
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Ecol. Evol. , 09 June 2022
Sec. Evolutionary Developmental Biology
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.850817
 Wanna Chetruengchai1,2,3†
Wanna Chetruengchai1,2,3† Worapong Singchat4,5†Chalurmpon Srichomthong1,2Adjima Assawapitaksakul1,2Kornsorn Srikulnath4,5
Worapong Singchat4,5†Chalurmpon Srichomthong1,2Adjima Assawapitaksakul1,2Kornsorn Srikulnath4,5 Syed Farhan Ahmad4,5*
Syed Farhan Ahmad4,5* Chureerat Phokaew1,2,6*Vorasuk Shotelersuk1,2
Chureerat Phokaew1,2,6*Vorasuk Shotelersuk1,2Water monitors (Varanus salvator macromaculatus, Deraniyagala, 1944) are the largest monitor in Southeast Asia and the second largest lizard globally after the Komodo dragon (Shine et al., 1996). They are a member of the family Varanidae within the genus Varanus. They live in semi-aquatic ecosystems inhabiting wetlands, hills, mangroves, and canals. During the past few decades of increased human invasion of their habitats, they have shown excellent adaptation to polluted urban environments (Uyeda, 2015). As a prolific scavenger, the water monitor consumes any animal, carrion, or food waste. This results in many beneficial effects including removing many infection sources from the environment, helping to clean surroundings (Research_and_Innovation_for_Sustainability_Center, 2021), maintaining the ecosystem, and balancing the food chain. The gut microbiota of V. salvator reveals the enrichment of bactericidal activity components (Akbar et al., 2019). Although V. salvator lives in a contaminated environment and consumes decaying carcasses, it still survives without any noticeable effects, suggesting its strong immunity. While mammals rely more on adaptive or acquired immunity, these reptiles rely heavily on innate immunities to combat infections (Rios and Zimmerman, 2015). Komodo dragon have a positive advantage of a blood-clotting mechanism, which is a part of their innate immune system (van der Poll and Herwald, 2014) helping them to survive in extreme environments (Lind et al., 2019). The transposable elements (TEs) within the vertebrate genome shaped innate immune evolution by triggering the antiviral innate immune pathway (Gazquez-Gutierrez et al., 2021). Reptiles' innate immunity also helps fight against microbe invasion and supports their adaptive immune response. Even though its immunity system is quite remarkable, only limited knowledge about its workings is known.
A high-quality reference genome is an excellent resource in comparative genomics. It emphasizes the importance of conservation biology and the discovery of potential molecular genetic mechanisms. Herein, we generated a first draft reference genome for the Varanus salvator macromaculatus using a hybrid approach of linked-read sequencing (10× Genomics and short-read sequencing) and PacBio's long-read sequencing platform. We have performed comparative genomics within and between species to understand the relationship between non-avian reptiles concerning evolution. We also identified positively selected genes that are associated with V. salvator-specific traits.
High molecular weight (HMW) genomic DNA from a female V. salvator's white blood cell (WBC) was corrected (Chaiprasertsri et al., 2013) and extracted using the Gentra Puregene blood kit (Qiagen). DNA quantity and quality examination were performed using a Qubit fluorometer and a 16 h pulse-field gel electrophoresis. For Pacbio sequencing, we generated >30 kb SMRTbell (insert hairpin loops) library using 20 μg of HMW DNA. With the PacBio Sequel sequencing system (Pacific Biosciences), 8 pM of SMRTbell templates with an expected size of 30 kb were loaded into 1,000,000 Zero Mode Waveguides SMRT cells. Pacbio SMRT sequencing was performed yielding 150 GB (16 SMRT cells). The average insert length and N50 read length were 18,294 bp and 29,450 bp, respectively. Given the expected genome size of 1.5 Gb similar to that of the Komodo dragon, PacBio sequencing data represented 100× depth coverage. For the linked-read sequencing (10× Genomics), we started with one ng of template HMW DNA. Whole-genome sequencing libraries were prepared using the Chromium Genome Library and Gel Bead Kit v.2 (10× Genomics). The barcoded DNA was sequenced by the Illumina NovaSeq6000 sequencer (Macrogen) and ~400 Gb raw data were obtained.
We utilized DEXTRACTOR (https://github.com/thegenemyers/DEXTRACTOR) to correct the Pacbio long-reads bam file (pulls sequence, Quiver, and/or Arrow information) and convert it to FASTA format files. Sequences shorter than 1,000 bp and quality (QV) <0.8 were filtered out. We obtained 135 Gb of raw PacBio long reads and the remaining 111 Gb of clean read data. The clean reads were used for de novo assembly with CANU v.1.84 (Koren et al., 2017) pipeline to generate the initial draft assembly comprised of 3 steps: correction, trimming, and assembly (using default parameters). It resulted in an assembly of ~1.89 Gb containing a total of 4,006 contigs with an N50 contigs length of 8.8 Mb. All 403 GB raw data of linked-read sequencing data were then assembled using version 2.1 of the Supernova Assembler (https://github.com/10XGenomics/supernova-chili-pepper). The Jellyfish software (Marcais and Kingsford, 2011) was performed to obtain a frequency distribution of 31-mers of the raw reads. The Jellyfish histogram output was uploaded to GenomeScope (Ranallo-Benavidez et al., 2020) to estimate genome size, repeat content, and heterozygosity (Supplementary Figure S1). The results suggested that the genome size of V. salvator is 1.5 Gb with 0.185% heterozygosity and 4.4% of duplication. The 10× linked-reads were assembled with Supernova software (version 2.1) by default parameters. The result demonstrated 1,443 scaffolds with an N50 contigs length of 43 Mb and a total sequence length of 1.57 Gb. Additionally, the supernova scaffolds and contigs from Pacbio were integrated with the default settings by Quickmerge (Chakraborty et al., 2016). Redundancy reduction, scaffolding, and gap closing were then carried out using Redundans (Pryszcz and Gabaldon, 2016) pipeline. The draft assembly was polished by aligning 10× Genomics Illumina reads to the genome using bwa-mem v0.7.5 (Li and Durbin, 2009) and Pilon v1.22 (Walker et al., 2014) resulting in the final assembly. To assess the qualities of the genome assemblies, descriptive measurement results including numbers of contigs, total number of assembled bases, and completeness were estimated using QUAST software (Gurevich et al., 2013).
The final draft genome assembly contained 858 scaffolds (>10 kb) with an N50 scaffold length of 71 Mb (longest scaffold: 121 Mb). Gaps comprised 1% of the assembly. The GC content of the water monitor is 44.0%. Detailed genome statistics are presented in Figure 1A and Supplementary Table S1. The Benchmarking Universal Single-Copy Orthologs pipeline [BUSCO v4.0.2] was used to evaluate the completeness of the genome and obtain the percentage of single-copy orthologous with vertebrata_odb10 BUSCO set (3,354 gene vertebrata gene set), 87.5% were found to be complete (83.3% as single genes and 4.2% as duplicated genes). We further validated with a Sauropsida BUSCO database for confirmation of completeness. Of this total, 91.6% were identified as complete including 86.2% single genes and 5.4% duplicated genes (Supplementary Table S1). To test for correct haplotype phasing and validate the haplotype retention during the assembly, a Merqury test (Rhie et al., 2020) and purge_dups analysis (Guan et al., 2020) were performed. The Merqury consensus quality value score before and after purge_dups were 43.16 (92.90% k-mer completeness) and 43.27 (79.68% k-mer completeness), respectively. The Merqury plot before and after purge_dups (Supplementary Figure S2) suggested a haplotype retention in our draft genome. This demonstrates that our V. salvator draft genome have some limitation according to our study's de novo assembly pipeline, however its sufficient as an essential resource in genome reference.
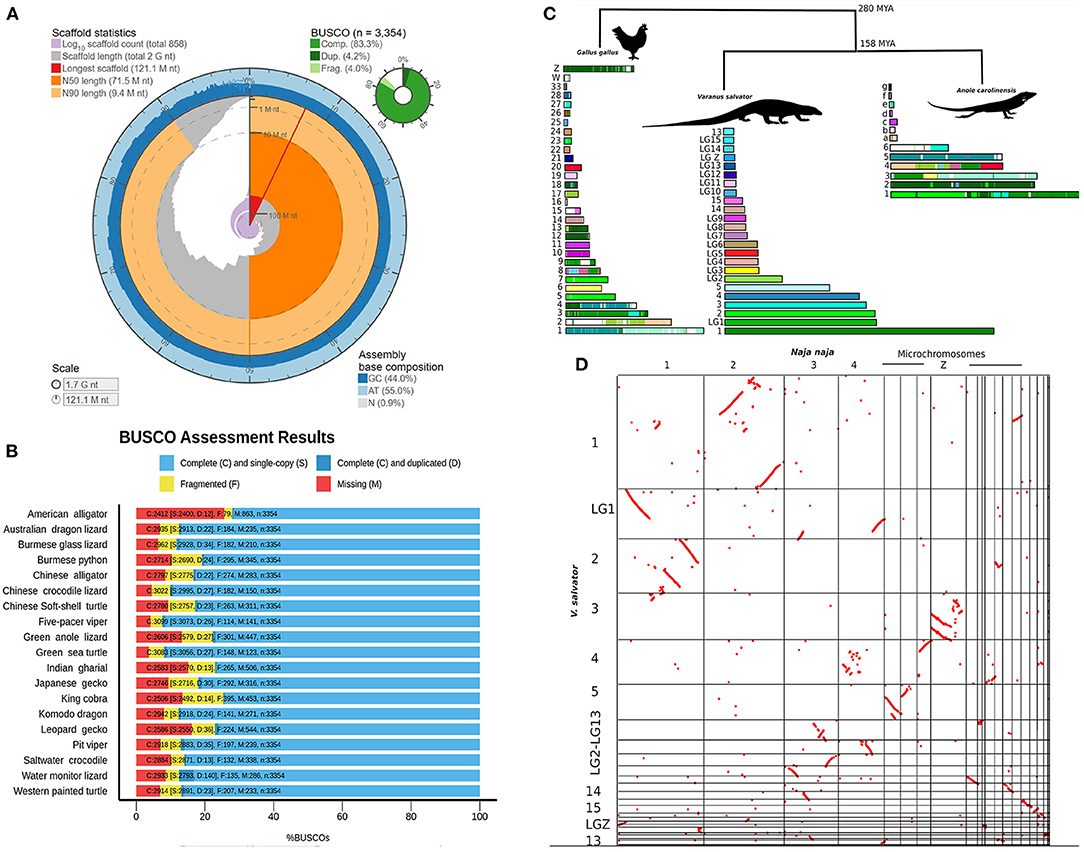
Figure 1. Varanus salvator genome completeness detail and comparative analysis. (A) Genome assembly statistics visualization of V. salvator. (B) Bar chart with summary assessments for the proportion of genes present in 19 non-avian reptile genomes. The summary assessment shows the proportions classified as complete (C, blues), complete single-copy (S, light blue), complete duplicated (D, dark blue), fragmented (F, yellow), and missing (M, red). (C) The cross-species syntenic comparison among V. salvator, G. gallus, and A. carolinensis genomes. The same color indicates a syntenic pattern between chromosome and V. salvator pseodchromosomes. (D) Dot plot showing patterns of chromosomal rearrangement between V. salvator and N. naja species.
The Varanus salvator genome assembly is 1.70 Gb in size, which is ~12% bigger than the genome of the Komodo dragon (Varanus komododensis) from the same genus and ~5% smaller than the green anole (Anolis carolinensis), a squamate lizard model (Supplementary Table S2). The completeness of the V. salvator assembly was comparable to other published reptile genome assemblies (Supplementary Table S2). BUSCO was also run with the same parameters on 18 reptile genomes for comparative analyses (Alligator mississippiensis, Alligator sinensis, Anolis carolinensis, Chelonia mydas, Chrysemys picta, Crocodylus porosus, Deinagkistrodon acutus, Dopasia gracilis, Eublepharis macularius, Gavialis gangeticus, Gekko japonicas, Ophiophagus hannah, Pelodiscus sinensis, Pogona vitticeps, Protobothrops mucrosquamatus, Python bivittatus, Shinisaurus crocodilurus, and Varanus komodoensis) (Supplementary Table S3). The result showed that our V. salvator draft genome has good quality and a high completeness level. Other previously reported standard genomes are presented (Figure 1B and Supplementary Table S3).
We anchored the V. salvator scaffolds into pseudochromosomes and linkage groups by RaGOO (Alonge et al., 2019) assembler using the retrieved chromosome level assembly of V. komodoensis (Lind et al., 2019) as the reference genome. This reference-guided assembly enabled us to compare the genome organization at the chromosome level among V. salvator, A. carolinensis, and G. gallus and determine their chromosomal homologies. The RaGOO assembly reduced the total number of scaffolds from 858 to 100 and increased the scaffold N50 from 71 MB to 178.2 MB with the longest scaffold of 178 Mb, producing a reference-guided assembly with a total of 8 pseudochromosomes and 16 linkage groups. For each pseudochromosome and linkage group, we determined a chromosomal equivalent to A. carolinensis and G. gallus (chicken) genomes (Supplementary Table S4, Figure 1C). Synteny analysis was carried out using Satsuma v3.1.0 (Grabherr et al., 2010) with the default parameters. Satsuma2 output of synteny blocks was plotted across pseudochromosomes using the ChromosomePaint tool to generate pseudochromosomes painted by synteny. Whole-genome synteny comparison between the V. salvator and G. gallus revealed large syntenic blocks among the micro-, macro-, and Z-chromosomes (Figure 1C). For instance, sequences equivalent to V. salvator pseudochromosome 2 are primarily on G. gallus chromosomes 3, 5, 7, and 23 (Figure 1C). Comparison of the water monitor lizard with the anole lizard also revealed numerous syntenic regions and chromosomal rearrangements. Our synteny analysis indicated that the anole chromosome 2 is equivalent to pseudochromosome 1 of the water monitor lizard (Figure 1C). We found a significant homology between the linkage group sequences of both anole and water monitor lizard genomes.
In addition, we utilized the CoGE SynMap (Lyons and Freeling, 2008) for comparative genomic analysis between V. salvator and N. naja genomes. With this analysis, we can identify the ancestral patterns of rearrangements in squamate genomes. The homologous scaffold in the Komodo dragon assembly was then assigned to the Z-linked region. Interestingly, we found several signatures of segmental duplications between pseudochromosome 1 of the monitor lizard and chromosome 2 of cobra genomes (Figure 1D). The Z chromosome of a snake was remarkably equivalent to pseudochromosome 3 of the monitor lizard. In comparison, the microchromosomes of the snake were highly syntenic to pseudochromosomes 4 and 5 of the monitor lizard. The synteny chromosome plot between V. salvator and N. naja genomes revealed a widespread inversion pattern. The large number of inversions suggest a genome reorganization based on a huge systematic difference between two species via genome evolution perspective (Kirkpatrick, 2010). To validate the gene order, we compared the 85 cDNAs of the V. salvator's pseudochromosomes and linkage groups with the previously reported V. salvator' cytogenetics map (Srikulnath et al., 2013) using BLAST (Altschul et al., 1990) (Supplementary Table S5). We found that the largest conserved gene order is a set of six genes (ATP5A1, GHR, CHD1, DMRT1, RPS6, and ACO1), which are located on chromosome 1p and pseudochromosome 1 according to the previous cytogenetic map Srikulnath et al. (2013) and this study, respectively. The linkage group 1, linkage group 5 and linkage group 8 are linked to the V. salvator chromosome 8 of Srikulnath et al. (2013). However, not all the genes were allocated in the corresponding chromosome, suggesting possible errors during the assembly or reference-guide scaffolding.
We utilized the RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) for run de novo repeat annotation and constructed species-specific repeat sequence libraries for the V. salvator. We then used the sequence libraries as a query to mask repetitive elements with RepeatMasker (http://www.repeatmasker.org). We found that repetitive elements accounted for 37% of the V. salvator genome (Figure 2A). The Interspersed Repeat Landscape suggested two peaks which indicated at least two transposition/TE burst events along with the V. salvator genome evolution. The first peak, the younger TE, is mostly from LINEs categories, including L1, RTE, CR1, and L2. The second peak indicates older TE activity with LINEs as the dominant group, followed by SINEs, LTRs, and DNAs. The proportion of LINEs in the V. salvator genome is 16.76% (the total TE is 35.99%) (Supplementary Table S6) which is higher than other squamate relatives such as V. komodoensis (LINE; 13.43% of the 32.18%) and A. carolinensis (LINE; 12.19% of the 33.82%).
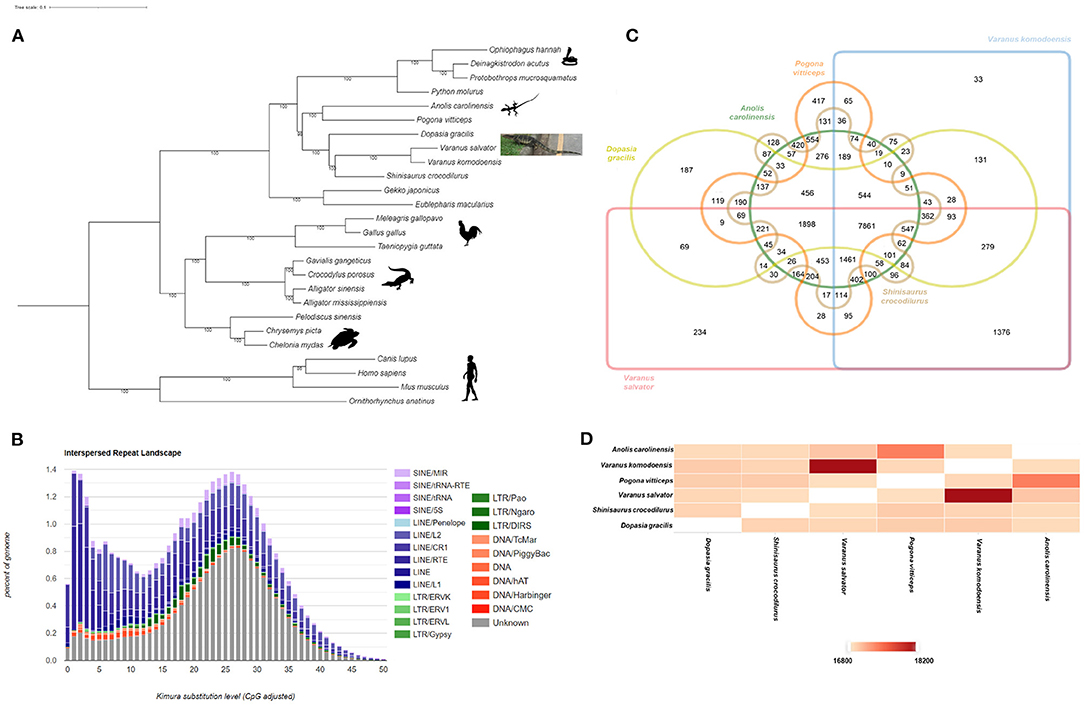
Figure 2. Phylogenetics, Interspersed Repeat Landscape and gene family analysis of V. salvator. (A) Phylogenetic relationships of Varanus salvator with 21 non-avian reptiles and 4 outgroups, inferred by IQ-TREE from 1,339 one-to-one orthologous proteins. Bootstrap values (10,000 pseudoreplicates) are shown at the nodes. All silhouettes reproduced from PhyloPic. Credits: Ophiophagus hannah silhouette, V. Deepak under a Creative Commons license CC BY 3.0. (B) Interspersed Repeat Landscape. The y axis shows the genome percent of each repeat families, the x-axis shows the level of kimura substitution of each repeat families. (C) Venn diagram showing the distribution of gene families (orthologous clusters) among six species including two members of the order Squamata (A. carolinensis and P. vitticeps) and four closely related members of the suborder Anguimorpha (V. salvator, V. komodoensis, S. crocodilurus, D. gracilis) (D) The heatmap of overlapping cluster numbers of protein that were shared each pair of genomes. Each cell indicates the overlapping cluster numbers between each pair of species.
For protein-coding gene analysis, we identified 21,937 protein-coding genes based on the combination of ab initio gene prediction by MAKER version 2.31.10 (Cantarel et al., 2008) pipeline and homology search by SNAP and Augustus version 3.3.1. Using InterProScan v5.30-69.0 (Quevillon et al., 2005), orthology assignments and predictions of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were carried out through the KEGG Automatic Annotation Server (KAAS) using bi-directional best hit (BBH) BLAST methods (https://www.genome.jp/kegg/kaas/). We were able to get functional annotations from 14,668 of 21,937 protein-coding genes. All functional annotated genes could be grouped into 32 KEGG pathways and 5 KEGG classifications (Supplementary Figure S3). According to the KEGG pathway analysis, the endocrine system (921 genes) and the immune system (919 genes) were the two dominant pathways in the organismal system.
We constructed a phylogenetic tree to validate the genome evolution of V. salvator macromaculatus (Figure 2B) by IQ-TREE version 2.0.3 (Nguyen et al., 2015) (bootstrap value set at 10,000). The IQ-TREE input data was the multiple sequence alignment from the PRANK v.170427 (Loytynoja and Goldman, 2005) using 1,339 single-copy orthologous genes, which were analyzed with OrthoFinder version 2.3.12 (Emms and Kelly, 2015) by comparing the whole genome sequences of 19 non-avian reptile species, four mammals (M. musculus, C. familiaris, H. sapiens, and O. anatinus), and three avian species (T. guttata, M. gallopavo, and G. gallus). According to the tree, V. salvator was most closely related to V. komodoensis with high bootstrap support (100%); both are the Varanus genus of the Varanidae family. In addition, a phylogenetic tree (Supplementary Figure S4) with evolution time between species studied was obtained from TimeTree (Kumar et al., 2017). Toxicofera is a clade of squamata that includes the Ophidia, Anguimorpha, and Iguania suborders. The time of divergence between V. salvator and V. komodoensis was estimated to be around 55 MYA (Supplementary Figure S4).
We use Orthovenn2 (Xu et al., 2019) to identify species-specific genes, presence/absence of gene families, and pathway enrichment among the six genomes of the same clade as V. salvator including four genomes of the Anguimorpha suborder (V. salvator, V. komodoensis, S. crocodilurus, and D. gracilis), and two members of the squamata order (A. carolinensis and P. vitticeps). The selected genome formed 21,221 clusters of 18,687 orthologous clusters (containing two species at least) and 2,534 single-copy gene clusters. The Venn diagram shows that 7,861 gene families were shared among the six genomes supporting their conservation in the lineage (Figure 2C). A total of 234 out of 7,861gene families are specific to V. salvator. We were able to annotate 122 out of 234 clusters according to the Swiss Protein Database, including 94 clusters (Supplementary Table S7) in biological processes (BP), 19 in molecular functions (MF), and 9 in cellular components (CC). For pathway enrichment analysis, the gene family encoding vomeronasal type 2 receptors (V2Rs) played a significant role in response to pheromones (GO:0019236, GO level: BP, P = 1.37e−11) (Supplementary Table S8). The histocompatibility antigen is essential for immune response (GO:0006955, GO level: BP, P = 3.89e−09) (Supplementary Table S8). Figure 2D shows the pairwise heatmap based on the similarity matrix, which illustrates the overlapping cluster numbers for the six genomes. Both V. salvator and V. komodoensis shared the highest number of ortholog clusters (18,199), with evidence of functional enrichment shared gene families between both genomes (Supplementary Table S9). All results emphasized the close relationship between V. salvator and V. komodoensis.
To determine the protein evolution in the V. salvator genome, we validated the positive selection with 1,339 one-to-one orthologs from one the six genomes of the same clade as V. salvator. Protein sequences were aligned using PRANK (Loytynoja and Goldman, 2005), and codon alignments were generated using PAL2NAL (Suyama et al., 2006). To obtain the ratio of non-synonymous and synonymous mutations (dN/dS ratio) and its associated p-value, the aligned 1,339 one-to-one orthologs from the six genomes were analyzed through the v2.5.15 Hyphy package's aBSREL model (adaptive Branch-Site Random Effects Likelihood) which fixes branch lengths based on the constructed phylogeny. A gene would be considered under positive selection for the Varanus salvator lineage if a p-value was lower than 0.05. Our results found 262 genes with a positive selection signal in V. salvator (Supplementary Table S10). To determine the Gene Ontology (GO) and gene set enrichment analysis of the metabolic pathway, the 262 positive selection genes were analyzed through PANTHER database (Thomas et al., 2003). Pathways with p <0.05 from Fisher's exact tests were then selected as enriched pathways in V. salvator. We found evidence for pathway enrichment across the regulators of blood coagulation with a raw p = 0.027 from the PANTHER pathway analysis (Thomas et al., 2003) and a raw value = 0.0426 from the Toll receptor signaling pathway (Supplementary Table S11). For the blood coagulation pathway genes F3, PLAU, and PLAT genes, the dN/dS ratios were 5.26, 4.19, and 9.89, respectively. Within the Toll receptor signaling pathway, which is in a family of evolutionarily strong conserved innate immune receptors, there were three genes (TAB1, CHUNK (IKK-α), and NFKBIE) with dN/dS ratios of 1.09, 1.29, and 1.53, respectively. The coagulation was part of the innate immune system which is the most critical pathway during activation of the early host response mechanism (van der Poll and Herwald, 2014). We also found a positive selection in V. salvator genome within the genes controlling blood clotting and innate immunity genes. Our findings support the development of a robust immune system in V. salvator via positive selection.
From our knowledge, this is only the second draft genome of the Varanus genus published. We generated the high-quality draft genome assembly from Pacbio long reads and the 10× Linked-Read Illumina short reads. The final size of the reference genome assembly of V. salvator was 1.7 Gb with an N50 scaffold size of 71 Mb featuring 91.6% completeness of the genome assembly. As expected, our comparative genomic analysis revealed that V. salvator is closely related to V. komodoensis. We also found a positive selection in V. salvator genome within the genes controlling blood clotting and innate immunity genes. Our findings support the development of a robust immune system in V. salvator via positive selection. In conclusion, these V. salvator draft genome data are expected to provide helpful information for future study and contribute significantly to understanding the evolution and adaptation of the Varanus salvator.
All data have been deposited at NCBI under BioProject PRJNA766478, biosample SAMN21848819. Raw reads (Illumina and Pacbio) are available in the Sequence Read Archive database under SRR16080541 (Pacbio data) and SRR16080542 (10X genomic data) accordingly. The genome assembly has been deposited at NCBI under Accession No. JAIXND000000000.
The animal study was reviewed and approved by the Sri Nakhon Khuean Khan Park (Royal Forest Department, Ministry of Natural Resources and Environment) and Kasetsart University (0909.6/15779).
WC performed analysis and drafted the manuscript. WS prepared specimens and contributed content on the manuscript. CS and AA performed the DNA extraction and sequencing. KS provided a specimen, participated in its design, and helped to draft the manuscript. SA participated in the analysis design, performed the chromosomal analysis and coordination, and helped to draft the manuscript. CP participated in its design, performed analysis and coordination, and final edit of the manuscript. VS conceived the study, participated in its design and coordination, and helped draft the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by Health Systems Research Institute (65-040), TSRI Fund (CU_FRB640001_01_30_10), and the Second Century Fund (C2F), Chulalongkorn University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.850817/full#supplementary-material
Akbar, N., Siddiqui, R., Sagathevan, K., Iqbal, M., and Khan, N. A. (2019). Gut bacteria of water monitor lizard (Varanus salvator) are a potential source of antibacterial compound(s). Antibiotics. 8, 164. doi: 10.3390/antibiotics8040164
Alonge, M., Soyk, S., Ramakrishnan, S., Wang, X., Goodwin, S., Sedlazeck, F. J., et al. (2019). RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20, 224. doi: 10.1186/s13059-019-1829-6
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Cantarel, B. L., Korf, I., Robb, S. M., Parra, G., Ross, E., Moore, B., et al. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Chaiprasertsri, N., Uno, Y., Peyachoknagul, S., Prakhongcheep, O., Baicharoen, S., Charernsuk, S., et al. (2013). Highly species-specific centromeric repetitive DNA sequences in lizards: molecular cytogenetic characterization of a novel family of satellite DNA sequences isolated from the water monitor lizard (Varanus salvator macromaculatus, Platynota). J. Hered. 104, 798–806. doi: 10.1093/jhered/est061
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D., and Emerson, J. J. (2016). Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic. Acids Res. 44, e147. doi: 10.1093/nar/gkw654
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Gazquez-Gutierrez, A., Witteveldt, J., R Heras, S., and Macias, S. (2021). Sensing of transposable elements by the antiviral innate immune system. RNA. 27, 735–752. doi: 10.1261/rna.078721.121
Grabherr, M. G., Russell, P., Meyer, M., Mauceli, E., Alfoldi, J., Di Palma, F., et al. (2010). Genome-wide synteny through highly sensitive sequence alignment: satsuma. Bioinformatics. 26, 1145–1151. doi: 10.1093/bioinformatics/btq102
Guan, D., McCarthy, S. A., Wood, J., Howe, K., Wang, Y., and Durbin, R. (2020). Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898. doi: 10.1093/bioinformatics/btaa025
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Kirkpatrick, M. (2010). How and why chromosome inversions evolve. PLoS Biol. 8, e1000501. doi: 10.1371/journal.pbio.1000501
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome. Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lind, A. L., Lai, Y. Y. Y., Mostovoy, Y., Holloway, A. K., Iannucci, A., Mak, A. C. Y., et al. (2019). Genome of the Komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nat. Ecol. Evol. 3, 1241–1252. doi: 10.1038/s41559-019-0945-8
Loytynoja, A., and Goldman, N. (2005). An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. U S A. 102, 10557–10562. doi: 10.1073/pnas.0409137102
Lyons, E., and Freeling, M. (2008). How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 53, 661–673. doi: 10.1111/j.1365-313X.2007.03326.x
Marcais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770. doi: 10.1093/bioinformatics/btr011
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pryszcz, L. P., and Gabaldon, T. (2016). Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44, e113. doi: 10.1093/nar/gkw294
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Ranallo-Benavidez, T. R., Jaron, K. S., and Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432. doi: 10.1038/s41467-020-14998-3
Research_and_Innovation_for_Sustainability_Center (2021). Water Monitor a Scavenger for Environment. Available online at: https://risc.in.th/knowledge/water-monitor-a-scavenger-for-environment (accessed Febuary 13, 2022).
Rhie, A., Walenz, B. P., Koren, S., and Phillippy, A. M. (2020). Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245. doi: 10.1186/s13059-020-02134-9
Rios, F. M., and Zimmerman, L. M. (2015). “Immunology of Reptiles,” in Encyclopedia of Life Sciences (John Wiley & Sons, Ltd).
Shine, R., Harlow, P. S., Keogh, J. S., and Boeadi (1996). Commercial harvesting of giant lizards: the biology of water monitors Varanus salvator in southern Sumatra. Biol. Conservat. 77, 125–134. doi: 10.1016/0006-3207(96)00008-0
Srikulnath, K., Uno, Y., Nishida, C., and Matsuda, Y. (2013). Karyotype evolution in monitor lizards: cross-species chromosome mapping of cDNA reveals highly conserved synteny and gene order in the Toxicofera clade. Chromosome Res. 21, 805–819. doi: 10.1007/s10577-013-9398-0
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–612. doi: 10.1093/nar/gkl315
Thomas, P. D., Kejariwal, A., Campbell, M. J., Mi, H., Diemer, K., Guo, N., et al. (2003). PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31, 334–341. doi: 10.1093/nar/gkg115
Uyeda, L. T. (2015). The Water Monitor Lizard Varanus salvator: Behavior, Ecology, and Human Dimensions in Banten, Indonesia. Seattle: University of Washington.
van der Poll, T., and Herwald, H. (2014). The coagulation system and its function in early immune defense. Thromb. Haemost. 112, 640–648. doi: 10.1160/TH14-01-0053
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 9, e112963. doi: 10.1371/journal.pone.0112963
Keywords: Varanus salvator, Asian water monitor lizard, draft genome assembly and annotation, PacBio sequencing, 10× linked-read sequencing
Citation: Chetruengchai W, Singchat W, Srichomthong C, Assawapitaksakul A, Srikulnath K, Ahmad SF, Phokaew C and Shotelersuk V (2022) Genome of Varanus salvator macromaculatus (Asian Water Monitor) Reveals Adaptations in the Blood Coagulation and Innate Immune System. Front. Ecol. Evol. 10:850817. doi: 10.3389/fevo.2022.850817
Received: 08 January 2022; Accepted: 12 May 2022;
Published: 09 June 2022.
Edited by:
Iker Irisarri, University of Göttingen, GermanyReviewed by:
José Ramón Pardos-Blas, National Museum of Natural Sciences (CSIC), SpainCopyright © 2022 Chetruengchai, Singchat, Srichomthong, Assawapitaksakul, Srikulnath, Ahmad, Phokaew and Shotelersuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Syed Farhan Ahmad, c3llZGZhcmhhbi5hQGt1LnRo; Chureerat Phokaew, Y2h1cmVlcmF0LnBAY2h1bGEuYWMudGg=
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.