
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ecol. Evol. , 11 April 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.871791
This article is part of the Research Topic Population Genomics and Adaptation to Novel Environments: Challenges and Opportunities View all 6 articles
 Gina F. Lamka1*
Gina F. Lamka1* Avril M. Harder1
Avril M. Harder1 Mekala Sundaram2
Mekala Sundaram2 Tonia S. Schwartz3
Tonia S. Schwartz3 Mark R. Christie4,5
Mark R. Christie4,5 J. Andrew DeWoody4,5
J. Andrew DeWoody4,5 Janna R. Willoughby1
Janna R. Willoughby1Epigenetic variation is often characterized by modifications to DNA that do not alter the underlying nucleotide sequence, but can influence behavior, morphology, and physiological phenotypes by affecting gene expression and protein synthesis. In this review, we consider how the emerging field of ecological epigenetics (eco-epi) aims to use epigenetic variation to explain ecologically relevant phenotypic variation and predict evolutionary trajectories that are important in conservation. Here, we focus on how epigenetic data have contributed to our understanding of wild populations, including plants, animals, and fungi. First, we identified published eco-epi literature and found that there was limited taxonomic and ecosystem coverage and that, by necessity of available technology, these studies have most often focused on the summarized epigenome rather than locus- or nucleotide-level epigenome characteristics. We also found that while many studies focused on adaptation and heritability of the epigenome, the field has thematically expanded into topics such as disease ecology and epigenome-based ageing of individuals. In the second part of our synthesis, we discuss key insights that have emerged from the epigenetic field broadly and use these to preview the path toward integration of epigenetics into ecology. Specifically, we suggest moving focus to nucleotide-level differences in the epigenome rather than whole-epigenome data and that we incorporate several facets of epigenome characterization (e.g., methylation, chromatin structure). Finally, we also suggest that incorporation of behavior and stress data will be critical to the process of fully integrating eco-epi data into ecology, conservation, and evolutionary biology.
When critical environmental shifts occur that make a habitat unsuitable, the common view is that species must either move to different habitats or adjust to the new conditions to avoid extirpation. One way that organisms can survive in situ is via adaptive evolution. Genetic adaptation to novel conditions occurs via a response to natural selection, where individuals with particular genotypes survive at higher rates than individuals with alternative genotypes, leading to an increase in the frequency of beneficial alleles in the population (Jones et al., 2012). By contrast, epigenetic modifications to DNA, which chemically alter the expression of genes but do not alter the underlying DNA sequence, can also influence how organisms respond to environmental change (Baerwald et al., 2016; Ibrahim et al., 2021). Because portions of the epigenome can be transmitted between generations (i.e., are heritable), epigenetic variants may play a critical role as a mechanism of adaptation (Kronholm and Collins, 2016).
Epigenetic variation can explain a portion of phenotypic variance that is due to alterations in gene expression; when epigenome variants change this alters gene expression, and these expression changes can lead to lasting phenotypic changes. Such alterations are typically moderated by epigenetic marks or chemical tags associated with DNA that include changes in histone modification, microRNA regulation, and DNA methylation (Oliver et al., 2016). Histone modifications and DNA methylation interact to regulate which areas of the genome are open for transcription. In this way, environmentally induced alterations of these epigenetic modifications can alter gene expression (Skvortsova et al., 2018). Critically, epigenetic modifications can be ephemeral in comparison to DNA sequence changes, meaning that these marks can be reverted and thus provide flexibility in how organisms react to environmental changes (Kronholm and Collins, 2016; Verhoeven et al., 2016).
Histone modifications and microRNAs have been understudied relative to DNA methylation in the context of ecology and evolutionary biology. Instead, the focus of epigenetic inheritance in ecology and evolution has centered around how DNA methylation changes are passed from parent to offspring. Methylation typically occurs at cytosine-phosphate-guanine (CpG) dinucleotides, but CpG islands – stretches of DNA with an abundance of CpG repeats – show more variable methylation, particularly when comparing CpG islands located in promoters (generally unmethylated if the gene is transcriptionally active) and those that are intragenic (generally methylated) (Figure 1). Gene expression can be altered through differential methylation of associated CpG islands, where increased methylation of promoters typically leads to decreased transcription, providing an avenue for the epigenome to influence phenotype (Jones, 2012). As a result of this variability and the influences these sites have on traits and fitness, these islands are considered the primary landmarks of the epigenome (Sleutels and Barlow, 2002; Angeloni and Bogdanovic, 2021).
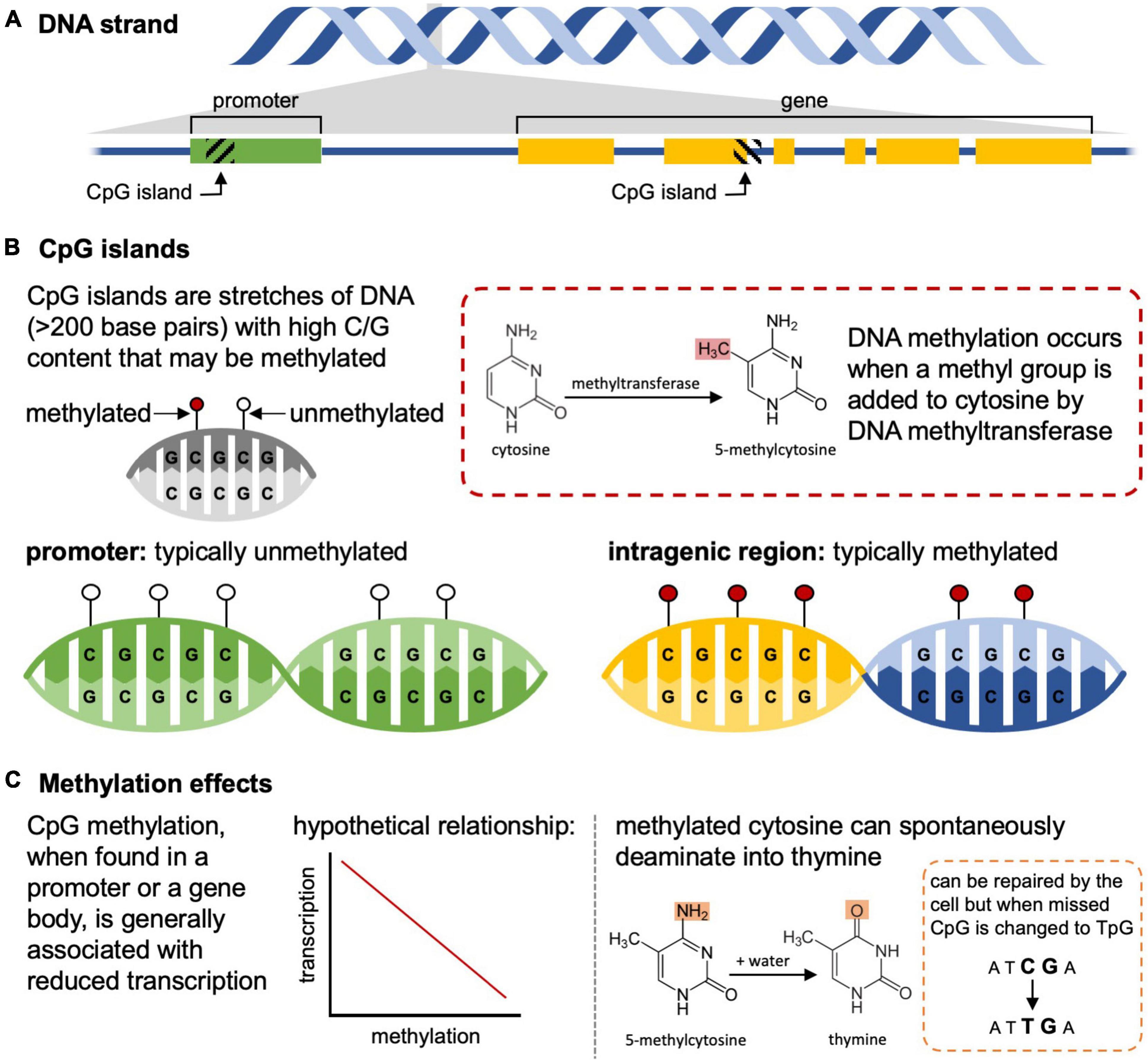
Figure 1. Methylation of cytosine-phosphate-guanine (CpG) islands influences transcription and can result in spontaneous mutations. (A) CpG islands can be found within promoters (left) or in coding and non-coding regions (right). (B) In mammals, the CpG islands in promoters of genes that are transcriptionally active are unmethylated, while those in gene bodies are often methylated. (C) Methylation of CpG islands in promoters is generally associated with reduced transcription relative to unmethylated regions, and methylated cytosines can spontaneously deaminate into thymine resulting in changes to the underlying DNA sequence.
Despite the recent surge in interest in epigenetic investigations, mechanisms of epigenetic inheritance among plants and animals are still being refined. In addition, the interactions of the effects of hard inheritance (i.e., characteristics are passed to offspring through DNA sequences that are not affected by parental environment) and soft inheritance (i.e., inherited offspring characteristics that are a result of parental behavior or environment) are not completely understood (Jablonka and Lamb, 2002; Noble, 2015). Part of the confusion comes from imprecise language around some epigenetic terms and how they are used in different fields of research. Here we use intergenerational inheritance to refer to epigenetic marks that are consistent between parent and the offspring who were exposed to a particular environment or stressor as embryos or embryonic germ cells. We use the term transgenerational inheritance to refer to epigenetic marks that are transmitted to offspring who were never exposed as embryos (F1) or even as germ cells (F2) to that environment or stressor of interest — in females this is typically the F3 generation removed from the stressor (see Figure 2). In addition to this confusion, epigenetic activity and the degree of transmission of epigenetic marks vary widely between plants and animals as well as between vertebrate and invertebrate animals; plants tend to be much more permissive of transgenerational epigenetics whereas vertebrates, particularly placental mammals, are much more restrictive (Morgan et al., 2005; Mounger et al., 2021). Today, the overall body of evidence supports the idea that individual phenotypic trait values are determined by both genetic and environmental effects, with epigenetics being a mediator for the short-term through regulation of phenotypic plasticity and intergenerational inheritance and in some cases long-term (transgenerational) effects of the environment on phenotype.
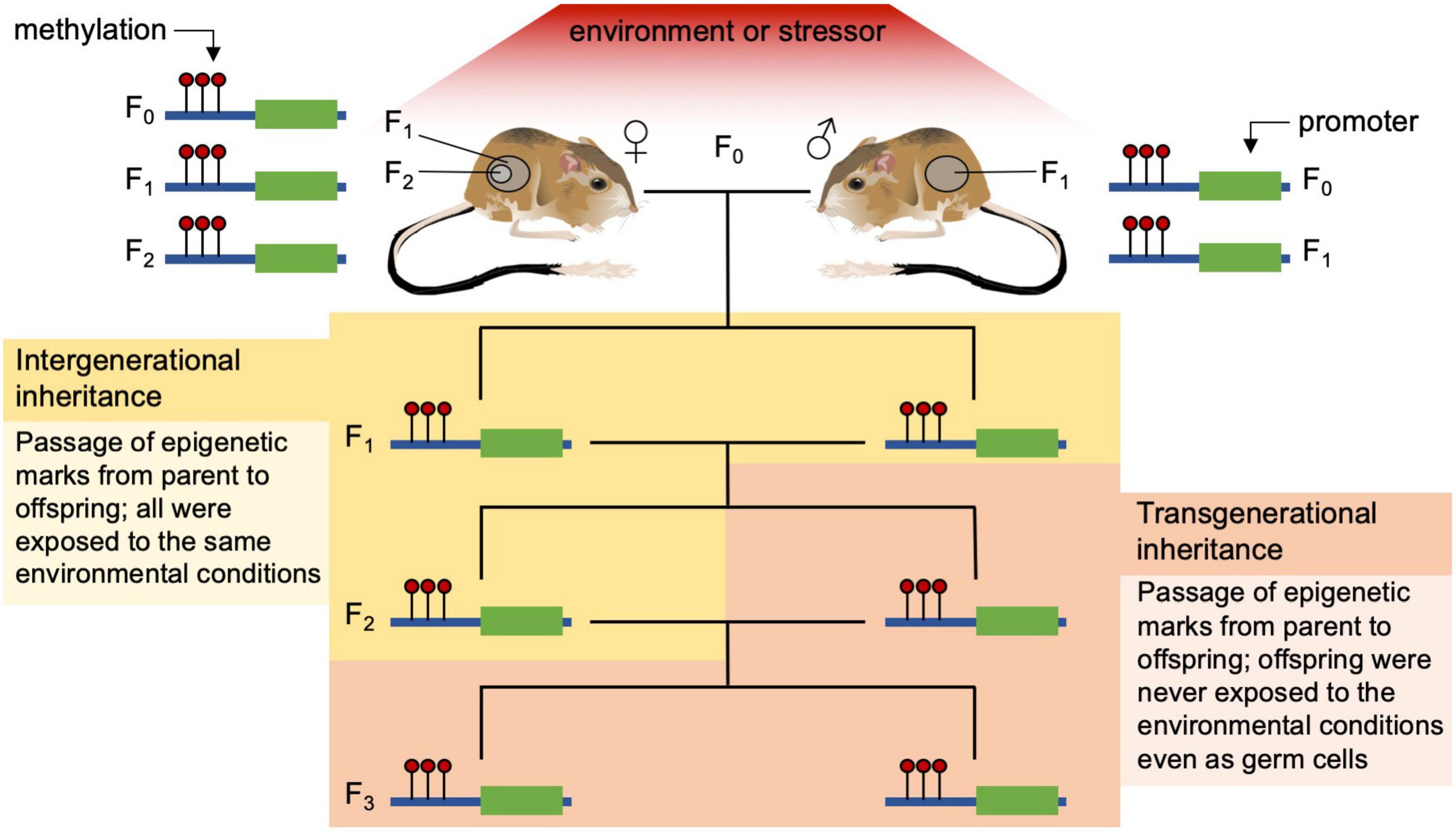
Figure 2. Examples of intergenerational and transgenerational inheritance of epigenetic marks (methylation) in a vertebrate. Intergenerational inheritance tracks epigenetic modifications through parent and offspring that were all exposed to the same environment or stressor. On the left a female kangaroo rat (F0), her developing embryo (F1), and the embryo’s germ cells (F2) are all exposed to the current environmental stressor that can alter the epigenome directly; on the right the male rat (F0) and his germ cells (sperm, F1) are being exposed to the same stressor. Transgenerational inheritance occurs when offspring (F3 from females, and F2 from males) never exposed to the stressor (even as germ cells) have the same methylation pattern as their parents that were exposed to the stressful environment.
Epigenetic differences between individuals can also be influenced by many biological factors including an individual’s age, sex, and the type of tissue profiled (Brunet and Berger, 2014; Martínez et al., 2014), and comparing epigenomes among these groups can be misleading if these differences are not included in data analysis (Teschendorff et al., 2017). These differences can also manifest through various environmental or ecological characteristics such as social system, parasite load, disease, climate, nutrition, and exposure to toxins (Verhoeven et al., 2016; Herrel et al., 2020; Rey et al., 2020). This means that within a single individual and tissue type, the epigenome is dynamic and changing over time making it difficult to link a single set of gene expression changes to a single phenotypic difference (Favé et al., 2018). For example, in monozygotic twins that begin genetically identical, a lifetime of behavior and environmental differences (e.g., diet, smoking habits, pharmaceutical use) results in disparate epigenomic patterns that have effects on the metabolism and health of an individual, some of which are indirect (Kanherkar et al., 2014). Additionally, these epigenetic modifications have the potential to permanently alter the genome. Methylated cytosines are prone to spontaneous mutation into thymine residues and, although this error can be detected by cellular repair mechanisms, the mutation nevertheless can be incorporated into the genome and thus leads to an increase in TpG sequences over an organisms’ lifetime (Figure 1; Chahwan et al., 2010). Finally, there are probably biological connections between the epigenome and the transcriptome in most species, but technical factors (e.g., ability to house and care for captive populations in large or long-lived organisms) have so far limited our ability to comprehensively study these connections in wild organisms. Overall, this leads to a complex relationship between the composition of the epigenome we can detect in a snapshot of sampling and the fitness, phenotype, or behaviors observed in any one tissue sampled from an individual at a particular time.
Just as additive genetic variation in populations can result in adaptation via a response to selection, epigenetic variation within and among genomes can lead to differential inheritance by selection on epigenetic variants and thus also provides an opportunity for adaptation. As a growing field of biology, ecological epigenetics (eco-epi) aims to use epigenetic variation to explain ecologically relevant phenotypic variation and predict evolutionary trajectories, both of which are relevant to modern conservation efforts. In this review, we consider how epigenetic data have contributed to our understanding of the ecology and evolution of wild populations, including plants, animals, and fungi. In part one of this review and synthesis, we identify and collate eco-epi literature published by the end of April 2021 and use these studies to summarize: (a) the distribution of eco-epi studies conducted to date, from both a taxonomic and ecosystem perspective; (b) eco-epi methodologies and data requirements; and (c) ecologically relevant questions addressed by eco-epi studies. In part two, we highlight what is needed to push forward and integrate epigenetics in ecological and evolutionary biology, including more focus on locus-level differences in the epigenome, measurement of the epigenome that is beyond the methylome, and integration of behaviors and stressors into epigenome experiments.
We used the ISI Web of Science to identify eco-epi studies published through April 2021. We searched “(ecolog* OR evolu* OR conserv*) AND (epigenetic* OR epigenome* OR methylome OR methylation)” and curated the resulting manuscripts by hand to include only those that used epigenetic data in an ecological or evolutionary context (Supplementary Appendix 1). We excluded review and opinion manuscripts and studies that reused existing epigenetic data to avoid incorporating the same data and interpretations multiple times. In total we identified 206 eco-epi manuscripts from which we extracted shared data including: focal species, geographic origin of samples analyzed, number of epigenomes assessed, the epigenetic methodology employed, as well as the general topics, questions, and conclusions reached by the authors (Supplementary Appendix 2). While these are unlikely to have captured all of the relevant literature, we interpret this set of manuscripts as representative of the work in the eco-epi field to date.
Eco-epi investigations that we identified through our search terms included research on a total of 184 different species, including 101 in Animalia, 78 in Plantae, 4 in Fungi, and 1 Chromista (single-celled eukaryote) species. However, these studies were not evenly distributed with respect to species richness, phyla, or high-order clades (Zhang, 2013). Instead, research was disproportionately focused on chordates and angiosperms, accounting for 38 and 41% of all studied species, respectively (Table 1). Similarly, sample collection for all studies occurred primarily in Europe (37%) and North America (24%), meaning that only 39% of eco-epi research effort was distributed across remaining global ecosystems found outside these two areas (Supplementary Appendix 3). Clearly, the future inclusion of more diverse focal species will provide additional insight into the broader mechanistic and systematic effects of epigenetic inheritance given global environmental variability.

Table 1. Taxonomic breakdown of the 184 unique species studied in the existing ecological epigenetic literature.
In epigenetic research, early research focused on model species and has only recently begun to extend to non-model species. We examined our data to identify emerging eco-epi model species by looking for multiple studies focused on the same species. We found only four species with five or more studies (Apis mellifera, honeybee; Gasterosteus aculeatus, three-spined stickleback; Helleborus foetidus, stinking hellebore; and Taraxacum officinale, dandelion), suggesting these are emerging eco-epi model species. However, including additional species from other groups with significant existing genomic resources (e.g., Salmo salar, Atlantic salmon; Taeniopygia guttata, zebra finch) may be beneficial because eco-epi research is more tractable with well assembled and annotated reference genomes that can be used for mapping high throughput sequence data from epigenomics studies (Le et al., 2020). Future work would be well suited in species with available high quality reference genome assemblies because these are required in the most cost-efficient and data-rich epigenetic methodologies. Recent interest in generating genome assemblies for diverse species (e.g., Vertebrate Genomes Project, Bird 10K Genomes Project, DNA Zoo) means that future epigenomic work will be possible on diverse species, which may be particularly important for those species that must adapt to changing environments.
Bias in conservation research effort is, at least in part, due to variation in funding, which hinders the development of genomic resources in non-model species (Donaldson et al., 2017). With limited resources in mind, we examined the available data to understand how existing eco-epi research may have been used to inform on-going conservation need or intervention. Our dataset contained 100 species for which extinction risk has been evaluated by the International Union for the Conservation of Nature (IUCN) and found that only 13 eco-epi focal species were included in the IUCN threatened categories (i.e., critically endangered, endangered, vulnerable). Because epigenetic marks can be modified in response to biological stressors—and because threatened species are likely to be under stress—we expect that future eco-epi studies that compare threatened and non-threatened populations of the same species might prove enlightening in the same sense that historic comparisons have illustrated that additive genetic variation is often reduced in threatened populations (Chapman et al., 2009). We expect that epigenetic modifications could affect fitness and influence future evolutionary potential for adaptation (Kardos et al., 2021).
The published eco-epi studies we evaluated used a variety of epigenetic methods that target DNA methylation, each of which provide very different views into an individual’s epigenome (Table 2). The most commonly used method in the studies we reviewed was methylation-sensitive amplified fragment length polymorphism. Because this procedure is methodologically simple, it was one of the first epigenetic methods applied to ecological questions. This method results in information about methylation status for the locus (or loci) targeted by the initial PCR amplification (Yamamoto et al., 2001). For example, Schulz et al. (2014) investigated the floodplain herb Viola elatior and found divergence in methylation status between populations with different light conditions using data from eight loci. Because this method can only generate data for a small proportion of the entire epigenome (humans have an estimated 30,000 CpG islands, or 28 million CpG sites, for example; Jeziorska et al., 2017), a large proportion of the epigenome variation is missed. Newer approaches that build on whole genome sequencing advances may provide more detailed insight into epigenome variation.

Table 2. Overview of epigenetic methods, including a brief description and a description of the generated data, used in the ecological epigenetic studies we reviewed.
Reduced representation epigenetic sequencing attempts to overcome the limitations inherent when considering only a few loci by generating data for many more loci in a cost-efficient manner. Reduced representation epigenetic bisulfite sequencing, which works similarly to RADseq, uses restriction enzymes to cut genomic DNA at enzyme-specific cut sites when the region is methylated, providing information on the methylation status of the cytosines in the restriction enzyme recognition site across the genome (Schield et al., 2016). One drawback to this process is that this method may introduce biases in the epigenetic data due to allelic drop-out caused by sequence variation at the restriction sites among individuals (Cariou et al., 2016) thereby resulting in missing information for individuals in the dataset (Crotti et al., 2019); another is the template DNA required, which may be difficult to obtain in sufficient quality, quantity, or both when studying species of conservation concern. Nevertheless, reduced representation epigenetic sequencing are cost-efficient methods for generating epigenetic data (Werner et al., 2020). For example, Platt et al. (2015) investigated the pattern of genetic and epigenetic variants across populations of valley oaks (Quercus lobata) from disparate climatic regions. The results of this research suggested that there were very large sections of the epigenome that were different between populations, and that the epigenome, as a whole, may influence patterns of local adaptation. In this case, the use of genome-wide data made it possible to detect regions that would have been missed with a lower density of epigenetic markers.
In contrast to these methods, bisulfite sequencing provides single-nucleotide resolution of methylation status (Frommer et al., 1992). When using a bisulfite sequencing approach, DNA sequences are treated with bisulfite, causing unmethylated cytosine to be converted to thymine, whereas methylated cytosines are protected. In this way, the unmethylated cytosines can then be detected by comparing the untreated sequence to the bisulfite converted sequence and identifying the single nucleotide variants (SNPs) (Frommer et al., 1992; Li and Tollefsbol, 2011). In addition, bisulfite sequencing can be conducted on a locus-by-locus comparison (using locus-specific primers) or for whole genomes using high throughput sequencing. For example, Laine et al. (2016) used whole genome bisulfite sequencing in great tits (Parus major) and identified increased CpG methylation near neural genes relative to the rest of the genome, suggesting epigenetic regulation of learning and memory. Similarly, whole genome bisulfite sequencing in three-spined stickleback (Gasterosteus aculeatus) showed hypermethylation of X chromosomes in females relative to males, providing a possible mechanism for the observed heteromorphic sex chromosomes in this species (Metzger and Schulte, 2018).
We also identified a subset of studies that measured global methylation across the genome using methods that rely on liquid chromatography. These methodologies quantify the proportion of the genome that is methylated by separating and quantifying genomic cytosine, providing information on the total methylation quantity in an individual sample (Roberts et al., 2018). This approach can be effective for considering the effects of widespread environmental change. In one example, Volkova et al. (2018) showed that Scots pine (Pinus sylvestris) that were exposed to chronic radiation as a result of the Chernobyl disaster tended to be significantly hypermethylated. Although the precise genomic location of the induced methylation was unknown, the authors suggest that the hypermethylation has prevented genome instability and reshuffling, thereby providing a mechanism to withstand the environmental stressors.
Very few of the eco-epi studies captured in our review (n = 5) relied on experimental manipulation of methylation patterns to directly assess the effects of methylation on individual phenotypes, but such investigations would provide the most direct evidence of mechanistic links between epigenetic variation and differential individual fitness. In all the studies that used this approach included in our analyses, individuals in an experimental group were treated with a demethylating agent and phenotypes were compared between treatment and control groups. For example, Vergeer et al. (2012) measured fitness-related traits in inbred and outbred dwarf pincushion flower (Scabiosa columbaria) in the presence and absence of experimental demethylation and determined that demethylation treatment of inbred individuals led to trait values resembling control outbred individuals. However, like the other experimental studies in our analysis, the effects of demethylation agents on global methylation levels were not measured directly. Treatment with demethylation agents, such as 5-azacytidine or zebularine, can result in variable degrees of demethylation across species and application methods, limiting inferences that can be drawn in the absence of direct quantification (Münzbergová et al., 2019). Nevertheless, experimental demethylation represents a clear approach to describe the effects of specific methylation patterns on phenotypes, particularly if paired with a high-resolution approach such as whole genome bisulfite sequencing, in systems and species where experimental manipulation is tractable.
The extent to which epigenetic variation in wild populations, in addition to genetic variation, contributes to the adaptability of populations is an important topic in ecology due to concerns related to global climate change, invasive species, and other large-scale concerns. As a result of this interest in the field, adaptation was a common theme in the eco-epi literature we surveyed (58% of studies). In these contexts, adaptation can be inferred—either exclusively or partially—to explain the differences in fitness or fitness-related traits as a result of environmental differences (n = 84 out of 121 adaptation-related articles) by studying the relationship between the epigenome and putative fitness-related traits (e.g., body size, genomic variability, seed production, parasite load; n = 75) or fitness directly (n = 10) across natural environmental clines or gradients in herbivory intensity, salinity, and temperature (Supplementary Appendix 2; note that one study considered fitness-related traits as well as fitness directly). In 86% of studies that we surveyed that had some focus on adaptation of the focal species in response to environmental change (n = 105), epigenetic variation was at least partially attributed to the environmental differences between study groups. However, inferences were limited when specific epigenetic markers could not be tied to phenotypic outcomes due to methodology. In addition, the limited taxonomic and geographical sampling means that it is difficult to assess how consistently these eco-epi/adaptation patterns hold across species and ecosystems.
One conservation-relevant issue that is likely to become important is the increased need for captively supported populations (Allentoft and O’Brien, 2010), and the adaptation of captive populations to the novel and predator free environments is one alluring system for eco-epi investigation (Williams and Hoffman, 2009). Furthermore, some species may require ex situ captive breeding efforts to prevent extinction. While such efforts can result in genetic adaptation to captive environments (Willoughby and Christie, 2019), it remains unknown whether epigenetic modifications can also decrease the fitness of captively-reared individuals released or reintroduced back into the wild (Le Luyer et al., 2017) or if epigenome modifications decreased overall fitness (irrespective of captive or wild status) in these populations (Kronholm and Collins, 2016). As we continue to amass studies comparing epigenomes and fitness, these analyses will provide an additional backdrop for characterizing the relationship between the epigenome and population trajectories under changing environmental conditions, with obvious applications to conservation biology.
From a population persistence point of view, and more broadly considering ecology and evolution, epigenetic changes that are heritable may be more relevant than those that are ephemeral because heritable changes can influence long term population fitness and evolution. Because epigenomes can change even over an individual’s lifetime, it is not clear how heritability may differ among populations and species. In the eco-epi literature, heritability of epigenetic modifications can be quantified by measuring methylation over at least two consecutive generations or by considering similarities and differences among families. Although heritability values were not directly estimated in the literature we surveyed, at least a portion of the studied epigenome was identified as heritable in 81% of the studies surveyed (25 out of 31 studies that addressed heritability). However, uncertainty about the heritability of methylation remains due to the variety of definitions of inheritance in combination with the variety of life history traits that contribute to differences in germ cell reprogramming and epigenome transmission (see Figure 2 for explanation of inter- and transgenerational inheritance).
The way we quantify epigenome heritability, either using intergenerational or transgenerational inheritance, can influence how we understand inheritance of these genomic markers. For example, in the shrub Lavandula latifolia, Herrera et al. (2018) compared the correlation between greenhouse-grown maternal and F1 offspring global cytosine methylation and found that maternal methylation explained 28% of the variation in offspring methylation across the genome. In this example, the proportion of this intergenerational inheritance that would also be inherited into further generations (i.e., transgenerational inheritance) is not clear. By contrast, Zheng et al. (2017) reported drought-induced epigenetic changes in wild rice that were detected up to 11 generations later, identifying potentially important epigenetic variants that were heritably transmitted into generations not subjected to drought conditions.
Experiments where an environmental stressor was experimentally applied suggests that even a relatively modest transmission between generations may be ecologically relevant; when sperm was collected from Atlantic salmon (Salmo salar) fathers and offspring after being exposed to hatchery conditions, six differentially methylated regions were passed to future generations, suggesting that these epigenetic modifications that arose during captive-rearing could result in differential fitness when hatchery-reared males interbreed with wild females (Rodriguez Barreto et al., 2019). It seems possible that the multi-generational transmission of the epigenome may provide important avenues for adaptation to new and stressful environmental conditions by preparing future generations to deal appropriately with those stressors. However, detecting these transgenerational traits can be problematic when epigenome patterns co-vary with other traits such as behaviors or microbiome communities (Heard and Martienssen, 2014). For example, when parental nurturing triggers a persistent methylation change in offspring, the observed methylation pattern would appear to be transgenerationally inherited even though it is acquired as a result of maternal actions after birth (Champagne and Curley, 2009). In addition, epigenomic heritability is not evidence of adaptation; epigenetic marks may be neutral with respect to phenotype and fitness or represent a stressed or disease-state particularly if the subsequent generations are exposed to different environments than their previous generations where the epigenetic marks originated.
The correlation between DNA sequence diversity (i.e., genetic diversity) and epigenome diversity may be an important aspect of integrating epigenetic data into ecological projects because this would allow us to apply existing genetic diversity theories to epigenomic data. However, the extent to which a relationship exists between these two estimates is unclear: among the eco-epi studies we reviewed that assessed (quantitatively or qualitatively) a relationship between DNA sequence and epigenetic diversity, 22 reported a correlation between genetic and epigenetic diversity (four negatively, 16 positively, and two with no correlation direction) and 24 found no correlation between genetic and epigenetic variation. For example, in a recent analysis of 10 populations of a clonal herb (Hydrocotyle vulgaris), Wang et al. (2020) show that epigenetic variation was correlated with genetic variation across and among populations, and that particular phenotypes were associated with particular epigenetic variants. Although this relationship has not yet been examined in most species, these and similar data suggest that the epigenome influences phenotype independently of genetic variation. However, the relationship between DNA sequence variation and epigenome variation is likely complicated by variants that are difficult to detect or quantify. For example, copy number variants and transposable element insertions may be important and inheritable DNA sequence variants, but detecting these remains difficult particularly in non-model organisms (Caballero-Lopez et al., 2021). When these undetected sequence variants are important to a measured phenotype and co-vary with epigenome variants, this may give the false impression that the trait is solely epigenetically inherited; if this happens frequently across the genome this will, perhaps falsely, suggest that sequence variation is not strongly related to epigenetic variation (Heard and Martienssen, 2014). Therefore, understanding the theoretical concepts that underpin the relationship between genetic and epigenetic variation in wild populations of diverse species is critical to the continued progress of the eco-epi field.
The effects of epigenetic variants and changes on disease or parasite susceptibility are important to wild population management because understanding how and when parasites invade new areas informs conservation and management actions to limit disease spread. For example, Hu et al. (2018) investigated populations of Trinidadian guppies (Poecilia reticulata) and showed that there were distinct epigenetic modifications associated with the progression of infection of an ectoparasite. Infected, symptomatic, and recovered individuals exhibited different suites of methylated regions. Interestingly, many of these methylated sites were associated with CpG islands and several overlapped genes associated with immune response. Current literature also suggests that internal parasites can mediate epigenetic changes; in red grouse (Lagopus lagopus), gastrointestinal parasites seemed to induce specific host epigenetic responses that were associated with different parasite loads (Wenzel and Piertney, 2014). These studies suggest that it is possible to develop epigenome panels for screening disease states in a target species or population, making it feasible to implement targeted disease treatment or strategies to limit spread of diseases in wild populations.
The ability to accurately know the age of a wild individual without lethal sampling is important for management and represents an additional exciting application of epigenetic data. Epigenetic clocks can be used to estimate an individual’s age by quantifying methylation level across a suite of loci that are known to correlate with age in that species (Bell et al., 2019). Although we identified very few studies (n = 9) with the explicit goal of evaluating epigenetic marker changes over a portion of an individual’s life, all of these studies reported positive and often predictable associations between age and methylation in at least some loci (Polanowski et al., 2014; Ito et al., 2018; Bicho et al., 2020; Tanabe et al., 2020). Although environmental effects (e.g., sun UV exposure) can alter an individual’s epigenome, environmental and age-related methylation changes often occur among distinct loci (Grönniger et al., 2010; Polanowski et al., 2014). Unfortunately, species- and perhaps population-specific epigenome models for estimating age are needed as it does not appear that markers identified in one species can be applied to others (Tanabe et al., 2020), although candidate loci identified in closely-related species may provide a head start (Mayne et al., 2021). Marker identification is additionally complicated by the paucity of the age-related regions, which are thought to occur in only ∼1% of the genome (Grönniger et al., 2010). However, the application of established protocols for identifying appropriate epigenetic markers in new species is promising (Polanowski et al., 2014) and has the potential to transform the way we study and manage wild populations.
We are interested in understanding how the field of eco-epi needs to progress if we are going to be able to take advantage of these data and insights in ecology and evolution. To characterize the eco-epi trajectory, we summarized the broader field of epigenetic research over the past 20 years. We searched ISI Web of Science using the topic search “epigenetic” and identified all studies published in the 20 journals with the most epigenetics articles published from 2000 to 2020. We used the abstracts from these 12,765 studies in an automated text analysis (Roberts et al., 2019) to identify the common themes and how these themes have changed in the last 20 years (see appendix for specific methods). We identified the top 20 topics, and here expand on those topics that relate to ecology (Figure 3; see Supplementary Appendix 6 for a complete list of the top 20 topics).
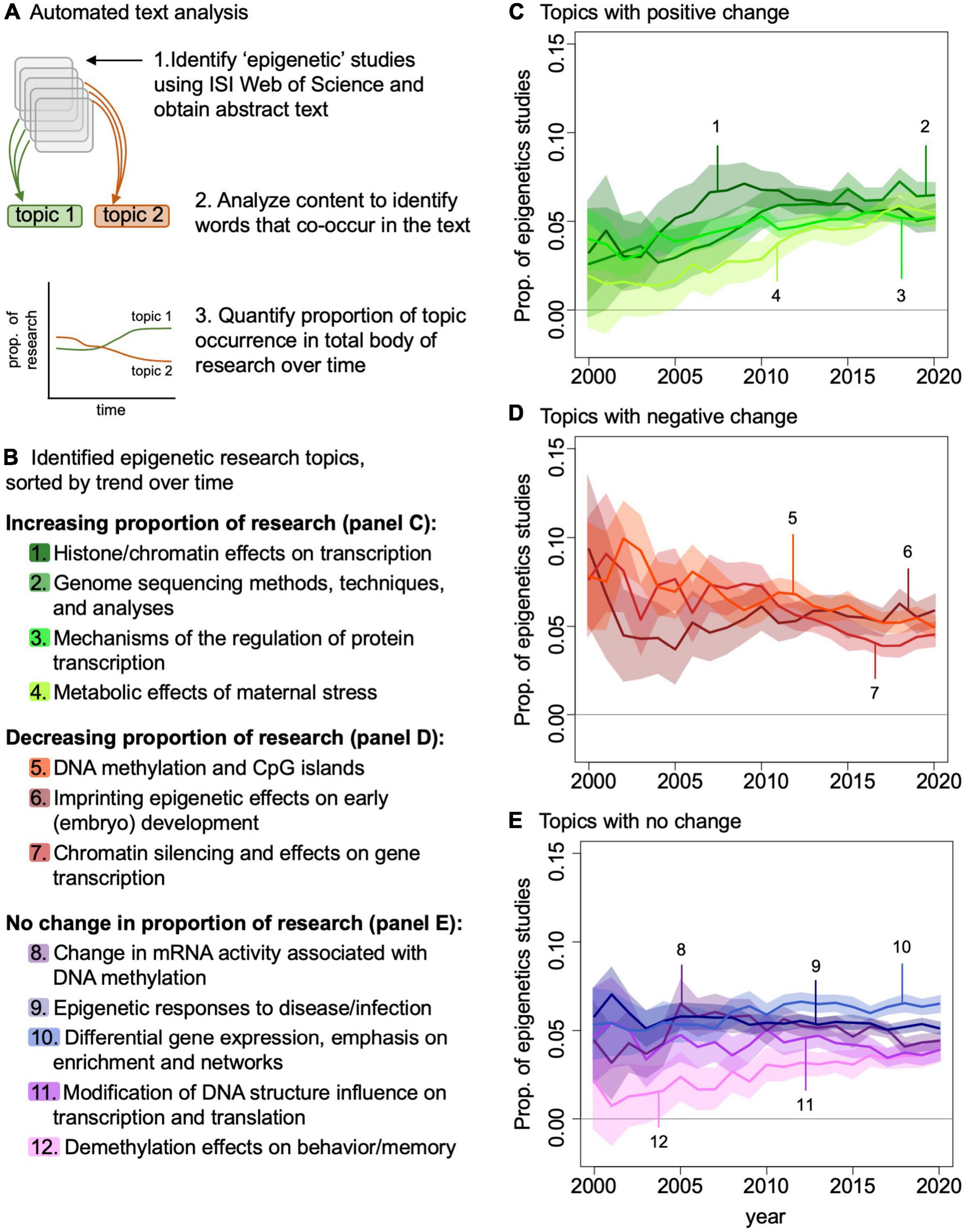
Figure 3. Automated text analysis of epigenetics literature. (A) Identification of literature; (B) top ecology-relevant topics contained in the epigenetics literature. Topic interest is plotted over time for those topics that increased (C), decreased (D), or remained constant (E) from 2000 to 2020.
One common theme in the epigenetics field over the past 20 years was research focused on genome sequencing methods, techniques, and data science (Figure 3). We identified substantial interest in analysis techniques associated with interpreting epigenetic data, such as the use of enrichment networks in conjunction with epigenetic data (Wang et al., 2021). The development of these methodologies and associated analysis techniques is critically important for how we assess the epigenomes of wild-living populations and individuals. Moving forward, we will need to build on these methods of epigenomic data that provide nucleotide-level insights across the genome, as these data provide the most utility for interpreting how epigenomes evolve (Paun et al., 2019).
Although we have largely considered epigenetics from a methylation perspective, epigenetic modifications can also occur via histone modifications (e.g., methylation, acetylation, and phosphorylation). Together histone modifications and DNA methylation alter the structure of chromatin and alter transcriptional regulation, ultimately influencing protein synthesis (Dai et al., 2020). We found that research in histone modifications and the effects of DNA structure modification on transcription/translation has been another core research area in the epigenetics field as a whole (Figure 3). However, we did not identify substantial interest in these topics in our eco-epi literature survey (Supplementary Appendix 2). While this may be due to practical limitations, for example the use of tissues that may be difficult to obtain from wild-living individuals (Buenrostro et al., 2013), understanding the full breadth of epigenetic responses in wild populations is desirable. For example, histone modifications in experimental populations of Arabidopsis can be related to environmental stress and ultimately confer survival benefits relative to individuals that lack these modifications (Kim et al., 2012). Future research in the field should include the use of methodologies such as ATAC-seq (Buenrostro et al., 2013) and genome structure analyses (Buitrago et al., 2021; Kumar et al., 2021) that are agnostic to the type of epigenetic modification, but rather can provide a more comprehensive picture of epigenetic state of the genome by identifying chromatin regions that are open or closed for transcriptional regulation. Advances of these chromatin-type methods require very little DNA (e.g., 50,000 cells) and can use snap frozen and preserved tissue as long as the nuclei are still intact (Corces et al., 2017; Peng et al., 2021). Similarly, quantifying the abundance of RNA variants via RNA profiling, which can also be accomplished with few cells, could be deployed to measure the functional output of the epigenome characters (Kostich, 2017). Understanding the full breadth of organism responses and adaptations will require consideration of expression changes irrespective of the type of epigenome modification.
We also identified substantial interest in methylation at CpG islands in the epigenetics field over the last 20 years (Figure 3). As demonstrated by the existing epigenetic framework laid down by the early CpG island studies, eco-epi has the opportunity to make quick gains in epigenetic data applications and contribute significantly to the broader field. For example, integration of epigenetic data with DNA sequencing data may provide necessary insight into the non-genetic components of phenotypic variation by identifying when transcription of specific genomic variants leads to observed phenotypes. This is likely to be more complicated than a single genotype-epigenome status-phenotype relationship and may require complex network analysis (e.g., weighted correlation network analysis) to identify genotype-epigenome status complexes of interest (Figure 4). This analysis works by identifying modules of genes that are correlated with each other and to the phenotype of interest and can be adapted to epigenome data by comparing genome and epigenome networks to each other and identifying overlapping genes and gene complexes (van Eijk et al., 2012). The increasing accessibility of genomic resources are likely to provide sufficient background for assessing suites of data such as these to understand how transcriptional regulation and phenotypes vary across temperatures, predator-prey environments, source populations, and other ecologically important variables. Epigenetic studies focused on these areas are poised to provide important ecological insights while simultaneously contributing to the broader discussion of gene regulation across additional (i.e., non-model) species and environmental conditions. An important caveat to this functional application of epigenetic data to understand phenotype and its response to environmental conditions is the need to sample the tissue that is responsible for the phenotypes since every tissue type will have a different epigenomic profile relevant to the function of that tissue (see “Outstanding Issues” below).
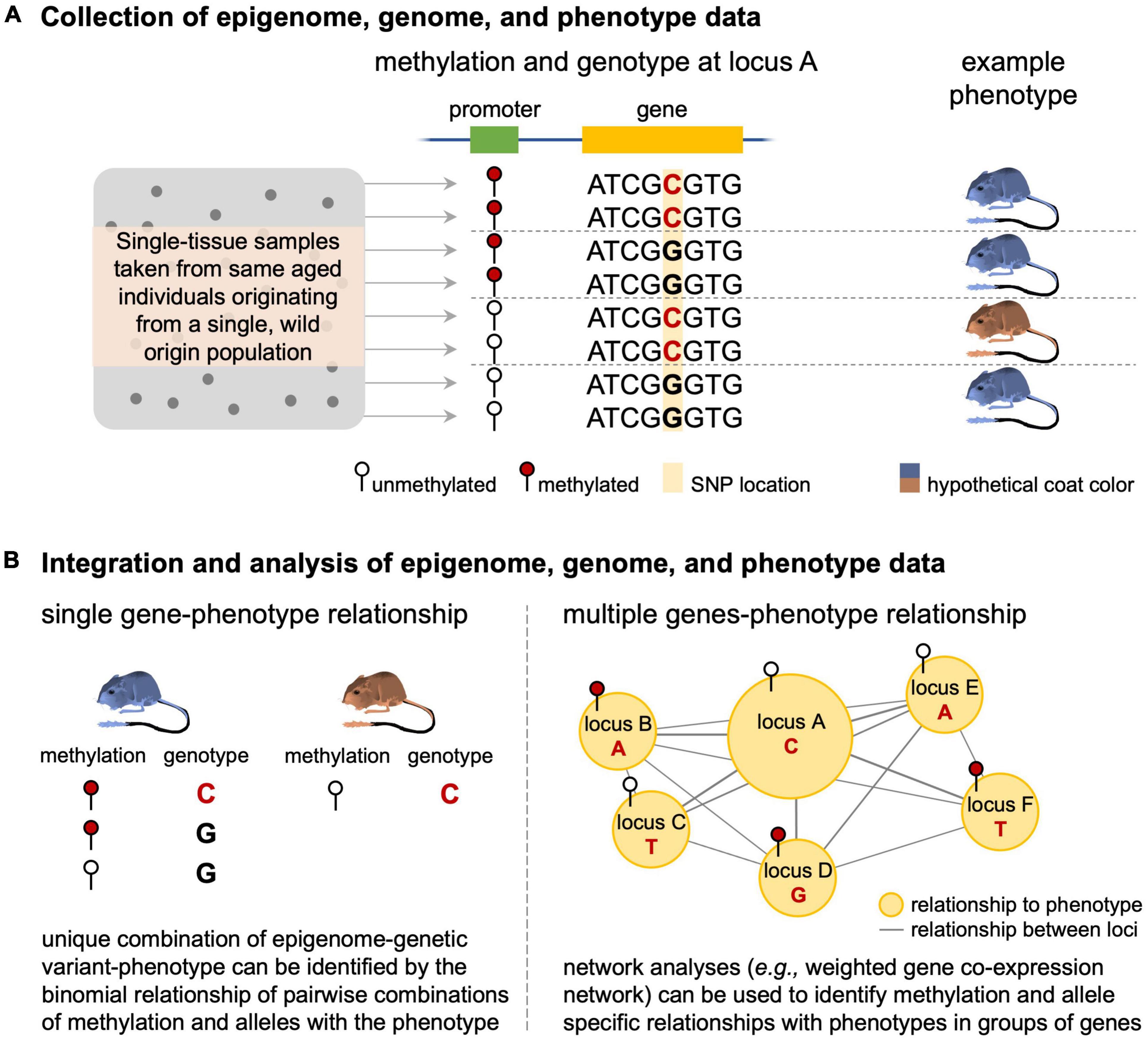
Figure 4. Linking epigenome differences, genome sequence variation, and phenotypic variation. The process starts by (A) collecting tissue from same age-individuals sampled from the same, natural population and generating epigenome, genome, and phenotypic data for each individual. (B) These data can then be analyzed to identify relationships where unique epigenome-DNA sequence variation relationships result in particular phenotypes (here illustrated with a locus of large effect) or by using network analyses when the relationship involves numerous genes.
Finally, studies on the effects of behaviors and stress on epigenomes has been of interest in the general epigenetic field and holds substantial promise for eco-epi growth (long-term metabolic effects of maternal stress on offspring and effects of imprinting on embryo development; Figure 3). These topics seem particularly important to reproduction generally and fitness specifically, and as such may be important considerations for eco-epi work. For example, the effects of environmental stress may be particularly influential during embryogenesis because this is often when epigenetic modifications are most dynamic over an individual’s lifetime (Bogdanović and Gómez-Skarmeta, 2014); stressful or otherwise unfavorable conditions that occur during embryogenesis but that are also atypical may, therefore, cause a mismatch between the environment and the individual later in life. The same logic also applies to the effects of epigenetic modifications on behavior, memory, and disease/infection. Thus, considering the epigenome when investigating population responses to environmental change, fragmentation, or any number of other perturbations may be particularly informative.
Although application of epigenetic methodologies across diverse ecological contexts is promising, current eco-epi investigations are limited by the extensive variation in epigenetic modifications that exist among and within individuals. Within an individual, patterns of epigenetic variation differ between somatic and germ cells, across tissue types (e.g., blood, liver, and skin), and even across cell types within a tissue (Gutierrez-Arcelus et al., 2015; Blake et al., 2020). Among individuals, epigenetic patterns can vary between sexes, age groups, and individuals sampled in different locations (Schulz et al., 2014; Ito et al., 2018; Metzger and Schulte, 2018). Combined, these factors suggest that carefully designed experiments and sampling regimes with large sample sizes are required to accurately identify meaningful biological patterns such as those that are expected to arise due to local adaptation or in response to novel disease-causing agents.
Furthermore, there are some species for which it is not possible to invasively sample relevant tissues from large numbers of individuals (e.g., endangered species). This effectively limits sampling options to the collection of blood from living individuals. On the one hand, epigenetic data from blood samples can be informative; in blood samples collected from shortfin molly (Poecilia mexicana) the methylation patterns suggested a mechanism by which this species has adapted to sulfidic springs (Kelley et al., 2021). On the other hand, methylation patterns found in blood may not be well correlated with those in other tissues because some methylation patterns are confined to particular tissues (Siller and Rubenstein, 2019; but see Lindner et al., 2021b). Furthermore, tissues are heterogeneous, and measured changes in epigenetic patterns may be due to this variability (e.g., sampling multiple cell types; Husby, 2020). Methods for accounting for and managing this variability should be incorporated into any eco-epi study relying on blood samples for methylation analysis (e.g., centrifugation; Viitaniemi et al., 2019; Kelley et al., 2021). Alternatively, newer methodologies that require far fewer cells than have previously been required (e.g., CUT&Tag, Kaya-Occur et al., 2019; transcription profiling, Orchard et al., 2018; RNA sequencing, Kelsey et al., 2017) could also be used, providing an avenue to gain insight into the epigenomes of the highest conservation priority species.
Another important consideration for eco-epi sampling design is the temporal variability of epigenetic variation (e.g., by age, life stage, season, environmental conditions, infection status, etc.). One way to account for this variation is by repeatedly sampling the same individuals and tissues over time. Recently, Lindner et al. (2021a) identify changes in methylation associated with reproductive status in great tit over a 6-month period, effectively using these data to understand the stability of methylation over this time period. However, this kind of experimental control is limited to species and tissue types that can be sampled multiple times (e.g., nucleated red blood cells in non-mammalian vertebrates) and may be logistically difficult to apply to wild populations. Because epigenetic responses to stress are known to vary among developmental stages (Anastasiadi et al., 2017), it will be important to assess methylation stability within control individuals not exposed to new stressors before considering how the epigenome is responding to new conditions. Until we know more about the temporal lability of epigenetic variation across taxa, individuals, and tissues, great care should be taken to minimize variability introduced by sampling design; large sample sizes and statistical approaches that account for inter-individual variation are recommended to control for these differences.
Finally, the link between DNA sequence variability (neutral or otherwise) and epigenetic variability is yet unknown, and we lack even theoretical models that would link effective population size change with methylation changes. This is at least partly because the relationship is complex; epigenetic variation and DNA sequence variation can covary (Zhang et al., 2013) and this alters the strength of the effect of methylation on phenotypes. Quantifying relationships between genetic variation and expression of highly polygenic phenotypes or even individual fitness outcomes has recently become easier through collections of genomic data (Perrier et al., 2018; Harrisson et al., 2019). Such analytical advancements in linking genetic variation to phenotypes can pave the way for incorporation of epigenetic information into models of phenotypic expression. For many traits across varying environments, consideration of epigenetic markers is likely to increase the proportion of trait variation explained beyond what genetic data can accomplish alone (Groot et al., 2018). Disentangling the complexities of epigenetic effects on phenotypes and fitness is likely to significantly enhance our understanding of ecology, evolution, and their interaction.
Epigenetic patterns and their underlying mechanisms have great potential to further contextualize ecological, evolutionary, and conservation biology. For example, as environments continue to experience globally shifting climate regimes, rapid adaptation will be required of many species and we expect this will often involve epigenetic responses. A more comprehensive understanding of epigenetic mechanisms, as well as their evolutionary rates and trajectories, may help us to identify species in need of management and suggest potential intervention strategies. Eco-epi investigation into these and other species and populations will provide unique insights into the interplay between epigenetic variation, genetic adaptation, and the conservation of populations.
GL, JD, and JW conceptualized and developed methodology for the project. GL, AH, and JW conducted data curation. GL, AH, MS, and JW conducted formal analysis and visualization. GL and JW wrote the original manuscript draft. GL, AH, MS, TS, MC, JD, and JW contributed to the investigation, provided critical review and edits to the final manuscript, and approved the submitted version.
This work was supported in part by USDA National Institute of Food and Agriculture support to JD, USDA National Institute of Food and Agriculture Hatch project 1025651 to JW, and by a Purdue University Elevating the Visibility of Agricultural Research award to JD and JW.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the editor, reviewers, and members of the DeWoody and Willoughby labs for constructive comments on earlier versions of this manuscript. We appreciate CLF Charlie and FC Euchre for support in the development of this manuscript. We also thank J. McLaughlin for the creation of the original kangaroo rat images.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.871791/full#supplementary-material
Allentoft, M., and O’Brien, J. (2010). Global amphibian declines, loss of genetic diversity and fitness: A review. Diversity (Basel) 2, 47–71.
Anastasiadi, D., Díaz, N., and Piferrer, F. (2017). Small ocean temperature increases elicit stage-dependent changes in DNA methylation and gene expression in a fish, the European sea bass. Sci. Rep. 7:12401. doi: 10.1038/s41598-017-10861-6
Angeloni, A., and Bogdanovic, O. (2021). Sequence determinants, function, and evolution of CpG islands. Biochem. Soc. Trans. 49, 1109–1119. doi: 10.1042/BST20200695
Baerwald, M. R., Meek, M. H., Stephens, M. R., Nagarajan, R. P., Goodbla, A. M., Tomalty, K. M. H., et al. (2016). Migration-related phenotypic divergence is associated with epigenetic modifications in rainbow trout. Mol. Ecol. 25, 1785–1800. doi: 10.1111/mec.13231
Bell, C. G., Lowe, R., Adams, P. D., Baccarelli, A. A., Beck, S., Bell, J. T., et al. (2019). DNA methylation aging clocks: challenges and recommendations. Gen. Biol. 20:249. doi: 10.1186/s13059-019-1824-y
Bicho, R. C., Scott-Fordsmand, J. J., and Amorim, M. J. B. (2020). Developing an epigenetics model species - From blastula to mature adult, life cycle methylation profile of Enchytraeus crypticus (Oligochaete). Sci. Total Environ. 732:139079. doi: 10.1016/j.scitotenv.2020.139079
Blake, L. E., Roux, J., Hernando-Herraez, I., Banovich, N. E., Perez, R. G., Hsiao, C. J., et al. (2020). A comparison of gene expression and DNA methylation patterns across tissues and species. Genom. Res. 30, 250–262. doi: 10.1101/gr.254904.119
Bogdanović, O., and Gómez-Skarmeta, J. L. (2014). Embryonic DNA methylation: insights from the genomics era. Brief. Funct. Genomics 13, 121–130. doi: 10.1093/bfgp/elt039
Brunet, A., and Berger, S. L. (2014). Epigenetics of aging and aging-related disease. J. Gerontol. A Biol. Sci. Med. Sci. 69(Suppl. 1), S17–S20. doi: 10.1093/gerona/glu042
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin. DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. doi: 10.1038/nmeth.2688
Buitrago, D., Labrador, M., Pablo Arcon, J., Lema, R., Flores, O., Esteve-Codina, A., et al. (2021). Impact of DNA methylation on 3D genome structure. Nat. Comm. 12:3243. doi: 10.1038/s41467-021-23142-8
Caballero-Lopez, V., Lundberg, M., Sokolovskis, K., and Bensch, S. (2021). Transposable elements mark a repeat-rich region associated with migratory phenotypes of willow warblers (Phylloscopus trochilus). Mol. Ecol. 31, 1128–1141. doi: 10.1111/mec.16292
Cariou, M., Duret, L., and Charlat, S. (2016). How and how much does RAD-seq bias genetic diversity estimates? BMC Evol. Biol. 16:240. doi: 10.1186/s12862-016-0791-0
Chahwan, R., Wontakal, S. N., and Roa, S. (2010). Crosstalk between genetic and epigenetic information through cytosine deamination. Trends Genet. 26, 443–448. doi: 10.1016/j.tig.2010.07.005
Chapman, J. R., Nakagawa, S., Coltman, D. W., Slate, J., and Sheldon, B. C. (2009). A quantitative review of heterozygosity-fitness correlations in animal populations. Mol. Ecol. 18, 2746–2765. doi: 10.1111/j.1365-294X.2009.04247.x
Champagne, F. A., and Curley, J. P. (2009). Epigenetic mechanisms mediating the long-term effects of maternal care on development. Neurosci. Biobehav. Rev. 33, 593–600. doi: 10.1016/j.neubiorev.2007.10.009
Corces, M. R., Trevino, A. E., Hamilton, E. G., Greenside, P. G., Sinnott-Armstrong, N. A., Vesuna, S., et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. doi: 10.1038/nmeth.4396
Crotti, M., Barratt, C. D., Loader, S. P., Gower, D. J., and Streicher, J. W. (2019). Causes and analytical impacts of missing data in RADseq phylogenetics: Insights from an African frog (Afrixalus). Zool. Scr. 48, 157–167.
Dai, Z., Ramesh, V., and Locasale, J. W. (2020). The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 21, 737–753.
Donaldson, M. R., Burnett, N. J., Braun, D. C., Suski, C. D., Hinch, S. G., Cooke, S. J., et al. (2017). Taxonomic bias and international biodiversity conservation research. Facets (Ott) 1, 105–113.
Favé, M.-J., Lamaze, F. C., Soave, D., Hodgkinson, A., Gauvin, H., Bruat, V., et al. (2018). Gene-by-environment interactions in urban populations modulate risk phenotypes. Nat. Comm. 9:827. doi: 10.1038/s41467-018-03202-2
Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., et al. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U. S. A. 89, 1827–1831. doi: 10.1073/pnas.89.5.1827
Grönniger, E., Weber, B., Heil, O., Peters, N., Stäb, F., Wenck, H., et al. (2010). Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 6:e1000971. doi: 10.1371/journal.pgen.1000971
Groot, M. P., Wagemaker, N., Ouborg, N. J., Verhoeven, K. J. F., and Vergeer, P. (2018). Epigenetic population differentiation in field- and common garden-grownScabiosa columbariaplants. Ecol. Evol. 8, 3505–3517.
Gutierrez-Arcelus, M., Ongen, H., Lappalainen, T., Montgomery, S. B., Buil, A., Yurovsky, A., et al. (2015). Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 11:e1004958. doi: 10.1371/journal.pgen.1004958
Harrisson, K. A., Magrath, M. J. L., Yen, J. D. L., Pavlova, A., Murray, N., Quin, B., et al. (2019). Lifetime fitness costs of inbreeding and being inbred in a critically endangered bird. Curr. Biol. 29, 2711.e–2717.e. doi: 10.1016/j.cub.2019.06.064
Heard, E., and Martienssen, R. A. (2014). Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 157, 95–109. doi: 10.1016/j.cell.2014.02.045
Herrel, A., Joly, D., and Danchin, E. (2020). Epigenetics in ecology and evolution. Funct. Ecol. 34, 381–384.
Herrera, C. M., Alonso, C., Medrano, M., Pérez, R., and Bazaga, P. (2018). Transgenerational epigenetics: Inheritance of global cytosine methylation and methylation-related epigenetic markers in the shrub Lavandula latifolia. Am. J. Bot. 105, 741–748. doi: 10.1002/ajb2.1074
Hu, J., Pérez-Jvostov, F., Blondel, L., and Barrett, R. D. H. (2018). Genome-wide DNA methylation signatures of infection status in Trinidadian guppies (Poecilia reticulata). Mol. Ecol. 27, 3087–3102. doi: 10.1111/mec.14771
Husby, A. (2020). On the use of blood samples for measuring DNA methylation in ecological epigenetic studies. Integr. Comp. Biol. 60, 1558–1566. doi: 10.1093/icb/icaa123
Ibrahim, A., Papin, C., Mohideen-Abdul, K., Le Gras, S., Stoll, I., Bronner, C., et al. (2021). MeCP2 is a microsatellite binding protein that protects CA repeats from nucleosome invasion. Science 372:eabd5581. doi: 10.1126/science.abd5581
Ito, H., Udono, T., Hirata, S., and Inoue-Murayama, M. (2018). Estimation of chimpanzee age based on DNA methylation. Sci. Rep. 8:9998. doi: 10.1038/s41598-018-28318-9
Jablonka, E., and Lamb, M. J. (2002). The changing concept of epigenetics. Ann. N. Y. Acad. Sci. 981, 82–96. doi: 10.1111/j.1749-6632.2002.tb04913.x
Jeziorska, D. M., Murray, R. J. S., De Gobbi, M., Gaentzsch, R., Garrick, D., Ayyub, H., et al. (2017). DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. U. S. A. 114, E7526–E7535. doi: 10.1073/pnas.1703087114
Jones, F. C., Grabherr, M. G., Chan, Y. F., Russell, P., Mauceli, E., Johnson, J., et al. (2012). The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61. doi: 10.1038/nature10944
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi: 10.1038/nrg3230
Kanherkar, R. R., Bhatia-Dey, N., and Csoka, A. B. (2014). Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2:49. doi: 10.3389/fcell.2014.00049
Kardos, M., Armstrong, E. E., Fitzpatrick, S. W., Hauser, S., Hedrick, P. W., Miller, J. M., et al. (2021). The crucial role of genome-wide genetic variation in conservation. Proc. Natl. Acad. Sci. U. S. A. 118, e2104642118. doi: 10.1073/pnas.2104642118
Kaya-Occur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., et al. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Comm. 10:1930.
Kelley, J. L., Tobler, M., Beck, D., Sadler-Riggleman, I., Quackenbush, C. R., Arias Rodriguez, L., et al. (2021). Epigenetic inheritance of DNA methylation changes in fish living in hydrogen sulfide-rich springs. Proc. Natl. Acad. Sci. U. S. A. 118:e2014929118. doi: 10.1073/pnas.2014929118
Kelsey, G., Stegle, O., and Reik, W. (2017). Single-cell epigenomics: Recording the past and predicting the future. Science 358, 69–75. doi: 10.1126/science.aan6826
Kim, J.-M., To, T. K., and Seki, M. (2012). An epigenetic integrator: new insights into genome regulation, environmental stress responses and developmental controls by histone deacetylase 6. Plant Cell Physiol. 53, 794–800. doi: 10.1093/pcp/pcs004
Kostich, M. S. (2017). A statistical framework for applying RNA profiling to chemical hazard detection. Chemosphere 188, 49–59. doi: 10.1016/j.chemosphere.2017.08.136
Kronholm, I., and Collins, S. (2016). Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 25, 1856–1868. doi: 10.1111/mec.13296
Kumar, S., Kaur, S., Seem, K., Kumar, S., and Mohapatra, T. (2021). Understanding 3D Genome organization and its effect on transcriptional gene regulation under environmental stress in plant: a chromatin perspective. Front. Cell Dev. Biol. 9:774719. doi: 10.3389/fcell.2021.774719
Laine, V. N., Gossmann, T. I., Schachtschneider, K. M., Garroway, C. J., Madsen, O., Verhoeven, K. J. F., et al. (2016). Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 7:10474. doi: 10.1038/ncomms10474
Le, N. T., Harukawa, Y., Miura, S., Boer, D., Kawabe, A., and Saze, H. (2020). Epigenetic regulation of spurious transcription initiation in Arabidopsis. Nat. Commun. 11:3224. doi: 10.1038/s41467-020-16951-w
Le Luyer, J., Laporte, M., Beacham, T. D., Kaukinen, K. H., Withler, R. E., Leong, J. S., et al. (2017). Parallel epigenetic modifications induced by hatchery rearing in a Pacific salmon. Proc. Natl. Acad. Sci. U. S. A. 114, 12964–12969. doi: 10.1073/pnas.1711229114
Li, Y., and Tollefsbol, T. O. (2011). DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol. Biol. 791, 11–21. doi: 10.1007/978-1-61779-316-5_2
Lindner, M., Laine, V. N., Verhagen, I., Viitaniemi, H. M., Visser, M. E., van Oers, K., et al. (2021a). Rapid changes in DNA methylation associated with the initiation of reproduction in a small songbird. Mol. Ecol. 30, 3645–3659. doi: 10.1111/mec.15803
Lindner, M., Verhagen, I., Viitaniemi, H. M., Laine, V. N., Visser, M. E., Husby, A., et al. (2021b). Temporal changes in DNA methylation and RNA expression in a small song bird: within- and between-tissue comparisons. BMC Genom. 22:36. doi: 10.1186/s12864-020-07329-9
Martínez, J. A., Milagro, F. I., Claycombe, K. J., and Schalinske, K. L. (2014). Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv. Nutr. 5, 71–81. doi: 10.3945/an.113.004705
Mayne, B., Espinoza, T., Roberts, D., Butler, G. L., Brooks, S., Korbie, D., et al. (2021). Nonlethal age estimation of three threatened fish species using DNA methylation: Australian lungfish, Murray cod and Mary River cod. Mol. Ecol. Resour. 21, 2324–2332. doi: 10.1111/1755-0998.13440
Metzger, D. C. H., and Schulte, P. M. (2018). The DNA methylation landscape of stickleback reveals patterns of sex chromosome evolution and effects of environmental salinity. Genome Biol. Evol. 10, 775–785. doi: 10.1093/gbe/evy034
Morgan, H. D., Santos, F., Green, K., Dean, W., and Reik, W. (2005). Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14, R47–R58.
Mounger, J., Ainouche, M. L., Bossdorf, O., Cavé-Radet, A., Li, B., Parepa, M., et al. (2021). Epigenetics and the success of invasive plants. Philos. Trans. R. Soc. Lond. B Biol. Sci. 376:20200117. doi: 10.1098/rstb.2020.0117
Münzbergová, Z., Latzel, V., Šurinová, M., and Hadincová, V. (2019). DNA methylation as a possible mechanism affecting ability of natural populations to adapt to changing climate. Oikos 128, 124–134.
Noble, D. (2015). Conrad Waddington and the origin of epigenetics. J. Exp. Biol. 218, 816–818. doi: 10.1242/jeb.120071
Oliver, V. F., van Bysterveldt, K. A., Merbs, S. L., and Tollefsbol, T. O. (2016). “Epigenetics in ocular medicine,” in Medical Epigenetics, ed. T. O. Tollefsbol (Amsterdam: Elsevier), 391–412. doi: 10.1016/B978-0-12-803239-8.00022-3
Orchard, P., White, J. S., Thomas, P. E., Mychalowych, A., Kiselva, A., Hensley, J., et al. (2018). Genome-wide chromatin accessibility and transcriptome profiling show minimal epigenome changes and coordinated transcriptional dysregulation of hedgehog signaling in Danforth’s short tail mice. Human Mole. Gen. 28, 736–750. doi: 10.1093/hmg/ddy378
Paun, O., Verhoeven, K. J. F., and Richards, C. L. (2019). Opportunities and limitations of reduced representation bisulfite sequencing in plant ecological epigenomics. New Phytol. 221, 738–742. doi: 10.1111/nph.15388
Peng, S., Bellone, R., Petersen, J. L., Kalbfleisch, T. S., and Finno, C. J. (2021). Successful ATAC-seq from snap-frozen equine tissues. Front. Genet. 12:641788. doi: 10.3389/fgene.2021.641788
Perrier, C., Lozano, del Campo, A., Szulkin, M., Demeyrier, V., Gregoire, A., et al. (2018). Great tits and the city: Distribution of genomic diversity and gene-environment associations along an urbanization gradient. Evol. Appl. 11, 593–613. doi: 10.1111/eva.12580
Platt, A., Gugger, P. F., Pellegrini, M., and Sork, V. L. (2015). Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations. Mol. Ecol. 24, 3823–3830. doi: 10.1111/mec.13230
Polanowski, A. M., Robbins, J., Chandler, D., and Jarman, S. N. (2014). Epigenetic estimation of age in humpback whales. Mol. Ecol. Resour. 14, 976–987. doi: 10.1111/1755-0998.12247
Rey, O., Eizaguirre, C., Angers, B., Baltazar-Soares, M., Sagonas, K., Prunier, J. G., et al. (2020). Linking epigenetics and biological conservation: Towards a conservation epigenetics perspective. Funct. Ecol. 34, 414–427.
Roberts, C. E., Raner, G. M., and Isaacs, G. D. (2018). High Performance Liquid Chromatography Separation of Epigenetic Cytosine Variants. Methods Protoc. 1:10. doi: 10.3390/mps1020010
Roberts, M. E., Stewart, B. M., and Tingley, D. (2019). Stm: An R package for structural topic models. J. Stat. Softw. 91, 1–40. doi: 10.18637/jss.v091.i02
Rodriguez Barreto, D., Garcia, de Leaniz, C., Verspoor, E., Sobolewska, H., Coulson, M., et al. (2019). DNA methylation changes in the sperm of captive-reared fish: A route to epigenetic introgression in wild populations. Mol. Biol. Evol. 36, 2205–2211. doi: 10.1093/molbev/msz135
Schield, D. R., Walsh, M. R., Card, D. C., Andrew, A. L., Adams, R. H., and Castoe, T. A. (2016). Epi RAD seq: scalable analysis of genomewide patterns of methylation using next-generation sequencing. Methods Ecol. Evol. 7, 60–69.
Schulz, B., Eckstein, R. L., and Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 23, 3523–3537. doi: 10.1111/mec.12835
Siller, S. J., and Rubenstein, D. R. (2019). A tissue comparison of DNA methylation of the glucocorticoid receptor gene (Nr3c1) in European starlings. Integr. Comp. Biol. 59, 264–272. doi: 10.1093/icb/icz034
Skvortsova, K., Iovino, N., and Bogdanović, O. (2018). Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 19, 774–790. doi: 10.1038/s41580-018-0074-2
Sleutels, F., and Barlow, D. P. (2002). The origins of genomic imprinting in mammals. Adv. Genet. 46, 119–163.
Tanabe, A., Shimizu, R., Osawa, Y., Suzuki, M., Ito, S., Goto, M., et al. (2020). Age estimation by DNA methylation in the Antarctic minke whale. Fish. Sci. 86, 35–41.
Teschendorff, A. E., Breeze, C. E., Zheng, S. C., and Beck, S. (2017). A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association studies. BMC Bioinfo. 18:105. doi: 10.1186/s12859-017-1511-5
van Eijk, K. R., de Jong, S., Boks, M. P. M., Langeveld, T., Colas, F., Veldink, J. H., et al. (2012). Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genom. 13:636. doi: 10.1186/1471-2164-13-636
Vergeer, P., Wagemaker, N. C. A. M., and Ouborg, N. J. (2012). Evidence for an epigenetic role in inbreeding depression. Biol. Lett. 8, 798–801. doi: 10.1098/rsbl.2012.0494
Verhoeven, K. J. F., vonHoldt, B. M., and Sork, V. L. (2016). Epigenetics in ecology and evolution: what we know and what we need to know. Mol. Ecol. 25, 1631–1638. doi: 10.1111/mec.13617
Viitaniemi, H. M., Verhagen, I., Visser, M. E., Honkela, A., van Oers, K., and Husby, A. (2019). Seasonal variation in genome-wide DNA methylation patterns and the onset of seasonal timing of reproduction in great tits. Genome Biol. Evol. 11, 970–983. doi: 10.1093/gbe/evz044
Volkova, P. Y., Geras’kin, S. A., Horemans, N., Makarenko, E. S., Saenen, E., Duarte, G. T., et al. (2018). Chronic radiation exposure as an ecological factor: Hypermethylation and genetic differentiation in irradiated Scots pine populations. Environ. Pollut. 232, 105–112. doi: 10.1016/j.envpol.2017.08.123
Wang, M.-Z., Li, H.-L., Li, J.-M., and Yu, F.-H. (2020). Correlations between genetic, epigenetic and phenotypic variation of an introduced clonal herb. Heredity 124, 146–155. doi: 10.1038/s41437-019-0261-8
Wang, X., Dong, Y., Zheng, Y., and Chen, Y. (2021). Multi-omics metabolic and epigenetics regulatory network in cancer: A systems biology perspective. J. Genet. Genom. 48, 520–530. doi: 10.1016/j.jgg.2021.05.008
Wenzel, M. A., and Piertney, S. B. (2014). Fine-scale population epigenetic structure in relation to gastrointestinal parasite load in red grouse (Lagopus lagopus scotica). Mol. Ecol. 23, 4256–4273. doi: 10.1111/mec.12833
Werner, O., Prudencio, ÁS., de la Cruz-Martínez, E., Nieto-Lugilde, M., Martínez-Gómez, P., and Ros, R. M. (2020). A cost reduced variant of epi-genotyping by sequencing for studying DNA methylation in non-model organisms. Front. Plant Sci. 11:694. doi: 10.3389/fpls.2020.00694
Williams, S. E., and Hoffman, E. A. (2009). Minimizing genetic adaptation in captive breeding programs: A review. Biol. Conserv. 142, 2388–2400. doi: 10.1111/j.1365-294X.2007.03399.x
Willoughby, J. R., and Christie, M. R. (2019). Long-term demographic and genetic effects of releasing captive-born individuals into the wild. Conserv. Biol. 33, 377–388. doi: 10.1111/cobi.13217
Yamamoto, F., Yamamoto, M., Soto, J. L., Kojima, E., Wang, E. N., Perucho, M., et al. (2001). Notl-Msell methylation-sensitive amplied fragment length polymorhism for DNA methylation analysis of human cancers. Electrophoresis 22, 1946–1956. doi: 10.1002/1522-2683(200106)22:10<1946::AID-ELPS1946>3.0.CO;2-Y
Zhang, Y.-Y., Fischer, M., Colot, V., and Bossdorf, O. (2013). Epigenetic variation creates potential for evolution of plant phenotypic plasticity. N. Phytol. 197, 314–322. doi: 10.1111/nph.12010
Zhang, Z.-Q. (2013). Animal biodiversity: An update of classification and diversity in 2013. In : Zhang, Z.-Q. (Ed.) Animal Biodiversity: An Outline of Higher-level Classification and Survey of Taxonomic Richness (Addenda 2013). Zootaxa 3703, 5–11. doi: 10.11646/zootaxa.3703.1.1
Keywords: eco-epi, methylation, epigenome, DNA modification, phenotypic variation, adaptation
Citation: Lamka GF, Harder AM, Sundaram M, Schwartz TS, Christie MR, DeWoody JA and Willoughby JR (2022) Epigenetics in Ecology, Evolution, and Conservation. Front. Ecol. Evol. 10:871791. doi: 10.3389/fevo.2022.871791
Received: 08 February 2022; Accepted: 16 March 2022;
Published: 11 April 2022.
Edited by:
Ferhat Matur, Dokuz Eylül University, TurkeyReviewed by:
Alexey Yanchukov, Bülent Ecevit University, TurkeyCopyright © 2022 Lamka, Harder, Sundaram, Schwartz, Christie, DeWoody and Willoughby. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gina F. Lamka, Z2ZsMDAwM0BhdWJ1cm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.