
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Ecol. Evol., 02 August 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.953131
This article is part of the Research TopicRecent Advances in Museomics: Revolutionizing Biodiversity ResearchView all 16 articles
 Kelly A. Speer1,2*
Kelly A. Speer1,2* Melissa T. R. Hawkins3
Melissa T. R. Hawkins3 Mary Faith C. Flores3
Mary Faith C. Flores3 Michael R. McGowen3Robert C. Fleischer1
Michael R. McGowen3Robert C. Fleischer1 Jesús E. Maldonado1
Jesús E. Maldonado1 Michael G. Campana1
Michael G. Campana1 Carly R. Muletz-Wolz1
Carly R. Muletz-Wolz1Animal specimens in natural history collections are invaluable resources in examining the historical context of pathogen dynamics in wildlife and spillovers to humans. For example, natural history specimens may reveal new associations between bat species and coronaviruses. However, RNA viruses are difficult to study in historical specimens because protocols for extracting RNA from these specimens have not been optimized. Advances have been made in our ability to recover nucleic acids from formalin-fixed paraffin-embedded samples (FFPE) commonly used in human clinical studies, yet other types of formalin preserved samples have received less attention. Here, we optimize the recovery of RNA from formalin-fixed ethanol-preserved museum specimens in order to improve the usability of these specimens in surveys for zoonotic diseases. We provide RNA quality and quantity measures for replicate tissues subsamples of 22 bat specimens from five bat genera (Rhinolophus, Hipposideros, Megareops, Cynopterus, and Nyctalus) collected in China and Myanmar from 1886 to 2003. As tissues from a single bat specimen were preserved in a variety of ways, including formalin-fixed (8 bats), ethanol-preserved and frozen (13 bats), and flash frozen (2 bats), we were able to compare RNA quality and yield across different preservation methods. RNA extracted from historical museum specimens is highly fragmented, but usable for short-read sequencing and targeted amplification. Incubation of formalin-fixed samples with Proteinase-K following thorough homogenization improves RNA yield. This optimized protocol extends the types of data that can be derived from existing museum specimens and facilitates future examinations of host and pathogen RNA from specimens.
Natural history collections are an essential and underused resource for emerging infectious disease research (Talley et al., 2015; Schmitt et al., 2018; Colella et al., 2021; Thompson et al., 2021). These collections preserve snapshots of animal and plant populations and their associated parasites and pathogens through time. This quality has been used to track the spread of invasive parasites and pathogens in wildlife (Kleindorfer and Sulloway, 2016) and emergence of human pathogens (Childs et al., 1994; Yates et al., 2002). Natural history collections also maintain voucher specimens that can be revisited and compared between projects and institutions, a feature that can make pathogen surveillance more effective and reproducible (Colella et al., 2021; Thompson et al., 2021). Lastly, these collections maintain multiple specimen types that can be analyzed in new ways as new technology is developed, enabling novel data to be derived from existing resources. For example, DNA sequencing revolutionized our understanding of the information stored within a natural history specimen. Now, with the development of more sensitive and accurate sequencing and imaging technologies, we can also detect the community of pathogens associated with a specimen.
The spillover of SARS-CoV-2 from wildlife to humans has led to increased screening for coronaviruses, a highly diverse family (Coronaviridae) of positive-sense single-stranded RNA viruses, in bats globally (Valitutto et al., 2020; Becker et al., 2022). There are also efforts to revisit bat specimens housed in natural history collections to examine the evolution and host associations of coronaviruses. However, few protocols are available for extracting RNA from museum specimens (although see Fanning et al., 2002), limiting the use of museum specimens in viral screening efforts. RNA is a rapidly deteriorating molecule that requires specialized stabilization (Camacho-Sanchez et al., 2013), and many museum specimens are not preserved with RNA in mind. While RNA is less stable than DNA, RNA can persist even in ancient plant and animal tissues (Fordyce et al., 2013; Shaw et al., 2019; Smith et al., 2019) and ancient RNA methods have been used to examine viruses (Castello et al., 1999; Fanning et al., 2002; Smith et al., 2014; Düx et al., 2020). The persistence of RNA in historical and ancient tissues supports the value of natural history specimens in examining viral pathogens through time and other downstream uses, including host gene expression profiles.
Here, we examine the quality and quantity of RNA that can be extracted from bat specimens ranging in age (19–136 years old) and varying in preservation method (i.e., formalin-fixed ethanol-preserved at room temperature, ethanol-preserved and stored at room temperature, ethanol-preserved and frozen, and flash-frozen without buffer; Figure 1). We present an optimized protocol for extracting RNA from formalin-fixed specimens that is refined from existing protocols developed for extracting RNA from formalin-fixed paraffin-embedded (FFPE) tissues (Krafft et al., 1997; Fanning et al., 2002; Sharma et al., 2012). We used a suite of tools to confirm the success of RNA extractions and examine the downstream usability of these extractions, including Qubit, Bioanalyzer, qPCR, and RNA-seq. This research builds on the growing body of evidence that natural history specimens capture an extended suite of data that can be used beyond the original intent for which that specimen was vouchered, reinforcing the value of natural history collections.
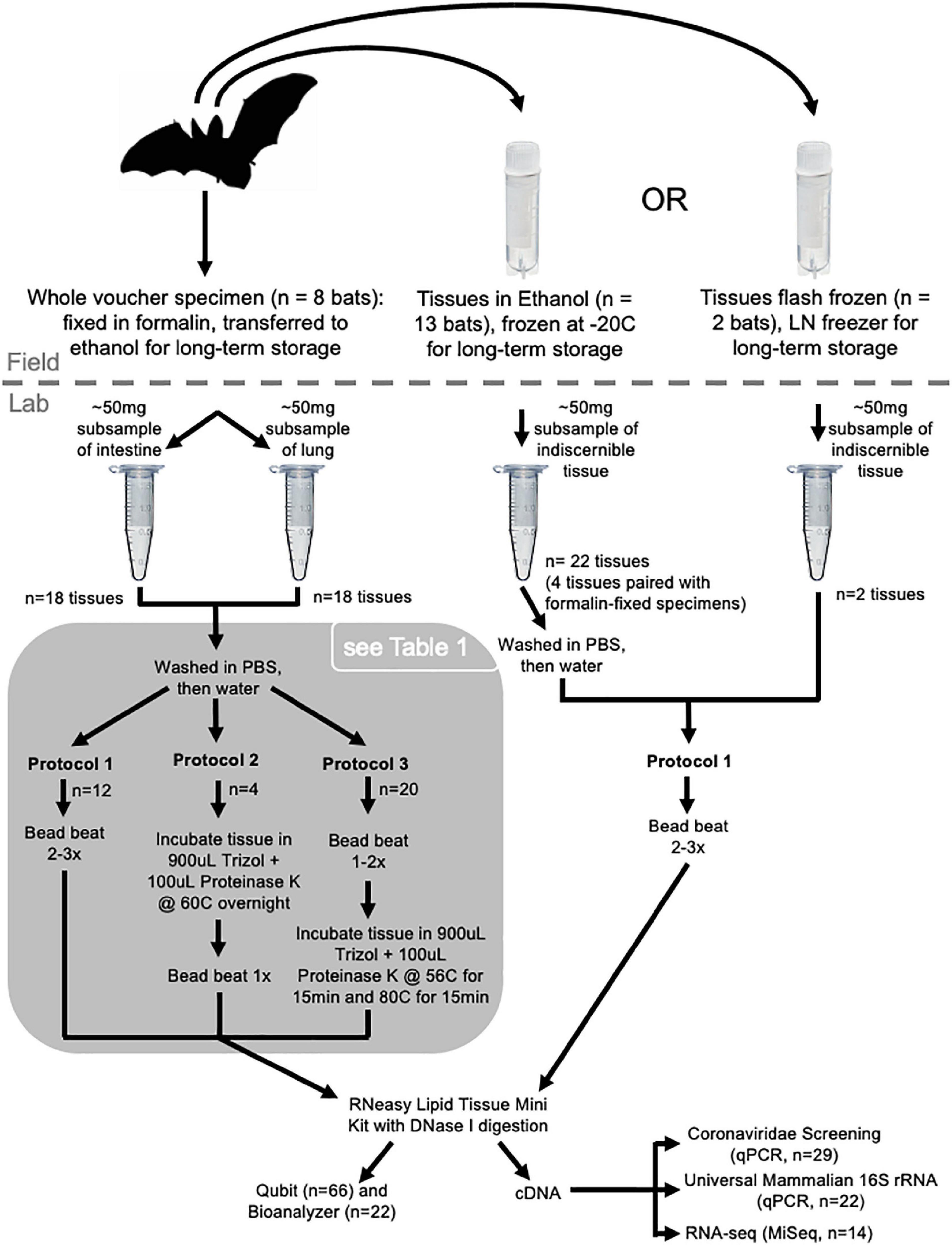
Figure 1. Sampling and processing design. Whole voucher specimens collected in 1886 and 1888 (n = 3 bats, n = 6 tissues; ethanol-RT) are not shown in the figure.
We sampled bat specimens and tissues (n = 22 unique bats) housed in the Smithsonian National Museum of Natural History (NMNH) that represented species from five genera (Rhinolophus, Hipposideros, Megareops, Cynopterus and Nyctalus) collected in 1886, 1888, and 2002–2003 in Myanmar, and in 1989 in China (Supplementary Table 1). Whole voucher specimens were either preserved in ethanol at room temperature (1886, 1888: ethanol-RT) or were fixed in formalin and transferred to ethanol for long-term storage (2002–2003: formalin-fixed). From some of these whole specimens (n = 4), organ and/or muscle tissue was sampled in the field and preserved in ethanol and then frozen for long-term storage at –20°C (ethanol-F). For other bats, only organ and muscle tissues were available. Some tissues sampled in the field were flash frozen using liquid nitrogen and stored in vapor phase liquid nitrogen freezers (1989: flash-frozen). As there are multiple sample types taken from the same bat individual, we use paired tissue subsamples to examine the impact of preservation method on RNA (Supplementary Table 1 and Figure 1).
We collected lung and small intestine tissue samples from formalin-fixed bat vouchers (n = 8 bats; Figure 1). Specimens were removed from their jars and blotted dry to remove excess 70% ethanol. Next, thoracic and abdominal cavities were dissected using sterilized instruments (forceps, scissors, hemostats and scalpels) treated with RNase AWAY™ (Thermo Fisher Scientific, Waltham, MA, United States). Approximately 50 mg subsamples of lung and small intestine were weighed and then placed in a 1.5 mL tube containing PBS buffer (to remove remaining ethanol), shaken for approximately 5 s, moved to a 1.5 mL tube containing ddH20, again shaken for 5 s, and finally transferred to a 1.5 mL tube containing Trizol™ buffer (Invitrogen, Waltham, MA, United States). After dissections were completed, all tubes were transferred to a –20°C freezer for temporary storage and then transferred to a –80°C freezer until extraction occurred. Ethanol-RT bat vouchers pre-dated the use of formalin in museums (collected in 1886 and 1888). Tissues from these specimens were sampled in the same way as formalin-fixed vouchers.
A subsample of frozen tissue samples preserved in ethanol or flash frozen were loaned from the NMNH Biorepository and stored at –80°C until extraction. During subsampling, all instruments were treated with RNase AWAY™. Frozen tissues were moved from –80°C to a –20°C freezer for approximately 1 h prior to subsampling. Flash-frozen tissues were then stored at 4°C and processed individually. Tubes containing ethanol-F tissues were removed from the –20°C freezer one at a time and stored on ice during subsampling. It was not possible to discern the tissue type of these subsamples as multiple organ types (usually heart, liver, lung, kidney, spleen) and muscle are frequently sampled in the field and put in the same tube for long-term storage. These tissues do not always maintain diagnostic morphology during long-term storage and become indiscernible from each other. Prior to extractions tissues were weighed to confirm they did not exceed ∼50 mg and washed in 1× nuclease-free PBS and nuclease-free water (all except flash-frozen samples) as described for tissues sampled from fluid vouchers. Following this washing step, samples were transferred to Trizol™ and extracted immediately.
Following the PBS and water washes for samples in ethanol (all except flash-frozen samples), all tissues were transferred to Trizol™ and either processed immediately or stored frozen until RNA extraction. We tested three protocols for extracting RNA from the tissues (Figure 1). Six negative controls used during extraction yielded no measurable RNA.
For protocol 1, tissues were homogenized manually or by bead-beating and then RNA was extracted using the RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany). Ethanol-RT samples were collected in 1886 and 1888, and therefore were processed in the Smithsonian National Zoo and Conservation Biology Institute’s ancient DNA laboratory. These samples were homogenized in 40μL Trizol using BioMashers II™ (Kimble, Rockwood, TX, United States) until no chunks of tissue were visible. Then 960μL of Trizol was added, the pestle was removed, and the tube was centrifuged for 1 min. Supernatant was transferred from the BioMasher tube to a screw cap tube before proceeding with the RNeasy Lipid Tissue Mini Kit extraction protocol, beginning after the homogenization steps in the Kit handbook (start at step 12, the first step under Preparation of Total RNA, handbook v. 07/2018). Ethanol-F samples were homogenized using a Mini-BeadBeater-96 (BioSpec, Bartlesville, OK, United States) and one 3 mm chrome steel bead. Prior to use, we soaked beads in RNase AWAY™ for 5 min and washed them twice with RNase-free water. We poured off the water washes and irradiated the beads with UV light for 5 min (UV Clave, Benchmark Scientific, Sayreville, NJ, United States) before transferring one bead to a screw-cap tube containing tissue sample and 1 mL Trizol buffer. Each sample was bead beat two times at maximum speed (40 oscillations/second) for 30 s and incubated at –20°C for 2 min following each bout of bead beating. If large chunks of tissue were visible, we repeated bead beating and incubation once more. Supernatant was transferred to a new tube and RNA extraction proceeded using the RNeasy Lipid Tissue Mini Kit (step 12 as above). Kit extraction followed the manufacturer’s protocol and included DNase I digestion (RNase-free DNase Set, Qiagen). RNA was eluted in 40 μL of RNase-free water and the elution was repeated using the original eluate to re-wet the filter as recommended to increase RNA concentration. Extractions were split into two aliquots to reduce freeze-thaw cycles and stored at –80°C.
Protocols 2 and 3 were optimized from Protocol 1 and Sharma et al. (2012) to improve RNA yield from formalin-fixed samples. For protocol 2, tissues in 900 μL Trizol and 100 μL Proteinase K (Qiagen) were incubated overnight at 60°C with agitation and then bead beat once as described above. Following bead beating, the supernatant was moved to a new tube and RNA extraction proceeded using the RNeasy Lipid Tissue Mini Kit as described in protocol 1. To improve access of Proteinase K to tissues, we switched the order of the homogenization and digestion steps in protocol 3. For protocol 3, tissues in 900μL Trizol and 100 μL Proteinase K were bead beat 1–2 times as described in protocol 1, followed by incubation with agitation at 56°C for 15 min, then 80°C for 15 min. Following incubation, the supernatant was moved to a new tube for extraction with the RNeasy Lipid Tissue Mini Kit as described in protocol 1.
To examine the quality and downstream use of RNA derived from museum specimens, we estimated RNA yield for all samples using Qubit (n = 66; Invitrogen, RNA HS or BR assay; Supplementary Table 1), the RNA Integrity Number (RIN) and DV200 (proportion of RNA fragments > 200 nucleotides in length) for 22 representative samples, and 260/280 and 260/230 ratios of RNA purity using NanoDrop, Thermo Fisher Scientific, Waltham, MA, United States for 56 representative samples. RIN and DV200 are a measures of RNA degradation and were quantified using the Bioanalyzer RNA 6000 Pico (Agilent, Santa Clara, CA, United States) Eukaryote Total RNA analysis following the manufacturer’s instructions (Supplementary Figures 1, 2). We compared RNA purity (i.e., 260/280 and 260/230 ratios) across preservation methodologies and, within formalin-fixed samples, across extraction protocols using one-way ANOVA. We used Tukey’s HSD to compare all groups to each other if a significant difference was detected. Friedman’s test was used to compare RNA purity between intestine samples used in optimization of the RNA extraction protocol from formalin-fixed samples.
For qPCR, we synthesized cDNA from RNA extractions using the ProtoScript II First Strand cDNA Synthesis Kit (New England Biolabs, Ipswich, MA, united States) using the Randomized Primer Mix and following the manufacturer’s instructions. We confirmed the presence of mammalian RNA in 22 representative samples by targeting a 100 bp region of the 16S rRNA gene using universal mammalian primers (Tillmar et al., 2013) using the SsoAdvanced SYBR Green Supermix (BioRad, Hercules, CA, United States), following the manufacturer’s instructions for a 20 μL final reaction volume. Reactions were incubated at 95°C for 30 s followed by 40 cycles of 95°C for 15 s and 58°C for 30 s. We included negative and positive controls (Leontopithecus rosalia, Callithrix geoffroyi, Choloepus didactylus, and Desmodus rotundus) with each assay.
We screened 29 samples (derived from 15 unique bats) for viruses in the subgenus Sarbecoronavirus by targeting the N gene region (HKU-N; primers and probe from Chu et al., 2020) and more broadly for alpha- and betacoronaviruses by targeting the RdRp gene region (RdRP; primers and probe I and probe III originally developed by Muradrasoli et al., 2009 and modified by Joffrin et al., 2020). Our sample size is smaller than what has previously been used for screening bats for coronaviruses (Joffrin et al., 2020). For the HKU-N assay, each 25 μL reaction contained 12.5 μL KlearKall Hot Start 2× Master Mix (LGC, Biosearch Technologies, Hoddeston, United Kingdom), 0.5 μM of each forward and reverse primer, 0.2 μM of Cy5-labeled probe, 20 μg BSA, and 2.5 μL cDNA. Reactions were incubated at 95°C for 15 min per manufacturer’s instructions, followed by 50 cycles of 95°C for 15 s then 58°C for 45 s. For the RdRp assay, 20 μL were used with each reaction containing 10 μL Luna Universal Probe qPCR 2x Master Mix (New England Biolabs), 0.4 μM of each forward and reverse primer, 0.2 μM FAM-labeled probe I, 0.2 μM HEX-labeled probe III, 20 μg BSA, and 2.5 μL cDNA. Thermal conditions followed Joffrin et al. (2020), with an initial incubation at 95°C for 1 min, followed by 2 cycles of 95°C for 15 s and 56°C for 30 s, 2 cycles of 95°C for 15 s and 54°C for 30 s, 2 cycles of 95°C for 15 s and 52°C for 30 s, and 50 imaged cycles of 95°C for 15 s and 50°C for 30 s. All virus-screening assays were performed in duplicate and included negative and positive controls (IDT Gblocks) with each assay.
We sequenced a subset of cDNA libraries to evaluate the composition of extracted products. Library preparation was performed following Hawkins et al. (2016) with a KAPA 528 Biosystems LTP Library Preparation kit Roche, Basel, Switzerland, and with UGA iTru style dual indices (Glenn et al., 2016). Due to the low input amount ∼10 μL and low concentration, libraries were amplified for 30 cycles instead of 14 as described in Hawkins et al. (2016). Following amplification, a 1 × SPRI purification (Rohland and Reich, 2012) was performed to remove primer and adapter dimer. Qubit fluorometry and TapeStation, Agilent, Santa Clara, CA, United States traces were completed for each sample to recover both the concentration and size distribution of each library. An Illumina MiSeq 2 × 150 PE v2 run was performed on 14 samples and two controls (Supplementary Table 1). Due to the insert length, the run was limited to 75 cycles.
Samples were demultiplexed by MiSeq Reporter software and adapters were trimmed using cutadapt v.2.4 (Martin, 2011). Sequence quality was assessed before and after trimming using fastqc v.0.11.8 (Andrews, 2010). Trimmed reads were mapped to GenBank reference genomes using STAR v.2.7.10a (Dobin et al., 2013) and Bowtie2 v.2.3.5 (Langmead and Salzberg, 2012) using default parameters. Reads were also mapped to reference transcriptomes when available using Bowtie2. For samples in the family Pteropodidae, reads were mapped to the Cynopterus brachyotis genome (GCA_009793145.1; Chattopadhyay et al., 2020) and the Rousettus aegyptiacus genome and transcriptome (GCF_014176215.1; Jebb et al., 2020). For samples in the genus Hipposideros, reads were mapped to the reference genome and transcriptome of Hipposideros armiger (GCF_001890085.1; Dong et al., 2017). For samples in the genus Rhinolophus, reads were mapped to the reference genome and transcriptome of Rhinolophus ferrumequinum (GCF_004115265.1; Jebb et al., 2020). In instances where multiple libraries were prepared for the sample bat individual (i.e., from replicate tissues), reads were concatenated for mapping. The function featureCounts in the Subread package was used to examine the genes to which reads mapped (Liao et al., 2013, 2014).
Metagenomic analysis was performed on sequenced reads to evaluate content using the software MEGAN6 Community Edition (Huson et al., 2007; Bağcı et al., 2021). Prior to taxonomic assignment from MEGAN, the DIAMOND (Buchfink et al., 2015) protein BLAST method was performed on the Smithsonian High Performance Computing Cluster using the Genbank NR database to compare all sequenced reads. Following DIAMOND, the .daa files were imported to MEGAN using the February 2022 database “MEGAN map.” All individual files were imported from DIAMOND input and “MEGANIZED” to make RMA6 files. Comparisons were performed between samples where replicates were sequenced as well as across individuals. Sample preservation, tissue type, and specimen were all used in comparisons.
We optimized our RNA extraction protocol using replicate lung and intestine tissues sampled from four formalin-fixed bat individuals (Table 1). Extractions from formalin-fixed tissues that were homogenized by bead beating twice prior to Proteinase K digestion more reliably yielded measurable RNA via Qubit quantification than samples homogenized with only one round of bead beating or those extracted without Proteinase K (Table 1). No tissue subsamples yielded measurable RNA when we extracted following Protocol 1, and two tissues that yielded measurable RNA when bead beat twice did not yield measurable RNA when bead beat once. In all cases, lung tissues did not yield measurable RNA. This is likely due to variation in how quickly formalin was able to penetrate these tissues compared to the intestine when the bat was originally preserved. We detected RNA from other formalin-fixed lung tissue (i.e., 583861c,f,g; Supplementary Table 1), suggesting that the specific preservation protocol used in the field may have substantial impact on resulting RNA preservation. We found that Proteinase K incubation and one additional bead-beating step did not impact RIN quality estimates.
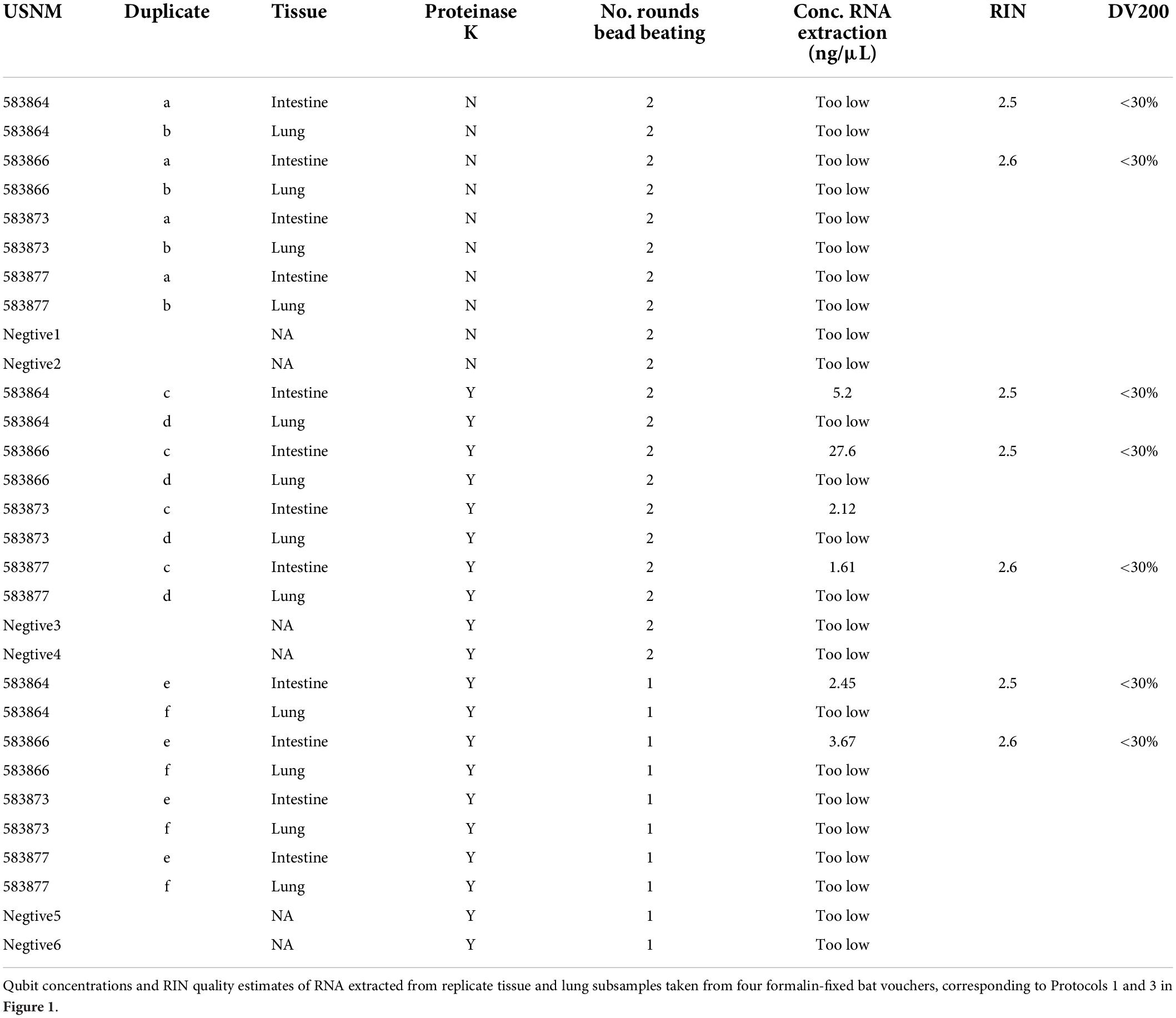
Table 1. Optimization of RNA extraction from formalin-fixed tissues.
The quality and quantity of RNA extracted from museum specimens varied and was related to preservation method (Figure 2). While there was no significant difference in the 260/280 ratio between preservation methods [one-way ANOVA: F(2, 47) = (0.226), p = 0.799], formalin-fixed samples typically had a 260/280 ratio lower than the target of 2, likely indicating protein contamination (Figure 2C). The 260/280 ratio got closer to the target of 2 when Proteinase K was used in the extraction [i.e., protocols 2 and 3; Figure 2D; one-way ANOVA: F(1, 31) = (3.659), p = 0.0654]. Estimates of RNA purity using 260/230 ratios are largely consistent with evidence from 260/280 ratios, except that there is a significant difference between the mean 260/230 ratios observed in formalin-fixed and ethanol-F samples [Supplementary Figure 3; one-way ANOVA: F(2, 47) = (8.037), p = 0.001; Tukey’s HSD: p = 0.0014, 95% C.I. = (–2.05, –0.44)]. There was no significant difference in RNA purity measured from repeated extractions of intestine tissue from the same formalin-fixed bat individuals [Friedman’s test: χ2(2) = 3.4545, p = 0.178]. Flash-frozen samples yielded the highest RIN values (4.5, 4.7) and high RNA quantity, while ethanol-F and formalin-fixed samples typically yielded lower RIN values (1.6–2.7) and low RNA quantity (Figure 2A). However, six ethanol-F samples yielded high RNA quantity (>1 μg/μL). All ethanol-F samples yielded detectable RNA, while many formalin-fixed samples did not. No ethanol-RT samples yielded measurable RNA; these individuals were collected in 1886 and 1888 and are much older than the rest of our samples. RNA quantity varied by individual bat, again suggesting a strong impact of the specific field preservation protocol on RNA persistence (Figure 2B). Tissue type did not influence RNA yield (Wilcoxon signed rank test, p = 0.078). Six negative controls yielded no detectable RNA, suggesting lab precautions were sufficient to protect even poorly preserved tissues from contamination during extraction.
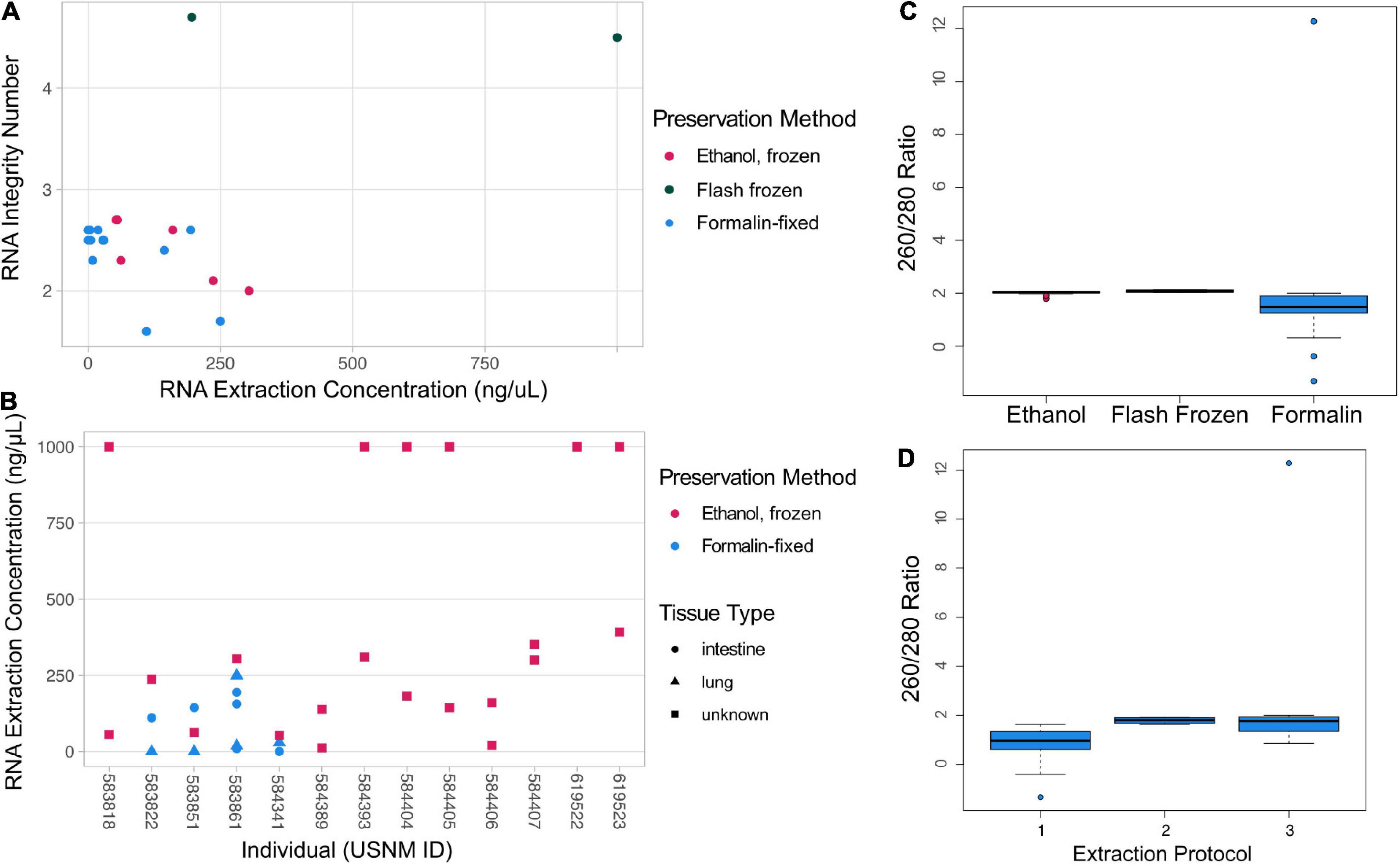
Figure 2. RNA Quality and Quantity. (A) RNA extraction concentration by RIN for different sample types with preservation method indicated by color. (B) Plot of the RNA extraction concentrations for individual bats (NMNH identification number, USNM ID) that had duplicate tissue samples taken. In this plot preservation is mapped as color and tissue type is indicated by shape. In both (A,B) plots, samples that have an indicated concentration of 1,000 ng/μL were above the detection threshold of the Qubit HS Kit and should be interpreted as having at least 1,000 ng/μL concentrations. (C) Estimate of RNA purity using Nanodrop 260/280 ratio compared to preservation of tissues used for extractions. (D) Comparison of Nanodrop 260/280 ratio across protocols used to extract RNA from formalin-fixed tissues. A 260/280 ratio of ∼2 is considered pure RNA.
All samples screened using mammalian universal 16S rRNA primers showed successful amplification with Cq values comparable to those of positive controls (i.e., modern mammal DNA). There was no impact of preservation on qPCR amplification. We did not detect any coronaviruses using our targeted qPCR assays (n = 15 bats; n = 29 tissues).
A small proportion of the RNA-seq data was mappable to bat genomic/transcriptomic references, as is expected for highly degraded libraries. A total of 34,484,466 reads passed sequencing quality filters. Of the reads passing filters 79.3% were demultiplexed (20.7% undetermined reads); the high proportion of undetermined reads is likely from excess sequencing adapters forming dimers. Following adapter trimming, the number of reads was reduced (ranging from 4,867 to 36,790 remaining per sample). Endogenous RNA content, estimated by mapping reads to annotated genomes, ranged from 1.1 to 8.71% using splice-aware mapping (i.e., STAR). Estimates of endogenous content were slightly higher when reads were mapped to reference genomes using Bowtie2, ranging from 3.1 to 18.86%. Overall alignment rate varied less when reads were mapped to transcriptomes, but was less successful (0.73–4.75%). Of the uniquely mapped reads for Hipposideros armiger and H. bicolor, the most well-represented species in our data, most aligned to the MAT1A gene (Supplementary Table 2). Reads were mapped to other genes, but coverage was shallow across the board.
Metagenomic analysis revealed high representation of bacteria in sequenced reads, with some reads mapping to mammals and viruses (Figure 3). There was large variation in the representation of different bacterial taxa between replicates, which did not correspond to preservation (Figure 3A) or tissue type (Figure 3B). Ordination of metagenome communities indicated differentiation between bat species (Figure 3D). A low proportion of reads mapped to taxa not likely represented in our sample, possibly as a consequence of the short read length or biases from the GenBank NR database.
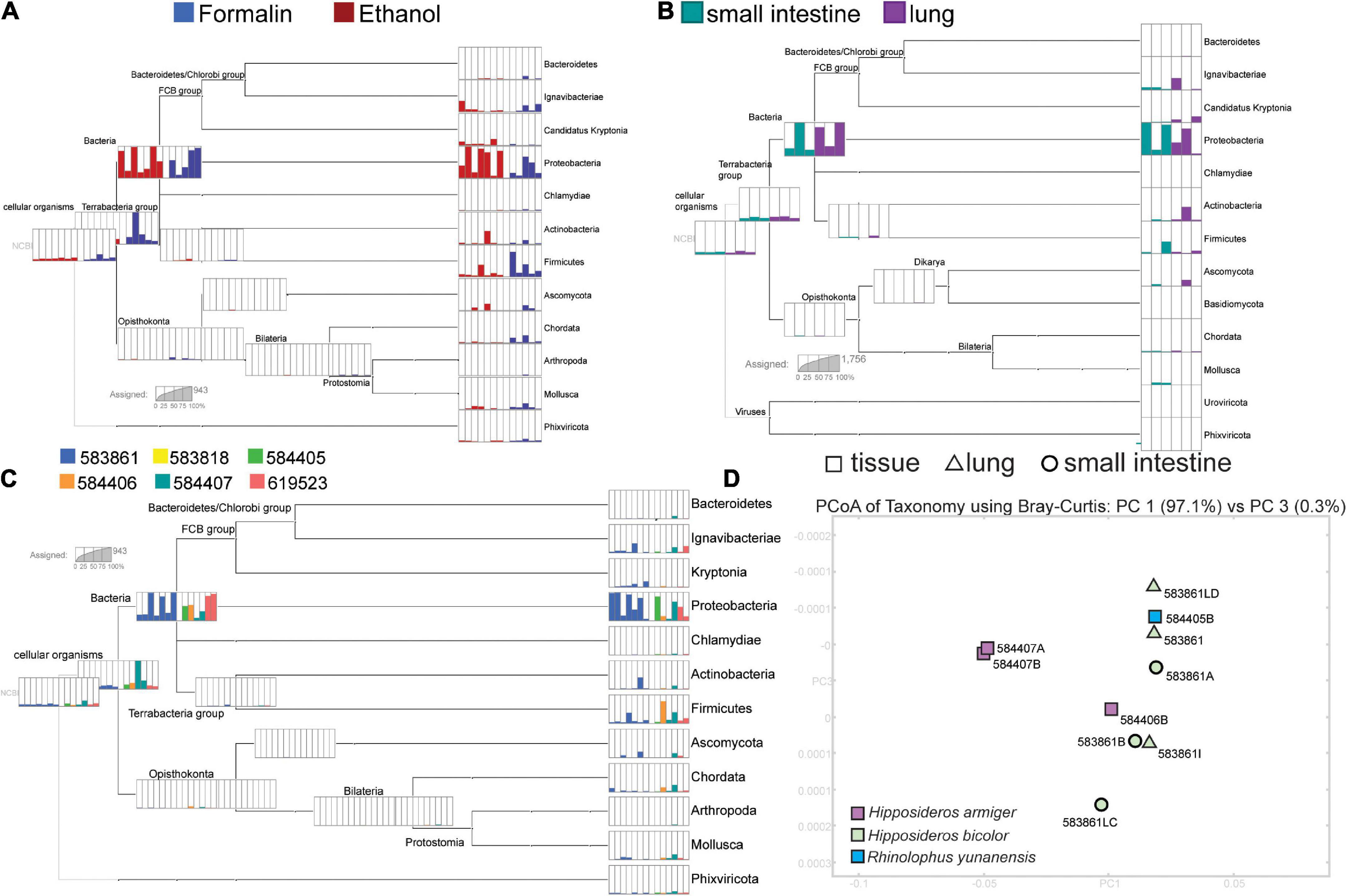
Figure 3. Results from the MEGAN analysis. Parts A-C each have separate scales inset in each section to indicate read counts. (A) The relative composition of phyla detected from of each sequenced library separated by preservation type. (B) Variation in phyla recovered across lung and small intestine replicates from specimen USNM 583861 (Hipposideros bicolor). (C) The relative proportions of reads identified to phylum from each sequenced library, colored by individual bat (USNM ID). (D) A PCoA of all sequenced insectivorous bats, with PC’s 1 and 3 shown. Tissue type is indicated by the shape (corresponding to Figure 2), and color indicates the species as shown in the bottom left corner of the PCoA.
Museum specimens are an underutilized resource in building foundational knowledge of zoonotic viruses (Colella et al., 2021; Thompson et al., 2021), other emerging infectious diseases (Talley et al., 2015; Byrne et al., 2019), and host gene expression responses to environmental change. These specimens can be used to screen a broad range of host species for pathogens that would be difficult to sample in the wild, track the host and geographic occurrence of pathogens through time, and gain historical snapshots of host and pathogen evolution (Colella et al., 2021; Thompson et al., 2021). However, methods have not been optimized for deriving RNA from natural history specimens, limiting the use of these specimens in RNA virus screening. Here, we present an optimized protocol for extracting RNA from formalin-fixed specimens and explore the quality and quantity of RNA that can be derived from museum specimens, extending the value and possible uses of these specimens.
The RNA extracted from museum specimens is highly degraded, but usable for downstream applications, including qPCR and sequencing (Figure 3 and Supplementary Table 2). Following recommendations from FFPE protocols (Krafft et al., 1997; Sharma et al., 2012), we found that incubation of formalin-fixed samples with Proteinase K following thorough homogenization improves RNA yield (Table 1). RNA from formalin-fixed and ethanol-F samples is highly degraded, but may include persistent mRNA as shown by our recovery of bat gene transcripts in RNA-seq data. We did not detect viral RNA in RNA-seq data or targeted qPCR methods, possibly due to the small number of bat individuals screened for viruses in our study. We found that a high proportion of RNA is of bacterial origin, which is likely due to persistence of rRNA in these degraded samples and may also be reflective of database bias, as bacterial rRNA is well-represented on GenBank. We suggest that highly degraded samples may be better suited for targeted approaches, like RT-PCR and qPCR (Castello et al., 1999; Fanning et al., 2002; Worobey et al., 2016).
Age and field preservation are the most important factors influencing the quantity and quality of RNA derived from museum specimens (Figure 2). Flash-frozen samples had high RIN values compared to ethanol-F and formalin-fixed samples. While these RIN values are lower than typically targeted for contemporary tissues (RIN > 7), these samples are likely still valuable for RNA-seq applications or other more targeted approaches. Within the ethanol-F and formalin-fixed samples, there was variation in RNA quantity between individual bats, which may indicate lasting impact of field-based preservation protocol. For example, the amount of time between when a bat was euthanized and when its tissues were sampled and preserved has a large impact on quality and quantity of RNA remaining in those tissues (Camacho-Sanchez et al., 2013). The details of in-the-field preservation method matter for the quantity of RNA derived from these samples and should be viewed as valuable specimen metadata. However, this type of metadata is not often recorded in enough detail to tease apart the preservation, storage, and handling of a specimen.
The practice of storing multiple tissue types within one sample tube, a common practice in field mammalogy, is not ideal for viral zoonotic disease screening and gene expression studies. In many instances, viruses are known to aggregate differentially across tissue types and gene expression studies often seek to compare expression profiles between tissues. Long-term storage of different tissue types in the same vial can make it difficult to separate and differentiate them as tissues do not maintain distinct morphology through long periods of storage, even at cryogenic temperatures. While separating tissues into individual tubes has its own limitations (i.e., space, sample tracking), we suggest that, when possible, storing each tissue type in an individual tube may improve the value of these tissues for pathogen screening and gene expression studies.
We find that museum specimens are a valuable source of RNA, even in cases where tissues have not been preserved with RNA in mind. This finding broadens the use of historical specimens in pathogen detection to include viruses. Further work is needed to examine the persistence of mRNA compared to rRNA in these specimens. However, findings from aRNA research provide evidence that mRNA may be maintained under specific conditions through thousands of years (Schmitt et al., 2018). Through efforts to derive new information from existing specimens, we continue to reaffirm the value of natural history collections and the necessity of expanding and maintaining these critical scientific resources.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s. Raw sequence data has been uploaded to NCBI SRA database. Available at: https://www.ncbi.nlm.nih.gov/sra/PRJNA838638.
KS, MH, CM-W, MM, RF, JM, and MC designed the study. MH, CM-W, MM, RF, JM, and MC secured funding. KS, MH, MF, and CM-W collected and analyzed the data. All authors contributed to writing and revising the manuscript.
KS was funded by the Smithsonian Institution Biodiversity Genomics Postdoctoral Fellowship and George E. Burch Postdoctoral Fellowship in Theoretical Medicine. CM-W was funded by the Robert and Arlene Kogod Secretarial Scholar’s donation.
We thank Suzan Murray for input early in the project and Smithsonian Channel’s Mission Critical program for featuring this research in the film “Virus Hunting: Caves to COVID. We also thank the Smithsonian Institution’s Office of the Undersecretary of Science and Research and the Smithsonian Channel’s Mission Critical program for research funding. Gratitude is extended to Nancy McInerney (Smithsonian Conservation Biology Institute, Center for Conservation Genomics), Katie Murphy (NMNH Laboratory of Analytical Biology), Chris Huddleston and Daniel DiMichele (NMNH Biorepository), and Darrin Lunde (NMNH Division of Mammals) for assistance in tissue acquisition and data generation. Some of the computations in this manuscript were conducted on the Smithsonian High Performance Cluster (SI/HPC; https://doi.org/10.25572/SIHPC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.953131/full#supplementary-material
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed February 1, 2022).
Bağcı, C., Patz, S., and Huson, D. H. (2021). DIAMOND+MEGAN: fast and easy taxonomic and functional analysis of short and long microbiome sequences. Curr. Protoc. 1:e59. doi: 10.1002/cpz1.59
Becker, D. J., Albery, G. F., Sjodin, A. R., Poisot, T., Bergner, L. M., Chen, B., et al. (2022). Optimising predictive models to prioritise viral discovery in zoonotic reservoirs. Lancet Microbe [Epub ahead of print]. doi: 10.1016/S2666-5247(21)00245-7
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using Diamond. Nat. Methods 12, 59–60.
Byrne, A. Q., Vredenburg, V. T., Martel, A., Pasmans, F., Bell, R. C., Blackburn, D. C., et al. (2019). Cryptic diversity of a widespread global pathogen reveals expanded threats to amphibian conservation. Proc. Natl. Acad. Sci. U. S. A. 116, 20382–20387. doi: 10.1073/pnas.1908289116
Camacho-Sanchez, M., Burraco, P., Gomez-Mestre, I., and Leonard, J. A. (2013). Preservation of RNA and DNA from mammal samples under field conditions. Mol. Ecol. Resour. 13, 663–673.
Castello, J. D., Rogers, S. O., Starmer, W. T., Catranis, C. M., Ma, L., Bachand, G. D., et al. (1999). Detection of tomato mosaic tobamovirus RNA in ancient glacial ice. Polar Biol. 22, 207–212.
Chattopadhyay, B., Garg, K. M., Ray, R., Mendenhall, I. H., and Rheindt, F. E. (2020). Novel de Novo Genome of Cynopterus brachyotis Reveals Evolutionarily Abrupt Shifts in Gene Family Composition across Fruit Bats. Genome Biol. Evol. 12, 259–272. doi: 10.1093/gbe/evaa030
Childs, J. E., Ksiazek, T. G., Spiropoulou, C. F., Krebs, J. W., Morzunov, S., Maupin, G. O., et al. (1994). Serologic and genetic identification of Peromyscus maniculatus as the primary rodent reservoir for a new hantavirus in the southwestern United States. J. Infect. Dis. 169, 1271–1280. doi: 10.1093/infdis/169.6.1271
Chu, D. K. W., Pan, Y., Cheng, S. M. S., Hui, K. P. Y., Krishnan, P., Liu, Y., et al. (2020). Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 66, 549–555.
Colella, J. P., Bates, J., Burneo, S. F., Camacho, M. A., Carrion Bonilla, C., Constable, I., et al. (2021). Leveraging natural history biorepositories as a global, decentralized, pathogen surveillance network. PLoS Pathog. 17:e1009583. doi: 10.1371/journal.ppat.1009583
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dong, D., Lei, M., Hua, P., Pan, Y.-H., Mu, S., Zheng, G., et al. (2017). The Genomes of Two Bat Species with Long Constant Frequency Echolocation Calls. Mol. Biol. Evol. 34, 20–34. doi: 10.1093/molbev/msw231
Düx, A., Lequime, S., Patrono, L. V., Vrancken, B., Boral, S., Gogarten, J. F., et al. (2020). Measles virus and rinderpest virus divergence dated to the sixth century BCE. Science 368, 1367–1370. doi: 10.1126/science.aba9411
Fanning, T. G., Slemons, R. D., Reid, A. H., Janczewski, T. A., Dean, J., and Taubenberger, J. K. (2002). 1917 avian influenza virus sequences suggest that the 1918 pandemic virus did not acquire its hemagglutinin directly from birds. J. Virol. 76, 7860–7862. doi: 10.1128/jvi.76.15.7860-7862.2002
Fordyce, S. L., Ávila-Arcos, M. C., Rasmussen, M., Cappellini, E., Romero-Navarro, J. A., Wales, N., et al. (2013). Deep sequencing of RNA from ancient maize kernels. PLoS One 8:e50961. doi: 10.1371/journal.pone.0050961
Glenn, T. C., Nilsen, R. A., Kieran, T. J., Sanders, J. G., Bayona-Vásquez, N. J., and Finger, J. W. Jr., et al. (2016). Adapterama I: universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). bioRxiv [Preprint]. doi: 10.1101/049114
Hawkins, M. T. R., Hofman, C. A., Callicrate, T., McDonough, M. M., Tsuchiya, M. T. N., Gutiérrez, E. E., et al. (2016). In-solution hybridization for mammalian mitogenome enrichment: pros, cons and challenges associated with multiplexing degraded DNA. Mol. Ecol. Resour. 16, 1173–1188. doi: 10.1111/1755-0998.12448
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386.
Jebb, D., Huang, Z., Pippel, M., Hughes, G. M., Lavrichenko, K., Devanna, P., et al. (2020). Six reference-quality genomes reveal evolution of bat adaptations. Nature 583, 578–584. doi: 10.1038/s41586-020-2486-3
Joffrin, L., Goodman, S. M., Wilkinson, D. A., Ramasindrazana, B., Lagadec, E., Gomard, Y., et al. (2020). Bat coronavirus phylogeography in the Western Indian Ocean. Sci. Rep. 10:6873. doi: 10.1038/s41598-020-63799-7
Kleindorfer, S., and Sulloway, F. J. (2016). Naris deformation in Darwin’s finches: experimental and historical evidence for a post-1960s arrival of the parasite Philornis downsi. Glob. Ecol. Conserv. 7, 122–131.
Krafft, A. E., Duncan, B. W., Bijwaard, K. E., Taubenberger, J. K., and Lichy, J. H. (1997). Optimization of the isolation and amplification of RNA from formalin-fixed, paraffin-embedded tissue: the armed forces institute of pathology experience and literature review. Mol. Diagn. 2, 217–230. doi: 10.1054/MODI00200217
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Liao, Y., Smyth, G. K., and Shi, W. (2013). The subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41:e108. doi: 10.1093/nar/gkt214
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.1089/cmb.2017.0096
Muradrasoli, S., Mohamed, N., Hornyák, A., Fohlman, J., Olsen, B., Belák, S., et al. (2009). Broadly targeted multiprobe QPCR for detection of coronaviruses: coronavirus is common among mallard ducks (Anas platyrhynchos). J. Virol. Methods 159, 277–287. doi: 10.1016/j.jviromet.2009.04.022
Rohland, N., and Reich, D. (2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946.
Schmitt, C. J., Cook, J. A., Zamudio, K. R., and Edwards, S. V. (2018). Museum specimens of terrestrial vertebrates are sensitive indicators of environmental change in the Anthropocene. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374:20170387. doi: 10.1098/rstb.2017.0387
Sharma, M., Mishra, B., Vandana Saikia, U. N., Bahl, A., Ratho, R. K., et al. (2012). Ribonucleic acid extraction from archival formalin fixed paraffin embedded myocardial tissues for gene expression and pathogen detection. J. Clin. Lab. Anal. 26, 279–285. doi: 10.1002/jcla.21518
Shaw, B., Burrell, C. L., Green, D., Navarro-Martinez, A., Scott, D., Daroszewska, A., et al. (2019). Molecular insights into an ancient form of Paget’s disease of bone. Proc. Natl. Acad. Sci. U. S. A. 116, 10463–10472. doi: 10.1073/pnas.1820556116
Smith, O., Clapham, A., Rose, P., Liu, Y., Wang, J., and Allaby, R. G. (2014). A complete ancient RNA genome: identification, reconstruction and evolutionary history of archaeological Barley Stripe Mosaic Virus. Sci. Rep. 4:4003. doi: 10.1038/srep04003
Smith, O., Dunshea, G., Sinding, M.-H. S., Fedorov, S., Germonpre, M., Bocherens, H., et al. (2019). Ancient RNA from Late Pleistocene permafrost and historical canids shows tissue-specific transcriptome survival. PLoS Biol. 17:e3000166. doi: 10.1371/journal.pbio.3000166
Talley, B. L., Muletz, C. R., Vredenburg, V. T., Fleischer, R. C., and Lips, K. R. (2015). A century of Batrachochytrium dendrobatidis in Illinois amphibians (1888–1989). Biol. Conserv. 182, 254–261.
Thompson, C. W., Phelps, K. L., Allard, M. W., Cook, J. A., Dunnum, J. L., Ferguson, A. W., et al. (2021). Preserve a Voucher Specimen! The Critical Need for Integrating Natural History Collections in Infectious Disease Studies. MBio 12, e2698–e2620. doi: 10.1128/mBio.02698-20
Tillmar, A. O., Dell’Amico, B., Welander, J., and Holmlund, G. (2013). A universal method for species identification of mammals utilizing next generation sequencing for the analysis of DNA mixtures. PLoS One 8:e83761. doi: 10.1371/journal.pone.0083761
Valitutto, M. T., Aung, O., Tun, K. Y. N., Vodzak, M. E., Zimmerman, D., Yu, J. H., et al. (2020). Detection of novel coronaviruses in bats in Myanmar. PLoS One 15:e0230802. doi: 10.1371/journal.pone.0230802
Worobey, M., Watts, T. D., McKay, R. A., Suchard, M. A., Granade, T., Teuwen, D. E., et al. (2016). 1970s and “Patient 0” HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 539, 98–101. doi: 10.1038/nature19827
Keywords: Coronaviridae, Chiroptera (bats), natural history collection, historical specimens, RNA
Citation: Speer KA, Hawkins MTR, Flores MFC, McGowen MR, Fleischer RC, Maldonado JE, Campana MG and Muletz-Wolz CR (2022) A comparative study of RNA yields from museum specimens, including an optimized protocol for extracting RNA from formalin-fixed specimens. Front. Ecol. Evol. 10:953131. doi: 10.3389/fevo.2022.953131
Received: 25 May 2022; Accepted: 13 July 2022;
Published: 02 August 2022.
Edited by:
Jonathan J. Fong, Lingnan University, ChinaReviewed by:
Bryan Stuart, North Carolina Museum of Natural Sciences, United StatesCopyright © 2022 Speer, Hawkins, Flores, McGowen, Fleischer, Maldonado, Campana and Muletz-Wolz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly A. Speer, c3BlZXJrQHNpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.