 Peter Kaňuch
Peter Kaňuch Anna Cassel-Lundhagen
Anna Cassel-Lundhagen Sonja Preuss
Sonja Preuss Göran Nordlander
Göran Nordlander Åsa Berggren
Åsa Berggren
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 22 March 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.812079
To understand colonization success of an invasive species we need to know the origin of the founders, where and when they were introduced, and how they spread from the introduction site(s) through the landscape. Admixture of different genetic lineages from multiple introductions is generally hypothesized to be beneficial to invasive species thanks to adaptive variation and heterozygosity-fitness correlations. In this study, population genetic and landscape data was gathered for Roesel’s bush-cricket, Roeseliana roeselii a small bush-cricket common in central and eastern Europe that currently is expanding its range in northern Europe. We examined how colonization history and landscape structure affect the spread of the species and its population genetic structure, as a consequence of multiple introductions. Using comprehensive information of the species ecology and dispersal, together with genetic structure inferred from samples from 29 locations in central Sweden (we employed data published by Preuss et al., 2015), we found that two parapatric founding lineages have coexisted with very little gene flow during a long time span. An isolation-by-distance pattern and a decrease of genetic diversity toward marginal areas were more pronounced in the lineage situated in forest dominated landscapes. Our findings are in strong contrast to the hypothesis that different genetic lineages will admix when introduced to the same area. The presence of the separate lineages decades after introduction and without physical barriers for gene flow shows that some mechanism prevents them from admixture. One possibility is that the lineages with different genetic setups have adapted independently to local conditions and their admixture resulted in loss of locally adapted genotypes and hybrid offspring, less viable than the respective ancestral genotypes. However, an alternative post-mating reproductive barrier and hybrid breakdown phenomenon should also be considered. Our data indicate that besides landscape characteristics, human transportation of agricultural goods may play an important role for the overall spatial genetic pattern of the species in the study area by aiding the spread of the species.
Different management strategies are used to prevent invasive species from entering new regions, to hinder them from establishing viable populations and expand within an area (Baker, 2017; Liebhold and Kean, 2019). For all these actions, there is a need to understand the ecology of species; e.g., how different traits may increase invasiveness and how the composition of the landscape affects establishment and spread (Beckmann et al., 2015; Fidler et al., 2018; Williams et al., 2021; Wyse and Hulme, 2021). Previous studies have found that species with low movement ability and/or with low adaptation capacity are generally less successful in establishing in new areas (Wamser et al., 2012; Medley et al., 2019). Fragmented landscapes where habitat patches are far apart and dispersal barriers are common, also impact the success of colonization and can hinder or slow down the spread of species (Greenwald et al., 2009; MacDonald et al., 2018; Melero et al., 2020). In addition to a species’ own active dispersal, humans can facilitate their movement into and within a region (Di Castri, 1989). This facilitation can occur from intentional or accidental transportations of individuals, or materials that harbor individuals (Kaňuch et al., 2013; Lanner et al., 2021). Complicating the picture of establishment and spread, is that founding individuals may enter regions multiple times from different sources and at a number of locations, before successful colonization occurs (Di Castri, 1989). Thus, to fully understand the ecology of an invasive species colonization and spread we need to know where the founders originated from (i.e., their genetic origin), where and when they were introduced, and how they spread from the introduction sites through the landscape.
It is generally hypothesized that admixture of different genetic lineages from multiple introductions is beneficial to invasive species thanks to adaptive variation and heterozygosity-fitness correlations, although it remains unclear whether admixture has a causal role in population expansion (Rius and Darling, 2014; Dlugosch et al., 2015). However, genetic interactions between isolated populations selected for locally adapted genotypes, may become unfavorable with genetic distance of different lineages. This may be particularly true in later generations due to outbreeding depression and hybrid breakdown—the poor performance of certain hybrids that leads to extinction (Brideau et al., 2006; Novicic et al., 2011). Most often, it is defined as a reduction in fitness below the midparent but studies are too few to have converged on a standard definition of outbreeding depression (see Edmands, 2007). This explains the low representation of admixture in introductions with high levels of source divergence (Ordóñez et al., 2013; Dlugosch et al., 2015), and it is difficult to examine whether admixed populations are more likely to become successful as colonizers than unmixed ones (Rius and Darling, 2014). One way to study dispersal and colonization pattern is to measure the level of gene flow between populations using neutral molecular markers (Parker et al., 1998). If a species disperses primarily over short distances and in a stepping-stone manner, the genetic structure is expected to follow an isolation-by-distance pattern (Kimura and Weiss, 1964). On the contrary, if there is no significant relationship between genetic and geographic distances a species likely disperses with no or few barriers in the landscape. Also, if dispersal is mediated via a vector, it is expected to be reflected in the genetic patterns across populations.
The Roesel’s bush-cricket, Roeseliana roeselii (Hagenbach, 1822) (Orthoptera: Tettigoniidae, synonym: Metrioptera roeselii) is a small (12–18 mm) bush-cricket common in central and eastern Europe (Bellmann, 2006). It is currently expanding its range in Sweden and other northern European countries (Poniatowski et al., 2012; Eriksson et al., 2013; Preuss et al., 2014, 2015; Beckmann et al., 2015). Passive dispersal of individuals by human transportation of grass-stems (hay) used for egg deposition may facilitate the colonization of distant habitat patches (De Jong and Kindvall, 1991; Wagner, 2004; Kaňuch et al., 2013), and consequently affect the spatial population genetic pattern. In the time of this study, the species distribution in central Sweden predominantly extended across an area of approximately 120 × 140 km in the Lake Mälaren region.1 The core area of this isolated population has the highest local genetic diversity and its location suggest that R. roeselii have been introduced via sea cargo to the banks of the westernmost bay of lake Mälaren (De Jong and Kindvall, 1991; Preuss et al., 2015). This region, with a historical homestead established by the Swedish king Gustav Vasa and his descendants in the sixteenth and seventeenth century, have well documented and long-running extensive horse and cattle imports where R. roeseli could have entered in with cargo (Bäckström, 1924; Montelius, 1993). A royal stud farm and military riding school at the town of Strömsholm in the area has previously been identified as a potential introduction site (Preuss et al., 2015). The nearby grasslands at Kungsör are also potential introduction sites, as large farms in the area have been active for very long time periods (Strengbom, 2019). In line with these theories, molecular analyses have suggested multiple introductions of the species and that founding individuals originate from different regions, most likely from the harbors in the Gulf of Finland or from the southern Baltic coast (Kaňuch et al., 2013).
Detailed studies on the species’ ecology (e.g., Ingrisch, 1984; Poniatowski and Fartmann, 2005; Holzhauer et al., 2006; Berggren, 2009), movement behavior (Berggren et al., 2002; Berggren, 2004, 2005; Poniatowski and Fartmann, 2011) and population genetics (Kaňuch et al., 2014, 2021) have increased the understanding of how R. roeselii responds to environmental factors during the colonization process and how it successfully colonizes uninhabited areas even with very small propagule sizes. As an omnivorous generalist, that preferentially colonizes tall grassland habitats in agricultural landscapes, it is found in various meadows, leys, grassy field margins around crop fields, ditches, and road verges (Marshall and Haes, 1988; Berggren et al., 2001; Preuss et al., 2011). Forests and intensively grazed pastures are considered to be unsuitable habitats (De Jong and Kindvall, 1991; Ingrisch and Köhler, 1998; Berggren, 2004), and large water bodies are expected to be strong barriers for gene flow for this mostly flightless species (98–99% of the population; Vickery, 1965; Wissmann et al., 2009). The minority, long-winged individuals, is capable to disperse over longer distances (Hochkirch and Damerau, 2009; Poniatowski and Fartmann, 2011), and thus may play a significant role for local genetic variation (Kaňuch et al., 2021).
Existing knowledge of the likely initial introduction area of the species (Preuss et al., 2015), and that those introductions have occurred from at least two different but adjacent regions within the continuous species range (Kaňuch et al., 2013), offer a very interesting opportunity to further explore ecological aspects of introduced species’ colonization and spread. In this study, we particularly aimed to move one step further in understanding the spatial genetic patterns associated with successful colonization. We examined how colonization history and landscape structure affect the spread of the species and its population genetic structure, as a consequence of multiple introductions. To do this we used a set of neutral genetic markers to assign individuals in the study area to their potential founders from the species main range and estimated the levels of admixture across different sites in a fragmented landscape (microsatellite data published previously by Preuss et al., 2015 were thus reused and reanalyzed). We tested three hypotheses: (i) Genetic lineages originating from different sources show an admixed pattern in areas where they meet. (ii) Pair-wise genetic differentiation between populations will be positively correlated with the least cost path distance, if dispersal is limited to movement through suitable habitat and different lineages are not admixed; but (iii) the decrease of genetic diversity from central toward marginal populations will be more pronounced in a lineage that colonize areas with less suitable habitats, if species disperses naturally without significant effect of human-mediated transport.
During July and August 2010, we sampled 27–30 short-winged adult individuals of R. roeselii from 29 locations in the Lake Mälaren region (Figure 1). Altogether 837 individuals were genotyped using a set of eight microsatellite markers developed for this species (MR2-42, 3-24, 3-34, Holzhauer and Wolff, 2005; Metroe08, 16, 19, 24, 27, Kaňuch et al., 2010). These are the same eight markers that were used in Preuss et al. (2015). All details on specimens’ collection, DNA extraction from muscle tissue, multiplex PCR protocols with fluorescently labeled primers, conditions for reactions and scoring of alleles are described in Preuss et al. (2015). Therein are reported also basic population-genetics characteristics, including sample size, allelic richness, unique alleles, heterozygosity, fixation index, and results of Hardy-Weinberg tests for each sampling site. All samples were in Hardy-Weinberg equilibrium and the pairs of microsatellites did not exhibit significant linkage disequilibrium. Three loci (Metroe19, MR2-42, and MR3-24) exhibited some elevated frequency of null alleles (20–25%) across all locations when tested using the Chakraborty et al.’s (1994) and Brookfield’s (1996) methods in the package “PopGenReport” 3.0.4 (Gruber and Adamack, 2019) of the R 3.6.3 software (R Core Team, 2020). Aware of possible bias that might result from this effect we kept these markers for further analyses to not reduce available dataset and to lower resolution of determined genetic structure.
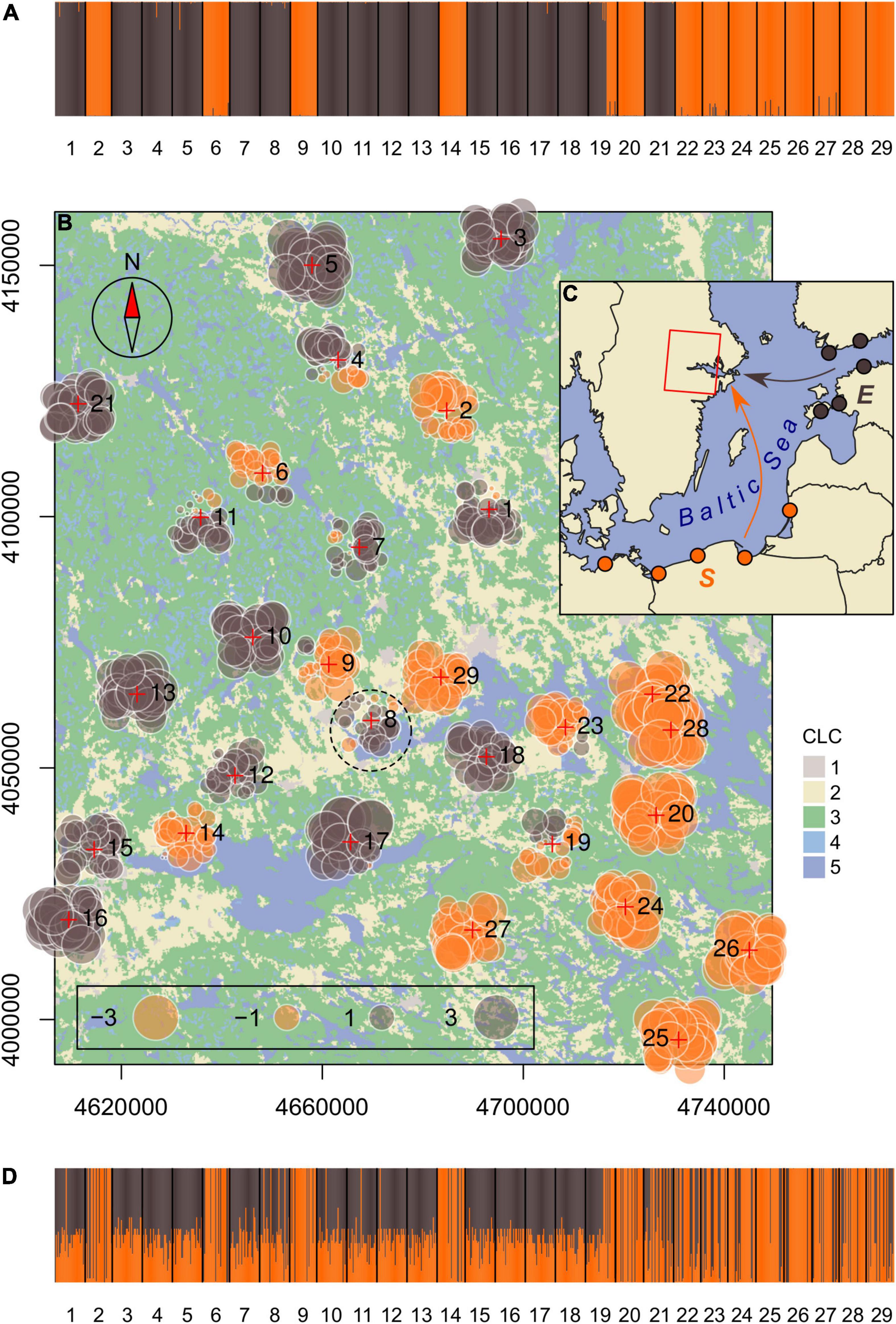
Figure 1. Map of the study area with locations of the 29 sampling sites of Roeseliana roeselii (crosses) in the Lake Mälaren region in central Sweden. The main land-cover classes categorized by the Corine Land Cover inventory (1, artificial surfaces; 2, agricultural areas; 3, forest and seminatural areas; 4, wetlands; 5, water bodies) and suggested area of introduction (dashed circle) are depicted. The coordinate reference system is ETRS89/LAEA Europe (meters). (A) Genetic structure of R. roeselii samples in the study area as inferred by the Structure admixture analysis. (B) Spatial MEMGENE analysis of the samples (MEM1 axis). Individuals with similar MEM scores (circles of similar size and color) suggest spatially homogenous genetic clusters. (C) Potential source populations (full circles) represent two distinct genetic lineages (E, eastern; S, southern) and are most likely co-founders of the populations in the study area (Kaňuch et al., 2013). (D) Membership probability plot from the assignment test. Individuals in the stacked bar plots are represented by vertical bars divided into parts proportional to their proposed ancestry in determined genetic clusters or membership to source population, respectively.
To obtain a robust support for identification of genetically homogeneous clusters of individuals in our samples we applied three different approaches. Firstly, we used an individual based clustering method implemented in the software Structure 2.3.4 (Pritchard et al., 2000; Hubisz et al., 2009). We ran the admixture model with correlated allele frequencies without the prior population information and degree of admixture α = 1. For each value of K (range 1–29), we conducted 20 independent runs with uniform priors using a burn-in of 100,000 iterations followed by 100,000 Markov chain Monte Carlo iterations. We discarded all individuals with > 20% missing genotypes (i.e., six individuals) and there were no identical multilocus genotypes present in the dataset. The number of genetic clusters K in the data set was inferred by the ΔK method (Evanno et al., 2005), which finds the breakpoint in the slope of the likelihood distribution for different K values, using the Structure Harvester Web 0.6.94 (Earl and von Holdt, 2012), and also through Q-matrix correlations, which identified the stable K solutions (identify anomalous runs), implemented in the R package “CorrSieve” 1.6–8 (Campana et al., 2011). Outputs of the Structure analysis were visualized with the Clumpak program (Kopelman et al., 2015). Secondly, to identify possibly weak spatial genetic patterns, we analyzed spatial autocorrelation in the genetic data using MEMGENE analysis by the R package “memgene” 1.0.1 (Galpern et al., 2014). This approach uses a regression framework which visualizes only statistically significant spatial patterns. Predictors are generated using Moran’s eigenvectors maps (MEM) from sampled locations (pairwise Euclidean distances among individuals) and the response variables are individual genetic distances (proportion of shared alleles, DPS). Thirdly, we calculated pairwise estimates of genetic distances among sampled locations and between determined genetic clusters using DPS (Bowcock et al., 1994) and G’ST (Hedrick, 2005), thus an individual- and a population-based estimator, respectively. These two different measures of genetic distance were explored using heatmaps and dendrograms of hierarchical clustering in the R package “adegenet” 2.1.3 (Jombart, 2008).
The assignment of samples collected in the Lake Mälaren region to potential founder populations was performed using a machine-learning framework implemented in the R package “assignPOP” 1.2.0 (Chen et al., 2018). A naïve Bayes classification method (a simple probabilistic classifier with strong independence assumptions between the features) was used as a default setting of this package to build predictive model which estimated individual membership probabilities in the study area using baseline microsatellite data that represented two distinct genetic lineages along the Baltic coast and likely co-founders of populations in central Sweden. Thus, five samples (120 individuals) from Finland and Estonia are hereafter called the “eastern” lineage, while five other samples (120 individuals) from Lithuania, Poland and Germany represent the “southern” lineage (Kaňuch et al., 2013). Alleles of four loci (Metroe07, Metroe08, Metroe19, and Metroe27) sized in both datasets were available for this assignment test.
Information on landscape composition was extracted from the Corine Land Cover data (100 m GeoTiff raster, CLC 2018 version 20).2 We classed the land-cover categories according to the assumed cost of dispersal that individuals experience when moving through different types of habitats (Sawyer et al., 2011). Based on previous studies on the species presence in the landscape (Preuss et al., 2011), agricultural areas were expected to be positively correlated with presence of the species (i.e., least costly for the species to move through, weight = 1). To other habitats more dispersal cost was attributed: weight = 2 for artificial surfaces as rural settlements and road networks, weight = 3 for wetlands as marshes and peat bogs. Forests and seminatural areas were considered unsuitable habitat (weight = 5), and lakes and waterways were classed as being most costly to move across (weight = 10). Using of Least-Cost Path plugin of the QGIS 3.8.1 software3 we created a cost surface map of the classified land-cover categories and calculated least cost path distances between all sampled sites (Supplementary Figure 1). Besides the least cost path distance, we measured also Euclidean distance (the geographic distance between sites without additional effects of landscape elements on gene flow). The isolation-by-distance (IBD) pattern in sampled populations was tested using Mantel test between matrices of genetic and both geographic distances in the R package “adegenet.” For genetic matrix we used chord distance DC (Cavalli-Sforza and Edwards, 1967).
Finally, to test the central-marginal hypothesis we employed a generalized additive model (GAM) for geospatial interpolation of the genetic diversity of R. roeselii in the study area. Using the previous knowledge about correlations between different estimates of genetic diversity (Preuss et al., 2015), we selected mean allelic richness per locus and sampling site as a suitable representative of genetic diversity. Smooth functions of predictor variables (x, y coordinates of sampling sites) were estimated and the model was visualized using the R packages “mgcv” 1.8-28 (Wood, 2019) and “raster” 3.0–12 (Hijmans, 2020). To test whether the proportion of land-cover classes differed between the areas colonized by different genetic introduction lineages, we fitted a model of independence for two-way tables that was visualized in mosaic plot using the R package “vcd” 1.4–6 (Meyer et al., 2020).
In the outputs of the admixture model simulated in the Structure analysis, both methods ΔK and Q-matrix correlations detected stable genetic structure at K = 2 (Figure 1A and Supplementary Figures 2, 3). Interestingly, all sampling sites except one was represented by individuals that belonged to only one of the two genetic clusters. Sampling site number 19 was the only location where individuals of both genetic clusters occurred together. This pattern was seen also in the MEMGENE analysis where the MEM variables indicated higher level of admixture of the two genetic clusters around the area of initial introduction (smaller absolute MEM scores), and higher genetic differentiation toward marginal zones of the current species distribution (greater absolute MEM scores; Figure 1B). The proportion of genetic variation along MEM axes (R2adj = 0.28) was sufficient to identify the spatial genetic structure. MEM1 axis explained most, 65%, of the spatial genetic variation (MEM2 and MEM3 explained 7 and 5%, respectively). The genetic structure of samples matched perfectly to the results of the assignment test. Most individuals of one genetic cluster in the lake Mälaren region were assigned to the southern genetic lineage and the second cluster appears to have been established from founders of the eastern lineage (Figures 1C,D). Contrary to the southern lineage, eastern lineage individuals in stacked barplots were assigned with a lower 70% membership probability which appears to result from a more limited set of available microsatellite loci which did not allow for better resolution in this analysis (see section “Materials and Methods”). Possibly the same technical restraint (low number of alleles that were identical between baseline and test data) assigned a number of individuals of the southern (orange) genetic cluster to eastern (brown) founder lineage.
Genetic differentiation of the inferred genetic clusters was very high (mean ± SD; DPS = 0.624 ± 0.055, G’ST = 0.605 ± 0.079) suggesting no or very little gene flow between the main clusters and two different estimators of genetic distance revealed consistent pattern (Figure 2). Therefore, testing of spatial correlations between matrices of genetic and geographic distances was meaningful only within each genetic lineage. Individuals from sampling site 19 were excluded from further spatial analyses as a consequence of the significant proportion of both genetic lineages. The estimated least cost path distances, representing the optimal dispersal routes between populations, were on average 38 and 29% longer than the Euclidean distance in the eastern and the southern lineage, respectively. In contrast to the eastern genetic lineage, neither using Euclidean distance nor least cost path distance resulted in a significant IBD pattern for the local populations of the southern lineage (Figure 3 and Supplementary Figure 4). Differences in the IBD pattern were similar to the modeled spatial patterns of genetic diversity in these two lineages. The decrease of genetic diversity from the introduction sites toward the marginal areas was more pronounced in the eastern lineage situated in more forest dominated landscapes, than in the southern lineage situated in landscapes dominated by arable land (Figure 4).
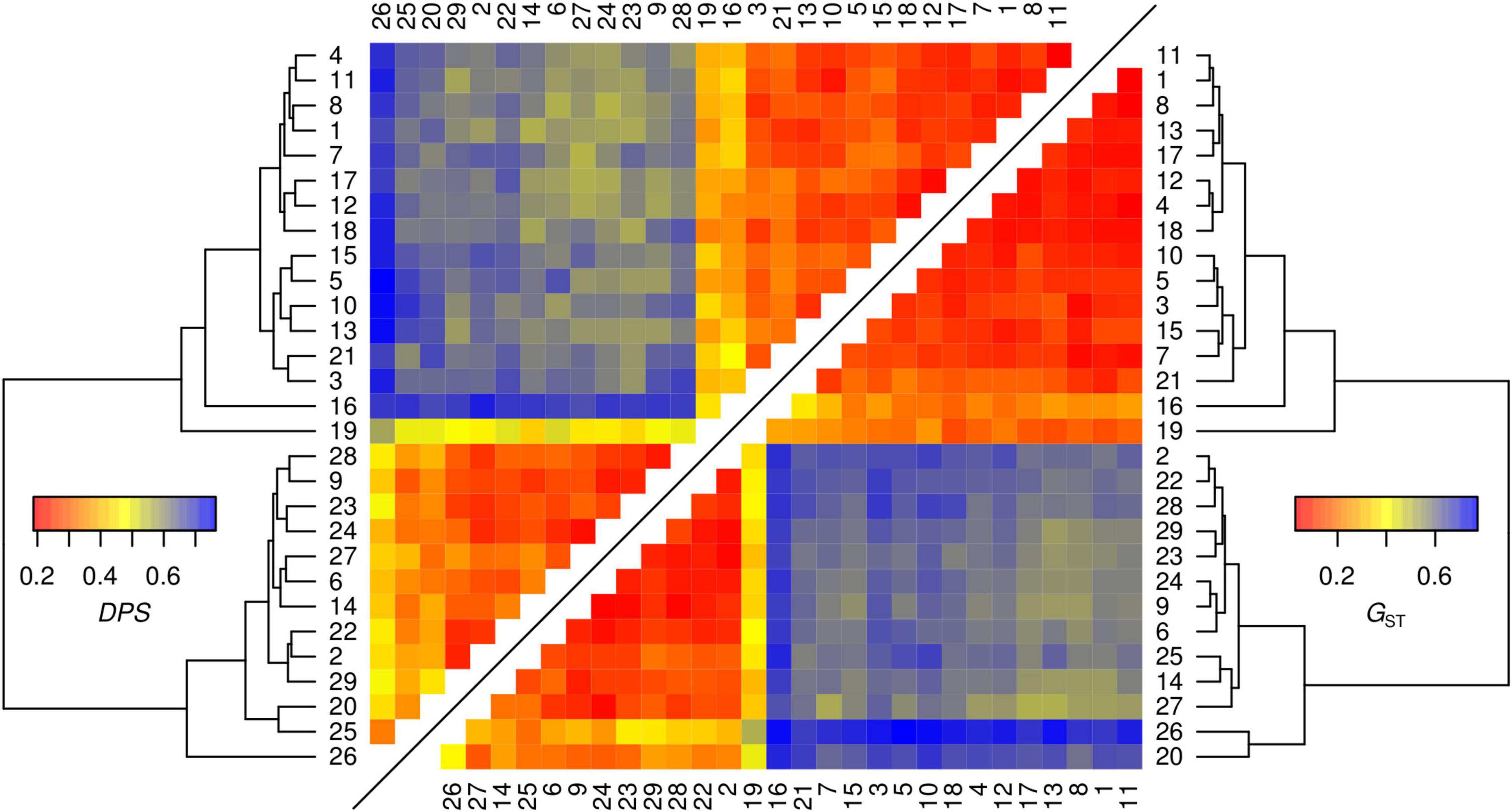
Figure 2. Pairwise heatmaps of genetic distances between the 29 sampled sites with hierarchical clustering dendrograms calculated by two different estimators (proportion of shared alleles DPS and Hedrick’s G’ST) based on genotypes determined on eight microsatellites loci.
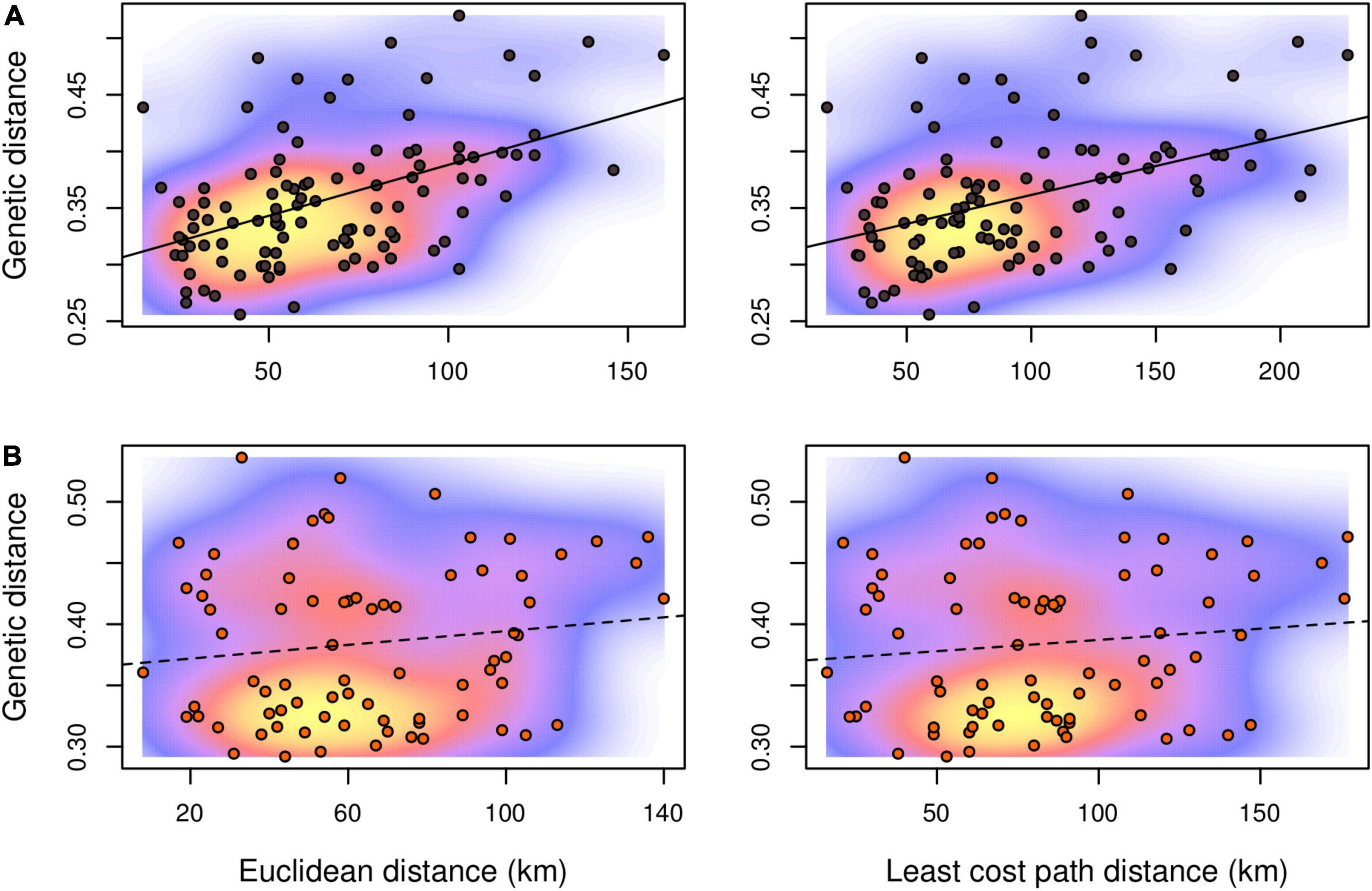
Figure 3. Isolation-by-distance scatterplots with kernel density estimations for pairwise values between R. roeselii local populations sampled in the Lake Mälaren region. (A) Samples assigned to the eastern and (B) to the southern genetic lineage, respectively. Mantel correlations are indicated by full (significant, p < 0.05) or dashed (non-significant, p > 0.05) lines.
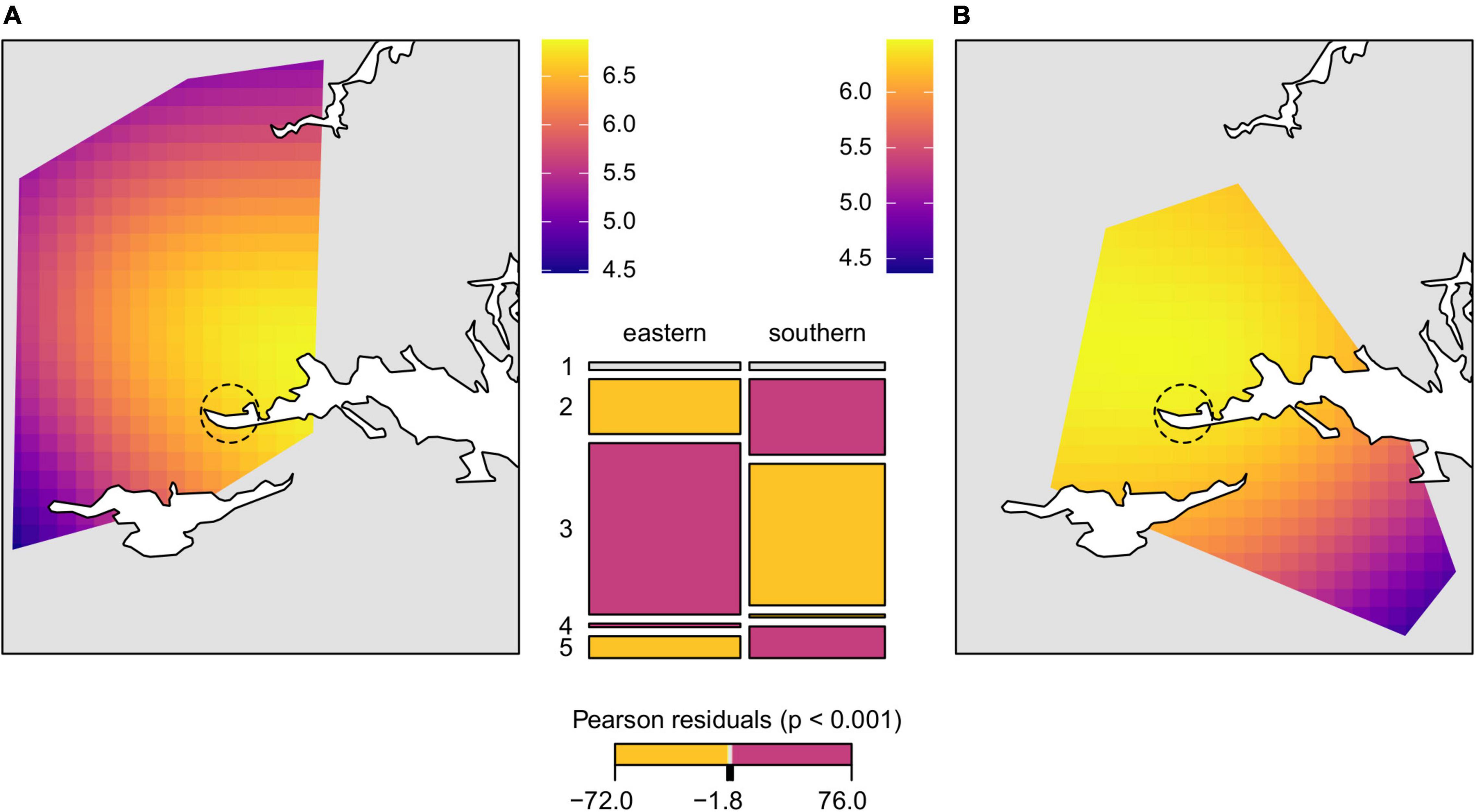
Figure 4. Geospatial interpolation surfaces fitted to the GAM model of allelic richness (colored raster polygons) in (A) the eastern and (B) the southern genetic lineage with superimposed large water bodies in the study area (white). Dashed circle marks suggested area of introduction. Mosaic plot (mid figure) presents the proportion of the main Corine Land Cover classes (1, artificial surfaces; 2, agricultural areas; 3, forest and seminatural areas; 4, wetlands; 5, water bodies) in the areas (minimum convex polygons) colonized by the different lineages. Each tile is colored to show the deviation from the expected frequency (residuals) from a Pearson χ2-test (red—significantly more, yellow—significantly less than expected, gray—non-significant).
We found a striking spatial genetic pattern consisting of two lineages which originate from two parapatric founding populations that today coexist in a large and naturalized population of the Roesel’s bush-cricket outside the native species range. Despite that gene flow creates a stepping stone like pattern of genetic distances among local populations, and that there is a decrease of genetic diversity from central toward marginal areas, there was almost no gene flow between these lineages. Some kind of mechanism, which is not due to physical distance, appears to exist that keeps genetic lineages separated. We have earlier shown that the colonization of R. roeselii in central Sweden is a result of more than one introduction event (Kaňuch et al., 2013). The fact that lineages have continued to stay separated truly was a surprise, especially considering the fact that the species has had a very long time to admix. The two lineages were introduced from the Baltic coast independently and we expect that propagules from the eastern genetic lineage (Gulf of Finland) were introduced more recently, maybe 5–10 decades ago since the species range is still progressing northwards in the source area (Karjalainen, 2009). While it is likely that in the southern lineage populations were established before the nineteenth century from the former Kingdom of Prussia (nowadays Poland and Germany). The current and isolated range of both lineages is similar, but they thrive genetically unmixed. This in spite of that they have been spatially more or less mixed around the Lake Mälaren for many dozens of generations. This finding is in strong contrast to the hypothesis that different genetic lineages will admix when introduced to the same area.
One possible mechanism behind this pattern can be that the lineages (Figure 2) were introduced at sites with some distance between them (e.g., terrestrial distance between potential introduction sites Strömsholm and Kungsör is 25 km) and that they, in combination with two different genetic setups from the start, adapted independently to the local conditions (Ma et al., 2020; see also Cassel-Lundhagen et al., 2011; Kaňuch et al., 2020) before colonizing the larger area. The diversified original gene pool and local adaptation hypothesis would explain the observed pattern if the admixture of alleles resulted in loss of locally adapted genotypes, and a hybrid offspring less viable than the respective ancestral genotypes (e.g., Brideau et al., 2006; Novicic et al., 2011; Rius and Darling, 2014). Rapid evolution of genotypes adapted to local conditions accords well with the fact that there is no evidence that populations of this species suffer from recent bottlenecks and/or high inbreeding level (Kaňuch et al., 2014). Thus, one could expect that there is a substantial genetic capacity to adapt to new environments and little effect of isolation on heterozygosity-fitness correlations. This hypothesis could be further tested with controlled mating experiments. What this finding also raises, is a potentially negative impact that such adaptive process could have for conservation introductions. Conservation management focused on increasing population sizes and species’ distributions by the addition of new individuals, could be less successful if existing adaptions are already present in the target species.
An alternative mechanism that would produce this genetic pattern could be the presence of a post-mating or post-zygotic reproductive barrier and hybrid breakdown phenomenon. It can be caused on one hand by mito-nuclear incompatibilities lowering the fitness of hybrids carrying different lineages’ mitochondrial haplotypes and nuclear genotypes, what has been seen also in related insect species (e.g., Marchant, 1988; Morgan-Richards and Wallis, 2003). On the other hand, Wolbachia bacterial infection manipulates the physiology and reproduction of its hosts and can induce cytoplasmic incompatibility. For example, if uninfected females of one lineage mate with infected males of the other lineage, hybrid offspring or hybrid F2 males can suffer increased mortality (Vala et al., 2000; Bordenstein et al., 2001). Because the frequency of Wolbachia infection is lower toward higher latitudes (Ahmed et al., 2015), this mechanism has an increased likelihood to exist in situations where lineages originate from latitudinally different regions, as in this study (Figure 1C). Due to the lack of biological material from specimens that were used in our study, these hypotheses could not be tested.
To a large extent the least cost pathway analysis did reflect what is known about the species’ movement behavior and its dispersal pattern in previous landscape studies (Berggren et al., 2001, 2002). Even though the dispersal of short-winged forms is tightly linked to movement through suitable habitat and linear elements, this may not be the same for individuals that are accidentally transported by humans. Human-aided dispersal may considerably affect the genetic structure of a population (Hochkirch and Damerau, 2009), similar to the effect of long-winged individuals. Several studies have found that least cost pathways can be poor in estimating the dispersal of individuals that have a different type of dispersal behavior (Simmons and Thomas, 2004; Poniatowski and Fartmann, 2011). Due to the very low proportion of long-winged individuals in the populations (own data) we assume that besides that landscape characteristics predict the species’ occurrence (Preuss et al., 2011), the high frequency of human transportation of agricultural goods and animals among local farms play an important role for the overall spatial genetic pattern of the Roesel’s bush-cricket population in the study area. From the genetic pattern seen in the southern lineage, these local populations may be more affected by this mechanism as neither Euclidean nor least cost path distance resulted in a significant IBD pattern (Figure 3B). This also makes it clear that if reproductively separated genetic clusters are combined in an IBD analysis, this can produce fallacious pattern due to that genetic distances are calculated also for pairwise combinations without possible gene flow (see Figure 3 in Preuss et al., 2015).
To understand the role of humans’ actions in facilitating species spread through the landscape, detailed knowledge not only about the species’ ecology but also the opportunities offered by land managers is needed. To be able to analyze the human-invasive species interactions in more detail, comprehensive data of detailed land use management activities and a knowledge of how these relate to opportunities of spread (e.g., by mediation of transport) are required. Human aided colonization and spread of species often go unnoticed when they happen (Hammer and Jensen, 2019; Sherpa et al., 2020). This is partly due to a lack of awareness on how one’s actions link to likelihood of invasions (Shannon et al., 2019, 2020). With an increasing trade of agricultural goods and organisms both within and between countries, people actively managing the land such as farmers may increasingly act as facilitators of invasive species spread. This will not always directly affect the farmers themselves when it comes to some organisms (which primarily constitute a threat to natural communities), but for other organisms, this may be a direct threat to domestic species important to these land managers (Bajwa et al., 2019; Pardo et al., 2020).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
AC-L and ÅB conceived the project. SP collected and genotyped the samples. PK analyzed the data and prepared the figures. PK, ÅB, and AC-L wrote the manuscript. SP and GN reviewed drafts of the manuscript. All authors approved the final draft.
This research was funded by the Swedish research Council for Sustainable Development (FORMAS) grant no. 2006-01869 to ÅB. PK was supported by the Slovak Scientific Grant Agency VEGA (2/0107/21). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Carin Eriksson for assistance in the field.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.812079/full#supplementary-material
Ahmed, M. Z., Araujo-Jnr, E. V., Welch, J. J., and Kawahara, A. Y. (2015). Wolbachia in butterflies and moths: geographic structure in infection frequency. Front. Zool. 12:16. doi: 10.1186/s12983-015-0107-z
Bajwa, A. A., Farooq, M., Nawaz, A., Yadav, L., Chauhan, B. S., and Adkins, S. (2019). Impact of invasive plant species on the livelihoods of farming households: evidence from Parthenium hysterophorus invasion in rural Punjab, Pakistan. Biol. Invasions 21, 3285–3305. doi: 10.1007/s10530-019-02047-0
Baker, C. M. (2017). Target the source: optimal spatiotemporal resource allocation for invasive species control. Conserv. Lett. 10, 41–48. doi: 10.1111/conl.12236
Beckmann, B. C., Purse, B. V., Roy, D. B., Roy, H. E., Sutton, P. G., and Thomas, C. D. (2015). Two species with an unusual combination of traits dominate responses of British grasshoppers and crickets to environmental change. PLoS One 10:e0130488. doi: 10.1371/journal.pone.0130488
Bellmann, H. (2006). Der Kosmos Heuschreckenfürer – Die Arten Mitteleuropas sicher bestimmen. Stuttgart: Franckh-Kosmos Verlags-Gmbh and Co.
Berggren, Å (2004). Impact of grazing on individual male movement in Roesel’s bush-cricket Metrioptera roeseli: One possible clue to species range expansion. J. Insect Behav. 17, 419–429. doi: 10.1023/B:JOIR.0000042531.27859.ac
Berggren, Å (2005). The effect of conspecifics on individual male movement in Roesel’s bush cricket, Metrioptera roeseli. Ecol. Entomol. 30, 480–483. doi: 10.1111/j.0307-6946.2005.00709.x
Berggren, Å (2009). Effects of landscape and population variables on immune response in experimentally introduced bush-cricket populations. Landsc. Ecol. 24, 749–757. doi: 10.1007/s10980-009-9348-6
Berggren, Å, Birath, B., and Kindvall, O. (2002). Effect of corridors and habitat edges on dispersal behavior, movement rates, and movement angles in Roesel’s bush-cricket (Metrioptera roeseli). Conserv. Biol. 16, 1562–1569. doi: 10.1046/j.1523-1739.2002.01203.x
Berggren, Å, Carlson, A., and Kindvall, O. (2001). The effect of landscape composition on colonization success, growth rate and dispersal in introduced bush-crickets Metrioptera roeseli. J. Anim. Ecol. 70, 663–670. doi: 10.1046/j.1365-2656.2001.00525.x
Bordenstein, S., O’Hara, F., and Werren, J. (2001). Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature 409, 707–710. doi: 10.1038/35055543
Bowcock, A. M., Ruiz-Linares, A., Tomfohrde, J., Minch, E., Kidd, J. R., and Cavalli-Sforza, L. L. (1994). High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368, 455–457. doi: 10.1038/368455a0
Brideau, N. J., Flores, H. A., Wang, J., Maheshwari, S., Wang, X., and Barbash, D. A. (2006). Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science 314, 1292–1295. doi: 10.1126/science.1133953
Brookfield, J. F. Y. (1996). A simple new method for estimating null allele frequency from heterozygote deficiency. Mol. Ecol. 5, 453–455. doi: 10.1111/j.1365-294x.1996.tb00336.x
Campana, M. G., Hunt, H. V., Jones, H., and White, J. (2011). CorrSieve: software for summarising and evaluating STRUCTURE output. Mol. Ecol. Res. 11, 349–352. doi: 10.1111/j.1755-0998.2010.02917.x
Cassel-Lundhagen, A., Kaňuch, P., Low, M., and Berggren, Å (2011). Limited gene flow may enhance adaptation to local optima in isolated populations of the Roesel’s bush cricket (Metrioptera roeselii). J. Evol. Biol. 24, 381–390. doi: 10.1111/j.1420-9101.2010.02174.x
Cavalli-Sforza, L. L., and Edwards, A. W. F. (1967). Phylogenetic analysis. Models and estimation procedures. Am. J. Hum. Genet. 19, 233–257.
Chakraborty, R., Zhong, Y., Jin, L., and Budowle, B. (1994). Nondetectability of restriction fragments and independence of DNA fragment sizes within and between loci in RFLP typing of DNA. Am. J. Hum. Genet. 55, 391–401.
Chen, K. Y., Marschall, E. A., Sovic, M. G., Fries, A. C., Gibbs, H. L., and Ludsin, S. A. (2018). assign POP: An R package for population assignment using genetic, non-genetic, or integrated data in a machine-learning framework. Methods Ecol. Evol. 9, 439–446. doi: 10.1111/2041-210X.12897
R Core Team (2020). R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing
De Jong, J., and Kindvall, O. (1991). Cikadavårtbitaren Metrioptera roeselii – nykomling eller hotad relikt? Fauna och Flora 86, 215–221.
Di Castri, F. (1989). “History of biological invasions with emphasis on the Old World,” in Biological Invasions: a Global Perspective, eds J. Drake, F. di Castri, and R. Groves (New York, NY: Wiley), 1–30.
Dlugosch, K. M., Anderson, S. R., Braasch, J., Cang, F. A., and Gillette, H. D. (2015). The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Mol. Ecol. 24, 2095–2111. doi: 10.1111/mec.13183
Earl, D. A., and von Holdt, B. M. (2012). Structure harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv. Genet. Res. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Edmands, S. (2007). Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 16, 463–475. doi: 10.1111/j.1365-294x.2006.03148.x
Eriksson, A., Low, M., and Berggren, Å (2013). Influence of linear versus network corridors on the movement and dispersal of the bush-cricket Metrioptera roeseli (Orthoptera: Tettigoniidae) in an experimental landscape. Eur. J. Entomol. 110, 81–86. doi: 10.14411/eje.2013.010
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Fidler, A. E., Bacq-Labreuil, A., Rachmilovitz, E., and Rinkevich, B. (2018). Efficient dispersal and substrate acquisition traits in marine invasive species via transient chimerism and colony mobility. PeerJ 6:e5006. doi: 10.7717/peerj.5006
Galpern, P., Peres-Neto, P. R., Polfus, J., and Manseau, M. (2014). MEMGENE: Spatial pattern detection in genetic distance data. Methods Ecol. Evol. 5, 1116–1120. doi: 10.1111/2041-210X.12240
Greenwald, K. R., Purrenhage, J. L., and Savage, W. K. (2009). Landcover predicts isolation in Ambystoma salamanders across region and species. Biol. Conserv. 142, 2493–2500. doi: 10.1016/j.biocon.2009.05.021
Gruber, B., and Adamack, A. (2019). PopGenReport: A Simple FrameWork to Analyse Population and Landscape Genetic Data. R package version 3.04. https://cran.r-project.org/package=popgenreport (accessed April 1, 2021).
Hagenbach, J. J. (1822). Symbola Faunae Insectorum Helvetiae Exhibentia vel Species Novas vel Nondum Depictas. Basel: Typis J. Georgii.
Hammer, S., and Jensen, J.-K. (2019). The invasion of two species of social wasps (Hymenoptera, Vespidae) to the Faroe Islands. BioInv. Rec. 8, 558–567. doi: 10.3391/bir.2019.8.3.11
Hedrick, P. W. (2005). A standardized genetic differentiation measure. Evolution 59, 1633–1638. doi: 10.1111/j.0014-3820.2005.tb01814.x
Hijmans, R. J. (2020). Raster: Geographic Data Analysis and Modeling. R package Version 3.0-12. https://cran.r-project.org/package=raster (accessed April 15, 2021).
Hochkirch, A., and Damerau, M. (2009). Rapid range expansion of a wing-dimorphic bush-cricket after the 2003 climatic anomaly. Biol. J. Linn. Soc. 97, 118–127. doi: 10.1111/j.1095-8312.2008.01199.x
Holzhauer, S., Ekschmitt, K., Sander, A.-C., Dauber, J., and Wolters, V. (2006). Effect of historic landscape change on the genetic structure of the bush-cricket Metrioptera roeseli. Landsc. Ecol. 21, 891–899. doi: 10.1007/s10980-005-0438-9
Holzhauer, S. I. J., and Wolff, K. (2005). Polymorphic microsatellite loci in the bush-cricket Metrioptera roeseli. Mol. Ecol. Notes 5, 502–503. doi: 10.1111/j.1471-8286.2005.00970.x
Hubisz, M. J., Falush, D., Stephens, M., and Pritchard, J. K. (2009). Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Res. 9, 1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x
Ingrisch, S. (1984). The influence of environmental factors on dormancy and duration of egg development in Metrioptera roeseli (Orthoptera: Tettigoniidae). Oecologia 61, 254–258. doi: 10.1007/bf00396769
Jombart, T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Kaňuch, P., Berggren, Å, and Cassel-Lundhagen, A. (2013). Colonization history of Metrioptera roeselii in northern Europe indicates human-mediated dispersal. J. Biogeogr. 40, 977–987. doi: 10.1111/jbi.12048
Kaňuch, P., Berggren, Å, and Cassel-Lundhagen, A. (2014). Genetic diversity of a successful colonizer: isolated populations of Metrioptera roeselii regain variation at an unusually rapid rate. Ecol. Evol. 4, 1117–1126. doi: 10.1002/ece3.1005
Kaňuch, P., Cassel-Lundhagen, A., and Berggren, Å (2021). A clue to invasion success: genetic diversity quickly rebounds after introduction bottlenecks. Biol. Invasions 23, 1141–1156. doi: 10.1007/s10530-020-02426-y
Kaňuch, P., Kiehl, B., Cassel-Lundhagen, A., Laugen, A. T., Low, M., and Berggren, Å (2020). Gene flow relates to evolutionary divergence among populations at the range margin. PeerJ 8:e10036. doi: 10.7717/peerj.10036
Kaňuch, P., Pfunder, M., Berggren, Å, and Cassel-Lundhagen, A. (2010). Description of nine new microsatellite loci in Metrioptera roeselii (Orthoptera:Tettigoniidae) and their multiplex PCR protocols. Mol. Ecol. Res. 10, 404–408.
Kimura, M., and Weiss, G. H. (1964). Stepping stone model of population structure and decrease of genetic correlation with distance. Genetics 49, 561–576.
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., and Mayrose, I. (2015). CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 15, 1179–1191. doi: 10.1111/1755-0998.12387
Lanner, J., Gstöttenmayer, F., Curto, M., Geslin, B., Huchler, K., Orr, M. C., et al. (2021). Evidence for multiple introductions of an invasive wild bee species currently under rapid range expansion in Europe. BMC Ecol. Evol. 21:17. doi: 10.1186/s12862-020-01729-x
Liebhold, A. M., and Kean, J. M. (2019). Eradication and containment of non-native forest insects: successes and failures. J. Pest Sci. 92, 83–91. doi: 10.1007/s10340-018-1056-z
Ma, L., Cao, L.-J., Hoffmann, A. A., Gong, Y.-J., Chen, J.-C., Chen, H.-S., et al. (2020). Rapid and strong population genetic differentiation and genomic signatures of climatic adaptation in an invasive mealybug. Divers. Distrib. 26, 610–622. doi: 10.1111/ddi.13105
MacDonald, Z. G., Anderson, I. D., Acorn, J. H., and Nielsen, S. E. (2018). Decoupling habitat fragmentation from habitat loss: butterfly species mobility obscures fragmentation effects in a naturally fragmented landscape of lake islands. Oecologia 186, 11–27. doi: 10.1007/s00442-017-4005-2
Marchant, A. D. (1988). Apparent introgression of mitochondrial DNA across a narrow hybrid zone in the Caledia captiva species-complex. Heredity 60, 39–46. doi: 10.1038/hdy.1988.7
Marshall, J. A., and Haes, E. C. M. (1988). Grasshoppers and Allied Insects of Great Britain and Ireland. Colchester: Harley Books.
Medley, K. A., Westby, K. M., and Jenkins, D. G. (2019). Rapid local adaptation to northern winters in the invasive Asian tiger mosquito Aedes albopictus: a moving target. J. Appl. Ecol. 56, 2518–2527. doi: 10.1111/1365-2664.13480
Melero, Y., Stefanescu, C., Palmer, S. C. F., and Travis, L. M. L. (2020). The role of the urban landscape on species with contrasting dispersal ability: insights from greening plans for Barcelona. Landsc. Urban Plan. 195:103707. doi: 10.1016/j.landurbplan.2019.103707
Meyer, D., Zeileis, A., and Hornik, K. (2020). vcd: Visualizing Categorical Data. R package version 1.4–6. https://cran.r-project.org/package=vcd (accessed March 25, 2021).
Morgan-Richards, M., and Wallis, G. P. (2003). A comparison of five hybrid zones of the weta Hemideina thoracica (Orthoptera: Anostostomatidae): degree of cytogenetic differentiation fails to predict zone width. Evolution 57, 849–861. doi: 10.1111/j.0014-3820.2003.tb00296.x
Novicic, Z. K., Stamenkovic-Radak, M., Pertoldi, C., Jelic, M., Veselinovic, M. S., and Andjelkovic, M. (2011). Heterozygosity maintains developmental stability of sternopleural bristles in Drosophila subobscura interpopulation hybrids. J. Insect Sci. 11:113. doi: 10.1673/031.011.11301
Ordóñez, V., Pascual, M., Rius, M., and Turon, X. (2013). Mixed but not admixed: a spatial analysis of genetic variation of an invasive ascidian on natural and artificial substrates. Mar. Biol. 160, 1645–1660. doi: 10.1007/s00227-013-2217-5
Pardo, G., Gómez, M. I., Cirujeda, A., and Martínez, Y. (2020). Economic cost of sharing the harvester in the control of an invasive weed. Sustainability 12:9046. doi: 10.3390/su12219046
Parker, P. G., Snow, A. A., Schug, M. D., Booton, G. C., and Fuerst, P. A. (1998). What molecules can tell us about populations: choosing and using molecular marker. Ecology 79, 361–382. doi: 10.2307/176939
Poniatowski, D., and Fartmann, T. (2005). Die Ökologie von Roesels Beißschrecke (Metrioptera roeselii) im Feuchtgrünland der Medebacher Bucht (Südwestfalen). Articulata 20, 85–111.
Poniatowski, D., and Fartmann, T. (2011). Does wing dimorphism affect mobility in Metrioptera roeselii (Orthoptera: Tettigoniidae)? Eur. J. Entomol. 108, 409–415. doi: 10.14411/eje.2011.052
Poniatowski, D., Heinze, S., and Fartmann, T. (2012). The role of macropters during range expansion of a wing-dimorphic insect species. Evol. Ecol. 26, 759–770. doi: 10.1007/s10682-011-9534-2
Preuss, S., Berggren, Å, and Cassel-Lundhagen, A. (2015). Genetic patterns reveal an old introduction event and dispersal limitations despite rapid distribution expansion. Biol. Invasions 17, 2851–2862. doi: 10.1007/s10530-015-0915-2
Preuss, S., Cassel-Lundhagen, A., and Berggren, Å (2011). Modelling the distribution of Roesel’s bush-cricket (Metrioptera roeselii) in a fragmented landscape. NeoBiota 11, 33–49. doi: 10.3897/neobiota.11.2060
Preuss, S., Low, M., Cassel-Lundhagen, A., and Berggren, Å (2014). Evaluating range-expansion models for calculating non-native species expansions. Ecol. Evol. 4, 2812–2822. doi: 10.1002/ece3.1106
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Rius, M., and Darling, J. A. (2014). How important is intraspecific genetic admixture to the success of colonising populations? Trends Ecol. Evol. 29, 233–242. doi: 10.1016/j.tree.2014.02.003
Sawyer, S. C., Epps, C. W., and Brashares, J. S. (2011). Placing linkages among fragmented habitats: do least-cost models reflect how animals use landscapes? J. Appl. Ecol. 48, 668–678. doi: 10.1111/j.1365-2664.2011.01970.x
Shannon, C., Quinn, C. H., Sutcliffe, C., Stebbing, P. D., Dally, T., Glover, A., et al. (2019). Exploring knowledge, perception of risk and biosecurity practises among researchers in the UK: a quantitative survey. Biol. Invasions 21, 303–314. doi: 10.1007/s10530-018-1837-6
Shannon, C., Stebbing, P. D., Quinn, C. H., Warren, D. A., and Dunn, A. M. (2020). The effectiveness of e-learning on biosecurity practice to slow the spread of invasive alien species. Biol. Invasions 22, 2559–2571. doi: 10.1007/s10530-020-02271-z
Sherpa, S., Renaud, J., Gueguen, M., Besnard, G., Mouyon, L., Rey, D., et al. (2020). Landscape does matter: disentangling founder effects from natural and human-aided post-introduction dispersal during an ongoing biological invasion. J. Anim. Ecol. 89, 2027–2042. doi: 10.1111/1365-2656.13284
Simmons, A. D., and Thomas, C. D. (2004). Changes in dispersal during species’ range expansions. Am. Nat. 164, 378–395. doi: 10.1086/423430
Strengbom, A. (2019). Förundersökning vid Kungörs kungsgård. Rapport 2019:48. Örebro: Arkeologgruppen AB.
Vala, F., Breeuwer, J. A. J., and Sabelis, M. W. (2000). Wolbachia-induced ‘hybrid breakdown’ in the two-spotted spider mite Tetranychus urticae Koch. Proc. R. Soc. Lond. B 267, 1931–1937. doi: 10.1098/rspb.2000.1232
Vickery, V. R. (1965). Factors governing the distribution and dispersal of the recently introduced grasshopper, Metrioptera roeseli (Hgb.) (Orthoptera: Ensifera). Annales de la Societe Entomologique du Quebec 10, 165–171.
Wagner, C. (2004). Passive dispersal of Metrioptera bicolor (Phillipi 1830) (Orthopteroidea: Ensifera: Tettigoniidae) by transfer of hay. J. Insect Conserv. 8, 287–296. doi: 10.1007/s10841-004-0404-x
Wamser, S., Diekötter, T., Boldt, L., Wolters, V., and Dauber, J. (2012). Trait-specific effects of habitat isolation on carabid species richness and community composition in managed grasslands. Insect Conserv. Diver. 5, 9–18. doi: 10.1111/j.1752-4598.2010.00110.x
Williams, H. E., Brockerhoff, E. G., Liebhold, A. M., and Ward, D. F. (2021). Mechanisms driving component Allee effects during invasions; using a biological control agent as model invader. Ecol. Entomol. 46, 1205–1214. doi: 10.1111/een.13068
Wissmann, J., Schielzeth, H., and Fartmann, T. (2009). Landscape-scale expansion of Roesel’s bush-cricket Metrioptera roeselii at the north-western range limit in central Europe (Orthoptera: Tettigoniidae). Entomol. Gen. 31, 317–326.
Wood, S. (2019). Mgcv: Mixed GAM Computation Vehicle with Automatic Smoothness Estimation. R package version 1.8-28. https://cran.r-project.org/package=mgcv (accessed April 15, 2021).
Keywords: human assisted dispersal, landscape connectivity, land-use, microsatellites, Orthoptera
Citation: Kaňuch P, Cassel-Lundhagen A, Preuss S, Nordlander G and Berggren Å (2022) Parapatric Genetic Lineages Persist in a Multiply Introduced Non-native Bush-Cricket. Front. Ecol. Evol. 10:812079. doi: 10.3389/fevo.2022.812079
Received: 09 November 2021; Accepted: 10 February 2022;
Published: 22 March 2022.
Edited by:
Matthew B. Hamilton, Georgetown University, United StatesReviewed by:
Ana-Maria Krapal, Grigore Antipa National Museum of Natural History, RomaniaCopyright © 2022 Kaňuch, Cassel-Lundhagen, Preuss, Nordlander and Berggren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Åsa Berggren, YXNhLmJlcmdncmVuQHNsdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.