
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 27 July 2022
Sec. Behavioral and Evolutionary Ecology
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.967692
This article is part of the Research TopicSeed Behavior in Response to Extreme EnvironmentsView all 7 articles
 Dong Liu1†
Dong Liu1† Liang Lin1†
Liang Lin1† Tie Zhang2
Tie Zhang2 Qian Xu1
Qian Xu1 Mulan Wang1
Mulan Wang1 Minghai Gao3
Minghai Gao3 Parag Bhople4
Parag Bhople4 Hugh W. Pritchard1
Hugh W. Pritchard1 Xiangyun Yang1*
Xiangyun Yang1* Fuqiang Yu1*
Fuqiang Yu1*The seed microbiome of crop wild relatives is a potential reservoir of beneficial traits that potentially improve their host plant resilience to fluctuating environments and pathogenic threats. Herein, we studied the seed microbiome of three species of the medicinal genus Panax (P. vietnamensis, P. japonicas, and P. stipuleanatus) collected from seven locations in Southwest China. We used qPCR and metabarcoding high-throughput sequencing to target both endophytic bacteria and fungi. Seed bacterial absolute abundance (1.1 × 109∼1.0 × 107 gene copy numbers per gram seed) was substantially higher than that of fungi (7.6 × 105∼3.7 × 102). Host plant genotype was the main driver of seed microbiome composition for both bacteria and fungi. Panax growing hypothermal environments significantly shaped their seed endophytic bacterial but not fungal microbiota. The three Panax species’ seeds harbored unique microbes [averaged ∼150 amplicon sequence variants (ASVs)], sharing only 12 bacterial ASVs (half affiliated to Halomonas) and four fungal ASVs. Network analysis showed that the Panax seed endophytic bacteria tend to form inter-weaved functional modules that are majorly connected by core members from the genus Halomonas, Pseudomonas, and Pantoea. These genera have been associated with nutrient cycling, plant, disease suppression, and tolerance to environmental fluctuation. Together, these novel insights may shade light on the ecological strategies of wild Panax plants adaptation to their thermal environment by possessing abundant beneficial seed endophytic bacteria.
The genus Panax is widely known for its medicinal plants that benefit human health (Wang et al., 2006; Kochkin et al., 2013). Panax consists of 19 species, distributed mainly in eastern Asia (17 species) and also in North America (two species) (Pandey and Ali, 2012). Among them, P. quinquefolium and P. ginseng are distributed at high latitudes while P. notoginseng grows at low latitudes. The species P. vietnamensis, P. japonicas, and P. stipuleanatus (ripe for harvest in August) are wild relatives of P. notoginseng (that ripens in December). P. notoginseng has a therapeutic effect on cardiovascular and coronary artery diseases, and a long history of cultivation (Christensen, 2008; Yang et al., 2015). To meet increasing consumer demand and improve yield, the intensive cultivation of Panax has diminished soil quality as a result of the excessive application of fertilizers, soil nutrient imbalance, a decrease in microbial diversity and the development of diseases (Singh and Gupta, 2018). Therefore, efforts have been made to shift plant cultivation from the field to the forest (Shi et al., 2021), and to apply bio-organic fertilizers (Shi et al., 2022). Previous studies on Panax microbes have focused on the rhizosphere (Shi et al., 2021, 2022), taxa differences in the bacterial and fungal endophytes among plant tissues (Hong et al., 2018a), as well as growing stages (Hong et al., 2018b). In contrast, little attention has been paid to Panax seed microbes.
Spermatophytes dominate the plant kingdom and have developed a special strategy of adapting to harsh environments such as evolving low seed mass with wind or water dispersal mode, or transmitting beneficial seed microbes from plant generation to another (Shade et al., 2017; Liu et al., 2022). Plentiful studies have shown that there are a great diversity of endophytic microorganisms, some of which stimulated seed germination, seedling health and growth (Shade et al., 2017; Orozco-Mosqueda and Santoyo, 2020; Liu et al., 2022). Understanding the factors that affect the presence of key microbial taxa can improve our ability to modify the plant microbiome and improve host plant fitness (Abdullaeva et al., 2020; Rego et al., 2020; Kusstatscher et al., 2021; Shao et al., 2021; Walsh et al., 2021). Seed endophytes are dynamic and highly diverse (Shade et al., 2017; Liu et al., 2022). The identification of shared and keystone microbes could shed light on microbial consortia and their interactive roles (Orgiazzi et al., 2013; Bjork et al., 2018). Apart from traditional Venn (shared/unique) microbe analysis, network analysis has been widely-used to (i) identify core microbial members that are essential in maintaining microbiome community structure and function (Pold and Deangelis, 2013; Banerjee et al., 2018); and (ii) visualize highly associated microbes that occur as functional modules, such as for carbon degradation or nitrogen fixation (Zhou et al., 2010; Turner et al., 2013).
The seed microbiome is normally acquired by vertical transmission between plant generations, and from the soil environment (Gundel et al., 2011). Seed endophytic bacterial microbiota are relatively conservative and change with its host plant genotype (Oehrle et al., 2000; Bacilio-Jiménez et al., 2001; Cottyn et al., 2001; Johnston-Monje and Raizada, 2011). In contrast, the seed endophytic fungi are more susceptible and mainly influenced by host plant growing soil conditions (Haimin et al., 2018; Hill et al., 2021). An assessment of the effect of the environment and plant genotype on the seed microbiome of eight Brassica napus lines, revealed that seed microbial communities were predominantly shaped by the environment, while the genotype only accounted for ∼10% of seed microbiota variance (Moreira et al., 2021). Therefore, plants of the same genus probably harbor larger differences in seed fungi as a consequence of the diverse geographic localities of their host plants.
Microbial influence is of great importance during the earliest phases of plant development, adaptive evolution, and fitness. Thus, the main objectives of this study were to (i) explore the absolute abundance, diversity, composition and core taxa of seed bacteria and fungi in Panax seeds, of species able to survive in harsh hypothermal environment; (ii) reveal the relative importance of geographic location and host plant genotype on the seed microbiome. To do so, we collected three wild Panax species from seven localities, including P. vietnamensis (four sites), P. japonicus (two sites), and P. stipuleanatus (1 site), used quantitative real-time PCR, 16S and ITS rRNA gene sequencing, and network analysis, to investigate comprehensive variations in the seed microbiome. We hypothesized that: (i) the Panax seed microbiome would be similar due to an affinity with their host plant (H1); and (ii) that wild Panax plants would share a portion of core/keystone microbes regardless of variations from localities due to the vertical transmission of seed bacterial microbiota and similarity in their host plants (H2).
Three wild relatives of ginseng, viz. P. vietnamensis (Pv), P. japonicus (Pj), and P. stipuleanatus (Ps), were collected from seven localities (Malipo County, YuanYang County, Jinping County, Longling County, Pingbian County, and two sites in Wenshan County) in Yunnan province, China (see Supplementary Table 1 for location code details). The studied Pv, Pj, and Ps are the only wild relatives of P. notoginseng. The distribution area of Ps and Pv is close to Wenshan county, the main cultivation area of P. notoginseng. Pj has a wider growing area, and sparse distributed in Wenshan. Therefore, Pj was collected one nearby and one relatively far from the local (Longling). Fresh infructescences were sampled during early to mid-August in the field and transferred in ice box to the lab within 24 h. For each Panax species, four infructescences were collected containing c. 50 single-seeded berries. Of the c. 200 seeds removed from each plant, 50 seeds were used for surface disinfection. The method followed that used for wild plant seeds using ethanol and bleach (Liu et al., 2022). Then, the seeds were surface dried under aseptic conditions and stored at –20°C prior to DNA extraction.
Panax seeds were homogenized and pulverized using a MM400 Mixer Mill (Retsch, Germany) and DNA extracted with a Power Soil DNA kit (12888, MoBio®, Carlsbad, CA, United Sates). The PCR amplification of the V5–V7 (F:AACMGGATTAGATACCCKG R:ACGTCA TCCCCACCTTCC; bacterial 16S rRNA) and ITS regions was carried out using primer pairs 799F-1193R (F:CTTGGTCATTTAGAGGAAGTAA R:GCTGCGTTCTTC ATCGATGC; for seed endophytic bacteria) (Horton et al., 2014) and ITS1F-ITS1R (for seed endophytic fungi) (Orgiazzi et al., 2012). PCR amplicons were purified using a gel extraction kit (OMEGA Bio-Tek, Doraville, GA, United Sates) and quantified with NanoDrop 2000 (Thermo Fisher Scientific, Wilmington, DE, United Sates). Then, the paired-end sequencing of the bacterial and fungal amplicons was conducted on the Illumina Miseq-PE250 platform (Personalbio®, Shanghai, China).
Raw bacterial and fungal reads were processed under the QIIME 2 pipeline. Briefly, the reads were quality-trimmed and assigned based on their barcodes. Chimeric sequences and singletons were all deleted. Filtered reads were then inferred into amplicon sequence variants (ASVs) at ≥ 99% similarity, via the taxonomically-annotated Greengenes (for 16S rRNA) (Yu et al., 2021) and UNITE (for ITS) databases, respectively (Nilsson et al., 2019; Perez-Moreno et al., 2021). All sequences were deposited in the SRA (Sequence Read Archive) under accession number SUB10550428.
The Panax seed endophytic microbiomes were quantified using the same primer pairs as used for amplicon sequencing via real-time RT-PCR. PCR reaction mix and calculating details are described elsewhere in Liu et al. (2022).
One-way analysis of variance (ANOVA) followed by Tukey HSD was used to compare significant variation of the microbial abundance (bacterial and fungal gene copy numbers), diversity (Shannon index) and richness (observed species) among Panax seeds from various geographic locations. Variance in the Panax seed microbiome structure difference was assessed using PERMANOVA (Permutational multivariate analysis of variance) and visualized using non-metric multidimensional scaling (NMDS) plots derived from pairwise Bray–Curtis distances (Bray and Curtis, 1957). Core and unique microbes were evaluated against a table of seed endophytic bacterial and fungal ASVs and visualized using a petal diagram. Keystone microbes were identified by network analysis that was constructed from the microbial correlation matrix. Highly connected microbes were gathered together as individual function modules and the module hub connectors were calculated based on their topological roles. The value of among-module connectivity was higher than 0.62, as previously recommended (Olesen et al., 2007; Liu et al., 2021).
For individual species’ seed microbiomes, bacterial abundances were predominantly higher than those of fungi, ranging from 2.7 × 102 (in Pj6) to 5.6 × 104 times (in Ps7) (Table 1). The abundance of Panax seed microbiome showed significant differences among geographic locations for the two Panax species: Pv and Pj (Table 1).

Table 1. Panax seed microbial gene copy numbers determined by qPCR.
Among the Panax species, considerable differences in microbial abundances were observed, with the highest values observed in Ps7 (Table 1). For seed bacteria, 16S rRNA gene copies varied > 100-fold, from 1.1 × 109 (Ps7) to 1.1 × 107 (Pv1, Pv3). For seed endophytic fungi, ITS gene abundance differed over 1,000-fold, from 3.7 × 102 (Pv1) to 7.6 × 105 (Pj6). The gene copy no. based abundance of Panax seed microbiome showed significant difference among geographic locations for Panax species (Pv and Pj) with more than one provenance.
After quality-filtering and removing singleton and chimeric sequences, the clear high-quality 16S rRNA reads (length of 370–380 bp) obtained from seeds ranged from 84,152 to 126,462 per sample (mean = 103,145). The filtered clear ITS dataset reads (length of 180–362 bp) ranged from 40,468 to 84,432 per sample (mean = 61,940). ASVs tables were rarefied to 48,680 bacterial and 3,766 fungal ASVs per sample, according to the samples with minimum sequences (Supplementary Table 2).
The major endophytic bacterial taxa across all Panax species’ seeds belonged to Proteobacteria, with an average of 89% of the total bacterial sequences obtained. Firmicutes (7%), Actinobacteria (2%), and Bacteroidetes (0.4%) were less abundant. The relative abundance of seed Proteobacteria was significantly lower in the species Ps7 (57%) compared to the others samples (averaged 91%, range 89–96%). In contrast, the species Ps7 had more Firmicutes than the other species (42% relative abundance vs. 1%) (Supplementary Figure 1).
At the genus level, the mean relative abundance was dominated by Halomonas (45%), followed by Pantoea (6.4%), Pseudomonas (4.0%), Lactobacillus (2.4%), Serratia (2.2%), Lactococcus (1.9%), Nesterenkonia (1.4%), Weissella (1.3%), Stenotrophomonas (0.7%), and Aliihoeflea (0.6%) (Figure 1). Among the species, Halomonas was most abundant in the species Pj6 (80%) and least abundant in the species Pv2 (19%). At the species level, we found that each Panax species harbored its own unique microbial taxa, as shown by the selective existence of the top 10 bacterial species in various Panax species (Supplementary Table 3). The identified Panax species’ endophytic fungi were significantly lower (4.5%, relative abundance) in terms of relative abundance compared to bacteria (98%). For the endophytic fungi sequences, 95.5% were assigned to uncultured and unclassified/unidentified taxa, the main fungal phyla of remaining 4.5% identified fungal sequences were Ascomycota (3.0%) and Basidiomycota (1.5%) (Supplementary Table 4). Among them, the most abundant fungal genera were Cladosporium (0.8%), Alternaria (0.7%), Malassezia (0.7%), and Fusarium (0.7%) (Supplementary Table 4).
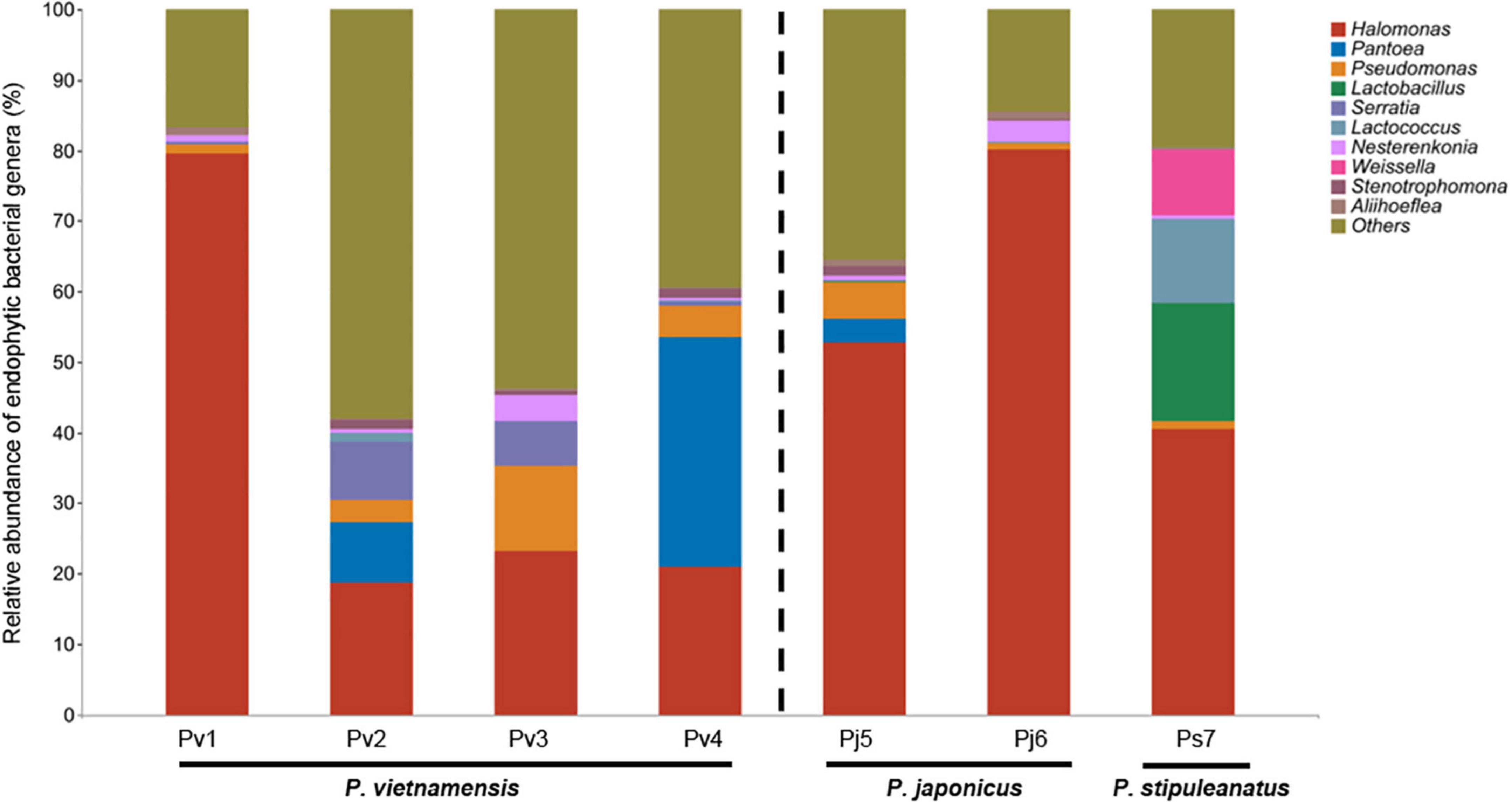
Figure 1. Relative abundance of top ten endophytic bacterial genera found in the seeds of three Panax species. Relative abundance of unclassified/unidentified taxa are grouped as “Others.” Values are the mean of three replicates. Abbreviations indicate three Panax species (Panax vietnamensis, Pv; Panax japonicus, Pj; Panax stipuleanatus, Ps) from seven provenances in Southwestern China (1, Malipo; 2, Wenshan; 3, Yuanyang; 4, Jinping; 5, Longling, 6, Wenshan; 7, Pingbian).
In order to identify the effects of species and geographic locations on the seed microbiome, their endophytic microbial diversity was compared using the Shannon diversity index and the number of observed bacterial/fungal species. In general, Panax seed endophytic fungal diversity (1.5 vs. 4.5, Shannon index) and richness (45 vs. 80, Observed in the evaluated species) were both significantly lower than the values obtained for bacteria. Since Ps was collected from only one place, the comparison of variations among different geographic locations was conducted for P. vietnamensis (among Pv1, Pv2, Pv3 and Pv4; Figures 2A,B) and P. japonicus (between Pj5 and Pj6; Figures 2C,D). Geographic factor did not significantly impact the Panax seed fungal microbiota (Supplementary Figure 2). However, Panax seed bacterial richness (Observed_species) and diversity (Shannon) were significantly affected by geographic locations in the case of Pv and Pj (Figures 2A–D).
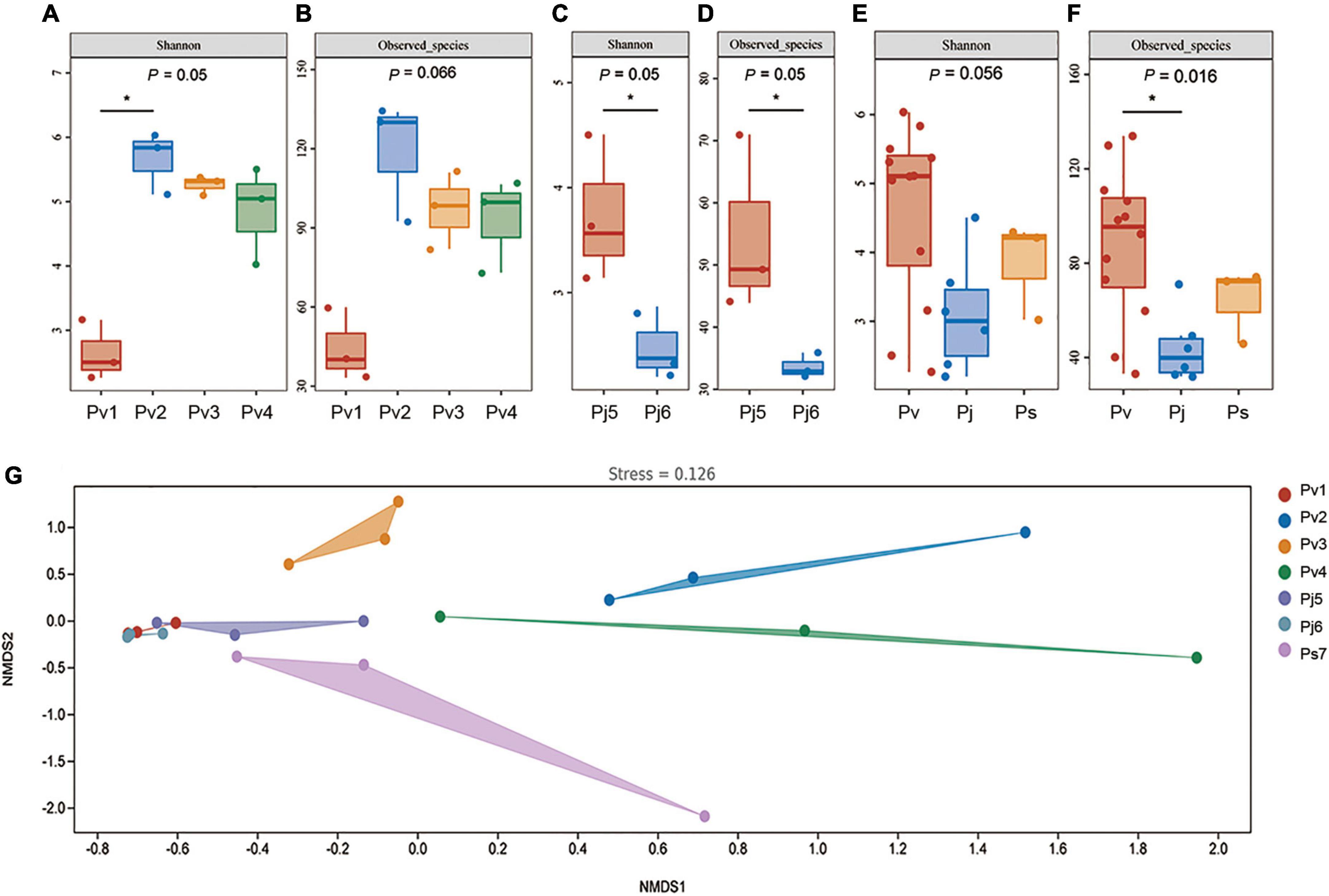
Figure 2. Panax seed endophytic bacterial diversity (A–F) and community changes (G), as affected by species’ geographic location (A–D) and host plant species (E,F). Seed endophytic bacterial community compositions were indicated by non-metric multidimensional scaling plots (NMDS) of the pairwise Bray–Curtis distance in the seven Panax species. Bacterial diversity and richness were evaluated by Shannon index and the number of Observed species, respectively. Asterisks (*) indicates significant differences at P < 0.05 (ANOVA) between means (Tukey’s HSD pairwise comparisons). Panax species abbreviations are described in Figure 1.
In order to evaluate the major drivers of Panax seed microbial community composition, beta-diversity analysis was conducted by non-metric multidimensional scaling (NMDS) (Figure 2) in combination with PERMANOVA test. There was strong variation in seed microbiomes. Among the grouping factors, host plant species was observed as the main driver of seed microbiome composition, which was the case for both bacteria (PERMANOVA test, sample size = 21, F = 2.73, P = 0.006) and fungi (PERMANOVA test, sample size = 21, F = 3.13, P = 0.001).
The comparison of seed endophytic community structure variations among different geographic locations were conducted only for Pv due to the fact that this species was sampled from four provenances (Pv1, Pv2, Pv3, and Pv4). Seed bacterial microbiota (PERMANOVA test, sample size = 12, F = 4.74, P = 0.001) but not fungal microbiota (PERMANOVA test, sample size = 12, F = 0.86, P = 0.579) were significantly affected by species’ location. This suggests that the three Panax species’ seed bacterial diversity and structure are shaped by the combination of plant species and their growing environments, In contrast, the Panax seed endophytic fungal microbiota were more influenced by plant genotype than by geographic factor.
Having explored variations in Panax seed microbiomes, we sought to understand the shared/core microbes that permanently exist regardless of changes in geographic conditions and host plants. For the same Panax species, seeds from different sites had greater numbers of their unique endophytic bacterial and fungi. For instance, the unique seed bacterial ASVs within Pv (Pv1, Pv2, Pv3, and Pv4) ranged from 38 (Pv1) to 117 (Pv2) for bacteria and from 55 (Pv3) to 96 (Pv4) for fungi (Figures 3A,B).
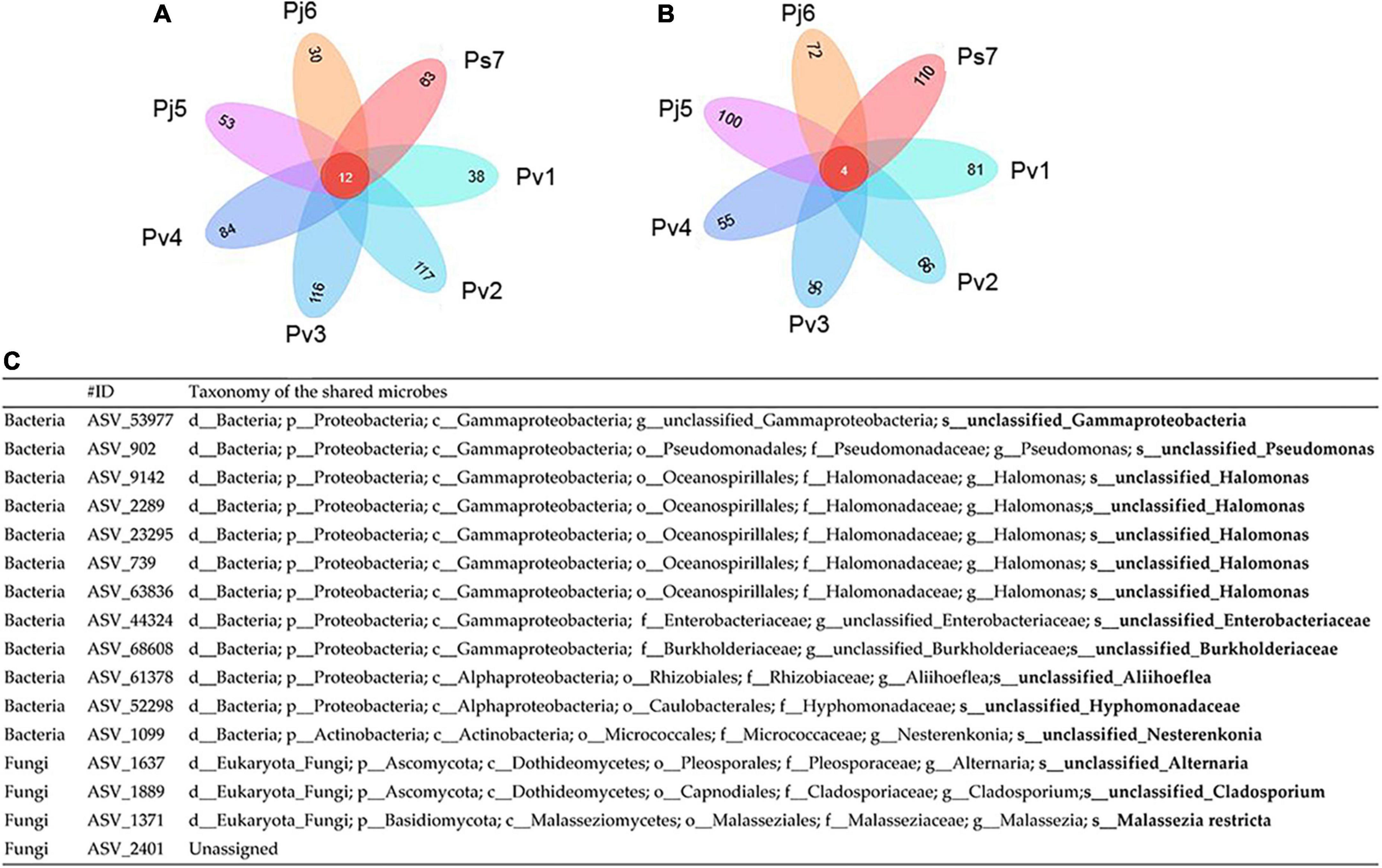
Figure 3. Petal diagram of the numbers of core and unique bacterial (A) and fungal (B) amplicon sequence variants (ASVs) (C) within seeds of three Panax species from various geographic localities (1–7): P. vietnamensis (Pv1, Pv2, Pv3, and Pv4), P. japonicus (Pj5, Pj6) and P. stipuleanatus (Ps7).
However, there were only 12 bacterial shared ASVs across the investigated seed microbiomes (Figure 3A). Among them, almost half (ASV_739, ASV_9142, ASV_2289, ASV_23295, and ASV_63836) were assigned to Halomonas (Figure 3A). Only four fungi ASVs were identified, including three from Alternaria (ASV_1637), Cladosporium (ASV_1889), and Malassezia (ASV_1371), and 1 unassigned fungi species (Figure 3C). Since the seeds investigated belonged to the same plant genus and were collected from one province, we conducted a microbial network analysis to gain more insight into the keystone taxa present (Figure 4). The seed bacterial network, which was constructed with whole seed samples, revealed that the microbes did not exist individually but co-occurred within tight (Module 1 and 3; Figure 4A) and loose (Module 2, 4, and 5; Figure 4B) functional modules. Modules 1 (yellow) and module 3 (red) were inter-woven and together constituted the largest module (as shown by light blue ellipse; Figure 4). This network space was dominated by the genera Pseudomonas, Halomonas, and Pantoea. In contrast to bacteria, the fungal network was less complex and featured only two positively connected functional modules (Supplementary Figure 3). Within the Panax seed microbe, there were 14 bacterial and eight fungal ASVs having high connectivity among-modules, serving as module connectors (Supplementary Table 5).
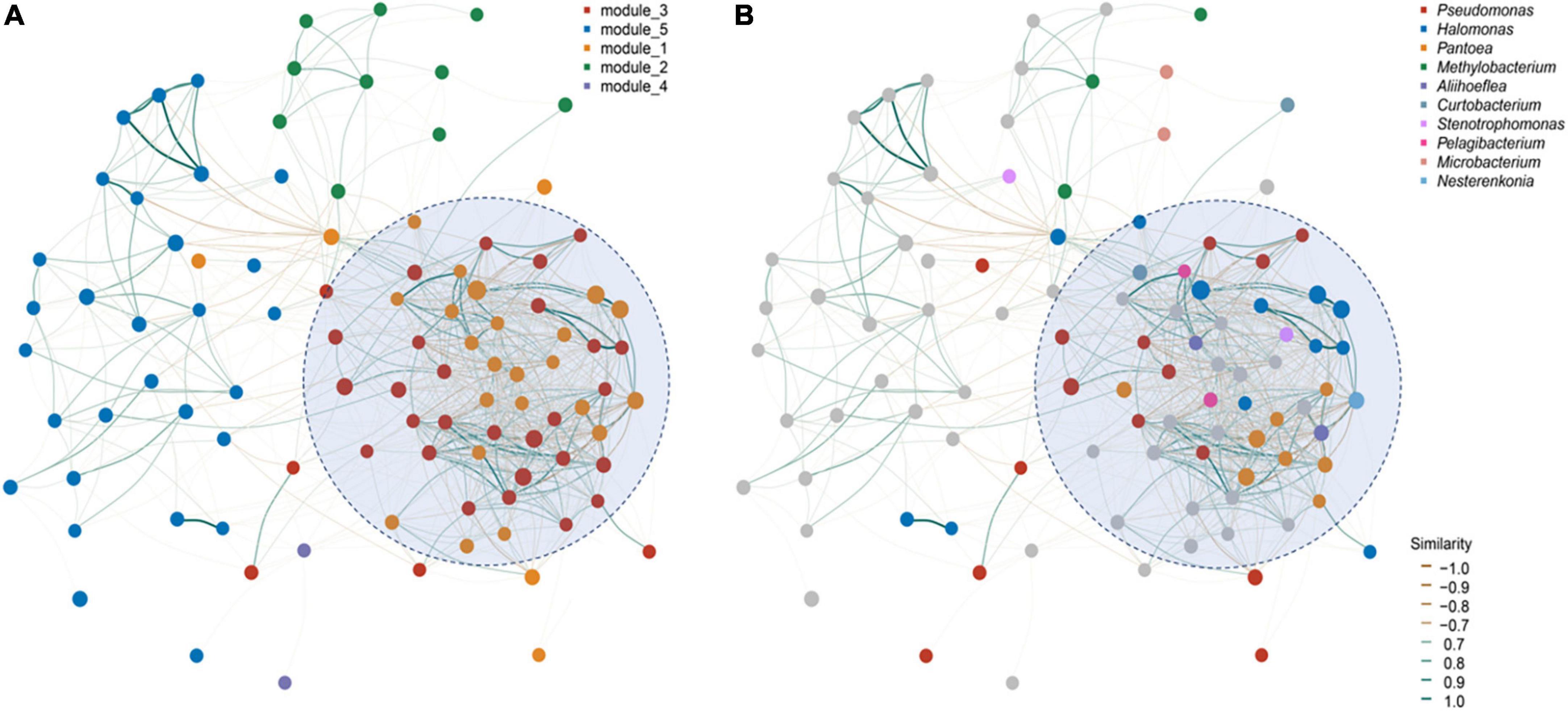
Figure 4. Panax seed endophytic bacterial networks based on (A) functional modules and (B) dominant bacterial genera. The networks were constructed based on seed microbial correlation matrices [bacterial ASVs tables for P. vietnamensis (n = 12), P. japonicus (n = 6) and P. stipuleanatus (n = 3)]. Nodes represent the top 50 dominant bacterial taxa. Line color refers to the correlation ingredient among nodes, being either positive (blue) or negative (orange). Modules within the networks were visualized by different colors and represent a cluster of highly interconnected nodes.
We initially hypothesized (H1) that due to the similarity of host plant species in a single genus, Panax, the abundance of seed endophytic microbiota would also be similar. However, based on the qPCR results, the abundance difference among host plants was 100-fold, which is much higher than the variation from geographic locations (∼10 times). On the other hand, the dominant bacterial abundance in the Panax seed microbiome was in agreement with the results from our previous study (16) on a series of wild plant seed microbes, where a high ratio of bacteria/fungi abundance was also found. The predominated bacterial endophytes have shown to have the ability to enhance seeding growth by increasing access to nutrients, such as by making iron more available, fixing nitrogen and solubilizing phosphate (Vendan et al., 2010; Hong et al., 2019; Liu et al., 2021; Gao et al., 2022; Goodwin, 2022). Such nutrients are essential to plant early-stage survival. In support of hypothesis 2, we observed that Panax plants shared a portion of core/keystone microbes regardless of any location-specific variation. Thus, the major endophytic bacterial taxa across all Panax species’ seeds belonged mainly to Proteobacteria (averaged ∼90%). The dominance of Proteobacteria likely relates to their many functional traits, including being involved in phosphate solubilization and indoleacetic acid production, and therefore contributing to early seed germination and later plant growth (Vendan et al., 2010; Chowdhury et al., 2017). For instance, a metagenomic analysis of bacterial endophytes in Panax ginseng plants of various ages indicated that after 2–6 years growth, Proteobacteria was still dominant (∼60%) (Goodwin, 2022). As found by Wei et al. (2021), Panax endophytes seem to be transferred via the seed and then variously dominate in plant tissues; for instance, Proteobacteria and Firmicutes are known to be abundant in stems and roots, respectively (Tian et al., 2014). Apart from Firmicutes, Actinobacteria and Bacteroidetes are also Panax root-aggregating bacterial phyla that show considerable variation in abundance among studies (Hong et al., 2018a; Wei et al., 2021; Gao et al., 2022).
At the phylum level, one of the biggest differences was that the relative abundance of Firmicutes in the seeds of P. stipuleanatus, reaching nearly 50%, compared with < 1% in the other two Panax species studied. Numerous members of Firmicutes produce spores, which are resistant to dehydration and extreme conditions, and could act as biocontrol agents of plant pathogens (Dong et al., 2018). These traits seems to be associated with the habitat of P. stipuleanatus. The species is confined to a narrow area in Southeastern Yunnan and northern Vietnam where the temperature and humidity are high (Hashmi et al., 2020).
Panax seed bacterial taxa with high relative abundance (such as Halomonas,Pseudomonas frederiksbergensis, Pseudomonas Poae, and Pantoea) are known to be beneficial to their hosts (Caspi et al., 2008; Wang and Ya-Qiong, 2018; Goodwin, 2022). A most recent study showed that a member of the genus Pantoea can inhibit fungal pathogenesis by targeting lipid rafts (Truyens et al., 2015). Based on microbial network analysis, we found that members from these dominant genera were highly-connected and formed the largest functional module (Figure 4). Pseudomonas tend to persist in Panax seedlings and inhibit plant pathogens, such as Cylindrocarpon destructans and Botrytis cinerea, by secreting antibiotics (Hong et al., 2018a). The most abundant Halomonas detected in Panax seeds are also known to be abundant in maize cultivars (Johnston-Monje and Raizada, 2011; Xu et al., 2022) and wild plant seeds (Liu et al., 2022). Moreover, based on the shared seed microbial analysis of Panax species and different geographies, we found that almost half of the core microbes were assigned to the genus Halomonas (Figure 3). We contend that the reason that Halomonas is widely spread and a dominating feature of Panax species relates to their tolerance of environmental temperature fluctuation, pH and salt levels. Consequently, Halomonas have of potential significance in plant stress resistance improvement programs (Rijavec et al., 2007).
For Panax seed’s fungal microbiota, the relative high abundance of Alternaria and Cladosporium, found in the present study corroborate studies on Panax notoginseng (Franzmann et al., 1988; Zheng et al., 2016). These are classified as dematiaceous fungi. Alternaria are capable of persisting in harsh ecological conditions and are also widely distributed in plants (Haimin et al., 2018; Jha, 2019; Liu et al., 2022; Simonin et al., 2022). In the present study, Cladosporium was presented in the seeds and Bensch et al. (2012) have shown that it is abundant in the rhizosphere. Such varied occupation is in agreement with Shade et al. (2017), stating that seeds are involved in the microbial transmission and act as the initial inoculum for the plant benificial microbiota. Moreover, the tissue-specific localization could be associated with their fungicidal activity against pathogenic yeasts, such as Candida albicans, C. glabrata, Cryptococcus neoformans, and Malassezia furfur (Xing et al., 2010). However, it is notable that some fungal isolates within Alternaria and Cladosporium are also known to be pathogens, such as Cladosporium oxysporum (Polonelli et al., 2012; Gao et al., 2022). Generally, plant-endophyte interactions are complex, diverse, and change over the different stages of plant development (Kim et al., 2018). For pathogenic fungi, commonly studied genera, such as Malassezia and Fusarium, tend to exist in many plants (Jha, 2019; Liu et al., 2022); however, their pathogenic features are not shown, probably because of their low abundance (as compared to predominant endophytic bacteria) or because of pathogen restrain by beneficial bacteria. A good example is the application of a Bacillus subtilis strain to suppress Fusarium verticillioides-caused diseases (Liu et al., 2022). All these findings indicate a complex picture for the role of Panax seed fungal endophytes that needs further clarification.
As a first study focusing on the seed microbiome of the selected wild Panax species’ surviving in harsh environments, we uncovered a predominant pattern of seed bacterial endophytes which can be closely associated with nutrient cycling during seedling growth and the attenuation of abiotic and biotic stresses in widely varying geographic conditions. The main key microbes found were also from bacterial microbiota such as Pseudomonas, Halomonas, and Pantoea. Panax seed bacterial richness and diversity were significantly affected by their host genotype and geographic locations. Geographic factor did not impose a significant effect on fungal microbiota, as shown by P. vietnamensis from four localities (in the case of the other two studied species there was a small number sampled locations). Panax species’ seeds endophytic fungi have a very low abundance but some beneficial taxa have a potential function helpful for pathogen resistance. Non-pathogenic fungi existing in seeds are thought to be suppressed by dominant bacterial network and therein antifungal taxa. Together, these novel insights provide a deeper understanding of highly diversified plant-specific seed bacterial microbiota and potential ecological strategies for plants in hypothermal environments.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/, PRJNA774071.
DL: manuscript writing, review, and editing. LL: experimental design, sample collection, manuscript revision, and concept development. QX and MW: sample preparation and experiment. TZ: and MG: sample collection pretreatment, data processing. PB: editing and critical review. FY, HP, and XY: experimental concept, discussions, and project management. All authors contributed to the article and approved the submitted version.
This work was funded by Grass Seed Industry Innovation Center Project of Invigorating Inner Mongolia through Science and Technology Key Program. MW was supported by Postdoctoral Research Funding Projects of Yunnan Province and Postdoctoral Directional Training Foundation of Yunnan Province. LL was supported by grants from the Key Basic Research program of Yunnan Province, China (grant no. 202101BC070003). This research was facilitated by the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences.
We are grateful to seed collectors during seed processing and the supply of background information. We thank the whole Seed Biology Research team’s valuable comments on the first draft. We are grateful to technicians in the high-throughput analyses center, and Zhonghua Li for technical assistance in laboratory and data interpretation.
MG was employed by the company Wenshan Huaxin Notoginseng Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.967692/full#supplementary-material
Supplementary Figure 1 | Relative abundance of endophytic bacterial phyla found in the seeds of three Panax species. Relative abundance of uncultured and unclassified/unidentified taxa are grouped as “Others.” Values are the mean of three replicates. Full names of species abbreviations are described in Supplementary Table 1.
Supplementary Figure 2 | Panax seed endophytic fungal microbiota changes. (A) Shannon diversity, (B) Observed species, and (C) Community structure. The abbreviations in the caption indicate the three Panax notoginseng (medicinal plant) wild relatives: (Panax vietnamensis, Pv; Panax japonicus, Pj; Panax stipuleanatus, Ps) from seven provenances in Southwestern China (1, Malipo; 2, Wenshan; 3, Yuanyang; 4, Jinping; 5, Longling, 6, Wenshan; 7, Pingbian).
Supplementary Figure 3 | Panax seed endophytic fungal network based on A) functional module and B) dominant fungal genus. The networks were constructed based on the seed microbial correlation matrix (fungal ASVs tables for Panax vietnamensis (n = 12), Panax japonicus (n = 6) and Panax stipuleanatus (n = 3). Nodes represent the top 50 dominant bacterial taxa. Line color refers to the correlation ingredient among nodes, either positive (blue) or negative (orange). Modules within the networks were visualized by different colors and represent a cluster of highly interconnected nodes.
Abdullaeva, Y., Binoy, A. M., Honermeier, B., Schnell, S., and Cardinale, M. (2020). Domestication affects the composition, diversity, and co-occurrence of the cereal seed microbiota. J. Adv. Res. 31, 75–78. doi: 10.1016/j.jare.2020.12.008
Bacilio-Jiménez, M., Aguilar-Flores, S., Valle, M. V. D., Pérez, A., Zepeda, A., and Zenteno, E. (2001). Endophytic bacteria in rice seeds inhibit early colonization of roots by Azospirillum brasilense. Soil Biol. Biochem. 33, 167–172. doi: 10.1016/S0038-0717(00)00126-7
Banerjee, S., Schlaeppi, K., and Van, D. (2018). Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576. doi: 10.1038/s41579-018-0024-1
Bensch, K., Braun, U., Groenewald, J. Z., and Crous, P. W. (2012). The genus Cladosporium. Stud. Mycol. 72, 1–401. doi: 10.3114/sim0003
Bjork, J. R., O’Hara, R. B., Ribes, M., Coma, R., and Montoya, J. M. (2018). The dynamic core microbiome: structure, dynamics and stability. bioRxiv [Preprint]. doi: 10.1101/137885
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Caspi, R., Foerster, H., Fulcher, C. A., Kaipa, P., Krummenacker, M., Latendresse, M., et al. (2008). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 36:D623–D631. doi: 10.1093/nar/gkr1014
Chowdhury, E. K., Jeon, J., Rim, S. O., Park, Y.-H., Lee, S. K., and Bae, H. (2017). Composition, diversity and bioactivity of culturable bacterial endophytes in mountain-cultivated ginseng in Korea. Sci. Rep. 7:10098. doi: 10.1038/s41598-017-10280-7
Christensen, L. P. (2008). Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv. Food Nutr. Res. 55, 1–99. doi: 10.1016/S1043-4526(08)00401-4
Cottyn, B., Regalado, E., Lanoot, B., Cleene, M. D., Mew, T. W., and Swings, J. (2001). Bacterial populations associated with rice seed in the tropical environment. Phytopathology 91, 282–292. doi: 10.1094/PHYTO.2001.91.3.282
Dong, L., Cheng, R., Xiao, L., Wei, F., Wei, G., Jiang, X., et al. (2018). Diversity and composition of bacterial endophytes among plant parts of Panax notoginseng. Chin. Med. 13:41. doi: 10.1186/s13020-018-0198-5
Franzmann, P. D., Wehmeyer, U., and Stackebrandt, E. (1988). Halomonadaceae fam. nov., a new family of the class Proteobacteria to accommodate the genera Halomonas and Deleya. Syst. Appl. Microbiol. 11, 16–19. doi: 10.1016/S0723-2020(88)80043-2
Gao, Y., Ma, J., Chen, J., Xu, Q., Jia, Y., Chen, H., et al. (2022). Establishing tetraploid embryogenic cell lines of Magnolia officinalis to facilitate tetraploid plantlet production and phenotyping. Front. Plant Sci. 13:900768. doi: 10.3389/fpls.2022.900768
Goodwin, P. H. (2022). The endosphere microbiome of ginseng. Plants 11:415. doi: 10.3390/plants11030415
Gundel, P. E., Rudgers, J. A., and Ghersa, C. M. (2011). Incorporating the process of vertical transmission into understanding of host–symbiont dynamics. Oikos 120, 1121–1128. doi: 10.1111/j.1600-0706.2011.19299.x
Haimin, C., Hongxia, W., Bin, Y., Hongguang, Z., Fenghua, L., Haihua, Z., et al. (2018). Core microbiome of medicinal plant Salvia miltiorrhiza seed: a rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 19:672. doi: 10.3390/ijms19030672
Hashmi, I., Bindschedler, S., and Junier, P. (2020). Firmicutes. in Beneficial Microbes in Agro-Ecology: Bacteria and Fung, eds N. Amaresan, M. Senthil Kumar, K. Annapurna, K. Kumar, A. Sankaranarayanan (Cambridge: Elsevier, Academic Press), 363–396. doi: 10.1016/b978-0-12-823414-3.00018-6
Hill, R., Llewellyn, T., Downes, E., Oddy, J., Macintosh, C., Kallow, S., et al. (2021). Seed banks as incidental fungi banks: fungal endophyte diversity in stored seeds of banana wild relatives. Front. Microbiol. 12:508. doi: 10.3389/fmicb.2021.643731
Hong, C. E., Kim, J. U., Woo, L. J., Lee, S. W., and Jo, I. H. (2018a). Diversity of bacterial endophytes in Panax ginseng and their protective effects against pathogens. 3 Biotech 8:397. doi: 10.1007/s13205-018-1417-6
Hong, C. E., Jo, S. H., Jo, I. H., and Park, J. M. (2018b). Diversity and antifungal activity of endophytic bacteria associated with Panax ginseng seedlings. Plant Biotechnol. Rep. 12, 409–418. doi: 10.1007/s118116-018-0504-9
Hong, C. E., Kim, J. U., Lee, J. W., Bang, K. H., and Jo, I. H. (2019). Metagenomic analysis of bacterial endophyte community structure and functions in Panax ginseng at different ages. 3 Biotech 9:300. doi: 10.1007/s13205-019-1838-x
Horton, M. W., Bodenhausen, N., Beilsmith, K., Meng, D., Muegge, B. D., Subramanian, S., et al. (2014). Genome-wide association study of Arabidopsis thaliana leaf microbial community. Nat. Commun. 5:532. doi: 10.1038/ncomms6320
Jha, S. (2019). Endophytes and Secondary Metabolites. Cham: Springer, 1–22. doi: 10.1007/978-3-319-90484-9
Johnston-Monje, D., and Raizada, M. N. (2011). Conservation and diversity of seed associated endophytes in Zea across boundaries of evolution, ethnography and ecology. PLoS One 6:e20396. doi: 10.1371/journal.pone.0020396
Kim, H., Rim, S. O., and Bae, H. (2018). Antimicrobial potential of metabolites extracted from ginseng bacterial endophyte Burkholderia stabilis against ginseng pathogens. Biol. Control 128, 24–30. doi: 10.1016/j.biocontrol.2018.08.020
Kochkin, D. V., Kachala, V. V., Shashkov, A. S., Chizhov, A. O., Chirva, V. Y., and Nosov, A. M. (2013). Malonyl-ginsenoside content of a cell-suspension culture of Panax japonicus var. repens. Dokl. Biochem. Biophys. 93:18. doi: 10.1016/j.phytochem.2013.03.021
Kusstatscher, P., Adam, E., Wicaksono, W. A., Bernhart, M., and Berg, G. (2021). Microbiome-assisted breeding to understand cultivar-dependent assembly in Cucurbita pepo. Front. Plant Sci. 12:642027. doi: 10.3389/fpls.2021.642027
Liu, D., Cai, J., He, H., Yang, S., Chater, C. C. C., and Yu, F. (2022). Anemochore seeds harbor distinct fungal and bacterial abundance, composition, and functional profiles. J. Fungi 8:89. doi: 10.3390/jof8010089
Liu, D., Perez-Moreno, J., He, X., Garibay-Orijel, R., and Yu, F. (2021). Truffle microbiome is driven by fruit body compartmentalization rather than soils conditioned by different host trees. mSphere 6:e0003921. doi: 10.1128/mSphere.00039-21
Moreira, Z. M., Helgason, B., and Germida, J. (2021). Environment has a stronger effect than host plant genotype in shaping spring seed microbiomes. Phytobiomes J. 5:2. doi: 10.1094/PBIOMES-08-20-0059-R
Nilsson, R. H., Larsson, K.-H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nuclc Acids Res. 47:D259–D264. doi: 10.1093/nar/gky1022
Oehrle, N. W., Karr, D. B., Kremer, R. J., and Emerich, D. W. (2000). Enhanced attachment of Bradyrhizobium japonicum to soybean through reduced root colonization of internally seedborne microorganisms. Can. J. Microbiol. 46, 600–606. doi: 10.1139/w00-030
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2007). The modularity of pollination networks. Proc. Natl. Acad. Sci. U.S.A. 104, 19891–19896. doi: 10.1073/pnas.0706375104
Orgiazzi, A., Bianciotto, V., Bonfante, P., Daghino, S., Ghignone, S., Lazzari, A., et al. (2013). 454 Pyrosequencing analysis of fungal assemblages from geographically distant, disparate soils reveals spatial patterning and a core mycobiome. Diversity 5, 73–98. doi: 10.3390/d5010073
Orgiazzi, A., Lumini, E., Nilsson, R. H., Girlanda, M., Vizzini, A., Bonfante, P., et al. (2012). Unravelling soil fungal communities from different Mediterranean land-use backgrounds. PLoS One 7:e34847. doi: 10.1371/journal.pone.0034847
Orozco-Mosqueda, M. D. C., and Santoyo, G. (2020). Plant-microbial endophytes interactions: scrutinizing their beneficial mechanisms from genomic explorations. Curr. Plant Biol. 25, 100–189. doi: 10.1016/j.cpb.2020.100189
Pandey, A. K., and Ali, M. A. (2012). Intraspecific variation in Panax assamicus Ban. populations based on internal transcribed spacer (ITS) sequences of nrDNA. Indian J. Biotechnol. 11, 30–38.
Perez-Moreno, J., Yu, F., Liu, D., He, X., and Chater, C. C. (2021). Microbiome community structure and functional gene partitioning in different micro-niches within a sporocarp-forming fungus. Front. Microbiol. 12:629352. doi: 10.3389/fmicb.2021.629352
Pold, G., and Deangelis, K. (2013). Up against the wall: the effects of climate warming on soil microbial diversity and the potential for feedbacks to the carbon cycle. Diversity 5, 409–425. doi: 10.3390/d5020409
Polonelli, L., Ciociola, T., Magliani, W., Zanello, P. P., D’Adda, T., Galati, S., et al. (2012). Peptides of the constant region of antibodies display fungicidal activity. PLoS One 7:e34105. doi: 10.1371/journal.pone.0034105
Rego, C., Frana-Silva, F., Gomes-Junior, F. G., Moraes, M., Medeiros, A., and Silva, C. (2020). Using multispectral imaging for detecting seed-borne fungi in Cowpea. Agriculture 10:361. doi: 10.3390/agriculture10080361
Rijavec, T., Lapanje, A., Dermastia, M., and Rupnik, M. (2007). Isolation of bacterial endophytes from germinated maize kernels. Can. J. Microbiol. 53, 802–808. doi: 10.1139/W07-048
Shade, A., Jacques, M. A., and Barret, M. (2017). Ecological patterns of seed microbiome diversity, transmission, and assembly. Curr. Opin. Microbiol. 37, 15–22. doi: 10.1016/j.mib.2017.03.010
Shao, J., Miao, Y., Liu, K., Ren, Y., Xu, Z., Zhang, N., et al. (2021). Rhizosphere microbiome assembly involves seed-borne bacteria in compensatory phosphate solubilization. Soil Biol. Biochem. 159:108273. doi: 10.1016/j.soilbio.2021.108273
Shi, R., Gu, H., He, S., Xiong, B., Huang, Y., Horowitz, A. R., et al. (2021). Comparative metagenomic and metabolomic profiling of rhizospheres of Panax notoginseng grown under forest and field conditions. Agronomy 11:2488. doi: 10.3390/agronomy11122488
Shi, R., Wang, S., Xiong, B., Gu, H., Wang, H., Ji, C., et al. (2022). Application of bioorganic fertilizer on Panax notoginseng improves plant growth by altering the rhizosphere microbiome structure and metabolism. Microorganisms 10:275. doi: 10.3390/microorganisms10020275
Simonin, M., Briand, M., Chesneau, G., Rochefort, A., and Barret, M. (2022). Seed microbiota revealed by a large-scale meta-analysis including 50 plant species. New Phytol. 234, 1448–1463. doi: 10.1101/2021.06.08.447541
Singh, J. S., and Gupta, V. K. (2018). Soil microbial biomass: a key soil driver in management of ecosystem functioning. Sci. Total Environ. 634, 497–500. doi: 10.1016/j.scitotenv.2018.03.373
Tian, L., Jiang, Y., Chen, C., Zhang, G., Li, T., Tong, B., et al. (2014). Screening and identification of an endophytic bacterium with 1-aminocyclopropane-1-carboxylate deaminase activity from Panax ginseng and its effect on host growth. Acta Microbiol. Sin. 54, 760–769. doi: 10.13343/j.cnki.wsxb.2014.07.006
Truyens, S., Weyens, N., Cuypers, A., and Vangronsveld, J. (2015). Bacterial seed endophytes: genera, vertical transmission and interaction with plants. Environ. Microbiol. Rep. 7, 40–50. doi: 10.1111/1758-2229.12181
Turner, R. D., Hurd, A. F., Cadby, A., Hobbs, J. K., and Foster, S. J. (2013). Cell wall elongation mode in Gram-negative bacteria is determined by peptidoglycan architecture. Nat. Commun. 4:1496. doi: 10.1038/ncomms2503
Vendan, R. T., Yj, Lee, S. H., and Rhee, Y. H. (2010). Diversity of endophytic bacteria in ginseng and their potential for plant growth promotion. J. Microbiol. 48, 559–565. doi: 10.1007/s12275-010-0082-1
Walsh, C. M., Becker-Uncapher, I., Carlson, M., and Fierer, N. (2021). Variable influences of soil and seed-associated bacterial communities on the assembly of seedling microbiomes. ISME J. 15, 2748–2762. doi: 10.1038/s41396-021-00967-1
Wang, C. Z., Mcentee, E., Wicks, S., Wu, J. A., and Yuan, C. S. (2006). Phytochemical and analytical studies of Panax notoginseng (Burk.) F.H. Chen. J. Nat. Med. 60, 97–106. doi: 10.1007/s11418-005-0027-x
Wang, Z. H., and Ya-Qiong, L. I. (2018). Geographical distribution and growth pattern of Panax stipuleanatus, an anti-cancer plant. Lishizhen Med. Materia Medica Res. 29:11. doi: 10.3969/j.issn.1008-0805.2018.11.067
Wei, G., Chen, Z., Wang, B., Wei, F., Zhang, G., Wang, Y., et al. (2021). Endophytes isolated from Panax notoginseng converted ginsenosides. Microb. Biotechnol. 14, 1730–1746. doi: 10.1111/1751-7915.13842
Xing, X., Guo, S., and Fu, J. (2010). Biodiversity and distribution of endophytic fungi associated with Panax quinquefolium L. cultivated in a forest reserve. Symbiosis 51, 161–166. doi: 10.1007/s13199-010-0062-6
Xu, S., Liu, Y. X., Cernava, T., Wang, H., Zhou, Y., Xia, T., et al. (2022). Fusarium fruiting body microbiome member Pantoea agglomerans inhibits fungal pathogenesis by targeting lipid rafts. Nat. Microbiol. 7, 831–843. doi: 10.1038/s41564-022-01131-x
Yang, M., Zhang, X., Xu, Y., Mei, X., Jiang, B., Liao, J., et al. (2015). Autotoxic ginsenosides in the rhizosphere contribute to the replant failure of Panax notoginseng. PLoS One 10:e0118555. doi: 10.1371/journal.pone.0118555
Yu, X. Y., Zhu, Y. J., Wang, B., Liu, D., and Yang, N. (2021). Effects of nitrogen addition on rhizospheric soil microbial communities of poplar plantations at different ages. For. Ecol. Manag. 494:119328. doi: 10.1016/j.foreco.2021.119328
Zheng, Y. K., Miao, C. P., Chen, H. H., Huang, F. F., Xia, Y. M., Chen, Y. W., et al. (2016). Endophytic fungi harbored in Panax notoginseng: diversity and potential as biological control agents against host plant pathogens of root-rot disease. J. Ginseng Res. 41, 353–360. doi: 10.1016/j.jgr.2016.07.005
Keywords: hypothermal environment, bacterial endophytes, ginseng, seed microbiome, crop wild relatives, microbial community, network analysis
Citation: Liu D, Lin L, Zhang T, Xu Q, Wang M, Gao M, Bhople P, Pritchard HW, Yang X and Yu F (2022) Wild Panax plants adapt to their thermal environment by harboring abundant beneficial seed endophytic bacteria. Front. Ecol. Evol. 10:967692. doi: 10.3389/fevo.2022.967692
Received: 13 June 2022; Accepted: 05 July 2022;
Published: 27 July 2022.
Edited by:
Jinlei Zhu, University of Hohenheim, GermanyReviewed by:
Jesus Perez-Moreno, Colegio de Postgraduados (COLPOS), MexicoCopyright © 2022 Liu, Lin, Zhang, Xu, Wang, Gao, Bhople, Pritchard, Yang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangyun Yang, eXh5QG1haWwua2liLmFjLmNu; Fuqiang Yu, ZnF5dUBtYWlsLmtpYi5hYy5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.