
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Ecol. Evol., 07 July 2023
Sec. Evolutionary and Population Genetics
Volume 11 - 2023 | https://doi.org/10.3389/fevo.2023.1218690
 Jacopo D’Ercole1,2*
Jacopo D’Ercole1,2* Roger Vila3
Roger Vila3 Leonardo Dapporto4
Leonardo Dapporto4 Mikko Pentinsaari1
Mikko Pentinsaari1 Gerard Talavera5Vlad Dincă6Paul D. N. Hebert1,2
Gerard Talavera5Vlad Dincă6Paul D. N. Hebert1,2Introduced species provide the opportunity to study evolutionary change on short time scales—a key first step to understand and manage the ecosystem-level impact of invasions. This study examined mitochondrial DNA sequence variation in cytochrome c oxidase subunit 1 (COI) for 26 insect species—Coleoptera (9), Hymenoptera (9), and Lepidoptera (8)—introduced to the Nearctic from the Palearctic. A total of 6,302 barcode records were retrieved from BOLD (boldsystems.org) to compare sequence diversity between the native and introduced range. As expected, genetic variation averaged nearly an order of magnitude lower in introduced populations (2.19 × 10−5 substitutions per nucleotide) than in the native range (1.48 ×10−4 substitutions per nucleotide). Nonsynonymous and synonymous changes had a similar incidence in the introduced populations (p-value = 0.83, averaging respectively 1.08 × 10−5 and 1.11 × 10−5 substitutions). By contrast, nonsynonymous changes were ten-fold less frequent than synonymous changes in the native populations (p-value < 0.001, averaging 1.74 × 10−5 and 1.3 × 10−4 substitutions, respectively). Patterns of sequence variation in the introduced range were largely congruent across the three insect orders which suggests that they are produced by general processes. This study explores the molecular evolution of introduced species, a fundamental aspect to improve understanding of their biology and manage their impact on ecosystems.
The molecular clock hypothesis (Zuckerkandl and Pauling, 1962; Margoliash, 1963) was a key advance in evolutionary biology because it enabled estimation of the age of lineages. However, it is now known that rates of molecular evolution vary among lineages due to different biological attributes (e.g., generation length, metabolic rate), demographic fluctuations, and natural selection (Langley and Fitch, 1974; Felsenstein, 1981). In addition, these rates also show variation within lineages through time, from high values in short-term pedigree studies to the much lower long-term phylogenetic rate (Ho et al., 2005; Ho et al., 2007; Henn et al., 2009).
Studying sequence divergence among breeding lines exposed to population bottlenecks is a common method for examining evolutionary rates over short intervals. This approach fosters the fixation of newly arisen variants via genetic drift, allowing detailed examination of the nature and incidence of spontaneous mutations. However, the insights obtained by this method have limited applicability to natural populations (Konrad et al., 2017) where substitution rates are determined by demographic history (Otto and Whitlock, 1997; Cabrera, 2021) and by the interplay of genetic drift and natural selection (Woolfit, 2009). Introduced species represent natural experiments unfolding in historical times, which provide the opportunity to compare the evolution of populations in their native and introduced ranges.
The present study compares rates and patterns of nucleotide substitution among populations of 26 species of Coleoptera, Hymenoptera, and Lepidoptera introduced to the Nearctic from the Palearctic within the last 20–200 years. Each of these species undoubtedly experienced a population bottleneck during its introduction, but then experienced exponential population growth, first at its site of introduction and subsequently as it expanded its range in the Nearctic. In fact, some of these species are still expanding their ranges (e.g., Sweeney et al., 2020; Strange et al., 2011). This work examines the molecular evolution of introduced species and addresses the following questions:
1. Is sequence variation in the natural and invaded range different?
2. Are there differences in the occurrence of nonsynonymous and synonymous mutations?
3. Are patterns taxon-specific or similar among insect orders?
This study examined sequence variation in the 5` end of the cytochrome c oxidase 1 gene for 6,302 specimens of 26 insect species introduced to the Nearctic from the Palearctic (Supplementary Table 1). Nine species were Coleoptera (1,954 records), nine were Hymenoptera (2,045 records), and eight were Lepidoptera (2,303 records) (Supplementary Table 2). The average number of records per species for Coleoptera was 161 (33–487) and 56 (21–136) respectively in the Nearctic and in the Palearctic. Hymenoptera averaged per species 203 (3–1,237) and 24 (11–81) records respectively in the Nearctic and Palearctic. The average records per species for Lepidoptera was 146 (11–428) and 142 (29–577) respectively in the Nearctic and Palearctic. Sequence records and associated metadata are available on BOLD (boldsystems.org) in the public dataset “DS-MTEVOL” (dx.doi.org/10.5883/DS-MTEVOL).
Sequences were available for 4,451 Nearctic and 1,851 Palearctic specimens. Most were obtained with Sanger sequencing, but some (Nearctic—2.4%, Palearctic—3.8%) were obtained using Sequel SMRT sequencing (Pacific Biosciences). All sequence records included a minimum of 500 unambiguous base pairs. DNA sequences for each species were aligned in Geneious Prime (version 2022.1.1) employing MUSCLE (Edgar, 2004) and the consensus sequence was used as the reference to assess sequence diversity. Rates of sequence variation were standardised with respect to the number of nucleotide sites examined. Because the termini of sequences generated by Sanger analysis are more subject to uncertainty, ~15 bp were trimmed from both the 5’ and 3’ ends of the alignment. The present analysis focused on sequence variation among singleton haplotypes which represented 228 of the total 1,062 haplotypes. They were targeted for analysis because we aimed to characterize sequence diversity that was likely to have arisen after introduction. Under this condition, exposure to genetic drift is maximized, while natural selection will only have removed the most disadvantageous mutations.
Mann-Whitney U tests were used to compare rates of sequence variation between Nearctic and Palearctic populations, as well as the ratio of nonsynonymous to synonymous mutations between Nearctic and Palearctic populations. ANOVA was performed at two levels of analysis. The first examined differences between two categories of substitution (i.e., nonsynonymous, synonymous) in the Nearctic and in the Palearctic. The second examined differences among insect orders (Coleoptera, Hymenoptera, Lepidoptera) for both substitution categories in the Nearctic and in the Palearctic. Statistical tests were performed using R studio (ver. 2022.7.1) (https://www.rstudio.com).
Variation in DNA barcode sequences for Nearctic specimens of the 26 introduced species (Figure 1A) was low, averaging just 2.19 × 10−5 substitutions per site (range: 1.4 × 10−5–2.85 × 10−5). By comparison, variation was almost an order of magnitude higher (U = 32; p-value < 0.001) (Figure 1A; Supplementary Table 3) in their Palearctic populations, averaging 1.48 ×10−4 (range: 1.01 × 10−4–2 × 10−4) substitutions per site.
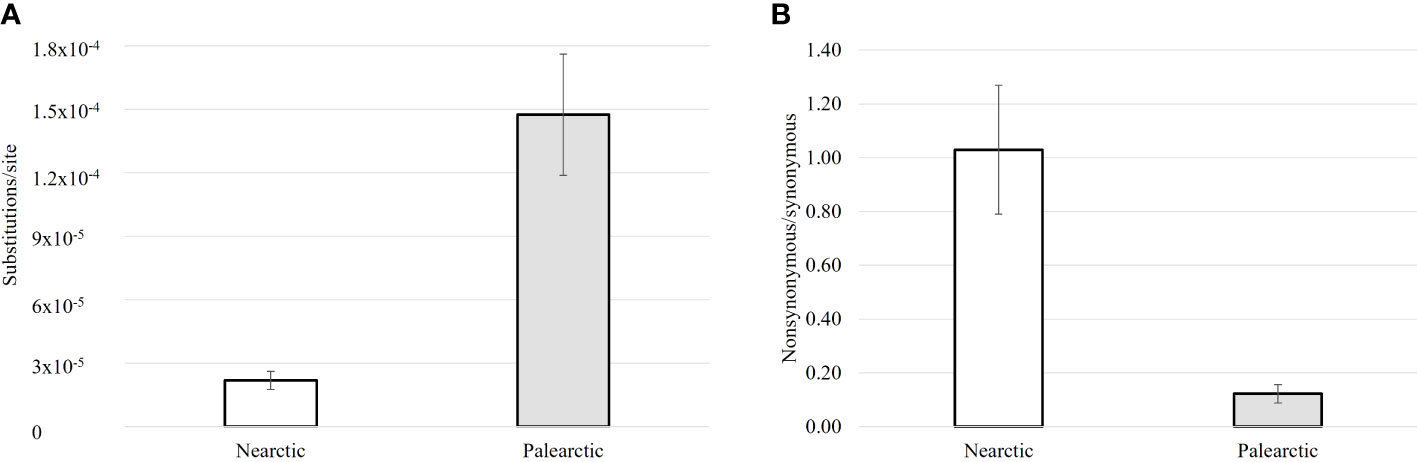
Figure 1 Comparison of the levels of DNA barcode sequence variation per nucleotide site (A), and mean ratio of nonsynonymous to synonymous changes (B) for populations of 26 insect species belonging to three insect orders introduced to the Nearctic from the Palearctic; bars show the standard error.
Analysis also revealed that nonsynonymous variants were, on average, slightly commoner than synonymous variants in Nearctic specimens (mean ratio = 1.03, range: 0.71–1.50) (Figure 1B). By contrast, the nonsynonymous to synonymous ratio was much lower in Palearctic specimens (mean = 0.12, range: 0.08–0.19) (Figure 1B). This nearly 10-fold difference was highly significant (U = 117; p-value < 0.001) (Supplementary Table 3).
The nonsynonymous and synonymous changes for all species among the three insect orders in the Nearctic averaged among the three insect orders respectively 1.08 × 10−5 (range: 6.51 × 10−6–1.4 × 10−5) and 1.11 × 10−5 (range: 7.44 × 10−6–1.6 × 10−5) substitution per site (Figure 2), with no significant differences among the two categories of substitution (F = 0.047; p-value = 0.829; Supplementary Table 4). However, analysis of Palearctic populations showed that nonsynonymous changes per site averaged 1.74 × 10−5 (range: 7.19 × 10−6–3.17 × 10−5), while synonymous changes were nearly 10-fold more frequent (F = 21.81; p-value < 0.001; Supplementary Table 4) with an average of 1.3 × 10−4 (range: 9.34 × 10−5–1.68 × 10−4) (Figure 2). Levels of sequence variation within each category of substitution (i.e., nonsynonymous and synonymous) across the three insect orders were similar and therefore non-significant in the Nearctic (Figure 2; Supplementary Table 4). Levels of sequence variation were also similar among taxa at the nonsynonymous changes in the native populations (F = 2.34; p-value = 0.12; Supplementary Table 4), but it revealed high variation among taxa at the synonymous changes in the native populations (F = 13.14; p-value < 0.001; Supplementary Table 4; Figure 2).
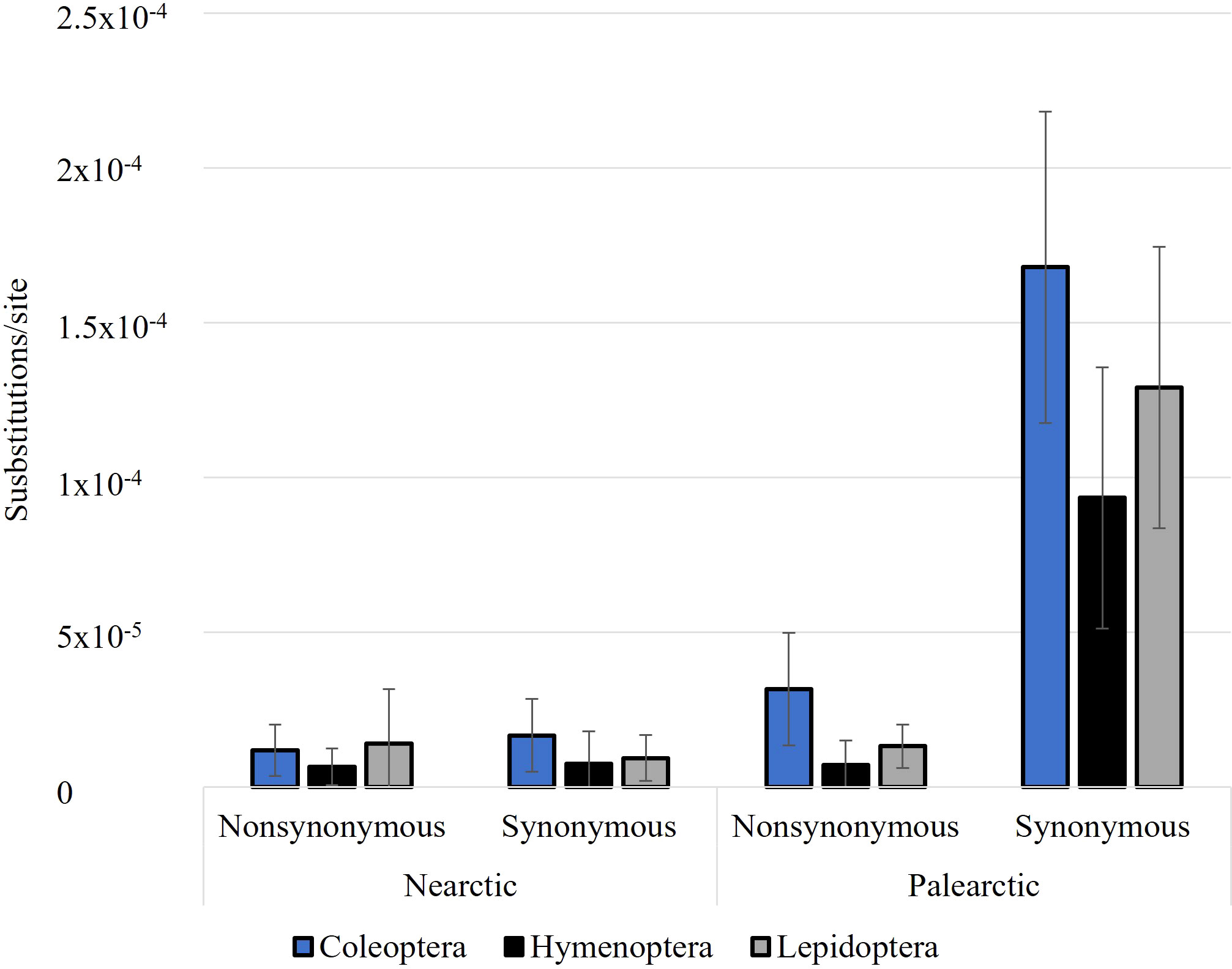
Figure 2 Comparison of the levels of sequence variation per nucleotide site in the barcode region of COI in Nearctic and Palearctic populations of 26 species belonging to three insect orders. Bars show the standard error.
In long-established populations such as those in the Palearctic, which served as the source for introduction to the Nearctic, intraspecific sequence variation has accumulated for hundreds of thousands of years across multiple glacial cycles (Hewitt, 2004). The much lower levels of variation detected in their Nearctic counterparts is an expected result of population bottlenecks during introduction. Genetic impoverishment is inevitable because they derive from just a few individuals from one or few populations from the native range—a pattern commonly observed in introduced species (e.g., Colautti and Lau, 2015; D’Ercole et al., 2022; North et al., 2021). While this work considers species introduced to different bioregions, it is worth noting that other mechanisms could produce similar patterns at smaller geographic scale. Not only adaptive mutations could deplete genetic diversity at linked sites through selective sweeps (e.g., Lattorff et al., 2015; VanBuren et al., 2016), but geographic isolation following founder effect (e.g., Barros et al., 2022) and anthropic pressure can also produce local loss of diversity (e.g., Robin et al., 2022). Given the severe bottlenecks experienced by introduced species, one might expect they only include haplotypes prevalent in the native range. However, they included a considerable number of low frequency haplotypes, variants which might have arisen after introduction. Human pedigree studies have revealed mutation rates up to 100-fold higher than phylogenetic rates and (Sigurdardóttir et al., 2000; Howell et al., 2003) with no bias towards synonymous changes (Denver et al., 2000). This time-dependent rate variation has been linked to transient polymorphisms through three main processes. The first is the limited efficiency of purifying selection over short intervals. While moderately deleterious haplotypes will ultimately be purged by natural selection, their fate is mainly determined by genetic drift over short intervals (Ho et al., 2005; Ho et al., 2007). Second, the apparent mutation rate is elevated by population expansion (Cabrera, 2021), and introduced populations typically experience exponential growth. When population size is stable through time (N0 = N1), the probability of transmission of a newly arisen mutation to the next generation remains constant (1/N0) (Kimura, 1991). By contrast, if population size doubles each generation (2N0 = N1), the probability of transmission also doubles (2/N0). This raises the number of both neutral and deleterious mutations, and extends their persistence in the population (Waxman, 2012; Gazave et al., 2013; Peischl et al., 2013)—a phenomenon termed “expansion load”. Lastly, effective population size can influence substitution rates because it regulates the balance between genetic drift and selection (Ohta, 1992; Woolfit, 2009). The small size of populations at the leading edge of the species distribution reduces the efficacy of selection so slightly deleterious mutations can increase in frequency by genetic drift. Although the retention of ancestral polymorphisms is often used to explain the occurrence of transient polymorphisms (Henn et al., 2009; Peterson and Masel, 2009), most introductions involve few individuals, making it unlikely that the diversity present in invasive species predates their introduction.
Multiple factors can explain the elevated ratio of nonsynonymous to synonymous changes in the Nearctic. First, the low occurrence of synonymous changes in the introduced populations is likely a consequence of their young age and their resulting lack of genetic structure. Second, because transient polymorphisms often involve slightly deleterious mutations (as described above), this higher ratio can, at least in part, be explained by the prevalence of such mutations. Third, selection could enhance the incidence of nonsynonymous mutations. While functional constraints generally impose purifying selection, which limit amino acid substitutions (Ballard and Whitlock, 2004; Meiklejohn et al., 2007), a shift in selection pressures can favour substitutions. Aside from occupying a novel physical environment, introduced populations are typically exposed to fewer parasites, parasitoids, and predators than their ancestral populations (Torchin et al., 2003; Perkins et al., 2008). Because of this relaxed selection, they can invest less in defensive mechanisms (Phillips et al., 2010). Hence, it is possible that reduced purifying selection (rather than positive selection) underlies the observed pattern. Lastly, while operational issues such as PCR errors, editing errors, and amplification of non-target sequences should equally affect specimens in the native and in the invaded range, their overall impact merits examination. Past studies revealed that the transition from T/A → C/G contributes to ~70% of all polymerase errors (McInerney et al., 2014; Potapov and Ong, 2017). Analysis of the nonsynonymous changes detected in this study showed that PCR errors have played a negligible role as T/A → C/G transitions only account for ~10% of all changes in Lepidoptera and in Coleoptera, and none in Hymenoptera. Similarly, editing errors have likely marginal effect on sequence variation. To confirm this fact, trace files were manually examined whenever available, and ~15 bp from the termini of each Sanger sequence were trimmed to minimize such errors. The incidence of heteroplasmy was never examined on introduced species, but the high spontaneous mutation rate and the presence of multiple copies of mitochondrial DNA per cell set the condition for the proliferation of such variants. Mutation accumulation studies support this theoretical prediction as they reveal that most mutations are in fact heteroplasmic when few generations are considered (Haag-Liautard et al., 2008; Howe et al., 2010; Konrad et al., 2017). While Sanger-based sequencing enables only a coarse investigation of sequence heterogeneity (Just et al., 2015), analysis of trace files in our dataset showed that about 25% of the nonsynonymous mutations in our sequence dataset are associated with double peaks. Future work could involve use of high throughput sequencing to allow a complete characterization of the spectrum of gene copies present in an individual, enabling detection of variants at frequency lower than 2% (White et al., 2017; González et al., 2020).
Mutations introduce genetic variation, enabling organisms to adapt to environmental change. Past studies have examined mutational patterns in the mitochondrial genome through short-term laboratory experiments, but similar studies have rarely been conducted in natural settings. In this study, we show how introduced species can address this gap. Our analysis shows that introduced populations show depleted levels of sequence variation as expected because of bottlenecking during population establishment. However, our results indicate that the constellation of newly arisen mutations in these introduced populations is characterized by an unusually high incidence of non-synonymous variants, a pattern consistent among the species in three insect orders. This pattern may reflect a response to varying selective pressure following the occupancy of new environments, or it may simply reflect that natural selection has not yet had sufficient time to excise weakly disadvantageous mutations in a scenario of demographic growth.
Publicly available datasets were analyzed in this study. The 6,302 barcode records and associated metadata are available in the BOLD dataset DS-MTEVOL (dx.doi.org/10.5883/DS-MTEVOL).
JD contributed to conception of the study, analysed the data, prepared figures and tables, reviewed drafts of the paper, and approved the final draft. RV contributed to conception of the study, reviewed drafts of the paper, and approved the final draft. LD contributed to conception of the study, reviewed drafts of the paper, and approved the final draft. GT contributed to conception of the study, reviewed drafts of the paper, and approved the final draft. VD contributed to conception of the study, reviewed drafts of the paper, and approved the final draft. PH contributed to conception of the study, reviewed drafts of the paper, and approved the final draft. All authors contributed to the article and approved the submitted version.
This work was supported by a NSERC Discovery grant to PDNH. RV was supported by project PID2019- 107078GB- I00/MCIN/AEI/10.13039/501100011033. VD was supported by the Academy of Finland (Academy Research Fellow, decisions 328895 and 352652).
We thank Sean Prosser and Kevin Kerr for their suggestions on earlier versions of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1218690/full#supplementary-material
Ballard J. W. O., Whitlock M. C. (2004). The incomplete natural history of mitochondria. Mol. Ecol. 13, 729–744. doi: 10.1046/j.1365-294x.2003.02063.x
Barros T., Fernandes J. M., Ferreira E., Carvalho J., Valldeperes M., Lavín S., et al. (2022). Genetic signature of blind reintroductions of Iberian ibex (Capra pyrenaica) in Catalonia, northeast Spain. PloS One 17, e0269873. doi: 10.1371/journal.pone.0269873
Cabrera V. M. (2021). Human molecular evolutionary rate, time dependency and transient polymorphism effects viewed through ancient and modern mitochondrial DNA genomes. Sci. Rep. 11, 5036. doi: 10.1038/s41598-021-84583-1
Colautti R. I., Lau J. A. (2015). Contemporary evolution during invasion: evidence for differentiation, natural selection, and local adaptation. Mol. Ecol. 24, 1927–2297. doi: 10.14411/eje.2022.039
D’Ercole J., Dapporto L., Schmidt B. C., Dincă V., Talavera G., Vila R., et al (2022). Patterns of DNA barcode diversity in butterfly species (Lepidoptera) introduced to the Nearctic. Eur. J. Entomol. 119, 379–387. doi: 10.14411/eje.2022.039.
Denver D. R., Morris K., Lynch M., Vassilieva L. L., Thomas W. K. (2000). High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science 289, 2342–2344. doi: 10.1126/science.289.5488.2342
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Felsenstein J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Gazave E., Chang D., Clark A. G., Keinan A. (2013). Population growth inflates the per-individual number of deleterious mutations and reduces their mean effect. Genetics 195, 969–978. doi: 10.1534/genetics.113.153973
González M. D. M., Ramos A., Aluja M. P., Santos C. (2020). Sensitivity of mitochondrial DNA heteroplasmy detection using next generation sequencing. Mitochondrion 50, 88–93. doi: 10.1016/j.mito.2019.10.006
Haag-Liautard C., Coffey N., Houle D., Lynch M., Charlesworth B., Keightley P. D. (2008). Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PloS Biol. 6, e204. doi: 10.1371/journal.pbio.0060204
Henn B. M., Gignoux C. R., Feldman M. W., Mountain J. L. (2009). Characterizing the time dependency of human mitochondrial DNA mutation rate estimates. Mol. Biol. Evol. 26, 217–230. doi: 10.1093/molbev/msn244
Hewitt G. M. (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. B: Biol. Sci. 359, 183–195. doi: 10.1098/rstb.2003.1388
Ho S. Y. W., Phillips M. J., Cooper A., Drummond A. J. (2005). Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22, 1561–1568. doi: 10.1093/molbev/msi145
Ho S. Y. W., Shapiro B., Phillips M. J., Cooper A., Drummond A. J. (2007). Evidence for time dependency of molecular rate estimates. Syst. Biol. 56, 515–522. doi: 10.1080/10635150701435401
Howe D. K., Baer C. F., Denver D. R. (2010). High rate of large deletions in Caenorhabditis briggsae mitochondrial genome mutation processes. Genome Biol. Evol. 2, 29–38. doi: 10.1093/gbe/evp055
Howell N., Smejkal C. B., Mackey D. A., Chinnery P. F., Turnbull D. M., Herrnstadt C. (2003). The pedigree rate of sequence divergence in the human mitochondrial genome: there is a difference between phylogenetic and pedigree rates. Am. J. Hum. Genet. 72, 659–670. doi: 10.1086/368264
Just R. S., Irwin J. A., Parson W. (2015). Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci. Int. Genet. 18, 131–139. doi: 10.1016/j.fsigen.2015.05.003
Kimura M. (1991). The neutral theory of molecular evolution: a review of recent evidence. J. J. H. G. 66, 367–386. doi: 10.1266/jjg.66.367
Konrad A., Thompson O., Waterston R. H., Moerman D. G., Keightley P. D., Bergthorsson U., et al. (2017). Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol. Biol. Evol. 34, 1319–1334. doi: 10.1093/molbev/msx051
Langley C. H., Fitch W. M. (1974). An examination of the constancy of the rate of molecular evolution. J. Mol. Evol. 3, 161–177. doi: 10.1007/BF01797451
Lattorff H., M. G., Buchholz J., Fries I., Moritz R., F. A. (2015). A selective sweep in a Varroa destructor resistant honeybee (Apis mellifera) population. Infect. Genet. Evol. 31, 169–176. doi: 10.1016/j.meegid.2015.01.025
Margoliash E. (1963). Primary structure and evolution of cytochrome c. Proc. Natl. Acad. Sci. U.S.A. 50, 672–679. doi: 10.1073/pnas.50.4.672
McInerney P., Adams P., Hadi M. Z. (2014). Error rate comparison during polymerase chain reaction by DNA polymerase. Mol. Biol. Int. 14, 1–8. doi: 10.1155/2014/287430
Meiklejohn C. D., Montooth K. L., Rand D. M. (2007). Positive and negative selection on the mitochondrial genome. Trends Genet. 23, 259–263. doi: 10.1016/j.tig.2007.03.008
North H. L., McGaughran A., Jiggins C. D. (2021). Insights into invasive species from whole-genome resequencing. Mol. Ecol. 30, 6289–6308. doi: 10.1111/mec.15999
Ohta T. (1992). The nearly neutral theory of molecular evolution. Annu. Rev. Eco. Syst. 23, 263–286. doi: 10.1146/annurev.es.23.110192.001403
Otto S. P., Whitlock M. C. (1997). The probability of fixation in populations of changing size. Genetics. 146, 723–733. doi: 10.1093/genetics/146.2.723
Peischl S., Dupanloup I., Kirkpatrick M., Excoffier L. (2013). On the accumulation of deleterious mutations during range expansions. Mol. Ecol. 22, 5972–5982. doi: 10.1111/mec.12524
Perkins S. E., Altizer S., Bjornstad O., Burdon J. J., Clay K., Gómez-Aparicio L., et al. (2008). “Invasion biology and parasitic infections,” in Infectious disease ecology: the effects of ecosystems on disease and disease on ecosystems. Eds. Ostfeld R. S., Keesing F., Eviner V. T. (New Jersey: Princeton University Press), 179–204.
Peterson G. I., Masel J. (2009). Quantitative prediction of molecular clock and Ka/Ks at short timescales. Mol. Biol. Evol. 26, 2595–2603. doi: 10.1093/molbev/msp175
Phillips B. L., Brown G. P., Shine R. (2010). Life-history evolution in range-shifting populations. Ecology 91, 1617–1627. doi: 10.1890/09-0910.1
Potapov V., Ong J. L. (2017). Examining sources of error in PCR by single-molecule sequencing. PloS One 12, e0169774. doi: 10.1371/journal.pone.0169774
Robin M., Ferrari G., Akgül G., Münger X., von Seth J., Schuenemann V. J., et al. (2022). Ancient mitochondrial and modern whole genomes unravel massive genetic diversity loss during near extinction of alpine ibex. Mol. Ecol. 31, 3548–3565. doi: 10.1111/mec.16503
Sigurdardóttir S., Helgason A., Gulcher J. R., Stefánsson K., Donnelly P. (2000). The mutation rate in the human mtDNA control region. Am. J. Hum. Genet. 66, 1599–1609. doi: 10.1086/302902
Strange J., Koch J., Gonzalez V. H., Nemelka L., Griswold T. (2011). Global invasion by Anthidium manicatum (Linnaeus) (Hymenoptera: Megachilidae): assessing potential distribution in North America and beyond. Biol. Invasions. 13, 2115–2133. doi: 10.1007/s10530-011-0030-y
Sweeney J., Hughes C., Zhang H., Hillier N. K., Morrison A., Johns R. (2020). Impact of the invasive beech leaf-mining weevil, Orchestes fagi, on American beech in Nova Scotia, Canada. Frontiers in Forests and Global Change 3, 1–11. doi: 10.3389/ffgc.2020.00046
Torchin M. E., Lafferty K. D., Dobson A. P., McKenzie V. J., Kuris A. M. (2003). Introduced species and their missing parasites. Nature 421, 628–630. doi: 10.1038/nature01346
VanBuren R., Wai C. M., Zhang J., Han J., Arro J., Lin Z., et al. (2016). Extremely low nucleotide diversity in the X-linked region of papaya caused by a strong selective sweep. Genome Biol. 17. doi: 10.1186/s13059-016-1095-9
Waxman D. (2012). Population growth enhances the mean fixation time of neutral mutations and the persistence of neutral variation. Genetics 191, 561–577. doi: 10.1534/genetics.112.139220
White E. J., Ross T., Lopez E., Nikiforov A., Gault C., Batorsky R., et al. (2017). Chasing a moving target: detection of mitochondrial heteroplasmy for clinical diagnostics. bioRxiv, 222109. doi: 10.1101/222109
Woolfit M. (2009). Effective population size and the rate and pattern of nucleotide substitutions. Biol. Let. 5, 417–420. doi: 10.1098/rsbl.2009.0155
Keywords: Coleoptera, Hymenoptera, Lepidoptera, DNA barcoding, mitochondrial DNA
Citation: D’Ercole J, Vila R, Dapporto L, Pentinsaari M, Talavera G, Dincă V and Hebert PDN (2023) Molecular evolution in introduced insect species—a mitochondrial perspective. Front. Ecol. Evol. 11:1218690. doi: 10.3389/fevo.2023.1218690
Received: 07 May 2023; Accepted: 21 June 2023;
Published: 07 July 2023.
Edited by:
Hovirag Lancioni, University of Perugia, ItalyReviewed by:
Irene Cardinali, University of Perugia, ItalyCopyright © 2023 D’Ercole, Vila, Dapporto, Pentinsaari, Talavera, Dincă and Hebert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacopo D’Ercole, amRlcmNvbGVAdW9ndWVscGguY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.