
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 11 July 2023
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 11 - 2023 | https://doi.org/10.3389/fevo.2023.1215319
This article is part of the Research Topic Biology, Systematics, Taxonomy and Evolution of Insect Vectors - Volume II View all 8 articles
 Tiago Belintani1,2*
Tiago Belintani1,2* Carlos Congrains3,4
Carlos Congrains3,4 Heloisa Pinotti2Samira Chahad-Ehlers4
Heloisa Pinotti2Samira Chahad-Ehlers4 Reinaldo A. de Brito4
Reinaldo A. de Brito4 Jader Oliveira5
Jader Oliveira5 Daniel Frias-Lasserre6Felipe Mendes Fontes7
Daniel Frias-Lasserre6Felipe Mendes Fontes7 João Aristeu da Rosa2
João Aristeu da Rosa2Introduction: The Mepraia genus is composed of three endemic species in Chile, namely Mepraia spinolai, Mepraia gajardoi, and Mepraia parapatrica, all capable of transmitting the causative agent of Chagas disease. Although species divergence within this genus is supported by morphological, molecular, and geological evidence, it has never been validated by genome-wide data.
Methods: In this sense, a pioneering phylogenomic study was conducted using transcriptome data of the three species, aiming to infer robust phylogenies and evaluate the impact of interspecific gene flow on the evolution of these insects. Our transcriptomes were used to robustly infer the phylogenetic relationships, employing multi-species coalescent and supermatrix approaches, as well as to investigate the occurrence of introgression.
Results and discussion: The inferred phylogenies support the monophyly of the three described taxa and infer divergence times, although gene trees display high levels of discordance. Our results indicate widespread introgression among Mepraia lineages. Thus, our results support that Mepraia diversification occurred in a short period resulting in reticulate phylogenies and low genetic diversity. Additionally, we obtained evidence of introgression suggesting that ancient hybridization has had a major role in the evolution of the three Mepraia species.
The Andes Mountains are a continuous chain of mountains along the western coast of South America with average altitudes of ~4,000 meters above sea level, whose formation has had a profound impact on the geography and climate of this continent (Hartley, 2003; Moreira-Muñoz, 2011). For instance, in Chile, these mountains define a unique, long, and narrow coastal environment, which exhibits a highly diverse climate due to the wide latitudinal variation (Moreira-Muñoz, 2011). The uplift of the Andes has had a profound effect on the climate of the Southern Hemisphere, creating an extensive barrier to atmospheric circulation and promoting biogeographic changes that have affected the evolutionary history of local fauna and flora (Pirie et al., 2006; Antonelli et al., 2009). The uplift of the Andes has exerted a significant influence on the dispersal and diversification of a variety of South American organisms, such as plants, mammals, insects and specifically in triatomines (Mauseth, 1996; Doan, 2003; Struwe et al., 2009; Monteiro et al., 2018; Pavan et al., 2021).
The genus Mepraia Mazza, Gajardo and Jörg, 1940 is a Triatominae Jeannel, 1919 endemic to arid and semi-arid regions (Frías-Lasserre et al., 2019). This previously considered monotypic genus (Lent et al., 1994), comprises three distinct species: Mepraia spinolai (Porter, 1933) Mepraia gajardoi (Frías-Lasserre et al., 1998), and Mepraia parapatrica (Frías-Lasserre, 2010). These species are of significant public health importance as they are involved in the sylvatic cycle of the etiologic agent (Chacón et al., 2016; Lambarri et al., 2018). M. gajardoi depends on seabirds as its main food source, as well as on lizards that inhabit coastal regions. M. parapatrica also feeds on these species but has been observed to include domesticated mammals in its diet when it invades human settlements (Frías-Lasserre, 2010). In contrast, M. spinolai prefers wild and domesticated mammals and exhibits a strong tendency to invade human habitats in Andean valleys, although it never feeds on seabirds (Frías-Lasserre, 2010).
Mepraia has unique attributes among the triatomines, such as diurnal feeding habits and wing polymorphism (Faúndez and Carvajal, 2012; Frías-Lasserre et al., 2019). In summary, all Mepraia females are micropterous (with reduced wings), which may be a new or apomorphic characteristic of Triatominae (Frías-Lasserre, 2010). On the other hand, all M. gajardoi males are brachypterous (with short wings) (Frías-Lasserre et al., 1998), and although M. parapatrica males are mostly brachypterous, some are macropterous (with long wings) (Frías-Lasserre, 2010). In contrast, M. spinolai has both macropterous and brachypterous males (Frías-Lasserre, 2010; Frías-Lasserre et al., 2019).
The studies on geographical distribution (Campos-Soto et al., 2020), molecular data (Campos-Soto et al., 2013; Campos-Soto et al., 2015; Campos-Soto et al., 2022), morphology (Campos-Soto et al., 2011; Frías-Lasserre et al., 2018; Frías-Lasserre et al., 2019), and cytogenetics (Frías-Lasserre et al., 1998; Frías-Lasserre et al., 2019) provide support for the three species of Mepraia. However, although phylogenies with few genes support the described taxa (Calleros et al., 2010; Campos-Soto et al., 2013), there is no consensus on the genus diversification process.
Two hypotheses have been proposed to explain the diversification of Mepraia. The first suggests that the genus has originated due to a vicariant process followed by dispersion and isolation, which was potentiated by the arid climate of the region (Frías-Lasserre, 2010). This hypothesis is based on morphological and cytogenetic data which suggest that after the emergence of the Andes, and their separation from their ancestor, a process of dispersion and speciation occurred in a North–South direction (Frías-Lasserre, 2010). According to this hypothesis, M. gajardoi is considered the most ancestral species, while M. parapatrica gave rise to M. spinolai after some cytogenetic alterations (Frías-Lasserre, 2010). This latter study suggests that the wing morphology supports the hypothesis that the macropterous wing structure of M. spinolai is derived from M. parapatrica and M. gajardoi, which have winged males with ancestral characteristics (Frías-Lasserre et al., 1998; Frías-Lasserre and Atria, 1998; Frías-Lasserre, 2010).
The second and most recent proposal suggests that the diversification of Mepraia was also driven by geological changes and climatic fluctuations over the past 5 million years (Campos-Soto et al., 2022). In this model, there were two geographically isolated clades (Campos-Soto et al., 2022). The northern group gave rise to M. parapatrica, M. gajardoi, and a new lineage, while the southern group formed M. spinolai. However, this hypothesis is based only on data from three mtDNA genes (Campos-Soto et al., 2022), and further studies are needed to confirm its validity.
Although there is still no definitive consensus on the processes that led to the diversification of Mepraia, recent research indicates that it likely occurred rapidly (Justi et al., 2016; Campos-Soto et al., 2020; Campos-Soto et al., 2022). Several lines of evidence, such as genetic and phenotypic differences between populations, geographic and reproductive barriers, and molecular dating, suggest that there was rapid species diversification (Calleros et al., 2010; Campos-Soto et al., 2022), though, currently, there are only a few studies that investigate levels of population isolation and gene flow between them. Resolving the phylogenetic relationships of groups that diverged recently can be challenging (Seehausen et al., 2014; Marques et al., 2016; Weng et al., 2020; Congrains et al., 2021) due to the retention of ancestral polymorphisms causing incomplete lineage sorting (ILS), introgression, and/or gene duplication (Mallet, 2005). This scenario can become even more complex if there has been ancient gene flow, which can produce conflicting phylogenetic signals throughout the genome (Zhou et al., 2016; Edelman et al., 2019). Multi-species coalescent methods have been useful in clarifying evolutionary relationships and mitigating the impact of deep coalescence (Degnan and Rosenberg, 2009; Young and Gillung, 2020).The combination of next-generation sequencing technologies and robust phylogenetic methods has effectively resolved ambiguous phylogenetic relationships for a variety of organisms (Rokas et al., 2003; Dunn et al., 2008; Regier et al., 2010; Irisarri et al., 2017) and may be useful in the study of Mepraia.
This study aimed to investigate phylogenomic relationships in the Mepraia genus for the first-time using transcriptome data from the heads and salivary glands of their three species. To do so, we also evaluated the impact of gene flow on the genus’ speciation by using different methodological approaches, which increases the reliability of the results. Concatenation-based approaches and multispecies coalescent methods were employed for this purpose, and the study also assessed the impact of ILS and introgression events on the inferred phylogenies.
The specimens used for this study were collected from six localities in Chile (Table 1), being two different localities per taxa (Figure 1). The specimens were deposited in the Triatominae Collections of the School of Pharmaceutical Sciences, São Paulo State University (Unesp), Araraquara, SP, Brazil. The species-level identification was carried out by Dr. Daniel Frías-Lasserre based on morphological characters (Frías-Lasserre, 2010).
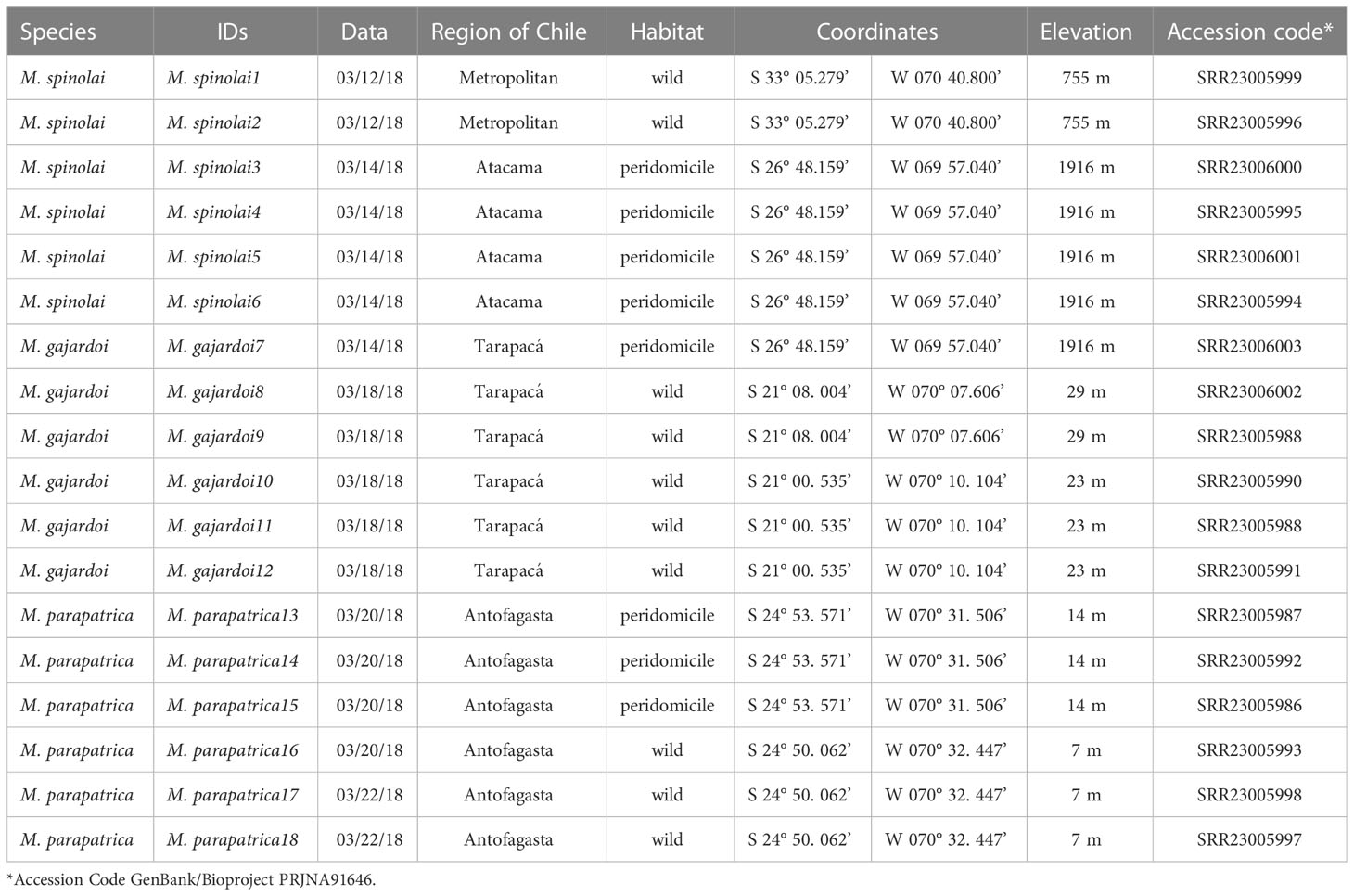
Table 1 Specimens used in the study and SRA accession code on GenBank.
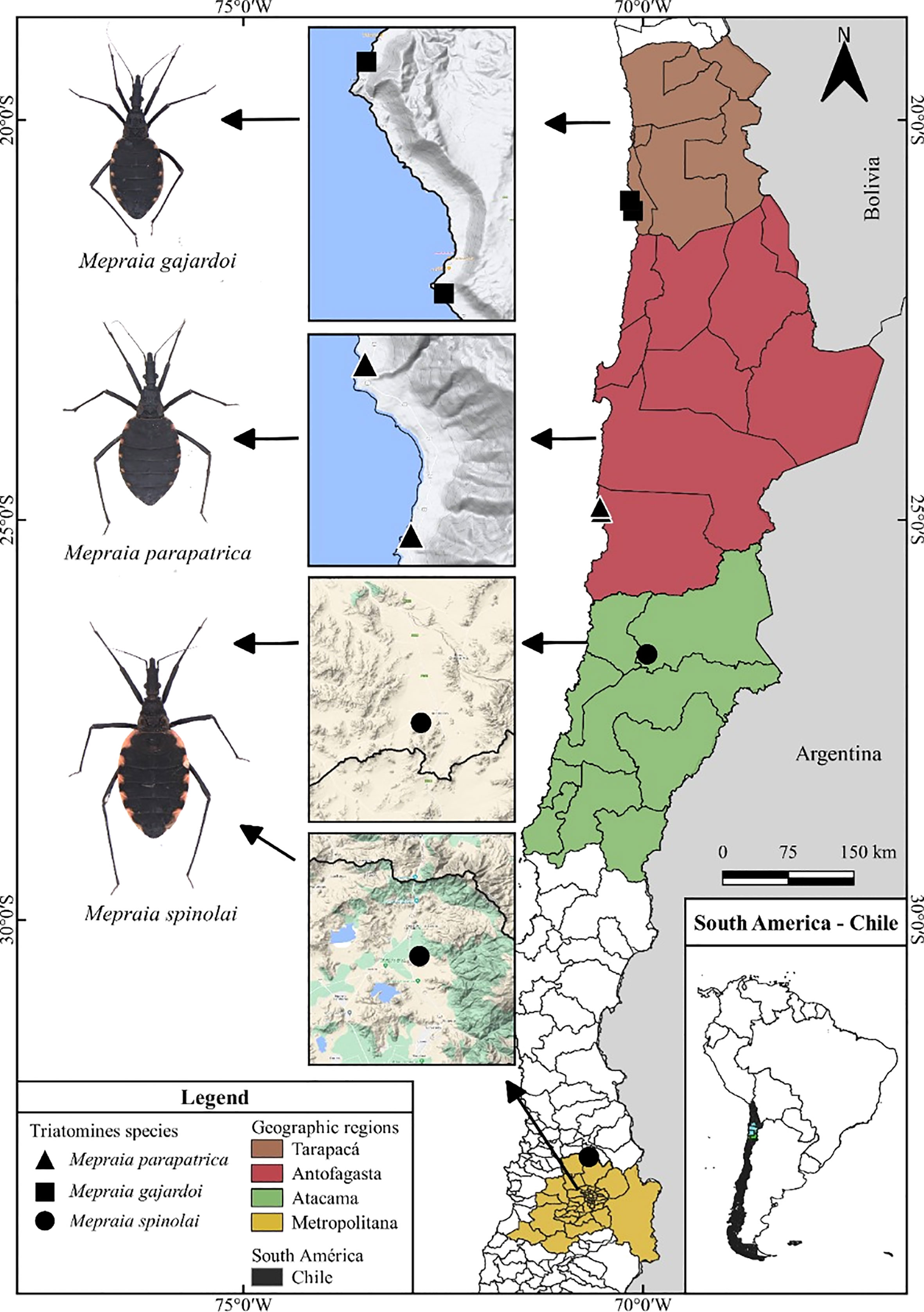
Figure 1 Map of Chile showing triatomine collection localities of M. gajardoi, M. parapatrica, and M. spinolai. The images of the specimens on the map do not represent the actual size proportion of the insect.
Head and salivary glands of 5th instar male nymphs of M. garjadoi (n = 6), M. spinolai (n = 6), and M. parapatrica (n = 6) were carefully removed to preserve the integrity of the salivary glands. Total RNA of head and salivary glands of each specimen was extracted following a Trizol/Chloroform protocol (Chomczynski and Mackey, 1995). RNA integrity was checked visually on agarose gel and quantified using Qubit fluorometer, while NanoDrop measurements ensured the selection of samples with absorbance indices of 260/230 and 260/280 close to 2.0. In addition, we obtained the RNA integrity number (RIN) using capillary electrophoresis on an Agilent 2100 Bioanalyzer System (Agilent Technologies®, Standard Protocol for RNA). The value obtained from the RIN was 7.1 ± 0.3 in a sample group of 18. Libraries were constructed using the TruSeq® RNA Sample Prep kit v2 (Illumina) following the protocol suggested by the manufacturer and were ran in an Illumina HiSeq2500 platform with 2 × 100 bp paired-end reads in the Laboratory for Functional Genomics Applied to Agriculture and Bioenergy of the Luiz de Queiroz College of Agriculture (ESALQ) at the University of São Paulo (USP) in Brazil.
Raw reads were quality-trimmed using Trimmomatic v.0.39 (Bolger et al., 2014); setting the parameters: LEADING:5 TRAILING:5 SLIDINGWINDOW:5:20 MINLEN:50. All Mepraia assemblies were performed using a de novo strategy using the Trinity package v.2.9.1 (Grabherr et al., 2011). To optimize contig assembly, the filtered reads were normalized using a maximum depth of 60 using the insilico_read_normalization.pl tool, which is included in the Trinity v.2.9.1 package (Grabherr et al., 2011), retaining only contigs with minimum size of 200 bp. Assembly statistics were computed using the TrinityStats.pl Perl script contained in the Trinity package. To mitigate the likelihood of including false and redundant transcripts, only the most expressed isoform per Trinity cluster was retained using the perl script align_and_estimate_abundance.pl available in Trinity. Redundant transcripts with ≥95% of identity was filtered out using CD-HIT-EST v. 4.7 (Fu et al., 2012). The filtered transcriptomes were used for downstream analyses. The completeness of the transcriptome assembly was assessed using BUSCO v. 4.0.2 (Simão et al., 2015) which was used to estimate the proportion of all single copy, duplicate and missing orthologs that are represented in the arthropod dataset (Kriventseva et al., 2019).
We used the software Transdecoder 5.5.0 (Haas et al., 2013) to identify putative coding sequences (CDSs). First, TransDecoder.LongOrfs was used to retain CDSs encoding peptides greater than 100aa. In the next step, inferred peptides were submitted to the tool HMMER 3.1b2 package (Eddy, 2011) to search for conserved domains using PFAM-A (Mistry et al., 2021) database. We also aligned the putative peptides against the GenBank non-redundant (nr) protein database using BLAST+ 2.10.1 (Camacho et al., 2009). The TransDecoder.Predict utility fetched information generated in previous steps to predict CDSs.
We used BLASTp (2.10.1) (Camacho et al., 2009) to search for regions with significant similarity to the putative CDSs against the Genbank (nr) Arthropoda v5 non-redundant protein database. Hits with e-value lower than 10−6 were considered significant. Putative CDSs were also scanned for the presence of conserved protein domains using InterProScan v. 5.24 (Jones et al., 2014). Blast2GO program (Conesa et al., 2005) was used to associate each CDS with gene ontology (GO) terms (Harris et al., 2004). We used the WEGO tool (Ye et al., 2006; 2018) to plot mapped GO terms.
We estimated a species-level phylogenetic analysis using SNP data under the coalescent framework model with three species of Mepraia and T. infestans as an outgroup. T. infestans is the closest species to Mepraia for which there are large-scale data available. For this, inferred CDSs from each sample were mapped against the genome of T. infestans (GCA_011037195.1) using the –nofw and –very-sensitive options in Bowtie2 (Langmead and Salzberg, 2012). Duplicate PCR reads were removed using MarkDuplicates in the Picard tools available in https://broadinstitute.github.io/picard/. SNP calling was performed using the mpileup tool from the Samtools package (Li et al., 2009) and mpileup2snp from VarScan2 (Koboldt et al., 2009), using the following parameters for the latter: a mapping quality of 20 and average Phred quality of 30, minimum coverage of 6 and strand filter (removed variants with more than 90% supported by only one strand). We also applied a high-filtered approach in SNPs to retain only the most suitable for phylogenetic analysis using Bcftools 1.9 (Danecek et al., 2021). For that, we excluded sites where no alternative alleles were called in one of the samples, all sites where only alternative alleles are called, and sites where the proportion of missing data is greater than 20% (“F_MISSING > 0.2”). We also ensured the presence of only biallelic SNPs in the dataset using the -m and -M commands in Bcftools 1.9 (Danecek et al., 2021).
The unlinked biallelic SNPs detected in all samples (excluding sites with missing data) were used to construct an extensive matrix of SNPs (60,685 characters), which was used to infer species trees using SVDquartets (Chifman and Kubatko, 2015) and SNAPP (Bryant et al., 2012). Using both SVDquartets and SNAPP techniques can improve the accuracy of inferring phylogenetic trees from biallelic SNPs by overcoming the limitation of few SNPs and providing a more complex evolutionary model to consider mutation and recombination rates.
The species tree inferred on SVDquartets (Chifman and Kubatko, 2015) was implemented in PAUP* (Phylogenetic Analysis Using Parsimony) (Swofford, 2002). We evaluated all possible quartets, both with and without prior assignment using non-parametric methods generated from 100,000 quartets and 1,000 bootstrap replicates. In the Bayesian coalescent multispecies analysis in SNAPP (Bryant et al., 2012), implemented in BEAST2.5 (Bouckaert et al., 2019), we performed two independent runs with a chain length of 109 generations, sampling every 1,000 generations. We checked for convergence (ESS>200) and determined the burnin (10%) using TRACER v1.6 (Rambaut and Drummond, 2007). A maximum clade credibility (MCC) tree was summarized with TreeAnnotator and visualized in FigTree. Additionally, we generated a density tree using Densitree (Bouckaert, 2010) to describe alternative topologies.
To predict orthologs from transcriptomes of non-model organisms, we used a pipeline developed by Yang and Smith (2014) using the inferred CDS sequences as input. This analysis was performed on the 18 transcriptome assemblies produced in this study, along with T. infestans (Accession code: SRR4427079) as an outgroup. The SRR442779 data was processed along with the samples collected in this study as described above. The CDSs predicted from the 19 transcriptomes were submitted to all-by-all BLAST using BLASTn (2.12.0) (Camacho et al., 2009) setting relaxed parameters (hit_fraction_cutoff = 0.4, inflation = 1.4) and filtered by the MLC program (Van Dongen, 2008). Clusters were used to estimate trees of putative homologs as described in Yang and Smith (2014). We used the script python “filter_1to1_orthologs” described by Yang and Smith (2014) to retain only putative orthologs to infer phylogenetic relationships, as described below. We combined putative orthologs in a supermatrix, which were aligned with MAFFT v.7.305 (Katoh and Toh, 2008). Phyutility v.2.2.6 (Smith and Dunn, 2008) was used to trim the aligned ortholog clusters, with a configuration of 0.3 for MIN_COLUMN_OCCUPANCY. The alignments were concatenated using the python scripty “concatenate_matrices” (Yang and Smith, 2014) and trimmed using trimAl v.1.2 (Capella-Gutiérrez et al., 2009). The orthologous inference resulted in a total of 3,245 orthologous cluster that were used in the downstream analyses. We constructed a supermatrix with 3,245 regions aligned orthologs to infer phylogenetic relationships using maximum likelihood (ML) in RAxML v.8.2.9 (Stamatakis, 2006). Evolutionary models for each orthologous cluster partition were evaluated independently under the GTRCAT model with sampling of 200 bootstrap replicates. The tree obtained was visualized in FigTree.
We used the 3,245 gene trees estimated in RaxML as input to estimate species trees in ASTRAL-III 5.7.8 (Zhang et al., 2018). ASTRAL-III 5.7.8 uses the quartet trees of the maximum likelihood phylogenies of each gene to produce the species tree topology and calculates the quartet support, which is the percentage of quartets that agree on a specific branch in the species tree. Our focus is on Mepraia species, so we inferred the coalescent tree of species in this group with T. infestans as an outgroup. The inferred species tree in newick format was visualized in FigTree (Rambaut, 2014).
We used SODA (Rabiee and Mirarab, 2021) implemented in ASTRAL-III 5.7.8 to narrow down candidate putative species for testing hybridization in a downstream analysis. SODA uses quartet topology frequencies to determine whether each branch in a guide tree inferred from gene trees is likely to have a positive length and uses the results to infer a new species tree that defines species boundaries. The analysis was performed with a cut-off p-value of 0.001. We used the species tree obtained by ASTRAL-III 5.7.8 and the 3,245 gene trees obtained by RAxML to characterize the potential conflict between the gene trees using PhyParts (Smith et al., 2015). A bipartition analysis was performed using the default bootstrap filter, ignoring edges with bootstrap values < 50% (Smith et al., 2015).
The phylogenetic relationship among Mepraia was also inferred using COI mtDNA. For this, we accessed Mepraia sequences available on the GenBank (Table S5). We accessed data of T. breyeri (Del Ponte, 1929) and T. eratyrusiformis (Del Ponte, 1929), which were used as outgroups because of their phylogenetic proximity to the Mepraia genus (Hypša et al., 2002; Justi et al., 2014). Triatoma is a paraphyletic genus of Triatominae, which makes some of their species phylogenetically closer to Mepraia than others (Justi et al., 2014). BLASTn (Camacho et al., 2009) was used to compare the set of transcriptomes against Mepraia COI sequences. The COI sequences were aligned using MAFFT v.7.305 (Katoh and Toh, 2008) and manually trimmed. Phylogenetic trees were inferred by Bayesian inference using MrBayes v3.2.6 (Ronquist et al., 2012) and the most likely evolutionary models selected by jModelTest2 (Darriba et al., 2012). In MrBayes v3.2.6 Markov chain Monte Carlo (MCMC) was used to approximate the posterior probability (Metropolis et al., 1953) with two independent runs of four MCMC chains for 1 x 107 generations performed with sampling of every 5,000 generations. Phylogenetic tree parameters reached stationarity after a burning period of 250,000 generations. Optimal trees were then sampled every 1,000 generations to obtain the final consensus tree and associated subsequent probabilities, which was visualized in FigTree (Rambaut, 2014). The COI sequences were used to estimate genetic distances, according to the Kimura 2-parameter model (Kimura, 1980) using MEGAX 10.2.6 (Kumar et al., 2018).
We used 3,245 concatenated orthologs to estimate the divergence time. For this, we used a stochastic branching process and relaxed clock model in a Bayesian framework using BEAST v2.6.1 (Bouckaert et al., 2019) under the Fossilized Birth-Death (FBD model) (Heath et al., 2014). The FBD model has become the most appropriate way to calibrate divergence time estimates when calibration dates represent fossil occurrence times (Heath et al., 2014). Thus, the time of occurrence of the fossil triatomine Panstrogylus hispaniolae (Ponair, 2013) was integrated, which is dated between ~20–15 million years ago (Mya). We integrated the species T. infestans with dates between 10 and 2 Mya to represent the uplift of the Andes (5 to 2 Mya) that supports the separation of Mepraia from other Argentine triatomines (Justi et al., 2016; Campos-Soto et al., 2022). Independent analyses of MCMC were performed with sampling every 5,000 generations. The convergence (ESS >200) evaluated by Tracer v1.7 (Rambaut et al., 2018) occurred after 5x109 generations. The first 50% tree topologies sampled were discarded as burn-in and the remaining trees were summarized in TreeAnnotator v2.5 (Bouckaert et al., 2019). The generated trees and the credibility intervals were visualized in FigTree (Rambaut, 2014).
ABBA-BABA tests were conducted (Patterson et al., 2012) to investigate potential introgression between Mepraia species. All reads were merged with the SamTools “merge” option (Li et al., 2009). The tests were performed using the Abbababa script implemented by ANGSD (Korneliussen et al., 2014). For this, the T. infestans transcriptome (GenBank: SRR4427079) was used as an outgroup and a reference for mapping. The ABBA-BABA test compares the segregation of the biallelic polymorphism of four samples (H1, H2, H3 and H4), where H1, H2 and H3 represent the inner group and H4 the outer group. Introgression was estimated using the D statistics, which is calculated from the deviations in the proportions of alleles shared between H2 and H3 (ABBA) and H1 and H2 (BABA). A jack-knife approach was also used to calculate the D corrected for bias, standard error D-statistics and Z-test (Soraggi et al., 2017).
HyDe (Blischak et al., 2018) was also used to detect introgression using the supermatrix with 3,245 aligned orthologous clusters of Mepraia. To detect phylogenetic invariants, HyDe uses a rooted network of four taxa, one of which is the outgroup and three triplets (two possible progenitors and one hybrid). For all analyses, we incorporated a Bonferroni correction and considered significance at α <0.05, with γ estimates between 0 and 1. Z-scores greater than 3 were interpreted as evidence of introgression.
As we found evidence of gene flow between Mepraia species, we inferred phylogenetic networks to detect hybridization events, visualized as reticulations in the network (Huson and Scornavacca, 2011), inferred from 3,245 clusters of Mepraia orthologs using pseudo-maximum likelihood approach (Yu and Nakhleh, 2015) implemented in PhyloNet v. 3.6.1 (Than et al., 2008). PhyloNet estimates the topology of the network and the probability of inheritance (γ) which is the probability of hybridization considering the population of two parents (Yu et al., 2012). We built four networks varying the reticulation number from 0 to 4 and defining taxa groups corresponding to each clade of the species-tree. The optimal network for each run was selected based on the highest log probability after 500 searches. The probability of the best network for each run was also compared to choosing the optimal number of crosslinks reticulations in the network.
We examined estimated coalescent branch lengths using ASTRAL III v. 5.7.3 to quantify the expectations of incomplete lineage sorting (ILS) along the tree. For the incomplete lineage sorting (ILS) test with the best ML nuclear gene trees from RAxML and 200 bootstraps (BS), we used ASTRAL III v 5.7.3. We also applied the JML test (Joly, 2012) with all 3,245 orthologous clusters concatenated as input to test introgression based on genetic distances (Joly, 2012). The input posterior distributions of the species trees were generated with *BEAST (Heled and Drummond, 2010) implemented in BEAST v.2.5 (Bouckaert et al., 2019) for 5 × 109 generations of tree sampling, which were tested with JML.
The 18 RNA libraries from head and salivary glands of the three Mepraia species generated a total of 267,766,381 reads of length of 100 bp, with an average of 15 million reads per library. The six libraries per species generated 83,257,292, 91,093,659 and 93,415,430 reads for M. gajardoi, M. parapatrica, and M. spinolai, respectively (Table S1). After trimming, a total of 239,843,935 reads were obtained, with an average of 14 million reads per library (Table S1). After trimming, libraries by species retained approximately 83, 81 and 73 million reads of M. gajardoi, M. parapatrica, and M. spinolai, respectively (Table S1). Transcriptome assemblies had an average of 49,925 unigenes for M. spinolai, 54,553 for M. garjadoi and 50,975 for M. parapatrica (Table S2). The BUSCO tool indicated that these unigenes were composed of 71–92% complete orthologs and 2–7% of fragmented groups for M. gajardoi, 88–91.9% and 3–4% for M. parapatrica, and 83.4–90.1% and 2–8% for M. spinolai, respectively (Table S3). All assemblies had an average of 10% of missing orthologs.
The CDS predictions resulted in an average of 22,955, 22,028 and 20,785 CDS in the transcriptomes of M. gajardoi, M. parapatrica, and M. spinolai, respectively (Table S4). The search against the NCBI’s non-redundant protein database using BLASTP resulted in between 9–17 thousand matches with the M. spinolai data, from 7 to 19 thousand with M. garjadoi and from 13 to 17 thousand with the M. parapatrica data (Table S4). We found on average 14,145, 12,885 and 12,038 proteins with InterPro signatures in the transcriptomes of M. garjadoi, M. spinolai and M. parapatrica, respectively (Table S4). The functional annotation through Blast2GO retrieved an average of 10,200 annotated genes in the 18 transcriptome assemblies. On average, we found 8,684 GO terms related to Molecular Function, 5,139 GO terms for Biological Process terms and 2,736 GO terms for Cellular Component (Table S4). WEGO predictions in this study were distributed into 45 ontological categories from the three classes: Biological Process (n=16), Molecular Function (n=14), and Cellular Component (n=15) (Figure S1).
The SNP calling performed on the dataset containing 19 samples, including six samples from each Mepraia species and one external group, resulted in the identification of 60,085 biallelic unlinked SNPs. The SNP matrix analyzed under the SVD quartet method resulted in a phylogeny with local posterior probability with robust support (LPP ≥ 1) (Figure 2A). The consensus tree generated by the SNAPP coalescent model is consistent with the quartet results, both inferences recovering M. gajardoi and M. parapatrica as sister species, and M. spinolai separated from the other species. On the other hand, density trees illustrate a high number of alternative topologies (Figure 2B). The agreement between the SVDquartets results and other methods reinforces confidence in the most supported topology. However, the presence of many alternative topologies in density trees suggests that some regions of the genome may have conflicting signals or incomplete lineage sorting.
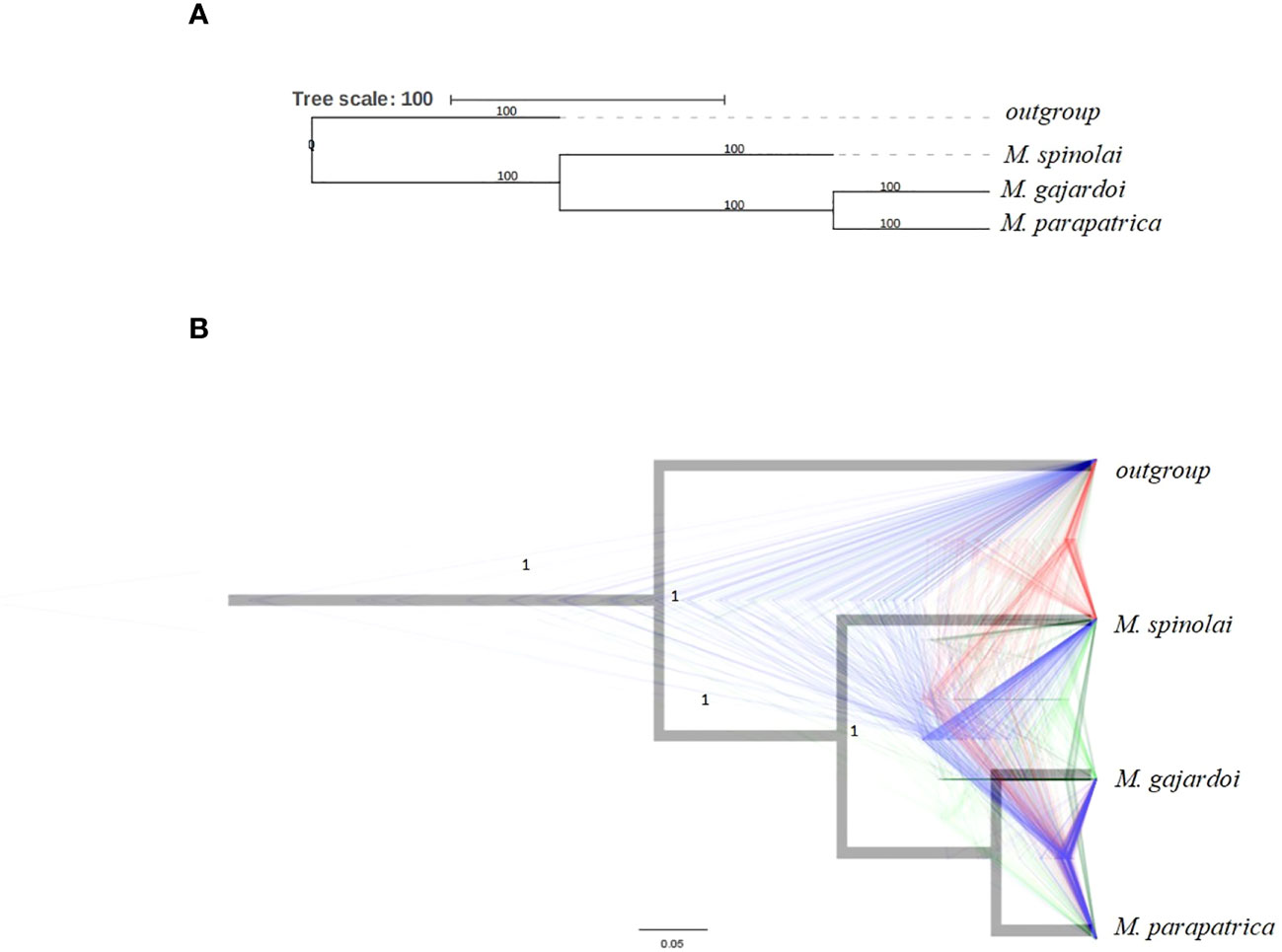
Figure 2 Inferred species trees with over 60,685 unliked biallelic SNPs under two different coalescent models. T. infestans species was used as an outgroup. (A) Overlapping species tree and density inferred from SNPs data in the SNAPP. The clade’s maximum credibility tree is in black, and alternative topologies are drawn in different colors, where blue represents the most favorable, followed by red and green. (B) Species tree inferred with SNPs with SVDquartets.
The study resulted in the identification of 3,245 putative orthologous clusters which were used for downstream analyses. The orthologs were concatenated to generate a matrix with 19 taxa, 4,105,693 characters and 72% occupation with approximately 28% of missing data. The concatenated-based phylogenetic inference using Maximum Likelihood resulted in three well-supported clades that separated the species of the genus, with the species M. gajardoi and M. parapatrica as sister taxa (Figure S2).
The species tree estimated by the coalescent model implemented in ASTRAL-III 5.7.8 with 3,245 gene trees generated 2,903,954 quartets, of which 72% are congruent with the main topology (Figure S3). The greedy consensus of the 100 bootstrap replicates of the phylogenetic trees provided strong support for species delimitation, showing that M. gajardoi and M. parapatrica are phylogenetically close, while M. spinolai is divergent in relation to these two species (Figure S3). The contrasting analysis of the results of the concatenated data was useful to show that a high percentage of gene trees have incongruent topologies with the inferred species tree for Mepraia (Figure 3A). SODA species delimitation analysis supports three monophyletic lineages consistent with the three described taxa (Figure 3A).
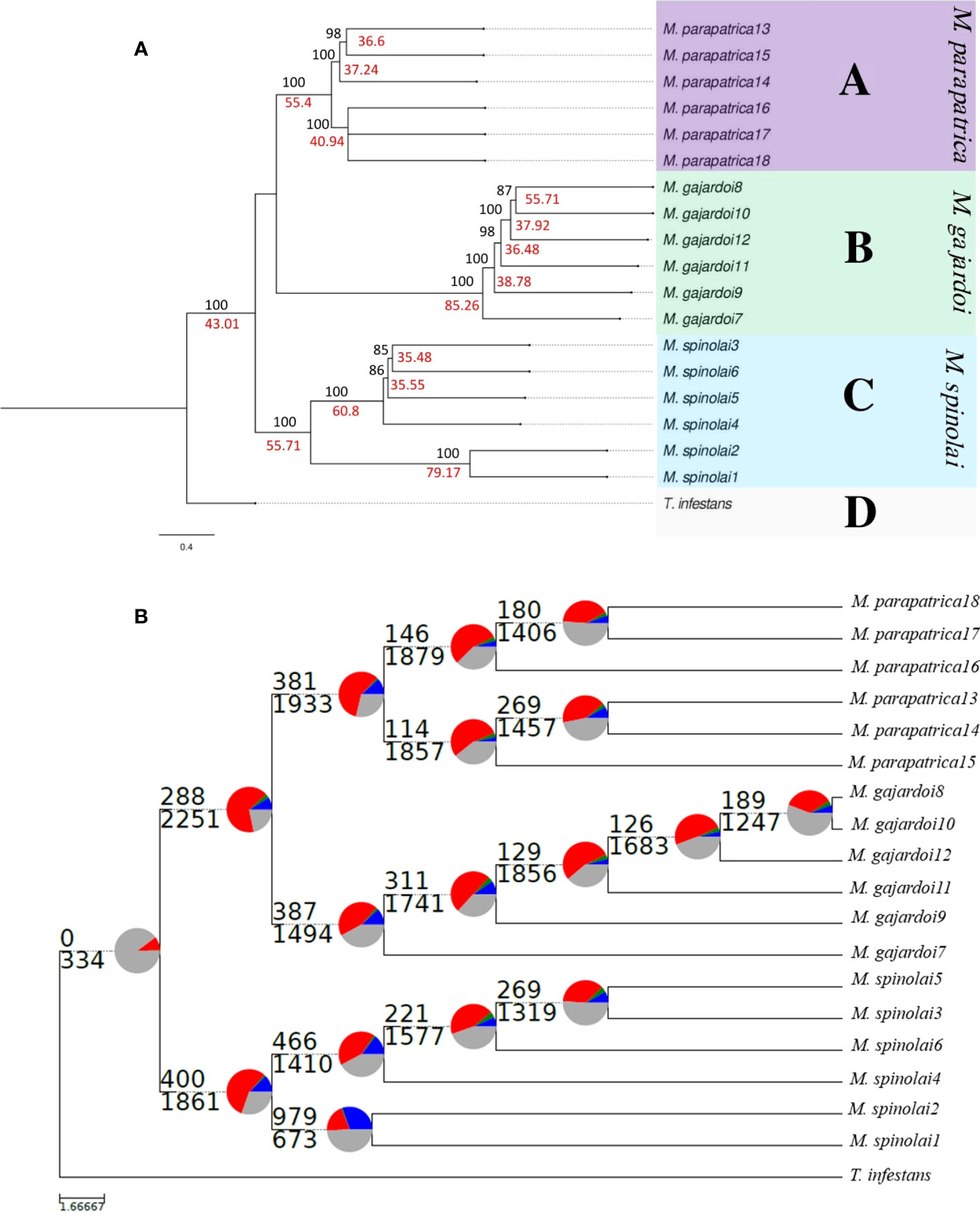
Figure 3 (A) Species tree generated using ASTRAL-III based on 3,245 single copy gene trees and delimitation species with SODA. Values above branches (in black) are branch support values obtained by ML with RAxML with 200 bootstrapping analysis and below branches (in red) are local posterior probabilities values for ASTRAL-III 5.7.8 with best maximum probability trees, respectively. The delimitation of species obtained with SODA is represented by the letters: A: M. parapatrica, B: M. gajardoi, C: M. spinolai and D: T. infestans. (B) Astral species tree based on 3,245 gene trees with a summary of conflict and concordant homologs. For each branch, the number of homologs concordant (top) or in conflict (bottom) with the species tree at each node is indicated. Blue: support the shown topology, Green: conflict with the shown topology (most common conflicting bipartition), Red: conflict with the shown topology (all other supported conflicting bipartitions), and have no support for conflicting bipartition).
Despite the high support for branching species in our analyses, the support values for quartets reveal a high level of discordance among gene trees. With PhyParts it was possible to analyze the 3,245 trees for the presence of conflict, agreement, and gene duplications in individual homologs throughout the phylogeny. The bipartition analyses were performed on modified phylogenetic trees that retained only branches with at least 50% bootstrap support. Although we inferred a dominant topology for the 3,245 orthologs clusters, our results indicate widespread discordance among gene trees (Figure 3B). However, the proportion of concordant trees shows little conflict with the dominant topology (Figure 3B), and the small proportion of conflicting bipartitions (green slice) supports a low number of alternative topologies. We also observe a high number of low-frequency topologies (red slice).
Based on mtDNA and Bayesian theory, the phylogeny inferred by Bayesian inference (BI) of the mitochondrial COI gene (57 taxa, 509 bp) identified three clades with high support (BPP 100), which are congruent with the Mepraia species (Figure S3). This analysis also supported the monophyly of the genus Mepraia and indicated M. garjadoi and M. parapatrica as sister species (BPP 0.97) separated from M. spinolai (BPP 100) (Figure S3). The sequences obtained from the transcriptomes of this study were also grouped into clades consistent with the species tree of the genus (Figure S3). Our values estimated under the Kimura 2-parameter model (K2P) suggest a low to moderate divergence between the compared species (Table S6).
The FBD-based divergence dating analysis estimated the origin of Mepraia to be between 1.21 and 2.31 Mya, with 2.31 being that of the stem group and 1.21 being the most recent possible (95% HPD, PP=1.0) (Figure 4). The analysis indicates that the separation of M. spinolai and the ancestor of the two other species happened between 0.07 and 1.61 Mya (PP=1.0), whereas the clades for the other taxa have more recent estimated dates, of 0.05 and 1.18 Mya for M. parapatrica (PP=1.0) and 0.04–0.36 Mya for M. gajardoi (PP=1.0) (Figure 4).
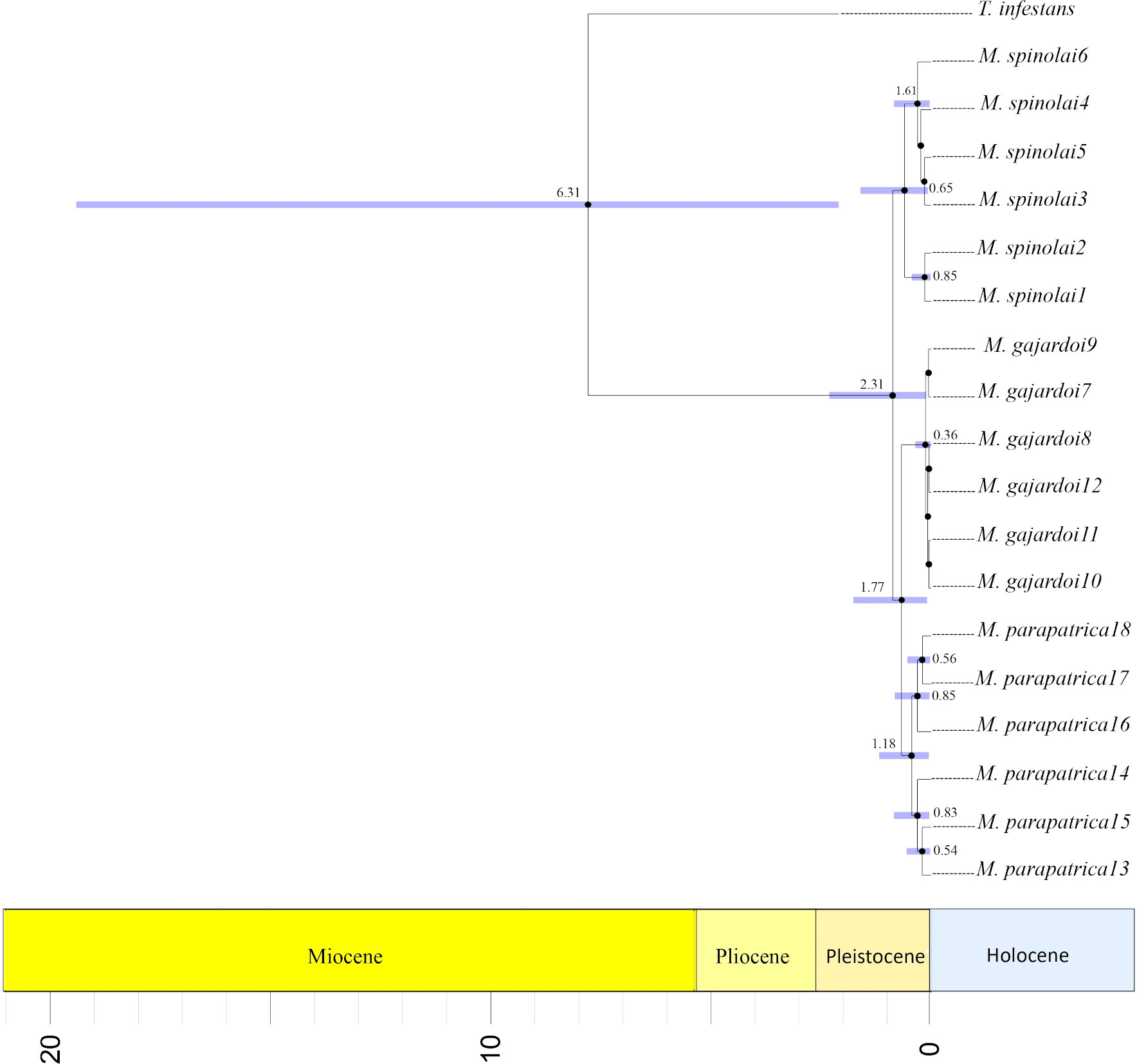
Figure 4 Phylogenetic tree base on Fossilized Birth-Death process representing the divergence time estimates of Mepraia species. The phylogeny was generated with a set of 3,245 orthologs. The calibration used the estimated date for dating the triatomine fossil Panstrongylus hispaniolae (~20–15 Mya). The horizontal blue bars at the nodes represent the 95% highest posterior density (HPD) intervals of the estimated node ages. The numbers on the main nodes represent ages of lineage divergence as well as the confidence intervals.
The ABBA-BABA test showed significant signs of introgression between M. parapatrica and M. spinolai (Table S7) (D-statistics = 0.158, Z-score 4.43 (Figure 5). The HyDe analysis also showed signs of significant hybridization for the M. parapatrica clade treated as for all triplets evaluated as species and individual (Tables S7–S11).
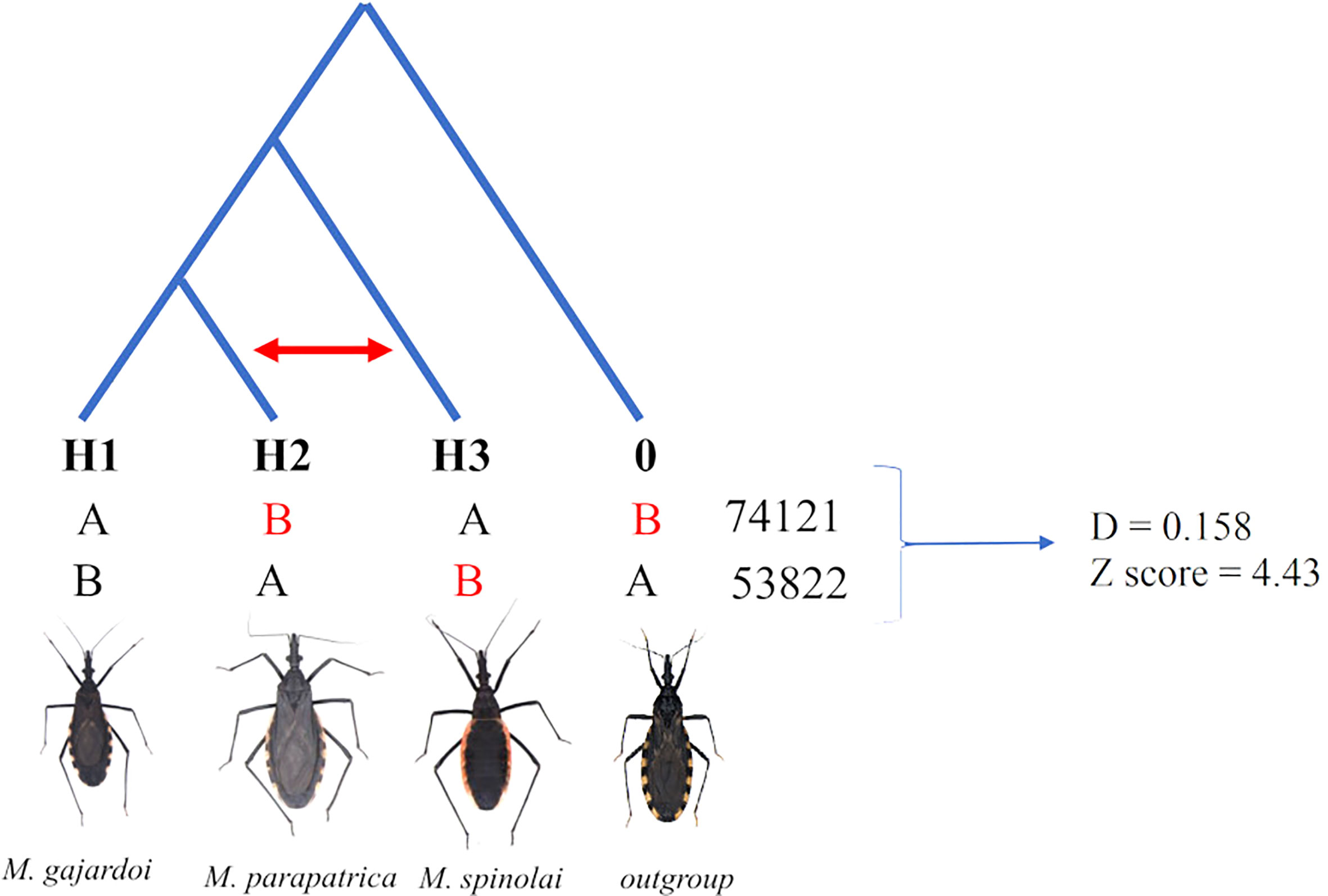
Figure 5 Results of the four-taxa ABBA-BABA test used to detect introgression among three species of Mepraia. The topology shows two possible discordant gene tree patterns, ABBA, and BABA, which normally occur in equal proportions under incomplete lineage classification, with A and B denoting the ancestral and derived allele states, respectively.
The maximum pseudo-likelihood Inference (MPL) model implemented in PhyloNet provided support for introgression among Mepraia species (Figure 6). We tested up to four reticulation events and the plateau was reached after two reticulations (Figure S4). The network with zero reticulations was consistent with the phylogenies inferred through different methodologies in this study, and the network with the highest probability showed introgression between the ancestral of M. parapatrica and M. gajardoi (Figure S5).
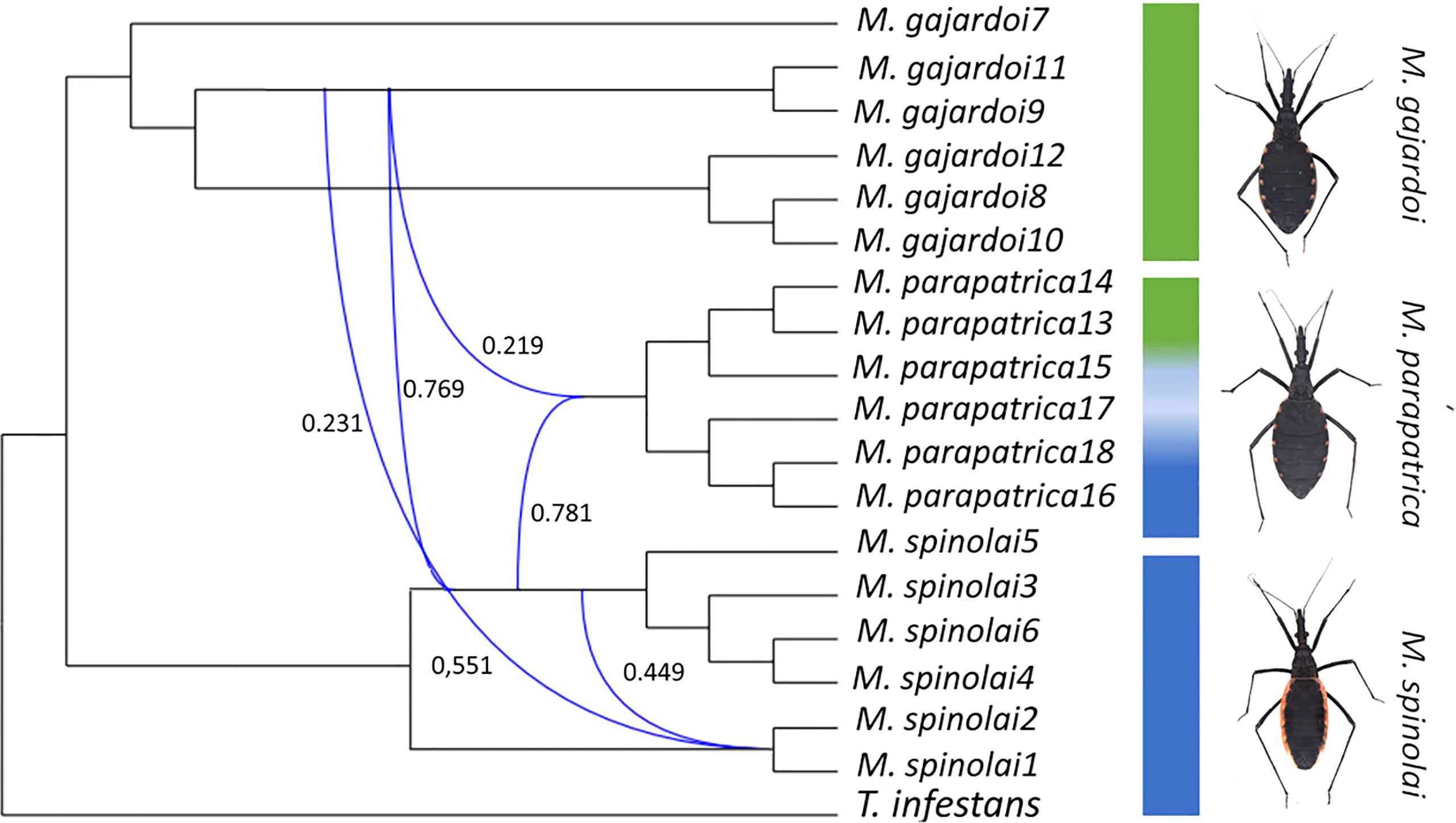
Figure 6 Pseudo-maximum-likelihood species networks of lineages showing signs of reticulation in Mepraia. Networks were inferred using a set of 3,245 orthologs. The network with the optimum number of reticulations is shown. Inheritance probabilities (γ) are shown in blue. .
ASTRAL-III showed that short branches were recovered for both main clades (Figure S5), indicating the presence of high levels of gene tree discordance that may be explained by ILS. However, the JML analysis using our cluster of 3,245 orthologs indicated that all comparisons were significant (p < 0.1) and provide evidence of hybridization between the compared sequences (Table S12).
This study contributed important genetic resources for the subfamily Triatominae, generating transcriptome of heads and salivary glands of M. gajardoi, M. parapatrica and M. spinolai. These species have epidemiological importance, as they are part of the wild cycle of T. cruzi in Chile (Campos-Soto et al., 2016). The 18 RNA libraries (six samples per species) generated transcriptome assemblies with similar quality metrics to other available Triatominae transcriptomes (Assumpção et al., 2011; Marchant et al., 2015; Carvalho et al., 2017; Brito et al., 2019). Transcriptome assemblies from the three species of Mepraia showed similar metrics, with N50 on average of 1,300 bp, which is in the range of N50 metrics of other available. Completeness and redundancy metrics are crucial for genomic studies, and in addition to N50, we used the BUSCO tool to estimate completeness using the Arthropoda database. Our transcriptomes contained between 71% and 90% complete orthologs, indicating a high percentage of complete copies for most predicted genes. The number of duplications and CDS annotations in downstream analysis correlated with the transcriptome quality.
Our transcriptomes provided a vast amount of data that allowed us to investigate the phylogenetic relationships of Mepraia. To identify strictly orthologous nuclear gene clusters, we employed the strategy described by Yang and Smith (2014) for non-model organisms, resulting in a total of 3,245 gene clusters. Our supermatrix has an overall occupancy of 72%, with about 28% of data missing from the matrix. Although the occupancy rate varied for each taxon, with some having a higher occupancy rate than others, there are an average of 3,200 loci per sample. The maximum likelihood (ML) tree inferred from the supermatrix produced a well-resolved topology with high support (bootstrap values >90), which is consistent with other studies (Justi et al., 2014; Campos-Soto et al., 2015; Campos-Soto et al., 2020), and corroborates the three lineages described in Mepraia, as well as infers M. gajardoi and M. parapatrica as sister groups and M. spinolai as more distant.
Based on mitochondrial genes, Bayesian analysis, and the Kimura two-parameter (K2P) model, we found low intra- and interspecific variability among the sequences, which was expected due to the close relationships among the species. The average genetic diversity rate for the 57 sequences was 0.20. It is worth noting that mitochondrial genes, such as COI, are widely used for species delimitation in triatomines (Hypša et al., 2002; Monteiro et al., 2003; Justi et al., 2014). However, some species, such as Triatoma maculata (Erichson, 1848), exhibit high levels of genetic distance, as evidenced by the CytB and ND4 genes (Gómez-Palacio et al., 2022). On the other hand, our findings are consistent with the low genetic distance observed in mtDNA between closely related species of the T. sordida subcomplex, as reported by Belintani et al. (2020).
The FBD method to estimate diversification time showed that the genus underwent recent divergence 2.3–1.2 Mya. Although the lack of morphological data on the fossil species P. hispaniolae used for calibration may have influenced our analysis, our findings are consistent with other available dating methods (Campos-Soto et al., 2020; Campos-Soto et al., 2022). Our dating also supports the hypothesis that Mepraia diverged from its Triatoma ancestors after the uprisal of the Andes mountains, which was estimated to have occurred around 5–2 Mya (Gregory-Wodzicki, 2000; Frías-Lasserre, 2010). The rise of the Andes and the resulting changes in climate have been identified as crucial factors that contributed to the diversification of the Triatomini and Rhodniini tribes in South America (Justi et al., 2016), which may have also influenced the diversification of Mepraia.
The phylogenetic analysis of Mepraia based on over 60,000 unlinked biallelic SNPs produced consistent phylogenies. Both support the relationships among species, with M. garjadoi and M. parapatrica inferred as sister taxa and M. spinolai as more distantly related. Although the phylogeny constructed with the quartet model has robust support, the Bayesian method used in the SNAPP software revealed a significant number of alternative topologies. The inferred alternative topologies may indicate that the evolutionary relationships between the species in question are uncertain or that there is a complex evolutionary history for these species that cannot be fully resolved with the available data set (Leaché and Oaks, 2017; McLean et al., 2022).
The coalescent-based species tree inferred using quartet inference in ASTRAL-III resulted in a topology consistent with the one produced by the concatenation method, as well as to that produced by mtDNA. However, despite the high bootstrap support values across the inferred tree, the normalized quartet scores were relatively low. The exact threshold for robust quartet support may vary depending on the study and the objectives of the analysis (Whidden and Burch, 2015). In general, quartet scores above 0.5 are considered to provide moderate support, while scores above 0.7 provide strong support for the topology. In our analysis, the inferred clades in the species tree have quartet values ranging from 43% to 56%, which prompted us to perform exploratory analyses to assess the potential conflict among gene trees across the Mepraia phylogeny.
A bipartition analysis of trees among different parts of the genome using PhyParts showed strong support for the general topology of species trees, while still indicating alternative topologies. This analysis is especially valuable as it offers a counterpoint to tree species, revealing that while we can identify a major topology, we can also clearly infer the existence of a significant number of incongruent topologies. Is worth noting that high posterior probability values obtained in ASTRAL-III can be misleading when evaluated alone and may obscure the underlying phylogenetic signal (Maddison and Knowles, 2006; Zhou et al., 2016).
Our results suggest that there are only a small number of concordant trees, indicating that several alternative topologies are being supported. This pattern is expected, as gene trees are rarely entirely concordant with species trees (Vargas et al., 2019; Meng et al., 2021; Xiao et al., 2022), especially in recently diverged taxa. Incongruence in the phylogenetic signal across distinct parts of the genome can be the result of various evolutionary phenomena, including incomplete lineage sorting, gene flow, hybridization, lateral gene transfer, recent divergence, convergent evolution, different evolutionary rates, or loss of information during evolution (Betancur et al., 2019). These phenomena shape the relationships among species, which, depending on the degree of conflict, can make phylogenetic inferences incredibly challenging. The conflict between the inferred species tree and the underlying gene trees indicates that some of the above-mentioned processes could have influenced the differentiation of Mepraia, especially incomplete lineage sorting and/or introgression.
Although phylogenies based on coalescent models at the species and gene level have produced similar relationship among Mepraia species, our data suggest a reticulated evolutionary history. As indicated above, several factors, especially ILS and introgression, can produce reticulate phylogenies, although distinguishing between the latter two can be challenging (Schumer et al., 2014; Wang et al., 2022). As ILS can occur even in the absence of introgression events, it is important to include it as null hypotheses when testing for introgression to properly investigate the causes of reticulate phylogenies (Mendes and Hahn, 2018; Hibbins and Hahn, 2022), which we did using different strategies.
Our study provides robust evidence of introgression between Mepraia species using four different methods. Firstly, an ABBA-BABA test considering only the most likely topology estimated a significant value of the D statistic (D = 0.158, Z-score 4.43), indicating introgression. Secondly, the HyDe test also revealed significant introgression between the three species, providing further support for introgression. Additionally, PhyloNet analyses showed strong evidence that M. parapatrica may result from introgression between M. gajardoi and M. spinolai. Finally, the JML test allowed identification of hybridization events in all comparisons (p-value <0.1).
Knowledge of current gene flow between Mepraia species is limited (Campos-Soto et al., 2013). Although hybridization has been observed in other Triatominae species (Panzera et al., 2021), there is still much to be discovered about the extent and effects of hybridization in Triatominae (Costa et al., 2009; Panzera et al., 2021; Pavan et al., 2021; Pinotti et al., 2021). To date, the only study that has investigated inter-specific crossing ability in the Mepraia genus, particularly between M. gajardoi and M. spinolai, found evidence of hybrid incompatibilities in their second backcross (Campos-Soto et al., 2016). Hybrid speciation involves the mixing of different lineages and gene flow through hybridization and backcrossing, resulting in the formation of a new species (Mallet, 2005; Mallet, 2007; Abbott et al., 2013). This process was previously thought to be rare in animals, but recent methodological and technical advances in the study of introgression (Guo et al., 2018; Lamichhaney et al., 2018) have shown its importance in evolution and prevalence in closely related species of several different organisms, such as plants, birds, fishes, and butterflies (Schwarz et al., 2005; Lamichhaney et al., 2018; Zhang et al., 2019).
The parapatric evolution model proposed for Mepraia suggests that cytogenetic changes were responsible for the emergence of M. parapatrica and later M. spinolai from the ancestral M. gajardoi (Frías-Lasserre, 2010). According to this hypothesis, character displacement among the three species in tension zones indicates absence of gene flow (Frías-Lasserre, 2010). However, the more recent speciation proposal by Campos-Soto et al. (2022) advocates a speciation model that involves two geographically distinct groups: the North, composed of M. garjadoi and M. parapatrica, and the South, composed of M. spinolai. The data presented in that study suggests an alternative model that may be compatible with ancient hybridization events or continued gene flow. In this sense, it is important to consider that the evolution of these species may not have occurred in a linear and bifurcating manner, and that the phylogenetic relationships between the species may not be described in a simplified way, but rather by diversification patterns that have been shaped by historical events of hybridization and geographic segmentation. This broader view of evolution may help to better understand the complexity of evolutionary processes and to reconstruct the evolutionary history of species more accurately.
Hybridization occurs when certain conditions are met, including the mixing of two parental lines, followed by reproductive isolation and a viable, genetically distinct hybrid line (Schumer et al., 2014). Based on our data and the current knowledge about Triatominae speciation (Pavan et al., 2021), we can speculate that incongruity patterns in gene trees cannot be explained only by incomplete lineage classification and stochasticity. Rather, to some extent, it may have been modulated by introgression, potentially due to ancestral or continuous gene flow. As discussed earlier, geoclimatic changes in Chile over the past 5 million years may have been crucial for the evolution of these Triatomine bugs (Campos-Soto et al., 2022). Similar diversification events have been observed in North American Drosophila (Campo et al., 2013), Anolis lizards on Caribbean islands (Mahler et al., 2010), and the Heliconius butterfly genus (Mavárez et al., 2006; Jiggins et al., 2008). In the Andean region of Chile, it is known that the Bombus dahlbomii bumblebee hybridized with other Bombus bee species in Chile (Arbetman et al., 2013).
This study successfully obtained and analyzed the transcriptomes of the head and salivary glands of three species of Mepraia, shedding new light on the relationships between M. gajardoi, M. parapatrica, and M. spinolai. Analyses of a large transcriptome dataset provided valuable information about the evolutionary history of the genus. We showed that while all methods produced concurrent phylogenetic relationships among species of the genus, a considerable level of incongruence is observed across gene trees. We demonstrated through several tests that such incongruence is not compatible only with ILS, rather, is an indication of introgression between lineages due to past hybridization events. Therefore, our data suggests that the diversification of Mepraia occurred recently and was significantly influenced by interspecific gene flow during its early stages of divergence. This study highlights the importance of considering introgression and hybridization in the evolution of organisms, especially in Chagas disease vector triatomines.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA916468.
TB designed the study, wrote the entire text, discussed results, and performed all study analyses. CC assisted and reviewed all bioinformatics analyses, as well as reviewing the text, results, and discussion. HP and SC-E assisted in RNA extraction and reviewed the text. RB reviewed the text, assisted in the discussion, and provided servers for bioinformatics analyses. JO collected specimens, assisted in the discussion, and reviewed the text. DF-L collected specimens, assisted in the discussion, and reviewed the text. FF mapped and reviewed the test. JR designed the study, collected specimens, assisted in the discussion, reviewed, and obtained resources. All authors contributed to the article and approved the submitted version.
JR: Fundação de Amparo à Pesquisa do Estado de São Paulo, processo: 17/06460-4. JO: Fundação de Amparo à Pesquisa do Estado de São Paulo, processo: 19/02145-2. TB: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Financing Code 001. RB is a CNPq Productivity fellow (#315287/2020-9). Financial support had neither role in study design, collection, analysis, interpretation, and manuscript writing.
We thank Maria Isabel Camargo Isler, Department of Genetics and Evolution, Federal University of São Carlos (UFSCar), São Carlos, SP, Brazil. The authors are grateful to the Universidade Estadual Paulista (Unesp), Faculdade de Ciências Farmacêuticas and to its collaborators who help in the maintenance of the Triatominae insectarium, especially to Tacachi Hatanaka and Dr. Vinicius Fernandes Paiva. We are grateful for the support of Natália Almeida Frota Santos, Tiradentes University, and help with making the map here shown.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1215319/full#supplementary-material
Abbott R., Albach D., Ansell S., Arntzen J. W., Baird S. J. E., Bierne N., et al. (2013). Hybridization and speciation. J. Evol. Biol. 26, 229–246. doi: 10.1111/j.1420-9101.2012.02599.x
Antonelli A., Nylander J. A. A., Persson C., Sanmartín I. (2009). Tracing the impact of the Andean uplift on Neotropical plant evolution. Proc. Nat. Acad. Sci. U.S.A. 106, 9749–9754. doi: 10.1073/pnas.0811421106
Arbetman M. P., Meeus I., Morales C. L., Aizen M. A., Smagghe G. (2013). Alien para site hitchhikes to Patagonia on invasive bumblebee. Biol. Inv. 15 (3), 489–494. doi: 10.1007/s10530-012-0311-0
Assumpção T. C. F., Charneau S., Santiago P. B. M., Francischetti I. M. B., Meng Z., Araújo C. N., et al. (2011). Insight into the salivary transcriptome and proteome of Dipetalogaster maxima. J. Proteome. Res. 10, 669–679. doi: 10.1021/PR100866H
Astral III [software]. Available at: https://github.com/smirarab/ASTRAL/ (Accessed April 8, 2023).
Belintani T., Oliveira J., Pinotti H., Silva L. A., Alevi K. C. C., Galvão C., et al. (2020). Phylogenetic and phenotypic relationships of the Triatoma sordida subcomplex (Hemiptera: Reduviidae: Triatominae). Act. Trop 212, 105679. doi: 10.1016/j.actatropica.2020.105679
Betancur R., Arcila D., Vari R. P., Hughes L. C., Oliveira C., Sabaj M. H., et al. (2019). Phylogenomic incongruence, hypothesis testing, and taxonomic sampling: the monophyly of characiform fishes. Evolution 73, 329–345. doi: 10.1111/EVO.13649
Blischak P. D., Chifman J., Wolfe A. D., Kubatko L. S. (2018). HyDe: a Python package for genome-scale hybridization detection. Syst. Biol. 67, 821–829. doi: 10.1093/SYSBIO/SYY023
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114. doi: 10.1093/BIOINFORMATICS/BTU170
Bouckaert R. (2010). DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26 (10), 1372–1373. doi: 10.1093/bioinformatics/btq110
Bouckaert R., Vaughan T. G., Barido-Sottani J., Duchêne S., Fourment M., Gavryushkina A., et al. (2019). BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PloS Comput. Biol. 15. doi: 10.1371/journal.pcbi.1006650
Brito R. N., Geraldo J. A., Monteiro F. A., Lazoski C., Souza R. C. M., Abad-Franch F. (2019). Transcriptome-based molecular systematics: Rhodnius montenegrensis (Triatominae) and its position within the Rhodnius prolixus-rhodnius robustus cryptic-species complex. Parasitol. Vectors 12. doi: 10.1186/s13071-019-3558-9
Bryant D., Bouckaert R., Felsenstein J., Rosenberg N. A., Roychoudhury A. (2012). Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917–1932. doi: 10.1093/MOLBEV/MSS086
Calleros L., Panzera F., Bargues M. D., Monteiro F. A., Klisiowicz D. R., Zuriaga M. A., et al. (2010). Systematics of Mepraia (Hemiptera-Reduviidae): cytogenetic and molecular variation. Infect. Genet. Evol. 10, 221–228. doi: 10.1016/J.MEEGID.2009.12.002
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10, 421. doi: 10.1186/1471-2105-10-421
Campo D., Lehmann K., Fjeldsted C., Souaiaia T., Kao J., Nuzhdin S. V.. (2013). Whole-genome sequencing of two North American Drosophila melanogaster populations reveals genetic differentiation and positive selection. Mol. Eco. 20, 5084–5097. doi: 10.1111/mec.12468
Campos-Soto R., Botto-Mahan C., Coronado X., Catala S. S., Solari A. (2013). Phylogenetic relationships of the spinolai complex and other triatomini based on mitochondrial DNA sequences (Hemiptera: Reduviidae). J. Vector. Borne Dis. 13, 73–76. doi: 10.1089/vbz.2011.0954
Campos-Soto R., Botto-Mahan C., Coronado X., Jaramillo N., Panzera F., Solari A. (2011). Wing shape differentiation of Mepraia species (Hemiptera: Reduviidae). Infect. Genet. Evol. 11, 329–333. doi: 10.1016/J.MEEGID.2010.11.002
Campos-Soto R., Díaz-Campusano G., Rives-Blanchard N., Cianferoni F., Torres-Pérez F. (2020). Biogeographic origin and phylogenetic relationships of Mepraia (Hemiptera, Reduviidae) on islands of northern Chile. PLoS One 15 (6), e0234056. doi: 10.1371/journal.pone.0234056
Campos-Soto R., Ortiz S., Cordova I., Bruneau N., Botto-Mahan C., Solari A. (2016). Interactions between Trypanosoma cruzi the chagas disease parasite and naturally infected wild Mepraia vectors of Chile. J. Vector Borne Dis. Dis. 16, 165–171. doi: 10.1089/VBZ.2015.1850
Campos-Soto R., Rodríguez-Valenzuela E., Díaz-Campusano G., Boric-Bargetto D., Zúñiga-Reinoso Á., Cianferoni F., et al. (2022). Testing phylogeographic hypotheses in Mepraia (Hemiptera: Reduviidae) suggests a complex spatio-temporal colonization in the coastal atacama desert. Insects 13, 419. doi: 10.3390/INSECTS13050419
Campos-Soto R., Torres-Pérez F., Solari A. (2015). Phylogenetic incongruence inferred with two mitochondrial genes in Mepraia spp. and Triatoma eratyrusiformis (Hemiptera, Reduviidae). Genet. Mol. Biol. 38, 390. doi: 10.1590/S1415-475738320140301
Capella-Gutiérrez S., Silla-Martínez J. M., Gabaldón T. (2009). TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/BIOINFORMATICS/BTP348
Carvalho D. B., Congrains C., Chahad-Ehlers S., Pinotti H., de Brito R. A., da Rosa J. A. (2017). Differential transcriptome analysis supports Rhodnius montenegrensis and Rhodnius robustus (Hemiptera, Reduviidae, Triatominae) as distinct species. PLoS One 12 (4), e0174997. doi: 10.1371/JOURNAL.PONE.0174997
Chacón F., Bacigalupo A., Quiroga J. F., Ferreira A., Cattan P. E., Ramírez-Toloza G. (2016). Feeding profile of Mepraia spinolai, a sylvatic vector of Chagas disease in Chile. Acta Trop. 162, 171–173. doi: 10.1016/J.ACTATROPICA.2016.06.027
Chifman J., Kubatko L. (2015). Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes, site-specific rate variation, and invariable sites. J. Theor. Biol. 374, 35–47. doi: 10.1016/J.JTBI.2015.03.006
Chomczynski P., Mackey K. (1995). Short technical reports. modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. BioTechniques 19 (6), 942–945.
Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M. (2005). Blast2GO: a universal tool for annotation, visualization, and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/BIOINFORMATICS/BTI610
Congrains C., Zucchi R. A., de Brito R. A. (2021). Phylogenomic approach reveals strong signatures of introgression in the rapid diversification of neotropical true fruit flies (Anastrepha: tephritidae). Mol. Phy. Evol. 162, 107200. doi: 10.1016/J.YMPEV.2021.107200
Costa J., Peterson A. T., Dujardin J. P. (2009). Morphological evidence suggests homoploid hybridization as a possible mode of speciation in the Triatominae (Hemiptera, Heteroptera, Reduviidae). Infect. Genet. Evol. 9 (2), 263–270. doi: 10.1016/J.MEEGID.2008.12.005
Danecek P., Bonfield J. K., Liddle J., Marshall J., Ohan V., Pollard M. O., et al. (2021). Twelve years of SAMtools and BCFtools. GigaScience 10 (2). doi: 10.1093/GIGASCIENCE/GIAB008
Darriba D., Taboada G. L., Doallo R., Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9 (8), 772. doi: 10.1038/nmeth.2109
Degnan J. H., Rosenberg N. A. (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340. doi: 10.1016/J.TREE.2009.01.009
Doan T. M. (2003). A south-to-north biogeographic hypothesis for Andean speciation: evidence from the lizard genus Proctoporus (Reptilia, gymnophthalmidae). J. Biogeogr. 30 (3), 361–374. doi: 10.1046/J.1365-2699.2003.00833.X
Dunn C. W., Hejnol A., Matus D. Q., Pang K., Browne W. E., Smith S. A., et al. (2008). Broad phylogenomic sampling improves resolution of the animal tree of life. Nat 452, 745–749. doi: 10.1038/nature06614
Eddy S. R. (2011). Accelerated profile HMM searches. PloS Comput. Biol. 7, e1002195. doi: 10.1371/JOURNAL.PCBI.1002195
Edelman N. B., Frandsen P. B., Miyagi M., Clavijo B., Davey J., Dikow R. B., et al. (2019). Genomic architecture and introgression shape a butterfly radiation. Sci 366 (6465), 594–599. doi: 10.1126/science.aaw2090
Faúndez E. I., Carvajal M. A. (2012). Notas sobre Mepraia gajardoi frías, Henry and gonzález 1998 (Heterptera: Reduviidae: Triatominae). Boletín la S.E.A. 50, 495–497.
Frías-Lasserre D. (2010). A new species and karyotype variation in the bordering-distribution of Mepraia spinolai (Porter) and Mepraia gajardoi frías et al., (Hemiptera: Reduviidae: Triatominae) in Chile and its parapatric model of speciation. Neo. Entom. 39 (4), 572–583.
Frías-Lasserre D., Atria J. (1998). Chromosomal variation, macroevolution and possible parapatric speciation in Mepraia spinolai (Porter) (Hemiptera: Reduviidae). Genet. Mol. Biol. 21, (2). doi: 10.1590/S1415-47571998000200002
Frías-Lasserre D. F., de Oliveira J., Pinotti H., da Rosa J. A. (2019). Morphological description of Mepraia spp. females (Hemiptera: Reduviidae, Triatominae). Acta Trop. 190, 389–394. doi: 10.1016/J.ACTATROPICA.2018.11.028
Frías-Lasserre D., González C. R., Reyes C., Lecaros Alvarado A. (2018). The interspecific hemelytra differences among Mepraia species males (Hemiptera, Reduviidae:Triatominae) in Chile. J. Med. Entomol. 55, 1478–1485. doi: 10.1093/JME/TJY119
Frías-Lasserre D., Henry A. A., Gonzalez C. (1998). Mepraia gajardoi: a new species of Triatominae (Hemiptera: Reduviidae) from Chile and its comparison with Mepraia spinolai. Rev. Chil. Hist. 71, 177–188.
Fu L., Beifang N., Zhu Z., Wu S., Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gómez-Palacio A., Pita S., Abad-Franch F., Monsalve Y., Cantillo-Barraza O., Monteiro F. A., et al. (2022). Molecular and cytogenetic evidence for sibling species in the chagas disease vector Triatoma maculata. Med. Vet. Entomol. 2), 316–329. doi: 10.1111/MVE.12633
Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., et al. (20112011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi: 10.1038/nbt.1883
Gregory-Wodzicki K. M. (2000). Uplift history of the central and nothern Andes: a review. GSA Bull. 112 (7), 1091–1105. doi: 10.1130/0016-7606(2000)112<1091:UHOTCA>2.0.CO;2
Guo X., Thomas D. C., Saunders R. M. K. (2018). Gene tree discordance and coalescent methods support ancient intergeneric hybridisation between dasymaschalon and friesodielsia (Annonaceae). Mol. Phy. Evol. 127, 14–29. doi: 10.1016/j.ympev.2018.04.009
Haas B. J., Papanicolaou A., Yassour M., Grabherr M., Blood P. D., Regev A. (2013). De novo transcript sequence reconstruction from RNA-seq: reference generation and analysis with trinity. Nat. Protoc. 8 (8), 1494–1512. doi: 10.1038/NPROT.2013.084
Harris M. A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., et al. (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, 258–261. doi: 10.1093/NAR/GKH036
Hartley A. J. (2003). Andean Uplift and climate change. J. Geol. Soc London. 160, 7–10. doi: 10.1144/0016-764902-083
Heath T. A., Huelsenbeck J. P., Stadler T. (2014). The fossilized birth-death process for coherent calibration of divergence-time estimates. Proc. Natl. Acad. Sci. 111 (29), E2957–E2966. doi: 10.1073/pnas.1319091111
Heled J., Drummond A. J. (2010). Bayesian Inference of species trees from multilocus data. Mol. Bio. Evol. 27 (3), 570–580. doi: 10.1093/MOLBEV/MSP274
Hibbins M. S., Hahn M. W. (2022). Phylogenomic approaches to detecting and characterizing introgression. Genetics 220 (2), iyab173. doi: 10.1093/GENETICS/IYAB173
Huson D. H., Scornavacca C. (2011). A survey of combinatorial methods for phylogenetic networks. Genome Biol. Evol. 3 (1), 23. doi: 10.1093/GBE/EVQ077
Hypša V., Tietz D. F., Zrzavý J., Rego R. O. M., Galvao C., Jurberg J. (2002). Phylogeny and biogeography of Triatominae (Hemiptera: Reduviidae): molecular evidence of a new world origin of the Asiatic clade. Mol. Phy. Evol. 23, 447–457. doi: 10.1016/S1055-7903(02)00023-4
Irisarri I., Baurain D., Brinkmann H., Delsuc F., Sire J. Y., Kupfer A., et al. (2017). Phylotranscriptomic consolidation of the jawed vertebrate timetree. Nat. Ecol. Evol. 1, 1370–1378. doi: 10.1038/S41559-017-0240-5
Jiggins C. D., Salazar C., Linares M., Mavarez J. (2008). Hybrid trait speciation and heliconius butterflies. Philos. Trans. R. Soc B: Biol. Sci. 363 (1506), 3047. doi: 10.1098/RSTB.2008.0065
Joly S. (2012). JML: testing hybrizitation from species trees. Mol. Eco. Res. 12, 179–184. doi: 10.1111/j.1755-0998.2011.03065.x
Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi: 10.1093/bioinformatics/btu031
Justi S. A., Galvão C., Schrago C. G. (2016). Geological changes of the americas and their influence on the diversification of the Neotropical kissing bugs (Hemiptera: Reduviidae: Triatominae). PloS Negl. Trop. 10 (4), e0004527. doi: 10.1371/JOURNAL.PNTD.0004527
Justi S. A., Russo C. A. M., Mallet J. R. D. S., Obara M. T., Galvão C. (2014). Molecular phylogeny of Triatomini (Hemiptera: Reduviidae: Triatominae). Parasitol. Vectors. 7, 149. doi: 10.1186/1756-3305-7-149
Katoh K., Toh H. (2008). Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinform. 9, 212. doi: 10.1186/1471-2105-9-212
Kimura M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evo. 16, 111–120. doi: 10.1007/BF01731581
Koboldt D. C., Chen K., Wylie T., Larson D. E., McLellan M. D., Mardis E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283. doi: 10.1093/BIOINFORMATICS/BTP373
Korneliussen T. S., Albrechtsen A., Nielsen R. (2014). ANGSD: analysis of next generation sequencing data. BMC Bioinform. 15, 1–13. doi: 10.1186/S12859-014-0356-4/TABLES/4
Kriventseva E. V., Kuznetsov D., Tegenfeldt F., Manni M., Dias R., Simão F. A., et al. (2019). OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807. doi: 10.1093/NAR/GKY1053
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGAX: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lambarri M. C., Cifuentes A. C., Ayala S., Tapia-Garay V., Lillo D. C. (2018). Eco-epidemiology of chagas disease in Chile. Chagas Dis. - Basic Investigations Challenges. doi: 10.5772/INTECHOPEN.74744
Lamichhaney S., Han F., Webster M. T., Andersson L., Grant B. R., Grant P. R. (2018). Rapid hybrid speciation in darwin’s finches. Science 359 (6372), 224–228. doi: 10.1126/science.aao4593
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Leaché A. D., Oaks J. R. (2017). The utility of single nucleotide polymorphism (SNP) data in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 48, 69–84. doi: 10.1146/ANNUREV-ECOLSYS-110316-022645
Lent H., Jurberg J., Galvao C. (1994). Revalidation of the genus Mepraia, mazza, gajardo and jorg 1940 (Hemiptera, Reduviidae, Triatominae). Mem. Inst. Oswaldo Cruz. 89 (3), 347–352. doi: 10.1590/S0074-02761994000300008
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Maddison W., Knowles L. (2006). Inferring phylogeny despite incomplete lineage sorting. Systematic Biol. 55 (1), 21–30. doi: 10.1080/10635150500354928
Mahler D. L., Ingram T., Revell L. J., Losos J. B. (2010). Exceptional convergence on the macroevolutionary landscape in island lizard radiations. Science 329 (5998), 1521–1524. doi: 10.1126/science.1192212
Mallet J. (2005). Hybridization as an invasion of the genome. Trends. Ecol. Evol. 20 (5), 229–237. doi: 10.1016/j.tree.2005.02.010
Marchant A., Mougel F., Almeida C., Jacquin-Joly E., Costa J., Harry M. (2015). De novo transcriptome assembly for a non-model species, the blood-sucking bug Triatoma brasiliensis, a vector of chagas disease. Genetics 143, 225–239. doi: 10.1007/S10709-014-9790-5/FIGURES/5
Marques D. A., Lucek K., Meier J. I., Mwaiko S., Wagner C. E., Excoffier L., et al. (2016). Genomics of rapid incipient speciation in sympatric threespine stickleback. PloS Genet. 12 (2), e1005887. doi: 10.1371/JOURNAL.PGEN.1005887
Mauseth J. D. (1996). Cacti: biology and uses (Berkeley and Los Angeles, California: University of California Press).
Mavárez J., Salazar C. A., Bermingham E., Salcedo C., Jiggins C. D., Linares M. (2006). Speciation by hybridization in Heliconius butterflies. Nature 441, 7095. doi: 10.1038/nature04738
McLean B. S., Bell K. C., Cook J. A. (2022). SNP-based phylogenomic inference in holarctic ground squirrels (Urocitellus). Mol. Phy. Evo. 169, 107396. doi: 10.1016/J.YMPEV.2022.107396
Mendes F. K., Hahn M. W. (2018). Why concatenation fails near the anomaly zone. Syst. Bio. 67 (1), 158–169. doi: 10.1093/SYSBIO/SYX063
Meng R., Luo L. Y., Zhang J. Y., Zhang D. G., Nie Z. L., Meng Y. (2021). The deep evolutionary relationships of the morphologically heterogeneous nolinoideae (Asparagaceae) revealed by transcriptome data. Front. Plant Sci. 11. doi: 10.3389/FPLS.2020.584981
Metropolis N., Rosenbluth A. W., Rosenbluth M. N., Teller A. H., Teller E. (1953). Equation of state calculations by fast computing machines. J. Chem. Phys. 21 (6), 1087–1092. doi: 10.1063/1.1699114
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G. A., Sonnhammer E. L. L., et al. (2021). Pfam: the protein families database in 2021. Nucleic. Acids Res. 49, D412–D419. doi: 10.1093/NAR/GKAA913
Monteiro F. A., Barrett T. V., Fitzpatrick S., Cordon-Rosales C., Feliciangeli D., Beard C. B. (2003). Molecular phylogeography of the Amazonian chagas disease vectors Rhodnius prolixus and r. robustus. Mol. Ecol. Evo 12, 997–1006. doi: 10.1046/j.1365-294X.2003.01802.x
Monteiro F. A., Weirauch C., Felix M., Lazoski C., Abad-Franch F. (2018). Evolution, systematics, and biogeography of the Triatominae vectors of chagas disease. Adv. Parasitol. 99, 265–344. doi: 10.1016/bs.apar.2017.12.002
Moreira-Muñoz A. (2011). The extravagant physical geography of Chile (Netherlands: Springer), 3–45. doi: 10.1007/978-90-481-8748-5_1
Panzera F., Pita S., Lorite P. (2021). Chromosome structure and evolution of Triatominae: a review (Cham: Springer), 65–99. doi: 10.1007/978-3-030-64548-9_4
Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., et al. (2012). Ancient admixture in human history. Genetics 192, 1065–1093. doi: 10.1534/GENETICS.112.145037
Pavan M. G., Lazoski C., Monteiro F. A. (2021). Speciation processes in Triatominae (Cham: Springer), 39–64. doi: 10.1007/978-3-030-64548-9_3
Pinotti H., de Oliveira J., Ravazi A., Madeira F. F., dos Reis Y. V., de Oliveira A. B. B., et al. (2021). Revisiting the hybridization processes in the Triatoma brasiliensis complex (Hemiptera, Triatominae): interspecific genomic compatibility point to a possible recent diversification of the species grouped in this monophyletic complex. PLoS One 16 (10), e0257992. doi: 10.1371/JOURNAL.PONE.0257992
Pirie M. D., Chatrou L. W., Mols J. B., Erkens R. H. J., Oosterhof J. (2006). “Andean-centred” genera in the short-branch clade of Annonaceae: testing biogeographical hypotheses using brumfield phylogeny reconstruction and molecular dating. J. Bio. Geo. 33, 31–46. doi: 10.1111/J.1365-2699.2005.01388.X
Rabiee M., Mirarab S. (2021). SODA: multi-locus species delimitation using quartet frequencies. Bioinformatics 36, 5623–5631. doi: 10.1093/BIOINFORMATICS/BTAA1010
Rambaut A. (2014). FigTree v1.4.2: tree figure drawing tool (Edinburgh: Institute of Evolutionary Biology, University of Edinburgh). Available at: http://tree.bio.ed.ac.uk/software/figtree/.
Rambaut A., Drummond A. J. (2007). Tracer v1.6 and higher (2007-2013). Available at: http://tree.bio.ed.ac.uk/software/tracer/.
Rambaut A., Drummond A. J., Xie D., Baele G., Suchard M. A. (2018). Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/SYSBIO/SYY032
Regier J. C., Shultz J. W., Zwick A., Hussey A., Ball B., Wetzer R., et al. (2010). Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 463, 1079–1083. doi: 10.1038/nature08742
Rokas A., W. B., K. N., C. S. (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804. doi: 10.1038/nature02053
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 (3), 539–542. doi: 10.1093/sysbio/sys029
Schumer M., Rosenthal G. G., Andolfatto P. (2014). How common is homoploid hybrid speciation? Evolution 68 (6), 1553–1560. doi: 10.1111/evo.12399
Schwarz D., Matta B. M., Shakir-Botteri N. L., McPheron B. A. (20052005). Host shift to an invasive plant triggers rapid animal hybrid speciation. Nature 436, 7050. doi: 10.1038/nature03800
Seehausen O., Butlin R. K., Keller I., Wagner C. E., Boughman J. W., Hohenlohe P. A., et al. (2014). Genomics and the origin of species. Nat. Rev. Genet. 15 (3), 176–1192. doi: 10.1038/nrg3644
Siddiq M. A., Hua X., Liu G., Wang J. (2016). Rapid evolutionary divergence and ecotypic diversification of the Western north American Drosophila. Proc. R. Soc B: Biol. Sci. 283 (1837), 20160109.
Simão F. A., Waterhouse R. M., Ioannidis P., Kriventseva E., Zdobnov E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/BIOINFORMATICS/BTV351
Smith S. A., Dunn C. W. (2008). Phyutility: a phyloinformatics tool for trees, alignments, and molecular data. Bioinformatics 24 (5), 715–716. doi: 10.1093/bioinformatics/btn058
Smith S. A., Moore M. J., Brown J. W., Yang Y. (2015). Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15, 1–15. doi: 10.1186/S12862-015-0423-0/FIGURES/5
Soraggi S., Wiuf C., Albrechtsen A. (2017). Improved d-statistic for low-coverage data. BioRxiv, 127852. doi: 10.1101/127852
Stamatakis A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22 (21), 2688–2690. doi: 10.1093/bioinformatics/btl446
Struwe L., Haag S., Heiberg E., Grant J. R. (20092009). Andean Speciation and vicariance in neotropical macrocarpaea (GENTIANACEAE–HELIEAE). Ann. Mo. Bot. Gard 96 (3), 450–469. doi: 10.3417/2008040
Swofford D. L. (2002). PAUP: phylogenetic analysis using parsimony (and other methods), Version 4.0 Beta 10 (Sunderland, Massachusetts: Sinauer Associates).
Than C., Ruths D., Nakhleh L. (2008). PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinform. 9 (1), 1–16. doi: 10.1186/1471-2105-9-322/COMMENTS
Van Dongen S. (2008). Graph clustering Via a discrete uncoupling process. SIAM J. 30, 1. doi: 10.1137/040608635
Vargas O. M., Heuertz M., Smith S. A., Dick C. W. (2019). Target sequence capture in the Brazil nut family (Lecythidaceae): marker selection and in silico capture from genome skimming data. Mol. Phy. Evo. 135, 98–104. doi: 10.1016/J.YMPEV.2019.02.020
Wang Z., Kang M., Li J., Zhang Z., Wang Y., Chen C., et al. (2022). Genomic evidence for homoploid hybrid speciation between ancestors of two different genera. Nat. Commu 13 (1), 1–9. doi: 10.1038/s41467-022-29643-4
Weng Y. M., Veire B. M., Dudko R. Y., Medeiros M. J., Kavanaugh D. H., Schoville S. D. (2020). Rapid speciation and ecological divergence into north American alpine habitats: the Nippononebria (Coleoptera: carabidae) species complex. Bot. J. Linn. Soc 130 (1), 18–33. doi: 10.1093/BIOLINNEAN/BLAA014
Whidden C., Burch T. (2015). Quartet inference and the assessment of node support in large-scale multi-locus phylogenetic analyses. Syst. Biol. 64 (3), 421–432.
Xiao J., Lyu R., He J., Li M., Ji J., Cheng J., et al. (2022). Genome-partitioning strategy, plastid and nuclear phylogenomic discordance, and its evolutionary implications of clematis (Ranunculaceae). Front. Plant Sci. 13. doi: 10.3389/FPLS.2022.1059379
Yang Z., Smith S. A. (2014). Optimizing likelihood functions for phylogenetic inference: new algorithms and methods. Syst. Bio. 63 (2), 345–361.
Ye J., Zhang Y., Cui H., Liu J., Wu Y., Cheng Y., et al (2018). WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 46 (W1), W71–W75.
Ye J., Fang L., Zheng H., Zhang Y., Chen J., Zhang Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res., 34. doi: 10.1093/nar/gkl031
Young A. D., Gillung J. P. (2020). Phylogenomics – principles, opportunities, and pitfalls of big-data phylogenetics. Syst. Entomol. 45, 225–247. doi: 10.1111/SYEN.12406
Yu Y., Degnan J. H., Nakhleh L. (2012). The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 8, e1002660. doi: 10.1371/JOURNAL.PGEN.1002660
Yu Y., Nakhleh L. (2015). A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 16 (10), 1–10. doi: 10.1186/1471-2164-16-S10-S10/COMMENTS
Zhang C., Rabiee M., Sayyari E., Mirarab S. (2018). “ASTRAL-III: increased scalability and impacts of contracting low support branches,” in Proceedings of the 2018 ACM international conference on bioinformatics, computational biology, and health informatics, 53–62). doi: 10.1145/3233547.3233548
Zhang B. W., Xu L. L., Li N., Yan P. C., Jiang X. H., Woeste K. E., et al. (2019). Phylogenomics reveals an ancient hybrid origin of the Persian walnut. Mol. Biol. Evol. 36 (11), 2451–2461. doi: 10.1093/molbev/msz112
Keywords: interspecific gene flow, multi-species coalescent approach, introgression, ILS, reticulate phylogenies
Citation: Belintani T, Congrains C, Pinotti H, Chahad-Ehlers S, de Brito RA, Oliveira J, Frias-Lasserre D, Fontes FM and da Rosa JA (2023) Transcriptome-based phylogenomic analysis reveals evidence of ancient hybridization events in the Mepraia genus (Hemiptera: Reduviidae: Triatominae). Front. Ecol. Evol. 11:1215319. doi: 10.3389/fevo.2023.1215319
Received: 01 May 2023; Accepted: 19 June 2023;
Published: 11 July 2023.
Edited by:
Diego Baldo, CONICET Institute of Subtropical Biology (IBS), ArgentinaReviewed by:
Julieta Nattero, Universidad de Buenos Aires, ArgentinaCopyright © 2023 Belintani, Congrains, Pinotti, Chahad-Ehlers, de Brito, Oliveira, Frias-Lasserre, Fontes and da Rosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiago Belintani, dGlhZ29iZWxsaW50YW5pQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.