 Stefanie Agne
Stefanie Agne Michaela Preick1
Michaela Preick1 Nicolas Straube
Nicolas Straube
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Ecol. Evol., 17 June 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.909846
This article is part of the Research TopicRecent Advances in Museomics: Revolutionizing Biodiversity ResearchView all 16 articles
A growing number of publications presenting results from sequencing natural history collection specimens reflect the importance of DNA sequence information from such samples. Ancient DNA extraction and library preparation methods in combination with target gene capture are a way of unlocking archival DNA, including from formalin-fixed wet-collection material. Here we report on an experiment, in which we used an RNA bait set containing baits from a wide taxonomic range of species for DNA hybridisation capture of nuclear and mitochondrial targets for analysing natural history collection specimens. The bait set used consists of 2,492 mitochondrial and 530 nuclear RNA baits and comprises specific barcode loci of diverse animal groups including both invertebrates and vertebrates. The baits allowed to capture DNA sequence information of target barcode loci from 84% of the 37 samples tested, with nuclear markers being captured more frequently and consensus sequences of these being more complete compared to mitochondrial markers. Samples from dry material had a higher rate of success than wet-collection specimens, although target sequence information could be captured from 50% of formalin-fixed samples. Our study illustrates how efforts to obtain barcode sequence information from natural history collection specimens may be combined and are a way of implementing barcoding inventories of scientific collection material.
The growing interest in accessing DNA of natural history wet-collection specimens, which have long been recalcitrant regarding DNA analyses, is reflected in increasing numbers of publications reporting sequencing of this highly fragmented DNA (e.g., Lyra et al., 2020; Rancilhac et al., 2020; Scherz et al., 2020; Hahn et al., 2021; Straube et al., 2021a,b). Combining ancient DNA extraction methods, single stranded DNA library construction and short-read high throughput sequencing technology allows for obtaining DNA sequences of museum specimens at unprecedented scales (e.g., Hahn et al., 2021; Straube et al., 2021a). In taxonomy, unlocking DNA sequence information from rare and extinct species as well as type material is of particular interest. Numerous described species are only known from few, aged museum specimens and often re-collection efforts are hindered by several factors such as extensive sampling efforts, conservation concerns, politically instable situations in countries of origin or simply rareness of the species in question. However, rare species described from remote localities are of special concern in conservation, directing the attention to museum specimens as potential alternative DNA sources for taxonomic evaluation as basis for conservation efforts. Besides their undoubted importance for taxonomic research (e.g., Lyra et al., 2020; Rancilhac et al., 2020; Scherz et al., 2020; Straube et al., 2021b), type specimens may as well represent the only representatives of a rare or extinct species. Most of such specimens lack a phylogenetically close reference genome, but for taxonomy, barcode genes for species delimitation are generally sufficient as references for phylogenetic placement of species’ haplotypes. In these circumstances, DNA sequences from type material can play a key role.
Ancient DNA methods have paved the way for accessing DNA sequence information from archival samples, including formalin-fixed wet-collection samples (Stiller et al., 2016; Gansauge et al., 2017; Straube et al., 2021a), even on the genome level (Hahn et al., 2021). These approaches are laborious and time consuming, however. As shown previously in Straube et al. (2021a), the level of target DNA in initial test-sequencing datasets may be low. Shotgun sequencing of such DNA libraries then becomes inefficient in terms of associated costs necessary to attain coverage levels allowing for reconstructing specific barcode loci. Target gene capture as alternative can be an additional costly and time-intensive step, especially when a second round of capture is performed which has been shown to increase sequencing success (e.g., Li et al., 2013, 2015; Templeton et al., 2013; Springer et al., 2015; Paijmans et al., 2016). In an effort to increase efficiency and decrease overall costs for target capture of sample specific barcode markers in museum specimens, we report here on the design and successful application of an RNA bait set targeting taxonomically useful barcode markers in a variety of natural history collection samples of different phyla. Undergoing this process, we also aim to detect factors that may have an impact on the capture success such as different target regions, tissue type, fixation history, and genetic distance between bait and target sequences.
We obtained 37 samples including dried bone, teeth, and soft tissue samples as well as muscle and skin from wet-collection specimens. Representatives of the following classes were included: Demospongiae, Gastropoda, Polychaeta, Malacostraca, Insecta, Actinopterygii, Chondrichthyes, Amphibia, and Reptilia. The investigated samples range in age from 25 to 192 years (Supplementary Table 1). Along with the tissue samples, we obtained information on the samples using a standardised sample sheet (Supplementary Table 2). The requested information relates to the age, fixation, and preservation details as far as available, target barcode loci, bait sequences to capture specific barcode loci, reference genomes and taxonomic history of the sample. DNA was extracted from samples listed in Supplementary Table 1 following the different DNA extraction treatments described in Straube et al. (2021a) based on the ancient DNA extraction protocol specified in Dabney et al. (2013) using a GuSCN based extraction buffer (Rohland et al., 2004). Subsequently, single stranded DNA libraries were prepared for each sample following the protocol by Gansauge et al. (2017). For obtaining information on the presence of target DNA, test-sequencing as described in Straube et al. (2021a) was performed. Independent of presence of endogenous DNA, target capture was subsequently performed for all samples to test if the limited information of the test-sequencing data may fail to detect endogenous DNA even though it is present in the DNA library.
For target capture of barcode loci, specific bait sequences and reference genomes provided partially by our collaborators, but mostly obtained from public resources (Supplementary Table 3) were sent to Arbor Biosciences® and split into a mitochondrial and a nuclear bait set. For both sets of sequences, 80 nt, 3x tiled baits were designed. While the mitochondrial baits were not further processed bioinformatically, the nuclear baits were filtered in two steps. First, baits were blasted to reference genomes from available most closely related species (Supplementary Table 1). Any bait that had blast hits to a region of the genome that was greater than 25% soft-masked for repeats was removed. The second filtering step was based on the number of bait hits and the predicted melting temperatures between the bait and those blast hits to detect the number of binding sites a bait may have, which ultimately resulted in the exclusion of 97 nuclear baits. A final set of 2,492 mitochondrial and 530 nuclear RNA baits was produced. Target capture was performed for each sample listed in Supplementary Table 1 following the manufacturer’s protocol for N = 2 samples. For the remaining 35 samples a target-gene enrichment protocol based on the Mybaits-manual-v3 was used, which is cost-reducing and requires less of RNA baits per sample compared to the recommended amount but maintaining the same level of target capture success (Huang et al., 2021). For both protocols, we used an in-solution hybridisation temperature of 65°C for 24 h. The capture was performed twice including a second amplification of libraries after the first round of target capture. Optimal number of amplification cycles was estimated for each library by performing a qPCR. DNA libraries were double-indexed during amplification and sequenced as described in Paijmans et al. (2017). Sequencing was performed on an Illumina Nextseq 500 sequencing platform, using 500/550 High Output v2.5 (75 cycles, Illumina 20024906) kits (75 bp single-end reads). All laboratory steps as well as sequencing was conducted in the molecular laboratories of the AG Hofreiter at the University of Potsdam. At least three million sequencing reads were targeted for each sample to gain sufficient coverage of target markers. Sequencing reads available after target capture underwent quality checking and trimming as in Straube et al. (2021a) and were subsequently used to reconstruct the target barcode loci using mapping and consensus sequence generation in BWA-ALN v.0.7.17 (Li and Durbin, 2009) and Bcftools v.1.9 (Li, 2011). We used either the bait sequences or phylogenetically closer reference sequences which became available after bait production (Supplementary Table 4). Afterwards, consensus sequences were analysed for phylogenetic position and classification.
We tested for correlation between the completeness of target genes after hybridisation capture and the phylogenetic distance of RNA bait sequences to target consensus sequences (p-distances). Therefore, each target consensus sequence was aligned to the appropriate bait sequence as listed in Supplementary Table 1 using Mafft v.7.49 (Katoh et al., 2002) and resulting p-distances were calculated using MEGA v.11.0.10 (Kumar et al., 2016). If several bait sequences were available for aligning to a genetic locus of a species, the reference with the smallest p-distance to the consensus sequence was used. For correlation analysis, Pearson’s correlation coefficient was calculated, and a t-test was performed. Specimens with too low endogenous DNA content to create a consensus sequence after target capture were not included in the analysis. We further tested for correlation between sequencing depth and completeness as described above.
After test-sequencing, we detected endogenous DNA in most of our samples (91.9%; Supplementary Table 1). For samples that showed no endogenous DNA after test sequencing, target capture attempts failed. Available sequencing data after target capture ranged from 391,964 to 12,195,369 raw reads and 91,101 to 10,611,372 reads after trimming. Trimmed reads including PCR duplicates that mapped to the reference sequences ranged between 0 and 69.23% (Supplementary Table 4). We were able to capture DNA sequence information of target barcode loci from 84% of our samples (Figure 1), 73.52% for mitochondrial and 94.28% for nuclear target genes, respectively. The completeness of all nuclear barcode loci is 85.15% and higher than that of the mitochondrial loci, the completeness of which is 72.25% (Figure 1). The best results in terms of consistency and sequence completeness were obtained from the crocodilian bone and dry skin samples with an average consensus sequence completeness of 98.31%. For wet-collection material we obtained sequence information for 86.2% of the target genes and an average sequence completeness of 71.74%. Similar differences are observed when comparing the different materials of the Demospongiae samples, with an average consensus sequence completeness of 55.02% for the wet collection tissues and 74.38% for the dried tissues, respectively. Three of the ten specimens for which formalin fixation is assumed resulted in target gene completeness above 75% (Figure 1). The Mollusca samples in particular showed a high target sequence completeness with an average of 96.12% in all five target loci tested.
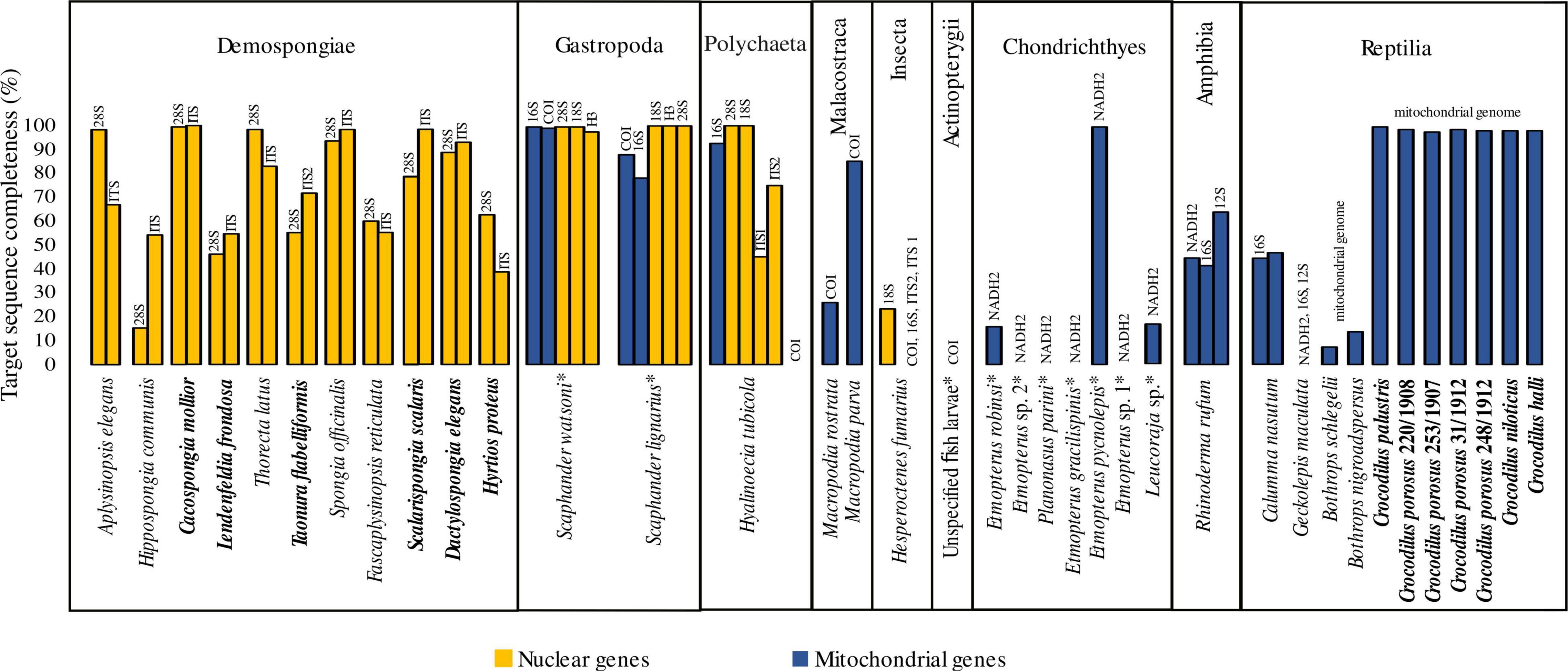
Figure 1. Completeness of target genes after hybridisation capture. Dry material is indicated in bold, all other samples originate from wet-collection specimens. Assumed formalin-fixation before wet-collection preservation of specimens is indicated by an asterisk.
The p-distance, defined as proportion of different nucleotides per total numbers of nucleotides compared, was on average 7.62% (range between 0 and 56.80%) and did not correlate with the target gene completeness (Pearson correlation coefficient: r = −0.20; p = 0.0). We found similar results when calculating the correlation coefficient for mitochondrial and nuclear data separately (Pearson correlation coefficient: r = −0.40; p = 0.49 for mitochondrial data; r = −0.22; p = 0.21 for nuclear data). A correlation between sequencing depth and target marker completeness was not detected (r = 0.29).
In this report, we present results from a target capture experiment using a mixed bait set covering specific taxa across several animal phyla (Porifera, Annelida, Mollusca, Arthropoda, and Chordata) set on a range of museum collection samples. We were able to obtain sequence information for 75% of all samples which is promising to be useful sequence information for phylogenetic placement of specimens. The obtained sequences will further be used for sample specific phylogenetic analyses. For the samples of the classes Demospongiae, Gastropoda, Polychaeta and Amphibia, we received consistently high sequence completeness (Figure 1 and Supplementary Table 4). Above all, dry crocodilian material (tooth and bone) that are up to 100 years old (Supplementary Table 1) have shown to be a reliable source of DNA. Several samples of the classes Malacostraca, Insecta and Chondrichthyes targeted for mitochondrial and nuclear loci show low capture success. The single actinopterygian sample failed, which may have been due to long-term formalin preservation (N. Schnell pers. comm.). Although our results imply that target capture of nuclear markers outperforms capture of mitochondrial markers, the differences are likely introduced by samples with a generally low completeness of target sequences. In cases where both nuclear and mitochondrial markers were captured, similar results regarding the target sequence completeness were obtained (Figure 1). In general, wet-collection specimens showed poorer results compared to dry material. Water in ethanol solutions used for long-term storage intensifies hydrolysis (Lindahl, 1993) and may have contributed to our results.
To overcome potential disadvantages of large phylogenetic distances between bait and target sequences, a second round of target capture, as performed herein, can increase capture efficiency (e.g., Li et al., 2013; Paijmans et al., 2016). In this study, the p-distances between the bait sequences and the completeness of the consensus sequences are not correlated, which might be different if all consensus sequences were complete and should be investigated in further studies. Further experimental optimisation such as hybridisation temperature and time may allow for increasing capture efficiency in samples with low or no target gene completeness. However, our study also includes samples that should have small phylogenetic distances between bait and target sequences (e.g., Etmopterus spp., Figure 1). We were able to recover the complete mitochondrial marker sequence from only a single of these specimens (E. pycnolepis). As insufficient sequencing effort can be ruled out, reasons for the failure of the remaining samples could be related to fixation and preservation induced DNA damage. Details on the fixation history of most samples are poorly known (Supplementary Table 1), however, formalin has severe DNA damaging effects (Hoffman et al., 2015). Different ways of formalin fixation can also play a role in the success of DNA recovery (e.g., Paireder et al., 2013) ultimately influencing the amount and complexity of available target DNA for the target capture experiment. Besides these factors degradation and associated short DNA fragment size may have impeded the mapping attempts (Huson et al., 2007).
An alternative to commercially purchased RNA baits as used in this study are home-made DNA baits using PCR products of amplified target markers for DNA bait library production (González Fortes and Paijmans, 2019). In general, bait production for a small sample number targeting a single or few barcode markers of phylogenetically close taxonomic units is costly and inefficient. A combination of taxon-specific bait sequences for target capturing widely different taxa can overcome these limitations and enables the simultaneous sequencing of several phylogenetically distant taxa of interest. Our approach allows for cost-sharing between collection subsections and paves the way for implementing barcoding inventories in natural history collections, for example barcoding inventories of type specimens.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: 10.6084/m9.figshare.19619052.
Ethical review and approval was not required for the animal study because no living animals were collected or examined. DNA samples were taken solely from museum specimens.
NS and MH designed the study. SA and NS performed the laboratory work under supervision of MP. SA analysed the data under supervision of NS and MH. NS and SA wrote the manuscript with contributions from all authors. All authors contributed to the article and approved the submitted version.
This work was funded by the German Research Foundation (DFG; project number 351649567 to NS and MH within the DFG SPP 1991 “Taxon-Omics”). The publication of this article was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; Project No. 491466077).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to sincerely thank our colleagues for entrusting us with their valuable samples and for their support with compiling the reference sequence set: Dirk Erpenbeck (LMU), Steffen Roth (UM), Justine Siegwald (UM), Nataliya Budaeva (UM), Frank Glaw (ZSM), Gavin Naylor (UF), Nalani Schnell (MNHN), Ralf Thiel (UH), Simon Weigmann (Elasmo-Lab), Michael Raupach (ZSM), Claudia Koch (ZFMK), Silke Schweiger (NHMW), and Axel Barlow (UWB). We would also like to thank the HPC team at the University of Potsdam for creating and maintaining the university’s server on which we ran some of our analyses.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.909846/full#supplementary-material
Dabney, J., Knapp, M., Glocke, I., Gansauge, M. T., Weihmann, A., Nickel, B., et al. (2013). Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. U.S.A. 110, 15758–15763. doi: 10.1073/pnas.1314445110
Gansauge, M. T., Gerber, T., Glocke, I., Korlević, P., Lippik, L., Nagel, S., et al. (2017). Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 45:e79. doi: 10.1093/nar/gkx033
González Fortes, G., and Paijmans, J. L. (2019). “Whole-genome capture of ancient DNA using homemade baits,” in Ancient DNA. eds S. Beth, B. Axel, D. H. Peter, H. Michael, L. A. P. Johanna, and E. R. S. André (New York: Humana Press), 93–105. doi: 10.1007/978-1-4939-9176-1_11
Hahn, E. E., Alexander, M. R., Grealy, A., Stiller, J., Gardiner, D. M., and Holleley, C. E. (2021). Unlocking inaccessible historical genomes preserved in formalin. Mol. Ecol. Resour. [Epub ahead of print]. doi: 10.1111/1755-0998.13505
Hoffman, E. A., Frey, B. L., Smith, L. M., and Auble, D. T. (2015). Formaldehyde crosslinking: a tool for the study of chromatin complexes. J. Biol. Chem. 290, 26404–26411. doi: 10.1074/jbc.R115.651679
Huang, J. M., Yuan, H., and Li, C. H. (2021). Protocol for Cross-species Target-gene Enrichment. Bio. 101:e1010606. doi: 10.21769/BioProtoc.1010606
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genom. Res. 17, 377–386. doi: 10.1101/gr.5969107
Katoh, K., Misawa, K., Kuma, K. I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Li, C., Corrigan, S., Yang, L., Straube, N., Harris, M., Hofreiter, M., et al. (2015). DNA capture reveals transoceanic gene flow in endangered river sharks. Proc. Natl. Acad. Sci. U.S.A. 112, 13302–13307. doi: 10.1073/pnas.1508735112
Li, C., Hofreiter, M., Straube, N., Corrigan, S., and Naylor, G. J. P. (2013). Capturing protein-coding genes across highly divergent species. BioTechniques 54, 321–326. doi: 10.2144/000114039
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature 362, 709–715. doi: 10.1038/362709a0
Lyra, M. L., Carolina, A., Lourenço, C., Pinheiro, P. D. P., Pezzuti, T. L., Baêta, D., et al. (2020). High-throughput DNA sequencing of museum specimens sheds light on the long-missing species of the Bokermannohyla claresignata group (Anura: Hylidae: Cophomantini). Zool. J. Linnean Soc. 190, 1235–1255. doi: 10.1093/zoolinnean/zlaa033
Paijmans, J. L. A., Baleka, S., Henneberger, K., Taron, U. H., Trinks, A., Westbury, M. V., et al. (2017). Sequencing single-stranded libraries on the Illumina NextSeq 500 platform. arXiv. [preprint]. doi: 10.48550/arXiv.1711.11004
Paijmans, J. L. A., Fickel, J., Courtiol, A., Hofreiter, M., and Förster, D. W. (2016). Impact of enrichment conditions on cross-species capture of fresh and degraded DNA. Mol. Ecol. Resour. 16, 42–55. doi: 10.1111/1755-0998.12420
Paireder, S., Werner, B., Bailer, J., Werther, W., Schmid, E., Patzak, B., et al. (2013). Comparison of protocols for DNA extraction from long-term preserved formalin fixed tissues. Anal. Biochem. 439, 152–160. doi: 10.1016/j.ab.2013.04.006
Rancilhac, L., Bruy, T., Scherz, M. D., Pereira, E. A., Preick, M., Straube, N., et al. (2020). Target-enriched DNA sequencing from historical type material enables a partial revision of the Madagascar giant stream frogs (genus Mantidactylus). J. Nat. Hist. 54, 87–118. doi: 10.1080/00222933.2020.1748243
Rohland, N., Siedel, H., and Hofreiter, M. (2004). Nondestructive DNA extraction method for mitochondrial DNA analyses of museum specimens. Biotechniques 36, 814–821. doi: 10.2144/04365ST05
Scherz, M. D., Rasolonjatovo, S. M., Köhler, J., Rancilhac, L., Rakotoarison, A., Raselimanana, A. P., et al. (2020). ‘Barcode fishing’ for archival DNA from historical type material overcomes taxonomic hurdles, enabling the description of a new frog species. Sci. Rep. 10:19109. doi: 10.1038/s41598-020-75431-9
Springer, M. S., Signore, A. V., Paijmans, J. L. A., Vélez-Juarbe, J., Domning, D. P., Bauer, C. E., et al. (2015). Interordinal gene capture, the phylogenetic position of Steller’s sea cow based on molecular and morphological data, and the macroevolutionary history of Sirenia. Mol. Phylogenet. Evol. 91, 178–193. doi: 10.1016/j.ympev.2015.05.022
Stiller, M., Sucker, A., Griewank, K., Aust, D., Baretton, G. B., Schadendorf, D., et al. (2016). Single-strand DNA library preparation improves sequencing of formalin-fixed and paraffin-embedded (FFPE) cancer DNA. Oncotarget 7:59115. doi: 10.18632/oncotarget.10827
Straube, N., Lyra, M. L., Paijmans, J. L. A., Preick, M., Basler, N., Penner, J., et al. (2021a). Successful application of ancient DNA extraction and library construction protocols to museum wet collection specimens. Mol. Ecol. Resour. 21, 2299–2315. doi: 10.1111/1755-0998.13433
Straube, N., Preick, M., Naylor, G. J. P., and Hofreiter, M. (2021b). Mitochondrial DNA sequencing of a wet-collection syntype demonstrates the importance of type material as genetic resource for lantern shark taxonomy (Chondrichthyes: Etmopteridae). R. Soc. Open Sci. 8:210474. doi: 10.1098/rsos.210474
Keywords: target capture, type specimens, molecular species identification, museum specimens, cross-species capture
Citation: Agne S, Preick M, Straube N and Hofreiter M (2022) Simultaneous Barcode Sequencing of Diverse Museum Collection Specimens Using a Mixed RNA Bait Set. Front. Ecol. Evol. 10:909846. doi: 10.3389/fevo.2022.909846
Received: 31 March 2022; Accepted: 17 May 2022;
Published: 17 June 2022.
Edited by:
Jonathan J. Fong, Lingnan University, ChinaReviewed by:
Daniel Janzen, University of Pennsylvania, United StatesCopyright © 2022 Agne, Preick, Straube and Hofreiter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefanie Agne, YWduZUB1bmktcG90c2RhbS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.