 Xiaoye Jin1,2†
Xiaoye Jin1,2† Zheng Ren1†
Zheng Ren1† Hongling Zhang1
Hongling Zhang1 Qiyan Wang1Yubo Liu1Jingyan Ji1
Qiyan Wang1Yubo Liu1Jingyan Ji1 Meiqing Yang1Han Zhang1Wen Hu3Ning Wang3Yicong Wang3*
Meiqing Yang1Han Zhang1Wen Hu3Ning Wang3Yicong Wang3* Jiang Huang1*
Jiang Huang1*- 1Department of Forensic Medicine, Guizhou Medical University, Guiyang, China
- 2Shanghai Key Lab of Forensic Medicine, Key Lab of Forensic Science, Ministry of Justice, Shanghai, China
- 3BGI-Shenzhen, Shenzhen, China
Multi-InDel, as the novel genetic markers, showed great potential in forensic research. Whereas, most scholars mainly focused on autosomal Multi-InDels, which might provide limited genetic information in some complex kinship cases. In this study, we selected 17 Multi-InDels on the X chromosome and developed a multiplex amplification panel based on the next-generation sequencing (NGS) technology. Genetic distributions of these 17 loci in Beijing Han, Chinese Southern Han, and the studied Guizhou Han populations revealed that most loci showed relatively high forensic application values in these Han populations. In addition, more allelic variations of some loci were observed in the Guizhou Han than those in Beijing Han and Southern Han populations. Pairwise FST values, multi-dimensional analysis, and phylogenetic tree of different continental populations showed that selected 17 loci generally could differentiate African, European, East Asian, and South Asian populations. To sum up, the developed panel in this study is not only viewed as the high-efficient supplementary tool for forensic individual identification and paternity analysis, but it is also beneficial for inferring biogeographical origins of different continental populations.
Introduction
Insertion/deletion (InDel) polymorphisms were the third genetic markers that displayed insertion or deletion of different deoxyribonucleic acid (DNA) fragments in the genome. As InDels were firstly identified by Weber et al. (2002), they have been paid a large number of attention by forensic researchers and human geneticists because they had low mutation rates and showed wide distributions in the human genome. Till now, a set of multiplex amplification panels have been developed for various forensic purposes (Chen L. et al., 2021; Jin et al., 2021; Zhang et al., 2021; Fan et al., 2022a). However, InDels commonly showed bi-allelic variations that result in their low genetic diversities in comparison with short tandem repeats (STRs). Therefore, there were defects of these bi-allelic InDels in forensic application. On the one hand, they were not conductive to mixture deconvolution; on the other hand, more InDels need to be incorporated into a multiplex amplification panel if forensic researchers wish to obtain comparable forensic efficiency of commonly used STR kits. To avoid the aforementioned shortcomings, a novel genetic marker (Multi-InDel) was proposed by Huang et al. (2014), formed by some closely linked InDels in the small molecular interval (200 bp). Multi-InDel could exhibit multiple allelic variations in the population and showed great application values in forensic individual identification and ancestry origin inferences (Sun et al., 2019; Qu et al., 2020). Nonetheless, the extant research mainly focused on Multi-InDel loci on the autosomes. These loci might provide relatively limited genetic information in some complex kinship cases like deficiency paternity cases. Hereto, genetic markers on the allosome may get more valuable insights into these complex kinship testing.
Genetic markers on the X chromosome possessed unique genetic patterns: for males, they were only transmitted from father to daughter; on the contrary, they showed a similar inheritance mode with genetic markers on the autosomes for females (Gomes et al., 2020). The specific genetic characteristics made genetic markers on the X chromosome play crucial roles in some complex kinship cases like grandparent-grandchild comparisons, half-sisters testing, paternity analyses in incest cases, and so on (Szibor, 2007). Currently, forensic workers generally employed X-STRs to paternity analysis. However, relatively high mutation rates of X-STRs might exert adverse effects on complex paternity analyses. Therefore, some X-InDel panels have been developed for forensic parentage testing (Zhang et al., 2015; Caputo et al., 2017; Chen L. et al., 2021) because InDels possess some advantageous features in comparison to STRs. In the subsequent studies, Fan et al. (2015, 2016) chose 13 Multi-InDels on the X chromosome and evaluated their forensic application values; obtained results revealed that these 13 loci were not only useful for personal identification and kinship testing, but they could also achieve ancestry resolutions of different continental populations. Even so, they proposed that more Multi-InDels need to select to obtain higher forensic effectiveness and discern genetic substructure of Chinese populations.
In the current study, we selected 17 Multi-InDels on the X chromosome. Next, genetic distributions and forensic efficiency evaluations of these loci in Chinese Beijing Han (CHB) and Southern Han (CHS) populations were conducted based on the previously reported data (Donnelly et al., 2015). Thirdly, a multiplex amplification system of these 17 loci was developed by the NGS technology and was used to genotype 217 Guizhou Han individuals in order to further evaluate its forensic application values. Finally, population genetic analyses of different continental populations were performed to assess the power of these loci to discriminate these continental populations.
Materials and methods
Sample information
We collected bloodstain samples of 217 unrelated healthy Guizhou Han individuals who have lived in Guizhou for at least three generations. All individual participants in this study provided their written informed consent. In addition, genetic data of selected loci in different continental populations were downloaded from 1000 Genome Project Phase III (Donnelly et al., 2015) to evaluate their genetic distributions. Only males were assembled because we could directly determine haplotype information of different InDels in the short DNA region. The general information of 26 reference population like CHB, CHS, and so on was given in Supplementary Table 1. This study was performed in line with the guidelines of the Guizhou Medical University ethics committee and warranted by the Guizhou Medical University ethics committee.
Selection of multi-insertion/deletion loci on the X chromosome
Multi-InDel loci on the X chromosome were selected based on previous reports (Donnelly et al., 2015; Fan et al., 2015) according to the following criteria: (1). physical distances between InDels on the short molecular interval were less than 200 bp; (2). fragment length of insertion/deletion was less than 30 bp; (3). the minor allelic frequencies of InDels in Chinese Han populations were greater than 0.1; (4). InDels on the short molecular interval (200 bp) showed different allelic frequency distributions. (5). Multi-InDel loci on the X chromosome were 1 Mb apart from each other.
Primer design and multiplex polymerase chain reaction of selected X-chromosomal multi-insertion/deletion
Primer sequences of selected X-chromosomal Multi-InDels were designed by the ATOPlex online tool.1 Detailed primer information was presented in Supplementary Table 2.
We conducted two steps of polymerase chain reaction (PCR) to construct the sequencing library. Firstly, one 1.2 mm bloodstain card from each individual was obtained by the punch and pro-processed with 25 μL Clean Buffer at 60°C for 10 min. After removing 16.5 μL Clean Buffer, we added 16.5 μL PCR cocktail including 12.5 μL PCR Enzyme Mix and 4 μL PCR Primer Pool. Next, the PCR mixture was conducted to thermal cycle reaction on the 9700 Thermal cycle PCR System (Thermo Fisher Scientific, MA, USA). The detailed reaction conditions were listed as follows: an initial pre-denaturation at 98°C for 5 min; 14 cycles of 98°C for 15 s, 64°C for 1 min, 60°C for 1 min, and 72°C for 30 s; 72°C for 2 min. And then we purified the amplified product by the MagBead DNA Purification kit (CWBIO, Beijing, China) according to the kit’s instructions. We used 6.5 μL TE Buffer to wash the purified DNA off and added second round PCR reagents comprising 12.5 μL PCR Enzyme Mix, 2 μL PCR Block, 2 μL PCR Dual Barcode Primer F, and 2 μL PCR Dual Barcode Primer R. The 9700 Thermal cycle PCR System was also used to conduct thermal cycle reaction according to the below parameters: 98°C for 5 min; 16 cycles of 98°C for 15 s, 64°C for 30 s, 60°C for 30 s, and 72°C for 30 s; 72°C for 2 min. Finally, the amplified product was also purified by the same method mentioned above.
Deoxyribonucleic acid sequencing and data analysis
DNA library was quantitated by the Quant-iT™ PicoGreen dsDNA Assay kit (Thermo Fisher Scientific, MA, USA). Based on DNA quantitation results, DNA libraries of all samples were mixed into a well (30 ng). We constructed the DNA Nano Ball (DNB) by the DNBSEQ OneStep DNB Make Reagent kit (MGI, Shenzhen, China) following its recommended specifications (Li et al., 2021; Fan et al., 2022b).
MGISEQ-2000RS sequencing platform was used to perform DNA sequencing of DNB with the mode of SE400 + 10 + 10. Raw data were processed to filter low-quality sequences and reads with multiple N bases by the Soapnuke software (Chen et al., 2018). Clean data were aligned to hs37d5 reference sequence by the Burrows-Wheeler Alignment software (Li and Durbin, 2009). Next, we used GATK HaplotypeCaller (Schmidt et al., 2010) with the -ERC GVCF parameter to generate GVCF files, and then used GATK CombineGVCFs with default parameters to combine the potential variants and joint genotyping. The mutations with depth more than 100 and frequency more than 0.2 were allowed. Analytical threshold and interpretation threshold were depth of coverage (DoC) × 1.5% and DoC × 4.5% if DoC were greater than 650; if not, they were 15 × and 30 ×, respectively.
Statistical analyses
Allelic frequencies and forensic related parameters of 17 Multi-InDels in CHB, CHS, and studied Guizhou Han populations were calculated by the StatsX v2.0 software (Lang et al., 2019). Further, the number of alleles and forensic parameters of these 17 loci in three Han populations were visually shown by R v4.1.0 and TBtools v1.0 software (Chen et al., 2020). Linkage disequilibrium analyses of 17 Multi-InDels in the studied Guizhou Han population were conducted by the STRAF online tool (Gouy and Zieger, 2017). Next, forensic parameters of two linkage groups were also estimated by the StatsX. Fixation index (FST) values of 17 Multi-InDels between different continental populations were calculated by the Arlequin v3.5.2.2 software (Excoffier and Lischer, 2010). Besides, we also estimated pairwise FST values of different continental populations by the Arlequin. Based on pairwise FST values, multi-dimensional analysis and phylogenetic tree of these continental populations were conducted by SPSS v18 and MEGA v11.0.9 (Kumar et al., 2018) software, respectively.
Results and discussion
Loci information
In this study, we screened 18 X chromosomal Multi-InDel loci based on established selection conditions. Even so, we found that two Multi-InDel loci (rs112111922_rs35401470 and rs201707878_rs71943052) showed complex sequences in their neighboring regions, which were hard to determine their genotype. In addition, rs58222634 locus of rs58222634_rs200362185 displayed a large number of di-nucleotide repeats, which brought about many noise reads. Thus, the rs58222634 locus and two Multi-InDels were eliminated from the developed multiplex panel. Moreover, we also added a multi-allelic InDel (rs78613336) locus into the developed system. Finally, we successfully developed the multiplex amplification system of 17 Multi-InDels. Loci information and physical locations of these loci were given in Table 1 and Figure 1A.
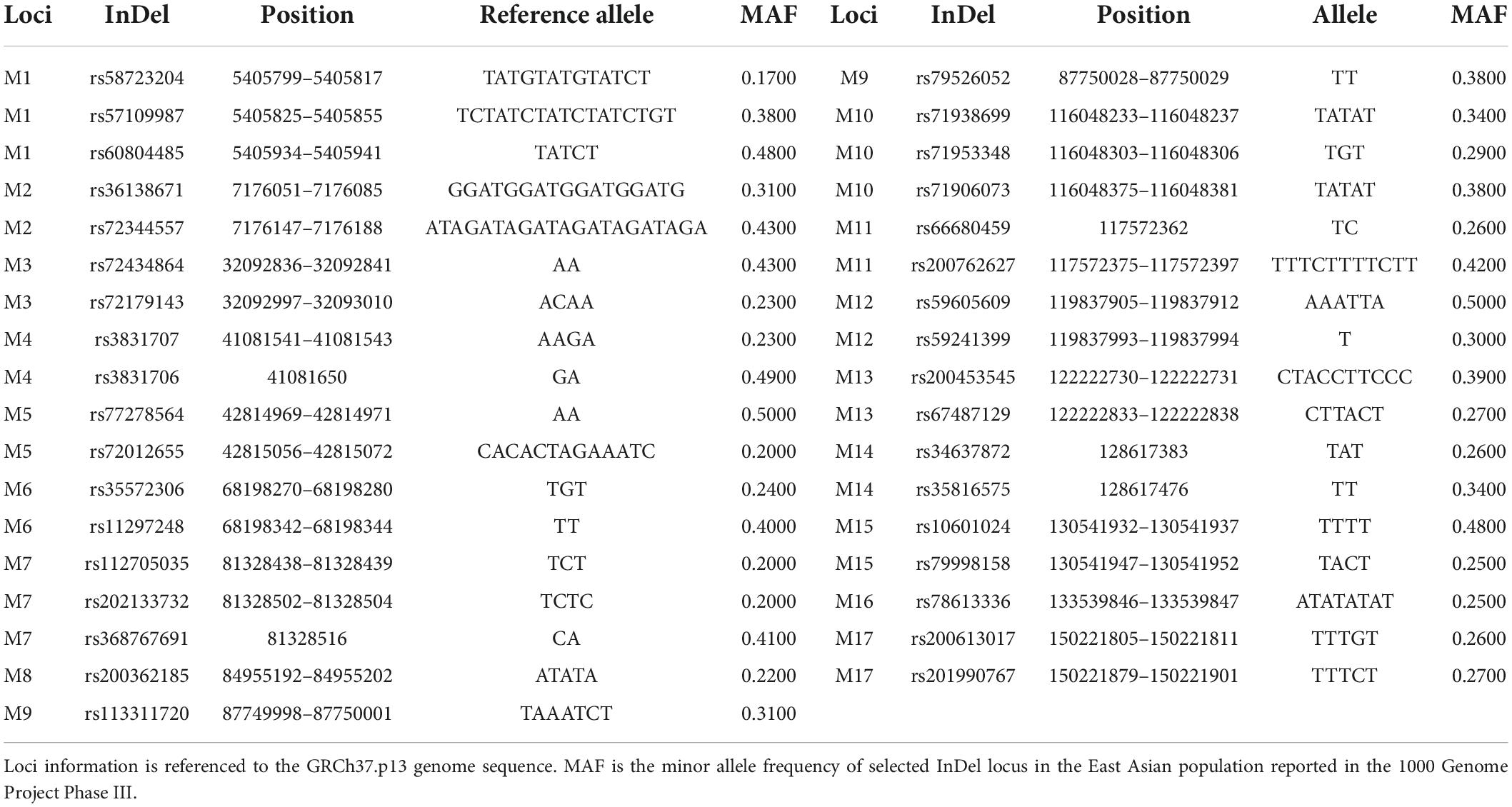
Table 1. General information of 17 selected Multi-InDel loci on the X chromosome.
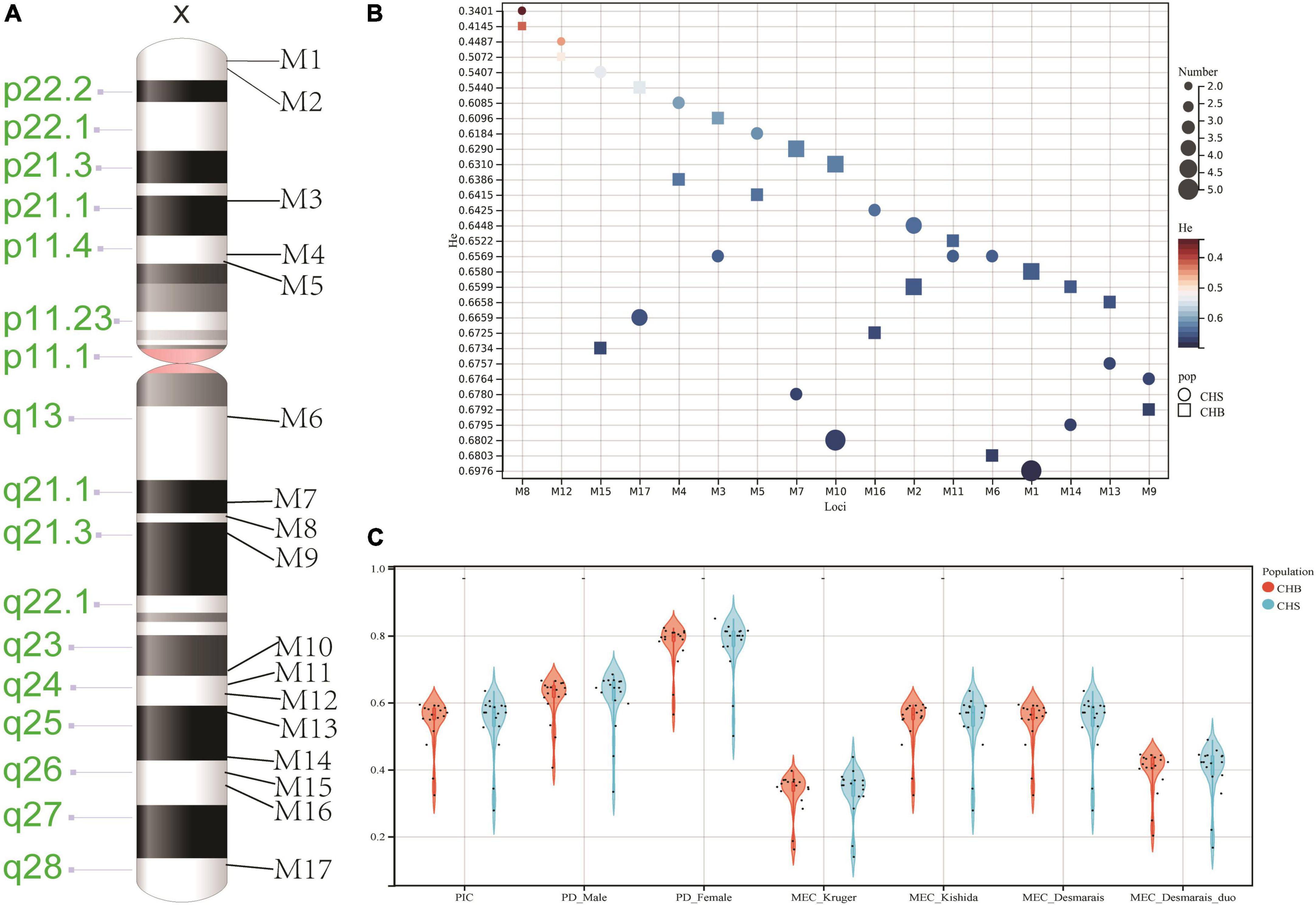
Figure 1. Physical locations (A), genetic diversities (B), and forensic efficiency evaluations (C) of 17 X-chromosomal Multi-InDels in Beijing Han (CHB) and Southern Han (CHS) populations. For genetic diversities, different colors represented different expected heterozygosity (He) values: red and blue denote small and large He values, respectively; the size of shape is proportional to the number of alleles observed in each locus.
Forensic efficiency evaluations of selected 17 loci in training data
Based on the selected 17 loci on the X chromosome, we firstly assessed the number of alleles and expected heterozygosities (He) of these loci in training data (CHB and CHS), as shown in Figure 1B. Not surprisingly, the M8 locus showed the lowest He and the least number of alleles in CHB and CHS populations since it only included one bi-allelic InDel (rs200362185). In addition, the rs59241399 locus of M12 was not reported in 1000 Genome Project Phase III. Therefore, we also observed that M12 locus showed two allelic variations in CHB and CHS populations. For the remaining 15 loci, more than two alleles could be observed in these loci, especially for M1 and M10. More importantly, most loci displayed relatively high He values (>0.5) in CHB and CHS populations, indicating they showed relatively high genetic diversities in these two Han populations.
Forensic-related parameters of these 17 loci in CHB and CHS populations were also estimated, as given in Figure 1C and Supplementary Table 3. In the CHB population, polymorphic information content (PIC) values of most loci were greater than 0.5 except M8, M12, and M17 loci. Power of discrimination in male (PD_M) and female (PD_F) for these 17 loci ranged from 0.4055 (M8) to 0.6655 (M6) and 0.5643 (M8) to 0.8231 (M1), respectively. Mean exclusion chance of these 17 loci in deficiency cases according to Krüger et al. (1968) (MEC_Krüger) and standard trios according to Kishida et al. (1997) (MEC_Kishida) distributed from 0.1616 (M8) to 0.3961 (M1) and 0.3233 (M8) to 0.5939 (M1), respectively. Further, we also calculated MEC in duos (MEC_Desmarais_duo) and trios (MEC_Desmarais) according to Desmarais’s study (Desmarais et al., 1998). The average MEC_Desmarais_duo and MEC_Desmarais values of 17 loci were 0.3927 and 0.5373, respectively. Cumulative PD_M, PD_F, MEC_Krüger, MEC_Kishida, MEC_Desmarais_duo, and MEC_Desmarais of these 17 loci in the CHB population were 0.9999999138, 0.99999999999236, 0.99897, 0.999998, 0.99981, and 0.999998, respectively. In the CHS population, similar results could be discerned from these forensic parameters. The average PIC, PD_M, PD_F, MEC_Krüger, MEC_Kishida, MEC_Desmarais_duo, and MEC_Desmarais values of these 17 loci were 0.5394, 0.6097, 0.7692, 0.3340, 0.5394, 0.3955, and 0.5394, respectively. The M1 locus showed the highest forensic application values; whereas, the M8 locus demonstrated the lowest forensic efficiencies. Cumulative PD_M, PD_F, MEC_Krüger, MEC_Kishida, MEC_Desmarais_duo, and MEC_Desmarais values of these 17 loci in the CHS population were 0.9999999212, 0.99999999999377, 0.99909, 0.99999856, 0.999833, and 0.99999856, respectively. In a nutshell, we proposed that these 17 loci could be used for forensic individual identification and paternity analyses in CHB and CHS populations since they showed high cumulative PD and MEC values.
Genetic distributions and forensic-related parameters of selected 17 loci in the studied Guizhou Han population
Even though capillary electrophoresis was widely used in forensic practice, it only detected length-based genetic variants that might reduce genetic diversities of studied genetic markers to some extent. Instead, not only can NGS detect length-based genetic variants, it can also discern sequence-based and additional genetic variants in surrounding regions of targeted markers (Børsting and Morling, 2015). In a previous study, Chen C. et al. (2021) investigated genetic polymorphisms of 231 genetic markers in the Chinese Hui group by the NGS, and they found that sequence-based genetic markers could show more allelic variations than length-based genetic markers. Thus, we developed the multiplex amplification system of 17 X chromosomal Multi-InDels based on the NGS technology. In this study, we chose the MGISEQ-2000RS sequencing platform to construct the multiplex amplification panel because previous studies pointed out that the MGISEQ-2000RS showed comparable or better performance than Illumina sequencing platforms (Jeon et al., 2021; Zhu et al., 2021).
Quality control results of 217 Guizhou Han individuals were presented in Supplementary Table 4. We found that Q20, Q30, and Coverage > = 100 of these individuals were greater than 0.89, 0.80, and 0.94, respectively. In addition, we also displayed DoC values of selected 17 loci in the Guizhou Han population (Figure 2A). Results revealed that most loci showed high DoC values, implying the developed panel possessed relatively balanced amplification performance. Even so, we found that the M10 locus displayed low DoC values in comparison to other loci.
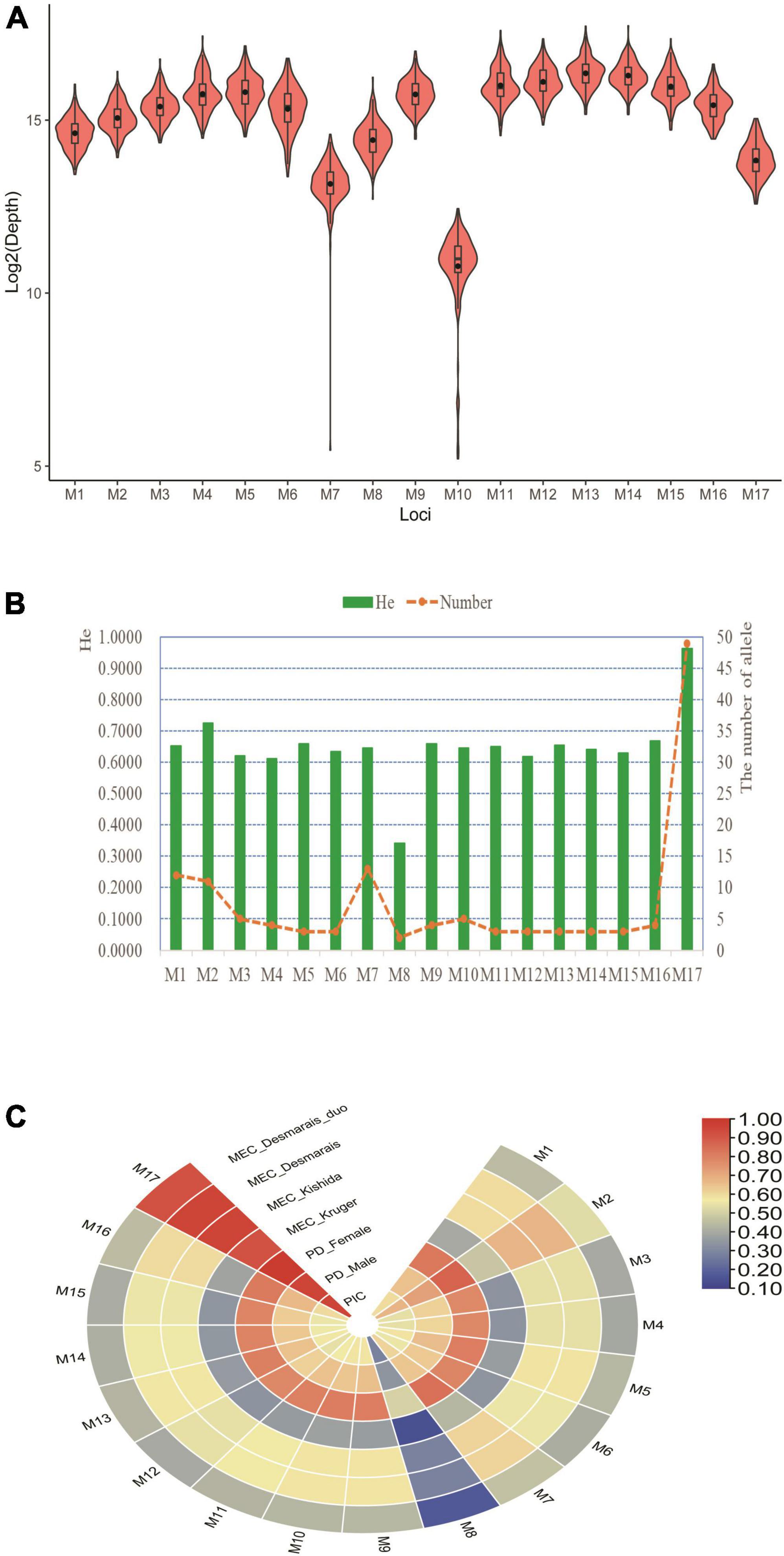
Figure 2. Depths of coverage (A), expected heterozygosities (B), and forensic parameters (C) of 17 X-chromosomal Multi-InDels in the studied Guizhou Han population.
The number of alleles and He values of these 17 loci in the Guizhou Han population were shown in Figure 2B and Supplementary Table 5. We found that most loci showed at least three allelic variations except the M8 locus. In addition, more than 10 alleles were observed at M1, M2, M7, and M17 loci. Compared to results from CHB and CHS populations, some loci showed more allelic variations in the studied population. We thought that NGS could detect potential sequence variations and other variations in the neighboring area, which made these loci possess higher genetic diversities. In addition, we observed that He values of 16 loci were larger than 0.5, implying that these loci exhibited relatively high genetic polymorphisms in the studied Han populations. Forensic-related parameters of these 17 loci were displayed in Figure 2C and Supplementary Table 5. The average PIC, PD_Male, PD_Female, MEC_Kruger, MEC_Kishida, MEC_Desmarais, and MEC_Desmarais_duo values of these 17 loci were 0.5828, 0.6449, 0.7999, 0.3875, 0.5827, 0.5828, and 0.4440, respectively. Besides, we also assessed linkage disequilibrium of these 17 loci in the Guizhou Han population (Supplementary Table 6). After applying Bonferroni correction (P = 0.05/136 = 0.0004), two linkage groups (LGs) were discerned from the studied Guizhou Han group. Among these two linkage groups, the number of haplotypes observed in LG1 (M2 and M3 loci) and LG2 (M10 and M17 loci) were 22 and 94, respectively. Haplotype diversities, PIC, PD_Male, PD_Female, MEC_Kruger, MEC_Kishida, MEC_Desmarais, and MEC_Desmarais_duo of LG1 were 0.8930, 0.8790, 0.8889, 0.9778, 0.7777, 0.8789, 0.8790, and 0.7936, respectively; and they were 0.9860, 0.9811, 0.9815, 0.9993, 0.9614, 0.9797, 0.9811, and 0.9634 at the LG2 locus, respectively. Next, we evaluated the cumulative forensic efficiency of two LGs and the remaining 13 X-chromosomal Multi-InDels in the Guizhou Han population. Accumulative PD_Male, PD_Female, MEC_Kruger, MEC_Kishida, MEC_Desmarais, and MEC_Desmarais_duo were 0.999999993691, 0.999999999999976, 0.999967, 0.999999936, 0.99999994, and 0.99999186, respectively. Compared to 17 autosomal Multi-InDels, 13 X chromosomal Multi-InDels, and 38 X-InDels (Fan et al., 2015; Qu et al., 2020; Chen L. et al., 2021), we found that these 17 loci presented in this study showed higher forensic application values in individual identification and paternity analysis, implying that the developed panel could be viewed as a more valuable tool for forensic research, especially for those complex paternity testing cases. Nonetheless, we found that the M8 locus showed relatively low genetic diversity in the Guizhou Han population. Besides, the M10 locus exhibited relatively low DoC values in comparison to other loci. Therefore, the developed system needs to be further improved in the future. Furthermore, developmental validation of the novel panel including mixture deconvolution, species specificity, stability, and concordance studies should be performed in the following study.
Population genetic analyses of different continental populations based on selected 17 loci
In a previous study, Fan et al. (2016) explored hierarchical genetic structure of different continental populations via 13 X-chromosomal Multi-InDels and found that these 13 loci could be used to differentiate East Asian, European, and African populations. Further, they also stated that Multi-InDels might be more appropriate for inferring biogeographical origins of different continental populations than multi-allelic InDels and STRs (Fan et al., 2016). Therefore, we also evaluated the power of these 17 loci to differentiate continental populations.
As shown in Figure 3A, we found that those populations from the same continent showed low FST values; whereas, populations from different continents had large FST values. Next, an MDS was also plotted based on pairwise FST values (Figure 3B). We found that these 26 populations formed four population clusters: five European populations located in the left bottom region; seven African populations positioned in the right area; five East Asian populations situated in the left top region; five South Asian populations clustered closely and distributed between East Asian and European populations. In addition, four American populations scattered among South Asian and East Asian populations. The phylogenetic tree of these 26 populations was given in Figure 3C. Similar to population distributions in MDS, we found that those populations from the same continent formed a branch except American populations. For American populations, abnormal population distribution patterns in MDS and phylogenetic tree might be related to their complex genetic components. On the one hand, the study about ancient DNA revealed that there were western Eurasian genetic signatures in modern-day Native Americans (Raghavan et al., 2014). On the other hand, four populations (MXL, PEL, PUR, and CLM) from American possessed different ancestral components from African, European, and indigenous American populations (Donnelly et al., 2015).
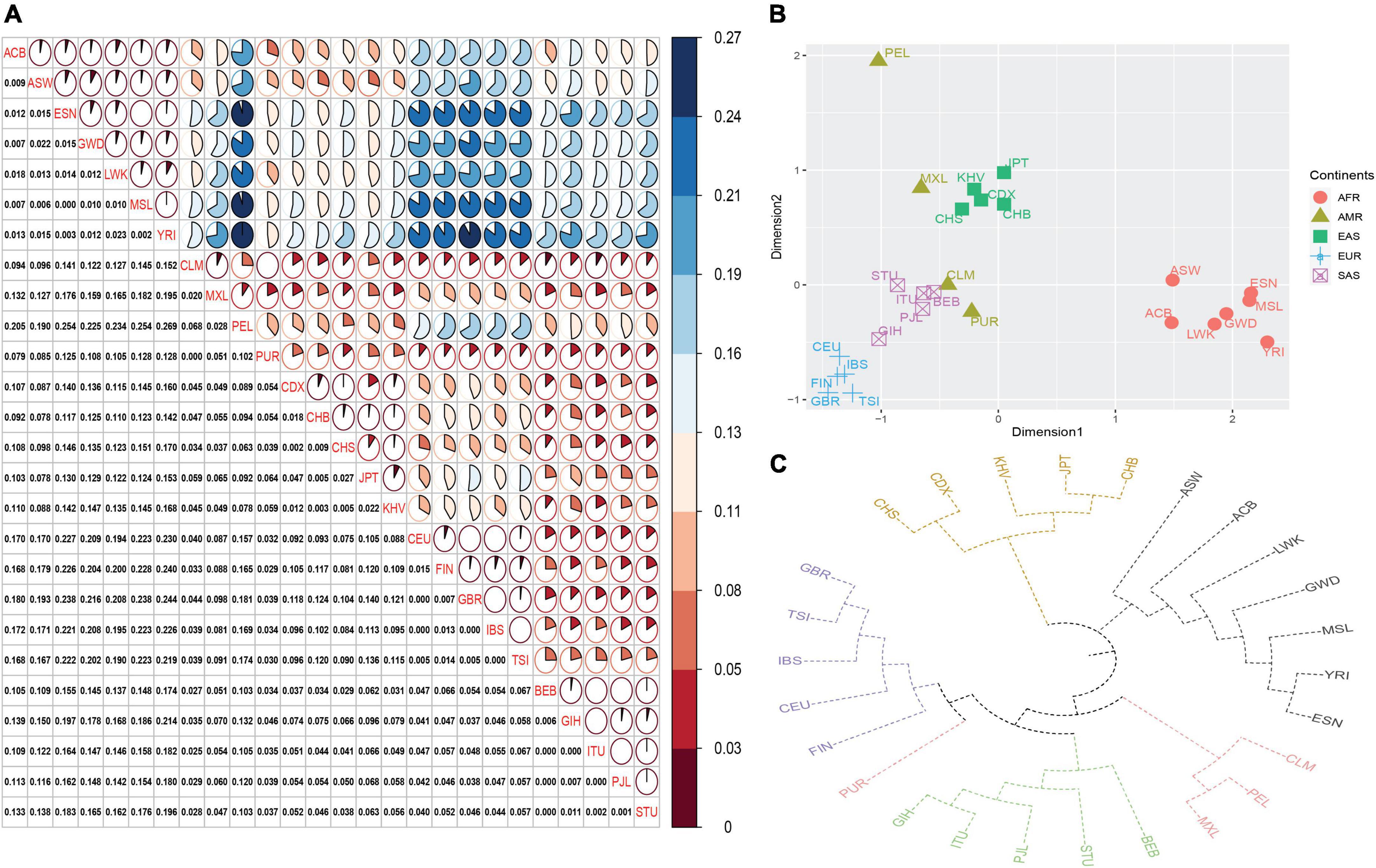
Figure 3. Population genetic analyses of different continental populations based on selected 17 loci. (A) Pairwise FST distances of different continental populations. Different colors represented different FST values: red denoted small FST values; blue denoted large FST values. (B) Multi-dimensional analysis of different continental populations. (C) The phylogenetic tree of different continental populations.
To further discern those loci showing large genetic differentiations among different continental populations, we also estimated pairwise FST values of each locus between different populations, as given in Supplementary Table 7. A previous study stated that genetic markers with high FST values between compared populations were conductive to differentiating these two populations (Phillips, 2015). We found that M2, M5, M6, M11, M14, and M15 loci displayed relatively high FST values (>0.1) between African population and other continental populations, implying that these loci could be viewed as potential ancestry informative markers for inferring biogeographical origins of African populations. Likewise, M14 locus showed relatively high FST values between East Asian and European, American, and South Asian populations; M13 locus displayed relatively high FST values between European and American and South Asian populations, which indicated that these loci could be utilized for differentiating these continental populations. In the following study, we need to further evaluate whether these loci can discern population substructure of Chinese different ethnic groups.
Conclusion
To conclude, we developed a novel multiplex amplification panel of 17 X-chromosomal Multi-InDels via the NGS platform. The majority of these loci showed relatively high genetic diversities in CHB, CHS, and studied Han populations, and they can be viewed as a valuable supplementary tool for forensic personal identification and paternity analyses, especially for those deficiency cases. In addition, we found that some out of these 17 loci were also beneficial to differentiating African, European, East Asian, and South Asian populations.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: CNGB Sequence Archive (CNSA) of the China National Genebank Database, accession number CNP0003564.
Ethics statement
The studies involving human participants were reviewed and approved by the Guizhou Medical University Ethics Committee. The patients/participants provided their written informed consent to participate in this study.
Author contributions
XJ and ZR wrote this manuscript. HLZ, QW, YL, and JJ collected samples and performed the experiment. MY, HZ, WH, and NW conducted data analysis. YW and JH designed the work and provided the conception. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Guizhou Provincial Science and Technology Projects (No. ZK [2022] General 355), the Guizhou Education Department Young Scientific and Technical Talents Project, Qian Education KY (No. [2022] 215), the Guizhou Scientific Support Project, Qian Science Support (No. [2021] General 448), the Shanghai Key Lab of Forensic Medicine, the Key Lab of Forensic Science, Ministry of Justice, China (Academy of Forensic Science), Open Project, (No. KF202207), the Guizhou Province Education Department, Characteristic Region Project, Qian Education KY (No. [2021] 065), the Guizhou “Hundred” High-level Innovative Talent Project, Qian Science Platform Talents (No. [2020] 6012), the Guizhou Scientific Support Project, Qian Science Support (No. [2020] 4Y057), the Guizhou Science Project, Qian Science Foundation (No. [2020] 1Y353), the Guizhou Scientific Support Project, Qian Science Support (2019) 2825, the Guizhou Scientific Cultivation Project, Qian Science Platform Talent (No. [2018] 5779-X), the Guizhou Engineering Technology Research Center Project, the Qian High-Tech of Development and Reform Commission (No. [2016] 1345), the Guizhou Innovation training program for college students (No. [2019] 5200926), and the National Natural Science Foundation of China (No. 82160324).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.985933/full#supplementary-material
Footnotes
References
Børsting, C., and Morling, N. (2015). Next generation sequencing and its applications in forensic genetics. Forensic Sci. Int. Genet. 18, 78–89. doi: 10.1016/j.fsigen.2015.02.002
Caputo, M., Amador, M. A., Santos, S., and Corach, D. (2017). Potential forensic use of a 33 X-InDel panel in the Argentinean population. Int. J. Legal Med. 131, 107–112. doi: 10.1007/s00414-016-1399-z
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, C., Jin, X., Zhang, X., Zhang, W., Guo, Y., Tao, R., et al. (2021). Comprehensive insights into forensic features and genetic background of chinese northwest Hui group using six distinct categories of 231 molecular markers. Front. Genet. 12:705753. doi: 10.3389/fgene.2021.705753
Chen, L., Pan, X., Wang, Y., Du, W., Wu, W., Tang, Z., et al. (2021). Development and validation of a forensic multiplex system with 38 X-InDel loci. Front. Genet. 12:670482. doi: 10.3389/fgene.2021.670482
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6. doi: 10.1093/gigascience/gix120
Desmarais, D., Zhong, Y., Chakraborty, R., Perreault, C., and Busque, L. (1998). Development of a highly polymorphic STR marker for identity testing purposes at the human androgen receptor gene (HUMARA). J. Forensic Sci. 43:14355J. doi: 10.1520/jfs14355j
Donnelly, P., Green, E. D., Knoppers, B. M., Mardis, E. R., Nickerson, D. A., Wilson, R. K., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Fan, G., Ye, Y., Luo, H., and Hou, Y. (2015). Use of multi-InDels as novel markers to analyze 13 X-chromosome haplotype loci for forensic purposes. Electrophoresis 36, 2931–2938. doi: 10.1002/elps.201500159
Fan, G. Y., Ye, Y., and Hou, Y. P. (2016). Detecting a hierarchical genetic population structure via Multi-InDel markers on the X chromosome. Sci. Rep. 6:32178. doi: 10.1038/srep32178
Fan, H., He, Y., Li, S., Xie, Q., Wang, F., Du, Z., et al. (2022a). Systematic evaluation of a novel 6-dye direct and multiplex PCR-CE-based InDel typing system for forensic purposes. Front. Genet. 12:744645. doi: 10.3389/fgene.2021.744645
Fan, H., Wang, L., Liu, C., Lu, X., Xu, X., Ru, K., et al. (2022b). Development and validation of a novel 133-plex forensic STR panel (52 STRs and 81 Y-STRs) using single-end 400 bp massive parallel sequencing. Int. J. Legal Med. 136, 447–464. doi: 10.1007/s00414-021-02738-1
Gomes, I., Pinto, N., Antão-Sousa, S., Gomes, V., Gusmão, L., and Amorim, A. (2020). Twenty years later: a comprehensive review of the X chromosome use in forensic genetics. Front. Genet. 11:926. doi: 10.3389/fgene.2020.00926
Gouy, A., and Zieger, M. (2017). STRAF—A convenient online tool for STR data evaluation in forensic genetics. Forensic Sci. Int. Genet. 30, 148–151. doi: 10.1016/j.fsigen.2017.07.007
Huang, J., Luo, H., Wei, W., and Hou, Y. (2014). A novel method for the analysis of 20 multi-Indel polymorphisms and its forensic application. Electrophoresis 35, 487–493. doi: 10.1002/elps.201300346
Jeon, S. A., Park, J. L., Park, S. J., Kim, J. H., Goh, S. H., Han, J. Y., et al. (2021). Comparison between MGI and Illumina sequencing platforms for whole genome sequencing. Genes Genom. 43, 713–724. doi: 10.1007/s13258-021-01096-x
Jin, R., Cui, W., Fang, Y., Jin, X., Wang, H., Lan, Q., et al. (2021). A novel panel of 43 insertion/deletion loci for human identifications of forensic degraded DNA samples: development and validation. Front. Genet. 12:610540. doi: 10.3389/fgene.2021.610540
Kishida, T., Wang, W., Fukuda, M., and Tamaki, Y. (1997). Duplex PCR of the Y-27H39 and HPRT loci with reference to japanese population data on the HPRT locus. Japanese J. Leg. Med. 51, 67–69.
Krüger, J., Fuhrmann, W., Lichte, K., and Steffens, C. (1968). On the utilization of erythrocyte acid phosphatase polymorphism in paternity evaluation. Dtsch Z Gesamte Gerichtl Med. 64, 127–146.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lang, Y., Guo, F., and Niu, Q. (2019). StatsX v2.0: the interactive graphical software for population statistics on X-STR. Int. J. Legal Med. 133, 39–44. doi: 10.1007/s00414-018-1824-6
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760.
Li, R., Shen, X., Chen, H., Peng, D., Wu, R., and Sun, H. (2021). Developmental validation of the MGIEasy signature identification library prep kit, an all-in-one multiplex system for forensic applications. Int. J. Legal Med. 135, 739–753. doi: 10.1007/s00414-021-02507-0
Phillips, C. (2015). Forensic genetic analysis of bio-geographical ancestry. Forensic Sci. Int. Genet. 18, 49–65. doi: 10.1016/j.fsigen.2015.05.012
Qu, S., Lv, M., Xue, J., Zhu, J., Wang, L., Jian, H., et al. (2020). Multi-indel: a microhaplotype marker can be typed using capillary electrophoresis platforms. Front. Genet. 11:567082. doi: 10.3389/fgene.2020.567082
Raghavan, M., Skoglund, P., Graf, K. E., Metspalu, M., Albrechtsen, A., Moltke, I., et al. (2014). Upper palaeolithic Siberian genome reveals dual ancestry of native Americans. Nature 505, 87–91. doi: 10.1038/nature12736
Schmidt, S., McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Sun, K., Yun, L., Zhang, C., Shao, C., Gao, T., Zhao, Z., et al. (2019). Evaluation of 12 Multi-InDel markers for forensic ancestry prediction in Asian populations. Forensic Sci. Int. Genet. 43:102155. doi: 10.1016/j.fsigen.2019.102155
Szibor, R. (2007). X-chromosomal markers: past, present and future. Forensic Sci. Int. Genet. 1, 93–99. doi: 10.1016/j.fsigen.2007.03.003
Weber, J. L., David, D., Heil, J., Fan, Y., Zhao, C., and Marth, G. (2002). Human diallelic insertion/deletion polymorphisms. Am. J. Hum. Genet. 71, 854–862. doi: 10.1086/342727
Zhang, S., Sun, K., Bian, Y., Zhao, Q., Wang, Z., Ji, C., et al. (2015). Developmental validation of an X-Insertion/Deletion polymorphism panel and application in HAN population of China. Sci. Rep. 5:18336. doi: 10.1038/srep18336
Zhang, X., Shen, C., Jin, X., Guo, Y., Xie, T., and Zhu, B. (2021). Developmental validations of a self-developed 39 AIM-InDel panel and its forensic efficiency evaluations in the Shaanxi Han population. Int. J. Legal Med. 135, 1359–1367. doi: 10.1007/s00414-021-02600-4
Keywords: Multi-InDel, X chromosome, NGS, forensic research, biogeographical origin inference
Citation: Jin X, Ren Z, Zhang H, Wang Q, Liu Y, Ji J, Yang M, Zhang H, Hu W, Wang N, Wang Y and Huang J (2022) Development and forensic efficiency evaluations of a novel multiplex amplification panel of 17 Multi-InDel loci on the X chromosome. Front. Ecol. Evol. 10:985933. doi: 10.3389/fevo.2022.985933
Received: 04 July 2022; Accepted: 15 September 2022;
Published: 21 October 2022.
Edited by:
Guanglin He, Sichuan University, ChinaCopyright © 2022 Jin, Ren, Zhang, Wang, Liu, Ji, Yang, Zhang, Hu, Wang, Wang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yicong Wang, d2FuZ3lpY29uZ0BnZW5vbWljcy5jbg==; Jiang Huang, bW1tX2hqQDEyNi5jb20=
†These authors have contributed equally to this work