holst-Jensen A., Scholtens I., Peelen T., et al. (2016). Advances in DNA metabarcoding for food and wildlife forensic species identification. Anal. Bioanal. Chem. 408, 4615–4630. doi: 10.1007/s00216-016-9595-8
Taberlet P., Coissac E., Pompanon F., Brochmann C., Willerslev E. (2012). Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21, 2045–2050. doi: 10.1111/j.1365-294X.2012.05470.x
Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tsuji S., Maruyama A., Miya M., Ushio M., Sato H., Minamoto T., et al. (2020). Environmental DNA analysis shows high potential as a tool for estimating intraspecific genetic diversity in a wild fish population. Mol. Ecol. Resour. 20, 1248–1258. doi: 10.1111/1755-0998.13165
Turon X., Antich A., Palacín C., Præbel K., Wangensteen O. S. (2020). From metabarcoding to metaphylogeography: separating the wheat from the chaff. Ecol. App. 30, e02036. doi: 10.1002/eap.2036
Vestheim H., Jarman S. N. (2008). Blocking primers to enhance PCR amplification of rare sequences in mixed samples - a case study on prey DNA in Antarctic krill stomachs. Front. Zool. 5, 1742–9994. doi: 10.1186/1742-9994-5-12
Vieira E. M., Port D. (2007). Niche overlap and resource partitioning between two sympatric fox species in southern Brazil. J. Zool. 272, 57–63. doi: 10.1111/j.1469-7998.2006.00237.x
Zhang J., Kobert K., Flouri T., Stamatakis A. (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Keywords: non-invasive genetic techniques, high-throughput sequencing (HTS), molecular ecology, environmental DNA, wild canids
Citation: Mannise N, Cosse M, Greif G, Bou N, Robello C, González S and Iriarte A (2024) Developing a DNA metabarcoding method to identify diet taxa in Neotropical foxes. Front. Ecol. Evol. 12:1360714. doi: 10.3389/fevo.2024.1360714
Received: 23 December 2023; Accepted: 29 April 2024;
Published: 30 May 2024.
Edited by:
Gabriela Paula Fernández, Universidad Nacional del Noroeste de la Provincia de Buenos Aires (UNNOBA), ArgentinaReviewed by:
Thales Renato Ochotorena De Freitas, Federal University of Rio Grande do Sul, BrazilCopyright © 2024 Mannise, Cosse, Greif, Bou, Robello, González and Iriarte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andres Iriarte, YWlyaWFydGVAaGlnaWVuZS5lZHUudXk=; Mariana Cosse, bWNvc3NlQGlpYmNlLmVkdS51eQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.