 Kunyuan Wanghe1†
Kunyuan Wanghe1† Chenguang Feng2†
Chenguang Feng2† Yongtao Tang3†
Yongtao Tang3† Delin Qi4
Delin Qi4 Shahid Ahmad5
Shahid Ahmad5 Ghulam Nabi6Xiaohui Li1Guojie Wang7Longsheng Jian7Sijia Liu1Kai Zhao1
Ghulam Nabi6Xiaohui Li1Guojie Wang7Longsheng Jian7Sijia Liu1Kai Zhao1 Fei Tian1*
Fei Tian1*- 1Key Laboratory of Adaptation and Evolution of Plateau Biota, Laboratory of Plateau Fish Evolutionary and Functional Genomics, Qinghai Key Laboratory of Animal Ecological Genomics, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, Qinghai, China
- 2School of Ecology and Environment, Northwestern Polytechnical University, Xi’an, Shaanxi, China
- 3College of Fisheries, Henan Normal University, Xinxiang, Henan, China
- 4State Key Laboratory of Plateau Ecology and Agriculture, College of Eco-environmental Engineering, Qinghai University, Xining, Qinghai, China
- 5School of Ecology and Environment, Hainan University, Haikou, Hainan, China
- 6Institute of Nature Conservation, Polish Academy of Sciences, Kraków, Poland
- 7Highland Hydrobiology and Hydroeco-environment Laboratory of Qinghai Fishery Environmental Monitoring station, Xining, Qinghai, China
Accurately delimiting phylogenetic relationships and taxonomic status is important for understanding species diversity and distributions and devising effective strategies for biodiversity conservation. However, species delimitation is controversial in Gymnocypris eckloni, a schizothoracine fish endemic to the Qinghai–Tibetan Plateau. The aim of this study is robustly identifying the phylogeny of G. eckloni in the Yellow River (YR) population and Qaidam basin (QB) population. The specific-locus amplified fragments sequencing (SLAF-seq) is employed with comprehensively sampling of schizothoracine fishes. In total, 350,181,802 clean reads and 5,114,096 SNPs are identified from SLAF-seq. Phylogenetic analysis recovers a non-monophyletic population of G. eckloni between YR and QB populations, representing an independent phylogenetic relationship between the two populations. Species delimitation analyses by SNAPPER and GMYC methods using the genome-wide SNP data confirm that their taxonomic statuses are separated. This study highlights the importance of further reconsidering clearer taxonomy, which would improve the genetic diversity conservation of Tibetan highland fishes.
Introduction
The Gymnocypris eckloni Herzenstein (Teleostei: Cyprinidae) is a schizothoracine fish endemic to the Qinghai–Tibetan Plateau. This species includes two geographical populations, the Yellow River population (YR) and the Qaidam basin (QB) population. The holotype of G. eckloni was described in 1891 by a specimen from the QB population (Herzenstein, 1888).
The phylogenetic relationship between the YR and QB populations was a controversial topic. Some studies reported that (Wu and Wu, 1992; Li et al., 2020) the morphological and phenotypic characteristics were generally the same between the YR and QB populations of G. eckloni. Based on a single mitochondrial cytochrome b gene, the previous phylogenetic study (He and Chen, 2007) indicated that the two populations formed monophyly (Wu and Wu, 1992; He and Chen, 2007; Li et al., 2020). On the contrary, by increasing species, sample sizes, and the number of informative genetic markers, our previous studies (Zhao et al., 2005, 2009; Zhang et al., 2020) accidentally found that YR and QB populations were clustered into two lineages. The opposite findings might result from the limited information provided by a single mitochondrial cytochrome b gene (Cummings et al., 1995; Zou and Ge, 2008).
Specific locus amplified fragment sequencing (SLAF-seq) is a newly developed simplified deep genome sequencing technology for large-scale single nucleotide polymorphism (SNP) discovery and genotyping, with advantages of low cost, high accuracy, specificity, and repeatability (Chen et al., 2013; Jiang et al., 2015; Liu et al., 2018; Jing et al., 2020). To illustrate the taxonomic status of YR and QB populations of G. eckloni, we thus adopted this advanced method with a comprehensive sampling of schizothoracine fishes (Figure 1), which reached a convincing result based on nuclear markers.
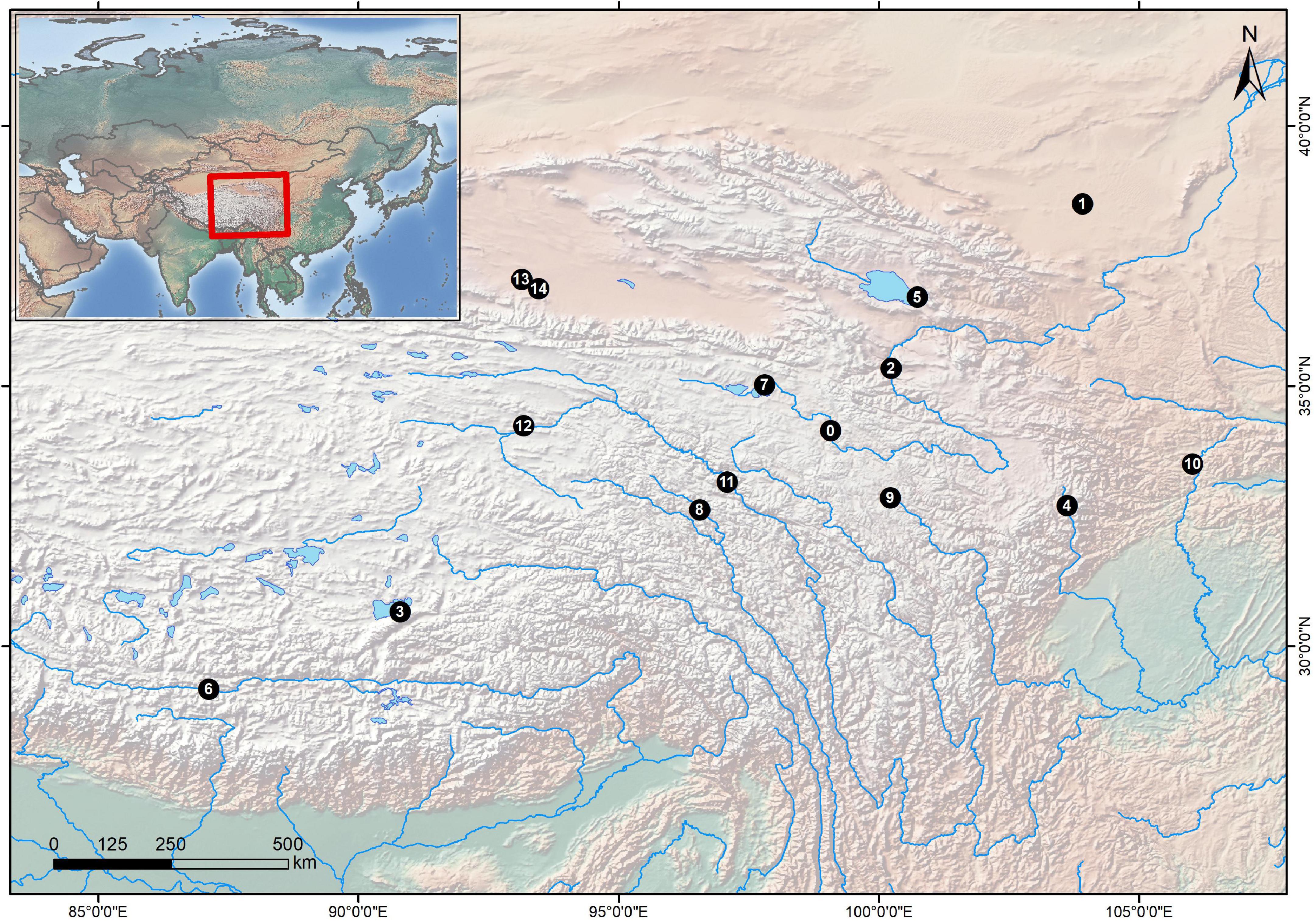
Figure 1. Sampling localities of the specimens. The black points are sampling sites, and the numbers in those points refer to the locality numbers in Table 1.
Materials and methods
Sample collection
The G. eckloni samples were collected in the Yellow River and Qaidam basin. Samples were net-captured to contain pelvic fin and stored immediately in liquid nitrogen for DNA extraction. The Qinghai Provincial Bureau of Fishery approved and supervised field investigations. All the animal experiments were conducted according to procedures described in “Guidelines for Animal Care and Use” and approved by the Animal Care and Use Committee at the Northwest Institute of Plateau Biology, Chinese Academy of Sciences.
Library construction and data processing
Genomic DNA was isolated from the fin using a standard phenol–chloroform method (Sambrook et al., 1989). DNA quality and concentration were assessed by a NanoDrop 2000 spectrophotometer (Nanodrop Technologies, Wilmington, NC, USA) and gel electrophoresis. The preparations of SLAF sequencing libraries were according to the description by Tang et al. (2019). Briefly, genomic DNA was digested by RsaI and HaeIII enzymes, respectively, and then ligated to Duplex Tag-labeled sequencing adapters with T4 DNA ligase (Life Technologies, Carlsbad, CA, USA) (Kozich et al., 2013). PCR reactions were performed using diluted restriction-ligation samples, dNTP, Q5® High-Fidelity DNA Polymerase (NEB China, Beijing, China), and then purified using Agencourt AMPure XP beads (Beckman Coulter, High Wycombe, UK). The 300–700 bp fragments were selected and purified with QIAquick Gel Extraction Kit. According to the manufacturer’s recommendations, the SLAF library was sequenced in the Illumina HiSeq 2500 system (Illumina, Inc., San Diego, CA, USA).
Raw reads of four samples of G. eckloni from two geographic populations, together with raw data from 14 schizothoracine fishes available in the NCBI SRA dataset (Table 1), formed the dataset for the current study.

Table 1. Taxonomic information, sampling sites, and GenBank accession numbers of all species used in phylogenetic analysis.
The raw data were filtered to generate high-quality clean reads, increasing the confidence of variant calling. This step was processed using the FASTQ preprocessor (Chen et al., 2018) in terms of three stringent filtering standards: (1) removing reads with ≥10% unidentified nucleotides; (2) removing low-quality reads (the percentage of bases with quality Phred-scaled quality score ≤ 20); and (3) removing reads aligned to the barcode adapter. Based on the genome size and guanine-cytosine content of G. eckloni, the whole genome sequencing of Gymnocypris przewalskii (NCBI SRA database Bioproject ID PRJNA664553) was used as the reference genome. The clean reads were aligned against that reference genome using the Burrows–Wheeler Aligner (Abuín et al., 2015; Wanghe et al., 2020) with the setting “mem 4 -k 32 –M”, where “k” was the minimum seed length and “-M” was an option used to mark shorter split alignment hits as secondary alignments (Li and Durbin, 2009). Variant calling was performed using the GATK’s Unified Genotyper. SNPs were filtered using GATK’s Variant Filtration with proper standards (-Window 4, -filter “QD < 2.0 || FS > 60.0 || MQ < 40.0”, -G_filter “GQ < 20”).
Phylogenetic analysis
The phylogenetic analysis by the maximum likelihood (ML) method was performed. The ML analyses were constructed using tree-building software raxmlGUI v1.3.1 (Silvestro and Michalak, 2012) under the substitution GTRGAMMAI model, a graphical interface, and a toolkit for phylogenetic analyses using RAxML (Silvestro and Michalak, 2012; Edler et al., 2021). The node support estimation was assessed by 1,000 bootstrap replicates, and the other parameters were set to default. The option of combining all ML trees into a single file was selected to generate a consensus tree (Edler et al., 2021). Oxygymnocypris stewartii was regarded as the out-group for the phylogenetic analysis. The consensus tree was viewed and visualized by FigTree v1.4.4 software (Rambaut, 2009), and the publication-ready figure of the tree was produced by the Adobe Illustrator CC 2020 software (Wang et al., 2019).
Species delimitation
Generalized mixed Yule coalescent model
The Generalized Mixed Yule Coalescent (GMYC) model (Fujisawa and Barraclough, 2013) was implemented to detect shifts in branching rates between intra- and interspecific relationships. The GMYC web server at the multiple-threshold version on https://species.h-its.org/gmyc/ was used to set up this model. We transformed the initial RAxML tree to a time-calibrated phylogenetic tree as the input of the GMYC model by the RelTime method (Tamura et al., 2018). Based on our previous studies (Wanghe et al., 2017; Tang et al., 2019), the most recent common ancestor between Schizopygopsis microcephalus and Schizopygopsis pylzovi set at 1.10–0.70 MA and between Gymnocypris przewalskii and G. eckloni (YR population) was set at 0.15 MA.
SNAPPER analyses
SNAPP (SNP and AFLP Package for Phylogenetic analysis) is a method applied to infer species trees and demographics from independent biallelic markers such as well-spaced SNPs in a full coalescent analysis (Bryant et al., 2012). SNAPPER is a computationally more efficient method compared to SNAPP (Stoltz et al., 2021). The SNAPPER v1.0.2 package in BEAST v2.6.7 (Stoltz et al., 2021) was used to implement this analysis. The parameter of path sampling was set as 48 steps, with MCMC length = 100,000 and pre-burnin = 1,000, following Andrea et al. (Quattrini et al., 2019). Samples were assigned to the following alternative species model (Figure 2): (1) run A, lumping by the current taxonomy delimited based on morphological discrimination, (2) run B, lumping by morphological discrimination but splitting the two populations of G. eckloni; (3) run C, lumping by genus; (4) run D, lumping by GMYC; (5) run E, lumping by basin/habitat; and (6) run F, lumping by basin but merging the two populations of G. eckloni. Marginal likelihood estimates (MLE) were obtained for each different model run in SNAPP analyses. The different species delimitation models were then ranked using the BFD* (Bayes factor delimitation with genomic data) methods (Leaché et al., 2014). Bayes factor (BF) was calculated between each alternative model by subtracting the MLE between two models, then multiplying the difference by two (Eq. 1). A positive BF value indicates support in favor of model 1, and a negative BF value indicates support in favor of model 2 (Kass and Raftery, 1995; Leaché et al., 2014). The strength of support from BF comparisons of competing models can be evaluated using the framework proposed by Kass and Raftery (1995). The BF scale is as follows: 0 < BF < 2 is not worth more than a bare mention, 2 < BF < 6 is positive evidence, 6 < BF < 10 is strong support, and BF > 10 is decisive:
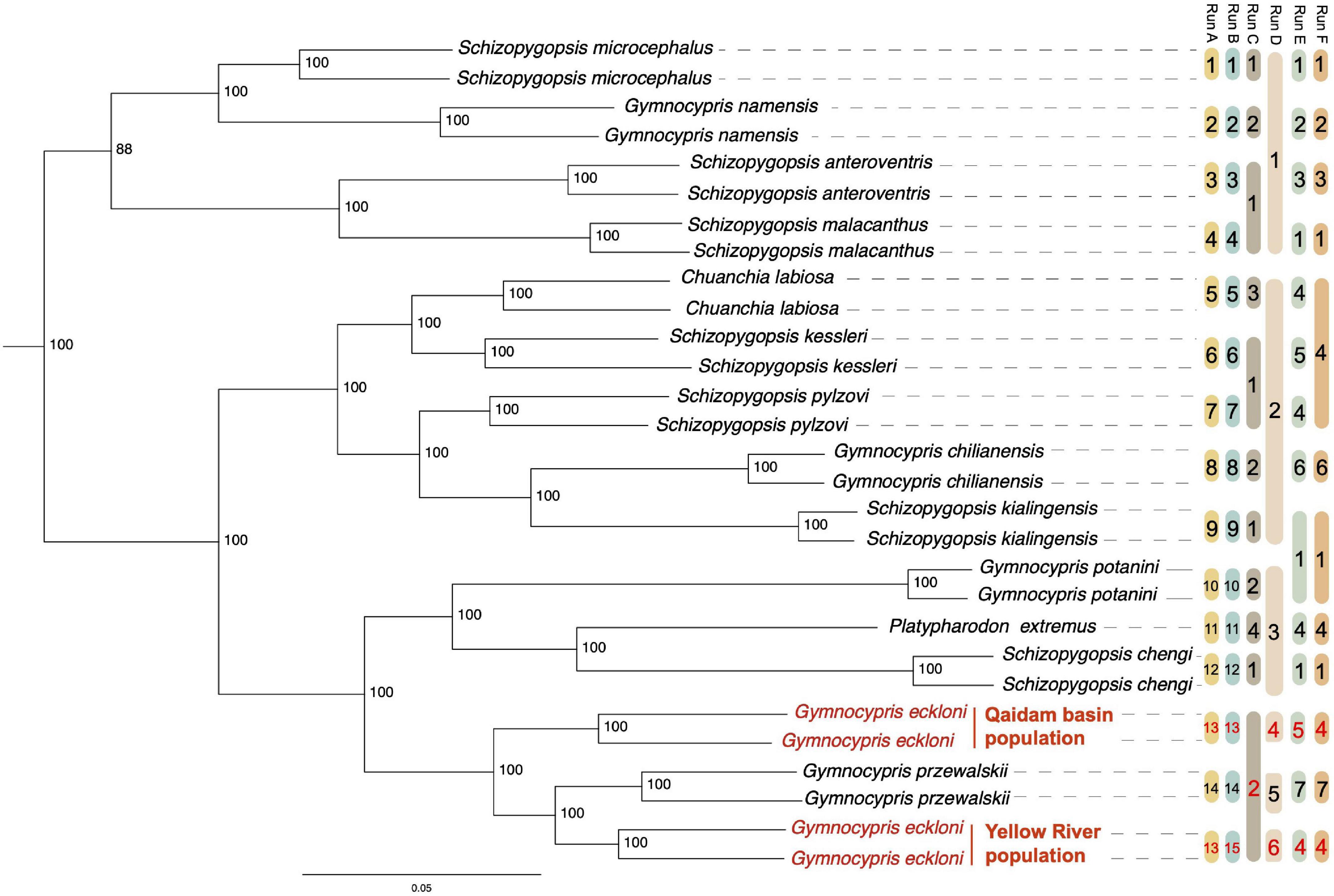
Figure 2. ML tree constructed by SALF-seq, plus a bar named Run D of GMYC results. Bootstrap support values were labeled in each node, and YR and QB populations of Gymnocypris eckloni were highlighted in red. Outgroup Oxygymnocypris stewartii was excluded from this tree. Remarks: The bars from Run A to Run F are the alternative species model for SNAPPER analyses. The same code in a bar refers to the same group for an alternative model. (1) Run A, lumping by the current taxonomy delimited based on morphological discrimination. (2) Run B, lumping by morphological discrimination but splitting the two populations of G. eckloni. (3) Run C, lumping by genus. (4) Run D, lumping by GMYC. (5) Run E, lumping by basin/habitat. (6) Run F, lumping by basin/habitat but merging the two populations of G. eckloni.
In Eq. 1, model 1 and model 2 are the MLE values obtained by two alternative runs of SNAPPER analyses.
Results
Specific-locus amplified fragment sequencing and single nucleotide polymorphism discovery
In total, 31 individuals (Figure 1 and Table 1) generated 350,181,802 clean reads, with an average quality score of 96.81%. We identified 5,114,096 SNPs with a MAF ≥ 0.05 and integrity ≥ 80%. The number of insertions and deletions were 261,371 and 312,411, respectively. All SLAF-seq raw data are available on NCBI SRA with accession numbers. The detailed information on the SNP number, index number, raw reads, clean reads, and mapping the ratio (Supplementary Table 1).
Phylogenetic analyses
Using 5,114,096 high-quality SNPs, 31 accessions from 15 species delimited based on morphological discrimination were classified into three major clades. A majority of the identified morphospecies formed well-supported monophyletic clades in the clades (Run A in Figure 2). While the same genus (Run C in Figure 2) and the fish species in the same basin/habitat (Run E in Figure 2) were not grouped with a monophyletic clade. Interestingly, we found that the YR and QB populations of G. eckloni were not grouped (Figure 2). YR population and G. przewalskii were clustered into one lineage and formed a paraphyletic relationship with the QB populations.
Species delimitation
Results of generalized mixed Yule coalescent
The multi-species coalescent thresholds of the GMYC model were 0.70 and 0.95 MA (Supplementary Figure 1A), indicating that the time before all nodes reflected speciation events and after which all nodes reflected coalescent events (Milan et al., 2020). The GMYC model delimited six primary lineages (Run D in Figure 2 and Supplementary Figure 1B) and recovered YR and QB populations of G. eckloni as separate individual groups.
Results of SNAPPER
The results of species delimitation by SNAPPER (Table 2) also decisively supported two separate individual groups between YR and QB populations of G. eckloni. The higher MLE value indicates a more likely alternative species model. Run A was the currently defined morphospecies with the second maximum MLE. The MLE of Run B was the maximum. Compared with Run A, Run B assumed that the YR and QB populations of G. eckloni were split into two independent species. The BF value between Run A and Run B was –644, which decisively supported in favor of Run B. Compared with Run E, Run F merged the two populations of G. eckloni, but the BF [value = (–2,743 – –2759) × 2 = 32] significantly supported that Run E (i.e., the two populations were separated) was the more likely alternative scenario.

Table 2. SNAPPER results for different species delimitation models.
Discussion
Gymnocypris eckloni is an essential freshwater germplasm species in the Tibetan Plateau (Li et al., 2020). Clarifying the phylogeny of this species would play a significant role in understanding the evolution of highland fishes and their biodiversity conservation. The previous studies (Zhao et al., 2005, 2009; He and Chen, 2007) did not agree on the phylogenetic relationship and taxonomic status of G. eckloni. In this study, we empirically confirmed that the phylogenetic relationship between the two populations was independent and that their taxonomic statuses were separated by applying the SLAF-seq of available schizothoracine fish. Those results accorded with our previous studies (Zhao et al., 2005, 2009), inferring that the YR population of G. eckloni was a substantially older divergence compared with the lineage of the QB population. Some related studies on fish species reported that (Cui et al., 2013; Tang et al., 2019) the convergent evolution, caused by dwelling in the same ecological environment, would produce an extensive reticulate evolution process, resulting in morphological similarity within some genetically close species. The above-discussed research would help explain the misled taxonomic definition of G. eckloni. Therefore, we suggested that a more definite taxonomy of G. eckloni in the Qaidam basin population should be reconsidered to improve the conservation of genetic diversity for this endemic fish.
In future studies, genome-wide SNP data would probably produce some direct evidence (Leaché et al., 2014; Kim and Roe, 2021). Additionally, the hidden morphological divergence between the two populations of G. eckloni needs to be examined by advanced approaches (Li et al., 2020), such as modern geometric morphometrics (Wang et al., 2017) and micro-computed tomography (Li et al., 2020), to test the unknown species taxon in the Qaidam basin population (Tang et al., 2016).
Conclusion
This study using SLAF-seq, a newly developed simplified deep genome sequencing technology for large-scale SNP discovery and genotyping, provide information for further understanding of the phylogenetics, adaptation, and evolution of G. eckloni. Our results emphasized that the phylogenesis between the YR and the QB population of G. eckloni was genetically independent. In summary, the current study underlines the great significance of G. eckloni in the Qaidam basin in protecting Tibetan highland fishes, laying the foundation for reconsidering a more straightforward taxonomy of Tibetan highland fishes and providing new insights for further taxonomic study.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the Table 1.
Ethics statement
All animal experiments were conducted according to procedures described in “Guidelines for Animal Care and Use” and approved by the Animal Care and Use Committee in Northwest Institute of Plateau Biology, Chinese Academy of Sciences.
Author contributions
KW, CF, and YT: conception, sampling, and writing – original draft. DQ and SA: analysis. GN, XL, GW, LJ, and SL: critical review. KZ and FT: funding acquisition and supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Natural Science Foundation of Qinghai province (2020-ZJ-912), National Natural Science Foundation of China under Grants (31870365 and 32071489), China Postdoctoral Science Foundation (No. 2021M693373), and CAS “Light of West China” Program and Joint Grant from Chinese Academy of Sciences–People’s Government of Qinghai Province on Sanjiangyuan National Park (No: LHZX-2020-01). The field works were supported by Sino BON-Inland Water Fish Diversity Observation Network and Investigation of Aquatic Biological Resources in the Aquatic Germplasm Resources Conservation Area of the Qinghai Section of the Yangtze River (No. E039831D01).
Acknowledgments
We thank members from Qinghai Provincial Key Laboratory of Animal Ecological Genomics for their help in sampling and experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.933632/full#supplementary-material
References
Abuín, J. M., Pichel, J. C., Pena, T. F., and Amigo, J. (2015). BigBWA: Approaching the Burrows-Wheeler aligner to Big Data technologies. Bioinformatics 31, 4003–4005. doi: 10.1093/bioinformatics/btv506
Bryant, D., Bouckaert, R., Felsenstein, J., Rosenberg, N. A., and Roychoudhury, A. (2012). Inferring species trees directly from biallelic genetic markers: Bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917–1932. doi: 10.1093/molbev/mss086
Chen, S., Huang, Z., Dai, Y., Qin, S., Gao, Y., Zhang, L., et al. (2013). The development of 7E chromosome-specific molecular markers for Thinopyrum elongatum based on SLAF-seq technology. PLoS One 8:e65122. doi: 10.1371/journal.pone.0065122
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cui, R., Schumer, M., Kruesi, K., Walter, R., Andolfatto, P., and Rosenthal, G. G. (2013). Phylogenomics reveals extensive reticulate evolution in xiphophorus fishes. Evolution (N Y) 67, 2166–2179. doi: 10.1111/evo.12099
Cummings, M. P., Otto, S. P., and Wakeley, J. (1995). Sampling properties of DNA sequence data in phylogenetic analysis. Mol. Biol. Evol. 12, 814–822. doi: 10.1093/oxfordjournals.molbev.a040258
Edler, D., Klein, J., Antonelli, A., and Silvestro, D. (2021). raxmlGUI 2.0: A graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol. Evol. 12, 1–5. doi: 10.1111/2041-210X.13512
Fujisawa, T., and Barraclough, T. G. (2013). Delimiting species using single-locus data and the generalized mixed yule coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 62, 707–724. doi: 10.1093/sysbio/syt033
He, D. K., and Chen, Y. F. (2007). Molecular phylogeny and biogeography of the highly specialized grade schizothoracine fishes (Teleostei: Cyprinidae) inferred from cytochrome b sequences. Chin. Sci. Bull. 52, 777–788. doi: 10.1007/s11434-007-0123-2
Herzenstein, S. M. (1888). Fische. In: Wissenschaftliche Resultate der von N. M. Przewalski nach Central-Asien unternommenen Reisen. Zool. Theil. 3, 1–91.
Jiang, B., Liu, W., Xie, D., Peng, Q., He, X., Lin, Y., et al. (2015). High-density genetic map construction and gene mapping of pericarp color in wax gourd using specific-locus amplified fragment (SLAF) sequencing. BMC Genomics 16:1035. doi: 10.1186/s12864-015-2220-y
Jing, H., Zhekui, O., Wenguang, L., and Maoxian, H. (2020). Genetic structure and diversity analysis of three natural populations of Tectus pyramis based on specific locus amplified fragment sequencing. J. Trop. Oceanogr. 39, 1–18. doi: 10.11978/2019136
Kass, R. E., and Raftery, A. E. (1995). Bayes factors. J. Am. Stat. Assoc. 90, 773–795. doi: 10.1080/01621459.1995.10476572
Kim, K. S., and Roe, K. J. (2021). Genome-wide SNPs redefines species boundaries and conservation units in the freshwater mussel genus Cyprogenia of North America. Sci. Rep. 11:10752. doi: 10.1038/s41598-021-90325-0
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Leaché, A. D., Fujita, M. K., Minin, V. N., and Bouckaert, R. R. (2014). Species delimitation using genome-wide SNP Data. Syst. Biol. 63, 534–542. doi: 10.1093/sysbio/syu018
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, X.-H., Tang, Y.-T., Tian, F., and Zhao, K. (2020). Morphological analysis used by geometric morphometrics combined with micro Ct among gymnocypris eckloni in two drainage (Teleostei: Cyprinidae). Acta Hydrobiol. Sin. 44, 853–861. doi: 10.7541/2020.102
Liu, L., Luo, Q., Teng, W., Li, B., Li, H., Li, Y., et al. (2018). Development of Thinopyrum ponticum-specific molecular markers and FISH probes based on SLAF-seq technology. Planta 247, 1099–1108. doi: 10.1007/s00425-018-2845-6
Milan, D. T., Mendes, I. S., Damasceno, J. S., Teixeira, D. F., Sales, N. G., and Carvalho, D. C. (2020). New 12S metabarcoding primers for enhanced Neotropical freshwater fish biodiversity assessment. Sci. Rep. 10:17966. doi: 10.1038/s41598-020-74902-3
Quattrini, A. M., Wu, T., Soong, K., Jeng, M. S., Benayahu, Y., and McFadden, C. S. (2019). A next generation approach to species delimitation reveals the role of hybridization in a cryptic species complex of corals. BMC Evol. Biol. 19:116. doi: 10.1186/s12862-019-1427-y
Rambaut, A. (2009). FigTree, a graphical viewer of phylogenetic trees. Edinburgh: Institute of Evolutionary Biology University of Edinburgh.
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular cloning: A laboratory manual. New York, NY: Cold Spring Harbor Laboratory Pressn, 146–157.
Silvestro, D., and Michalak, I. (2012). RaxmlGUI: A graphical front-end for RAxML. Org. Divers. Evol. 12, 1–3. doi: 10.1007/s13127-011-0056-0
Stoltz, M., Baeumer, B., Bouckaert, R., Fox, C., Hiscott, G., and Bryant, D. (2021). Bayesian inference of species trees using diffusion models. Syst. Biol. 70, 145–161. doi: 10.1093/sysbio/syaa051
Tamura, K., Tao, Q., and Kumar, S. (2018). Theoretical foundation of the reltime method for estimating divergence times from variable evolutionary rates. Mol. Biol. Evol. 35, 1770–1782. doi: 10.1093/molbev/msy044
Tang, Y., Li, C., Wanghe, K., Feng, C., Tong, C., Tian, F., et al. (2019). Convergent evolution misled taxonomy in schizothoracine fishes (Cypriniformes: Cyprinidae). Mol. Phylogenet. Evol. 134, 323–337. doi: 10.1016/j.ympev.2019.01.008
Tang, Y.-T., Feng, C.-G., Wanghe, K.-Y., Li, G.-G., and Zhao, K. (2016). Taxonomic status of a population of Gymoncypris waddelli Regan, 1905 (Cypriniformes: Schizothoracinae) distributed in Pengqu River, Tibet, China. Zootaxa 4126, 123–137. doi: 10.11646/zootaxa.4126.1.7
Wang, H. K. Y., Tang, Y. T., and Li, G. G. (2017). Geometric morphometrics of the cephalic contour and its morphological variations among Schizopygopsis stoliczkai (Teleostei: Cyprinidae). Acta Hydrobiol. Sin. 41, 182–193. doi: 10.7541/2017.23
Wang, W., Wang, W., Peng, B., and Yang, L. (2019). Simulating the surface of litchi grain leather by creating quadrilateral-continuous pattern in adobe illustrator CC. Leather Footw. J. 19, 1–92. doi: 10.24264/lfj.19.1.6
Wanghe, K., Guo, X., Wang, M., Zhuang, H., Ahmad, S., Khan, T. U., et al. (2020). Gravity model toolbox: An automated and open-source ArcGIS tool to build and prioritize ecological corridors in urban landscapes. Glob. Ecol. Conserv. 22:e01012. doi: 10.1016/j.gecco.2020.e01012
Wanghe, K., Tang, Y., Tian, F., Feng, C., Zhang, R., Li, G., et al. (2017). Phylogeography of Schizopygopsis stoliczkai (Cyprinidae) in Northwest Tibetan Plateau area. Ecol. Evol. 7, 9602–9612. doi: 10.1002/ece3.3452
Wu, Y., and Wu, C. (1992). The fishes of the Qinghai-Xizang plateau. Sichuan: Sichuan Publishing House of Science & Technology.
Zhang, Y., Li, X. H., Tian, F., Liu, S. J., Feng, C. G., and Zhao, K. (2020). Mitochondrial genome and phylogenetic relationship of Gymnocypris eckloni (Schizothoracinae) in Qaidam river basin. Genomics 112, 4316–4321. doi: 10.1016/j.ygeno.2020.07.030
Zhao, K., Duan, Z. Y., Peng, Z. G., Guo, S. C., Li, J. B., He, S. P., et al. (2009). The youngest split in sympatric schizothoracine fish (Cyprinidae) is shaped by ecological adaptations in a Tibetan Plateau glacier lake. Mol. Ecol. 18, 3616–3628. doi: 10.1111/j.1365-294X.2009.04274.x
Zhao, K., Li, J., Yang, G., Duan, Z., He, S., and Chen, Y. (2005). Molecular phylogenetics of Gymnocypris (Teleostei: Cyprinidae) in Lake Qinghai and adjacent drainages. Chin. Sci. Bull. 50, 1325–1333. doi: 10.1360/982005-223
Keywords: genetic diversity conservation, non-monophyletic, population phylogeny structure, species delimitation, SLAF-seq, species synonymy
Citation: Wanghe K, Feng C, Tang Y, Qi D, Ahmad S, Nabi G, Li X, Wang G, Jian L, Liu S, Zhao K and Tian F (2022) Phylogenetic relationship and taxonomic status of Gymnocypris eckloni (Schizothoracinae) based on specific locus amplified fragments sequencing. Front. Ecol. Evol. 10:933632. doi: 10.3389/fevo.2022.933632
Received: 01 May 2022; Accepted: 17 October 2022;
Published: 10 November 2022.
Edited by:
Liang Liu, University of Georgia, United StatesReviewed by:
Renyi Zhang, Guizhou Normal University, ChinaPervaiz Tariq, University of California, Riverside, United States
Copyright © 2022 Wanghe, Feng, Tang, Qi, Ahmad, Nabi, Li, Wang, Jian, Liu, Zhao and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Tian, dGlhbmZlaUBud2lwYi5jYXMuY24=
†These authors have contributed equally to this work