
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 01 April 2022
Sec. Population, Community, and Ecosystem Dynamics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.837381
This article is part of the Research Topic Insights into Bee Diseases and Bee Health View all 6 articles
 D. N. Ganeshprasad1
D. N. Ganeshprasad1 Jafar K. Lone1
Jafar K. Lone1 Kunal Jani2
Kunal Jani2 Yogesh S. Shouche2
Yogesh S. Shouche2 Khalid Ali Khan3Samy Sayed4Mustafa Shukry5Showket A. Dar6
Khalid Ali Khan3Samy Sayed4Mustafa Shukry5Showket A. Dar6 Muntazir Mushtaq7*
Muntazir Mushtaq7* A. H. Sneharani1*
A. H. Sneharani1*
Honeybees are eusocial insects with close interaction with their surrounding environment. Gut microbiota in honeybees play a significant role in host health, biology, and interaction behavior with the surrounding environment. Apis florea, a wild bee, is the most primitive among all honeybees and is indigenous to the Indian subcontinent. Previous reports on reared honeybee species provide information on the gut microbiome. No such studies are reported on the gut microbiota of the wild honeybee species. This study aimed at studying the gut microbiome of the wild honeybee species, A. florea. The study reports the analysis and the identification of gut bacteria in the wild honeybee species, A. florea, employing culture-based and culture-independent methods. Cultured bacteria were identified and characterized by MALDI-TOF MS and 16S rRNA sequencing. A comprehensive analysis and identification of non-culturable bacteria were performed by 16S rRNA amplicon next-generation sequencing. This approach splits gut bacteria into four bacterial phyla, four families, and 10 genera in major. The dominant taxa identified in A. florea belonged to the family Enterobacteriaceae (79.47%), Lactobacillaceae (12.75%), Oxalobacteraceae (7.45%), and Nocardiaceae (0.13%). The prevailing bacteria belonged to Enterobacter, Lactobacillus, Escherichia-Shigella, Massilia, Klebsiella, Citrobacter, Pantoea, Serratia, Rhodococcus, and Morganella genera, belonging to phyla Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes. This study observed the occurrence of a few bacteria that are not previously reported for their occurrence in other species of the Apis genus, making this investigation highly relevant with regard to the bee microbiome.
Honeybees are fascinating and vivid creatures with a complex social system that has close interaction with their surrounding environment making them superorganisms. Fossil records indicate that among any other living species of Apis, Apis florea is the most primitive. This is substantiated by their colony size, open nest, and behavioral pattern (Radloff et al., 2005; Kaspi and Shafir, 2013). The evolutionary divergence of A. florea from the common ancestor shows it to be much older than the remaining Apis clades (Biewer et al., 2016). These bees are prevalent and restricted to southern and south-eastern parts of Asia. A. florea is commonly known as dwarf bee or little bee and endures temperatures of up to 50°C (Biewer et al., 2016). These bees produce honey in smaller quantities and often consume it, and hence, they are not reared for commercial activity. Their contribution to pollination is agriculturally and ecologically significant rather than their role in producing honey. Depending upon forage availability, A. florea often migrates between plains and adjacent low-lying slopes during different seasons. The species prefers a warmer climate for their foraging activity (Balachandra et al., 1999) and functions as a significant pollinator.
In recent times, honeybees have experienced an uncommon decline in their population. Various reasons have been proposed for extreme honeybee losses. These include indiscriminate use of toxic pesticides, poor nourishment, genetic diversity, parasites, and microbial pathogens (Anjum et al., 2018). Ecosystems and species with greater biodiversity have more stability. For example, species with significant genetic diversity and diverse populations that are adapted to a wide range of environments are more likely to withstand weather disturbances, disease, and climate change (The Elements of Biodiversity, 2021). Accumulated evidence suggests a crucial role for host–microbe interactions (Saraithong et al., 2015). Microbes contribute to the functional competencies of the host by making essential nutrients available and confer immunological defense to the honeybee. Diversity and sociality have an effect on an organism’s physiological and behavioral adaptations. Gut microbiome in the honeybee protect against attacking pathogens (Kwong and Moran, 2016). Some studies showed that the relative abundance of core gut species has a direct impact on susceptibility to various pathogens (Raymann and Moran, 2018). The functions of gut bacteria are delineated by studying the interactions between gut bacteria and the host. The gut bacterial diversity of Apis mellifera (western honeybee) is well-demonstrated. Different social corbiculate bees possess highly characteristic gut communities. Invertebrate animal hosts provide models that are more tractable since their gut microbial populations are often simple. These microbial communities, however, are often variable in their composition and are dominated by opportunistic environmental microorganisms (Cilia et al., 2020b), rather than by specialized microbial symbionts that are specific to the host environment, like those found in mammalian guts (Chandler et al., 2011; Engel and Moran, 2013b; Kwong and Moran, 2016). These distinct gut communities inside the host are in large part independent of the geographical occurrence of the host (Martinson et al., 2011; Moran et al., 2012; Kwong et al., 2017). The composition of the microbiota in the adult gut changes little, despite seasonal variations and modifications in nutrition, behavior, and gene expression that occur when workers transition from nursing bees to foragers (Martinson et al., 2012; Corby-Harris et al., 2014). However, high strain-level diversity is reported within the gut microbiota of A. mellifera and Apis cerana, providing insights into the gut microbiota functioning and host-symbiont interactions (Ellegaard et al., 2020).
Unfortunately, the majority of microorganisms are unculturable and, hence, cannot be analyzed using culture-based techniques. Culture-based approaches have several drawbacks, particularly when it comes to determining the number of bacteria; they are known to be biased because bacteria can only be cultivated if their metabolic and physiological requirements can be mimicked in vitro (Nadkarni et al., 2009). Culture-independent techniques have recently been successful in gaining access to previously unexplored genomic resources. However, culture-based approaches cannot fully exploit such resources because the vast majority of microorganisms are unculturable in the laboratory (Zapka et al., 2017).
The microbiome of the majority of wild honeybee species has a complex composition and functions and must be elucidated (Engel et al., 2016). The demand for such a study emerges since the gut microbiome play a critical role in the host health, and they show the relationship between host health in both managed and wild pollinator bees (Engel et al., 2016). Despite surging reports on gut microbiota, there is a lack of information on the gut microbiota of A. florea. Since A. florea is an essential contributor to crop pollination, this study aimed to explore and characterize the gut microbiota in the whole alimentary canal of A. florea. For a culture-based approach, isolates were subjected to MALDI-TOF-MS and 16S rRNA sequencing, and to characterize the non-culturable organisms, a high-throughput sequencing technique was used. The study provides insights into this primitive clade of Apis. This investigation adds to the knowledge on bee microbiome to unravel the evolution and ecology of host–microbiome interactions (Engel et al., 2016).
To study the cultivable and uncultivable bacteria of dwarf bee gut, a total of one hundred twenty worker bees (A. florea) from three bee colonies were collected from Mavina Halla Forest, Kodagu, Karnataka (Latitude:12.55821 DMS N 12° 33′ 29.556″, Longitude:75.95338 DMS E 75° 57′ 12.167″ and Latitude:12.56614 DMS N 12° 33′ 58.104″, Longitude:75.94256 DMS E 75° 56′ 33.216″) and Ponnampet, Kodagu, Karnataka (Latitude:12.14455 DMS N 12° 8′ 40.38″, Longitude: 75.9382 DMS E 75° 56′ 17.519″) located in Western Ghats forest of India (Figure 1). The honeybees were identified and authenticated by Dr. R. N. Kencharaddi, Asst. Professor of Agricultural Entomology, Dept. of FBTI, College of Forestry, Ponnampet, Karnataka, India. Live bees were transported to the laboratory in small cages on the same day; the bees from all three colonies were pooled, and whole bees were cold-anesthetized and were surface-sterilized with 7% sodium hypochlorite and 70% ethanol in sterile falcon tubes (Inglis et al., 2012), followed by washing four times with sterile phosphate-buffered saline (PBS). They were subsequently dissected. The whole alimentary canals of bees were aseptically dissected on slides using normal saline (0.9%) by stabbing the bee thorax with a sterile needle tip, and with sterile microforceps, the complete alimentary canal was gently pulled off in a single motion (Coleman et al., 2007). The dissected guts were transferred to PBS and immediately stored at −20°C until further experiments (Anjum et al., 2018).
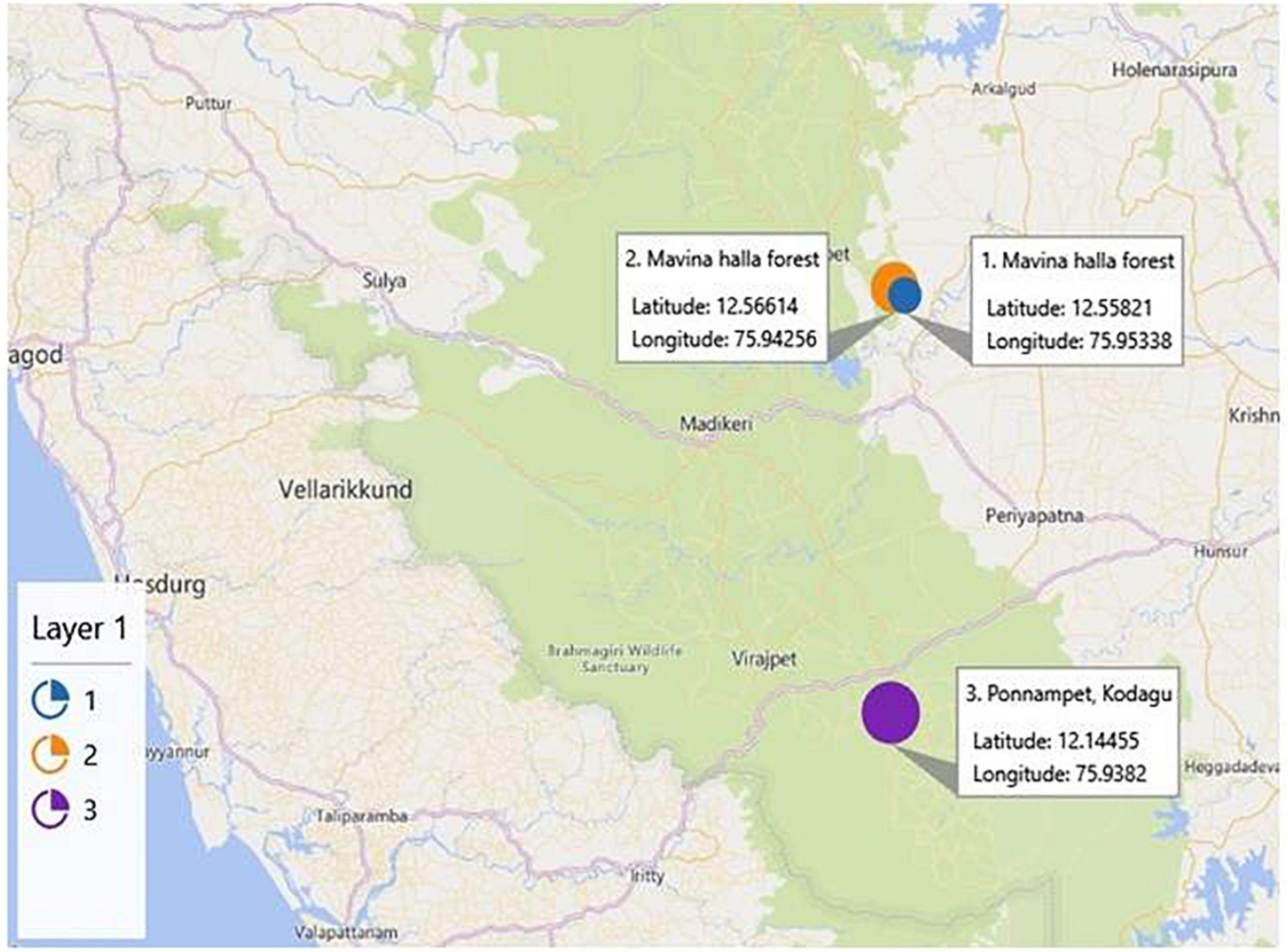
Figure 1. Representation of the geographical location of sampling sites, namely, Mavina Halla forest (1 and 2) and Ponnampet (3), of Kodagu district located in the Western Ghats forest of India (courtesy: Google maps).
The bee gut samples (n = 90) were homogenized in 1.0 ml PBS using a micropestle. Different dilutions (i.e., 1/10, 1/100, and 1/1,000) of this composite homogenate were made, and 100 μl aliquots each of the diluted samples were inoculated aerobically into six different media, namely, Nutrient agar (NA), De Man, Rogosa and Sharpe (MRS) agar, brain heart infusion (BHI) agar, eosin methylene blue (EMB) agar, Luria Bertani (LB) agar, and gluconobacter (GB) agar (HiMedia, Mumbai, India), and incubated for 24–72 h at 30°C. The bacterial colonies grown on the plates were enumerated and selected based on different morphologies. The separated colonies in master plates were repetitively subcultured to obtain pure colonies of the bacteria.
A simple extraction protocol was employed to analyze the bacterial sample. A loopful of actively grown bacterial cultures was mixed thoroughly with ethanol (70% v/v), and the suspended cells were centrifuged at 12,000 rpm for 5 min. The pellet was recovered by discarding the supernatant carefully. The pellet was air-dried at room temperature and resuspended in formic acid (70% v/v) by vigorous mixing followed by the addition of acetonitrile. The mixture was centrifuged at 12,000 rpm to separate the pellet, and 1 μl of clear supernatant was placed on a MALDI target plate. The bacterial smear was overlaid with 1 ml of saturated solution of alpha-cyano-4-hydroxycinnamic acid (HCCA) matrix prepared in acetonitrile (50%) and trifluoroacetic acid (2.5%) and was allowed to dry at room temperature. The extracted samples were analyzed using the Autoflex speed system (Bruker Daltonik GmbH, Germany). Mass spectra were obtained in a mode of linear positive ion extraction at a laser frequency of 1,000 Hz in a mass range of 2 k–20 kDa. The voltages of ion source 1, ion source 2, and lens were 19.5, 18.2, and 7 kV, respectively, and the extraction delay time was 240 ns. The calibration of the spectra was done externally by using the standard calibration mixture (Escherichia coli extracts including RNase A and myoglobin as additional proteins, Bruker Daltonics). MALDI Biotyper software 3.0 (Bruker Daltonik) was used to identify the bacterial isolates and to visualize the mass spectra. Species-level identity has been considered for the isolates with the biotyper score value ≥2.0, while the analysis for the isolates with score values ranging from 1.7 to 1.99 was repeated to achieve higher score values (Kurli et al., 2018). The isolates that were not identified up to the species level and with the biotyper score value <2.0 were subjected to 16S rRNA sequencing.
The 16S rRNA gene sequence was amplified using universal primers (8F: 5′-AGAGTTTGATCCTGGCTCAG-3′ and 1391R: 5′-GACGGGCGGTGTGTRCA-3′) according to the method of Turner et al. (1999). These primers are specific for conserved regions of bacterial 16S ribosomal RNA (Lane, 1991). The PCR conditions are as follows: Step 1: predenaturation: 94°C, 5 min; denaturation: 94°C, 1 min; annealing: 55°C, 1 min; extension: 72°C, 1.30 min; Step 2: final extension: 72°C, 7 min; stored at 4°C. PCR products (3 μl) were subjected to electrophoresis, and 22 μl was purified by the PEG-NaCl method (Sowani et al., 2016). The purified PCR products were sequenced using ABI PRISM Big Dye Terminator v3.1 Cycle Sequencing kit on a 3730xl Genetic Analyzer (Thermo Fisher Scientific®, United Kingdom).
The sequence data obtained were assembled and analyzed using the DNA sequence-assembling software Lasergene SeqMan Pro (DNASTAR Inc., Madison, WI, United States). The 16S rRNA sequence of each bacterial isolate was compared using BLAST (Camacho et al., 2009) against the 16S ribosomal RNA sequence (Bacteria and Archaea) database (a subdivision of GenBank).
The dwarf bees were surface-sterilized, and the alimentary tracts of the worker dwarf bees (30) were collected in 1.0 ml of PBS. To this, 200 μl of cell lysis buffer (ATL buffer, Genomic DNA extraction, Qiagen Tissue kit, United States) was added and subjected to homogenization in the presence of glass beads using the temperature-controlled (∼22°C) vortex shaker for 30 min. Of note, 20 μl of proteinase K were added, and further steps followed were as per the manufacturer’s protocol. The concentration of DNA was measured using a Nano-DropND-1000 spectrophotometer (Nano Drop Technologies, Willington, United States), and the quality was checked on 0.8% agarose gel, as described by Kumbhare et al. (2015). The DNA was kept at −20°C until it was further processed.
To investigate the bacterial diversity of the dwarf bee gut, next-generation sequencing (NGS) library preparation was carried out by targeting the V4 region of the 16S rRNA gene using primers 515F (F515: 5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (806R: 5′-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2010a; Kumbhare et al., 2015). The library preparation of 16S rRNA gene amplicons was performed by following the protocols by Illumina (United States). The resultant libraries were purified, pooled in equimolar concentration, and sequenced using 2 × 250 bp v2 Chemistry on Illumina MiSeq platform.
The raw reads obtained from the high-throughput sequencing were assembled using the Fast Length Adjustment of Short reads (FLASH) tool, a paired-end assembler for DNA sequences (Magoč and Salzberg, 2011), and the assembled reads were analyzed using Quantitative Insights into Microbial Ecology (QIIME v1.8) pipeline (Caporaso et al., 2010b). The operational taxonomic units (OTUs) picking was carried out by reference-based OTU picking method against SILVA (v132) (Edgar, 2010). The taxonomic assignment was performed using SILVA (v132) raw taxonomy. Alpha diversity indices were assessed by richness like Chao1 and ACE, and diversity (Shannon) was calculated via QIIME, which predisposes sample rarefaction to the same sequencing depth (Bokulich et al., 2012). In addition, the functional analysis of bacterial communities was performed using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) tool (Langille et al., 2013). It involves normalizing the 16S rRNA gene copy number followed by predicting the metagenome. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to analyze the resulting dataset. In addition, the functional contribution of each taxon was performed using a function of metagenomic contribution and quantified using a normalized dataset (Jani et al., 2018).
High-throughput sequencing and quality trimming of 16S rRNA gene yielded ∼0.121 million quality reads that were used for subsequent analysis. Taxonomic assignment of sequences with the reference database resulted in 589 OTUs. The average Good’s coverage of the sample of A. florea for 16S rRNA amplicon sequencing was found to be 99.09 ± 0.03% (mean ± SD). Alpha diversity estimation of the gut of A. florea using species richness and non-parametric Shannon index suggested higher bacterial diversity in A. florea worker bees (Table 1). Shannon index for bacterial communities was 3.121, suggestive of richness in bacterial diversity in the gut of A. florea.
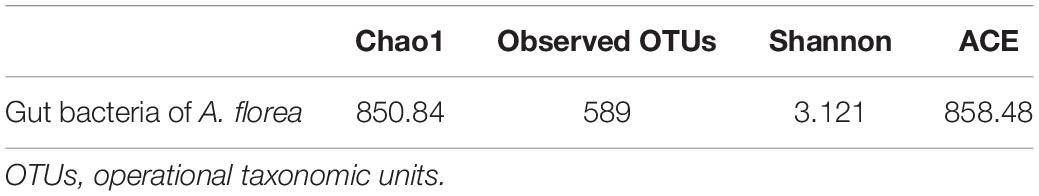
Table 1. Alpha diversity estimation and non-parametric alpha diversity in A. florea gut bacteria.
In the culture-dependent approach, the identification and characterization of the culturable diversity of bee gut bacteria were done by employing MALDI-TOF-MS and 16S rRNA gene-sequencing analysis by colony PCR. A total of 146 aerobic and facultative anaerobic bacteria were isolated from the guts of worker A. florae bees. Based on colony morphology and other bacteriological characteristics, 106 bacterial isolates were initially subjected to MALDI-TOF-MS. Seventy-one isolates were identified up to species level with the biotyper score value ≥2.0. However, isolates with the biotyper score value <2.0 could not be identified up to species level. The remaining thirty-five isolates were subjected to 16S rRNA sequencing using colony PCR. Colony PCR differs from standard PCR in the template preparation step. In addition, the primers are used specifically for colony PCR (Bergkessel and Guthrie, 2013; Kim et al., 2014). MALDI-TOF-MS and 16S rRNA sequencing of the culture-dependent analysis identified three bacterial phyla/classes: alpha-Proteobacteria (1%), Firmicutes (16%), and gamma-Proteobacteria (83%; Figure 2A). The percentages of isolates belonging to the genera Klebsiella, Enterobacter, Bacillus, Citrobacter, Staphylococcus, and Lactobacillus are represented in Figure 2B.
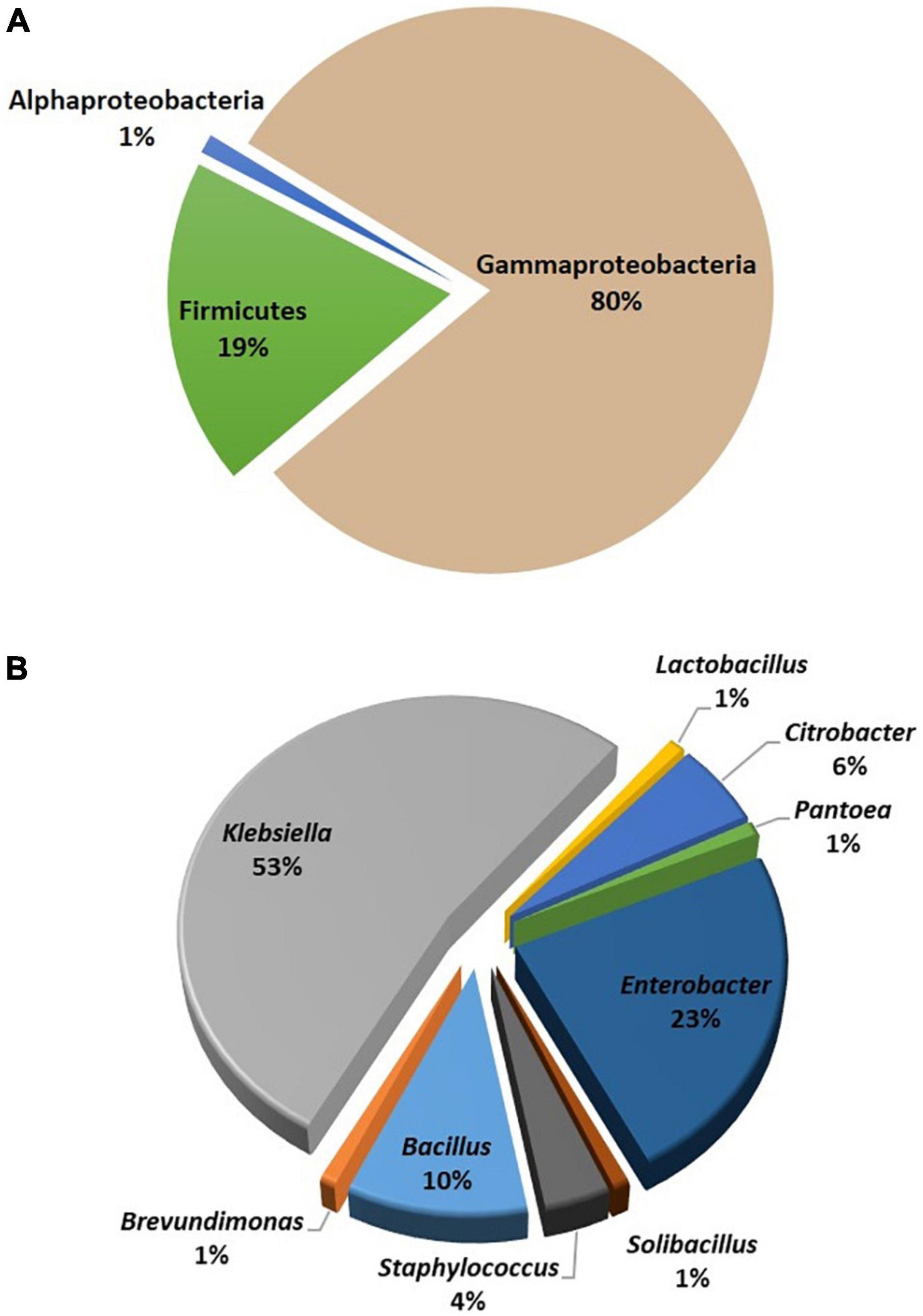
Figure 2. Classification of cultured gut bacterial flora of Apis florea. The gut bacterial isolates were classified based on MALDI-TOF-MS and 16S rRNA gene sequencing data. (A) Bacterial phyla and (B) bacterial genera.
In this culture-independent study, a comprehensive microbial diversity analysis using a high-throughput sequencing approach was carried out. The bacterial community profile of dwarf bee gut as revealed by 16S rRNA gene amplicon sequencing yielded 12 bacterial phyla representing collective phyla in the gut of A. florea (Figure 3A). The distribution of bacterial phyla is as follows: Proteobacteria (86.86%), Firmicutes (12.81%), Actinobacteria (0.27%), and Bacteroidetes (0.04%), and the rest (0.02%) constituted the minor phyla. The eight minor phyla, namely, Acidobacteria, Cyanobacteria, Chloroflexi, Verrucomicrobia, Planctomycetes, Nitrospirae, Lentisphaerae, and Tectomicrobia, were not significant in number; hence, they were not included in the percentage of composition shown in Figure 3A. Bacterial phylum Proteobacteria outnumbered other bacterial phyla. At the family level, OTUs with ≥0.1% abundance were filtered (Figure 3B), which retained eight differentially abundant families in the gut of A. florea. The family-level distribution of bacteria in the gut of A. florea is as follows: Enterobacteriaceae (79.47%), Lactobacillaceae (12.75%), Oxalobacteraceae (7.45%), Nocardiaceae (0.13%), Micrococcaceae (0.07%), Bifidobacteriaceae (0.05%), Staphylococcaceae (0.04%), and Prevotellaceae (0.04%). The composition percentage of the bacterial family, which was less than 1%, was not shown in Figure 3B.
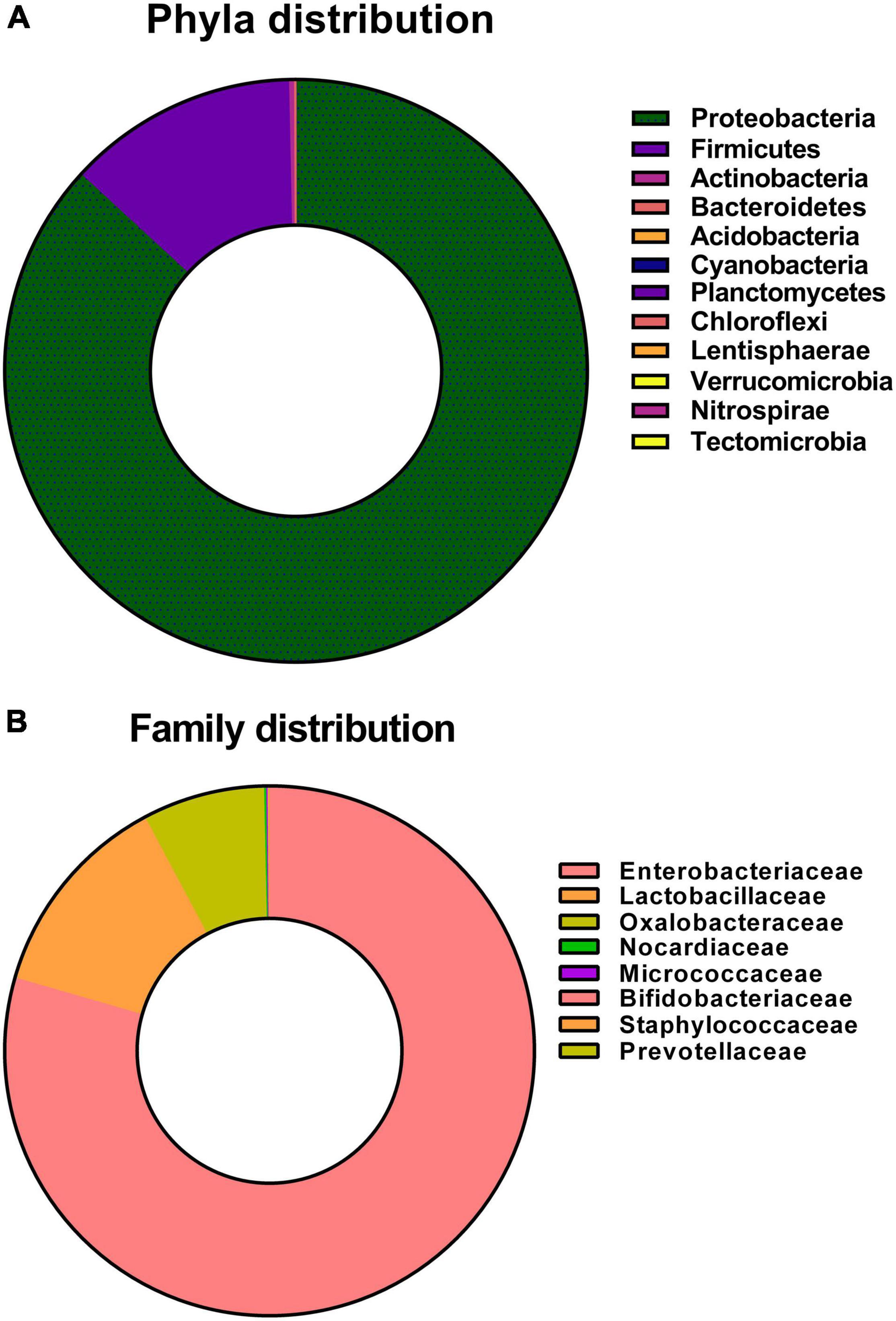
Figure 3. Next-generation sequencing of culture-independent gut bacterial flora of A. florea. Taxa distributions of (A) phylum and (B) family at different phylogenetic levels of A. florea gut bacterial flora.
At the genus level, OTUs with ≥0.1% abundance were filtered. The distribution of bacterial genera in the gut of A. florea is represented in Figure 4 and is as follows: Enterobacter (58.49%), Lactobacillus (14.47%), Escherichia-Shigella (13.29%), Massilia (8.45%), Klebsiella (1.71%), Citrobacter (1.35%), Pantoea (1.32%), Serratia (0.63%), Rhodococcus (0.15%), and Morganella (0.13%). Among these, Proteobacteria and Firmicutes were the predominant phyla. Actinobacteria and Bacteroidetes are highly diverse and are often found in the intestine, which are frequently mentioned in the literature on bee gut microbiota. In addition, we observed the presence of Tectomicrobia, Chloroflexi, Lentisphaerae, and Verrucomicrobia in the gut of A. florea.
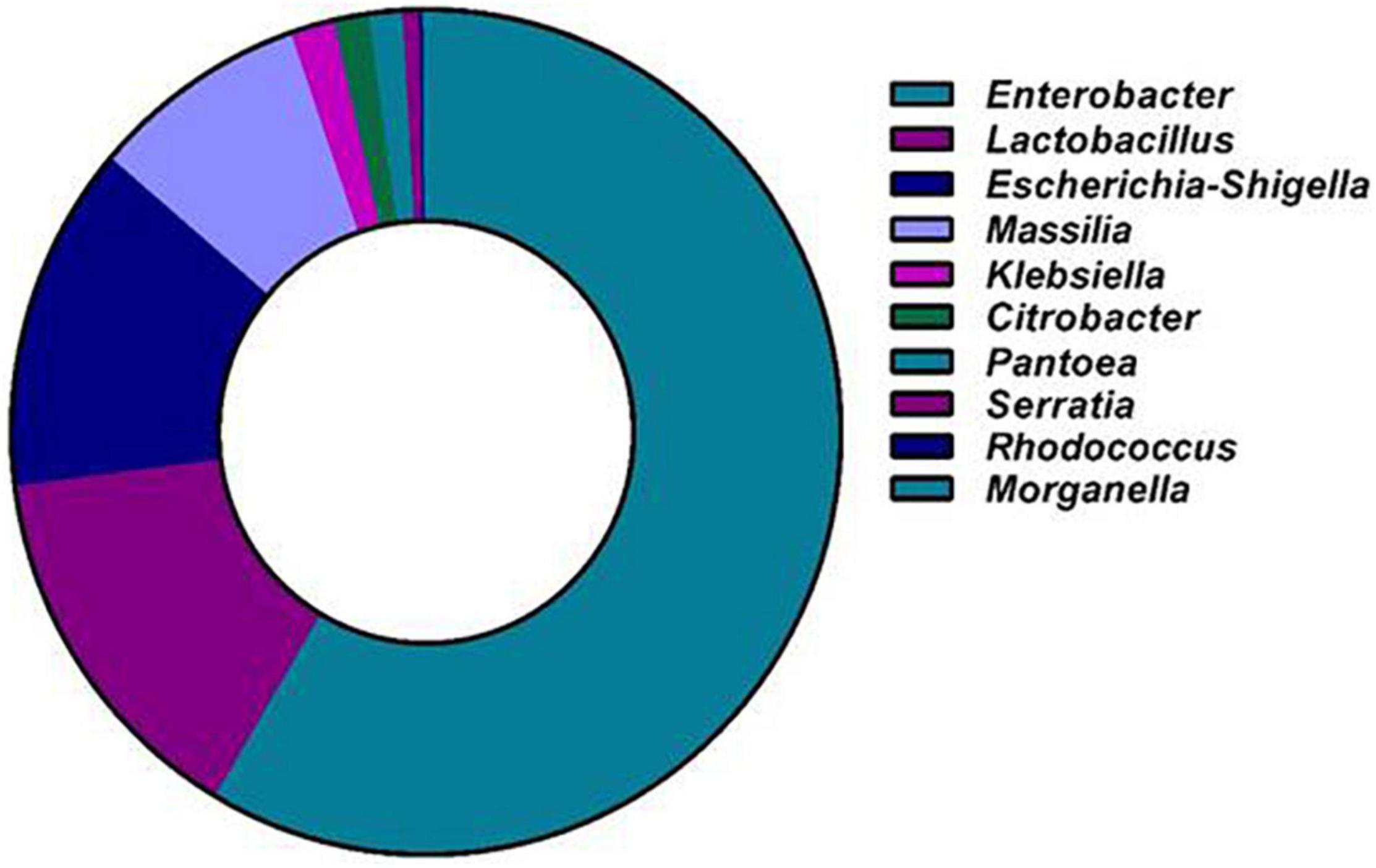
Figure 4. Next-generation sequencing of culture-independent gut bacterial flora of A. florea. Genus level distribution of A. florea gut bacterial taxa.
The 16S rRNA gene sequences of the bacterial isolates of A. florea were used to construct the phylogenetic tree by showing the relationship among the bacteria with reference strains of GenBank (Figure 5). The evolutionary history was inferred using the maximum likelihood method based on the Tamura-Nei model (Tamura and Nei, 1993). The tree with the highest log likelihood (-5509.8000) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach and then selecting the topology with a superior log likelihood value. The analysis involved 39 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 805 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 (Tamura et al., 2013). The bacterial populations in the gut were diverse among forager bees of A. florea, which belonged to the phyla Firmicutes and alpha- and beta-Proteobacteria.
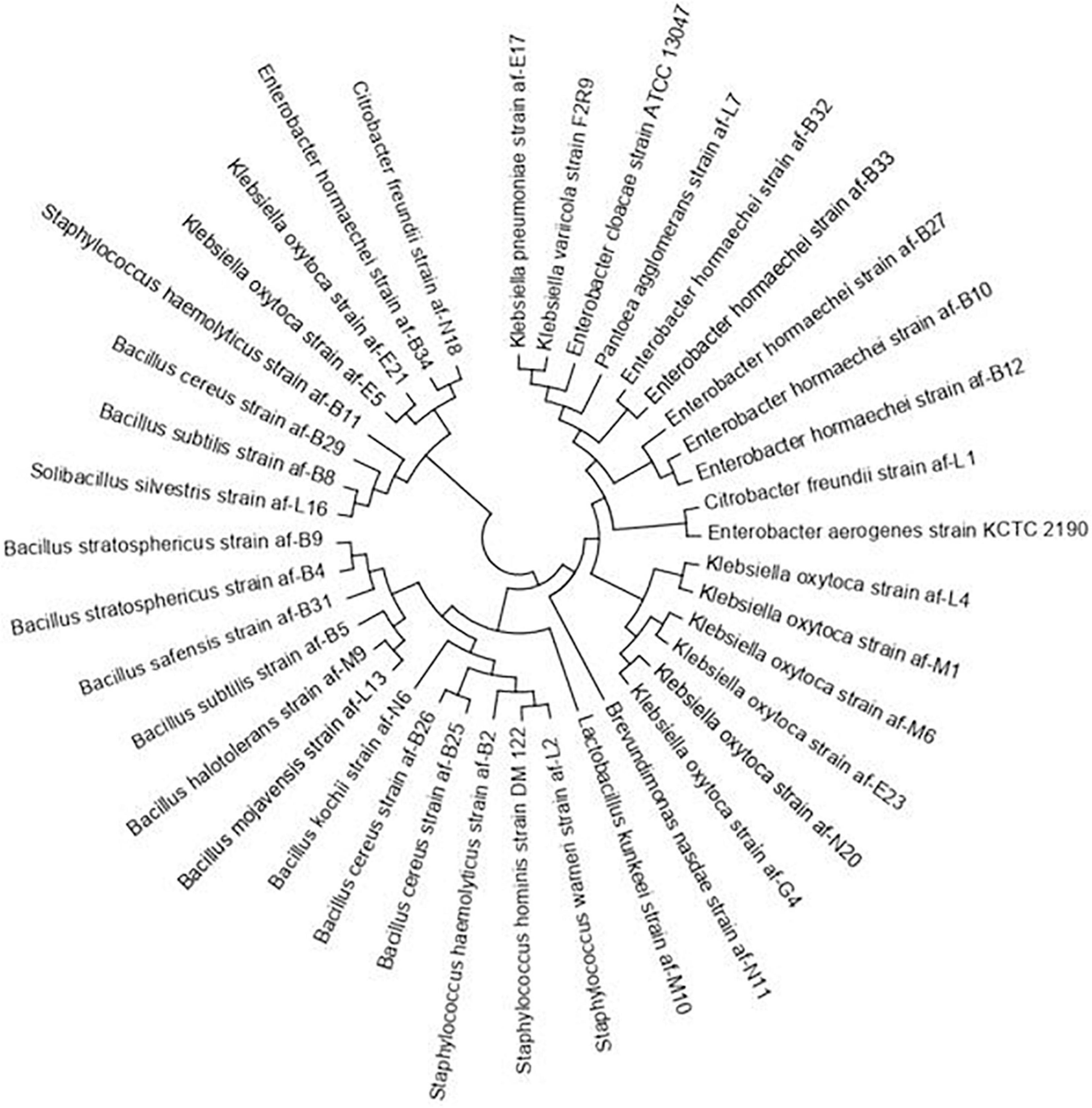
Figure 5. Phylogenetic dendrogram of the 16S rRNA sequences of A. florea gut bacteria. Evolutionary analyses were conducted in MEGA6 using the maximum likelihood method.
Functional predictions using PICRUSt2 (Langille et al., 2013) showed higher metabolism and environmental information processing in the gut of A. florea bacterial taxa, suggesting the significant contribution of these bacterial communities in the degradation of macromolecules. In addition, gene families associated with the genetic information processing and cellular processes were present (Figure 6).
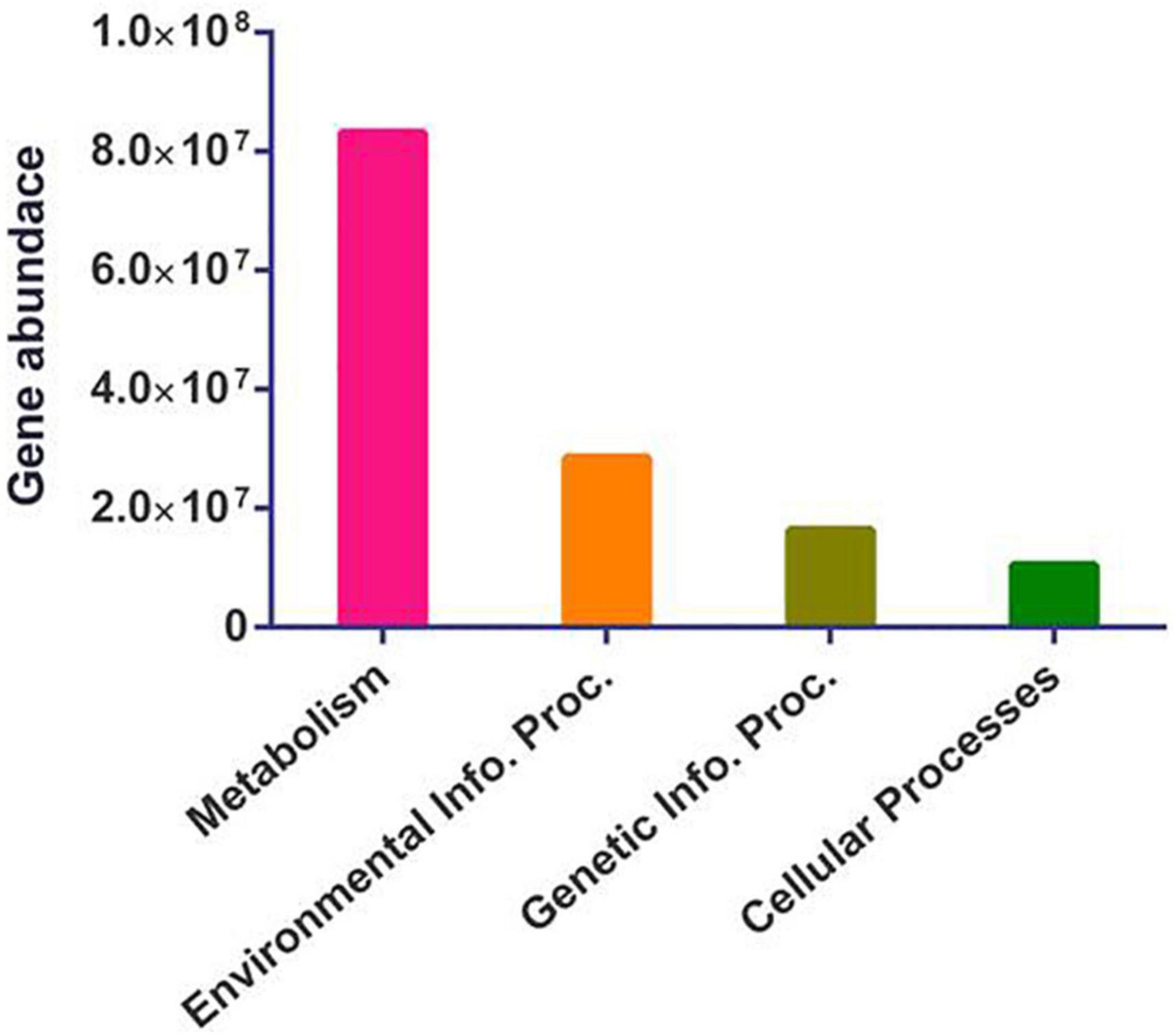
Figure 6. Functional imputations. It represents the differential abundance of metabolism, environmental information processing (Environmental Info. Proc.), genetic information processing (Genetic Info. Proc.), and cellular processes-related gene families in the gut bacterial communities of A. florea.
Diversification of individual functional modules such as metabolism represents proportionately higher carbohydrate metabolism (33.8%), followed by amino acid metabolism (15.85%), cofactor and vitamin metabolism (12.8%), nucleotide metabolism (10.05%), energy metabolism (9.5%), and lipid metabolism (5.33%). Similarly, membrane transport (62.17%), signal transduction (37.2%), translation (45.4%), replication (31%), and cellular motility (43.64%) were noted as the prominent contributing feature under environmental information processing, genetic information processing, and cellular processes.
The global decline in the population of pollinator bees (Potts et al., 2010) has attracted the attention of researchers for a comprehensive study of host-microbiome communities. As major contributors to pollination, honeybees are crucial organisms in securing agricultural produce and in maintaining the ecosystem. Unlike western bees, A. florea are not reared for commercial honey or wax; rather, they occur in the wild (Balachandra et al., 1999). The most important contribution of this dwarf honeybee is its valuable pollination activity of many flowering plants and diverse vegetation in tropical ecosystems (Soman and Chawda, 1996). These little dwarf bees differ from western bees and are known for their defense against pathogens (Suwannapong et al., 2012). The wild flies have distinct bacterial communities and have more diversification from those of reared species (Chandler et al., 2011), including bee gut bacteria (Engel et al., 2013; Kwong and Moran, 2016). Studies on the honeybee gut microbiota have found that they influence host nutrition, weight gain, endocrine signaling, immunological function, and pathogen resistance and that disrupting the microbiota can result in reduced host fitness (Zheng et al., 2018). The defensive and metabolic abilities of bees are significantly connected with the gut microbiome interactions and similarly add knowledge on the role and interaction of the microbial community with the host; hence, the gut bacteria of A. florea honeybees were analyzed. In this study, dwarf bees, A. florea, were selected to study and analyze their gut microbiome. A. florea (dwarf bee) is naturally distributed in the Indian subcontinent all through south-east Asia. Bees were obtained directly from three bee colonies across the Western Ghats forest area of Kodagu district, which is recognized as a global biodiversity hotspot in India. The complete alimentary canals of bees were processed for analysis, and the gut microbes and organisms attached to hemolymph or tissues were also analyzed. The gut bacteria of a total of 120 worker dwarf honeybees were analyzed by culture-dependent and culture-independent techniques. A culture-based approach was carried out to facilitate further characterization of cultivable bacteria involved in polysaccharide breakdown and by-products of fermentation and their possible application in pharmaceutical and nutraceutical industries and also focus on the comparison between bacterial populations that are cultivable and non-cultivable.
The bacterial isolates analyzed by the culture-dependent method are found to be common in the Apis clade (Yoshiyama and Kimura, 2009; Martinson et al., 2012; Khan et al., 2017; Cilia et al., 2020a). The presence of a diverse group of bacteria including the phyla Firmicutes and alpha- and beta-Proteobacteria suggests their ecological importance, such as antibiotic use, diet, and psychological stress (Kwong et al., 2017). Most of these bacteria belonging to the above phyla are facultative anaerobes. They ferment sugars and tolerate acidic environment. The bacterial isolates identified are common in soil and water, and a few of them are organisms of clinical relevance in humans. The occurrence of such bacteria in the insect gut is not uncommon and has been described earlier (Engel and Moran, 2013b). Staphylococcus, Enterobacter, and Klebsiella found in abundance in the gut of dwarf bee are typical inhabitants in humans too. These bacteria are considered as beneficial healthy gut microbiota in humans, insects, and other animals as they are the fermenters of sugar and are involved in the host defense mechanism (Anderson et al., 2011). Klebsiella oxytoca, a prominent organism considered as a probiotic, positively affects the health of the host by suppressing parasite colonization (Engel et al., 2013). In termite, Staphylococcus is probably involved in the degradation of cellulose (Paul et al., 1986). Klebsiella spp. (K. oxytoca) and Pantoea agglomerans were predominantly found in our analysis. P. agglomerans, significantly present in the desert locust, Schistocerca gregaria, are involved in the breakdown of dietary components, leading to the synthesis of aggregation pheromones that function in the swarming behavior of locusts (Dillon and Charnley, 2002). Bacteria gain entry into the bee gut during foraging activity (Loncaric et al., 2009). In this study, Bacillus safensis, Bacillus kochii, and Bacillus halotolerans were identified in the gut of dwarf bee. These organisms are reported to exist and to survive in extreme environments and under stress conditions (Seiler et al., 2012; Lateef et al., 2015; Zhang et al., 2018). In two separate studies that carried out profiling of bacterial community in the gut of flesh flies and floral nectar, B. safensis was identified and isolated (Fridman et al., 2012; Gupta et al., 2014). Desert locust, S. gregaria, inhabits B. safensis, which possesses high cellulolytic activity, indicating the metabolic significance of this bacterium (Nelson et al., 2021). Strains of B. safensis are reported to produce many industrially relevant enzymes, such as amylase, protease, lipase, inulinase, and chitinase (Lateef et al., 2015). The occurrence of Brevundimonas nasdae (1%) was prominent in our study. The pH of honeybee midgut is around 8, and this pH favors the optimal growth of Bacillus species and B. nasdae and may aid in degrading the carbohydrates fed by the bees. In addition to B. nasdae (Phylum: Proteobacteria), Bacillus species and Solibacillus silvestris (Phylum: Firmicutes) were predominant in the gut of A. florea. These bacterial species are hardly reported in Apis gut microbiota of other species, except for A. mellifera. The differences in the occurrence of these bacteria might be due to geographical location, might be due to existing characteristics of the environment, or may be due to the feature of A. florea species itself.
A total of one hundred twenty dwarf honeybees were included in this study. The phyla Proteobacteria was detected most frequently in both culture-dependent (84%) and culture-independent (87%) approaches followed by Firmicutes (culture-dependent approach 16% and culture-independent approach 13%). At the genus level, Klebsiella (53%) and Enterobacter (23%) were found to be predominant in the culture-dependent approach, whereas in the culture-independent approach, Enterobacter (59%) and Lactobacillus (14%) were found to be predominant. However, the composition of gut bacterial flora changes less within the species of habitat-specific bees, and in this study, the variation among individual bees could not be analyzed due to the pooling of samples. The bacteria found using culture-dependent and culture-independent approaches were significantly overlapped. The analysis of gut flora in A. florea was rather complementary in both approaches. Culture-independent approaches were found to be superior in detecting gut inhabitants of A. florea.
Enterobacter, Lactobacillus, Escherichia-Shigella, Massilia, Klebsiella, Citrobacter, Pantoea, Serratia, Rhodococcus, and Morganella were the dominant genera. The bacterial genera Prevotella and Massilia are uncultivable under typical laboratory conditions as they require strict anaerobic or haloalkaliphilic conditions. Prevotella, Massilia, Rhodococcus, and Morganella are not reported in the available literature of honeybee gut microbiota; however, Massilia and Prevotella are the predominant inhabitants of the gut in the Red Mason Bee Osmia bicornis (Keller et al., 2013) and bark beetle (Dendroctonus rhizophagus) (Briones-Roblero et al., 2017). The Enterobacter genus is mainly involved in lactose fermentation (Cabral, 2010), and the non-core gut Enterobacteriaceae family is more likely to be found in wild bees than in bees from indoor colonies (Kwong and Moran, 2016). The genera Lactobacillus and Bifidobacterium commonly found in A. florea gut can possess antimicrobial activity and ferment dietary carbohydrates (Kwong and Moran, 2016; Kwong et al., 2017). Species of Prevotella are non-cellulolytic carbohydrate-degrading bacteria, which bring about the digestion of cell wall polysaccharides, such as xylan (Flint et al., 2012). A relatively smaller proportion of Bifidobacterium, Brevundimonas, Staphylococcus, Streptococcus, Gluconobacter, and Gilliamella genera was observed in our culture-independent studies, whereas many of these are predominant in A. mellifera (Kwong and Moran, 2016). Some of these phylotypes are found in a diverse range of social bees, inferring that they coevolved with bee eusociality. The gut microbiota of social bees are an emerging model to study the genomic diversity and evolution of host-associated bacterial communities because of these properties (Kwong et al., 2017).
The results obtained in this study motivated a desire to know more about the habitat functional biodiversity. To gain insights into the potential of the bacterial communities investigated in this study, we employed an imputed metagenome approach using the PICRUSt2 tool to determine the bacterial gene pool in the gut of A. florea. The results of these efforts revealed a wide range of genetic diversity involved in various essential processes such as environmental information signaling, membrane transport, signal transduction; genetic information translation, replication, transcription; cellular processes; and metabolism of carbohydrates, proteins, and other biomolecules, including the synthesis of some secondary metabolites. We found ∼60% higher assemblage of gene families exhibiting metabolic functions in the bacterial communities of A. florea.
The bacterial phyla Firmicutes and Proteobacteria found in A. florea in our study were similar to previous reports in A. florea (Saraithong et al., 2015). In this study, A. florea encountered more environmental opportunistic bacterial taxa like Klebsiella and Enterobacter spp. in comparison with previous reports. The abundance of non-core gut bacteria in A. florea is due to exposure to a wide ecosystem and floral diversity, since they are wild pollinators and not domestic. The observed differences in bacterial diversity of these critical pollinator species could be due to differential ecological evolution or habitat size (Ellegaard et al., 2020).
Dominant gut inhabitants of A. mellifera belong to the phyla Firmicutes, Actinobacteria, and Proteobacteria (Ahn et al., 2012; Kwong and Moran, 2016; Romero et al., 2019). The bee gut microbial communities in A. mellifera are specific and are dominated by nine bacterial species clusters, namely, Bartonella apis, Parasaccharibacter apium, Frischella perrara, S. alvi, Gilliamella apicola, Bifidobacterium spp., Lactobacillus Firm-4, Lactobacillus Firm-5, and these are believed to impart social behavior among individuals (Kwong and Moran, 2016). Snodgrassella alvi and G. apicola are ubiquitous in the gut of A. mellifera (Kwong and Moran, 2012). In our study, S. alvi was not detected, whereas G. apicola was found in traces. Acinetobacter was found in our study similar to Kim et al.’s (2014) study. Acinetobacter apis spp. nov. was isolated from the intestinal tract of a honeybee, A. mellifera (Kim et al., 2014). In the metagenomic survey, the class alpha-Proteobacteria and gamma-Proteobacteria dominated the gut environment of A. mellifera (Engel and Moran, 2013a). Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Acidobacteria were the predominant phyla in A. florea in our study. Citrobacter spp., Providencia vermicola, Planomicrobiumo keanokoites, and Exiguobacterium acetylicum were reported for the first time in the genus Apis by culture-dependent 16S rRNA sequencing (Khan et al., 2017). Pyrosequencing analysis of the bacterial community structure was made in the midguts and hindguts of the adult honeybees of A. cerana and A. mellifera. Higher frequencies of Enterobacteriaceae-, Lactococcus-, Bartonella-, Spiroplasma-, and Flavobacteriaceae-related OTUs were found in the guts of A. cerana, while Bifidobacterium- and Lachnospiraceae-related OTUs were more abundant in the guts of A. mellifera (Ahn et al., 2012). Anjum et al. (2018) reported Firmicutes (60%), Proteobacteria (26%), and Actinobacteria (14%) in A. mellifera gut by 16S rDNA sequencing.
The dominant phyla, Proteobacteria and Firmicutes, are reported in eusocial wasps (Order: Hymenoptera) inhabiting the eastern and southern Asian regions (Suenami et al., 2019). A variety of bacterial phyla are commonly present in insect guts, including gamma-Proteobacteria, alpha-Proteobacteria, beta-Proteobacteria, Bacteroidetes, Firmicutes including Lactobacillus and Bacillus species, Clostridia, Actinomycetes, Spirochaetes, Verrucomicrobia, Actinobacteria, and others (Colman et al., 2012). Numerous non-culture-based studies show that dominant taxa in Drosophila melanogaster are influenced by diet and vary among laboratories; however, certain taxa recur (Broderick and Lemaitre, 2012). The gut inhabitants of honeybees coevolved with bumble bees and comprise a distinctive gut community (Martinson et al., 2011). Various studies indicate the contribution of insect gut flora in metabolism, nutrition, immune modulation, and protection against foreign entities (Engel and Moran, 2013b). Understanding the bacterial network offers knowledge into bee pathology and host−microbe interaction and aids in improving honeybee health and discovering new sources of biotechnologically potential molecules and enzymes (Romero et al., 2019).
The gut communities of A. florea are more diverse in composition compared with other Apis clades, and the sequence analysis approach classified gut bacteria into Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Acidobacteria as the major phyla. Most of the isolates observed are opportunistic and beneficial gut inhabitants. Prevalence of members of the genera Enterobacter (59%) is observed in our study, which is distinct in abundance in opposition to other species of Apis clade. Functional analysis revealed the highest abundance of metabolism-related genes in bacterial communities of A. florea. High-throughput sequencing of the whole gut using NGS has allowed us to analyze the importance of microbiome role in A. florea host to some extent. Similarly, with other microbiome projects (human, animal, and insects), this study provides additional information and data pertaining to the microbiome of A. florea, a primitive clade of honeybee, revealing some of the bacterial symbionts not found in earlier reports on honeybee microbiome project. However, further genomic studies are required to understand the significance of diverse microbial populations, and a comparative analysis of managed and wild bee species may offer insight into host-microbe interaction.
The sequences obtained from culture-dependent approach are available under NCBI BioProject SUB6349615, MN512276–MN512310. The sequencing reads generated during high-throughput sequencing are available under NCBI BioProject PRJNA777397.
DG conducted the research and wrote the manuscript. KJ performed the data analyses and reviewed the manuscript. JL, KK, and MM edited and reviewed the manuscript. YS provided experimental support. AS planned, supervised, and organized the experiment, wrote and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by Taif University Researchers Supporting Project Number (TURSP-2020/92), Taif University, Taif, Saudi Arabia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are thankful to the National Centre for Microbial Resource, Pune, India, for providing the laboratory facility to carry out the research work and to Sunil Kumar Dhar for his assistance.
Ahn, J. H., Hong, I. P., Bok, J. I., Kim, B. Y., Song, J., and Weon, H. Y. (2012). Pyrosequencing analysis of the bacterial communities in the guts of honey bees Apis cerana and Apis mellifera in Korea. J. Microbiol. 50, 735–745. doi: 10.1007/s12275-012-2188-0
Anderson, K. E., Sheehan, T. H., Eckholm, B. J., Mott, B. M., and DeGrandi-Hoffman, G. (2011). An emerging paradigm of colony health: microbial balance of the honey bee and hive (Apis mellifera). Insectes Sociaux 58, 431–444. doi: 10.1007/s00040-011-0194-6
Anjum, S. I., Shah, A. H., Aurongzeb, M., Kori, J., Azim, M. K., Ansari, M. J., et al. (2018). Characterization of gut bacterial flora of Apis mellifera from north-west Pakistan. Saudi J. Biol. Sci. 25, 388–392. doi: 10.1016/j.sjbs.2017.05.008
Balachandra, C., Subash Chandran, M. D., and Ramachandra, T. V. (1999). Honeybee Diversity, Role in Pollination and Beekeeping Scenario in South Indian Western Ghats. New Delhi: The Ministry of Environment Forests and Climate Change.
Biewer, M., Lechner, S., and Hasselmann, M. (2016). Similar but not the same: insights into the evolutionary history of paralogous sex-determining genes of the dwarf honey bee Apis florea. Heredity (Edinb) 116, 12–22. doi: 10.1038/hdy.2015.60
Bokulich, N. A., Joseph, C. M., Allen, G., Benson, A. K., and Mills, D. A. (2012). Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS One 7:e36357. doi: 10.1371/journal.pone.0036357
Briones-Roblero, C. I., Hernández-García, J. A., Gonzalez-Escobedo, R., Soto-Robles, L. V., Rivera-Orduña, F. N., and Zúñiga, G. (2017). Structure and dynamics of the gut bacterial microbiota of the bark beetle, Dendroctonus rhizophagus (Curculionidae: scolytinae) across their life stages. PLoS One. 12:e0175470. doi: 10.1371/journal.pone.0175470
Broderick, N. A., and Lemaitre, B. (2012). Gut-associated microbes of Drosophila melanogaster. Gut Microbes 3, 307–321. doi: 10.4161/gmic.19896
Cabral, J. (2010). Water microbiology. Bacterial pathogens and water. Int. J. Environ. Res. Public Health 7, 3657–3703. doi: 10.3390/ijerph7103657
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2010a). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1) 4516–4522. doi: 10.1073/pnas.1000080107
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chandler, J. A., Morgan Lang, J., Bhatnagar, S., Eisen, J. A., and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host–microbe model system. PLoS Genet. 7:e1002272. doi: 10.1371/journal.pgen.1002272
Cilia, G., Fratini, F., Tafi, E., Turchi, B., Mancini, S., Sagona, S., et al. (2020b). Microbial profile of the ventriculum of honey bee (Apis mellifera ligustica Spinola, 1806) fed with veterinary drugs, dietary supplements and non-protein amino acids. Vet. Sci. 7:76. doi: 10.3390/vetsci7020076
Cilia, G., Fratini, F., Tafi, E., Mancini, S., Turchi, B., Sagona, S., et al. (2020a). Changes of Western honey bee Apis mellifera ligustica (Spinola, 1806) ventriculus microbial profile related to their in-hive tasks. J. Apic. Res. 60, 198–202.
Coleman, J., Juhn, J., and James, A. A. (2007). Dissection of midgut and salivary glands from Ae. aegypti mosquitoes. J. Vis. Exp. 5:228. doi: 10.3791/228
Colman, D. R., Toolson, E. C., and Takacs-Vesbach, C. D. (2012). Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21, 5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x
Corby-Harris, V., Snyder, L. A., Schwan, M. R., Maes, P., McFrederick, Q. S., and Anderson, K. E. (2014). Origin and effect of Alpha 2.2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. nov. Appl. Environ. Microbiol. 80, 7460–7472. doi: 10.1128/AEM.02043-14
Dillon, R., and Charnley, K. (2002). Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res. Microbiol. 153, 503–509. doi: 10.1016/s0923-2508(02)01361-x
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Ellegaard, K. M., Suenami, S., Miyazaki, R., and Engel, P. (2020). Vast differences in strain-level diversity in the gut microbiota of two closely related honey bee species. Curr. Biol. 30, 2520.e7–2531.e7. doi: 10.1016/j.cub.2020.04.070
Engel, P., Kwong, W. K., McFrederick, Q., Anderson, K. E., Barribeau, S. M., Chandler, J. A., et al. (2016). The Bee microbiome: impact on bee health and model for evolution and ecology of host-microbe interactions. mBio 7, e2164–e2115. doi: 10.1128/mBio.02164-15
Engel, P., Kwong, W. K., and Moran, N. A. (2013). Frischella perrara gen. nov., sp. nov., a gammaproteobacterium isolated from the gut of the honey bee, Apis mellifera. Int. J. Syst. Evol. Microbiol. 63(Pt 10) 3646–3651. doi: 10.1099/ijs.0.049569-0
Engel, P., and Moran, N. A. (2013b). The gut microbiota of insects - diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Engel, P., and Moran, N. A. (2013a). Functional and evolutionary insights into the simple yet specific gut microbiota of the honey bee from metagenomic analysis. Gut Microbes 4, 60–65. doi: 10.4161/gmic.22517
Flint, H. J., Scott, K. P., Duncan, S. H., Louis, P., and Forano, E. (2012). Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306. doi: 10.4161/gmic.19897
Fridman, S., Izhaki, I., Gerchman, Y., and Halpern, M. (2012). Bacterial communities in floral nectar. Environ. Microbiol. Rep. 4, 97–104.
Gupta, A. K., Rastogi, G., Nayduch, D., Sawant, S. S., Bhonde, R. R., and Shouche, Y. S. (2014). Molecular phylogenetic profiling of gut-associated bacteria in larvae and adults of flesh flies. Med. Vet. Entomol. 28, 345–354. doi: 10.1111/mve.12054
Inglis, G. D., Enkerli, J., and Goettel, M. S. (2012). “Laboratory techniques used for entomopathogenic fungi. Hypocreales,” in Manual of Techniques in Invertebrate Pathology, 2nd Edn. ed. L. Lacey (Amsterdam: Elsevier Ltd).
Jani, K., Dhotre, D., Bandal, J., Shouche, Y., Suryavanshi, M., Rale, V., et al. (2018). World’s largest mass bathing event influences the bacterial communities of Godavari, a Holy River of India. Microb. Ecol. 76, 706–718. doi: 10.1007/s00248-018-1169-1
Kaspi, R., and Shafir, S. (2013). Associative olfactory learning of the red dwarf honey bee Apis florea. Apidologie 44, 100–109.
Keller, A., Grimmer, G., and Steffan-Dewenter, I. (2013). Diverse microbiota identified in whole intact nest chambers of the red mason bee Osmia bicornis (Linnaeus 1758). PLoS One 8:e78296. doi: 10.1371/journal.pone.0078296
Khan, K. A., Ansari, M. J., Al-Ghamdi, A., Nuru, A., Harakeh, S., and Iqbal, J. (2017). Investigation of gut microbial communities associated with indigenous honey bee (Apis mellifera jemenitica) from two different eco-regions of Saudi Arabia. Saudi J. Biol. Sci. 24, 1061–1068. doi: 10.1016/j.sjbs.2017.01.055
Kim, P. S., Shin, N. R., Kim, J. Y., Yun, J. H., Hyun, D. W., and Bae, J. W. (2014). Acinetobacter apis sp. nov., isolated from the intestinal tract of a honey bee, Apis mellifera. J. Microbiol. 52, 639–645. doi: 10.1007/s12275-014-4078-0
Kumbhare, S. V., Dhotre, D. P., Dhar, S. K., Jani, K., Apte, D. A., Shouche, Y. S., et al. (2015). Insights into diversity and imputed metabolic potential of bacterial communities in the continental shelf of Agatti Island. PLoS One 10:e0129864. doi: 10.1371/journal.pone.0129864
Kurli, R., Chaudhari, D., Pansare, A. N., Khairnar, M., Shouche, Y. S., and Rahi, P. (2018). Cultivable microbial diversity associated with cellular phones. Front. Microbiol. 9:1229. doi: 10.3389/fmicb.2018.01229
Kwong, W., and Moran, N. A. (2012). Cultivation and characterization of the gut symbionts of honey bees and bumble bees: snodgrassella alvi gen. nov., sp. nov., a member of the Neisseriaceae family of the Betaproteobacteria; and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the Enterobacteriales order of the Gammaproteobacteria. Intl. J. Syst. Evol. Microbiol. 63(Pt 6), 2008–2018. doi: 10.1099/ijs.0.044875-0
Kwong, W. K., Medina, L. A., Koch, H., Sing, K. W., Soh, E. J. Y., Ascher, J. S., et al. (2017). Dynamic microbiome evolution in social bees. Sci. Adv. 3:e1600513. doi: 10.1126/sciadv.1600513
Kwong, W. K., and Moran, N. A. (2016). Gut microbial communities of social bees. Nat. Rev. Microbiol. 14, 374–384. doi: 10.1038/nrmicro.2016.43
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: Wiley), 115–175.
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Lateef, A., Adelere, I., and Gueguim-Kana, E. B. (2015). The biology and potential biotechnological applications of Bacillus safensis. Biologia 70, 411–419.
Loncaric, I., Heigl, H., Licek, E., Moosbeckhofer, R., Busse, H., and Rosengarten, R. (2009). Typing of Pantoea agglomerans isolated from colonies of honey bees (Apis mellifera) and culturability of selected strains from honey. Apidologie 40, 40–54.
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Martinson, V. G., Danforth, B. N., Minckley, R. L., Rueppell, O., Tingek, S., and Moran, N. A. (2011). A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 20, 619–628. doi: 10.1111/j.1365-294X.2010.04959.x
Martinson, V. G., Moy, J., and Moran, N. A. (2012). Establishment of characteristic gut bacteria during development of the honeybee worker. Appl. Environ. Microbiol 78, 2830–2840. doi: 10.1128/AEM.07810-11
Moran, N. A., Hansen, A. K., Powell, J. E., and Sabree, Z. L. (2012). Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One 7:e36393. doi: 10.1371/journal.pone.0036393
Nadkarni, M. A., Martin, F. E., Hunter, N., and Jacques, N. A. (2009). Methods for optimizing DNA extraction before quantifying oral bacterial numbers by real-time PCR. FEMS Microbiol. Lett. 296, 45–51. doi: 10.1111/j.1574-6968.2009.01629.x
Nelson, K., Muge, E., and Wamalwa, B. (2021). Cellulolytic Bacillus species isolated from the gut of the desert locust Schistocerca gregaria. Sci. Afr. 11:e00665.
Paul, J., Sarkar, A., and Varma, A. (1986). In vitro studies of cellulose digesting properties of Staphylococcus-saprophyticus isolated from termite gut. Curr. Sci. India 55, 710–714.
Potts, S., Biesmeijer, J., Kremen, C., Neumann, P., Schweiger, O., and Kunin, W. (2010). Global pollinator declines: trends, impacts and drivers. Trends Ecol. Evol. 25, 345–353. doi: 10.1016/j.tree.2010.01.007
Radloff, S., Hepburn, H., Hepburn, C., Fuchs, S., Otis, G., Sein, M., et al. (2005). Multivariate morphometric analysis of Apis cerana of southern mainland Asia. Apidologie 36, 127–139.
Raymann, K., and Moran, N. A. (2018). The role of the gut microbiome in health and disease of adult honey bee workers. Curr. Opin. Insect. Sci. 26, 97–104. doi: 10.1016/j.cois.2018.02.012
Romero, S., Nastasa, A., Chapman, A., Kwong, W. L., and Foster, L. J. (2019). The honey bee gut microbiota: strategies for study and characterization. Insect Mol. Biol. 28, 455–472. doi: 10.1111/imb.12567
Saraithong, P., Li, Y., Saenphet, K., Chen, Z., and Chantawannakul, P. (2015). Bacterial community structure in Apis florea larvae analyzed by denaturing gradient gel electrophoresis and 16S rRNA gene sequencing. Insect Sci. 22, 606–618. doi: 10.1111/1744-7917.12155
Seiler, H., Schmidt, V., Wenning, M., and Scherer, S. (2012). Bacillus kochii sp. nov., isolated from foods and a pharmaceuticals manufacturing site. Int. J. Syst. Evol. Microbiol. 62, 1092–1097. doi: 10.1099/ijs.0.027771-0
Soman, A. G., and Chawda, S. S. (1996). A contribution to the biology and behavior of the dwarf bee, A. florea and its economic importance in Kutch, Gujarat, India. Ind. Bee J. 58, 81–88.
Sowani, H., Mohite, P., Munot, H., Shouche, Y., Bapat, T., Kumar, A., et al. (2016). Green synthesis of gold and silver nanoparticles by an actinomycete Gordonia amicalis HS-11: mechanistic aspects and biological application. Process Biochem. 51, 374–383.
Suenami, S., Konishi Nobu, M., and Miyazaki, R. (2019). Community analysis of gut microbiota in hornets, the largest eusocial wasps, Vespa mandarinia and V. simillima. Sci. Rep. 9:9830. doi: 10.1038/s41598-019-46388-1
Suwannapong, G., Benbow, M., and Nieh, J. (2012). “Biology of thai honeybees: natural history and threats,” in Bees: Biology, Threats and Colonies, ed. R. M. Florio (Hauppauge, NY: Nova Science).
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
The Elements of Biodiversity (2021). The Elements of Biodiversity. Available online at: https://www.biologicaldiversity.org/programs/biodiversity/elements_of_biodiversity/ (accessed June 1, 2021).
Turner, S., Pryer, K. M., Miao, V. P. W., and Palmer, J. D. (1999). Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J. Eukaryotic Microbiol. 46, 327–338. doi: 10.1111/j.1550-7408.1999.tb04612.x
Yoshiyama, M., and Kimura, K. (2009). Bacteria in the gut of Japanese honeybee, Apis cerana japonica, and their antagonistic effect against Paenibacillus larvae, the causal agent of American foulbrood. J. Invertebr. Pathol. 102, 91–96. doi: 10.1016/j.jip.2009.07.005
Zapka, C., Leff, J., Henley, J., Tittl, J., De Nardo, E., Butler, M., et al. (2017). Comparison of standard culture-based method to culture-independent method for evaluation of hygiene effects on the hand microbiome. mBio 8, e93–e17. doi: 10.1128/mBio.00093-17
Zhang, Z., Yin, L., Li, X., Zhang, C., Liu, C., and Wu, Z. (2018). The complete genome sequence of Bacillus halotolerans ZB201702 isolated from a drought- and salt-stressed rhizosphere soil. Microb. Pathog. 123, 246–249. doi: 10.1016/j.micpath.2018.07.019
Keywords: dwarf honeybee, Apis florea, gut microbiota, MALDI-TOF MS, 16S rRNA, next-generation sequencing
Citation: Ganeshprasad DN, Lone JK, Jani K, Shouche YS, Khan KA, Sayed S, Shukry M, Dar SA, Mushtaq M and Sneharani AH (2022) Gut Bacterial Flora of Open Nested Honeybee, Apis florea. Front. Ecol. Evol. 10:837381. doi: 10.3389/fevo.2022.837381
Received: 16 December 2021; Accepted: 31 January 2022;
Published: 01 April 2022.
Edited by:
Giovanni Cilia, Council for Agricultural and Economics Research (CREA), ItalyReviewed by:
Kilmer Soares, Federal University of Paraíba, BrazilCopyright © 2022 Ganeshprasad, Lone, Jani, Shouche, Khan, Sayed, Shukry, Dar, Mushtaq and Sneharani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muntazir Mushtaq, bXVudGF6aXJidEBnbWFpbC5jb20=; A. H. Sneharani, c25laGFyYW5pYWhAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.