 Ivan M. De-la-Cruz
Ivan M. De-la-Cruz Eunice Kariñho-Betancourt
Eunice Kariñho-Betancourt Juan Núñez-Farfán
Juan Núñez-Farfán Ken Oyama1
Ken Oyama1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 15 November 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.916762
Elucidating the diversification process of congeneric species makes it necessary to identify the factors promoting species variation and diversification. Comparative gene family analysis allows us to elucidate the evolutionary history of species by identifying common genetic/genomic mechanisms underlying species responses to biotic and abiotic environments at the genomic level. In this study, we analyzed the high-quality transcriptomes of four Datura species, D. inoxia, D. pruinosa, D. stramonium, and D. wrightii. We performed a thorough comparative gene family analysis to infer the role of selection in molecular variation, changes in protein physicochemical properties, and gain/loss of genes during their diversification processes. The results revealed common and species-specific signals of positive selection, physicochemical divergence and/or expansion of metabolic genes (e.g., transferases and oxidoreductases) associated with terpene and tropane metabolism and some resistance genes (R genes). The gene family analysis presented here is a valuable tool for understanding the genome evolution of economically and ecologically significant taxa such as the Solanaceae family.
One of the fundamental endeavors in biology is to unravel the process and mechanisms of species diversification (Savolainen et al., 2013). The development of phylogenetic theory (Hennig, 1950, 1966), followed by the generation of massive DNA sequence datasets, increased computing power, and the proliferation of analytical methods (e.g., maximum likelihood, Bayesian inference, Bayesian molecular dating), has led to the recognition of the temporal dynamics of evolutionary radiation (Magallon and Sanderson, 2001; Alfaro et al., 2009; Stadler, 2011; Nürk et al., 2020). In addition, recent advances in genomics and bioinformatics have allowed the elucidation of the transcriptome and genome assemblies of several plant species (Leebens-Mack et al., 2019), which constitute the basic resources to find candidate genes underlying physiological, developmental, and ecological traits involved in species diversification (Eckardt, 2001; Ehrenreich and Purugganan, 2006; Anderson et al., 2011; Chang et al., 2012; Savolainen et al., 2013; Flood and Hancock, 2017; Sork, 2018; Bamba et al., 2019; Kariñho-Betancourt et al., 2020; Rich-Griffin et al., 2020). Genomic approaches that unravel physicochemical divergence, natural selection, and/or gene expansions/contractions in gene families of related taxa have also contributed to enhanced understanding of the evolutionary trajectories of specific traits (Stone and Sidow, 2005; Arendt and Reznick, 2008; Han et al., 2013; Murrell et al., 2013; Emms and Kelly, 2019). For instance, it is possible to discover candidate genes involved in the production of secondary plant metabolites that could also play important roles in mediating plant-based interactions in the evolution of differentiated defensive mechanisms (Bergelson et al., 2001; Friedman and Baker, 2007; Ramakrishna and Ravishankar, 2011; Chang et al., 2012; Kariñho-Betancourt, 2018; Kourelis and van der Hoorn, 2018; Wink, 2018; Erb and Kliebenstein, 2020; Rich-Griffin et al., 2020).
The comparative genomics study (e.g., gene family analysis) of plants has provided a broad view of the mechanisms at the molecular level behind species evolution and diversification (e.g., genomic signatures of directional selection on particular genes) (Song and Mitchell-Olds, 2011; Savolainen et al., 2013; Flood and Hancock, 2017; Russell et al., 2017; Anderson and Song, 2020). Genomic studies have been central to investigating the evolution and diversification of ecologically and economically relevant plant species, such as the Solanaceae family (Soltis and Soltis, 2021). Several species of this plant family have been subjected to metabolomic and genomic studies on specialized secondary metabolites, such as terpenoids, tropane alkaloids, glycoalkaloids, and phenolics, implicated as defenses against natural enemies (Friedman, 2002, 2006; Cheng et al., 2007; Sierro et al., 2014; Akyol et al., 2016; Chowański et al., 2016; Xu et al., 2017; De-la-Cruz et al., 2021). However, this is not the case for nonmodel plant species in the Solanaceae family – such as Datura species – where few genomic studies examining the processes and candidate genes that underlie evolutionary diversification have been carried out.
Within the Solanaceae family, the species of the genus Datura produce high concentrations of tropane alkaloids (Jirschitzka et al., 2012; Kohnen-Johannsen and Kayser, 2019; Céspedes-Méndez et al., 2021), metabolites long used as medicines (Roddick, 1991; Geeta and Gharaibeh, 2007; Benítez et al., 2018; Sahu et al., 2022). Ecological evidence indicates that some of these chemical compounds, as well as some terpenoids, play central roles as defenses against herbivores and possibly against pathogens, viruses, and fungi in Datura (Shonle and Bergelson, 2000; Hare and Elle, 2001; Hare and Smith, 2005; Castillo et al., 2014; Kariñho-Betancourt et al., 2015; Miranda-Pérez et al., 2016; De-la-Cruz et al., 2020a,2021; Goldberg et al., 2020; Céspedes-Méndez et al., 2021; Velázquez-Márquez et al., 2021). Furthermore, there is evidence that these species produce different concentrations of total tropane alkaloids (Doncheva et al., 2006; Kariñho-Betancourt et al., 2015). The genus Datura includes 13–14 species of annual herbs and perennial shrubs native to dry, temperate, and subtropical regions of North America, distributed mainly in Mexico, which is considered its center of origin (Avery, 1959; Barclay, 1959; Symon and Haegi, 1991; Luna-Cavazos and Bye, 2011). Plant size ranges from 30 cm to 2 m in height (Avery, 1959). Flowers open at dusk and are pollinated by hawkmoths (Jiménez-Lobato and Núñez-Farfán, 2021). All species have tubular erect flowers with large variations in size and color (Matuda, 1952). While most ruderal species have small-sized white, yellow and purple flowers (3–8 cm), plants distributed in less disturbed environments have mostly large white flowers (10–17 cm). Nevertheless, the genomic mechanisms underlying the evolutionary divergence of Datura species are unknown.
In this study, we selected four Datura species of two different clades of the genus (Mace et al., 1999; Bye and Sosa, 2013; Kariñho-Betancourt et al., 2015), some of which also overlap with respect to their distribution ranges within Mexico (Supplementary Figure 1). Datura inoxia, D. wrightii, and D. pruinosa are nested within the section of nodding fruits and larger flowers (except for D. pruinosa). Datura inoxia occurs in central and northern Mexico, D. wrightii occurs in northern Mexico, and Datura pruinosa is distributed in southern Mexico. In contrast, D. stramonium is nested within the section of erect fruits and is distributed throughout North and South Mexico (Mace et al., 1999; Bye and Sosa, 2013; Kariñho-Betancourt et al., 2015). All Datura species studied here are summer annuals, except for D. wrightii, which is perennial. Hence, these species represent good systems for addressing their evolutionary divergence at the genomic level. To this end, we performed a thorough gene family analysis using de novo leaf transcriptomes of these four Datura species to find candidate genes evolving under positive selection, physicochemical divergence (changes in physicochemical properties of proteins) and expansion (gain of genes).
Our sample design consisted of nine plants per species (different maternal families) from the four Datura species (see above). Seeds were collected in the field (natural progenies; Supplementary Figure 1) and germinated in the glasshouse of the Institute of Ecology, UNAM (UTM 19.3218, −99.1922). Once true leaves appeared and seedlings reached 3–5 cm long, they were planted in 150 mL pots in sterilized soil, watered ad libitum, and grown under a 16:8 L:D cycle at 25:20°C (L:D). Then, we sampled fully expanded leaves from each plant of the four Datura species before and during flowering (dx.doi.org/10.17504/protocols.io.bx4zpqx6). All leaves were flash-frozen in liquid nitrogen and kept in a −80°C freezer. We extracted total RNA from the leaves of each plant species of Datura studied here using the TRIzol extraction method (doi.org/10.17504/protocols.io.bx4zpqx6) (Rio et al., 2010). RNA quality and quantity were determined using a Nanodrop 2000 instrument (Thermo Scientific) and Bioanalyzer Chip RNA 7500 series II (Agilent). Each sample was sequenced using an Illumina NextSeq 500 in paired-end mode (2 × 75 bp). The library preparation and sequencing were carried out at the Unidad Universitaria de Secuenciación Masiva y Bioinformática (UUSMB) of the Institute of Biotechnology, UNAM. The quality of the RNA sequences of each plant individual was verified using FastQC (Andrews, 2010). Trimmomatic (Bolger A. M. et al., 2014) was used to remove sequences below 20 Phred quality scores.
We pooled the sequences of all analyzed plants of each Datura species as input for a de novo assembly using Trinity v2.12.0 (Grabherr et al., 2011). After transcriptome assembly, we retained only the longest isoforms using TransDecoder v5.5.0.1 A further step was carried out using CD-HIT v4.8.1 to cluster similar proteins and remove their redundancy (Li and Godzik, 2006; Fu et al., 2012) to retain only “primary transcripts” (Li and Godzik, 2006; Fu et al., 2012). We evaluated the transcriptome assemblies using the standard metrics (total genes and transcripts, percent of GC, Nx length statistics and median and average contig) with the script “TrinityStats.pl” of Trinity v2.11.0 (Grabherr et al., 2011). We assessed transcriptome completeness using the Benchmarking Universal Single-Copy Orthologs (BUSCO), which determines completeness by determining the fraction of a lineage-specific set of conserved genes present in an assembly. We performed the analysis using BUSCO v.5.1.2 (Seppey et al., 2019) with the transcriptome mode option. The database “embryophyta_odb9” was used as a reference for searching BUSCOs in the four Datura transcriptomes (Simão et al., 2015). BUSCOs were classified as complete and single-copy (S), complete and duplicated (D), fragmented (F), or missing (M).
Four Datura and three Nicotiana transcriptomes were used for phylogenetic and gene family evolution analyses. Since we encountered logistic issues downloading other Solanaceae transcriptomes (e.g., Solanum transcriptomes), we decided to use these three Nicotiana species for gene family construction (see De-la-Cruz et al., 2021). Thus, protein-coding genes and CDS from the three Nicotiana transcriptomes were sourced from the Sol Genomics Network2 [N. tomentosiformis (Sierro et al., 2013), N. sylvestris (Sierro et al., 2013), and N. attenuata (Xu et al., 2017)]. We used the combined set of Datura and Nicotiana proteomes to infer orthogroups in the OrthoFinder v2.5.2 program (Emms and Kelly, 2019). Briefly, DIAMOND blast (Buchfink et al., 2015) was used in OrthoFinder (Emms and Kelly, 2019) for orthogroup inference (E-value < 1e–5) and the MCL algorithm was employed for clustering of similar sequences (Enright et al., 2002). MAFFT v7 was used as a multiple protein sequence aligner of each orthogroup/gene family (Katoh et al., 2002), and FastTree2 v2.1.10 (Price et al., 2010) was used for maximum likelihood gene tree inference. The species tree inference was constructed using a concatenated alignment of 3,219 single-copy orthogroups in OrthoFinder (Emms and Kelly, 2019). The species tree was inferred with FastTree2 (Price et al., 2010). Rooting was performed using the STRIDE algorithm (Species Tree Root Inference from Duplication Events; Emms and Kelly, 2019) of OrthoFinder, which selected N. sylvestris, N. attenuata, and N. tomentosiformis as outgroups of the Datura phylogeny.
To build an ultrametric phylogeny (i.e., estimating divergence times and rates of substitution in a phylogeny) for the analysis of gene family evolution (expansions and contractions in gene families; see below), we used the rooted species tree obtained from OrthoFinder to search the divergence times between the branches using the TimeTree webtool (Kumar et al., 2017). Then, the rooted species tree and the information of divergence times were used to create an input file using the script ‘‘cafetutorial_prep_r8s.py’’ from the CAFE tutorial webpage3 to run the r8 v1.5 program for ultrametric inference (Sanderson, 2003). The tip-to-root length was adjusted to match the approximately 35-million-year evolutionary history between Nicotiana and Datura species (Bombarely et al., 2016; Kumar et al., 2017; De-la-Cruz et al., 2021).
To evaluate the rate of gene gain and loss across the Datura and Nicotiana species studied here, we used only the gene families with more than three genes per family (24,884) and the ultrametric species tree as inputs to CAFE v4.2.1 (Han et al., 2013). CAFE (Computational Analysis of gene Family Evolution) estimates the birth and death parameter (λ) for the provided tree and gene family counts. The λ parameter describes the probability that any gene will be gained or lost (Han et al., 2013). First, the Python scripts provided by the CAFE pipeline were used to estimate the error in our dataset (Han et al., 2013). Then, CAFE was run using the mode in which the gain and loss rates were estimated together (λ) for the whole phylogeny (Han et al., 2013). The CAFE overall p value threshold was kept at its default value (0.01) for the entire analysis. We used the script “cafetutorial_report_analysis.py” from CAFE to obtain summary statistics. The script parseCafeResult.R from the SlydGeneFamsAnalyses R package (see De-la-Cruz et al., 2021) was used to parse the CAFE outputs for functional enrichment analysis (see below).
Multiple sequence alignments of the 24,884 protein families (families with more than three proteins) were used to carry out a multivariate analysis of protein polymorphisms (MAPP program) (Stone and Sidow, 2005). MAPP quantifies the physicochemical variation (hydropathy, polarity, charge, volume, free energy in alpha-helix conformation, and free energy in beta-strand conformation) in each column of a multiple sequence alignment and calculates the deviation of candidate amino acid replacements from this variation. The greater the deviation, the higher the probability that a replacement impairs the function of the protein and the greater its predicted effect on the protein’s function (Stone and Sidow, 2005). Therefore, MAPP was used to estimate the physicochemical divergence in each protein family. First, we used the script readAndParseOrthogroupsTxt.R from the SlydGeneFamsAnalyses R package (De-la-Cruz et al., 2021) to parse and create folders from each protein family and store the corresponding protein tree and multiple sequence alignment from OrthoFinder results. Then, the MAPP program was run with default parameters for each of the protein families (Stone and Sidow, 2005). The script readMappResults.R in the SlydGeneFamsAnalyses R package (De-la-Cruz et al., 2021) was used to parse and read all the MAPP results of gene families. This script reads the MAPP results for all gene families, adjusts the p value, finds genes of families with good multiple sequence alignments (Valdar Score > 0.6), and only retains the significant sites with physicochemical divergence that correspond with conserved domain proteins (De-la-Cruz et al., 2021). This method allows us to score residues in a multiple sequence alignment and assigns a score ranging from 0 for low conservation to 1 for high conservation (Valdar, 2002).
A codon-level analysis of positive natural selection was performed with FUBAR (Fast, Unconstrained Bayesian Approximation) (Murrell et al., 2013) on 24,884 gene families. FUBAR uses a Bayesian approach to infer nonsynonymous (dN) and synonymous (dS) substitution rates on a per-site basis for a given coding alignment and corresponding phylogeny (Murrell et al., 2013). This method assumes that the selection pressure on each site is constant along the entire phylogeny (Murrell et al., 2013). FUBAR reports evidence for positive selection using posterior probabilities (which range from 0 to 1), not p values. Generally, posterior probabilities > 0.9 strongly suggest positive selection (Murrell et al., 2013).
To run FUBAR, we first removed trailing stop codons from the CDS in each transcriptome and then applied PAL2NAL (Suyama et al., 2006) to produce a codon alignment for each gene family. PAL2NAL is a program that converts a multiple sequence alignment of proteins and the corresponding DNA (CDS) sequences into a codon alignment (Suyama et al., 2006). Therefore, we used the protein tree that we already had from each protein family to run PAL2NAL. FUBAR was run for each protein family’s codon alignments (De-la-Cruz et al., 2021). A custom Python script was used to transform the “.json” format from the FUBAR result to tabular format. Then, the R script “loadFubarResults.R” from the R package GeneFamilies (De-la-Cruz et al., 2021) was used to obtain a table with the significant posterior probabilities of a codon being subject to positive selection for each gene family (significant posterior probabilities ≥ 0.98; Bayes Factor > 100). As stated above, we only used alignments with a Valdar score > 0.6.
Fisher’s exact test (Fisher and Russell, 1922) was employed for functional enrichment/overrepresentation analyses to look for biological functions enriched in a gene list to a greater extent than expected by chance. In other words, the overrepresentation method compares the set of all genes annotated to pathways to a list of those genes that are significantly expanded, positively selected and which exhibit physicochemical divergence between species. To this end, we used all the proteins from the seven transcriptomes (262,248 proteins) as the background to detect overrepresented proteins that showed signals of expansion, physicochemical divergence (MAPP), and positively selected conserved amino acids (codons) (FUBAR) (De-la-Cruz et al., 2021). Functional annotation of all transcriptomes was performed using MapMan4 (Schwacke et al., 2019). The enrichment analysis was carried out using our custom script “enrichedAnnosInExpContrFams.R (CAFE)”, “identifyDomainsAtSelectedSites.R (FUBAR)” and “readMappResults.R (MAPP)” from the SlydGeneFamsAnalyses R package (De-la-Cruz et al., 2021). In addition, protein domains were annotated for the four complete Datura transcriptomes using Interproscan v.5.24 (Jones et al., 2014).
Illumina sequencing yielded between 1 and 14 M (million) paired-end reads per sequenced individual of every Datura species (Table 1 and Supplementary Table 2). This resulted in good-quality RNA-seq datasets containing between 86 and 104 M paired-end reads for each species (Table 1 and Supplementary Table 1). The raw read data from Illumina sequencing for each species are deposited in the NCBI under BioProject PRJNA669339. Trinity constructed 81,748 transcripts for D. inoxia, 103,954 for D. pruinosa, 113,343 for D. stramonium and 111,196 for D. wrightii (Table 1). After polishing with TransDecoder and CD-HIT, we retained only 26,112, 30,191, 33,851, and 30,385 transcripts for D. inoxia, D. pruinosa, D. stramonium, and D. wrightii, respectively, for subsequent analyses (see below and Table 1). The percentages of GC and N50 were relatively similar among the four Datura transcriptomes (Table 1). The Datura transcriptomes covered 86.1, 91.8, 90.3, and 92.8% complete single-copy BUSCOs for D. inoxia, D. pruinosa, D. stramonium, and D. wrightii, respectively (Supplementary Table 2). Overall, the four assemblies were relatively similar in gene completeness, with D. wrightii and D. inoxia having the highest and lowest BUSCO scores, respectively (Supplementary Table 2).

Table 1. RNA reads produced by the transcriptome assembly of four Datura species.
Protein coding genes from three Nicotiana species sourced from the Sol Genomics Network (see “Materials and methods” section) and proteomes/CDS of four Datura species were used to construct gene families/orthogroups. From 262,248 genes, OrthoFinder assigned 85.6% to 30,065 orthogroups/protein families. Fifty percent of all genes were in orthogroups with eight or more genes (G50 = 8; Supplementary Table 3). There were 3,219 orthogroups that consisted entirely of single-copy genes and 11,786 orthogroups with all species present (Supplementary Table 3). The two species sharing the most protein families were N. tomentosiformis and N. sylvestris. Likewise, compared to the other Nicotiana species, N. attenuata shares the highest number of orthogroups with the four Datura species (Supplementary Figure 2). Within the Datura genus, D. wrightii and D. pruinosa shared the highest number of orthogroups (protein families), followed by D. stramonium and D. pruinosa. Datura inoxia was the species that shared the lowest number of orthogroups with the other three Datura species (Supplementary Figure 2). This difference may be explained by the lower number of transcripts/genes assembled for D. inoxia.
According to the estimated calibration time, Nicotiana and Datura diverged ∼25 Mya (Figure 1). Our evidence indicated that D. inoxia, D. pruinosa, and D. wrightii diverged from D. stramonium ∼2.6 Mya. Datura inoxia and D. wrightii were the most recently derived species within the Datura group (Figure 1). The rate of gene gain and loss (λ) resulting from CAFE analysis was 0.039 per year for the whole tree (Figure 1). The internal branch with the largest number of protein gains (expansions) corresponded to the most recent common ancestor of N. tomentosiformis (Figure 1). The internal branch with the largest contractions corresponded to the most recent common ancestor of the Datura clade (Figure 1). Within the Datura group, there was no evidence of expansions and contractions for the most recent common ancestor of D. pruinosa, D. inoxia, and D. wrightii. The terminal branch with the largest number of expansions corresponded to N. sylvestris (Figure 1 and Table 2). Within the Datura clade, the terminal branch with the largest number of expansions was the branch leading to D. stramonium. The terminal branch with the greatest number of contractions was the branch leading to N. attenuata (Figure 1 and Table 2). Within the Datura group, the terminal branch with the largest contractions was the branch leading to D. inoxia, followed by D. stramonium (Figure 1 and Table 2). The internal branch with the largest rapidly evolving gene families corresponded to the most recent common ancestor of N. attenuata and N. sylvestris (Figure 1). The terminal branch with the most rapidly evolving gene families was the branch leading to N. attenuata (Figure 1 and Table 2). Within the Datura clade, the terminal branch with the most rapidly evolving gene families was the branch leading to D. inoxia, followed by D. wrightii (Figure 1 and Table 2). According to CAFE, the number of protein families that experienced neither expansion nor contraction in the genomes of all species tested here varies from 15,896 to 19,743 (Table 2).
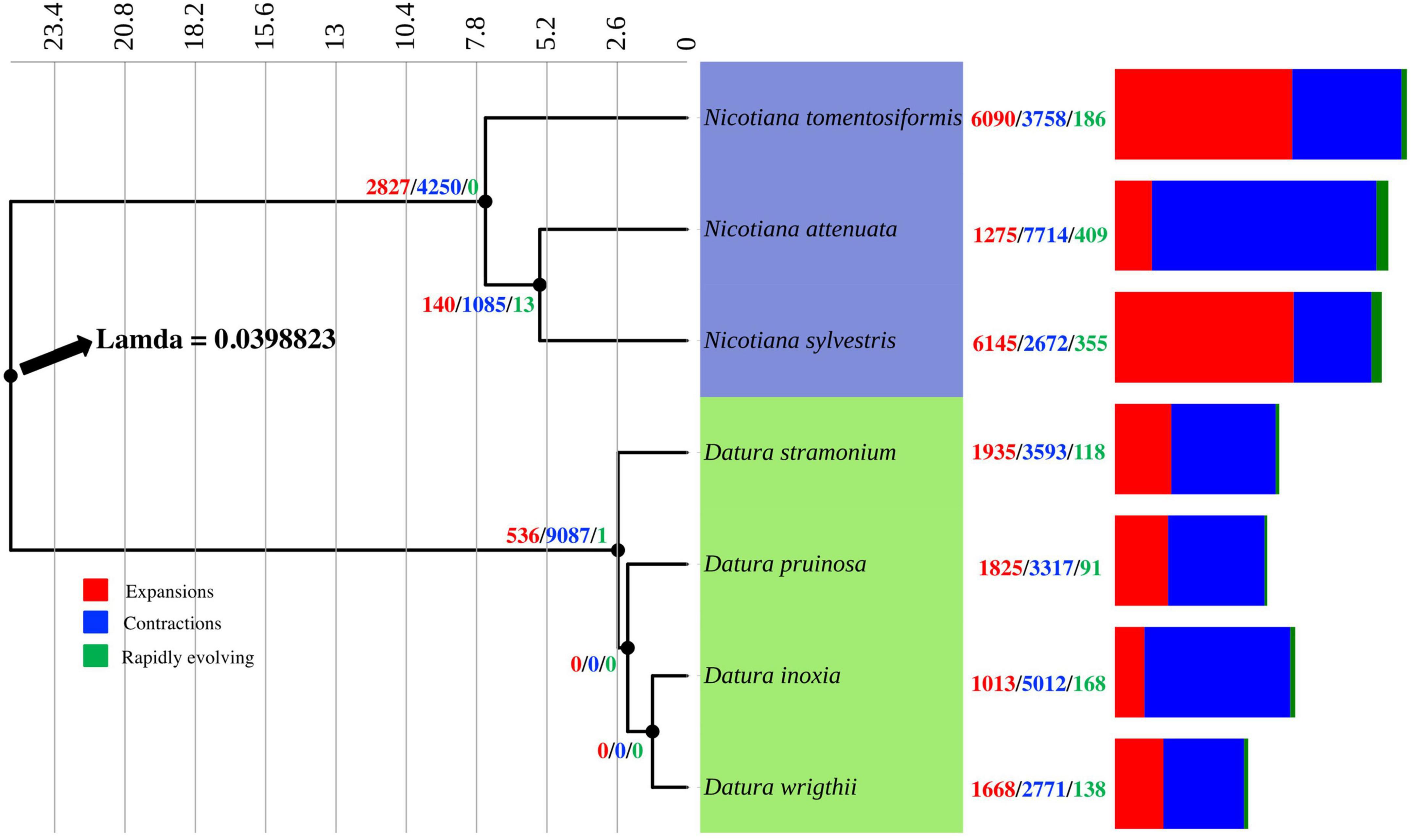
Figure 1. Phylogenetic time tree constructed from concatenated alignment of 3,219 orthogroups consistent entirely of single-copy genes from four Datura species and three Nicotiana species. Nicotiana and Datura species diverged approximately 25 Mya. Expansions (red), contractions (blue) and rapidly evolving gene families (green) are shown at nodes and terminal branches. The rate of gene gain and lost (λ) resulted from CAFE analysis was 0.039 for the whole tree.
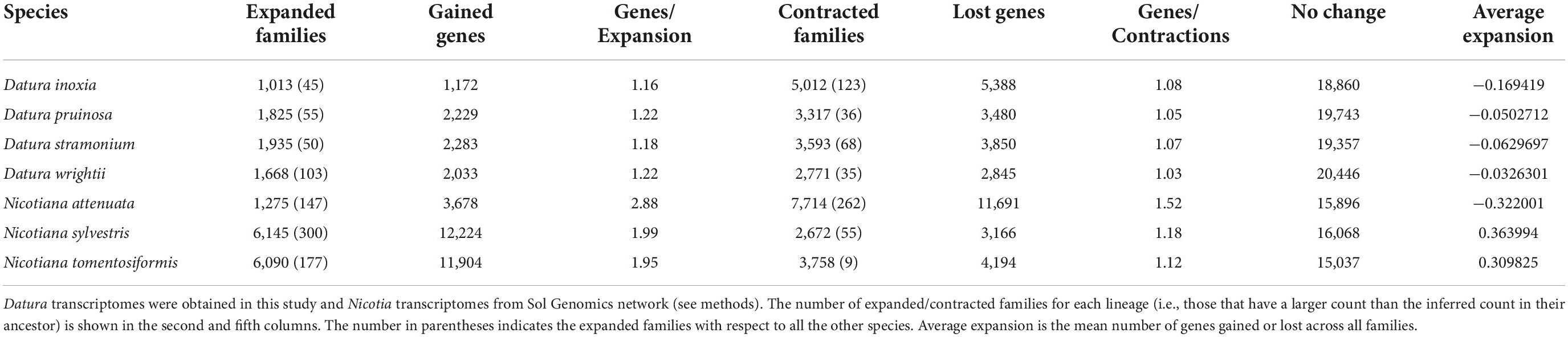
Table 2. Comparative computational analysis of gene family evolution (CAFE analysis) for seven Solanaceae transcriptomes.
We found a total of 11, 14, 10, and 11 enriched protein families (i.e., functionally overrepresented protein families) with a signal of expansion in D. inoxia, D. pruinosa, D. stramonium, and D. wrightii, respectively (Figure 2 and Supplementary Table 4). We also detected 22, 17, 12, and 24 enriched proteins with signals of physicochemical divergence (Figure 3 and Supplementary Table 5) and 51, 59, 51, and 54 enriched proteins with positively selected conserved amino acids (Figure 4 and Supplementary Table 6) for D. inoxia, D. pruinosa, D. stramonium, and D. wrightii, respectively. Several of these enriched family proteins with signals of expansion, positive selection, or physicochemical divergence have been related to responses to specific abiotic and biotic pressures, as discussed below. We found that one and 29 expanded and positively selected protein families were shared between all Datura species, while there was no protein family with significant physicochemical divergence shared between all Datura species (Figures 2–4).
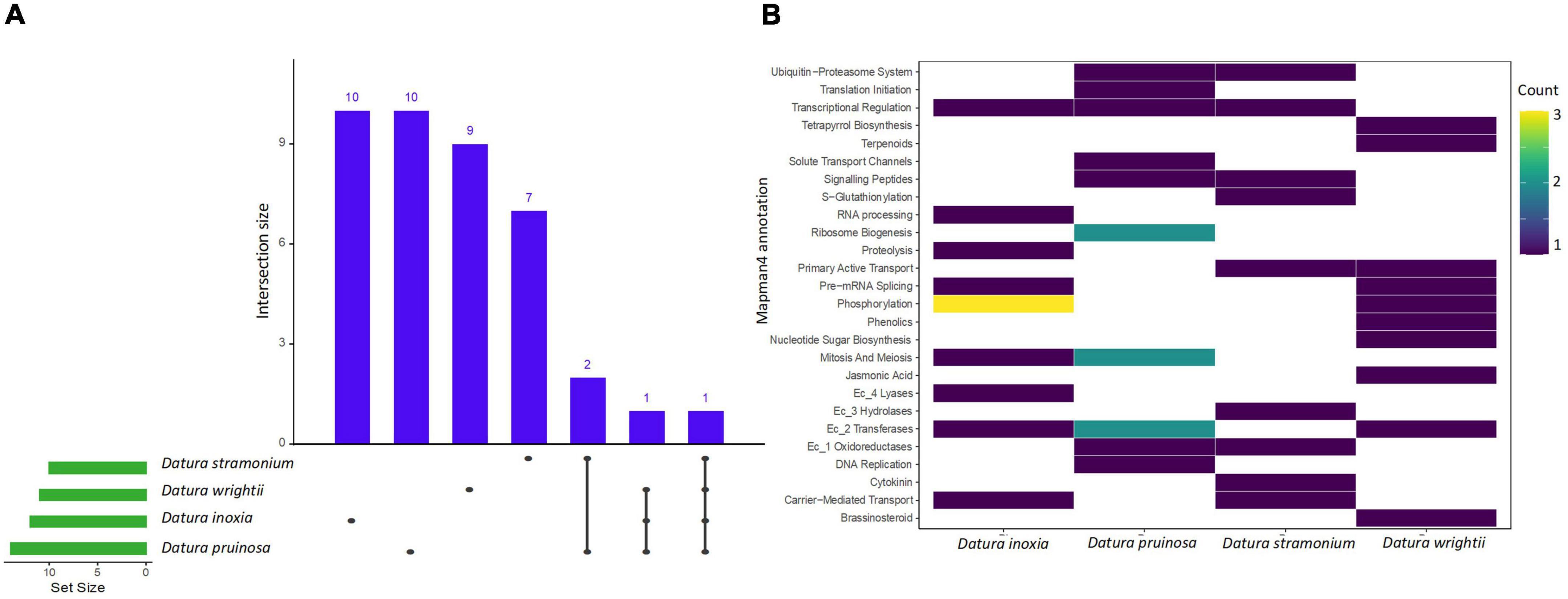
Figure 2. The UpSet plot shows the intersections of the set of a particular expanded protein family annotated with Mapman4 from the seven transcriptomes. Filled cells indicate that the set is participating in the intersection and that the species share that protein family with signal of expansion (A). Heatmap of the enriched expanded proteins of the four Datura species studied here (B). The brightness of color indicates the number (counts) of enriched protein families with signal of expansion that are shared between species. See also Supplementary Tables 4–6.
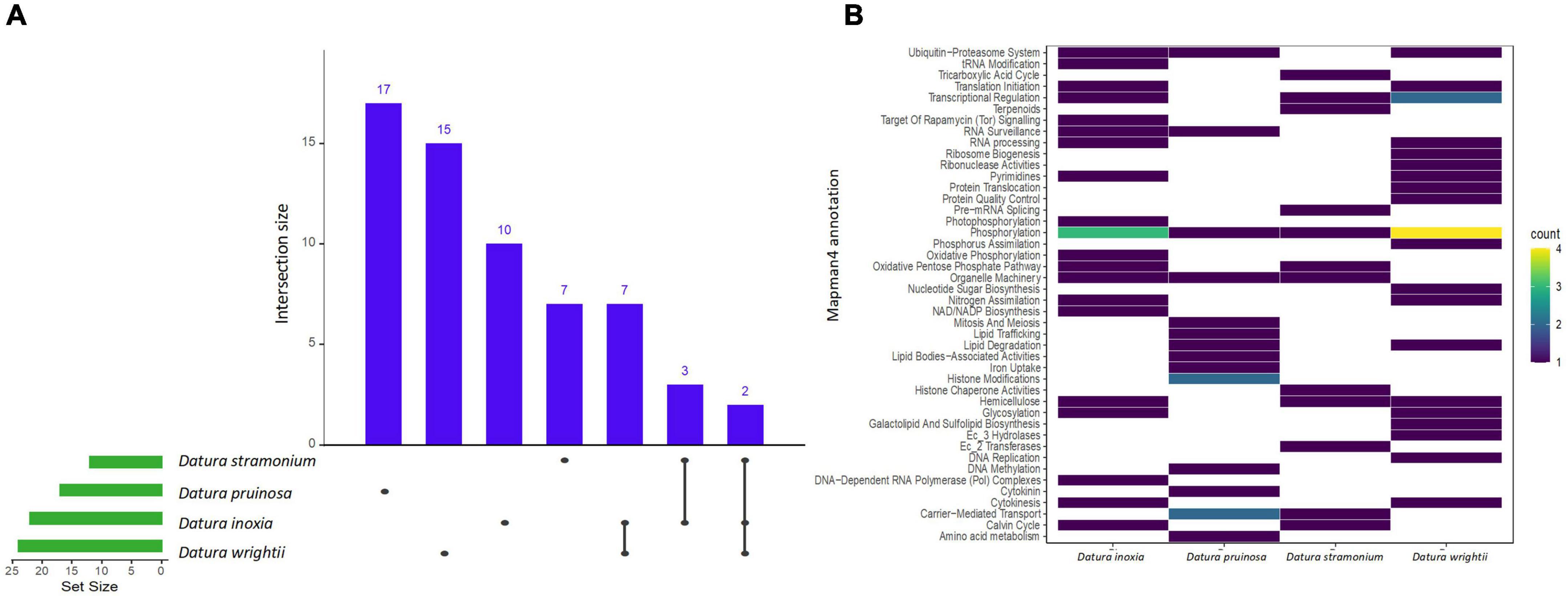
Figure 3. UpSet plot shows the intersections of the set of a particular protein family annotated Mapman4 with signal of physicochemical divergence from the seven transcriptomes. Filled cells indicate that the set is participating in the intersection and that the species share that protein family with signal of physicochemical divergence (A). Heatmap of the enriched proteins with signal of physicochemical divergence of the four Datura species studied here (B). The brightness of color indicates the number (counts) of enriched protein families with signal of physicochemical divergence that are shared between species. See also Supplementary Table 5.
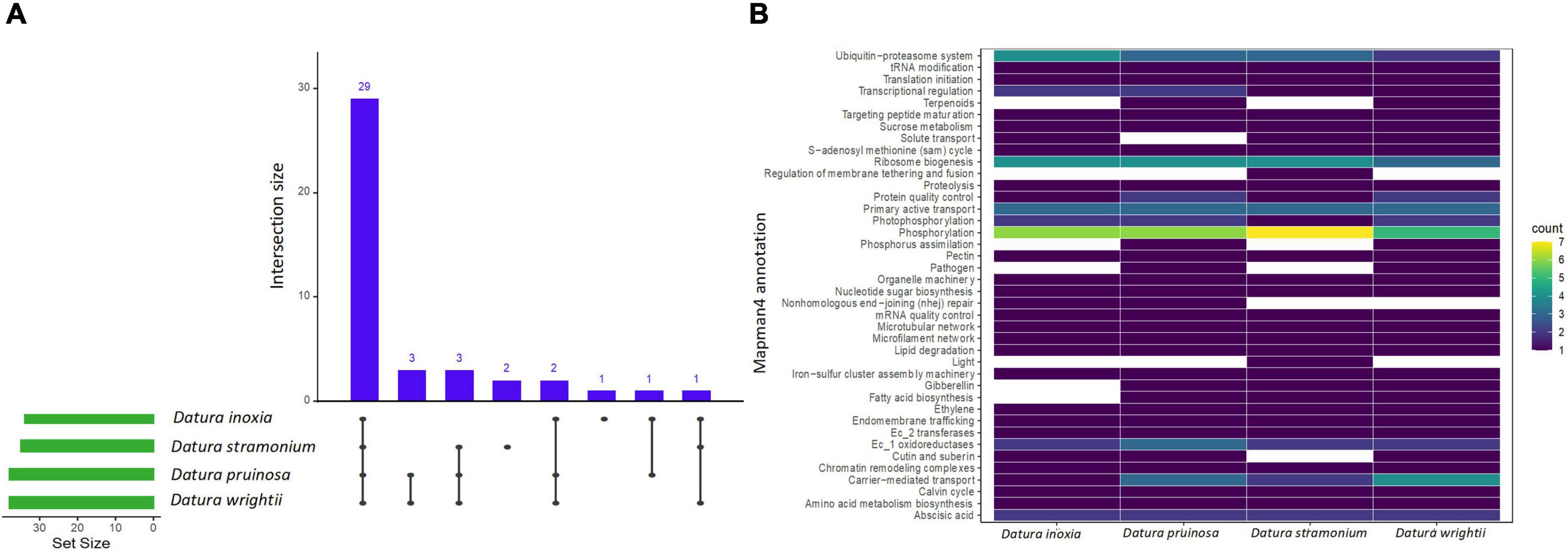
Figure 4. UpSet plot shows the intersections of the set of a particular protein family annotated with Mapman4 with signal of positive selection from the seven transcriptomes. Filled cells indicate that the set is participating in the intersection and that the species share that protein family with signal of positive selection (A). Heatmap of the enriched proteins with signal of positive selection of the four Datura species studied here (B). The brightness of color indicates the number (counts) of enriched protein families with signal of positive selection that are shared between species. See also Supplementary Table 6.
We detected several protein families related to plant resistance to pathogens, herbivores and viruses. For instance, the UDP-dependent glycosyl transferase protein family - related to cytokinin conjugation - was expanded and positively selected only in D. stramonium (Figure 2). We also detected enriched protein families with signals of positive selection related to glycosyltransferases and oxidoreductases in all Datura species. Expansion of oxidoreductases was also detected only in D. pruinosa and D. stramonium, while expansion of glycosyltransferases occurred in all the species except in D. stramonium. Moreover, we also observed positive selection for terpenoid proteins in D. pruinosa and D. wrightii, but not in D. inoxia and D. stramonium (Figure 4 and Supplementary Table 6). However, we also found physicochemical divergence with respect to 2-C-methyl-D-erythritol 4-phosphate (of the terpenoid family) only in D. stramonium (Figure 3 and Supplementary Table 5).
Genes related to plant immunity, such as phosphorylation-related proteins belonging to the large family of leucine-rich repeat receptor-like protein kinases (LRRs), showed expansion, physicochemical divergence, and positive selection in all Datura species (Supplementary Tables 4–6). We also found several enriched proteins belonging to the kinase superfamily with physicochemical divergence, expansion and positive selection (Supplementary Tables 4–6), such as the SD-1 kinase protein, which was positively selected in all Datura species (Figure 4). Plant nucleotide binding proteins, such as leucine-rich repeat (NLR) receptors, were only positively selected in D. pruinosa and D. wrightii (Figure 4), and proteins related to receptor-like protein kinase (RLCK-IXb) were expanded in D. inoxia (Figure 2 and Supplementary Table 4).
We also found different enriched proteins with signals of expansion, physicochemical divergence and positive selection in the Datura species studied here (Supplementary Tables 4–6) associated with abiotic stresses. For instance, we found physicochemical divergence in a protein family related to glutamate biosynthesis (the pyrroline-5-carboxylate synthase gene family) in D. pruinosa (Figure 3). In addition, we found physicochemical divergence in different protein families related to solute transport, such as the metal chelator transporter TCR and metal chelator nicotianamine transporters in D. pruinosa (Figure 4). Datura stramonium showed expansion in the class of Tau glutathione S-transferases (Figure 2 and Supplementary Table 4). In the case of D. inoxia, we detected a signal of expansion in receptor-like cytoplasmic kinases (RLCKs) (Figure 2 and Supplementary Table 4), while we did not find expansions in TCR, RLCKs, and Tau glutathione S-transferases in D. wrightii.
In this study, the completeness of single-copy orthologs and general statistics (N50, number of genes) suggest a good quality assembly for all de novo Datura transcriptomes (Simão et al., 2015). The number of complete BUSCOs for all transcriptome assemblies is very similar to that previously reported for D. stramonium and other Solanaceae species, such as tomato and potato, Nicotiana and its wild relatives, as well as Petunia inflata and Petunia axilaris (Xu et al., 2011, 2017; Sato et al., 2012; Sierro et al., 2013; Bolger A. et al., 2014; Bombarely et al., 2016; Hulse-Kemp et al., 2018; Razali et al., 2018; Barchi et al., 2019; De-la-Cruz et al., 2021). Our comparative gene family analysis also revealed signals of positive selection, physicochemical divergence and expansions for candidate genes possibly associated with species divergence.
Our results revealed that the divergence times reported here are consistent with other Solanaceae phylogenies (Särkinen et al., 2013; Bombarely et al., 2016; De-la-Cruz et al., 2021). We also found that N. attenuata shares the highest number of orthogroups with the four Datura species. This result could suggest a closer evolutionary history (i.e., genes forming orthogroups due to a shared origin from a single ancestral gene of the last common ancestor of the compared genomes) between these species (Koonin, 2005). Nevertheless, duplication events could also generate many of these genes forming orthogroups (Koonin, 2005). We detected positive selection of the same 29 protein families of all Datura species. This result could suggest parallel evolution of these genes among all Datura species.
As in many other plant species, we also found several enriched protein families (significantly, in more from the full protein set) in the Datura transcriptomes, with signals of expansion, physicochemical divergence and/or positive selection varying among species. For instance, UDP-dependent glycosyl transferase was expanded and positively selected only in D. stramonium. In plants, UDP-glycosyltransferases glycosylate various phytohormones and metabolites (e.g., triterpenoids) in response to biotic and abiotic stresses such as defenses to herbivores (Vogt and Jones, 2000; Rehman et al., 2018; Rahimi et al., 2019; Wilson and Tian, 2019). Indeed, we also detected enriched protein families with signals of positive selection related to glycosyltransferases and oxidoreductases in all Datura species. Transferases and oxidoreductases are enzymes directly involved in the biosynthesis of terpenes, tropane alkaloids (Kanehisa and Goto, 2000; Dräger, 2006; Jirschitzka et al., 2012; Kanehisa et al., 2016, 2017) and many other plant defensive compounds, such as phenolics, glucosinolates, salicylates, and anthocyanins, via glycosylation (Vogt and Jones, 2000; Bowles et al., 2005; Wilson and Tian, 2019). Expansion of oxidoreductases was also detected only in D. pruinosa and D. stramonium, while expansion of glycosyltransferases occurred in all the species except D. stramonium. Moreover, we also found positive selection of terpenoid proteins in D. pruinosa and D. wrightii, but not in D. inoxia and D. stramonium (Figure 4 and Supplementary Table 6). However, we also found physicochemical divergence in the protein family related to 2-C-methyl-D-erythritol 4-phosphate (terpenoids) only in D. stramonium (Figure 3 and Supplementary Table 5). This molecule is one of the two major building blocks of terpenoid compounds (Rohdich et al., 2000; Narayanasamy et al., 2010). Thus, it is likely that this divergence in the physical and chemical properties in this protein family could be related to changes in the production and/or concentration of specific terpenoid compounds. A recent study carried out in natural conditions with experimental plants of D. stramonium found that at least one triterpenoid compound was positively selected (Darwinian fitness) as a defense against the larvae of the specialized beetle Lema trilienata daturaphila, which is responsible for most of the leaf damage to plants (De-la-Cruz et al., 2020b). Thus, we speculate that terpenoid genes (and their products) could be involved in the production of defensive compounds to guard against natural enemies (e.g., as we have found in D. stramonium).
Terpenoids are involved in several biological functions related to development and defense against herbivores, fungi and pathogens (Degenhardt et al., 2009; Singh and Sharma, 2015; Boncan et al., 2020; Zhou and Pichersky, 2020). Moreover, several terpenoids are volatilized to attract predators and parasitoids of herbivores (Singh and Sharma, 2015; Lackus et al., 2018; Boncan et al., 2020). Since the four Datura species are attacked by the specialist herbivore L. trilineata daturaphila (Kogan and Goeden, 1970; Castillo et al., 2014; De-la-Cruz et al., 2020b; Goldberg et al., 2020), we speculate that the positive selection (D. pruinosa and D. wrightii), expansion (only in D. wrightii) and physicochemical divergence (only in D. stramonium) detected for terpenoids could be related how these species cope with their natural enemies such as herbivores, pathogens and viruses (Pichersky and Raguso, 2018; Karunanithi and Zerbe, 2019; Boncan et al., 2020; De-la-Cruz et al., 2020a,b; Goldberg et al., 2020, 2021; Zhou and Pichersky, 2020; Velázquez-Márquez et al., 2021). Thus, positive selection of terpenoid proteins in almost all Datura species could suggest species-specific evolutionary dynamics related to the different biotic and abiotic pressures faced by each Datura species in their natural environments (Castillo et al., 2014; Kariñho-Betancourt et al., 2015; Pichersky and Raguso, 2018; Karunanithi and Zerbe, 2019; De-la-Cruz et al., 2020b; Goldberg et al., 2020, 2021; De-la-Cruz et al., 2021).
We also found that the large family of leucine-rich repeat receptor-like protein kinases (LRRs) showed expansion, physicochemical divergence, and positive selection in all Datura species. LRR genes play a central role in plant innate immunity and are involved in the regulation of jasmonate/ethylene and salicylate hormones implicated in the defensive response against pathogens and herbivores (McHale et al., 2006; Padmanabhan et al., 2009; Magalhães et al., 2016; Tang et al., 2017). Notably, D. stramonium had the highest number of protein families related to phosphorylation under positive selection. We also found several enriched proteins belonging to the kinase superfamily with physicochemical divergence, expansion and positive selection, such as the SD-1 kinase protein, which was positively selected in all Datura species. Kinase superfamily proteins have been related to plant defense signaling in response to different stresses, including pathogen invasion (Stone and Walker, 1995; Afzal et al., 2008). Likewise, plant nucleotide binding and leucine-rich repeat (NLR) receptors are linked to defensive responses against pathogens triggering plant immunity effectors (Cui et al., 2015; Thordal-Christensen, 2020; van Wersch et al., 2020). In our study, NLR proteins were only positively selected in D. pruinosa and D. wrightii, and proteins related to the receptor-like protein kinase (RLCK-IXb) were expanded in D. inoxia.
On the other hand, we observed a signal of physicochemical divergence in genes related to transcriptional regulation (transcriptional corepressor LUG) among the three Datura species. It has been reported that LUG plays an important role in leaf blade outgrowth and flower development, also enhancing the polarity and growth defects in leaves of Arabidopsis thaliana (Stahle et al., 2009; Zhang et al., 2019). It could be possible that the physicochemical divergence detected in LUG could be involved in the mechanisms determining leaf shape and flower size of these Datura species. Interestingly, the transcriptional corepressor LUG is not enriched in D. pruinosa, which has smaller leaves and flowers than the four Datura species studied here (Figure 4; Matuda, 1952). Thus, LUG genes could be candidates for studying leaf and flower shape and size in Datura.
We detected physicochemical divergence in protein families related to hemicellulose function, such as xylosyltransferase IRX9 genes, among D. inoxia, D. stramonium, and D. wrightii, but not for D. pruinosa. It has been reported that mutations in IRX9 cause a collapsed xylem phenotype and decreases in xylose and cellulose in cell walls, suggesting that IRX9 is required for normal glucuronoxylan elongation (Peña et al., 2007; Wu et al., 2010). Glucuronoxylans occur in angiosperm secondary walls and are the main hemicellulosic polysaccharide in the secondary cell walls of plants, and it has been detected that IRX9 is specifically expressed in cells undergoing secondary wall thickening (Peña et al., 2007; Wu et al., 2010). Moreover, a study in A. thaliana revealed that IRX mutants had reduced cellulose content in the stems, resulting in decreased stiffness of stem material (Turner and Somerville, 1997). Thus, the IRX9 protein is also a candidate gene to study in terms of involvement in leaf thickness, stem stiffness, stem architecture and elongation differentiation in the Datura species studied here.
We also found different enriched proteins with signals of expansion, physicochemical divergence and positive selection in D. pruinosa that have been associated with abiotic stresses. For instance, we observed physicochemical divergence in a protein family related to glutamate biosynthesis (the pyrroline-5-carboxylate synthase gene family) in D. pruinosa. In Datura stramonium, De-la-Cruz et al. (2020b) found that pyrroline, a derivative compound from pyrroline-5-carboxylate synthase, is involved in defense against the folivore beetle Epitrix parvula (Chrysomelidae). This gene is also involved in proline accumulation, a widespread metabolic response of plants to osmotic stress (Pérez-Arellano et al., 2010). In plants, proline is useful not only during conditions of drought and high salt osmotic stress, but also as a response to environmental changes such as low temperature, nutritional deficit, heavy metals, UV radiation, and bacterial pathogens (Parvanova et al., 2004; Haudecoeur et al., 2009; Pérez-Arellano et al., 2010; Qamar et al., 2015). In some species, proline also accumulates during the development of flowers, pollen, ovules or fruits (Venekamp and Koot, 1988; Fujita et al., 1998; Pérez-Arellano et al., 2010).
In addition, we found physicochemical divergence in different protein families related to solute transport, such as the metal chelator transporter TCR and metal chelator nicotianamine transporters in D. pruinosa. Transition metals (e.g., iron, copper, and zinc) are essential for many physiological processes but can be toxic at elevated levels (Clemens, 2001). TCR proteins contribute to metal detoxification by buffering cytosolic metal concentrations (Clemens, 2001). In the same way, D. stramonium showed expansion in the Tau glutathione S-transferase class. The Tau and Phi classes of transferases are the largest groups in plants and play crucial roles in the remediation of environmental pollution by organic xenobiotics, including herbicides and industrial chemicals, as well as salinity, oxidative stress, and drought (Kilili et al., 2004; Benekos et al., 2010; Dixon and Edwards, 2010; Sharma et al., 2014; Gallé et al., 2019). In the case of D. inoxia, we detected a signal of expansion in receptor-like cytoplasmic kinases (RLCKs), which regulate plant cellular activities in response to biotic/abiotic stresses and endogenous extracellular signaling molecules (Liang and Zhou, 2018). Datura plants grow in highly human-disturbed, contaminated habitats and degraded soils, including landfills.
Interestingly, TCR, RLCKs, and Tau glutathione S-transferases are not expanded in D. wrightii, which occurs in less disturbed habitats. Thus, it could be possible that TCR, RLCKs and Tau glutathione S-transferases could be involved in the mechanisms related to the assimilation, long-distance transportation and detoxification of metal compounds in D. pruinosa, D. inoxia, and D. stramonium, respectively, to survive and grow in highly disturbed and polluted habitats. All of these hypotheses should be tested through functional analysis in further studies.
The transcriptome assemblies of these four Datura species provide a valuable molecular resource, as there are few sequenced transcriptomes of nonmodel species from the Solanaceae family. Our results provide new insights into the diversification among these four Datura species. Understanding the evolution of genes involved in the synthesis of secondary compounds and genes related to defense and other functions, such as development and growth, is essential to elucidating their roles in species divergence. Here, we described how the Datura gene families expanded and revealed positive selection and physicochemical divergence of proteins related to terpenoids, R genes, tropanes and other secondary metabolites putatively relevant to the diversification processes among these species. Gene family analysis is a useful tool for understanding the genomic evolution of economically and ecologically significant taxa. It is important to note that the overrepresented candidate genes with signals of positive selection and physicochemical divergence should be tested with respect to functional analysis to evaluate their ecological and evolutionary significance.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA669339.
ID-L-C and EK-B conceived and designed the study and wrote the first draft of the manuscript. EK-B designed and performed the experiments, RNA extraction, and data compilation. ID-L-C analyzed the data. KO, JN-F, EK-B, and ID-L-C contributed reagents, materials, and analysis tools, revised, and corrected the manuscript. All authors contributed to the article and approved the submitted version.
This work was provided by the Secretaría de Desarrollo Institucional Universidad Nacional Autónoma de México (UNAM) to EK-B and ID-L-C and from the CONACyT “Fronteras de la Ciencia” #1527 to JN-F. Postdoctoral research funds to EK-B were granted by the Direección General de Asuntos Academicos (DGAPA, UNAM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.916762/full#supplementary-material
Afzal, A. J., Wood, A. J., and Lightfoot, D. A. (2008). Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant Microbe Interact. 21, 507–517. doi: 10.1094/MPMI-21-5-0507
Akyol, H., Riciputi, Y., Capanoglu, E., Caboni, M. F., and Verardo, V. (2016). Phenolic compounds in the potato and its byproducts: An overview. Int. J. Mol. Sci. 17:835. doi: 10.3390/ijms17060835
Alfaro, M. E., Santini, F., Brock, C., Alamillo, H., Dornburg, A., Rabosky, D. L., et al. (2009). Nine exceptional radiations plus high turnover explain species diversity in jawed vertebrates. Proc. Natl. Acad. Sci. U.S.A. 106, 13410–13414.
Anderson, J. T., and Song, B.-H. (2020). Plant adaptation to climate change—Where are we? J. Syst. Evol. 58, 533–545. doi: 10.1111/jse.12649
Anderson, J. T., Willis, J. H., and Mitchell-Olds, T. (2011). Evolutionary genetics of plant adaptation. Trends Genet. 27, 258–266. doi: 10.1016/j.tig.2011.04.001
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed February 15, 2021).
Arendt, J., and Reznick, D. (2008). Convergence and parallelism reconsidered: What have we learned about the genetics of adaptation? Trends Ecol. Evol. 23, 26–32. doi: 10.1016/j.tree.2007.09.011
Avery, A. G. (1959). “Historical review,” in Blakeslee: The genus Datura, eds A. G. Avery, S. Satina, and J. Rietsma (New York, NY: Ronald Press), 3–15.
Bamba, M., Kawaguchi, Y. W., and Tsuchimatsu, T. (2019). Plant adaptation and speciation studied by population genomic approaches. Dev. Growth Differ. 61, 12–24. doi: 10.1111/dgd.12578
Barchi, L., Pietrella, M., Venturini, L., Minio, A., Toppino, L., Acquadro, A., et al. (2019). A chromosome-anchored eggplant genome sequence reveals key events in Solanaceae evolution. Sci. Rep. 9:11769. doi: 10.1038/s41598-019-47985-w
Barclay, A. S. (1959). Studies in the genus Datura (Solanaceae) I. Taxonomy of subgenus Datura. Unpublished Ph.D. Dissertation. Cambridge, MA: Harvard University, 221.
Benekos, K., Kissoudis, C., Nianiou-Obeidat, I., Labrou, N., Madesis, P., Kalamaki, M., et al. (2010). Overexpression of a specific soybean GmGSTU4 isoenzyme improves diphenyl ether and chloroacetanilide herbicide tolerance of transgenic tobacco plants. J. Biotechnol. 150, 195–201. doi: 10.1016/j.jbiotec.2010.07.011
Benítez, G., March-Salas, M., Villa-Kamel, A., Cháves-Jiménez, U., Hernández, J., and Montes-Osuna, et al. (2018). The genus Datura L.(Solanaceae) in Mexico and Spain–Ethnobotanical perspective at the interface of medical and illicit uses. J. Ethnopharmacol. 219, 133–151. doi: 10.1016/j.jep.2018.03.007
Bergelson, J., Kreitman, M., Stahl, E. A., and Tian, D. (2001). Evolutionary dynamics of plant R-genes. Science 292, 2281–2285. doi: 10.1126/science.1061337
Bolger, A., Scossa, F., Bolger, M. E., Lanz, C., Maumus, F., Tohge, T., et al. (2014). The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat. Genet. 46, 1034–1038. doi: 10.1038/ng.3046
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England) 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bombarely, A., Moser, M., Amrad, A., Bao, M., Bapaume, L., Barry, C. S., et al. (2016). Insight into the evolution of the solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2, 1–9. doi: 10.1038/nplants.2016.74
Boncan, D. A. T., Tsang, S. S. K., Li, C., Lee, I. H. T., Lam, H.-M., Chan, T.-F., et al. (2020). Terpenes and terpenoids in plants: Interactions with environment and insects. Int. J. Mol. Sci. 21:7382. doi: 10.3390/ijms21197382
Bowles, D., Isayenkova, J., Lim, E.-K., and Poppenberger, B. (2005). Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 8, 254–263. doi: 10.1016/j.pbi.2005.03.007
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Bye, R., and Sosa, V. (2013). Molecular phylogeny of the Jimsonweed genus Datura (Solanaceae). Syst. Bot. 38, 818–829. doi: 10.1600/036364413X670278
Castillo, G., Cruz, L. L., Tapia-López, R., Olmedo-Vicente, E., Carmona, D., Anaya-Lang, A. L., et al. (2014). Selection mosaic exerted by specialist and generalist herbivores on chemical and physical defense of Datura stramonium. PLoS One 9:e102478. doi: 10.1371/journal.pone.0102478
Céspedes-Méndez, C., Iturriaga-Vásquez, P., Hormazábal, E., Céspedes-Méndez, C., Iturriaga-Vásquez, P., and Hormazábal, E. (2021). Secondary metabolites and biological profiles of Datura genus. J. Chil. Chem. Soc. 66, 5183–5189. doi: 10.4067/S0717-97072021000205183
Chang, Y. M., Liu, W. Y., Shih, A. C. C., Shen, M. N., Lu, C. H., Lu, M. Y., et al. (2012). Characterizing regulatory and functional differentiation between maize mesophyll and bundle sheath cells by transcriptomic analysis. Plant Physiol. 160, 165–177. doi: 10.1104/pp.112.203810
Cheng, A.-X., Lou, Y.-G., Mao, Y.-B., Lu, S., Wang, L.-J., and Chen, X.-Y. (2007). Plant terpenoids: Biosynthesis and ecological functions. J. Integr. Plant Biol. 49, 179–186. doi: 10.1111/j.1744-7909.2007.00395.x
Chowański, S., Adamski, Z., Marciniak, P., Rosiñski, G., Büyükgüzel, E., and Büyükgüzel, et al. (2016). A review of bioinsecticidal activity of solanaceae alkaloids. Toxins 8:60. doi: 10.3390/toxins8030060
Clemens, S. (2001). Molecular mechanisms of plant metal tolerance and homeostasis. Planta 212, 475–486. doi: 10.1007/s004250000458
Cui, H., Tsuda, K., and Parker, J. E. (2015). Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 66, 487–511. doi: 10.1146/annurev-arplant-050213-040012
Degenhardt, J., Köllner, T. G., and Gershenzon, J. (2009). Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70, 1621–1637. doi: 10.1016/j.phytochem.2009.07.030
De-la-Cruz, I. M., Cruz, L. L., Martínez-García, L., Valverde, P. L., Flores-Ortiz, C. M., Hernández-Portilla, L. B., et al. (2020a). Evolutionary response to herbivory: Population differentiation in microsatellite loci, tropane alkaloids and leaf trichome density in Datura stramonium. Arthropod Plant Interact. 14, 21–30. doi: 10.1007/s11829-019-09735-7
De-la-Cruz, I. M., Hallab, A., Olivares-Pinto, U., Tapia-López, R., Velázquez-Márquez, S., Piñero, D., et al. (2021). Genomic signatures of the evolution of defence against its natural enemies in the poisonous and medicinal plant Datura stramonium (Solanaceae). Sci. Rep. 11:882. doi: 10.1038/s41598-020-79194-1
De-la-Cruz, I. M., Merilä, J., Valverde, P. L., Flores-Ortiz, C. M., and Núñez-Farfán, J. (2020b). Genomic and chemical evidence for local adaptation in resistance to different herbivores in Datura stramonium. Evolution 74, 2629–2643. doi: 10.1111/evo.14097
Dixon, D. P., and Edwards, R. (2010). Glutathione transferases. Arabidopsis Book. 8:e0131. doi: 10.1199/tab.0131
Doncheva, T., Berkov, S., and Philipov, S. (2006). Comparative study of the alkaloids in tribe datureae and their chemosystematic significance. Biochem. Syst. Ecol. 34, 478–488. doi: 10.1016/j.bse.2006.01.008
Dräger, B. (2006). Tropinone reductases, enzymes at the branch point of tropane alkaloid metabolism. Phytochemistry 67, 327–337. doi: 10.1016/j.phytochem.2005.12.001
Eckardt, N. A. (2001). Functional evolutionary genetics and plant adaptation: Linking phenotype and genotype. Plant Cell 13, 1249–1254. doi: 10.2307/3871291
Ehrenreich, I. M., and Purugganan, M. D. (2006). The molecular genetic basis of plant adaptation. Am. J. Bot. 93, 953–962. doi: 10.3732/ajb.93.7.953
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Erb, M., and Kliebenstein, D. J. (2020). Plant secondary metabolites as defenses, regulators, and primary metabolites: The blurred functional trichotomy. Plant Physiol. 184, 39–52. doi: 10.1104/pp.20.00433
Fisher, R. A., and Russell, E. J. (1922). On the mathematical foundations of theoretical statistics. Philos. Trans. A Math. Phys. 222, 309–368. doi: 10.1098/rsta.1922.0009
Flood, P. J., and Hancock, A. M. (2017). The genomic basis of adaptation in plants. Curr. Opin. Plant Biol. 36, 88–94. doi: 10.1016/j.pbi.2017.02.003
Friedman, A. R., and Baker, B. J. (2007). The evolution of resistance genes in multi-protein plant resistance systems. Curr. Opin. Genet. Dev. 17, 493–499. doi: 10.1016/j.gde.2007.08.014
Friedman, M. (2002). Tomato glycoalkaloids: Role in the plant and in the diet. J. Agric. Food Chem. 50, 5751–5780. doi: 10.1021/jf020560c
Friedman, M. (2006). Potato glycoalkaloids and metabolites: Roles in the plant and in the diet. J. Agric. Food Chem. 54, 8655–8681. doi: 10.1021/jf061471t
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Fujita, T., Maggio, A., Garcia-Rios, M., Bressan, R. A., and Csonka, L. N. (1998). Comparative analysis of the regulation of expression and structures of two evolutionarily divergent genes for Delta1-pyrroline-5-carboxylate synthetase from tomato. Plant Physiol. 118, 661–674. doi: 10.1104/pp.118.2.661
Gallé, Á, Czékus, Z., Bela, K., Horváth, E., Ördög, A., Csiszár, J., et al. (2019). Plant glutathione transferases and light. Front. Plant Sci. 9:1944. doi: 10.3389/fpls.2018.01944
Geeta, R., and Gharaibeh, W. (2007). Historical evidence for a pre-columbian presence of Datura in the old world and implications for a first millennium transfer from the new world. J. Biosci. 32, 1227–1244. doi: 10.1007/s12038-007-0132-y
Goldberg, J. K., Lively, C. M., Sternlieb, S. R., Pintel, G., Hare, J. D., Morrissey, M. B., et al. (2020). Herbivore-mediated negative frequency-dependent selection underlies a trichome dimorphism in nature. Evol. Lett. 4, 83–90. doi: 10.1002/evl3.157
Goldberg, J. K., Sternlieb, S. R., Pintel, G., and Delph, L. F. (2021). Observational evidence of herbivore-specific associational effects between neighboring conspecifics in natural, dimorphic populations of Datura wrightii. Ecol. Evol. 11, 5547–5561. doi: 10.1002/ece3.7454
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997. doi: 10.1093/molbev/mst100
Hare, J. D., and Elle, E. (2001). Geographic variation in the frequencies of trichome phenotypes of Datura wrightii and correlation with annual water deficit. Madroño 48, 33–37.
Hare, J. D., and Smith, J. L. (2005). Competition, herbivory, and reproduction of trichome phenotypes of Datura wrightii. Ecology 86, 334–339. doi: 10.1890/04-0972
Haudecoeur, E., Planamente, S., Cirou, A., Tannières, M., Shelp, B. J., Moréra, S., et al. (2009). Proline antagonizes GABA-induced quenching of quorum-sensing in Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. U.S.A. 106, 14587–14592. doi: 10.1073/pnas.0808005106
Hennig, W. (1950). Grundzuge einer theorie der phylogenetischen systema tik. Berlin: Deutscher Zentralveriag, 370. Reprinted 1981. Koenigstein: Koeltz.
Hulse-Kemp, A. M., Maheshwari, S., Stoffel, K., Hill, T. A., Jaffe, D., Williams, S. R., et al. (2018). Reference quality assembly of the 3.5-Gb genome of Capsicum annuum from a single linked-read library. Hortic. Res. 5, 1–13. doi: 10.1038/s41438-017-0011-0
Jiménez-Lobato, V., and Núñez-Farfán, J. (2021). Mating system of Datura inoxia: Association between selfing rates and herkogamy within populations. PeerJ 9:e10698. doi: 10.7717/peerj.10698
Jirschitzka, J., Schmidt, G. W., Reichelt, M., Schneider, B., Gershenzon, J., and D’Auria, J. C. (2012). Plant tropane alkaloid biosynthesis evolved independently in the Solanaceae and Erythroxylaceae. Proc. Natl. Acad. Sci. U.S.A. 109, 10304–10309. doi: 10.1073/pnas.1200473109
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kariñho-Betancourt, E. (2018). Plant-herbivore interactions and secondary metabolites of plants: Ecological and evolutionary perspectives. Bot. Sci. 96, 35–51. doi: 10.17129/botsci.1860
Kariñho-Betancourt, E., Agrawal, A. A., Halitschke, R., and Núñez-Farfán, J. (2015). Phylogenetic correlations among chemical and physical plant defenses change with ontogeny. New Phytol. 206, 796–806. doi: 10.1111/nph.13300
Kariñho-Betancourt, E., Soto, P. H., Cortés, N. C., Anaya, M. R., Estrella, A. H., and Oyama, K. (2020). “Ecological genomics of plant-insect interactions: The case of wasp-induced galls,” in Evolutionary ecology of plant-herbivore interaction, eds J. Núñez-Farfán and P. Valverde (Berlin: Springer Nature), 315–341. doi: 10.1007/978-3-030-46012-9_17
Karunanithi, P. S., and Zerbe, P. (2019). Terpene synthases as metabolic gatekeepers in the evolution of plant terpenoid chemical diversity. Front. Plant Sci. 10:1166. doi: 10.3389/fpls.2019.01166
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kilili, K. G., Atanassova, N., Vardanyan, A., Clatot, N., Al-Sabarna, K., Kanellopoulos, P. N., et al. (2004). Differential roles of tau class glutathione S-transferases in oxidative stress. J. Biol. Chem. 279, 24540–24551. doi: 10.1074/jbc.M309882200
Kogan, M., and Goeden, R. D. (1970). The host-plant range of Lema trilineata daturaphila (Coleoptera: Chrysomelidae). Ann. Entomol. Soc. Am. 63, 1175–1180. doi: 10.1093/aesa/63.4.1175
Kohnen-Johannsen, K. L., and Kayser, O. (2019). Tropane alkaloids: Chemistry, pharmacology, biosynthesis and production. Molecules 24:796. doi: 10.3390/molecules24040796
Koonin, E. V. (2005). Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 39, 309–338.
Kourelis, J., and van der Hoorn, R. A. L. (2018). Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for r protein function. Plant Cell 30, 285–299. doi: 10.1105/tpc.17.00579
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Lackus, N. D., Lackner, S., Gershenzon, J., Unsicker, S. B., and Köllner, T. G. (2018). The occurrence and formation of monoterpenes in herbivore-damaged poplar roots. Sci. Rep. 8:17936. doi: 10.1038/s41598-018-36302-6
Leebens-Mack, J. H., Barker, M. S., and Carpenter, E. J. (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685. doi: 10.1038/s41586-019-1693-2
Li, W., and Godzik, A. (2006). Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Liang, X., and Zhou, J.-M. (2018). Receptor-like cytoplasmic kinases: Central players in plant receptor kinase–mediated signaling. Annu. Rev. Plant Biol. 69, 267–299. doi: 10.1146/annurev-arplant-042817-040540
Luna-Cavazos, M., and Bye, R. (2011). Phytogeographic analysis of the genus Datura (Solanaceae) in continental Mexico. Rev. Mex. Biodivers. 82, 977–988. doi: 10.22201/ib.20078706e.2011.3.720
Mace, E. S., Gebhardt, C. G., and Lester, R. N. (1999). AFLP analysis of genetic relationships in the tribe datureae (Solanaceae). Theor. Appl. Genet. 99, 634–641. doi: 10.1007/s001220051278
Magalhães, D. M., Scholte, L. L. S., Silva, N. V., Oliveira, G. C., Zipfel, C., Takita, M. A., et al. (2016). LRR-RLK family from two Citrus species: Genome-wide identification and evolutionary aspects. BMC Genom. 17:623. doi: 10.1186/s12864-016-2930-9
Magallon, S., and Sanderson, M. J. (2001). Absolute diversification rates in angiosperm clades. Evolution 55, 1762–1780.
McHale, L., Tan, X., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: Adaptable guards. Genome Biol. 7:212. doi: 10.1186/gb-2006-7-4-212
Miranda-Pérez, A., Castillo, G., Hernández-Cumplido, J., Valverde, P. L., Borbolla, M., Cruz, L. L., et al. (2016). Natural selection drives chemical resistance of Datura stramonium. PeerJ 4:e1898. doi: 10.7717/peerj.1898
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L., et al. (2013). FUBAR: A Fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205. doi: 10.1093/molbev/mst030
Narayanasamy, P., Eoh, H., Brennan, P. J., and Crick, D. C. (2010). Synthesis of 4-Diphosphocytidyl-2-C-Methyl-D-Erythritol 2-Phosphate and kinetic studies of Mycobacterium tuberculosis IspF. Chem. Biol. 17, 117–122. doi: 10.1016/j.chembiol.2010.01.013
Nürk, N. M., Linder, H. P., Onstein, R. E., Larcombe, M. J., Hughes, C. E., Piñeiro Fernández, L., et al. (2020). Diversification in evolutionary arenas—Assessment and synthesis. Ecol. Evol. 10, 6163–6182.
Padmanabhan, M., Cournoyer, P., and Dinesh-Kumar, S. P. (2009). The leucine-rich repeat domain in plant innate immunity: A wealth of possibilities. Cell. Microbiol. 11, 191–198. doi: 10.1111/j.1462-5822.2008.01260.x
Parvanova, D., Popova, A., Zaharieva, I., Lambrev, P., Konstantinova, T., Taneva, S., et al. (2004). Low temperature tolerance of tobacco plants transformed to accumulate proline, fructans, or glycine betaine. variable chlorophyll fluorescence evidence. Photosynthetica 42, 179–185. doi: 10.1023/B:PHOT.0000040588.31318.0f
Peña, M. J., Zhong, R., Zhou, G.-K., Richardson, E. A., O’Neill, M. A., Darvill, A. G., et al. (2007). Arabidopsis irregular xylem8 and irregular xylem9: Implications for the complexity of glucuronoxylan biosynthesis. Plant Cell 19, 549–563. doi: 10.1105/tpc.106.049320
Pérez-Arellano, I., Carmona-Álvarez, F., Martínez, A. I., Rodríguez-Díaz, J., and Cervera, J. (2010). Pyrroline-5-carboxylate synthase and proline biosynthesis: From osmotolerance to rare metabolic disease. Protein Sci. 19, 372–382. doi: 10.1002/pro.340
Pichersky, E., and Raguso, R. A. (2018). Why do plants produce so many terpenoid compounds? New Phytol. 220, 692–702. doi: 10.1111/nph.14178
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Qamar, A., Mysore, K. S., and Senthil-Kumar, M. (2015). Role of proline and pyrroline-5-carboxylate metabolism in plant defense against invading pathogens. Front. Plant Sci 6:503. doi: 10.3389/fpls.2015.00503
Rahimi, S., Kim, J., Mijakovic, I., Jung, K.-H., Choi, G., Kim, S.-C., et al. (2019). Triterpenoid-biosynthetic UDP-glycosyltransferases from plants. Biotechnol. Adv. 37:107394.
Ramakrishna, A., and Ravishankar, G. A. (2011). Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. Behav. 6, 1720–1731. doi: 10.4161/psb.6.11.17613
Razali, R., Bougouffa, S., Morton, M. J. L., Lightfoot, D. J., Alam, I., Essack, M., et al. (2018). The genome sequence of the wild tomato Solanum pimpinellifolium provides insights into salinity tolerance. Front. Plant Sci 9:1402. doi: 10.3389/fpls.2018.01402
Rehman, H. M., Nawaz, M. A., Shah, Z. H., Ludwig-Müller, J., Chung, G., Ahmad, M. Q., et al. (2018). Comparative genomic and transcriptomic analyses of Family-1 UDP glycosyltransferase in three Brassica species and Arabidopsis indicates stress-responsive regulation. Sci. Rep. 8:1875.
Rich-Griffin, C., Stechemesser, A., Finch, J., Lucas, E., Ott, S., and Schäfer, P. (2020). Single-cell transcriptomics: A high-resolution avenue for plant functional genomics. Trends Plant Sci. 25, 186–197. doi: 10.1016/j.tplants.2019.10.008
Rio, D. C., Ares, M., Hannon, G. J., and Nilsen, T. W. (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, db–rot5439. doi: 10.1101/pdb.prot5439
Roddick, J. (1991). “The importance of the solanceae in medicine and drug therapy,” in Solanaceae III: Taxonomy, chemistry, evolution, eds G. Hawkes, R. N. Lester, M. Nee, and N. Estrada (Kew: Royal Botanic Garden Press), 7–23.
Rohdich, F., Wungsintaweekul, J., Luttgen, H., Fischer, M., Eisenreich, W., Schuhr, C. A., et al. (2000). Biosynthesis of terpenoids: 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase from tomato. Proc. Natl. Acad. Sci. U.S.A 97, 8251–8256. doi: 10.1073/pnas.140209197
Russell, J. J., Theriot, J. A., Sood, P., Marshall, W. F., Landweber, L. F., Fritz-Laylin, L., et al. (2017). Non-model model organisms. BMC Biol. 15:55. doi: 10.1186/s12915-017-0391-5
Sahu, P. K., Pradhan, S. P., and Kumar, P. S. (2022). Isolation, elucidation, and structure–activity relationships of phytoalkaloids from solanaceae. Stud. Nat. Prod. Chem. 72, 371–389. doi: 10.1016/B978-0-12-823944-5.00007-7
Sanderson, M. J. (2003). r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302. doi: 10.1093/bioinformatics/19.2.301
Särkinen, T., Bohs, L., Olmstead, R. G., and Knapp, S. (2013). A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): A dated 1000-tip tree. BMC Evol. Biol. 13:214. doi: 10.1186/1471-2148-13-214
Sato, S., Tabata, S., Hirakawa, H., Asamizu, E., Shirasawa, K., Isobe, S., et al. (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641. doi: 10.1038/nature11119
Savolainen, O., Lascoux, M., and Merilä, J. (2013). Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820. doi: 10.1038/nrg3522
Schwacke, R., Ponce-Soto, G. Y., Krause, K., Bolger, A. M., Arsova, B., Hallab, A., et al. (2019). MapMan4: A refined protein classification and annotation framework applicable to multi-omics data analysis. Mol. Plant 12, 879–892. doi: 10.1016/j.molp.2019.01.003
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). “BUSCO: Assessing genome assembly and annotation completeness,” in Gene prediction: Methods and protocols, ed. M. Kollmar (Berlin: Springer), 227–245. doi: 10.1007/978-1-4939-9173-0_14
Sharma, A., Pandey, A., Sharma, S., Chatterjee, I., Mehrotra, R., Sehgal, A., et al. (2014). Genetic polymorphism of glutathione S-transferase P1 (GSTP1) in Delhi population and comparison with other global populations. Meta Gene 2, 134–142. doi: 10.1016/j.mgene.2013.12.003
Shonle, I., and Bergelson, J. (2000). Evolutionary ecology of the tropane alkaloids of Datura stramonium l. (Solanaceae). Evolution 54, 778–788. doi: 10.1111/j.0014-3820.2000.tb00079.x
Sierro, N., Battey, J. N. D., Ouadi, S., Bakaher, N., Bovet, L., Willig, A., et al. (2014). The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5:3833. doi: 10.1038/ncomms4833
Sierro, N., Battey, J. N., Ouadi, S., Bovet, L., Goepfert, S., Bakaher, N., et al. (2013). Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 14:R60. doi: 10.1186/gb-2013-14-6-r60
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Singh, B., and Sharma, R. A. (2015). Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. 3 Biotech 5, 129–151. doi: 10.1007/s13205-014-0220-2
Stadler, T. (2011). Mammalian phylogeny reveals recent diversification rate shifts. Proc. Natl. Acad. Sci. U.S.A. 108, 6187–6192.
Soltis, P. S., and Soltis, D. E. (2021). Plant genomes: Markers of evolutionary history and drivers of evolutionary change. Plants People Planet 3, 74–82. doi: 10.1002/ppp3.10159
Song, B.-H., and Mitchell-Olds, T. (2011). Evolutionary and ecological genomics of non-model plants. J. Syst. Evol. 49, 17–24. doi: 10.1111/j.1759-6831.2010.00111.x
Sork, V. L. (2018). Genomic studies of local adaptation in natural plant populations. J. Hered. 109, 3–15. doi: 10.1093/jhered/esx091
Stahle, M. I., Kuehlich, J., Staron, L., von Arnim, A. G., and Golz, J. F. (2009). YABBYs and the transcriptional corepressors LEUNIG and LEUNIG_HOMOLOG maintain leaf polarity and meristem activity in Arabidopsis. Plant Cell 21, 3105–3118. doi: 10.1105/tpc.109.070458
Stone, E. A., and Sidow, A. (2005). Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 15, 978–986. doi: 10.1101/gr.3804205
Stone, J. M., and Walker, J. C. (1995). Plant protein kinase families and signal transduction. Plant Physiol. 108, 451–457. doi: 10.1104/pp.108.2.451
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Symon, D., and Haegi, L. (1991). “Datura (Solanaceae) is a new world genus,” in Solanaceae, eds J. G. Hawkes, R. N. Lester, M. Nee, and N. Estrada-R (Kew: Royal Botanic Gardens and London: Linnaean Society), 197–211.
Tang, D., Wang, G., and Zhou, J.-M. (2017). Receptor kinases in plant-pathogen interactions: More than pattern recognition. Plant Cell 29, 618–637. doi: 10.1105/tpc.16.00891
Thordal-Christensen, H. (2020). A holistic view on plant effector-triggered immunity presented as an iceberg model. Cell. Mol. Life Sci. 77, 3963–3976. doi: 10.1007/s00018-020-03515-w
Turner, S. R., and Somerville, C. R. (1997). Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant Cell 9, 689–701. doi: 10.1105/tpc.9.5.689
Valdar, W. S. J. (2002). Scoring residue conservation. Proteins Struct. Funct. Bioinf. 48, 227–241. doi: 10.1002/prot.10146
van Wersch, S., Tian, L., Hoy, R., and Li, X. (2020). Plant NLRs: The whistleblowers of plant immunity. Plant Commun. 1:100016. doi: 10.1016/j.xplc.2019.100016
Velázquez-Márquez, S., De-la-Cruz, I. M., Tapia-López, R., and Núñez-Farfán, J. (2021). Tropane alkaloids and terpenes synthase genes of Datura stramonium (Solanaceae). PeerJ 9:e11466. doi: 10.7717/peerj.11466
Venekamp, J. H., and Koot, T. M. (1988). The sources of free proline and asparagine in field bean plants, Vicia faba L., during and after a short period of water withholding. J. Plant Physiol. 132, 102–109. doi: 10.1016/S0176-1617(88)80192-5
Vogt, T., and Jones, P. (2000). Glycosyltransferases in plant natural product synthesis: Characterization of a supergene family. Trends Plant Sci. 5, 380–386. doi: 10.1016/S1360-1385(00)01720-9
Wilson, A. E., and Tian, L. (2019). Phylogenomic analysis of UDP-dependent glycosyltransferases provides insights into the evolutionary landscape of glycosylation in plant metabolism. Plant J. 100, 1273–1288. doi: 10.1111/tpj.14514
Wink, M. (2018). Plant secondary metabolites modulate insect behavior-steps toward addiction? Front. Physiol. 9:364. doi: 10.3389/fphys.2018.00364
Wu, A.-M., Hörnblad, E., Voxeur, A., Gerber, L., Rihouey, C., Lerouge, P., et al. (2010). Analysis of the Arabidopsis IRX9/IRX9-L and IRX14/IRX14-L pairs of glycosyltransferase genes reveal critical contributions to biosynthesis of the hemicellulose glucuronoxylan. Plant Physiol. 153, 542–554. doi: 10.1104/pp.110.154971
Xu, S., Brockmöller, T., Navarro-Quezada, A., Kuhl, H., Gase, K., Ling, Z., et al. (2017). Wild tobacco genomes reveal the evolution of nicotine biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 114, 6133–6138. doi: 10.1073/pnas.1700073114
Xu, X., Pan, S., Cheng, S., Zhang, B., Mu, D., Ni, P., et al. (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195. doi: 10.1038/nature10158
Zhang, F., Wang, H., Kalve, S., Wolabu, T. W., Nakashima, J., Golz, J. F., et al. (2019). Control of leaf blade outgrowth and floral organ development by LEUNIG, ANGUSTIFOLIA3 and WOX transcriptional regulators. New Phytol. 223, 2024–2038. doi: 10.1111/nph.15921
Keywords: comparative genomics, Datura, gene family analysis, positive selection, secondary metabolites, transcriptomics
Citation: De-la-Cruz IM, Kariñho-Betancourt E, Núñez-Farfán J and Oyama K (2022) Gene family evolution and natural selection signatures in Datura spp. (Solanaceae). Front. Ecol. Evol. 10:916762. doi: 10.3389/fevo.2022.916762
Received: 10 April 2022; Accepted: 24 October 2022;
Published: 15 November 2022.
Edited by:
Diego Baldo, CONICET Institute of Subtropical Biology (IBS), ArgentinaReviewed by:
Amy Litt, University of California, Riverside, United StatesCopyright © 2022 De-la-Cruz, Kariñho-Betancourt, Núñez-Farfán and Oyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eunice Kariñho-Betancourt, a2FyaW5oby5iZXRhbmNvdXJ0QGdtYWlsLmNvbQ==
†Present Address: Ivan M. De-la-Cruz, Department of Plant Protection Biology, Swedish University of Agricultural Sciences (SLU), Alnarp, Sweden
‡These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.