 Petr Kotlík
Petr Kotlík Silvia Marková
Silvia Marková Michaela Horníková
Michaela Horníková Marco A. Escalante
Marco A. Escalante Jeremy B. Searle2*
Jeremy B. Searle2*

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ecol. Evol., 28 April 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.866605
This article is part of the Research TopicGenomic Basis of Adaptations to New Environments in Expansive and Invasive SpeciesView all 7 articles
The legacy of climatic changes during the Pleistocene glaciations allows inferences to be made about the patterns and processes associated with range expansion/colonization, including evolutionary adaptation. With the increasing availability of population genomic data, we have the opportunity to examine these questions in detail and in a variety of non-traditional model species. As an exemplar, here we review more than two decades of work by our group and others that illustrate the potential of a single “non-model model” mammal species - the bank vole (Clethrionomys glareolus), which is particularly well suited to illustrate the complexities that may be associated with range expansion and the power of genomics (and other datasets) to uncover them. We first summarize early phylogeographic work using mitochondrial DNA and then describe new phylogeographic insights gained from population genomic analysis of genome-wide SNP data to highlight the bank vole as one of the most compelling examples of a forest mammal, that survived in cryptic extra-Mediterranean (“northern”) glacial refugia in Europe, and as one of the species in which substantial replacement and mixing of lineages originating from different refugia occurred during end-glacial colonization. Our studies of bank vole hemoglobin structure and function, as well as our recent ecological niche modeling study examining differences among bank vole lineages, led us to develop the idea of “adaptive phylogeography.” This is what we call the study of the role of adaptive differences among populations in shaping phylogeographic patterns. Adaptive phylogeography provides a link between past population history and adaptation that can ultimately help predict the potential of future species responses to climate change. Because the bank vole is part of a community of organisms whose range has repeatedly contracted and then expanded in the past, what we learn from the bank vole will be useful for our understanding of a broad range of species.
Climate change is impacting plant and animal communities around the world and represents one of the greatest challenges to biodiversity in the coming decades. As the global climate is projected to continue to change, species in many areas are likely to be pushed beyond their climatic tolerance limits. Range shifts, in which species track their climatic niches (the climatic conditions within which a species can survive and reproduce) in space, were a regular response to past climate changes during Pleistocene glacial cycles, particularly in the northern hemisphere (Hewitt, 2000; Williams and Blois, 2018), and they are also already widespread in the present (Parmesan and Yohe, 2003; Thomas, 2010). Therefore, range shifts are often considered the “first” response that species will show to changing climate in the future (Bradshaw and McNeilly, 1991).
However, the ability of species to track their niche in space has its limits, and there are situations where range shifts are limited or impossible, such as for terrestrial species on islands (Harter et al., 2015) or species in fragmented habitats (Jump and Peñuelas, 2005). If species are able to survive in such situations, it will be through their ability to adapt in place, either through new mutations (which are usually rare) or more likely by exploiting existing adaptive variation. In some situations, this may occur through utilizing allelic variation from connected populations along climatic gradients adapted to different local climatic conditions.
Here we will focus on past large-scale climate changes during Pleistocene glaciations to provide a window on the patterns and processes of species’ responses to climate change, including range shifts or expansions and evolutionary adaptation to new environments. We look through that window with the help of one particular species. It is not a traditional model species, and indeed this review will illustrate what can now be inferred from such a “non-model model” species (Russell et al., 2017) - chosen not for convenience and because of the depth of laboratory studies already available on them, but because they have the right natural history relevant to specific situations where a detailed understanding is needed to fully explain global biodiversity. Therefore, what we provide here is an overview of over two decades of work by our group and others that illustrates the potential of a single small mammal species - the bank vole (Figure 1) - in elucidating patterns and processes during the end-glacial colonization of Europe using a variety of datasets and approaches.

Figure 1. The bank vole Clethrionomys glareolus, an emerging “non-model model” species for evolutionary and ecological research. Image credit: PK.
The bank vole Clethrionomys glareolus (also known as Myodes glareolus; Kryštufek et al., 2020) is a widespread cricetid rodent that occurs in forests and various scrub habitats throughout much of Europe, and its current range includes all major glacial refugial areas for temperate species (Figure 2). The location of glacial refugia has long been of interest to researchers, and the bank vole, as one of the mammalian species ecologically closely associated with forest ecosystems, was therefore well suited to answer this question (Deffontaine et al., 2005). Through our work and others, the bank vole has become one of the best studied mammals used for genetic and genomic studies of the response of European fauna to climate change after the Last Glacial Maximum (LGM; e.g., Bilton et al., 1998; Deffontaine et al., 2005; Kotlík et al., 2006; Wójcik et al., 2010; Marková et al., 2020; Horníková et al., 2021). It is one of the most convincing examples of a forest mammal surviving in cryptic extra-Mediterranean (“northern”) refugia during the LGM in Europe, i.e., refugia further north than the traditionally recognized southern refugia in the Mediterranean region (Bilton et al., 1998; Deffontaine et al., 2005; Kotlík et al., 2006; Bhagwat and Willis, 2008), and one of the species showing substantial lineage replacement and mixing during end-glacial colonization (Searle et al., 2009; Kotlík et al., 2018; Marková et al., 2020; Horníková et al., 2021). The results of our work on bank vole hemoglobin (Hb) led us to develop the idea of “adaptive phylogeography” (Kotlík et al., 2014), i.e., the study of the role of adaptive genotype and phenotype differences between populations in shaping phylogeographic patterns (see below).
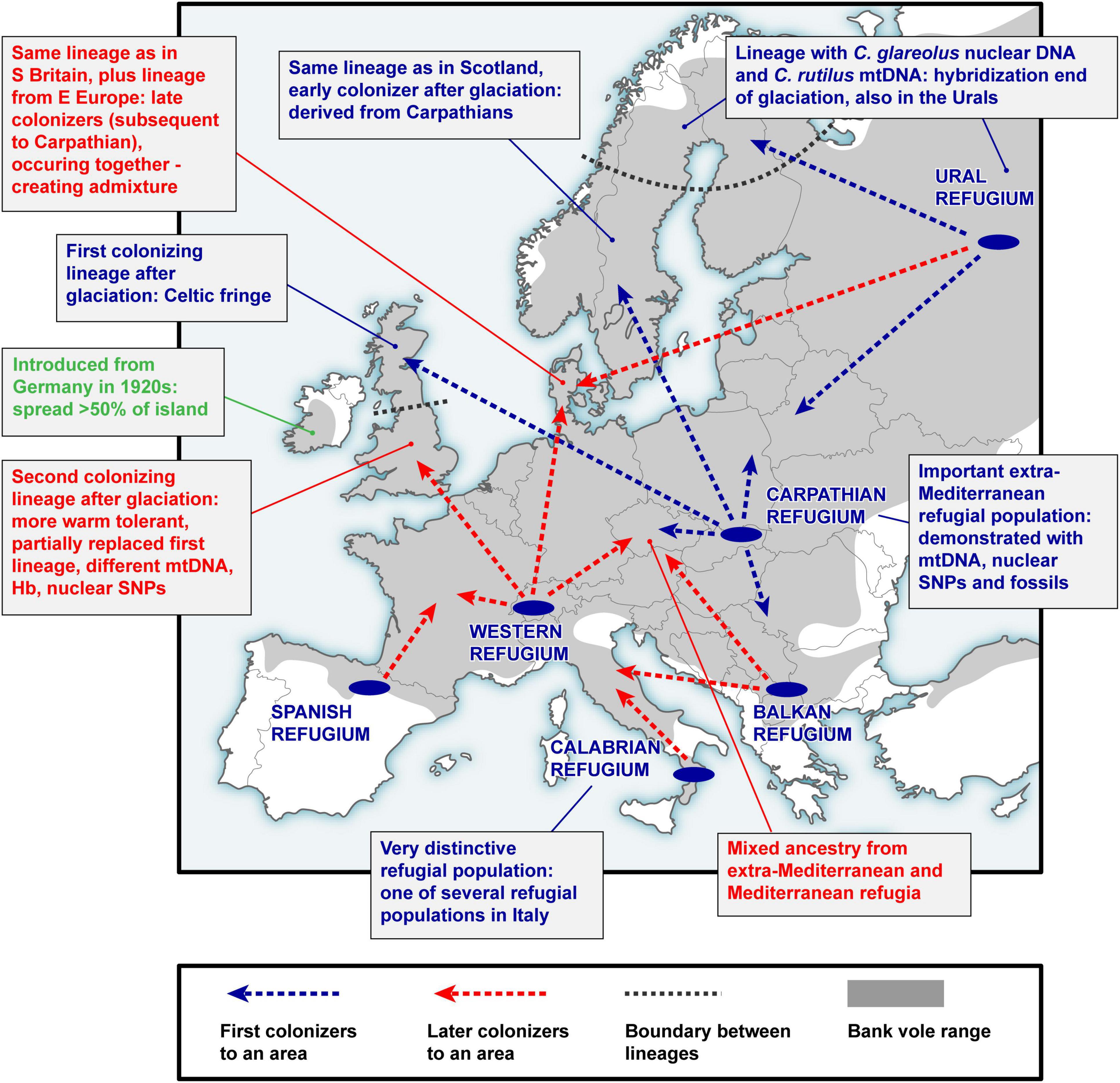
Figure 2. A simplified scheme of bank vole colonization from glacial refugia in Europe. This represents a summary of over two decades of research on bank vole phylogeography, reviewed in this article.
We believe that the bank vole is a particularly good example of the potential complexity of range expansion and the power of genomics to elucidate the details underlying this complexity. We first summarize early phylogeographic work using mitochondrial DNA (mtDNA) ranging from single genes to complete mitogenomes. Subsequently we describe new phylogeographic insights gained from population genomic analysis of single nucleotide polymorphism (SNP) data obtained first by RNA-Seq and later by genotyping-by-sequencing (GBS). Insights into adaptive differences are then illustrated by our Hb work, which combines sequencing of the bank vole globin gene repertoire with protein analysis (Kotlík et al., 2014). Complementary insights are provided by our recent ecological niche modelling (ENM) study (Escalante et al., 2021), in which we examine differences among bank vole lineages in environmental space and estimate the spatial distribution of suitable climatic conditions in the present and past (LGM).
The results that we describe highlight the importance of genomic data for phylogeographic interpretation, and provide evidence that different lineages evolved different physiological and ecological (e.g., thermal) tolerances. This suggests that dispersal of populations with different adaptive alleles may have played a role in determining phylogeographic patterns, which is the central idea of adaptive phylogeography (Kotlík et al., 2014). The findings from the bank vole provide insights relevant to other species during future rapid climate change.
The earliest studies relied on restriction fragment length polymorphism (RFLP) of mtDNA (Tegelström, 1987), but partial or complete mitochondrial gene sequences soon became the norm for bank vole phylogeography (Bilton et al., 1998; Deffontaine et al., 2005). Based on sequence data of the gene encoding cytochrome b (mt-Cytb), a number of divergent mtDNA clades were described from different parts of the bank vole distribution (Figure 3). In the first comprehensive study by Deffontaine et al. (2005), five bank vole clades were described - three in southern Europe on the Mediterranean peninsulas of Iberia (Spain), Italy, and the Balkans, and two widespread continental clades, a western European clade and an eastern clade whose distribution extends from eastern Europe to western Siberia (Figure 3). Later, another, highly divergent clade (Figure 3) was described from Calabria in the extreme south of the Italian peninsula (Colangelo et al., 2012) - the site of a glacial refugium - now characterized by distinct lineages in a variety of species (Vega et al., 2010; Schmitt et al., 2021). These results, refining the earlier study by Bilton et al. (1998) and confirmed with the phylogeny of complete mtDNA genomes obtained by Sanger and Illumina sequencing (Filipi et al., 2015), were some of the first published genetic arguments against the generality of the scenario postulating northward colonization from Mediterranean refugia as the main route of recolonization of the European continent at the end of the last glaciation, and thus for the existence of extra-Mediterranean refugia in the bank vole (and also in other small mammals; Bilton et al., 1998). Support for each Mediterranean clade (Spanish, Italian, and Balkan) and for the clade uniting the three clades (Figure 3) not only suggests that bank voles form a distinct maternal lineage on each peninsula, but also supports the idea that Mediterranean Europe is an area of long-term intraspecific endemism, without effective matrilineal gene exchange with more northern continental populations (Bilton et al., 1998; Deffontaine et al., 2005; Filipi et al., 2015).
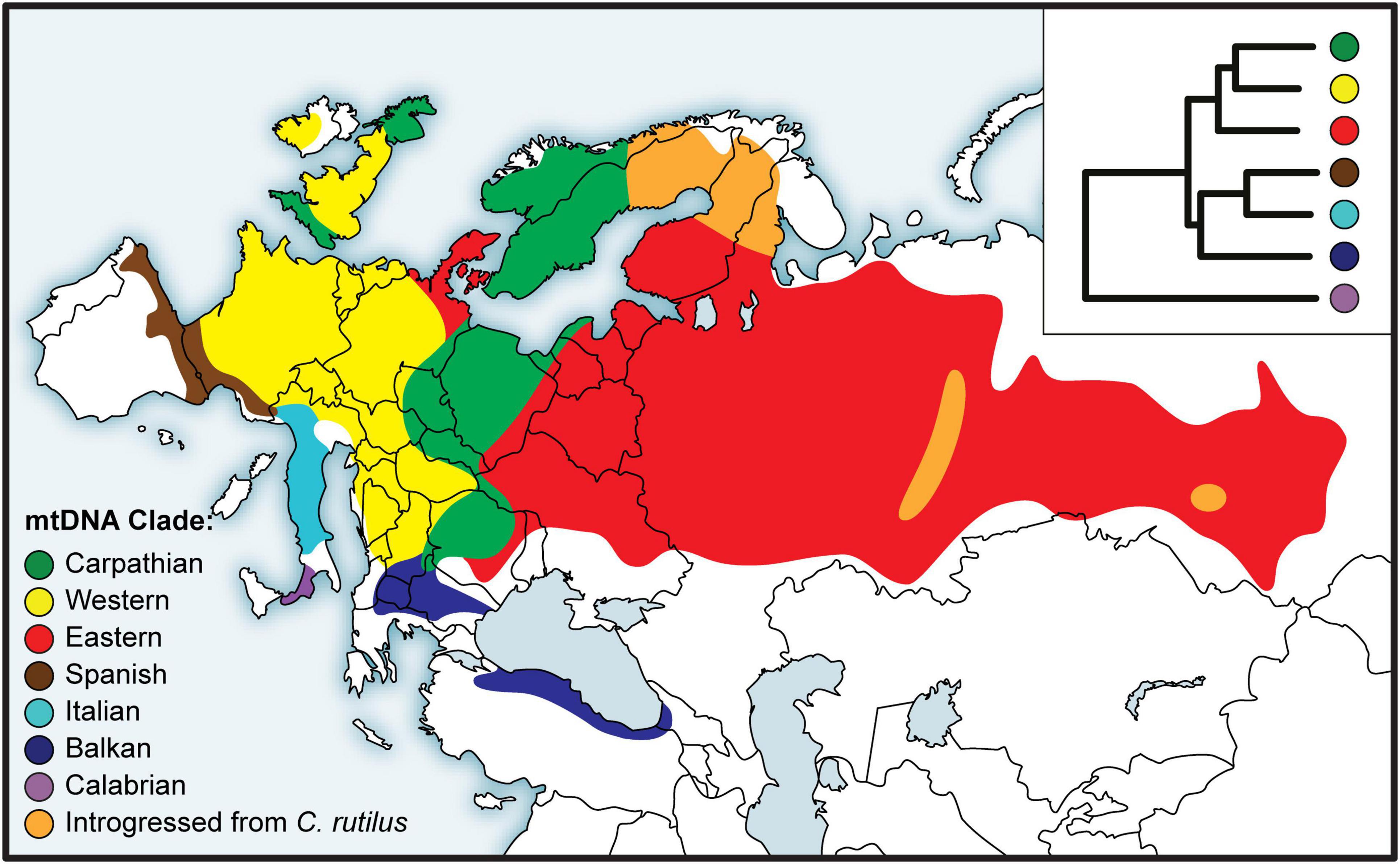
Figure 3. Distribution of bank vole phylogeographic clades inferred from mitochondrial (mt) DNA sequences, with a schematic representation of the phylogeny in the inset top right (adapted from Filipi et al., 2015).
A key argument for the existence and location of previously cryptic extra-Mediterranean refugia for bank voles was the discovery of the Carpathian clade (Figure 3) that was inferred to have survived the LGM near the Carpathians (Kotlík et al., 2006). This genetic evidence was consistent with the fossil record demonstrating the survival of bank voles and other forest mammals during the peak of the last glacial period in the Carpathians (Nadachowski, 1989; Horáček, 2000). By and large, the results are consistent with evidence suggesting that trees were part of the local glacial environment in various parts of the Carpathians (Jankovská et al., 2002; Willis and Van Andel, 2004). The later discovery (Wójcik et al., 2010) of the occurrence of the Carpathian clade over a large area north of the Carpathians to the Baltic coast (Figure 3) demonstrates an involvement of the refugium in the postglacial colonization of the deglaciated parts of Europe by bank voles (Figure 2).
The mitogenome phylogeny also assigns Britain, Sweden and Norway to the Carpathian clade (Figure 3). Thus, mtDNA links populations on the periphery of Europe in Britain and Fennoscandia to the Carpathian refugium (Figure 2). This was unexpected, as the Carpathian clade is found no further west in continental Europe than in what is now the Czech Republic and Germany, where it is replaced by the western clade in France, Belgium and the Netherlands, and by the eastern clade in northern Germany and Denmark (which lies on the southern postglacial expansion route into Scandinavia) (Figure 3). In Britain, the Carpathian clade is largely restricted to Scotland (Figure 3) and has been replaced by the western clade in most parts of England (Searle et al., 2009; see below). This discontinuous distribution of the Carpathian clade suggests a complex sequence of colonization events, with mtDNA signatures of more recent expansions replacing those of earlier expansions in different parts of Europe (Figure 2).
To assess whether there are differences among the bank vole clades that may have led to adaptive divergence and contributed to phylogeographic patterns in the bank vole, we compared the observed codon substitution patterns in the phylogeny of bank vole mtDNA genomes with expectations at neutrality and tested for possible functional consequences based on predicted changes in amino acid physicochemical properties (Filipi et al., 2015).
While analysis revealed no single branch and only a single site in a gene (mt-Cytb) with a dN/dS ratio greater than 1, the same substitution at an Ala codon by a Thr codon occurred a total of ten times in the phylogeny, at least once in almost every bank vole clade (Filipi et al., 2015). Analysis of amino acid properties also suggests that substitutions involving a significant change in amino acid side chain volume are more common at this site than expected by chance (Filipi et al., 2015). The site is located in the N-terminal region of the cytochrome b protein within the ubiquinone binding site (Xia et al., 2013), and the change may therefore be important for the reductase activity of the cytochrome bc1 complex (Atta-Asafo-Adjei and Daldal, 1991). While these results suggest an effect of selection on the physicochemical properties of the N-terminal region of the cytochrome b protein (Beckstead et al., 2009), potentially giving an advantage to individual haplotype lineages within different clades, the phylogenetic distribution of the substitution means that it is unlikely to have given a competitive advantage to a particular bank vole clade during end-glacial colonization (Filipi et al., 2015).
In addition, we found significant changes in amino acid properties along the branches defining three clades - the Calabrian, the Italian and the subclade of the Carpathian clade formed by genomes from Norway, but there was no common pattern in terms of protein evolution between clades (Filipi et al., 2015). Interestingly, the three clades are geographically restricted to the extreme latitudes of the bank vole’s distribution - two clades in Italy (in the south) and one in Norway (in the north). Populations at the extreme latitudes of a species’ distribution are likely to be exposed to more extreme climatic conditions and therefore are likely to be under greater selection pressure than populations at mid-latitudes (Mishmar et al., 2003; Pearson et al., 2009) - this then may be an explanation for the increased protein evolution rates found in Italian and Norwegian bank vole clades. Similar evidence for adaptive evolution of mtDNA genes was later found in geographically extreme populations of other rodents (Bartáková et al., 2021).
Overall, while there is evidence for adaptive evolution in bank vole mt-Cytb, possibly related to extreme latitudes, we found no evidence for a consistent role of functional divergence in bank vole clade evolution, suggesting that positive selection has not had widespread effects on bank vole mtDNA phylogeography.
The mtDNA pattern in Britain (Figure 4A) is a particularly well-documented example of lineage replacement (Figure 2), likely caused by climate change, that the bank vole shares with several other small mammals (Searle et al., 2009). Until after the end of the last glaciation, Britain and continental Europe were connected by a land bridge, and small mammals colonized Britain across the landmass “Doggerland” (Searle et al., 2009), which later got submerged beneath the North Sea (Spinney, 2008). The formation of the English Channel resulted in Britain being isolated from mainland Europe (Weninger et al., 2008), preventing further colonization by land (Searle et al., 2009). Thus, the replacement event (Figure 2) occurred prior to this separation of Britain. The mtDNA phylogeny revealed two distinct clades (and in one case chromosomal variants) in each of four small mammals in Britain, two shrews and three voles (Searle et al., 2009). Remarkably, in all of these species, one clade was found to have a peripheral distribution relative to the second clade (Figure 4A), with striking similarities to that of the Celtic people, hence the distributional pattern has been termed a “Celtic Fringe” (Searle et al., 2009). In all cases, both clades were also found in continental Europe (Searle et al., 2009). The phylogeny of bank vole mtDNA genomes (Filipi et al., 2015) clearly links the first colonization to the Carpathian clade and thus the Carpathian refugium, and the second colonization to the western clade, most likely originating from a forest refugium in the Alpine foothills (Kotlík et al., 2006; Magri, 2008; Figures 2, 3). This made the bank vole one of the few well-documented examples of mammalian species that have colonized Britain from both southern and northern refugia (Montgomery et al., 2014).
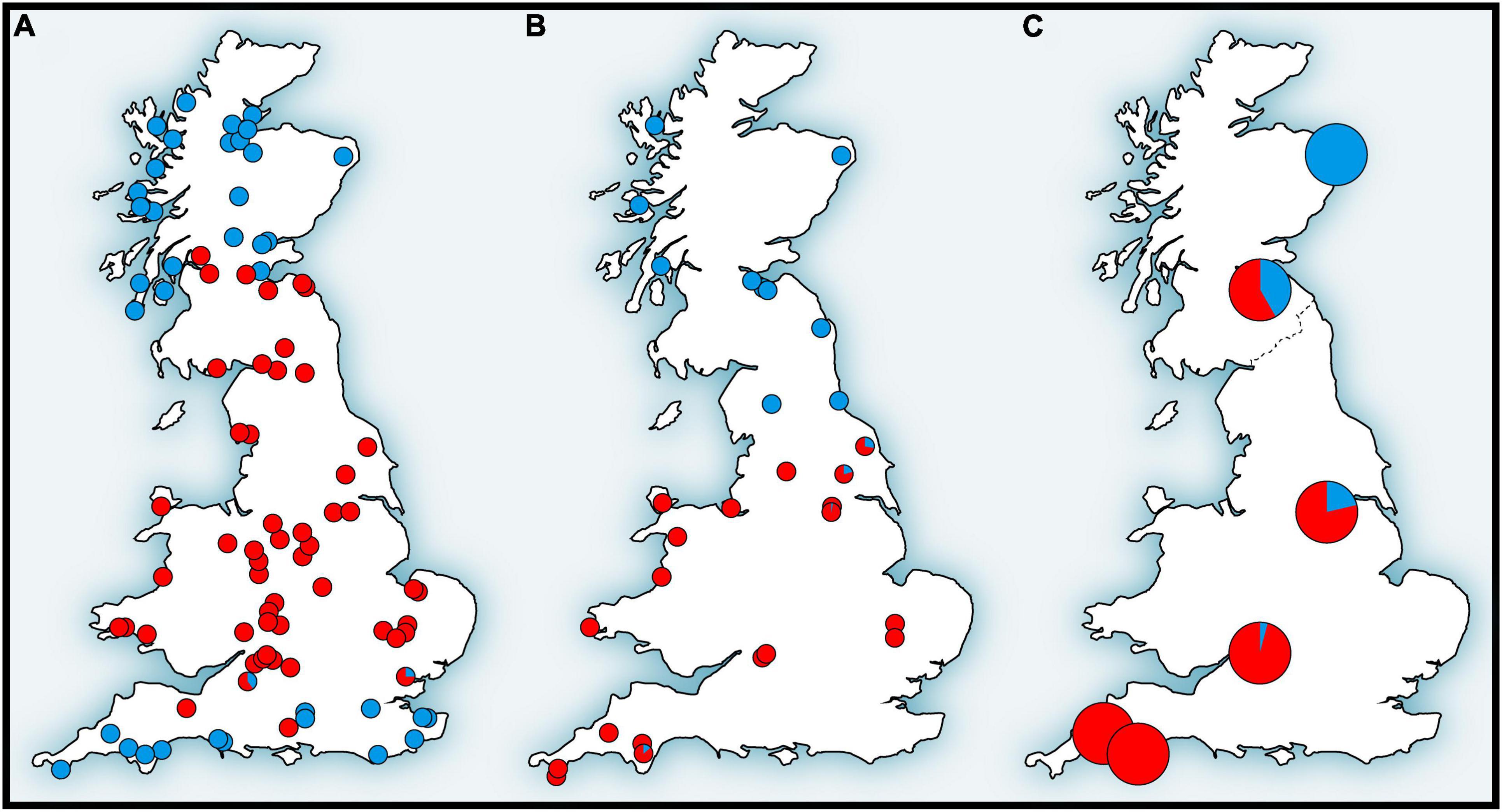
Figure 4. Distribution patterns of (A) mitochondrial DNA, (B) hemoglobin, and (C) single nucleotide polymorphism (SNP) variations of the bank vole in Britain that form the genetic “Celtic fringe” (for details, see text) (adapted from Kotlík et al., 2018).
The most likely explanation for this phylogeographic pattern is a two-stage colonization (Figure 2), with the first colonist already present in Britain during the Younger Dryas (YD; the final cold period of the last glaciation) and the second (replacing) colonist arriving across the land bridge from mainland Europe immediately after the YD (Searle et al., 2009; Brace et al., 2016) (see also below). The extreme climate change (from very cold to warm) that occurred over a very short time frame (perhaps only 50 years) at the end of the YD (Coope, 1979) probably forced this replacement (Searle et al., 2009). It is likely that the topography of Britain, with uplands in the margins to the north, west and south and several major rivers, played a role in the replacement (Searle et al., 2009).
Interestingly, there is no evidence that the functional divergence of proteins encoded in the mtDNA drove the replacement of the Carpathian clade mtDNA with the western clade mtDNA (Filipi et al., 2015; see above). However, there is evidence in the nuclear genome that the second colonists had a competitive advantage over the first colonists due to differences in Hb function (Kotlík et al., 2014; Figure 4B; see below).
The pioneering mtDNA studies of bank voles in Fennoscandia (Finland, Sweden, and Norway) by Tegelström (1987) based on RFLP data revealed considerable diversity and five distinct mtDNA groups. Three groups were found in southern Sweden and Norway, one in southern Finland, while a group closely resembling the mtDNA of the Holarctic northern red-backed vole (C. rutilus) was found in northern Sweden and Finland. The pattern of multiple bank vole clades as well as the presence of C. rutilus mtDNA in some bank vole populations (Figure 3) was later confirmed using sequences of mt-Cytb (Deffontaine et al., 2005) and complete mtDNA genomes (Filipi et al., 2015). This complex pattern is apparently the result of bidirectional colonization of Fennoscandia by bank voles at the end of the last glaciation (Figure 2) when there was a land bridge between Denmark and southern Sweden and a northeastern route via Russia (Tegelström and Jaarola, 1998). Colonization thus occurred via both the southern and northeastern routes, and a recent study (Filipi et al., 2015) has linked their origin to extra-Mediterranean glacial refugia in the Carpathian and Ural Mountains, respectively (Figure 2).
The presence of mtDNA derived from the northern red-backed vole in bank voles from northern Finland, Sweden and Norway (Figure 3) is clearly the result of unidirectional mtDNA introgression through hybridization (Tegelström, 1987; Deffontaine et al., 2005; Filipi et al., 2015). Using genome-wide SNP data (obtained by genotyping-by-sequencing, GBS; see below), we found evidence for a single colonization of Finland and northern Sweden and Norway via the northeastern route (Figure 2), but only some bank vole populations in Finland that originated from this colonization carried C. rutilus mtDNA (Figure 3), suggesting that hybridization occurred during colonization of Fennoscandia or shortly thereafter, after separation from Finnish populations not carrying C. rutilus mtDNA (Marková et al., 2020). In the same study, SNP clustering analyzes revealed no evidence of recent interspecific hybridization between bank and northern red-backed voles consistent with early work on protein electrophoresis (Tegelström et al., 1988). The lack of introgression in the nuclear genome, at least to the extent that it can be detected using GBS, suggests that many generations of backcrossing have occurred between hybrids and “pure” bank vole individuals. Taken together, these results suggest the rare occurrence of interspecific hybridization between these two species. This could be related to bank vole population dynamics in Fennoscandia, with occasional hybridization occurring in “crash years” of low vole density, a scenario considered to explain independent cases of mtDNA introgression in the Ural Mountains (Osipova and Soktin, 2006; Melnikova et al., 2012). Alternatively, as bank voles expanded their range into Fennoscandia, they may have regionally displaced C. rutilus in response to the changing climate, capturing its mtDNA through hybridization (Wielstra and Arntzen, 2012).
Interestingly, Boratyński et al. (2014) identified a possibly functional substitution within the ubiquinone binding site that distinguishes the cytochrome b proteins encoded in the mtDNA of C. glareolus and C. rutilus. This substitution, in conjunction with selection signatures from neutrality tests, suggests that introgressed mtDNA may have an advantage over bank vole mtDNA under local conditions, which is also supported by their climatic envelopes (Boratyński et al., 2014). Whether mt-Cytb itself or other linked genes were the target of selection remains unknown, but these results suggest that introgression of allospecific mtDNA in the northernmost bank vole population may have been favored by the selective advantage of rare hybrids over individuals not carrying C. rutilus mtDNA.
In the original mtDNA study of British bank voles (Searle et al., 2009) it was not possible to determine whether the replacement in Britain (Figure 2) involved a selective sweep of mtDNA (Figure 4A) or if other parts of the genome also show signatures of replacement. This could range from a selective sweep involving one or a few loci, to complete population replacement, involving a genome of one population at the cost of another. Therefore, we tested whether the phylogeographic pattern in Britain with a peripheral (early-colonizing) and a central (late-colonizing) mtDNA clade is reflected in the population structure of a set of > 10 thousand SNP loci obtained by RNA-Seq from the bank vole transcriptome (Kotlík et al., 2018). This study, made possible by the emerging availability of genomic data, provided the first genome-wide perspective of population divergence and admixture in a small mammal species showing the Celtic fringe. Analyses suggested that British bank voles are a mixture of two ancestral genomes, with proportions increasing toward the north and south, respectively (Figure 4C), similar to mtDNA (Figure 4A) and Hb (Figure 4B; see below), with pure genomes occurring at the northern and southern ends, respectively (Kotlík et al., 2018). This rejected the possibility that the Celtic fringe pattern in Britain was the result of an mtDNA-specific selection process. Scenario testing with approximate Bayesian computation and machine learning using random forest (ABC-RF) indicated that the northernmost site represented early colonization of Britain during a brief warming period shortly after the LGM (Kotlík et al., 2018). The first bank vole population in Britain would then likely have shrunk and/or become sparse during the YD cold snap, making it vulnerable to displacement by the second population that arrived during the final warming after the YD (see also above). This study, although carried out in a confined geographical area (Britain), has shown that it is possible to detect true population replacements and distinguish them from selective sweeps at one or a few loci by using large SNP datasets in elaborate tests of population scenarios. Although mtDNA turnovers have been shown to have occurred in other species elsewhere and at various times in the past (Barnes et al., 2002; Pergams et al., 2003; Dalén et al., 2012), the colonization of Britain by bank voles (and other small mammals) is one of the best-studied models of mtDNA replacement in association with recolonization at the Pleistocene–Holocene transition.
Fennoscandia seems to have undergone a similar history of multiple colonization and population replacement as Britain (Figure 2). The same (Carpathian) mtDNA clade found in the apex of the expansion into Britain (i.e., occupying the peripheral “Celtic Fringe”) is also found in the apex of the expansion into Fennoscandia from the south (Figure 3; see above). In Denmark (which is on the southern expansion route into Fennoscandia), as in central Britain, there is a separate lineage representing the second colonizers, in this case the eastern mtDNA clade (Figure 3). To infer the likely scenario of colonization of Fennoscandia and assess the impact of multiple colonization from different glacial refugia on genome-wide genetic diversity in Fennoscandia, we conducted a genotyping-by-sequencing (GBS) study of > 800 bank voles from across Europe, including a detailed sample from Fennoscandia (Marková et al., 2020). The population genomic analysis of genome-wide SNPs (> 6000 loci) using ABC-RF and other approaches revealed that bank voles colonized Fennoscandia multiple times via two different routes (Figure 2): three separated arrivals of colonists from the Carpathian refugium via a southern land bridge route and one via a northeastern route from an eastern glacial refugium near the Ural Mountains (Marková et al., 2020). As for Britain, temporal estimates revealed that the first of the Carpathian colonizations occurred before the YD, implying that the first colonists survived the YD in Fennoscandia. The ABC-RF analysis also suggested that introgression between bank and northern red-backed voles in northern Fennoscandia occurred shortly after end-glacial colonization from the eastern refugium (see above).
The best supported colonization scenario involved an origin of bank vole populations in Denmark through admixture among the ancestors of three refugial populations (eastern, Carpathian, and western) (Figure 2). This suggests a double genomic replacement in Denmark, where the genome of the original colonists from the Carpathians was partially replaced by the genome from the eastern refugium, including a complete mtDNA replacement by the eastern mtDNA clade (Figure 3; see above), and later by admixture with a genome from a western refugium near the Alps (Kotlík et al., 2006; Filipi et al., 2015; Figure 2). Thus, the study of bank voles in Fennoscandia shows that similar processes of genome mixing and replacement as originally described in British small mammals have also occurred in a continental setting, resulting in the extraordinary genetic patchwork we observed in the bank vole (Figure 2).
The important conclusion from the results of Marková et al. (2020) is that not only mixing of long isolated populations originating from different refugia can increase genetic diversity in a colonized area, but that increased diversity can also represent mixing from multiple colonizations originating from the same refugia. Genetic diversity generated by repeated founder events from the same Carpathian refugium, effectively nullifying the negative effects of the independent series of bottlenecks associated with a particular colonization, thus combined with diversity brought into Fennoscandia from other refugia to counteract the processes that would otherwise have resulted in low population genetic diversity in the north (Marková et al., 2020). The occurrence of repeated colonizations of Fennoscandia by bank voles from the Carpathian refugium in central Europe therefore not only argues against the universal importance of southern refugia for the genomic contribution to northern Europe after the LGM, but also illustrates why the “southern richness, northern purity” model (Hewitt, 2000) sometimes fits poorly (Marková et al., 2020).
The presence of distinct, divergent mtDNA haplotypes in Mediterranean bank voles led to the conclusion that these populations made little genetic contribution to central European diversity (Bilton et al., 1998; Deffontaine et al., 2005; see above). Mediterranean populations appeared isolated on separate peninsulas and evolved into endemic lineages (Figure 3; Bilton et al., 1998; Deffontaine et al., 2005; Filipi et al., 2015). Our recent population genomic study (Horníková et al., 2021), using the extensive SNP dataset of > 800 individuals (Marková et al., 2020; see above), provided the first genome-wide perspective on these questions, focusing on central Europe - a key geographic area when considering the colonization of the entire European continent. The results provided strong evidence that both extra-Mediterranean (Carpathians) and Mediterranean (Spain, Calabria and Balkans) refugia contributed to the ancestry and genomic diversity of bank vole populations in central Europe (Horníková et al., 2021; Figure 2). Although the genomic contribution of the centrally located Carpathian refugium predominates, populations in different parts of central Europe have mixed origins from Mediterranean and Carpathian sources (Figure 2). Complementing our other genomic studies on the bank vole (Kotlík et al., 2018; Marková et al., 2020), this study thus provided important details on the complex end-glacial colonization history of this well-studied species (Figure 2) and highlights the importance of genomic data for phylogeographic interpretation.
Understanding these historical patterns in the bank vole is important for evaluating their implications for evolutionary change throughout its range. For example, voles originating from different glacial refugia may have evolved different environmental tolerances (e.g., thermal) and may be adapted to local refugial conditions (see below). Therefore, we hypothesized that “warm-adapted” southern alleles with specific physiological functions were introduced into more northerly populations during the presumed admixture events, facilitating the Holocene dispersal of admixed populations throughout Europe (Horníková et al., 2021). In addition, the genomic contributions of Mediterranean populations (Figure 2) could also serve as sources of adaptive variation under future climate change. For example, adaptive alleles that evolved in the Mediterranean environment could be beneficial in central Europe under a future warmer climate (Rishishwar et al., 2015). These results thus opened new avenues for future research on evolutionary adaptation to physiological challenges during climate change, with the bank vole serving as an important model for understanding other species.
Unlike Britain and some small offshore islands, bank voles did not colonize Ireland naturally (Figure 2). There is convincing evidence that the species was accidentally introduced from Germany in the 1920s (Stuart et al., 2007). A joint study by Stuart et al. (2007) examined the genetic evidence for this introduction using mtDNA sequences of mt-Cytb from across the Irish range, from Britain and from two suspected areas of origin in southern Germany. The results showed a close relationship between the Irish and German sequences, suggesting that Irish voles are part of the western clade (Figure 3), but it was impossible to pinpoint the origin of the voles with certainty to either area in Germany (Stuart et al., 2007), indicating that larger and more widespread samples from Europe and evaluation of additional molecular markers are required to definitively determine the origin of Irish bank voles.
The bank vole in Ireland continues to expand without human intervention. Irish bank voles are therefore an excellent system to study processes associated with range expansion (McManus et al., 2021), including adaptation to the range expansion process itself. To this end, White et al. (2013) used the GBS approach to simultaneously identify and genotype a panel of SNPs for bank vole samples arranged in three transects that ran from the point of introduction to the wave front of expansion. They then searched for outlier loci with strong correlations between allele frequency and distance from the site of introduction, with the direction of correlation being the same in all three transects. The rationale was that outliers showing similar allele frequency clines in multiple transects were more likely to reflect selection than drift or allele surfing (White et al., 2013). Candidates for selection identified using this approach included genes with immunological functions and several potential candidates associated with differential reproductive investment on the expansion front (White et al., 2013). These results, which suggest that the bank vole was able to respond adaptively to range expansion despite the overall loss of genetic diversity, are likely to be relevant to many other species that are expanding their ranges, for example due to climate change.
The striking north-south pattern of two structurally and functionally distinct adult Hb types in British bank voles (Figure 4B) was first discovered by protein electrophoresis by Hall (1979), who designated the two types as HbS (slow) and HbF (fast). The pattern was later reinterpreted by Searle et al. (2009) from the perspective of the Celtic fringe and is most likely the result of the arrival of HbF across the land bridge connecting England to mainland Europe after the YD and the partial replacement of HbS, which was already present in Britain at that time (Figure 2). The fact that Hb in the bank vole (Figure 4B) has the same signature of replacement as mtDNA (Figure 4A; see above) gave us a starting point to investigate the possibility of adaptive processes to explain the replacement in Britain.
Using a combination of in vitro protein analysis, structural modelling, electrostatics calculation, and sequencing of the entire repertoire of globin genes in the bank vole, we demonstrated that HbS and HbF differ by a single amino acid substitution, in which a serine normally present on the outer surface of the beta subunit is replaced by a redox-active cysteine (Kotlík et al., 2014; Figure 5). Redox-active cysteines in vertebrate Hb are key elements of cellular antioxidant capacity (Rossi et al., 1998; Reischl et al., 2007; Vitturi et al., 2013; Petersen et al., 2018), and we have shown that bank vole HbF confers greater erythrocyte resistance to oxidative stress (Kotlík et al., 2014). Moreover, our gene expression studies indicate that duplication of the bank vole beta-globin gene, with a high and a low expressed paralog, allows fine-tuning of HbF concentration in erythrocytes (Strážnická et al., 2018; Dvořáková et al., 2020). Oxidative stress induced by an increase in environmental temperature is considered one of the most important proximate selection pressures expected to influence the physiology and longevity of organisms in the context of future climate change (Paital et al., 2016; Cassia et al., 2018; Jacobs et al., 2020), strongly suggesting that physiological variation mediated by variable cysteine content in bank vole Hb is relevant to tolerance to climate-related stress (Kotlík et al., 2014). This is supported by the finding that the distribution of HbF in Britain and mainland Europe is significantly associated with bioclimatic variables describing annual and seasonal climatic extremes (Strážnická et al., 2018). Consequently, we hypothesized that the displacement of HbS by HbF in southern Britain (Figure 2) was a response to an altered selection regime due to a temperature increase during the end-glacial climate warming (Searle et al., 2009), with the advantage of HbF providing greater tolerance to environmental stress (Kotlík et al., 2014).
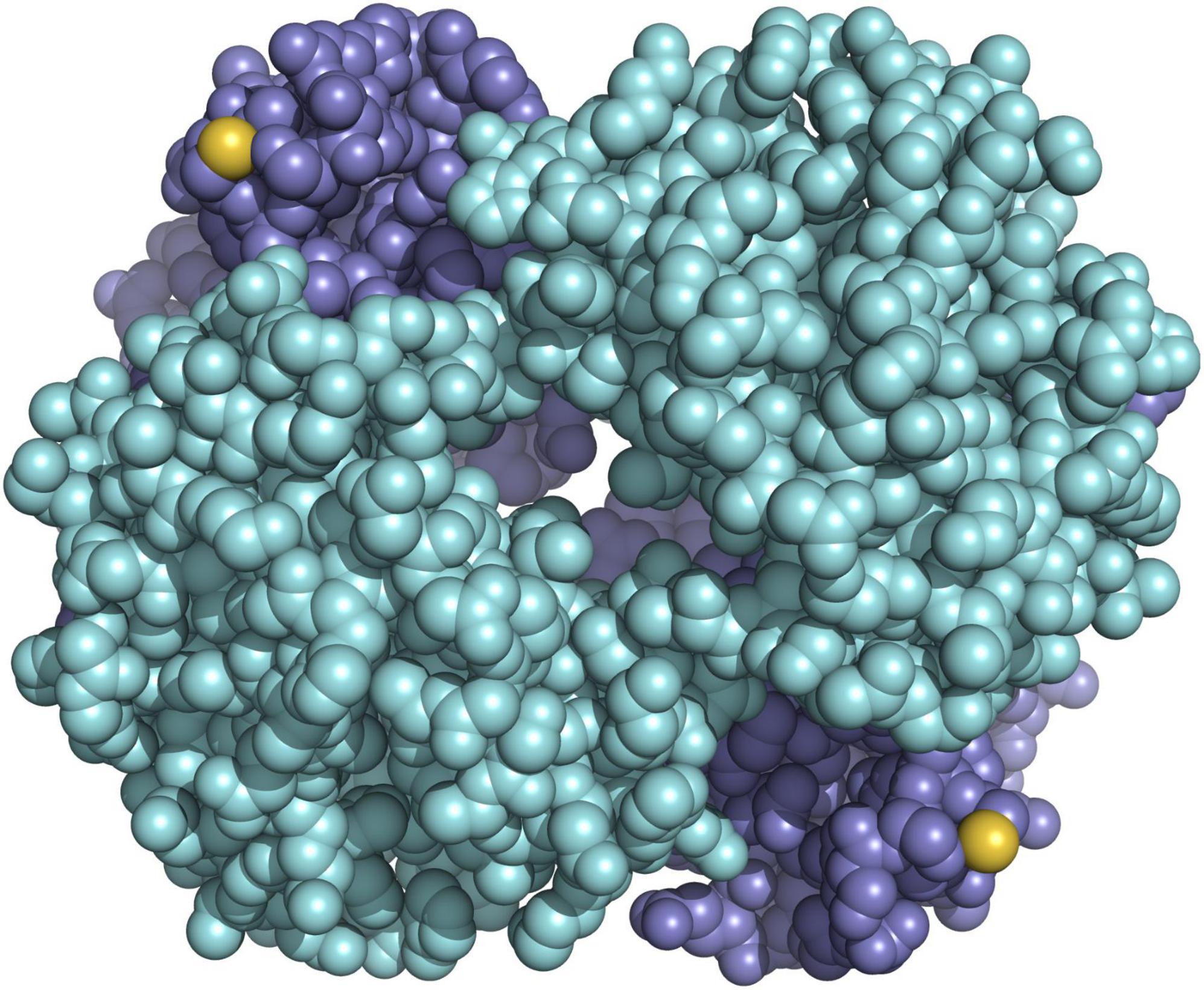
Figure 5. Model of hemoglobin HbF of the bank vole showing the positions of the reactive cysteines (sulfur atoms in yellow) on the surface of the molecule, which consists of the two subunits alpha (cyan) and the two subunits beta (blue).
Interestingly, Hb screening in mainland Europe using a pyrosequencing assay has shown that the bank vole population in western Europe, which owes a substantial part of its ancestry to the Spanish population (Horníková et al., 2021; Figure 2), shares HbF with the Spanish population (Strážnická et al., 2018). Therefore, the HbF allele may have been transferred from the Mediterranean (Spanish) to the western population and spread to western Europe and Britain (Horníková et al., 2021), where it contributed to genic and population replacement (Figure 2).
Physiological variation related to bank vole Hb was examined long ago (Kostelecka-Myrcha, 1967; Kostelecka-Myrcha et al., 1970), but only in our recent work has it become clear that different bank vole lineages have evolved different physiological tolerances that are likely climate relevant. Therefore, dispersal of populations carrying different Hb types appears to have played a role in the adaptive phylogeography of the bank vole, with gene flow and admixture between lineages carrying different Hb types facilitating adaptation of local populations.
As a complementary approach to assess the role of adaptive divergence in bank vole phylogeography, we used ENM in a recent study (Escalante et al., 2021). Most previous ENM studies assumed that niches are constant throughout the geographic range of a species (e.g., Fløjgaard et al., 2009). However, niches can vary among populations, and niche evolution is particularly likely in intraspecific evolutionary lineages (Schulte et al., 2012; Chardon et al., 2020). Therefore, we focused on the four major mtDNA lineages of the bank vole - the Carpathian, western, eastern, and southern lineages (the latter combines the three Mediterranean clades: Spanish, Italian, and Balkan; see above), created a separate ecological model for each lineage, and tested for niche differentiation among them. Niches differed significantly among all lineage pairs, and these differences could not be explained by climatic differences among their respective ranges (Escalante et al., 2021). Spatial separation of climatic suitability among lineages (Figures 6A,B) suggested that the distribution of a particular lineage is likely restricted because the niches of other lineages predict higher suitability in other parts of the bank vole range. Taken together, the ENM results provided strong arguments for intraspecific ecological differentiation in the bank vole and its role in bank vole phylogeography (Escalante et al., 2021).
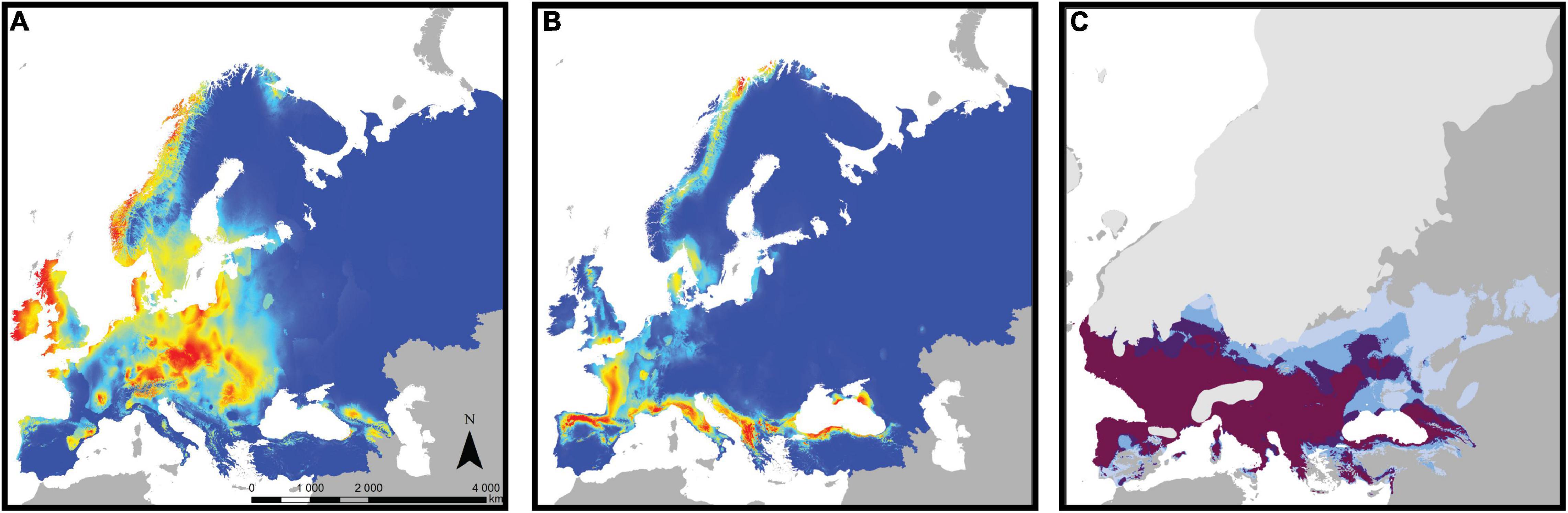
Figure 6. Climate suitability estimated by ecological niche modeling for the bank vole and its lineages. Heat maps of current climate suitability for (A) the Carpathian clade and (B) the southern clade (combining the three Mediterranean clades; see text for details), with warmer colors indicating higher suitability. (C) Areas where the climate of the Last Glacial Maximum is suitable for bank vole, with darker colors indicating support from an increasing number of models from a total of four models, each of which considered a different climate data set (adapted from Escalante et al., 2021).
When projected onto the LGM climate reconstruction (Figure 6C), the models showed that much of the Mediterranean and central and western Europe had climates favorable for bank vole survival, with several models predicting highly favorable conditions for the vicinity of the Carpathians (Escalante et al., 2021). As discussed by Escalante et al. (2021), forest-associated species such as the bank vole likely had more restricted and patchy distributions in areas where the LGM climate was suitable for them, e.g., due to low atmospheric CO2 concentrations leading to patchy forest cover, compared to climate alone (Fløjgaard et al., 2009). Therefore, bank voles have likely survived in geographically restricted refugia within those areas, in the form of isolated populations that have contributed to the genetic and phenotypic diversification we observe in the bank vole today.
We introduced the term “adaptive phylogeography” to refer to a phylogeographic study of the genes underlying functional phenotypic variation, with the goal of gaining insight into the role of adaptation and selection in observed phylogeographic patterns and ultimately the processes that shape the evolutionary history of the species (Kotlík et al., 2014). Applying this approach to bank vole Hb was the first direct attempt to investigate adaptive divergence in glacial refugia and the potential role of selection during end-glacial range expansion of the bank vole, and one of the first such attempts ever (de Lafontaine et al., 2018).
Phylogeographic data have been used to provide historical context for divergence through local adaptation, e.g., to provide information on how many independent times an adaptive phenotype has evolved, and whether it has the same or a different genetic basis, and to provide a time frame for adaptation (e.g., Aldenhoven et al., 2010). Because such studies use phylogeographic data to study adaptation, they reasonably fit within the concept of “adaptive phylogeography”, although we initially applied the term primarily to a phylogeographic study of the genes underlying the functional traits themselves (Kotlík et al., 2014).
In its most rigorous and comprehensive form, adaptive phylogeography seeks to establish links between locally adapted genotypes/phenotypes, population history, and environmental (e.g., climatic) selection, as recently reviewed by Zamudio et al. (2016) (but without explicitly using the term “adaptive phylogeography”). This involves identification and functional characterization of adaptive phenotypes, analyzing specimens that span the range of environmental and phenotypic variation across the species’ distribution, and inferring the demographic history of populations using neutral genetic variation (Zamudio et al., 2016). Because the genetic basis of ecologically relevant traits is unknown in most non-traditional model species, this approach has been applied mainly to model species with well-developed genomic resources (e.g., house mouse, Mus musculus Storz et al., 2007a) or to species in which a causal relationship between an adaptive phenotype and underlying genetic variation is already well established (e.g., rock pocket mice, Chaetodipus intermedius Hoekstra et al., 2004; deer mouse, Peromyscus maniculatus Storz et al., 2007b).
As identification of the genetic basis of phenotypic variation in non-traditional model species becomes increasingly feasible (see below), phylogeographic study of functional variation in phenotypes holds great promise for investigating relationships between genotypic and phenotypic diversity and adaptation in different environments (Zamudio et al., 2016). This is especially true in conjunction with the recent development of model-based approaches and the application of refined hypotheses based on taxon-specific attributes, e.g., their niche (termed “trait-based phylogeography”; Paz et al., 2015), including in a multitaxon comparative context (Papadopoulou and Knowles, 2016). In the bank vole, we have been iterative in our research, starting from basic phylogeography, but introducing elements that have developed into a fully adaptive phylogeographic approach. With other study species, the program of research could focus on adaptive phylogeography right from the start. As with basic phylogeography (e.g., Taberlet et al., 1998), there is a massive gain in understanding if an adaptive phylogeographic approach is applied to multiple species in the same geographic area, making use of modern comparative methodologies such as those discussed by Papadopoulou and Knowles (2016).
In the case of the European theater, there are already numerous species for which it would be worthwhile to incorporate them into that multi-species adaptive phylogeographic approach together with the bank vole, because of a strong foundation of relevant studies and interesting findings, e.g., for small mammals: common vole, Microtus arvalis (Martínková et al., 2013; Baca et al., 2020), field vole, Microtus agrestis (Herman et al., 2019), wood mouse, Apodemus sylvaticus (Herman et al., 2017; Martin Cerezo et al., 2020), stoat, Mustela erminea (Martínková et al., 2007), weasel, Mustela nivalis (McDevitt et al., 2012).
In this review, we have examined the work of our group and others, all of which use the bank vole as a study system. These studies decipher the complexity of the bank vole’s colonization pattern (Figure 2) and illustrate the power of genomic data combined with mtDNA phylogeography, functional analysis, and ecological modeling to illuminate the particular role of intraspecific variation and ultimately selection in shaping the species’ response to climate change after the end of the last glaciation.
The phylogeographic studies on the bank vole were one of the first genetic arguments for the existence of forest glacial refugia outside the Mediterranean region, namely in the Carpathians. These results were later confirmed in studies of other species, as was the pattern of the Celtic fringe in Britain, which is one of the best-studied examples of population replacement associated with range expansion at the Pleistocene-Holocene transition, with similar signatures in at least five species. Our study of the adaptive phylogeography of bank vole Hb was one of the first direct attempts to investigate adaptive divergence in glacial refugia and the potential role of selection during end-glacial range expansion.
Overall, the combined results suggest that end-glacial colonization was accompanied by the dispersal of populations that exhibited ecologically relevant functional differences, leading to an adaptive phylogeographic pattern in the bank vole. Variation in expanding populations was enhanced by mixing of ancestral populations adapted to the climate in their respective glacial refugia (Figure 2). We expect that similar patterns of mixing between populations from southern European refugia and refugia further north and east, as seen in the bank vole, may be observed in other species. This could be of particular importance in the event of future rapid climate change, where the emergence of adaptive variation through new mutations is likely to be less important than utilization of standing variation. These results open new avenues for future research on evolutionary adaptation to physiological challenges during climate change, with the bank vole serving as an important model for understanding other species. We propose that by examining variation in populations along climatic gradients from different refugia and identifying genes subject to selection and their physiological function, this adaptive phylogeographic approach can link past population history to adaptation and help predict the potential for future responses of temperate species to climate change. The predictive power of adaptive phylogeography is likely to increase substantially in the future with a multi-species adaptive phylogeographic approach, as identification of the underlying genetic basis of phenotypic variation within species becomes more feasible in non-traditional model species.
Although we have learned much about the patterns and processes associated with range expansion and colonization by the bank vole, our understanding of the mechanisms of adaptation of the species and its populations to climate and other aspects of the environment is still in its infancy. Future research should focus on how genetic and resulting phenotypic variation (e.g., in resistance to oxidative stress) influence individual fitness. This is a necessary next step in understanding the relationship between genetic and physiological variation, fitness, and environment in the bank vole. Low-throughput experimental effort will be required for this purpose (Storz and Wheat, 2010; Jacobs et al., 2021).
Further development of the bank vole as an emerging model system should be greatly facilitated by the availability of complete genome sequences. The bank vole mtDNA genome that we sequenced (Bendová et al., 2016) has provided a useful benchmark for other species (Donath et al., 2019), as it was one of the first complete mammalian mtDNA genomes to be annotated with RNA-Seq data (Marková et al., 2015). Recently, we started a whole genome sequencing project for the bank vole and obtained an assembly almost at the chromosome level (Kotlík et al., 2022). Together with the de novo transcriptome we previously generated (Kotlík et al., 2018), the new genome provides rich genetic resources for the bank vole. The availability of the reference genome allows resequencing of bank voles from different lineages and populations spanning climatic gradients (e.g., latitude) (Figure 4). We anticipate that increasing integration of genomic data with other data types (e.g., ecological, functional, and fitness data) will expand system-wide knowledge of the bank vole. This is important for further development of the bank vole as a model species not only for evolutionary and ecological studies, but also for other research areas such as rodent-borne zoonotic parasites (McManus et al., 2021) and pathogens (e.g., viruses: Ermonval et al., 2016; Rossi et al., 2021).
In this review, we have highlighted the bank vole as an emerging system that can be used to answer a wide range of questions in ecology and evolutionary biology (and beyond), with a precision that until recently was only possible with major model species. The advantage of systems that do not rely on established model organisms is that they can be chosen because they have the right natural history relevant to specific situations where a detailed understanding is needed to fully explain global biodiversity. The work examined here shows how the bank vole system can provide new insights into the patterns and processes associated with range expansion/colonization of Europe (Figure 2), including evolutionary adaptation to climate change, which are important for both understanding the past and predicting the future. Because the bank vole is part of a community of temperate organisms whose range has repeatedly contracted and then expanded in the past (Bhagwat and Willis, 2008), what we learn from the bank vole will be useful for our understanding of a broad spectrum of species, making the bank vole a true “non-model model” (Russell et al., 2017).
PK and JS conceived the manuscript. PK wrote the first draft, which was critically reviewed and amended by JS. SM, MH, and ME provided input in the preparation of the manuscript and prepared illustrations. All authors contributed to the article and approved the submitted version.
This review was supported by the Czech Science Foundation (Grant No. 20-11058S) and Ministry of Education, Youth and Sports of the Czech Republic (project EXCELLENCE CZ.02.1.01/0.0/0.0/15_003/0000460 OP RDE).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Figure 2 uses a modified map vector originally created by freepik - www.freepik.com.
Aldenhoven, J. T., Miller, M. A., Corneli, P. S., and Shapiro, M. D. (2010). Phylogeography of ninespine sticklebacks (Pungitius pungitius) in North America: glacial refugia and the origins of adaptive traits. Mol. Ecol. 19, 4061–4076. doi: 10.1111/j.1365-294X.2010.04801.x
Atta-Asafo-Adjei, E., and Daldal, F. (1991). Size of the amino acid side chain at position 158 of cytochrome b is critical for an active cytochrome bc1 complex and for photosynthetic growth of Rhodobacter capsulatus. Proc. Natl. Acad. Sci. U. S. A. 88, 492–496. doi: 10.1073/pnas.88.2.492
Baca, M., Popović, D., Baca, K., Lemanik, A., Doan, K., Horáček, I., et al. (2020). Diverse responses of common vole (Microtus arvalis) populations to Late Glacial and Early Holocene climate changes – Evidence from ancient DNA. Quat. Sci. Rev. 233:106239. doi: 10.1016/j.quascirev.2020.106239
Barnes, I., Matheus, P., Shapiro, B., Jensen, D., and Cooper, A. (2002). Dynamics of Pleistocene population extinctions in Beringian brown bears. Science 295, 2267–2270. doi: 10.1126/science.1067814
Bartáková, V., Bryjová, A., Nicolas, V., Lavrenchenko, L. A., and Bryja, J. (2021). Mitogenomics of the endemic Ethiopian rats: looking for footprints of adaptive evolution in sky islands. Mitochondrion 57, 182–191. doi: 10.1016/J.MITO.2020.12.015
Beckstead, W. A., Ebbert, M. T. W., Rowe, M. J., and McClellan, D. A. (2009). Evolutionary pressure on mitochondrial cytochrome b is consistent with a role of CytbI7T affecting longevity during caloric restriction. PLoS One 4:e5836. doi: 10.1371/JOURNAL.PONE.0005836
Bendová, K., Marková, S., Searle, J. B., and Kotlík, P. (2016). The complete mitochondrial genome of the bank vole Clethrionomys glareolus (Rodentia: Arvicolinae). Mitochondrial DNA 27, 111–112. doi: 10.3109/19401736.2013.873927
Bhagwat, S. A., and Willis, K. J. (2008). Species persistence in northerly glacial refugia of Europe: a matter of chance or biogeographical traits? J. Biogeogr. 35, 464–482. doi: 10.1111/J.1365-2699.2007.01861.X
Bilton, D. T., Mirol, P. M., Mascheretti, S., Fredga, K., Zima, J., and Searle, J. B. (1998). Mediterranean Europe as an area of endemism for small mammals rather than a source for northwards postglacial colonization. Proc. R. Soc. B Biol. Sci. 265, 1219–1226. doi: 10.1098/rspb.1998.0423
Boratyński, Z., Melo-Ferreira, J., Alves, P. C., Berto, S., Koskela, E., Pentikäinen, O. T., et al. (2014). Molecular and ecological signs of mitochondrial adaptation: consequences for introgression? Heredity 113, 277–286. doi: 10.1038/hdy.2014.28
Brace, S., Ruddy, M., Miller, R., Schreve, D. C., Stewart, J. R., and Barnes, I. (2016). The colonization history of British water vole (Arvicola amphibius (Linnaeus, 1758)): origins and development of the Celtic fringe. Proc. R. Soc. B Biol. Sci. 283:20160130. doi: 10.1098/rspb.2016.0130
Bradshaw, A. D., and McNeilly, T. (1991). Evolutionary response to global climatic change. Ann. Bot. 67, 5–14. doi: 10.1093/oxfordjournals.aob.a088209
Cassia, R., Nocioni, M., Correa-Aragunde, N., and Lamattina, L. (2018). Climate change and the impact of greenhouse gasses: CO2 and NO, friends and foes of plant oxidative stress. Front. Plant Sci. 9:273. doi: 10.3389/fpls.2018.00273
Chardon, N. I., Pironon, S., Peterson, M. L., and Doak, D. F. (2020). Incorporating intraspecific variation into species distribution models improves distribution predictions, but cannot predict species traits for a wide-spread plant species. Ecography 43, 60–74. doi: 10.1111/ecog.04630
Colangelo, P., Aloise, G., Franchini, P., Annesi, F., and Amori, G. (2012). Mitochondrial DNA reveals hidden diversity and an ancestral lineage of the bank vole in the Italian peninsula. J. Zool. 287, 41–52. doi: 10.1111/j.1469-7998.2011.00884.x
Coope, G. R. (1979). Late Cenozoic fossil Coleoptera: evolution, biogeography, and ecology. Annu. Rev. Ecol. Syst. 10, 247–267. doi: 10.1146/annurev.es.10.110179.001335
Dalén, L., Orlando, L., Shapiro, B., Brandström-Durling, M., Quam, R., Gilbert, M. T. P., et al. (2012). Partial genetic turnover in neandertals: Continuity in the east and population replacement in the west. Mol. Biol. Evol. 29, 1893–1897. doi: 10.1093/molbev/mss074
de Lafontaine, G., Napier, J. D., Petit, R. J., and Hu, F. S. (2018). Invoking adaptation to decipher the genetic legacy of past climate change. Ecology 99, 1530–1546. doi: 10.1002/ecy.2382
Deffontaine, V., Libois, R., Kotlík, P., Sommer, R., Nieberding, C., Paradis, E., et al. (2005). Beyond the Mediterranean peninsulas: evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol. Ecol. 14, 1727–1739. doi: 10.1111/j.1365-294X.2005.02506.x
Donath, A., Jühling, F., Al-Arab, M., Bernhart, S. H., Reinhardt, F., Stadler, P. F., et al. (2019). Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47, 10543–10552. doi: 10.1093/nar/gkz833
Dvořáková, V., Horníková, M., Němcová, L., Marková, S., and Kotlík, P. (2020). Regulatory variation in functionally polymorphic globin genes of the bank vole: a possible role for adaptation. Front. Ecol. Evol. 7:514. doi: 10.3389/fevo.2019.00514
Ermonval, M., Baychelier, F., and Tordo, N. (2016). What do we know about how hantaviruses interact with their different hosts? Viruses 8:223. doi: 10.3390/v8080223
Escalante, M. A., Horníková, M., Marková, S., and Kotlík, P. (2021). Niche differentiation in a postglacial colonizer, the bank vole Clethrionomys glareolus. Ecol. Evol. 11, 8054–8070. doi: 10.1002/ece3.7637
Filipi, K., Marková, S., Searle, J. B., and Kotlík, P. (2015). Mitogenomic phylogenetics of the bank vole Clethrionomys glareolus, a model system for studying end-glacial colonization of Europe. Mol. Phylogenet. Evol. 82, 245–257. doi: 10.1016/j.ympev.2014.10.016
Fløjgaard, C., Normand, S., Skov, F., and Svenning, J.-C. (2009). Ice age distributions of European small mammals: insights from species distribution modelling. J. Biogeogr. 36, 1152–1163. doi: 10.1111/j.1365-2699.2009.02089.x
Hall, S. J. G. (1979). Haemoglobin polymorphism in the bank vole, Clethrionomys glareolus, in Britain. J. Zool. 187, 153–160. doi: 10.1111/j.1469-7998.1979.tb03939.x
Harter, D. E. V., Irl, S. D. H., Seo, B., Steinbauer, M. J., Gillespie, R., Triantis, K. A., et al. (2015). Impacts of global climate change on the floras of oceanic islands – Projections, implications and current knowledge. Perspect. Plant Ecol. Evol. Syst. 17, 160–183. doi: 10.1016/j.ppees.2015.01.003
Herman, J. S., Jóhannesdóttir, F., Jones, E. P., McDevitt, A. D., Michaux, J. R., White, T. A., et al. (2017). Post-glacial colonization of Europe by the wood mouse, Apodemus sylvaticus: Evidence of a northern refugium and dispersal with humans. Biol. J. Linn. Soc. 120, 313–332. doi: 10.1111/bij.12882
Herman, J. S., Stojak, J., Paupério, J., Jaarola, M., Wójcik, J. M., and Searle, J. B. (2019). Genetic variation in field voles (Microtus agrestis) from the British Isles: Selective sweeps or population bottlenecks? Biol. J. Linn. Soc. 126, 852–865. doi: 10.1093/biolinnean/bly213
Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. doi: 10.1038/35016000
Hoekstra, H. E., Drumm, K. E., and Nachman, M. W. (2004). Ecological genetics of adaptive color polymorphism in pocket mice: Geographic variation in selected and neutral genes. Evolution 58, 1329–1341. doi: 10.1111/j.0014-3820.2004.tb01711.x
Horáček, I. (2000). Glacial cycles and mammalian biodiversity of central Europe: large scale migrations or vicariance dynamics? Geolines 11, 103–107.
Horníková, M., Marková, S., Lanier, H. C., Searle, J. B., and Kotlík, P. (2021). A dynamic history of admixture from Mediterranean and Carpathian glacial refugia drives genomic diversity in the bank vole. Ecol. Evol. 11, 8215–8225. doi: 10.1002/ece3.7652
Jacobs, P. J., Oosthuizen, M. K., Mitchell, C., Blount, J. D., and Bennett, N. C. (2020). Heat and dehydration induced oxidative damage and antioxidant defenses following incubator heat stress and a simulated heat wave in wild caught four-striped field mice Rhabdomys dilectus. PLoS One 15:e0242279. doi: 10.1371/JOURNAL.PONE.0242279
Jacobs, P. J., Oosthuizen, M. K., Mitchell, C., Blount, J. D., and Bennett, N. C. (2021). Oxidative stress in response to heat stress in wild caught Namaqua rock mice, Micaelamys namaquensis. J. Therm. Biol. 98:102958. doi: 10.1016/j.jtherbio.2021.102958
Jankovská, V., Chromy, P., and Niznianska, M. (2002). Šafárka - first palaeobotanical data of the character of Last Glacial vegetation and landscape in the West Carpathians (Slovakia). Acta Palaeobot. 42, 39–50.
Jump, A. S., and Peñuelas, J. (2005). Running to stand still: adaptation and the response of plants to rapid climate change. Ecol. Lett. 8, 1010–1020. doi: 10.1111/j.1461-0248.2005.00796.x
Kostelecka-Myrcha, A. (1967). Variation of morpho-physiological indices of blood in Clethrionomys glareolus (Schreber, 1780). Acta Theriol. 7, 191–222. doi: 10.4098/at.arch.67-20
Kostelecka-Myrcha, A., Gębczyński, M., and Myrcha, A. (1970). Some morphological and physiological parameters of mountain and lowland populations of the bank vole. Acta Theriol. 15, 133–142. doi: 10.4098/AT.arch.70-8
Kotlík, P., Deffontaine, V., Mascheretti, S., Zima, J., Michaux, J. R., and Searle, J. B. (2006). A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc. Natl. Acad. Sci. U. S. A. 103, 14860–14864. doi: 10.1073/pnas.0603237103
Kotlík, P., Marková, S., Konczal, M., Babik, W., and Searle, J. B. (2018). Genomics of end-Pleistocene population replacement in a small mammal. Proc. R. Soc. B Biol. Sci. 285:20172624. doi: 10.1098/rspb.2017.2624
Kotlík, P., Marková, S., Vojtek, L., Stratil, A., Šlechta, V., Hyršl, P., et al. (2014). Adaptive phylogeography: functional divergence between haemoglobins derived from different glacial refugia in the bank vole. Proc. R. Soc. B Biol. Sci. 281:20140021. doi: 10.1098/rspb.2014.0021
Kotlík, P., Marková, S., White, T. A., and Searle, J. B. (2022). Chromosome-Level de novo Genome Assembly of the Bank Vole.
Kryštufek, B., Tesakov, A. S., Lebedev, V. S., Bannikova, A. A., Abramson, N. I., and Shenbrot, G. (2020). Back to the future: the proper name for red-backed voles is Clethrionomys Tilesius and not Myodes Pallas. Mammalia 84, 214–217. doi: 10.1515/mammalia-2019-0067
Magri, D. (2008). Patterns of post-glacial spread and the extent of glacial refugia of European beech (Fagus sylvatica). J. Biogeogr. 35, 450–463. doi: 10.1111/j.1365-2699.2007.01803.x
Marková, S., Filipi, K., Searle, J. B., and Kotlík, P. (2015). Mapping 3’ transcript ends in the bank vole (Clethrionomys glareolus) mitochondrial genome with RNA-Seq. BMC Genomics 16:870. doi: 10.1186/s12864-015-2103-2
Marková, S., Horníková, M., Lanier, H. C., Henttonen, H., Searle, J. B., Weider, L. J., et al. (2020). High genomic diversity in the bank vole at the northern apex of a range expansion: The role of multiple colonizations and end-glacial refugia. Mol. Ecol. 29, 1730–1744. doi: 10.1111/mec.15427
Martin Cerezo, M. L., Kucka, M., Zub, K., Chan, Y. F., and Bryk, J. (2020). Population structure of Apodemus flavicollis and comparison to Apodemus sylvaticus in northern Poland based on RAD-seq. BMC Genomics 21:241. doi: 10.1186/s12864-020-6603-3
Martínková, N., Barnett, R., Cucchi, T., Struchen, R., Pascal, M., Pascal, M., et al. (2013). Divergent evolutionary processes associated with colonization of offshore islands. Mol. Ecol. 22, 5205–5220. doi: 10.1111/mec.12462
Martínková, N., McDonald, R. A., and Searle, J. B. (2007). Stoats (Mustela erminea) provide evidence of natural overland colonization of Ireland. Proc. R. Soc. B Biol. Sci. 274, 1387–1393. doi: 10.1098/rspb.2007.0334
McDevitt, A. D., Zub, K., Kawałko, A., Oliver, M. K., Herman, J. S., and Wójcik, J. M. (2012). Climate and refugial origin influence the mitochondrial lineage distribution of weasels (Mustela nivalis) in a phylogeographic suture zone. Biol. J. Linn. Soc. 106, 57–69. doi: 10.1111/j.1095-8312.2012.01840.x
McManus, A., Holland, C. V., Henttonen, H., and Stuart, P. (2021). The invasive bank vole (Myodes glareolus): A model system for studying parasites and ecoimmunology during a biological invasion. Animals 11:2529. doi: 10.3390/ani11092529
Melnikova, E. N., Kshnyasev, I. A., Bodrov, S. Y., Mukhacheva, S. V., Davydova, Y. A., and Abramson, N. I. (2012). Sympatric area of Myodes glareolus and M. rutilus (Rodentia, Cricetidae): historic and recent hybridization. Proc. Zool. Inst. RAS 316, 307–323.
Mishmar, D., Ruiz-Pesini, E., Golik, P., Macaulay, V., Clark, A. G., Hosseini, S., et al. (2003). Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. U. S. A. 100, 171–176. doi: 10.1073/pnas.0136972100
Montgomery, W. I., Provan, J., McCabe, A. M., and Yalden, D. W. (2014). Origin of British and Irish mammals: disparate post-glacial colonisation and species introductions. Quat. Sci. Rev. 98, 144–165. doi: 10.1016/J.QUASCIREV.2014.05.026
Nadachowski, A. (1989). Origin and history of the present rodent fauna in Poland based on fossil evidence. Acta Theriol. 34, 37–53. doi: 10.4098/at.arch.89-2
Osipova, O. V., and Soktin, A. A. (2006). Bank and red vole hybridization under experimental conditions. Dokl. Biol. Sci. 410, 381–383. doi: 10.1134/S0012496606050103
Paital, B., Panda, S. K., Hati, A. K., Mohanty, B., Mohapatra, M. K., Kanungo, S., et al. (2016). Longevity of animals under reactive oxygen species stress and disease susceptibility due to global warming. World J. Biol. Chem. 7, 110–127. doi: 10.4331/wjbc.v7.i1.110
Papadopoulou, A., and Knowles, L. L. (2016). Toward a paradigm shift in comparative phylogeography driven by trait-based hypotheses. Proc. Natl. Acad. Sci. U. S. A. 113, 8018–8024. doi: 10.1073/pnas.1601069113
Parmesan, C., and Yohe, G. (2003). A globally coherent fingerprint of climate change impacts across natural systems. Nature 421, 37–42. doi: 10.1038/nature01286
Paz, A., Ibáñez, R., Lips, K. R., and Crawford, A. J. (2015). Testing the role of ecology and life history in structuring genetic variation across a landscape: A trait-based phylogeographic approach. Mol. Ecol. 24, 3723–3737. doi: 10.1111/mec.13275
Pearson, G. A., Lago-Leston, A., and Mota, C. (2009). Frayed at the edges: selective pressure and adaptive response to abiotic stressors are mismatched in low diversity edge populations. J. Ecol. 97, 450–462. doi: 10.1111/J.1365-2745.2009.01481.X
Pergams, O. R. W., Barnes, W. M., and Nyberg, D. (2003). Rapid change in mouse mitochondrial DNA. Nature 423:397. doi: 10.1038/423397a
Petersen, A. G., Petersen, S. V., Frische, S., Drakulic, S., Golas, M. M., Sander, B., et al. (2018). Hemoglobin polymerization via disulfide bond formation in the hypoxia-tolerant turtle Trachemys scripta: implications for antioxidant defense and O2 transport. Am. J. Physiol. Regul. Integr. Comp. Physiol. 314, R84–R93. doi: 10.1152/ajpregu.00024.2017
Reischl, E., Dafre, A. L., Franco, J. L., and Wilhelm Filho, D. (2007). Distribution, adaptation and physiological meaning of thiols from vertebrate hemoglobins. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 146, 22–53. doi: 10.1016/J.CBPC.2006.07.015
Rishishwar, L., Conley, A. B., Wigington, C. H., Wang, L., Valderrama-Aguirre, A., and Jordan, I. K. (2015). Ancestry, admixture and fitness in Colombian genomes. Sci. Rep. 5:12376. doi: 10.1038/srep12376
Rossi, C., Zadra, N., Fevola, C., Ecke, F., Hörnfeldt, B., Kallies, R., et al. (2021). Evolutionary relationships of Ljungan virus variants circulating in multi-host systems across Europe. Viruses 13:1317. doi: 10.3390/v13071317
Rossi, R., Barra, D., Bellelli, A., Boumis, G., Canofeni, S., Di Simplicio, P., et al. (1998). Fast-reacting thiols in rat hemoglobins can intercept damaging species in erythrocytes more efficiently than glutathione. J. Biol. Chem. 273, 19198–19206. doi: 10.1074/jbc.273.30.19198
Russell, J. J., Theriot, J. A., Sood, P., Marshall, W. F., Landweber, L. F., Fritz-Laylin, L., et al. (2017). Non-model model organisms. BMC Biol. 15:55. doi: 10.1186/s12915-017-0391-5
Schmitt, T., Fritz, U., Delfino, M., Ulrich, W., and Habel, J. C. (2021). Biogeography of Italy revisited: genetic lineages confirm major phylogeographic patterns and a pre-Pleistocene origin of its biota. Front. Zool. 18:34. doi: 10.1186/s12983-021-00418-9
Schulte, U., Hochkirch, A., Lötters, S., Rödder, D., Schweiger, S., Weimann, T., et al. (2012). Cryptic niche conservatism among evolutionary lineages of an invasive lizard. Glob. Ecol. Biogeogr. 21, 198–211. doi: 10.1111/j.1466-8238.2011.00665.x
Searle, J. B., Kotlík, P., Rambau, R. V., Marková, S., Herman, J. S., and McDevitt, A. D. (2009). The Celtic fringe of Britain: insights from small mammal phylogeography. Proc. R. Soc. B Biol. Sci. 276, 4287–4294. doi: 10.1098/rspb.2009.1422
Storz, J. F., Baze, M., Waite, J. L., Hoffmann, F. G., Opazo, J. C., and Hayes, J. P. (2007a). Complex signatures of selection and gene conversion in the duplicated globin genes of house mice. Genetics 177, 481–500. doi: 10.1534/genetics.107.078550
Storz, J. F., Sabatino, S. J., Hoffmann, F. G., Gering, E. J., Moriyama, H., Ferrand, N., et al. (2007b). The molecular basis of high-altitude adaptation in deer mice. PLoS Genet. 3:0448–0459. doi: 10.1371/journal.pgen.0030045
Storz, J. F., and Wheat, C. W. (2010). Integrating evolutionary and functional approaches to infer adaptation at specific loci. Evolution 64, 2489–2509. doi: 10.1111/j.1558-5646.2010.01044.x
Strážnická, M., Marková, S., Searle, J. B., and Kotlík, P. (2018). Playing hide-and-seek in beta-globin genes: gene conversion transferring a beneficial mutation between differentially expressed gene duplicates. Genes 9:492. doi: 10.3390/genes9100492
Stuart, A. P., Mirimin, L., Cross, T. F., Sleeman, D. P., Buckley, N. J., Telfer, S., et al. (2007). The origin of Irish bank voles Clethrionomys glareolus assessed by mitochondrial DNA analysis. Irish Nat. J. 28, 440–446.
Taberlet, P., Fumagalli, L., Wust-Saucy, A. G., and Cosson, J. F. (1998). Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464. doi: 10.1046/J.1365-294X.1998.00289.X
Tegelström, H. (1987). Transfer of mitochondrial DNA from the northern red-backed vole (Clethrionomys rutilus) to the bank vole (C. glareolus). J. Mol. Evol. 24, 218–227. doi: 10.1007/BF02111235
Tegelström, H., and Jaarola, M. (1998). Geographic localization of a contact zone between bank voles Clethrionomys glareolus with distinctly different mitochondrial DNA. Acta Theriol. 43, 175–183. doi: 10.4098/AT.arch.98-13
Tegelström, H., Wyöni, P. I., Gelter, H., and Jaarola, M. (1988). Concordant divergence in proteins and mitochondrial DNA between two vole species in the genus Clethrionomys. Biochem. Genet. 26, 223–237. doi: 10.1007/BF00561462
Thomas, C. D. (2010). Climate, climate change and range boundaries. Divers. Distrib. 16, 488–495. doi: 10.1111/J.1472-4642.2010.00642.X
Vega, R., Fløjgaard, C., Lira-Noriega, A., Nakazawa, Y., Svenning, J.-C., and Searle, J. B. (2010). Northern glacial refugia for the pygmy shrew Sorex minutus in Europe revealed by phylogeographic analyses and species distribution modelling. Ecography 33, 260–271. doi: 10.1111/j.1600-0587.2010.06287.x
Vitturi, D. A., Sun, C. W., Harper, V. M., Thrash-Williams, B., Cantu-Medellin, N., Chacko, B. K., et al. (2013). Antioxidant functions for the hemoglobin β93 cysteine residue in erythrocytes and in the vascular compartment in vivo. Free Radic. Biol. Med. 55, 119–129. doi: 10.1016/J.FREERADBIOMED.2012.11.003
Weninger, B., Schulting, R., Bradtmöller, M., Clare, L., Collard, M., Edinborough, K., et al. (2008). The catastrophic final flooding of Doggerland by the Storegga Slide tsunami. Doc. Praehist. 35, 1–24. doi: 10.4312/dp.35.1
White, T. A., Perkins, S. E., Heckel, G., and Searle, J. B. (2013). Adaptive evolution during an ongoing range expansion: The invasive bank vole (Myodes glareolus) in Ireland. Mol. Ecol. 22, 2971–2985. doi: 10.1111/mec.12343
Wielstra, B., and Arntzen, J. W. (2012). Postglacial species displacement in Triturus newts deduced from asymmetrically introgressed mitochondrial DNA and ecological niche models. BMC Evol. Biol. 12:161. doi: 10.1186/1471-2148-12-161
Williams, J. E., and Blois, J. L. (2018). Range shifts in response to past and future climate change: Can climate velocities and species’ dispersal capabilities explain variation in mammalian range shifts? J. Biogeogr. 45, 2175–2189. doi: 10.1111/jbi.13395
Willis, K. J., and Van Andel, T. H. (2004). Trees or no trees? The environments of central and eastern Europe during the Last Glaciation. Quat. Sci. Rev. 23, 2369–2387. doi: 10.1016/j.quascirev.2004.06.002
Wójcik, J. M., Kawałko, A., Marková, S., Searle, J. B., and Kotlík, P. (2010). Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: a case study of bank voles using museum specimens. J. Zool. 281, 249–262. doi: 10.1111/j.1469-7998.2010.00699.x
Xia, D., Esser, L., Tang, W.-K., Zhou, F., Zhou, Y., Yu, L., et al. (2013). Structural analysis of cytochrome bc1 complexes: Implications to the mechanism of function. Biochim. Biophys. Acta - Bioenerg. 1827, 1278–1294. doi: 10.1016/j.bbabio.2012.11.008
Keywords: adaptation, climate change, cryptic refugia, genomics, last glacial maximum, Myodes glareolus, niche
Citation: Kotlík P, Marková S, Horníková M, Escalante MA and Searle JB (2022) The Bank Vole (Clethrionomys glareolus) as a Model System for Adaptive Phylogeography in the European Theater. Front. Ecol. Evol. 10:866605. doi: 10.3389/fevo.2022.866605
Received: 31 January 2022; Accepted: 28 March 2022;
Published: 28 April 2022.
Edited by:
Agnieszka Kloch, University of Warsaw, PolandReviewed by:
Mirosław Ratkiewicz, University of Białystok, PolandCopyright © 2022 Kotlík, Marková, Horníková, Escalante and Searle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petr Kotlík, a290bGlrQGlhcGcuY2FzLmN6; Jeremy B. Searle, amVyZW15LnNlYXJsZUBjb3JuZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.