 Zongfu Hu
Zongfu Hu Qing Tong
Qing Tong Jie Chang1
Jie Chang1 Junzhao Xu
Junzhao Xu Baiyila Wu
Baiyila Wu Huaxin Niu
Huaxin Niu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 02 February 2024
Sec. Coevolution
Volume 12 - 2024 | https://doi.org/10.3389/fevo.2024.1341359
Background: Freshwater snails are not only intermediate hosts for parasites but also an important part of the food chain as they convert plant biomass and humus into animal biomass. However, being widely distributed in freshwater environments, snails are highly affected by human activities, which makes their adaptation to altering environments challenging. The gut microbiome helps animals in their digestion, immune system, growth and adapting to changing environments. The effect of host species on intestinal microbial community has been poorly studied in snails.
Methods: In this study, single-molecule real-time sequencing technology (SMRT) was used to obtain full-length 16S rRNA genes to determine the intestinal microbiomes of three species of freshwater snails (SQ: Sinotaia quadrata, BU: Boreoelona ussuriensis, RP: Radix plicatula) with similar feeding habits in a same water environment.
Results: Unifrac PCoA (P<0.05), hierarchical cluster and Ternary analyses showed distinct and significant segregation of the intestinal microbiomes of the three hosts. The phyla Cyanobacteria, Proteobacteria, Firmicutes and Planctomycetota dominated snail guts, comprising 93.47%, 86.22%, and 94.34% of the total reads in SQ, BU and RP, respectively. Of these, only 25.26% of OTUs were identified up to species level, while 72.07% of OTUs were identified at the genus level. Although 72.94% of the total bacterial species (566) were common to three snails, significant differences were observed in terms of their abundance (P < 0.05). Several genera can help to determine the phenotype of the intestinal microbiota. In this case, Lelliottia contributed mainly to stress tolerance, biofilm formation, potential pathogenicity, mobile elements and facultatively anaerobic phenotypes in RP. Furthermore, Romboutsia and Clostridium_sensu_stricto_1 contributed to the anaerobic phenotype of SQ and RP, while Pirellula contributed to the aerobic phenotype in SQ. Moreover, PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) predicted 68 GH (glycoside hydrolase) genes, with these including monosaccharide-, disaccharide-, polysaccharide-, and starch-digesting enzyme genes as well as enzymes specific to aquatic plants. Many of the identified pathways were related to Infectious diseases and Xenobiotics biodegradation and metabolism, which expanded the resistance of freshwater snails.
Concludes: Lelliottia, Romboutsia, Clostridium_sensu_stricto_1, and Pirellula play an important role in the intestinal microbiota phenotype of the host snails. In general, the host species affects the structure of the gut microbial community, which in turn helps gastropods improve their environmental adaptability, but further study is still needed.
The intestinal microbiota, with long-term co-evolutionary associations with the host, plays a vital role in the survival and overall health of wild animals. Indeed, they significantly contribute to the process of digestion, especially the breaking down of complex plant materials that cannot be digested by the animal’s own enzymes (Pawar et al., 2015). In addition to aiding digestion, commensal intestinal bacteria also contribute to the immune defense of wild animals by producing bioactive compounds against potential pathogens (Liu et al., 2020). Furthermore, these microorganisms can help in detoxification and provide resistance against environmental pollutants, such as heavy metals and plant toxins (van den Bosch and Welte, 2017; Loh et al., 2020; Chen et al., 2022). The composition of the gut microbiota is dependent on the diet, habitat and species; therefore, it reflects the animal’s adaptation to its environment (Haworth et al., 2019). Based on the above literature, it can be inferred that preserving the diversity and gut microbial balance is vital for the well-being and survival of wild animals.
Freshwater snails, the most common mollusk found in freshwater environments, feed on algae, aquatic grasses, litter and humus (Thomas et al., 1985; Madsen, 1992). As such, they are an important link for converting plant biomass into animal biomass within freshwater ecosystems. Research on these snails is of great significance for public health as they host a number of parasites, such as flukes and nematodes (Dodangeh et al., 2019). These snails can also inhibit water eutrophication and absorb or decompose certain pollutants through their intestinal microbiota, thereby purifying and enhancing the water quality (Takougang et al., 2008).
A number of studies on the gut microbiota of humans and other animals have been performed. In comparison, only a few have focused on the intestinal microbiota of freshwater mollusks. Research data has shown that the intestinal microbiota of freshwater snails is influenced by many factors, such as diet composition, water quality factors, development and sex amongst others (Van Horn et al., 2012; Hu et al., 2018; Chalifour and Li, 2021; Hu et al., 2021; Kivistik et al., 2022; Lyu et al., 2022). For example, the number of OTUs in the intestinal microbiota of herbivorous freshwater snails reduced by 71% after they ate diets without fiber, indicating that the gut microbiome is highly dependent on the presence of cellulose-containing plant nutrients (Hu et al., 2021). Furthermore, a study on the intestinal bacterial communities of Bellamya aeruginosa showed that the microbiome did not strongly depend on the microbiota in food (Liu and Li, 2022). Therefore, when sampling freshwater snails from similar natural water bodies, slight food differences have limited effects on the intestinal microbiota composition. However, the stronger host effect on intestinal microbiota has not been reported.
Previous studies on the intestinal microbiota of mollusks were mostly focused on host species of commercial value or were conducted under artificial conditions. In contrast, only a few studies actually investigated the intestinal microbiota of freshwater snails within their natural environment (Chalifour and Li, 2021). Furthermore, the intestinal microorganisms in mollusks have been seldom studied by high-throughput analysis of a full-length 16S rRNA gene. Instead, research has been limited to fragments of the 16S rRNA gene, which may be less accurate at discriminating bacteria at the species level. Compared to short 16S rRNA gene fragments sequence, its long read sequencing can nearly cover the whole 16S rRNA gene, providing accurate identification of taxon at the species level. The near full-length reads also result in a finer phylogenetic resolution, and some further refined OTUs may sometimes reflect different strains of the same species (Koeppel and Wu, 2013; Gehrig et al., 2022). The Pacific Biosciences Sequel instrument used in this study not only produce near-full-length sequences at a relatively low cost, and it also produce a high accuracy (99.9%) of reads by sequenced them using a circular consensus mode (Marshall et al., 2012).
The three freshwater snail species used in this study, namely Boreoelona ussuriensis (BU), Sinotaia quadrata (SQ) and Radix plicatula (RP), with similar feeding habits, consume algae and aquatic grasses as well as various organic matter, such as dead leaves and decaying plants, thereby significantly contributing to the upward flow of primary production in freshwater ecosystems (Thomas et al., 1985; Madsen, 1992; Cao and Jiang, 1998; Wu et al., 2017; Zhou et al., 2023). To investigate and compare the gut microbiomes of these three freshwater snail species, a single-molecule real-time sequencing technique that captures the full length of the 16S rRNA gene was used. As in the previous studies, on Galerucella leaf beetles and Nasonia, the phylogeny of host species is closely related to their gut microbiota, indicating that gut microbiota has the potential of co-evolutionary processes with their host species (Brucker and Bordenstein, 2012; An et al., 2023). The question arises: do different species of freshwater snails also have intestinal microbiomes with interspecific differences? Unfortunately, the studies on intestinal microbiota of freshwater snails are lagging behind. The present study hypothesized that the intestinal microbiomes of freshwater snails were related to the host species and, therefore, differed structurally and functionally between different species. In addition, these microbial communities help the snails to adjust to different environmental conditions. The findings of this study will increase the understanding of the commonalities and differences in the composition and function of intestinal microbiomes of freshwater snails with similar feeding habits in the same water area.
Three species of freshwater snails, namely 40 RP (Radix plicatula), 40 BU (Boreoelona ussuriensis) and 36 SQ (Sinotaia quadrata), were collected from the West Liao River (43° 36’ 51” N; 122° 13’ 28” E) at Tongliao, Inner Mongolia, China. All samples were taken from shallow water along the waterline, with the main food source of these snails was benthic algae, such as diatoms, as well as humus from emergent plants, aquatic grasses and decaying plant matter. As Table 1 indicates, the average height of SQ, BU and RP shells was 24.78 ± 1.9 mm, 12.2 ± 0.6 mm and 21.3 ± 1.8 mm, respectively. After collection, the snails were quickly transported to the laboratory where their surfaces were scrubbed with 70% alcohol before extracting their intestines under sterile conditions. In this case, based on the size of the intestinal samples, they were combined in groups of five for RA and BU and groups of three for BP. Therefore, a total of 8 RP samples, 8 BU samples and 12 SQ samples were obtained. Species identification was confirmed by the mitochondrial cytochrome c oxidase subunit I (COI) gene, which was amplified with primers LCO1490 and HCO2198 (Folmer et al., 1994). The gene sequences were registered at NCBI GenBank (Boreoelona ussuriensis: OR990564, Sinotaia quadrata: OR994503 and Radix plicatula: OR994310) (Supplementary Figure S1). This study was approved by the ethics committee of Inner Mongolia Minzu University (IACUC-202211-073).
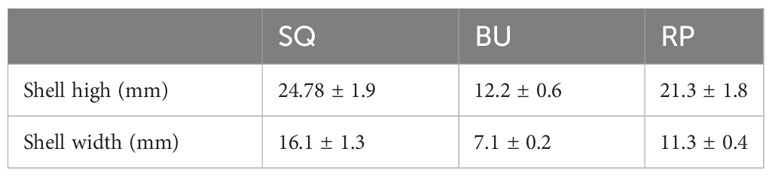
Table 1 The shell high and shell width of snails.
Microbial genomic DNA was extracted from snail gut samples using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to the manufacturer’s instructions. Then, the extracted DNA was subjected to 1% agarose gel separation before determining its concentration, and purity was assessed with a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA). For bacterial community analysis, the 16S rRNA genes were amplified using the universal bacterial primers 27F (5’-AGRGTTYGATYMTGGCTCAG-3’) and 1492R (5’-RGYTACCTTGTTACGACTT-3’) (Weisburg et al., 1991) to which PacBio barcode sequences were attached to distinguish between samples. In this case, the PCR reactions were performed in triplicate, and each reaction volume was 20-μL comprising 4 μL of 5 ×FastPfu buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu DNA Polymerase, 10 ng of template DNA and DNase-free water. The amplification program was then set up on a 9700 PCR thermocyclerm (ABI GeneAmp®, CA, USA) as follows: initial denaturation at 95°C for 3 min, followed by 27 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 45 s. The reaction ended with a final extension at 72°C for 10 min. Subsequently, the resulting PCR products were checked via electrophoresis prior to purification using AMPure® PB beads (Pacific Biosciences, CA, USA) and quantification with a Quantus™ Fluorometer (Promega, WI, USA).
After pooling purified DNA products in equimolar amounts, the DNA libraries were constructed using the SMRTbell® Express Template Prep Kit 2.0 (Pacifc Biosciences, CA, USA), as instructed by the manufacturer. These libraries were subsequently sequenced on the Pacbio Sequel II System (Pacific Biosciences, CA, USA) by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
PacBio raw reads were processed using the SMRTLink analysis software (version 8.0) to obtain demultiplexed circular consensus sequence (CCS) reads with a minimum of three full passes and 99% sequence accuracy. Then, based on the barcodes, these CCS reads were identified and filtered according to their length. Reads with sequences of <1,000 bp or >1,800 bp removed. Subsequently, the optimized-CCS reads were de-noised using the recommended parameters of the DADA2 (Callahan et al., 2016) plugin in the Qiime2 (Bolyen et al., 2019) (version 2020.2) pipeline to generate amplicon sequence variants (ASVs) at single nucleotide resolution. Furthermore, to minimize the effects of sequencing depth on the diversity metrics, the number of 16S rRNA gene sequences in each sample was rarefied to 13994, with this value yielding an average good coverage of 96.74%. Moreover, the taxonomic assignment of ASVs was performed using the Naive Bayes (Vsearch, or BLAST) consensus taxonomy classifier implemented in Qiime2 as well as the SILVA 16S rRNA database (v138).
Optimized-CCS reads were also clustered into operational taxonomic units (OTUs) at a 97% sequence similarity level using UPARSE 7.1 (Edgar, 2013). After selecting each OUT’s most abundant sequence as a representative sequence, those assigned to chloroplast and mitochondrial sequences were removed from all samples. Then, the taxonomy of the remaining OTU representative sequences was determined using the RDP Classifier version 2.2 (Wang et al., 2007) against the 16S rRNA gene database (Silva v138) at a confidence threshold of 0.7. Additionally, based on OTU representative sequences, the metagenomic function was predicted using PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) (Douglas et al., 2020) based on OTU representative sequences. In this case, BugBase (https://bugbase.cs.umn.edu/index.html) was also used to predict the microbiome phenotype of snails (Ward et al., 2017).
Bioinformatic analysis of the gut microbiota was performed using the Majorbio Cloud platform (https://cloud.majorbio.com). Rarefaction curves and alpha diversity indices, including Simpson index, Shannon index and Sobs index, were then calculated with Mothur v1.30.1 (Schloss et al., 2009) based on the ASVs’ information. Furthermore, based on Bray-Curtis dissimilarity, the similarity between the microbial communities of different samples was determined by principal coordinate analysis (PCoA) using the Vegan v2.5-3 package. For hierarchical cluster analysis, the beta diversity distance matrix was computed in Qiime before generating a corresponding tree using R language (version 3.3.1). The latter was also used for statistics, mapping Venn diagrams, generating bar charts of community composition based on taxonomy summaries as well as for heatmap and bubble map analysis with the Vegan package (version 2.4.3). Additionally, ternary analysis was performed by GGTERN (http://www.ggtern.com/). Lastly, the linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011) (http://huttenhower.sph.harvard.edu/LEfSe) was carried out to identify significantly abundant bacterial taxa (phylum to genera) among the different groups (LDA score > 4, P < 0.05).
Quality-filtering of reads generated a total of 1,030,244 sequences, with an average length of 1472 bases, for 28 samples of freshwater snails (8 samples for BU and RP, respectively, and 12 samples for SQ). Additional filtering was performed to remove sequences related to Chloroplast and Mitochondria as well as those from OTUs having a sequence count less than 5. This step yielded 721,211 sequences which were then clustered into 2,898 operational taxonomic units (OTUs) at a 97% sequence-identity threshold. Subsequent taxonomics classified these OTUs into 26 phyla, 50 classes, 124 orders, 203 families, 434 genera and 776 species.
The gut microbiota of the three species of freshwater snails were then analyzed based on the Sobs, Shannon, and Simpson indexes at the OTU level. The Shannon diversity index for SQ, BU and RP were 6.08, 6.24, and 5.72 respectively, while the observed species (Sobs) count was 1545, 1654, and 1500 respectively. Whereas the Simpson index, the values were 0.008, 0.007, and 0.016 respectively. The different diversity indices suggested that the alpha-diversity of BU was higher than RP (P < 0.05), while no significant differences were noted when comparing the Shannon and Simpson diversity metrics of SQ and BU or SQ and RP (P > 0.05) (Figure 1). The rarefaction and Shannon curves further showed that the sequencing depth was adequate for microbial diversity analysis (Supplementary Figure S2).
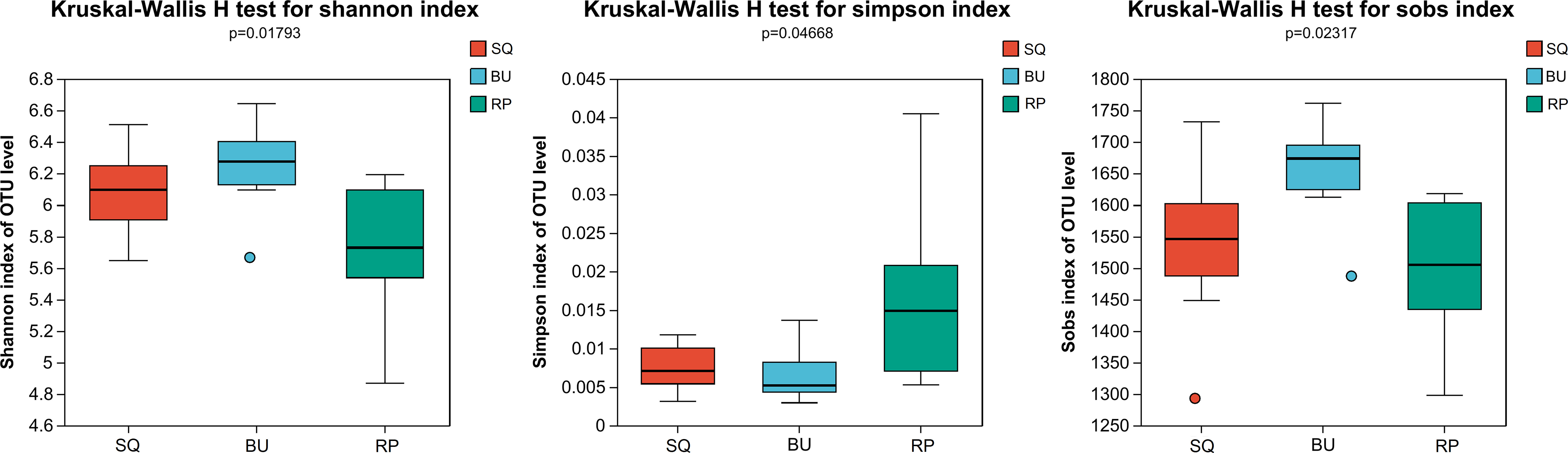
Figure 1 Significant changes were noted in the alpha-diversity metrics of the three freshwater snails’ intestinal communities. Red, blue and green columns represent SQ, BU and RP snails, respectively. SQ, Sinotaia quadrata; BU, Boreoelona ussuriensis; RP, Radix plicatula.
PCoA analysis showed significant differences between the intestinal bacterial communities of the three snail species (Figure 2A: unweighted unifrac analysis of variance, R = 0.99; P=0.001; Figure 2B: weighted unifrac analysis of variance, R = 0.93, P=0.001). Furthermore, the hierarchical clustering analysis divided the samples into three distinct groups that were significantly different based on host species (Figure 2C).
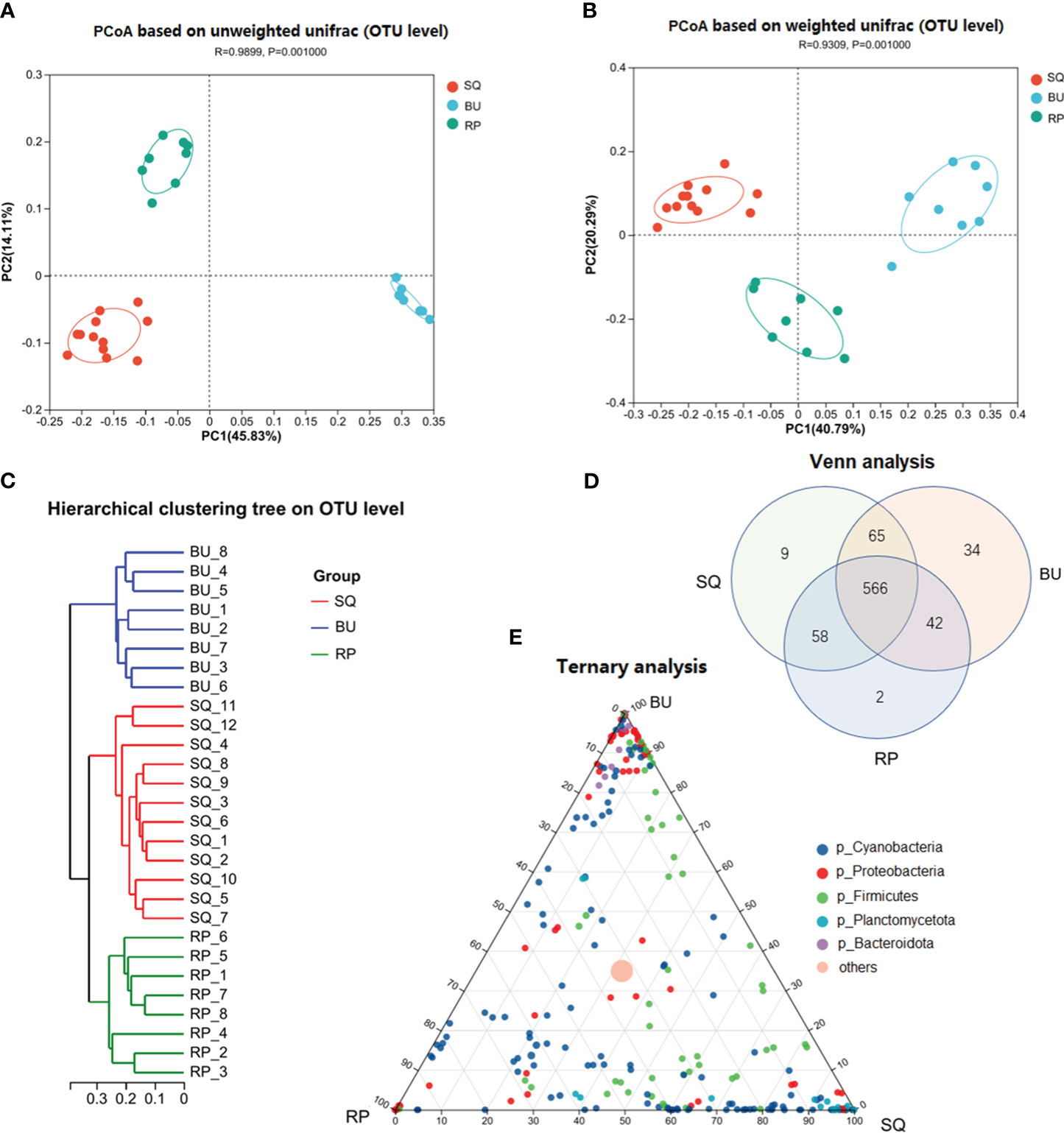
Figure 2 Beta-diversity analysis of the intestinal microbiome of the three snail species. (A) PCoA analysis at OTU level based on unweighted and (B) weighted unifrac. (C) Hierarchical cluster analysis using pairwise Bray-Curtis distances at OTU level. (D) VENN analysis of the gut microbiota revealed that 566 microbial species were common between the three snail species. (E) Ternary analysis based on the relative occurrence of the most abundant phyla (dot) in the gut samples of SQ, BU, and RP. The OTUs are colored according to their taxonomy at the phylum level, while the size of the circles is proportional to the mean abundance within the communities. SQ, Sinotaia quadrata; BU, Boreoelona ussuriensis; RP, Radix plicatula.
The VENN analysis revealed that 566 bacteria, accounting for 72.94% of the total bacteria, were common to the three snails. Of these, 145 were identified up to the species level (25.61%) and included Verrucomicrobia_bacterium_ADurb.Bin474, Limnoraphis_robusta_CS-951, Exiguobacterium_sp._MH3, Chroococcidiopsis_thermalis_PCC_7203 and Exiguobacterium_sp. ZWU0009 (Supplementary Figure S3, Supplementary Table S1). In addition, 165 bacteria were common to only two snail species; of these species, a level of 47 (28.48%) was identified. More specifically, the species that were common to SQ and BU included Lactococcus_garvieae_ATCC_419156, Lactobacillus_amylovorus, Lactobacillus_pontis, Acinetobacter_sp._CIP_64.7, Burkholderia_stabilis and Streptococcus_alactolyticus, while those common to SQ and RP consisted of cyanobacterium_scsio_7.2, Synechococcus_sp._WH__5701 and Exiguobacterium_sp. Similarly, Scytonematopsiscontorta__HA4292-MV4, Chroococcidiopsis sp. PCC 6712 and Parasegetibacter luojiensis were among the species that were common to BU and RP. Furthermore, 45 bacteria were only present in one snail species (Figure 2D). Of these, with only 4 identified up to the species level. For instance, Brevinema_andersonii and Synechocystis_aquatilis_SAG_90.79 were specific to the SQ snail, while Melainabacteria_bacterium_MEL.A1 and Pantoea_vagans_C9-1 were only identified in the BU snail (Supplementary Figure S4, Supplementary Table S1).
At the phylum level, 10 dominant phyla, with > 1% abundance, were identified, including Cyanobacteria, Proteobacteria, Firmicutes, and Planctomycetota, accounting for 93.47, 86.22, and 94.34% of the total reads of SQ, BU, and RP, respectively (Figure 3A). The distribution of microbes at the OTUs level between the three snail species was also determined based on ternary analysis. In this case, the OTUs of Proteobacteria were mostly present in SQ, while the OTUs of Planctomycetota were mainly found in SQ. Similarly, the OTUs of Firmicutes and Cyanobacteria were widely identified in all three snail species (Figure 2E).
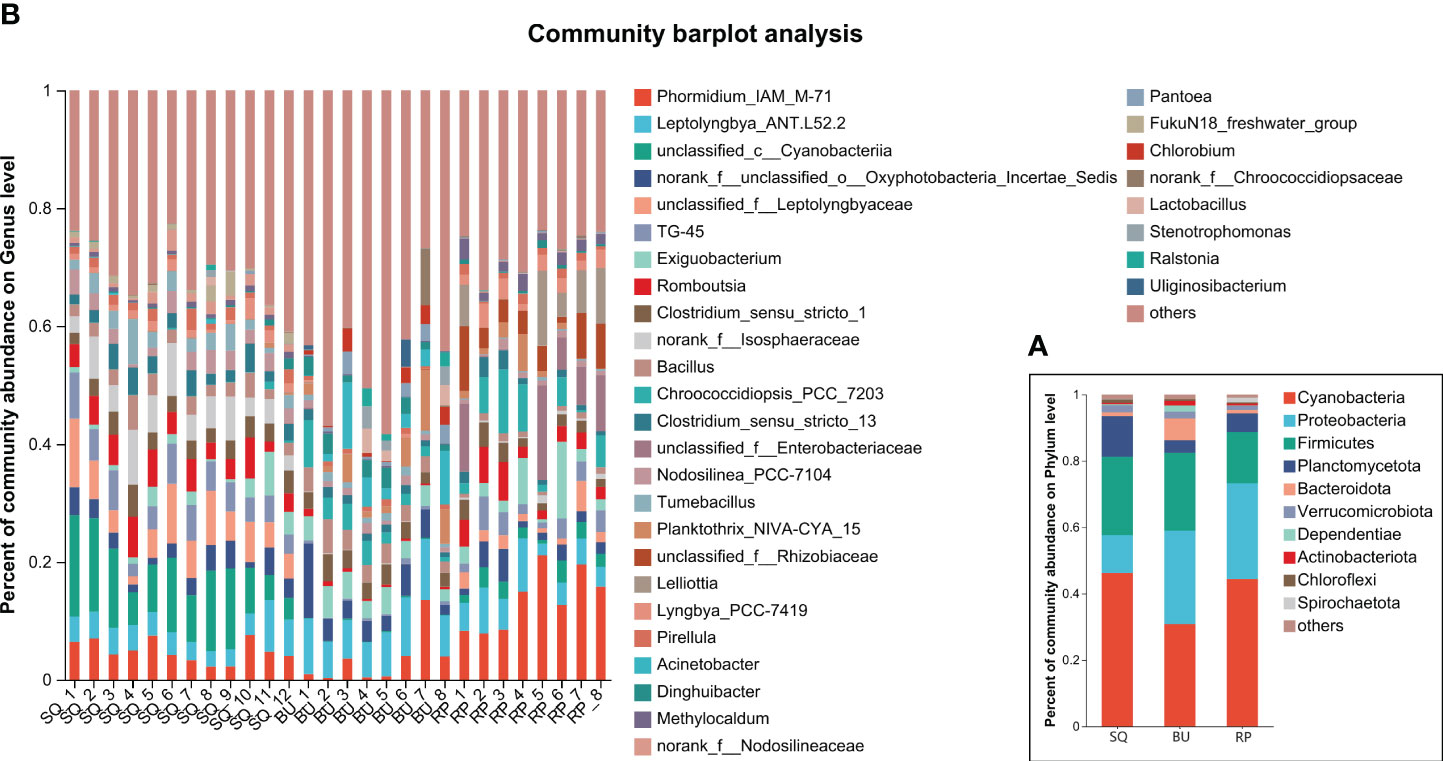
Figure 3 Intestinal microbiota composition of the three snail species. (A) phylum level, (B) genus level (relative abundance > 0.03).
At the genus level, 33 genera with an abundance of more than 3% were identified, with some examples being Phomidium_IAM_M-71, Leptolyngbya_ANT.L52.2, Exiguobacterium, Romboutsia and Clostridium_sensu_strito_1, Bacillus (Figure 3B).
The abundance of bacterial communities in the different snail species was analyzed and visualized using a heatmap and bubble map (Figure 4), with the results showing differences in the composition and distribution of bacteria across the samples.
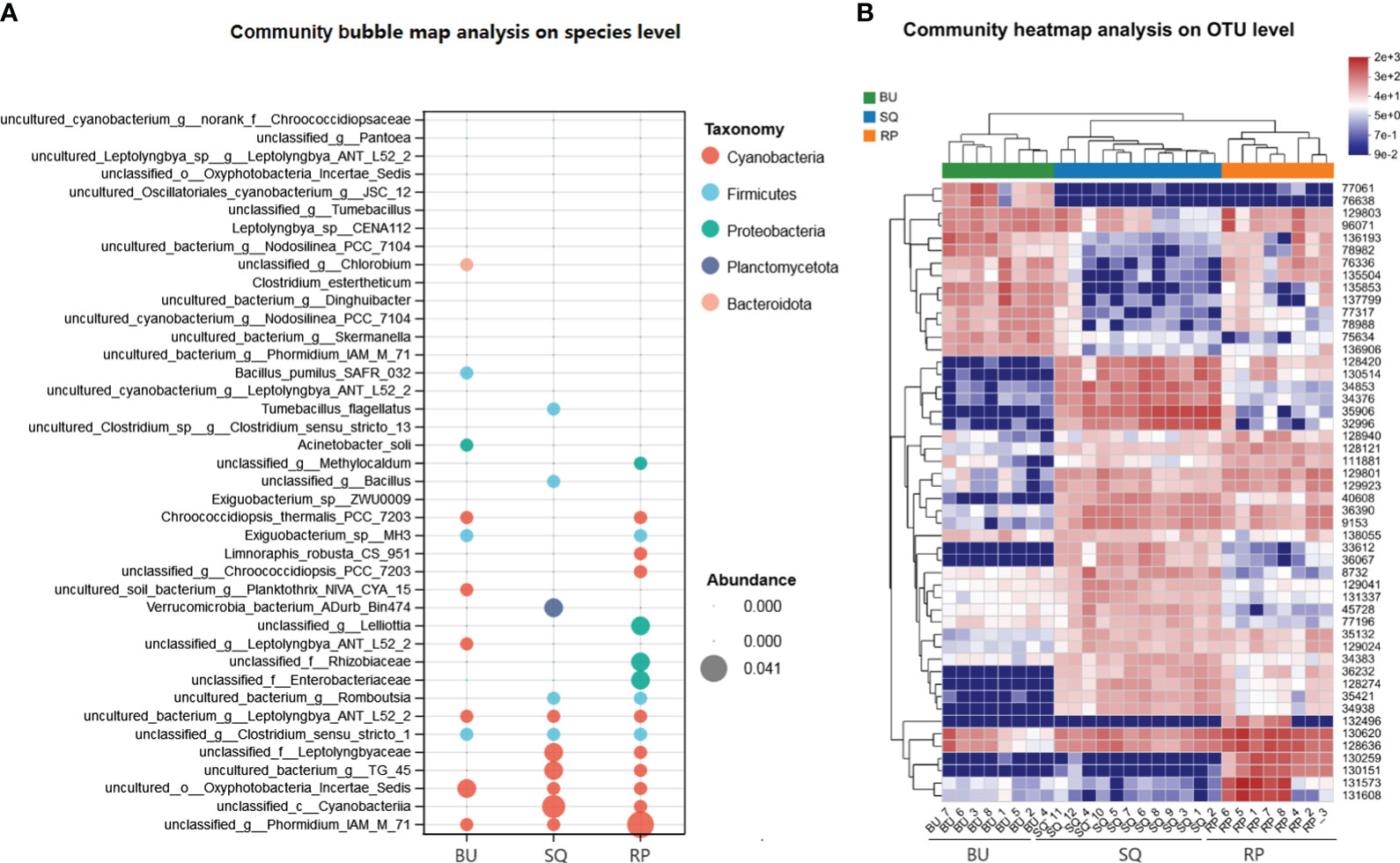
Figure 4 Abundance analysis of bacterial communities. (A) Bubble map showing the abundance of different phyla in the snails’ intestines. The size of the bubbles reflects the abundance, while each color corresponds to a phylum. Bacterial taxonomy is shown on the left, and the species of freshwater snails are at the bottom. (B) Heatmap of the top 50 most abundant gut bacteria present in the different snail species.
In particular, the bubble map analysis showed that the top five abundant species of Cyanobacteria mainly occurred in SQ and RP snails, while the top three abundant species from Proteobacteria distributed in RP (Figure 4A). Verrucomicrobia_bacterium_ADurb_Bin474, the largest abundant species in Planctomycetota, was only distributed in SQ. In the case of the heatmap which displayed the 50 most abundant OTUs, the results not only reflected the differences in the bacterial composition of the three groups of samples but also highlighted a close similarity between the intestinal microbiota of SQ and RP (Figure 4B). In addition, a number of lactic acid bacteria, such as Lactobacillus_rhamnosus_GG, Lactobacillus_amylovorus, Lactobacillus_mucosae_LM1 and Lactobacillus_pontis were found to be common to SQ and BU (Supplementary Table S1).
The intestinal microbiota of SQ, BU and RP were also compared to identify significant differences between them. The results showed that most of the microbiota were significantly different between the three snail species from phylum to the species level (Supplementary Figures S5, S6).
In particular, LEfSe analysis (LDA threshold > 4) revealed significant enrichment of specific bacterial species in each snail species (Figure 5). These included phyla Planctomycetota, class Planctomycetes, orders Leptolyngbyales, Isosphaeralesin and Alicyclobacillales in SQ samples, orders Rhizobiales and genus Phormidium_IAM_M-71 in RP samples, while class Bacilli, orders Chitinophagales, diplorickettsiaceae and Burkholderiales in BU (Figure 5).
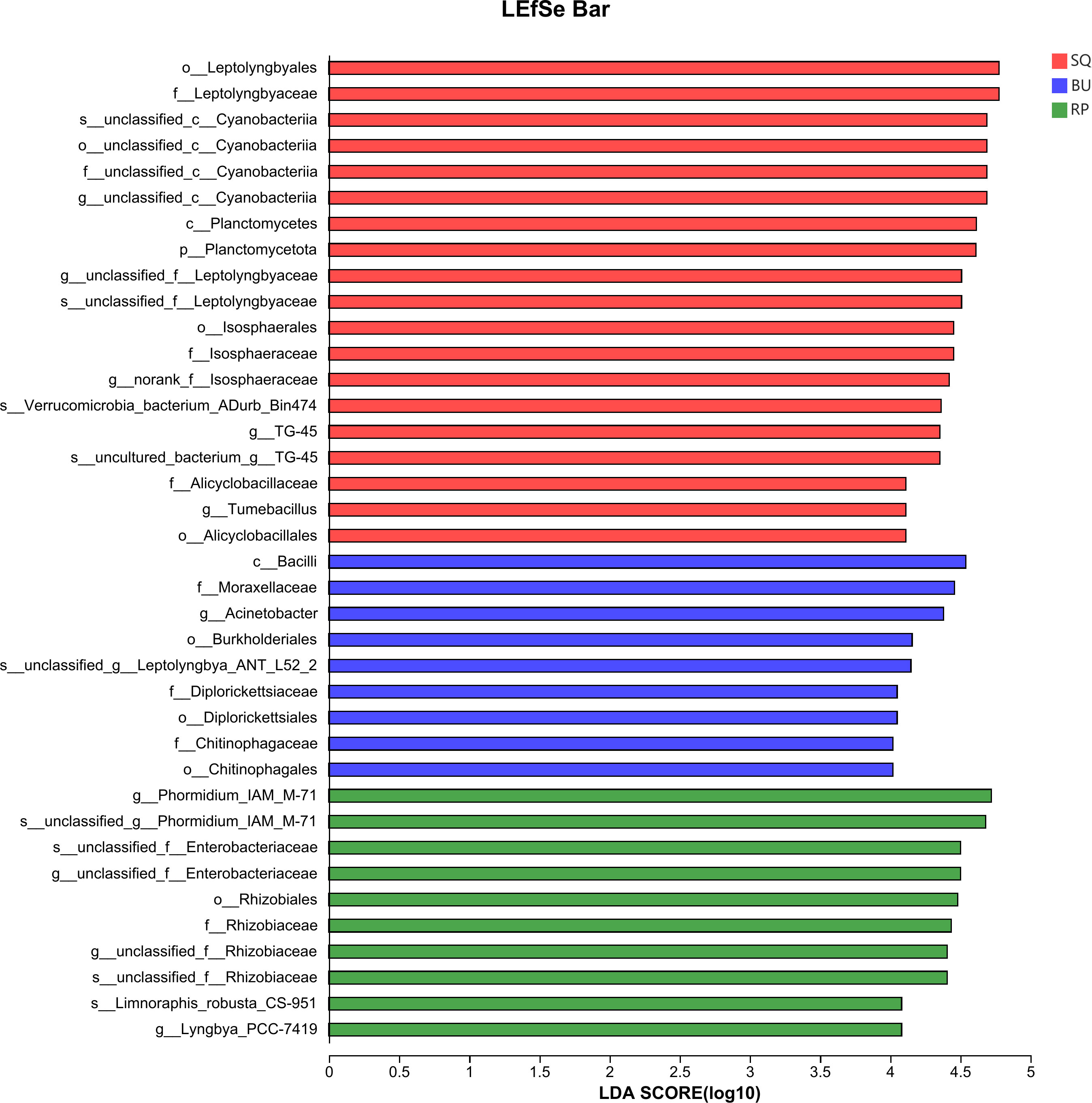
Figure 5 LEfSe analysis identified the markedly different species of gut microbiome in three species of snails (LDA threshold above 4).
Functional prediction of the intestinal bacterial communities of the three snail species was performed using PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) with the results shown in Figure 6. Overall, the abundance of the KEGG metabolic pathways was consistent across the three species, with 18 pathways having an abundance of more than 1%. Of these, Global and overview maps were the most abundant, followed by Carbohydrate metabolism, Amino acid metabolism and Energy metabolism, just to name a few. A number of these metabolic pathways, including Global and overview maps, Amino acid metabolism, Energy metabolism and Metabolism of cofactors and vitamin amongst others, were significantly different between the three snail species (P < 0.05).
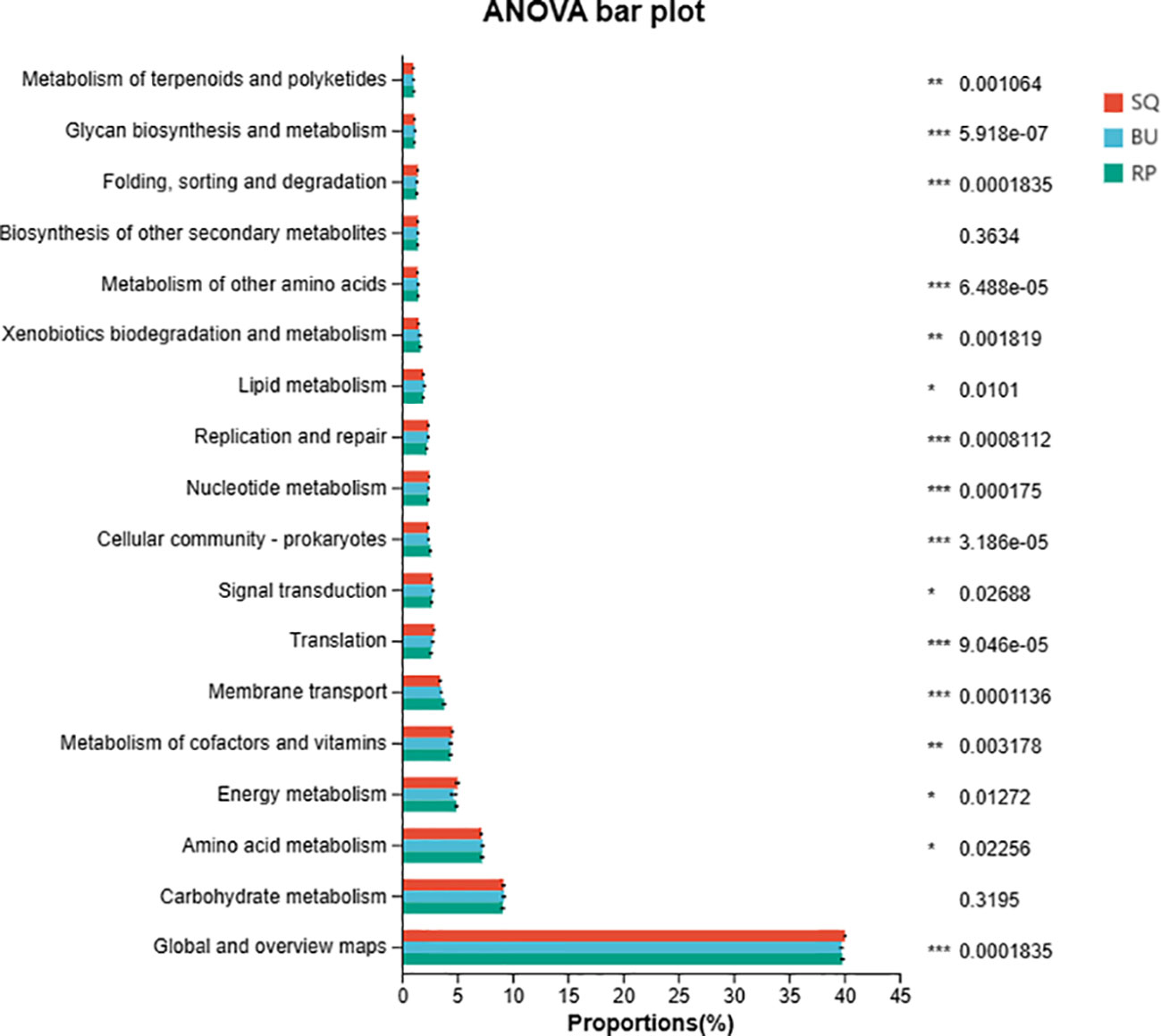
Figure 6 Functions of the intestinal bacterial communities of the three snail species as predicted by PICRUSt2. The figure shows second-level KEGG metabolic pathways with an abundance of > 1%. “*” 0.01 < P < 0.05; “**” 0.001 < P < 0.01; “***” P < 0.001.
BugBase predicted a number of microbiome phenotypes for the snails, including biofilm formation, pathogenicity, stress tolerance, aerobic, anaerobic or facultative anaerobic respiration, presence of mobile elements as well as gram-positive or gram-negative features which were all different between the three species (P < 0.05) (Figure 7A). More specifically, RP showed significant differences in the abundance of bacteria that contributed to stress tolerance, pathogenicity, biofilm formation, the presence of mobile elements and facultative anaerobic phenotypes (P < 0.05) (Figure 7A). Whereas bacteria that contributed to the aerobic phenotypes were significantly more abundant in BU than in RP, although no differences were noted when compared with SQ (P < 0.05) (Figure 7A). Finally, anaerobic phenotypes were significantly more abundant in SQ compared with BU and RP (P < 0.05) (Figure 7A).
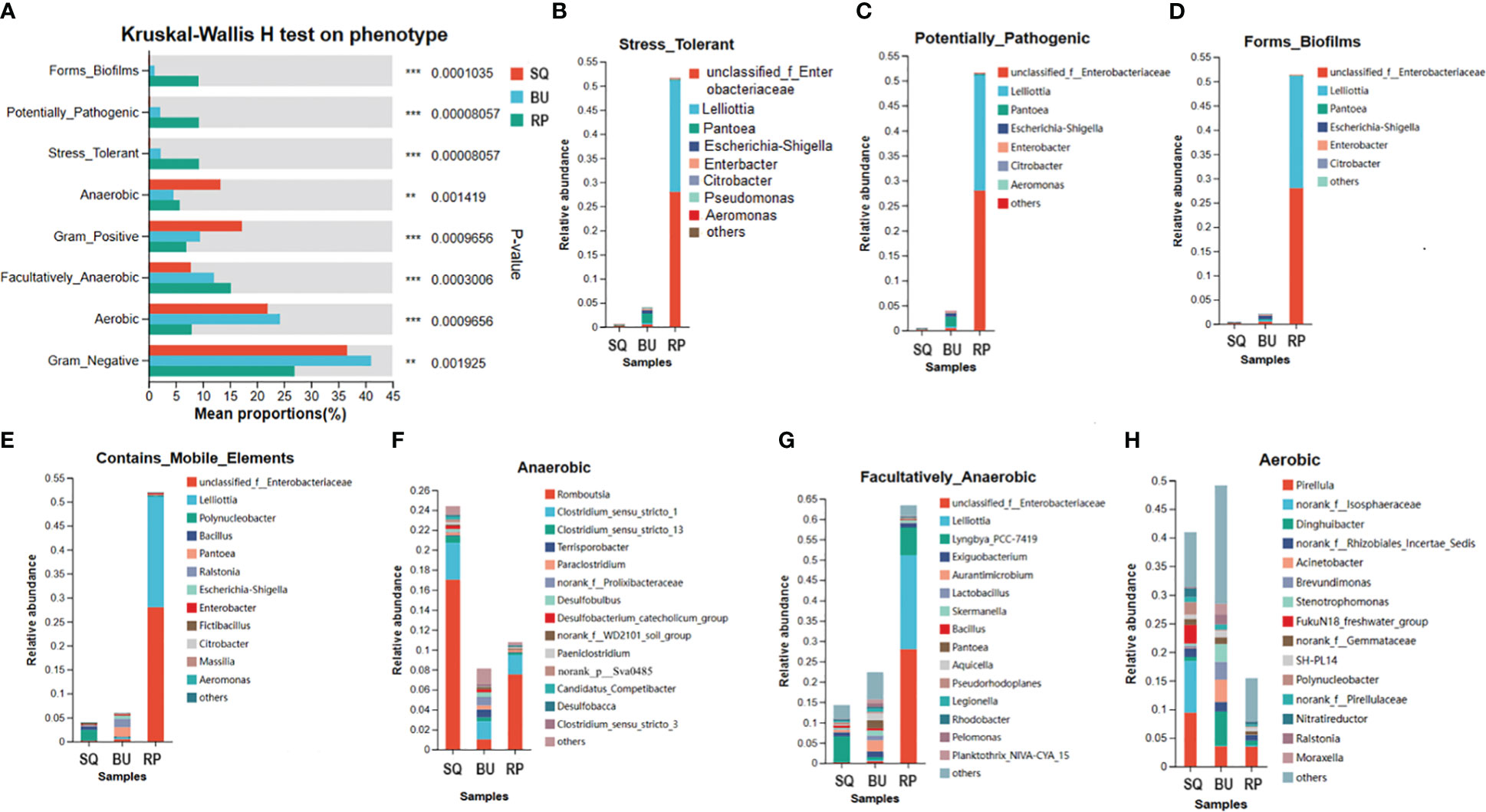
Figure 7 BugBase predicts the proportion of biological phenotype in gut microbiomes of SQ, BU, and RP snails. (A) Differences between phenogroups. The horizontal coordinate indicates the phenotype name, the vertical coordinate indicates the percentage value of the relative abundance of a phenotype in the sample, and different colors indicate different groups. On the far right is the p-value, **0.001 < P ≤ 0.01, ***P ≤ 0.001. (B-H) Analysis of the specie’s contribution to phenotype indicated the dominant species composition of a particular phenotype, that is, reflects the correspondence between species and phenotype. The horizontal coordinate is the group name, the different color legend represents different species, and the vertical coordinate is the contribution of different species in the sample to this phenotype.
Specific bacterial species were markedly correlated with the different phenotypes. For instance, in RP, unclassified_Enterobacteriaceae and Lelliottia were linked to stress tolerance, biofilm formation, potential pathogenicity, mobile elements, and facultatively anaerobic phenotypes, with a contribution of 28.01% and 23.06%, respectively (Figures 7B–G). Similarly, Romboutsia and Clostridium_sensu_stricto_1 respectively contributed 17.01% and 3.70% to the anaerobic phenotype predicted for SQ, while in the case of RP, the degree of contribution was 7.52% and 1.95%, respectively (Figure 7F). In addition, norank_f_Isosphaeraceae and Pirellula were correlated with the aerobic phenotype in SQ, contributing 9.43% and 9.01% respectively. In BU, the most significant contribution came from Pirellula, Dinghuibacter, Acinetobacter, Brevundimonas and Stenotrophomonas (Figure 7H).
A total of 68 GH (glycoside hydrolase) genes were predicted by PICRUSt, with their abundance accounting for 1.26%, 1.55%, and 1.50% of the total genes in SQ, BU and RP, respectively. Table 2 shows 31 highly abundant genes, of which 21 were significantly different between the three groups, while 10 indicated no significant differences.
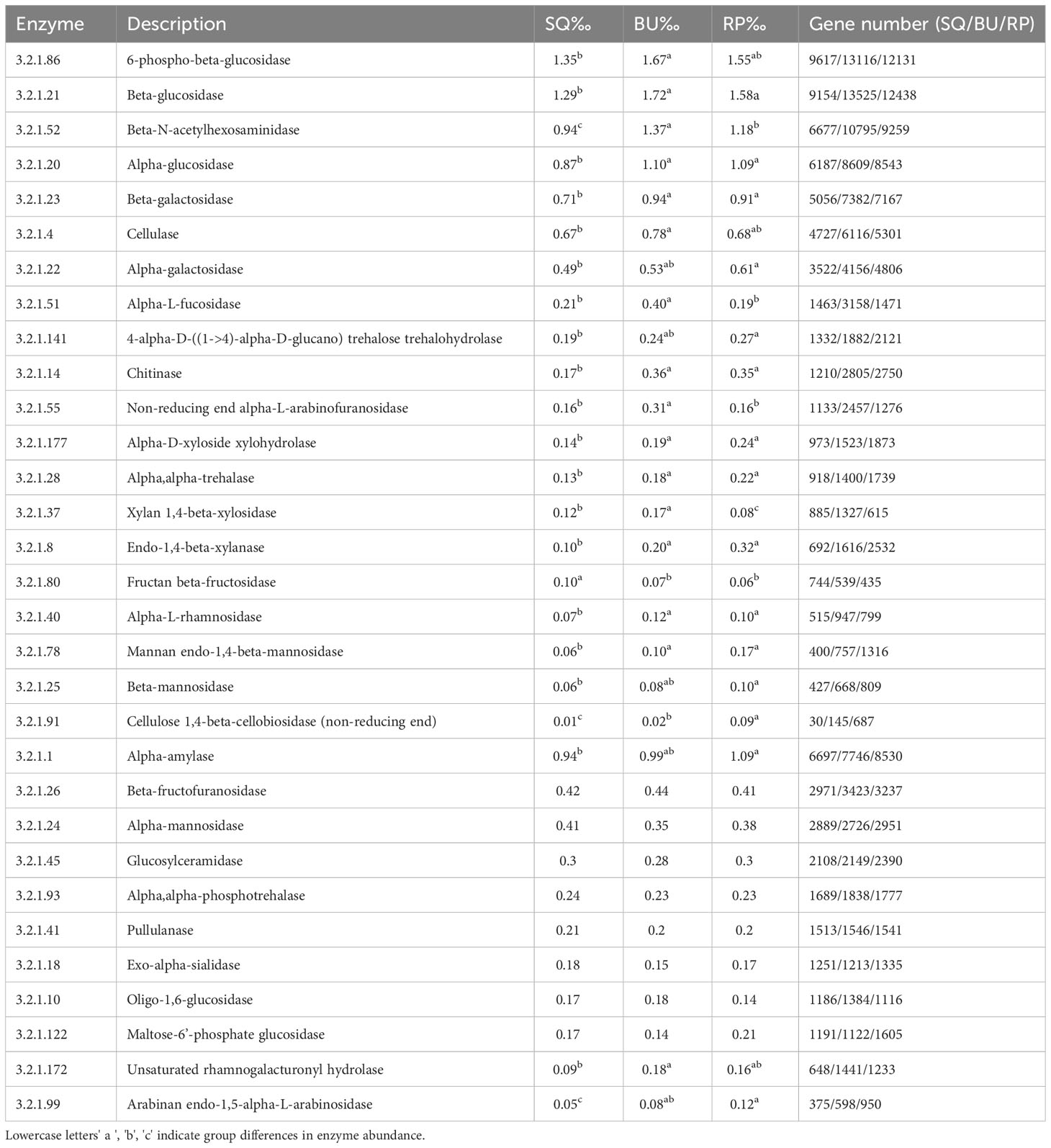
Table 2 Comparison of the glycoside hydrolase (GH, EC3.2.1) genes involved in lignocellulose degradation in the snail SQ, BU, RP metagenomes predicted genes by PICRUSt.
Of these, in BU, the relatively abundant genes included 6-phospho-beta-glucosidase (EC 3.2.1.86), Beta-glucosidase (EC 3.2.1.21), Beta-N-acetylhexosaminidase (EC 3.2.1.52), Alpha-glucosidase (EC 3.2.1. 20), Beta-galactosidase (EC 3.2.1. 23), Cellulase (EC 3.2.1.4), Alpha-galactosidase (EC 3.2.1. 22) and Alpha-L-fucosidase (EC 3.2.1. 51) (Table 2). Whereas, their abundance was relatively low in SQ, and intermediate in RP.
In this study, a number of genes encoding carbohydrate-digesting enzymes were found in the three species of snails. These enzymes included monosaccharide-digesting ones, such as Beta-glucosidase, Alpha-glucosidase, Beta-galactosidase, Alpha-galactosidase and Alpha-L-rhamnosidase (3.2.1.40), disaccharide-digesting ones, such as Alpha-mannosidase (3.2.1.24) and Beta-mannosidase (3.2.1.25) as well as those that act on polysaccharides, namely Cellulase (3.2.1.4), Cellulose 1,4-beta-Cellobiosidase (3.2.1.91) and Endo-1,4-beta-xylanase (3.2.1.8). In addition, genes of enzymes that digest starch, including Alpha-amylase (3.2.1.1) and Pullulanase (3.2.1.41), as well as for those that are specific to aquatic plants, such as alpha-trehalase (3.2.1.28) and Chitinase (3.2.1.14), were also present (Table 2).
The identification of pathways related to Xenobiotics biodegradation and metabolism indicated the presence of Drug metabolism, Benzoate degradation, Chloroalkane and chloroalkene degradation, Aminobenzoate degradation, Metabolism of xenobiotics by cytochrome P450 and Naphthalene degradation pathways in all three snail species (Supplementary Figure S7). Furthermore, the presence of pathways related to Infectious diseases suggested that Tuberculosis, Salmonella infection, Legionellosis, Pathogenic Escherichia coli infection and Pertussis could also be abundant in those snails (Supplementary Figure S8).
In this study, SMRT sequencing of full-length 16S rRNA genes was used to compare the gut microbiota of three species of wild herbivorous snails living in the same freshwater environment. Although three snail species, namely Sinotaia quadrata (SQ), Boreoelona ussuriensis (BU) and Radix plicatula (RP), are common in China (Wu et al., 2017; Ye et al., 2021); however, their intestinal microbial communities have not yet been reported. Through the successful identification of a number of bacteria up to the species level, this work yielded distinct gut microbial community profiles for the three snail species.
The sequencing results revealed a high diversity of intestinal microbes in the three snails, as evidenced by the abundance of operational taxonomic units (OTUs) and the alpha diversity metrics. Among the indices, the Shannon diversity index indicated a particular diversity of 6.08, 6.24, and 5.72 for SQ, BU, and RP, respectively. After clustering sequences at a 97% similarity-level threshold, a total of 2898 OTUs (SQ: 2532, BU: 2341, RP: 2443) were obtained for the intestinal microbiota of the three snail species. Furthermore, the alpha-diversity metrics results were higher than that of Pomacea canaliculate with the Shannon and Chao indices of 3.35 and 1106, respectively (Chen et al., 2021).
The complex food sources of herbivorous freshwater snails are rich in indigestible lignocelluloses; therefore, a large diversity of microbial species is required in their digestive tracts to efficiently digest and utilize these substances (Harris, 1993). For example, in cattle, a complex microbiota in the rumen helps to decompose plant fibers, with over 5000 OTUs in the rumen microbiota in second-aged cattle (Jami et al., 2013). The results of this study also demonstrated that the diversity of the gut microbiota was significantly higher in SQ compared with RP. This difference could, in fact, be related to the body size and intestinal morphology of freshwater snails, as SQ species have larger and longer intestines, which allow them to retain a higher diversity of microorganisms.
At the phylum level, Cyanobacteria, Proteobacteria, Firmicutes and Planctomycetota were common to all three snail species. In contrast, a study on the gut microbiota of R. auricularia (Shannon 5.12) reported Proteobacteria, Cyanobacteria, Chloroflexi, Firmicutes, and Actinobacteria as dominant phyla (Hu et al., 2018), while in another study on Planorbella trivolvis, Proteobacteria, Bacteroidetes and Actinobacteria were predominant (Hu et al., 2021). Nevertheless, the results of this study, along with the above reports, suggest that Proteobacteria tend to be a major bacterial group in the intestinal microbiota of freshwater snails (Walters et al., 2022; Zhou et al., 2022). However, this may not always be the case. Indeed, the intestinal microbiota of P. canaliculate, collected from culture ponds, had a high abundance of Tenericutes (45.7%) and Firmicutes (27.85%), while Proteobacteria (11.86%) ranked third in abundance (Chen et al., 2021). These differences could be attributed to variations in the food composition or water environment. Although Planctomycetota was found to be highly abundant in all three snail species and also found in another study of Planorbidae snails (Van Horn et al., 2012), it has rarely been reported in other freshwater snails (Hu et al., 2018; Hu et al., 2021; Liu and Li, 2022; Zhou et al., 2022). In fact, it was only reported in 1 study on the intestinal microbiota of terrestrial Hydrobiidae (Walters et al., 2022).
As far as Cyanobacteria was concerned, it accounted for 784 of the 2,898 total OTUs, and its abundance reached 42, 32, and 42% in SQ, BU, and RP, respectively. Cyanobacteria is a naturally prevalent microbe that is usually attached to fallen leaves, aquatic grasses, humus, and rocky soil. During ingestion, freshwater snails take in a large number of these Cyanobacteria, resulting in a high abundance of this phylum among the intestinal microorganisms of freshwater snails. However, this abundance is dependent on the environmental abundance of Cyanobacteria, with significant changes likely affecting the physiological functions of the host (Lyu et al., 2022).
Identifying the intestinal microbiota of freshwater snails is helpful to accurately understand their role. However, most previous studies were based on the high-throughput sequencing of partial 16S rRNA genes (e.g., the V3-V4 region), resulting in sequence lengths that did not allow accurate identification up to the species level. Here, full-length 16S rRNA genes were sequenced to determine the intestinal microorganisms of three freshwater snails. Yet, limitations in the amount of information available in the database restricted the identification of OTUs to the species level to only 25.62%. Therefore, the addition of comprehensive information in the database is expected to improve accuracy. In addition, the identification rate of bacterial species was also impacted by the availability of research data on freshwater snail intestinal microbiota, hence emphasizing the need for further research in this area.
A total of 196 species were identified in this study, and this provided an understanding of their roles in intestinal microbiota. For example, Tumebacillus_flagellatus, abundant in SQ snails (1.93%), is an alpha-amylase/pullulanase-producing bacterium that plays an important role in starch digestion (Wang et al., 2013). Similarly, Limnoraphis_robusta, a nitrogen-fixing and carotenoid-rich cyanobacterial species, is a common bloom species that does not produce cyanotoxins (Komarek et al., 2013), while Clostridium_estertheticum, common to all three snail species, can secrete a bacteriocin with potential antibacterial activity against Bacillus cereus, Staphylococcus aureus (methicillin-resistant), Escherichia coli and Pseudomonas aeruginosa (Wambui et al., 2022). Therefore, full-length 16S rRNA gene sequencing is crucial for identifying the role of strains in the gut.
Surprisingly, some Lactobacillus species were present in SQ and BU species but not in RP, such as Lactobacillus_rhamnosus_GG, Lactobacillus_amylovorus, Lactobacillus_mucosae_LM1 and Lactobacillus_pontis. Lactobacillus are considered as probiotics that are beneficial for animal digestion and health (Borchers et al., 2002). Lactobacillus rhamnosus, in particular, has been widely studied as a prebiotic for its positive effects on gut health and immune enhancement (Segers & Lebeer., 2014). Similarly, by producing lactate and acetate, Lactobacillus_amylovorus is also an important intestinal probiotic that contributes to gut health (Jing et al., 2022), especially in conferring health benefits, such as anti-hyperammonemia and antiviral properties (Singh et al., 2018; Sunmola et al., 2019). Furthermore, Lactobacillus_mucosae has also been studied for its immunomodulatory or nutritional effects (Ryan et al., 2019; Wang et al., 2022), while Lactobacillus_pontis is commonly found in fermentation processes, such as sourdough or silage (Wu et al., 2014; Wang et al., 2020). Therefore, the intestinal microbiota of freshwater snails acts as a reservoir for probiotic species.
Most of the studies on the intestinal microbiota of mollusks were mainly focused on host species of commercial value or were performed in artificial environments. In contrast, only few involved wild freshwater snails. Therefore, differences in the intestinal microbiota of freshwater snail hosts with similar feeding habits and from the same natural environment are yet to be understood.
The gut microbiome of animals is influenced by many factors, such as the host species, the water environment or food, and therefore, in the case of freshwater snails, much of the experimental data tend to come from artificially controlled environments (Chalifour and Li, 2021; Hu et al., 2021; Li et al., 2022; Zhou et al., 2022). This is because in natural water bodies, it is difficult to model the effects of host species on the microbiota due to a number of naturally occurring uncontrollable factors. Hence, in this work, freshwater snails with similar feeding habits were selected from the same water environment to reduce the influence of environmental and food factors. The results subsequently confirmed that, under similar natural conditions, the intestinal microbiota of freshwater snails was dependent on the host species.
The study of Walters et al. (2022) on the intestinal microbes of two snail species from the same family (Hydrobiidae) showed that both the surrounding water environment and host species influenced the gut microbiota. However, in addition to hosts’ influence, differences in food composition may also affect the gut microbiota, and in this context, studies have shown that the main food source of herbivorous freshwater snails was detritus, followed by decaying macrophytes, diatoms and filamentous algae (Madsen, 1992). For instance, Zhou et al. (2022) fed the same vegetable (Brassica rapa) to two freshwater snails, Pomacea canaliculata and Cipangopaludina chinensis (under laboratory rearing conditions) and studied their intestinal microbiota, which revealed differences between the intestinal microbiota of the two species. Here, although a number of bacteria were found to be common to the three snails, the findings of this study highlighted that their abundance was different. In addition, the dominant species and genera were also different. Thus, altogether, the results suggest that some microorganisms are shared by two or more hosts, while others are specific to a single host.
Bacterial phenotypes determine the functions of gut microorganisms. For instance, biofilms can improve the host’s resistance against pathogen invasion, while the presence of mobile elements can improve the host’s tolerance to drug resistance (Palmer et al., 2010; Stokes and Gillings, 2011). Stress tolerance can further improve adaptability to environmental changes, such as temperature, salinity and contaminants (Fahy et al., 2008; Li et al., 2022; Liu et al., 2023). In this study, significant differences were found between the intestinal microbiome phenotypes of the three snail species, as evidenced from the results of BugBase. In the case of RP, Lelliottia and the genus unclassified_Enterobacteriaceae contributed to phenotypes, such as biofilm formation, potential pathogenicity, stress tolerance, the presence of mobile elements as well as facultative anaerobic characteristics. Lelliottia is a widespread strain in natural environments such as water (Kämpfer et al., 2018) but can also be found in Giant pandas or as an endophyte of desert cactus (Eke et al., 2019; Zhang et al., 2022). Similarly, in addition to its wide distribution in soil and water, Enterobacteriaceae are also largely found in humans, animals and plants (Cohen et al., 2020). Pirellula, assigned to the phylum Planctomycetes, is a chemoheterotrophic aerobe that is partly responsible for the aerobic phenotype in snails. In fact, many Planctomycetes are aerobes and, as such, they may play a central role in the degradation and digestion of heteropolysaccharides from plants and algae (Fuerst, 2017). In contrast, Romboutsia contributed to the anaerobic phenotype of the three snails, especially in SQ and RP. The members of this genus are gram-positive and spore-forming obligate anaerobes that are present not only in human and animal guts but also in natural environments such as coastal estuarine mud and alkaline-saline lake sediment (Gerritsen et al., 2014; Wang et al., 2015; Ricaboni et al., 2016; Maheux et al., 2017).
The intestinal microbiota plays an important role in host metabolism, immunity, digestion, growth and development (Brestoff and Artis, 2013; Lin et al., 2017). In the current study, it was found that the intestinal microorganisms of freshwater snails involved in various important metabolic pathways, such as carbon metabolism, amino acid synthesis, energy metabolism, and metabolism of cofactors and vitamin, just to name a few. These metabolic functions enable microorganisms to participate in food digestion and absorption, protein synthesis, as well as in the supply of energy, vitamins and cofactors in the host’s gut. Consistent with this study’s findings, the microbial community associated with A. fulica was also highly enriched in Carbohydrates Metabolism, Amino Acids Metabolism, Cofactors and Vitamins Metabolism, Protein Metabolism and Membrane Transport (Cardoso et al., 2012).
The food of plant-eating gastropods is rich in cellulose that is difficult to digest. Hence, the gut microbiota secretes large amounts of enzymes to assist in the digestion process. In this study, a large number of genes encoding carbohydrate-digesting enzymes were found to be present in the three species of snails. These included monosaccharide-, disaccharide-, polysaccharide- and starch-digesting enzymes, along with those that are specific to aquatic plants. In addition, cellulase (β-1, 4-glucan-4-glucan hydrolase) (EC. 3.2.1.4) is important for converting cellulose into glucose, and interestingly, the gene encoding this enzyme was highly abundant in the intestinal microbiota of all three snail species, thereby highlighting the importance of these bacteria in helping the host to digest lignocellulose. It has been reported that the intestinal microbiota of the herbivorous snail Achatina fulica is rich in cellulase- and hemicellulose-coding sequences (Cardoso et al., 2012). These cellulose-degrading genes can actually be of value in the development of lignocellulose-derived biofuel.
The enrichment of pathways related to Xenobiotics biodegradation and metabolism indicated that the gut microbiota could play an important role in helping freshwater snails decompose foreign harmful substances such as drugs and pollutants that are discharged through industrial, agricultural or human activities (Liu et al., 2023). In addition, the enrichment of pathways related to Infectious diseases further suggested that some gut microbes could be pathogenic to humans and animals, thereby highlighting their significance for public health.
It should be pointed out that PICRUSt2 has certain limitations in the prediction of microbial function. PICRUSt2 predicts the approximate function of a community based on community membership identified by the 16S rRNA gene; however, it does not provide direct information on microbial genes and their functional composition. Furthermore, the prediction cannot fully recapitulate metagenomics-based functional metabolic profiles, distinguish strain-specific functionality, or identify rare environment-specific functions (Douglas et al., 2020; Gehrig et al., 2022). Due to constraints inherent with 16S sequencing, BugBase has the same limitations as the PICRUSt2, such as lower prediction accuracy, unlike to identify OTUs at the strain level and to distinguish horizontal gene transfer between OTUs (Ward et al., 2017).
This study indicates that the gut microbiome is different in different freshwater snails within the same habitats. Of the 776 species of intestinal bacteria, 566 (72.94%) were common to all three snail species, although significant differences were noted in their abundance. Furthermore, a high abundance of specific bacteria was also found to influence the biological phenotypes of snails. In addition, genes encoding a number of carbohydrate-digesting enzymes were present in the three snail species, and these included monosaccharide-, disaccharide-, polysaccharide- and starch-digesting enzymes as well as those that were specific to aquatic plants. The identification of many pathways related to Infectious diseases and Xenobiotics biodegradation and metabolism suggested that freshwater snails could harbor resistance to foreign substances. Overall, given that the influence of host species on the gut microbiome is scarcely explored in natural water environments, this research is expected to contribute to the understanding of the relationship between freshwater snails, their microbial communities and their functions.
The original contributions presented in the study are publicly available. Raw sequencing data of 16S rRNA have been deposited in the Sequence Read Archive (SRA) database of the National Center for Biotechnology Information (NCBI) with the Bioproject ID PRJNA1037673.
The animal study was approved by The ethics committee of Inner Mongolia Minzu University (IACUC-202111-073). The study was conducted in accordance with the local legislation and institutional requirements.
ZH: Data curation, Funding acquisition, Investigation, Software, Writing – original draft. QT: Methodology, Writing – review & editing. JC: Formal analysis, Funding acquisition, Writing – review & editing. JX: Data curation, Methodology, Writing – review & editing. BW: Conceptualization, Formal analysis, Writing – review & editing. YH: Methodology, Software, Writing – review & editing. JY: Data curation, Writing – review & editing. HN: Supervision, Validation, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Ph.D. start-up fund of Inner Mongolia Minzu University (BS635); The program for Young Talents of Science and Technology of the Universities of Inner Mongolia Autonomous Region (NJYT22054); Natural Science Foundation of Inner Mongolian (2022MD03074); National Natural Science Foundation of China (31860730).
We are also very grateful to the editors and reviewers for their valuable suggestions, which greatly improved the quality of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1341359/full#supplementary-material
An Y., Braga M. P., Garcia S. L., Grudzinska-Sterno M., Hambäck P. A. (2023). Host phylogeny structures the gut bacterial community within galerucella leaf beetles. Microb. Ecol. 86 (4), 2477–2487. doi: 10.1007/s00248-023-02251-5
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37 (8), 852–857. doi: 10.1038/s41587-019-0209-9
Borchers A. T., Keen C. L., Gershwin M. E. (2002). The influence of yogurt/Lactobacillus on the innate and acquired immune response. Clin. Rev. Allergy Immunol. 22 (3), 207–230. doi: 10.1007/s12016-002-0009-7
Brestoff J. R., Artis D. (2013). Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 14, 676–684. doi: 10.1038/ni.2640
Brucker R. M., Bordenstein S. R. (2012). The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution 66 (2), 349–362. doi: 10.1111/j.1558-5646.2011.01454.x
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Cao Z., Jiang X. (1998). The influence of environmental factors on Bellamya purificata. Shanghai Fish. Univ 7, 200–205.
Cardoso A. M., Cavalcante J. J., Vieira R. P., Lima J. L., Grieco M. A., Clementino M. M., et al. (2012). Gut bacterial communities in the giant land snail Achatina fulica and their modification by sugarcane-based diet. PloS One 7 (3), e33440. doi: 10.1371/journal.pone.0033440
Chalifour B., Li J. (2021). A review of the molluscan microbiome: ecology, methodology and future. Malacologia 63 (2), 285–304. doi: 10.4002/040.063.0208
Chen L., Li S., Xiao Q., Lin Y., Li X., Qu Y., et al. (2021). Composition and diversity of gut microbiota in Pomacea canaliculata in sexes and between developmental stages. BMC Microbiol. 21 (1), 200. doi: 10.1186/s12866-021-02259-2
Chen R., Tu H., Chen T. (2022). Potential application of living microorganisms in the detoxification of heavy metals. Foods 11 (13), 1905. doi: 10.3390/foods11131905
Cohen R., Paikin S., Rokney A., Rubin-Blum M., Astrahan P. (2020). Multidrug-resistant Enterobacteriaceae in coastal water: an emerging threat. Antimicrob. Resist. Infect. Control. 9 (1), 169. doi: 10.1186/s13756-020-00826-2
Dodangeh S., Daryani A., Sharif M., Gholami S., Kialashaki E., Moosazadeh M., et al. (2019). Freshwater snails as the intermediate host of trematodes in Iran: a systematic review. Epidemiol. Health 41, e2019001. doi: 10.4178/epih.e2019001
Douglas G. M., Maffei V. J., Zaneveld J. R., Yurgel S. N., Brown J. R., Taylor C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38 (6), 685–688. doi: 10.1038/s41587-020-0548-6
Edgar R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10 (10), 996–998. doi: 10.1038/nmeth.2604
Eke P., Kumar A., Sahu K. P., Wakam L. N., Sheoran N., Ashajyothi M., et al. (2019). Endophytic bacteria of desert cactus (Euphorbia trigonas Mill) confer drought tolerance and induce growth promotion in tomato (Solanum lycopersicum L.). Microbiol. Res. 228, 126302. doi: 10.1016/j.micres.2019.126302
Fahy A., Ball A. S., Lethbridge G., Timmis K. N., McGenity T. J. (2008). Isolation of alkali-tolerant benzene-degrading bacteria from a contaminated aquifer. Lett. Appl. Microbiol. 47 (1), 60–66. doi: 10.1111/j.1472-765X.2008.02386.x
Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3 (5), 294–299.
Fuerst J. A. (2017). Planctomycetes-new models for microbial cells and activities. Microbial. Resour., 1–27. doi: 10.1016/B978-0-12-804765-1.00001-1
Gehrig J. L., Portik D. M., Driscoll M. D., Jackson E., Chakraborty S., Gratalo D., et al. (2022). Finding the right fit: evaluation of short-read and long-read sequencing approaches to maximize the utility of clinical microbiome data. Microb. Genom. 8 (3), 794. doi: 10.1099/mgen.0.000794
Gerritsen J., Fuentes S., Grievink W., van Niftrik L., Tindall B. J., Timmerman H. M., et al. (2014). Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int. J. Syst. Evol. Microbiol. 64 (Pt5), 1600–1616. doi: 10.1099/ijs.0.059543-0
Harris J. M. (1993). The presence, nature, and role of gut micromicrobiota in aquatic invertebrates: A synthesis. Microb. Ecol. 25 (3), 195–231. doi: 10.1007/BF00171889
Haworth S. E., White K. S., Côté S. D., Shafer A. B. A. (2019). Space, time and captivity: quantifying the factors influencing the fecal microbiome of an alpine ungulate. FEMS Microbiol. Ecol. 95 (7), fiz095. doi: 10.1093/femsec/fiz095
Hu Z., Chen X., Chang J., Yu J., Tong Q., Li S., et al. (2018). Compositional and predicted functional analysis of the gut microbiota of Radix auricularia (Linnaeus) via high-throughput Illumina sequencing. PeerJ 6, e5537. doi: 10.7717/peerj.5537
Hu Z., Tong Q., Chang J., Yu J., Li S., Niu H., et al. (2021). Gut bacterial communities in the freshwater snail Planorbella trivolvis and their modification by a non-herbivorous diet. PeerJ 9, e10716. doi: 10.7717/peerj.10716
Jami E., Israel A., Kotser A., Mizrahi I. (2013). Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7 (6), 1069–1079. doi: 10.1038/ismej.2013.2
Jing Y., Mu C., Wang H., Shen J., Zoetendal E. G., Zhu W. (2022). Amino acid utilization allows intestinal dominance of Lactobacillus amylovorus. ISME J. 16 (11), 2491–2502. doi: 10.1038/s41396-022-01287-8
Kämpfer P., Glaeser S. P., Packroff G., Behringer K., Exner M., Chakraborty T., et al. (2018). Lelliottia aquatilis sp. nov., isolated from drinking water. Int. J. Syst. Evol. Microbiol. 68 (8), 2454–2461. doi: 10.1099/ijsem.0.002854
Kivistik C., Käiro K., Tammert H., Sokolova I. M., Kisand V., Herlemann D. P. R. (2022). Distinct stages of the intestinal bacterial community of Ampullaceana balthica after salinization. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.767334
Koeppel A. F., Wu M. (2013). Surprisingly extensive mixed phylogenetic and ecological signals among bacterial operational taxonomic units. NucleicAcids Res. 41, 5175–5188. doi: 10.1093/nar/gkt241
Komarek J., Zapomelova E., Smarda J., Kopecky J., Komarkova J. (2013). Polyphasic evaluation of limnoraphis robusta, awater-bloom forming cyanobacterium from lake atitlan, Guatemala, with adescription of limnoraphis. gen. nov. Fottea. 13, 1, 3952.
Li S., Qian Z., Gao S., Shen W., Li X., Li H., et al. (2022). Effect of long-term temperature stress on the intestinal microbiome of an invasive snail. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.961502
Lin R., Liu W., Piao M., Zhu H. (2017). A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids 49, 2083–2090. doi: 10.1007/s00726-017-2493-3
Liu F., Li K. (2022). Comparison of epiphytic and intestinal bacterial communities in freshwater snails (Bellamya aeruginosa) living on submerged plants. PeerJ 10, e14318. doi: 10.7717/peerj.14318
Liu H., Yang X., Yang W., Zheng Z., Zhu J. (2023). Gut microbiota of freshwater gastropod (Bellamya aeruginosa) assist the adaptation of host to toxic cyanobacterial stress. Toxins (Basel). 15 (4), 252. doi: 10.3390/toxins15040252
Liu C., Yao H., Chapman S. J., Su J., Wang C. (2020). Changes in gut bacterial communities and the incidence of antibiotic resistance genes during degradation of antibiotics by black soldier fly larvae. Environ. Int. 142, 105834. doi: 10.1016/j.envint.2020.105834
Loh Z. H., Ouwerkerk D., Klieve A. V., Hungerford N. L., Fletcher M. T. (2020). Toxin degradation by rumen microorganisms: A review. Toxins (Basel) 12 (10), 664. doi: 10.3390/toxins12100664
Lyu T., Zhu J., Yang X., Yang W., Zheng Z. (2022). Responses of gut microbial community composition and function of the freshwater gastropod bellamya aeruginosa to cyanobacterial bloom. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.906278
Madsen H. (1992). Food selection by freshwater snails in the Gezira irrigation canals, Sudan. Hydrobiologia 228, 203–217. doi: 10.1007/BF00006587
Maheux A. F., Boudreau D. K., Bérubé È., Boissinot M., Raymond F., Brodeur S., et al. (2017). Draft genome sequence of Romboutsia maritimum sp. nov. strain CCRI-22766T, isolated from coastal estuarine mud. Genome Announc. 5 (41), e01044–e01017. doi: 10.1128/genomeA.01044-17
Marshall C. W., Ross D. E., Fichot E. B., Norman R. S., May H. D. (2012). Electrosynthesis of commodity chemicals by an autotrophic microbial community. Appl. Environ. Microbiol. 78, 8412–8420. doi: 10.1128/aem.02401-12
Palmer R. J., Periasamy S., Jakubovics N. S., Kolenbrander P. E. (2010). Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 8 (7), 471–480. doi: 10.1038/nrmicro2381
Pawar K. D., Dar M. A., Rajput B. P., Kulkarni G. J. (2015). Enrichment and identification of cellulolytic bacteria from the gastrointestinal tract of Giant African snail, Achatina fulica. Appl. Biochem. Biotechnol. 175 (4), 1971–1980. doi: 10.1007/s12010-014-1379-z
Ricaboni D., Mailhe M., Khelaifia S., Raoult D., Million M. (2016). Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 12, 6–7. doi: 10.1016/j.nmni.2016.04.001
Ryan P. M., Stolte E. H., London L. E. E., Wells J. M., Long S. L., Joyce S. A., et al. (2019). Lactobacillus mucosae DPC 6426 as a bile-modifying and immunomodulatory microbe. BMC Microbiol. 19 (1), 33. doi: 10.1186/s12866-019-1403-0
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75 (23), 7537–7541. doi: 10.1128/AEM.01541-09
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12 (6), R60. doi: 10.1186/gb-2011-12-6-r60
Segers M.E., and Lebeer S. (2014). Towards a better understanding of Lactobacillus rhamnosus GG--host interactions. Microb Cell Fact 13 (Suppl 1(Suppl 1)), S7. doi: 10.1186/1475-2859-13-S1-S7
Singh P., Chung H. J., Lee I. A., D’Souza R., Kim H. J., Hong S. T. (2018). Elucidation of the anti-hyperammonemic mechanism of Lactobacillus amylovorus JBD401 by comparative genomic analysis. BMC Genomics 19 (1), 292. doi: 10.1186/s12864-018-4672-3
Stokes H. W., Gillings M. R. (2011). Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into gram-negative pathogens. FEMS Microbiol. Rev. 35 (5), 790–819. doi: 10.1111/j.1574-6976.2011.00273.x
Sunmola A. A., Ogbole O. O., Faleye T. O. C., Adetoye A., Adeniji J. A., Ayeni F. A. (2019). Antiviral potentials of Lactobacillus plantarum, Lactobacillus amylovorus, and Enterococcus hirae against selected Enterovirus. Folia Microbiol. (Praha). 64 (2), 257–264. doi: 10.1007/s12223-018-0648-6
Takougang I., Barbazan P., Tchounwou P. B., Noumi E. (2008). The value of the freshwater snail dip scoop sampling method in macroinvertebrates bioassessment of sugar mill wastewater pollution in Mbandjock, Cameroon. Int. J. Environ. Res. Public Health 5 (1), 68–75. doi: 10.3390/ijerph5020068
Thomas J. D., Nwanko D. I., Sterry P. R. (1985). The feeding strategies of juvenile and adult biomphalaria glabrata (say) under simulated natural conditions and their relevance to ecological theory and snail control. Proc. R. Soc. London 226 (1243), 177–209.
van den Bosch T. J. M., Welte C. U. (2017). Detoxifying symbionts in agriculturally important pest insects. Microb. Biotechnol. 10 (3), 531–540. doi: 10.1111/1751-7915.12483
Van Horn D. J., Garcia J. R., Loker E. S., Mitchell K. R., Mkoji G. M., Takacs-Vesbach C. D. (2012). Complex intestinal bacterial communities in three species of planorbid snails. J. Molluscan Studies volume 78 (1), 74–80(7). doi: 10.1093/mollus/eyr038
Walters A. D., Alex A., Cerbie G. M., Trujillo D. A., Kiss A. J., Berg D. J. (2022). Phylogenetic relationship and habitat both impact the gut microbiome in two microendemic gastropods. J. Mollus Stud. 88, eyac002. doi: 10.1093/mollus/eyac002
Wambui J., Stevens M. J. A., Sieber S., Cernela N., Perreten V., Stephan R. (2022). Targeted genome mining reveals the psychrophilic Clostridium estertheticum complex as a potential Source for novel bacteriocins, including Cesin A and Estercticin A. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.801467
Wang Q., Fang Z., Li L., Wang H., Zhu J., Zhang P., et al. (2022). Lactobacillus mucosae exerted different antiviral effects on respiratory syncytial virus infection in mice. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.1001313
Wang Q., Garrity G. M., Tiedje J. M., Cole J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73 (16), 5261–5267. doi: 10.1128/AEM.00062-07
Wang Y., Song J., Zhai Y., Zhang C., Gerritsen J., Wang H., et al. (2015). Romboutsia sedimentorum sp. nov., isolated from an alkaline-saline lake sediment and emended description of the genus Romboutsia. Int. J. Syst. Evol. Microbiol. 65 (Pt 4), 1193–1198. doi: 10.1099/ijs.0.000079
Wang Q., Xie N., Qin Y., Shen N., Zhu J., Mi H., et al. (2013). Tumebacillus flagellatus sp. nov., an α-amylase/pullulanase-producing bacterium isolated from cassava wastewater. Int. J. Syst. Evol. Microbiol. 63 (Pt9), 3138–3142. doi: 10.1099/ijs.0.045351-0
Wang X., Zhu X., Bi Y., Zhao R., Nie Y., Yuan W. (2020). Dynamics of microbial community and changes of metabolites during production of type I sourdough steamed bread made by retarded sponge-dough method. Food Chem. 330, 127316. doi: 10.1016/j.foodchem.2020.127316
Ward T., Larson J., Meulemans J., Hillmann B., Lynch J., Sidiropoulos D., et al. (2017). BugBase predicts organism level microbiome phenotypes. bioRxiv, 133462. doi: 10.1101/133462
Weisburg W. G., Barns S. M., Pelletier D. A., Lane D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173 (2), 697–703. doi: 10.1128/jb.173.2.697-703.1991
Wu J. J., Du R. P., Gao M., Sui Y. Q., Xiu L., Wang X. (2014). Naturally occurring lactic Acid bacteria isolated from tomato pomace silage. Asian-Australas J. Anim. Sci. 27 (5), 648–657. doi: 10.5713/ajas.2013.13670
Wu H., Guan Q., Lu X., Batzer D. P. (2017). Snail (Mollusca: Gastropoda) assemblages as indicators of ecological condition in freshwater wetlands of Northeastern China. Ecol. Indic. 75, 203–209. doi: 10.1016/j.ecolind.2016.12.042
Ye B., Hirano T., Saito T., Dong Z., Do V. T., Chiba S. (2021). Molecular and morphological evidence for a unified, inclusive Sinotaia quadrata (Caenogastropoda: Viviparidae: Bellamyinae). J. Mollus Stud. 87 (3), eyab013. doi: 10.1093/mollus/eyab013
Zhang X., Wang X., Ayala J., Liu Y., An J., Wang D., et al. (2022). Possible effects of early maternal separation on the gut microbiota of captive adult giant pandas. Anim. (Basel). 12 (19), 2587. doi: 10.3390/ani12192587
Zhou M., Hou Y., Jia R., Li B., Zhu J. (2023). Effects of Bellamya purificata Cultivation at Different Stocking Densities on the Dynamics and Assembly of Bacterial Communities in Sediment. Biomolecules 13 (2), 254. doi: 10.3390/biom13020254
Keywords: freshwater snails, Sinotaia quadrata, Boreoelona ussuriensis, Radix plicatula, single-molecule real-time sequencing technology (SMRT), microbial community, function prediction
Citation: Hu Z, Tong Q, Chang J, Xu J, Wu B, Han Y, Yu J and Niu H (2024) Host species of freshwater snails within the same freshwater ecosystem shapes the intestinal microbiome. Front. Ecol. Evol. 12:1341359. doi: 10.3389/fevo.2024.1341359
Received: 20 November 2023; Accepted: 17 January 2024;
Published: 02 February 2024.
Edited by:
Jingchun Li, University of Colorado Boulder, United StatesReviewed by:
Cheng Li, James Madison University, United StatesCopyright © 2024 Hu, Tong, Chang, Xu, Wu, Han, Yu and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaxin Niu, bml1aHhAaW11bi5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.