 Sean Obrochta
Sean Obrochta Maria Luisa Savo Sardaro
Maria Luisa Savo Sardaro Katherine R. Amato
Katherine R. Amato Maureen H. Murray
Maureen H. Murray- 1Department of Biology, Northwestern University, Evanston, IL, United States
- 2Department of Anthropology, Northwestern University, Evanston, IL, United States
- 3Department of Conservation and Science, Urban Wildlife Institute, Lincoln Park Zoo, Chicago, IL, United States
Microbiome analysis presents an opportunity to understand how urban environments affect avian physiology. For example, habitat use can affect microbiome diversity and composition, and hosts with more diverse gut microbiota are thought to be more resistant to pathogens and have increased fitness. However, the microbiome is an understudied aspect of avian ecology, particularly in the context of migration and urbanization in wild birds. For this study, we hypothesized that, within urban birds, migrants would exhibit greater microbial diversity and inter-individual variation in microbiome composition than residents because they are exposed to more diverse habitats. We focused on Canada geese (Branta canadensis), one of many migratory species that exhibit increasingly more year-round residency in cities. We used 16S rRNA gene amplicon sequencing to quantify microbiome taxonomic composition in fecal samples from 32 GPS-tracked Canada geese, 22 of which were year-round residents of the Chicago area and 10 of which were migrants. Similar to recent studies on wild species feeding near human habitation, urban resident geese had higher gut microbial diversity than migrants. They also had increased inter-individual variation in microbiome composition and, on average, lower relative abundances of bacteria in the phylum Firmicutes, and the genera Terrisporobacter, Turicibacter, and Cellulosilyticum, which all have metabolic functions that may aid in goose digestion. Therefore, the gut microbiome of resident geese may provide fewer potential health benefits. These patterns may be a result of anthropogenic influences on aspects of resident goose ecology, such as diet, as well the influence of migration on migrant goose ecology and biology. Overall, our results suggest that reduced migration for urban-adapted wildlife species may have important consequences for physiology and health.
Introduction
Migratory birds are increasingly exposed to urban environments as anthropogenic activities lead to altered land use practices. Understanding the impact of this environmental change on migratory birds is essential. Over 10% of migratory birds are currently threatened worldwide, and this percentage will likely increase due to habitat loss and climate change (Runge et al., 2015; Zurell et al., 2018). However, it can be difficult and costly to study migratory populations, as they inhabit different global regions and ecosystem types throughout the year. For migratory species that persist in urban environments, existing research demonstrates that living in urban areas can affect avian ecology and physiology through changes in habitat use, diet, exposure to pollution, and stress levels (Ruiz et al., 2002; Meillère et al., 2016; Murray et al., 2019). In addition, urbanization appears to contribute to changes in long-distance movements. Over the past decades, there has been an increasing prevalence of partially migratory species (i.e., species in which some populations undergo seasonal long-distance flights to and from specific regions while others do not). In many cases, the non-migratory individuals of these species exhibit year-round residency in urban areas, likely because of consistent resource availability and more moderate microclimates (Wiener and Tuljapurkar, 1994; Møller et al., 2014; Bonnet-Lebrun et al., 2020). One study found that living in urban areas increased the likelihood of residency in 9 of 12 partially migratory species (Bonnet-Lebrun et al., 2020). Another reported that shifts in the timing of food availability and breeding due to climate change favored shorter migrations and year-long residency in South German Blackcaps (Sylvia atricapilla) (Pulido and Berthold, 2010; Soriano-Redondo et al., 2020).
While some evidence indicates that resident birds have a larger average mass than their migratory counterparts, the impact of year-long urban residency on the health and physiology of partially migratory birds is not well-studied (Pérez-Tris and Tellería, 2002). Gut microbiome analysis offers a new non-invasive tool for studying these outcomes because it plays a critical role in host immune development, hormone regulation, and digestion in a variety of animal species (West et al., 2019; Mueller et al., 2020). The avian gut microbiome is strongly influenced by environmental factors such as habitat and diet (Hird et al., 2014; Waite and Taylor, 2015; Xie et al., 2016; Grond et al., 2017; Wu et al., 2018; Cao et al., 2020); therefore, changes in microbiome composition and/or diversity can have important consequences for avian health and fitness.
The effects of migration and urbanization on bird microbiomes has only been studied in a few populations. However, both migration and urbanization appear to affect the microbiome. Because migratory birds experience marked shifts in their environment, they are also more likely to exhibit shifts in the microbiome over time (Liu and Swanson, 2014). For instance, certain species of migratory birds change their diet preceding and during their migrations, which could result in a shift in the microbiome during migration (McLandress and Raveling, 1981). In migratory swan geese (Anser cygnoides), microbial communities shift between their breeding grounds and wintering area, with a core group of shared microbial taxa at both locations (Wu et al., 2018). Additionally, migratory birds often exhibit heightened stress responses during their migration due to the demanding physical toll of migration, and heightened stress has been linked to temporary changes in the microbiome in some animals (Liu and Swanson, 2014). The extent to which these dynamics are beneficial to hosts or induce negative health consequences is unclear (Risely et al., 2018). In contrast, urbanization appears to result in lower microbiome diversity in many current studies. Urban American white ibises (Eudocimus albus), house sparrows (Passer domesticus), and herring gulls (Larus argentatus) all exhibit lower microbial diversity (Fuirst et al., 2018; Teyssier et al., 2018; Murray et al., 2020), potentially due to the lower environmental diversity and smaller home ranges. In several bird species, the presence of Firmicutes, a key microbial taxon for dietary carbohydrate degradation, is negatively correlated with increased urban land cover (Drovetski et al., 2018; Wu et al., 2018; Murray et al., 2020). Therefore, year-long urban resident individuals of partially migratory species are likely to have distinct microbiomes compared to migratory individuals.
To begin to test this broad hypothesis, we assessed microbiome composition in Canada geese (Branta canadensis) in and around Chicago, Illinois, United States. The Canada goose is a large anatid waterfowl native to most of northern North America (Smith et al., 1999). Some populations of Canada geese migrate in fall and spring to their respective wintering and summering grounds, while others remain in one home range year-round (Conover, 1991). As a result, Canada geese have become common in urban areas and tend to be year-round residents, primarily feeding on lawn grasses, although migratory flocks can also be found in urban areas (Conover, 1991). The physiological differences between migratory and resident populations have been seldom studied. Although urban habitats may provide some benefits to geese, such as protection from predators and human hunting (Balkcom, 2010), urban-associated shifts in diet and movements might shift microbiome composition and diversity, affecting their health.
In this study, we assessed the microbiome composition of fecal samples from GPS-tracked individual Canada Geese categorized as either resident or migratory but occupying the same urban areas in Chicago, Illinois, United States. Based on the existing literature, we had two main predictions. First, we predicted that migrants would exhibit greater microbial diversity and inter-individual variation in microbiome composition compared to urban residents because they are exposed to more diverse habitats. Second, we predicted that migrants would have higher relative abundances of beneficial bacteria such as Firmicutes relative to residents because of their increased energetic demands. Given the current paucity of data in this area, our results will provide an important foundation for understanding the effects of year-long residency on avian microbiomes and, ultimately, physiology and health.
Materials and Methods
Fecal Sample Collection
We collected 36 fecal samples from 32 geese in spring and fall 2018. Of the 36 fecal samples used in this study, 29 were collected between September and November (henceforth referred to as the “fall” samples), and 7 were collected between April and August (henceforth referred to as the “spring” samples). The geese in this study were previously banded and attached with GPS transmitters as part of a concurrent project on goose movement in Chicago (Askren et al., 2019). Migrant geese were identified as those birds that were captured and collared in the Chicago area but spent the breeding season outside of Illinois, traveling up to 2,000 miles to Northern Canada (Figure 1). We collected fecal samples from migrant geese within 1 week of their return to the Chicago area in the fall for the non-breeding season. We collected 26 samples from 22 residents and 10 samples from 10 migrants. Of the resident geese, four were sampled twice (43R, 46R, 52R, and 54R). The geese were located via visual confirmation of neck band codes at their latest GPS location in Chicago. Once spotted, we conducted behavioral observations and collected a fecal sample within 5 min of deposition. Location data, designated quantitatively by latitude and longitude and qualitatively by the name of the site where a bird was sampled, was also recorded. In some cases, GPS data at the exact time of fecal sampling was unavailable, so the latitude and longitude were approximated by averaging out the GPS data from the sampling day while a goose was at the sampling site. The feces were collected in 5 mL tubes and were kept on ice in coolers before being placed in long-term storage at −80°C within 2 h of collection.
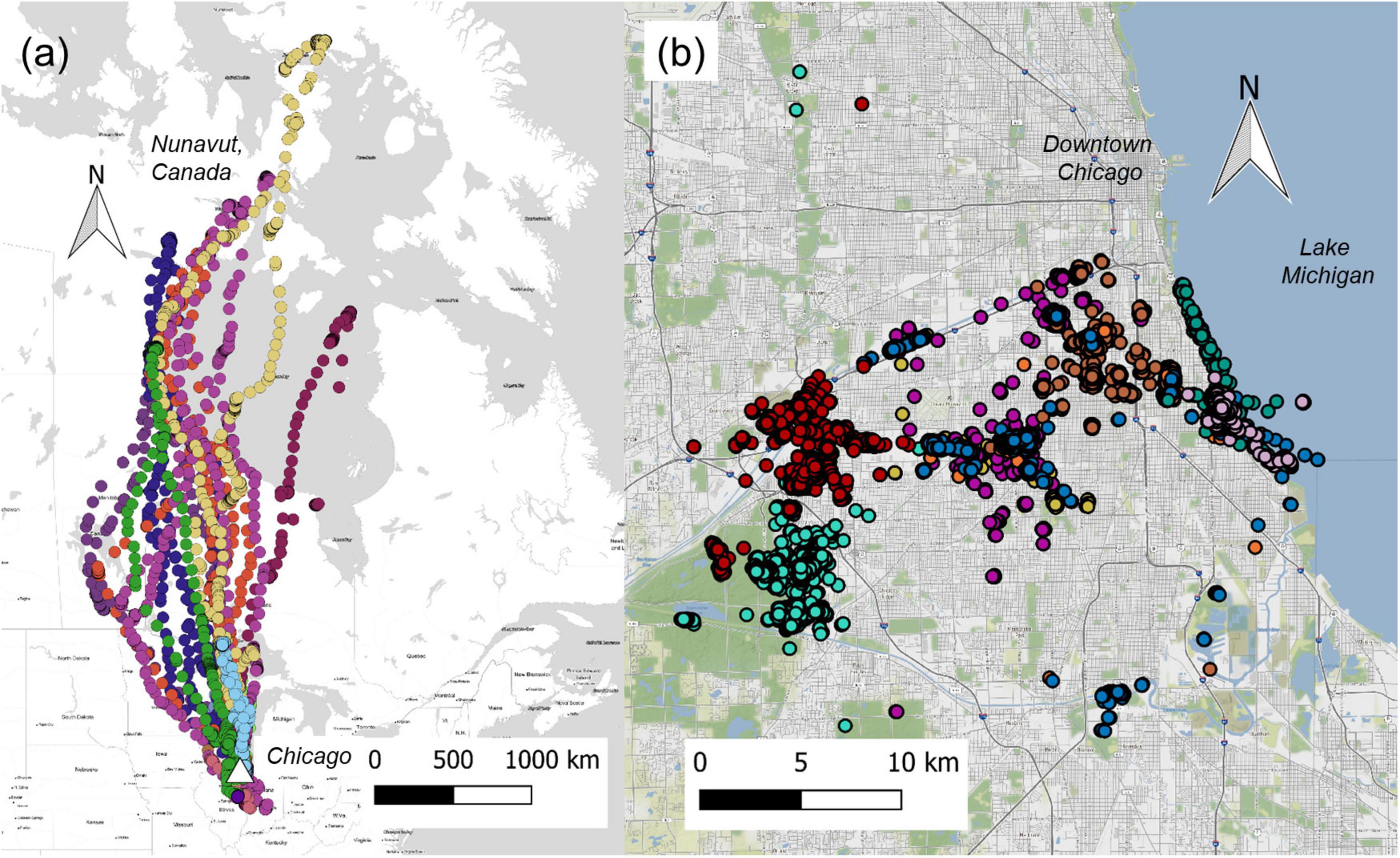
Figure 1. Map of GPS locations of Canada geese (n = 32) taken hourly during daytime hours from May 2018 to October 2019. We categorized geese as migrants if they left the state of Illinois for the breeding season in 2018 (a; n = 10) and as residents if they stayed in Chicago year-round (b; n = 22). Chicago is indicated as a white triangle in (a). Basemaps were created by OpenStreetMap under an open license.
Fecal Sample Processing and Sequencing
To describe the microbiome in these 36 fecal samples, we extracted DNA using the Qiagen Powersoil Kit with an incubation period of 10 min at 65°C added to the standard protocol to increase output. The fecal samples were extracted on three total plates, and a negative control containing no fecal matter was included in each session. Samples were extracted in order of priority, rather than randomly assigned to a plate, because lab access was uncertain due to COVID-19 protocols. As a consequence, almost all of the migrant samples were included in the same DNA extraction round. After extraction, the V4-V5 region of the 16S ribosomal RNA gene was amplified using a modified version of the PCR Earth Microbiome Project protocol (Thompson et al., 2017; Mallott and Amato, 2018) and the 515Fa/926R primer set (Walters et al., 2016). Negative controls were included in both rounds of PCR. After both rounds of PCR, gel electrophoresis was conducted to ensure that the negative controls contained no DNA. The DNA band during the gel electrophoresis for sample C195 appeared weak, so two separate PCR products were created from the original C195 extraction. The resulting amplicons were sequenced on the Illumina MiSeq V2 platform at the DNA Services Facility at the University of Illinois at Chicago. In total, 37 fecal PCR products and 5 negative controls (3 from DNA extraction and 2 from PCR) were sent in for sequencing.
Statistical Analysis
After sequencing, there were a total of 599,762 sequences with an average of approximately 14,600 sequences per sample, ranging between 46 sequences (the negative control for the PCR) to 28,198 sequences (goose 52R). These sequences were processed using QIIME2 (Bolyen et al., 2019). The DADA2 algorithm was used to filter the forward sequences to include only those between 20 and 260 base pairs, and for the reverse sequences, between 21 and 240 base pairs. A total of 362,508 sequences were retained across the 33 samples that were not excluded, with an average of approximately 11,000 sequences per sample, ranging from 6,410 sequences (goose 88C) to 16,996 (goose 46R3). After quality filtering, we used the DADA2 algorithm to cluster amplicon sequence variants (ASVs) and assigned taxonomy (at the phylum and genus level) using a pre-trained Bayesian classifier and the GreenGenes 13.8 reference database. ASVs from mitochondria and chloroplasts were excluded, and the data were rarified to 6,400 reads per sample. The rarifying step excluded four fecal samples (13Y, 85C, 31, 42R) that had less than 6,400 sequences.
Due to the contamination seen in the third negative DNA extraction control, we ran PERMANOVA and ANOVA tests for the alpha diversity and beta diversity metrics to discern whether the DNA extraction round affected the results. Ultimately, we determined that the DNA extraction round and associated contamination did not drive the trends in the data (Supplementary Tables A–E). Therefore, we used the full dataset for all subsequent analyses.
We generated alpha diversity (richness, Faith’s Phylogenetic, Shannon) and beta diversity indices (unweighted and weighted UniFrac) using the core diversity script in QIIME2. Analysis of variance (ANOVA) was conducted to determine whether there was significant variation between migrant and resident alpha diversity. The car (Fox and Weisberg, 2019), fdrtool (Strimmer, 2008), and tidyverse (Wickham et al., 2019) packages in R version 4.0.4 (R Core Team, 2019) were utilized for the ANOVA. Non-metric multidimensional scaling (NMDS) plots were constructed in R for data visualization. We used permutational analysis of variance (PERMANOVA) using the data.table and vegan packages in RStudio to test if the overall microbiome composition was significantly different between migratory and residential Canada goose populations or as a function of sampling location, We also conducted a beta dispersion test to determine whether there was a significant difference in the inter-individual variation of microbiome composition within the migrant and resident groups. We accounted for both season and individual identity in all of our models.
To further explore the effect of location on microbiome composition, we conducted Mantel tests to detect any correlation between the geographical distance between sampling sites (calculated using the latitude and longitude from the bird GPS data) and both the unweighted and weighted UniFrac distances using the ade4 package in R. We included both weighted and unweighted analyses because unweighted analyses only include the presence or absence of bacterial taxa and so rare taxa tend to have a disproportionate effect on the results while weighted analyses takes bacterial abundance into account and is biased toward more abundant taxa. We also ran our alpha and beta diversity analyses through 40 random subsamples of 9 residents and 9 migrants to control for biases due to uneven sample size.
Finally, we used a series of linear models (ANOVA) to determine if any specific microbial taxa were significantly overrepresented in residents or migrants at the ASV, genus, and phylum levels using nlme and car packages. Before running this analysis, we filtered out every taxon that was not present in at least four samples of either the resident or migrant group. We again controlled for the effects of season and individual and corrected p-values for multiple tests using the fdrtool package.
Results
Contrary to our predictions, microbial alpha diversity was higher in resident geese across all three metrics used (Figure 2, Table 1 and Supplementary Table F). Also counter to our predictions, inter-individual variance (beta diversity) was also significantly higher in residents than in migrants (unweighted UniFrac: F = 17.57, p = 0.00021, variance = 0.0075; weighted UniFrac: F = 8.05, p = 0.0080, variance = 0.014; Figure 3). Migrant status significantly explained overall community composition (PERMANOVA, unweighted UniFrac: r2 = 0.057, p < 0.05; weighted UniFrac: r2 = 0.081, p < 0.05) (Figure 4 and Table 2). Community composition did not significantly differ by sampling site (Table 2). Geographical distance and microbiome dissimilarity were not significantly correlated (Table 3). To ensure that our results were not a result of our unbalanced sample size between migrant and resident geese, we reran each of these analyses 40 times using a random subset of nine residents and nine migrants. All of our results remained consistent when we subsampled, except for the weighted UniFrac community composition (Supplementary Tables G–I).
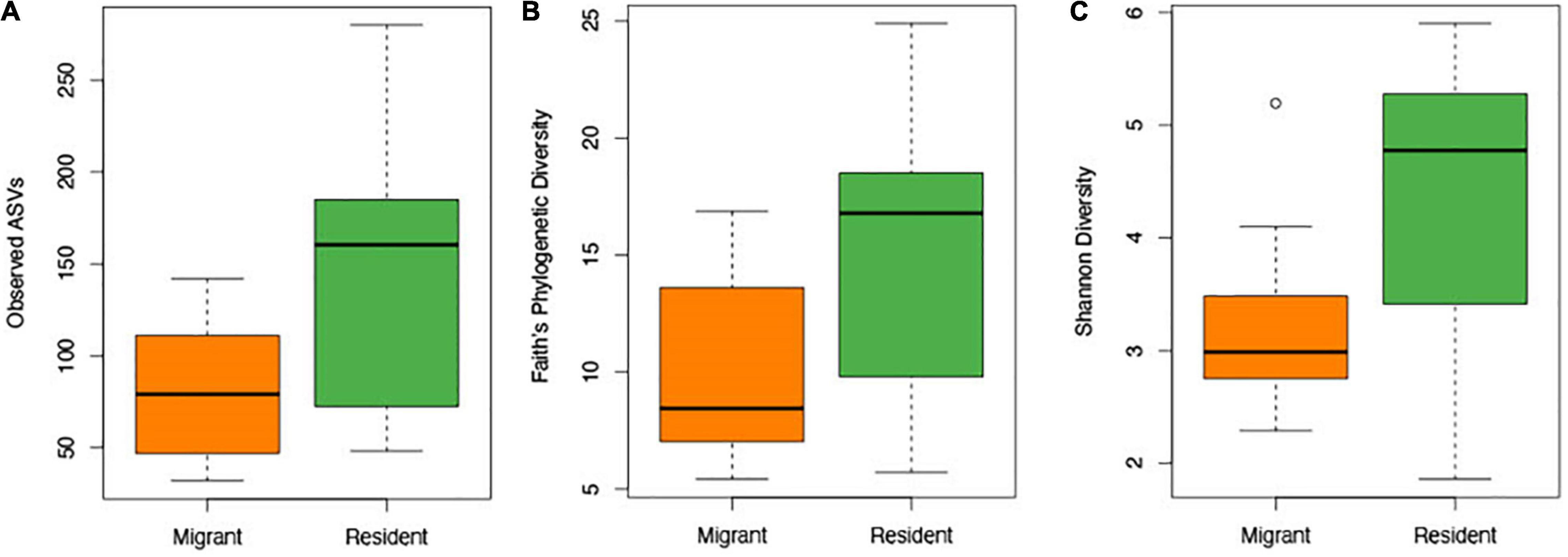
Figure 2. Box plot constructed using the number of observed ASVs (A), Faith’s phylogenetic diversity (B), and Shannon diversity data (C) to compare resident and migrant taxonomic richness and evenness.
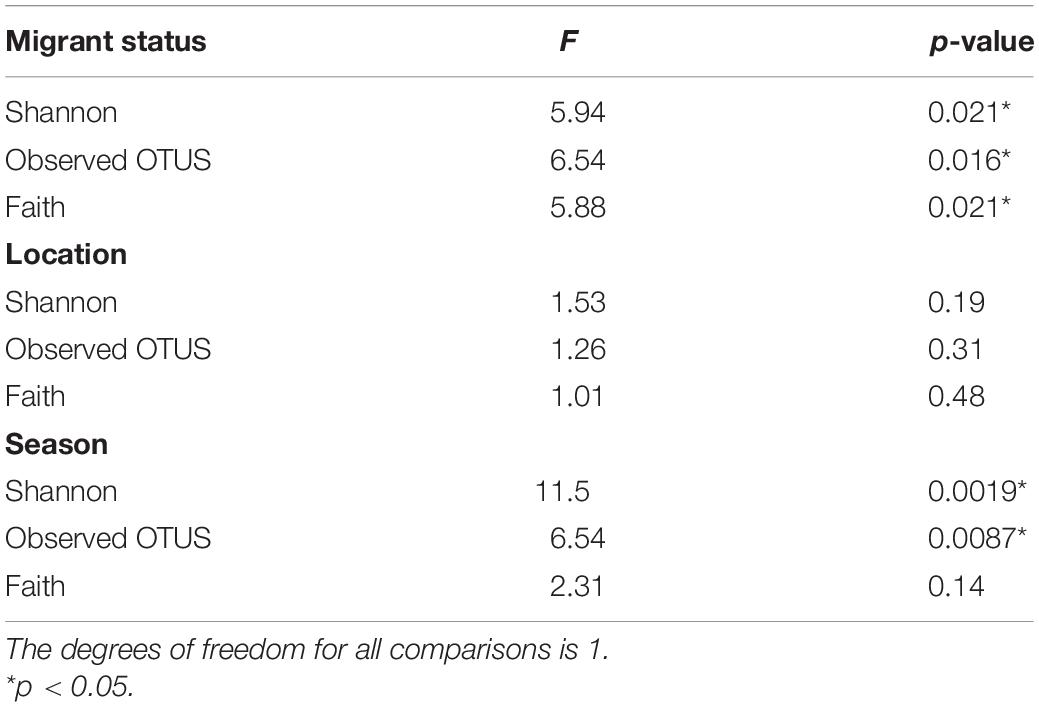
Table 1. Statistics from the ANOVA tests measuring the significance of the variation in average alpha diversity between migrants and residents.
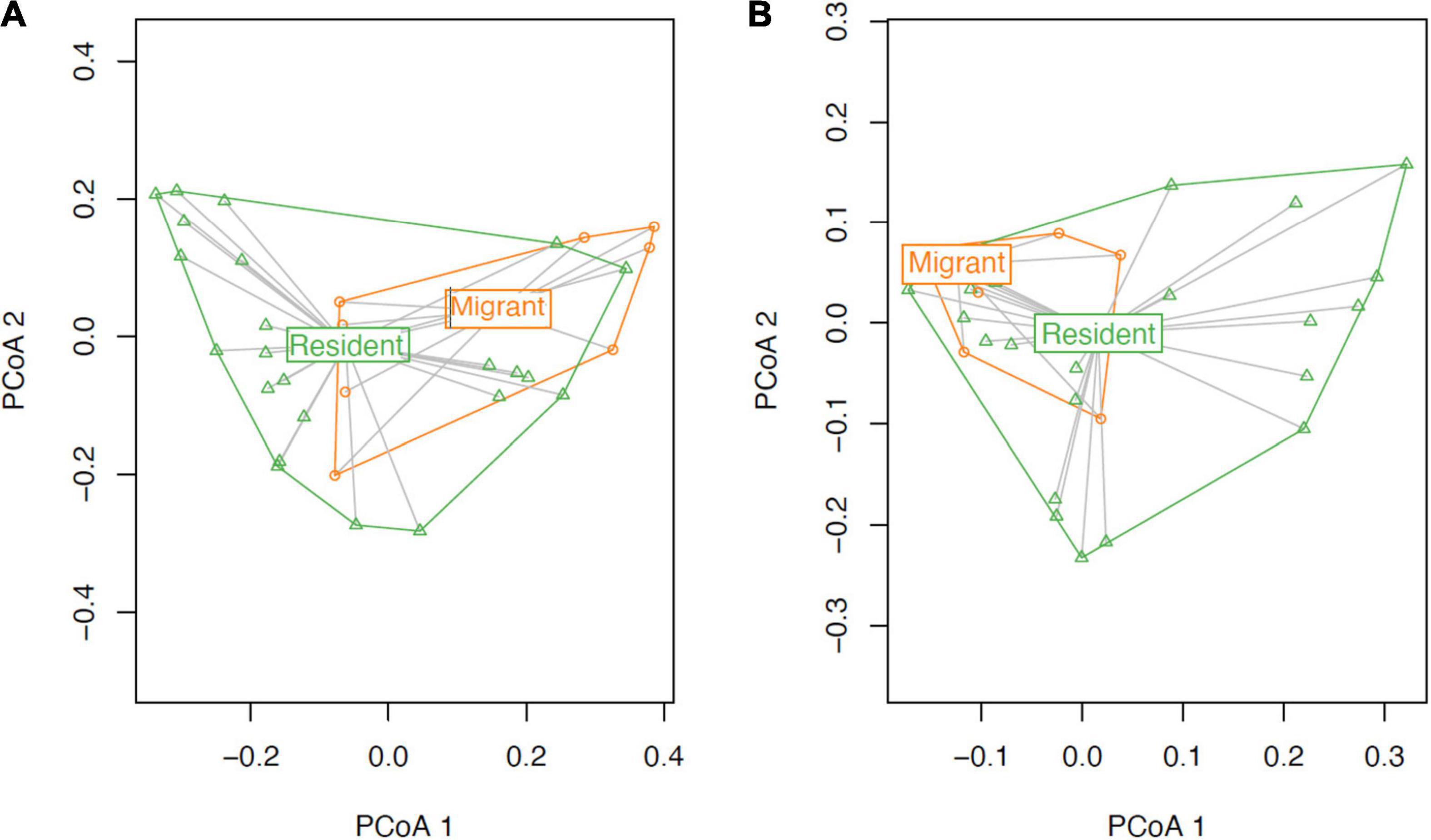
Figure 3. Beta dispersal ordination plot of the weighted and unweighted (A) and weighted (B) UniFrac distance matrices. The data for this figure is non-parametric, and the distance between the points represents the distance in the similarity of the microbiomes (i.e., close points have similar microbiomes). The ellipses represent a confidence interval of 95% (Friendly et al., 2013).
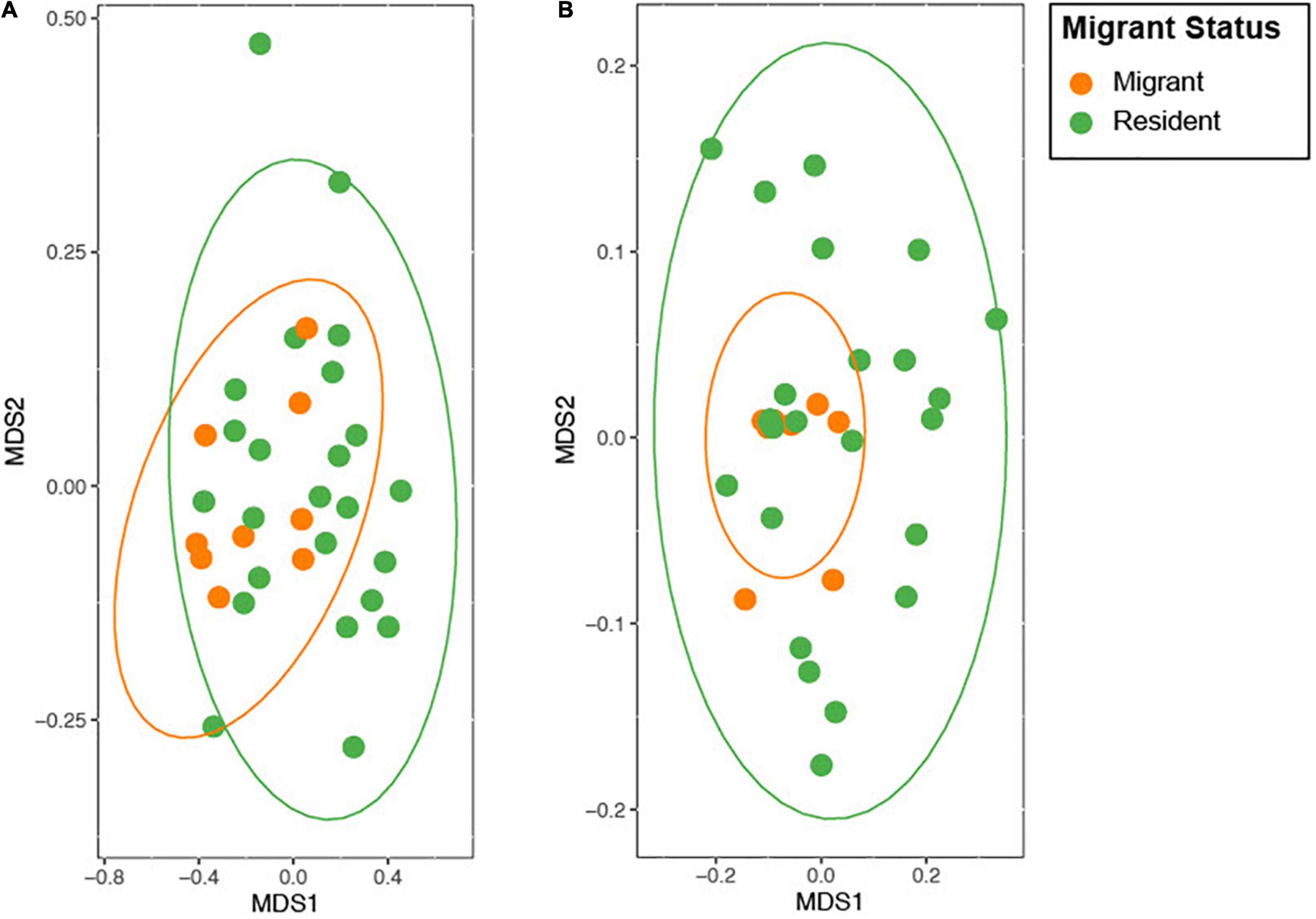
Figure 4. NMDS ordination plot of the weighted and unweighted (A) and weighted (B) UniFrac distance. The data for this figure is non-parametric, and the distance between the points represents the distance in the similarity of the microbiomes (i.e., close points have similar microbiomes). The ellipses represent a confidence interval of 95% (Friendly et al., 2013).
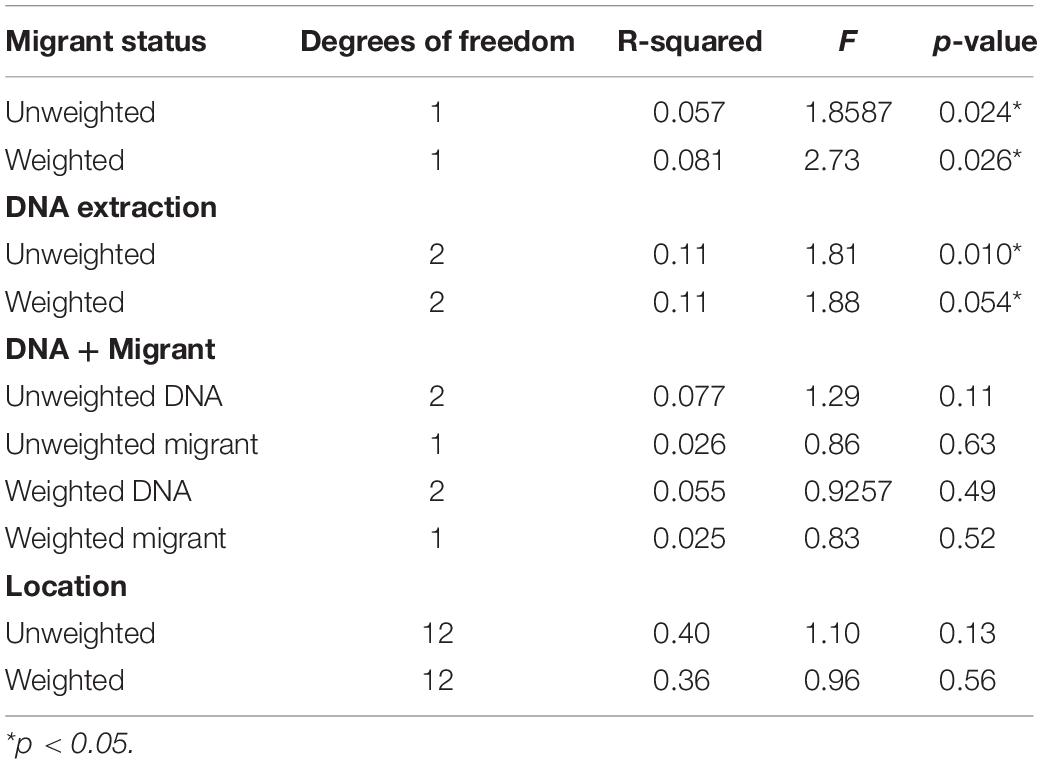
Table 2. PERMANOVA test output of the unweighted and weighted UniFrac distance matrices to quantify the extent to which migrant status, DNA extraction round, location, and season explain variance in microbiomes among individuals.
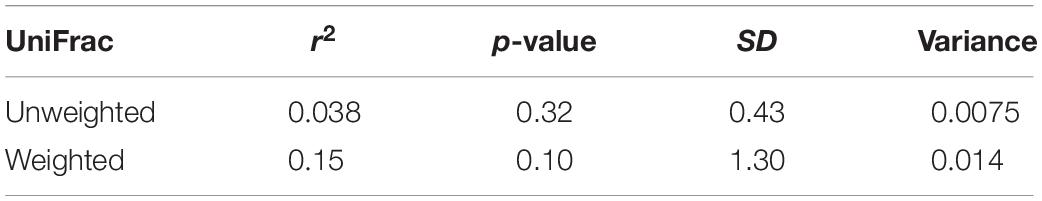
Table 3. Statistics from the Mantel tests comparing the unweighted and weighted UniFrac distances to the physical distances between sampling locations.
Several taxa exhibited significantly higher relative abundances (q < 0.05) in migrant geese compared to resident geese (Table 4). No taxa had significantly higher relative abundances in residents relative to migrants. Supporting our predictions, at the phylum level, there was a significantly higher average relative abundance of Firmicutes in migrants (approximately 81.36%) relative to residents (53.72%) (Figure 5). Firmicutes was the highest abundant phylum in 27 out of 33 samples (Figure 6). Within the phylum Firmicutes, four genera were significantly more relatively abundant in migrants than in residents: Terrisporobacter, Turicibacter, Cellulosilyticum, and Epulopiscium. Ten ASVs had significantly higher relative abundances in migrants compared to residents, eight of which were in the order Clostridiales (Table 4).
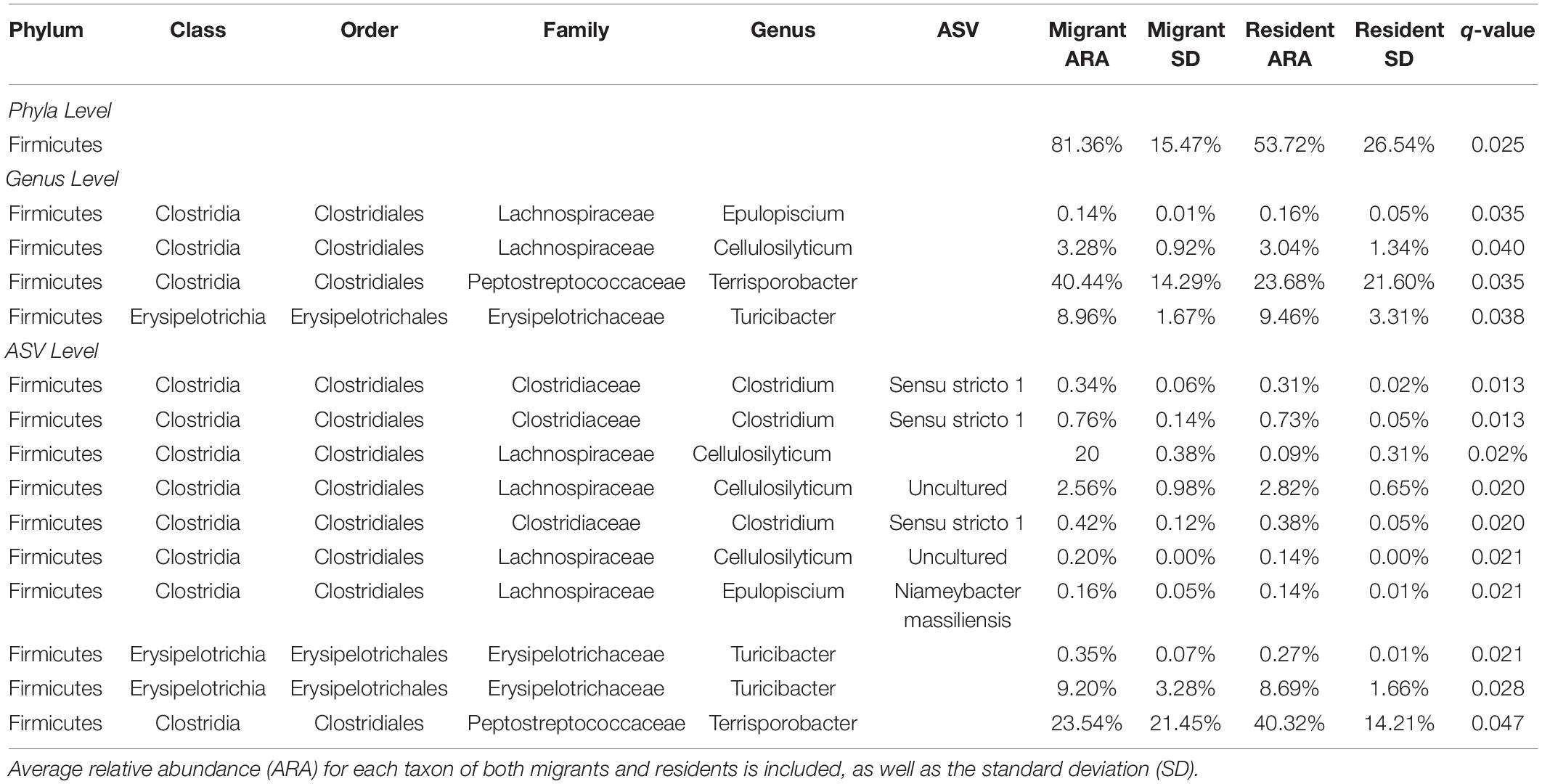
Table 4. The significant phylum, genera, and ASVs that had a higher abundance in migrants relative to residents.
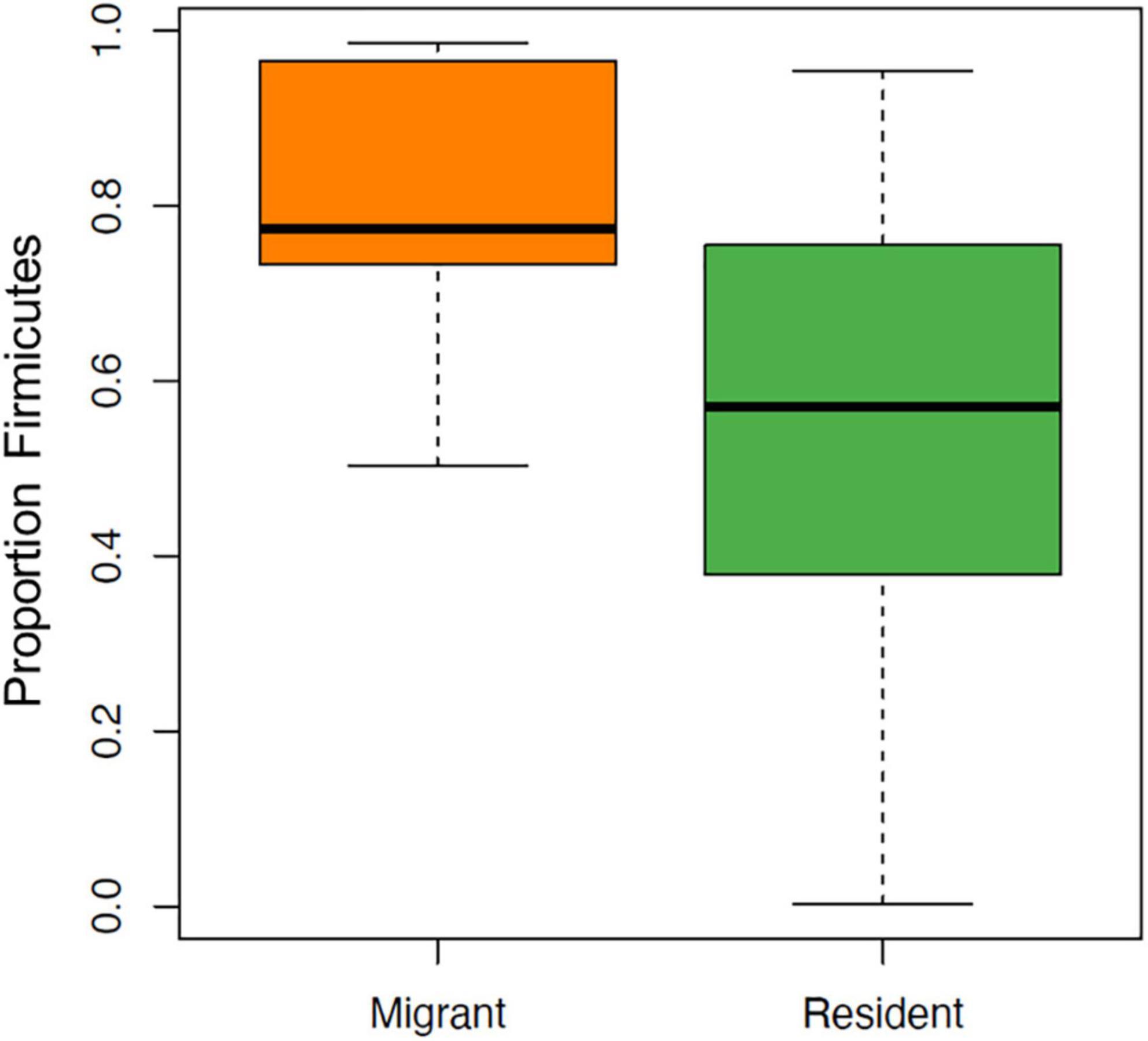
Figure 5. Boxplot showing the relative abundance of the phylum Firmicutes in migrant relative resident Canada geese, represented as the proportion of total sequences.
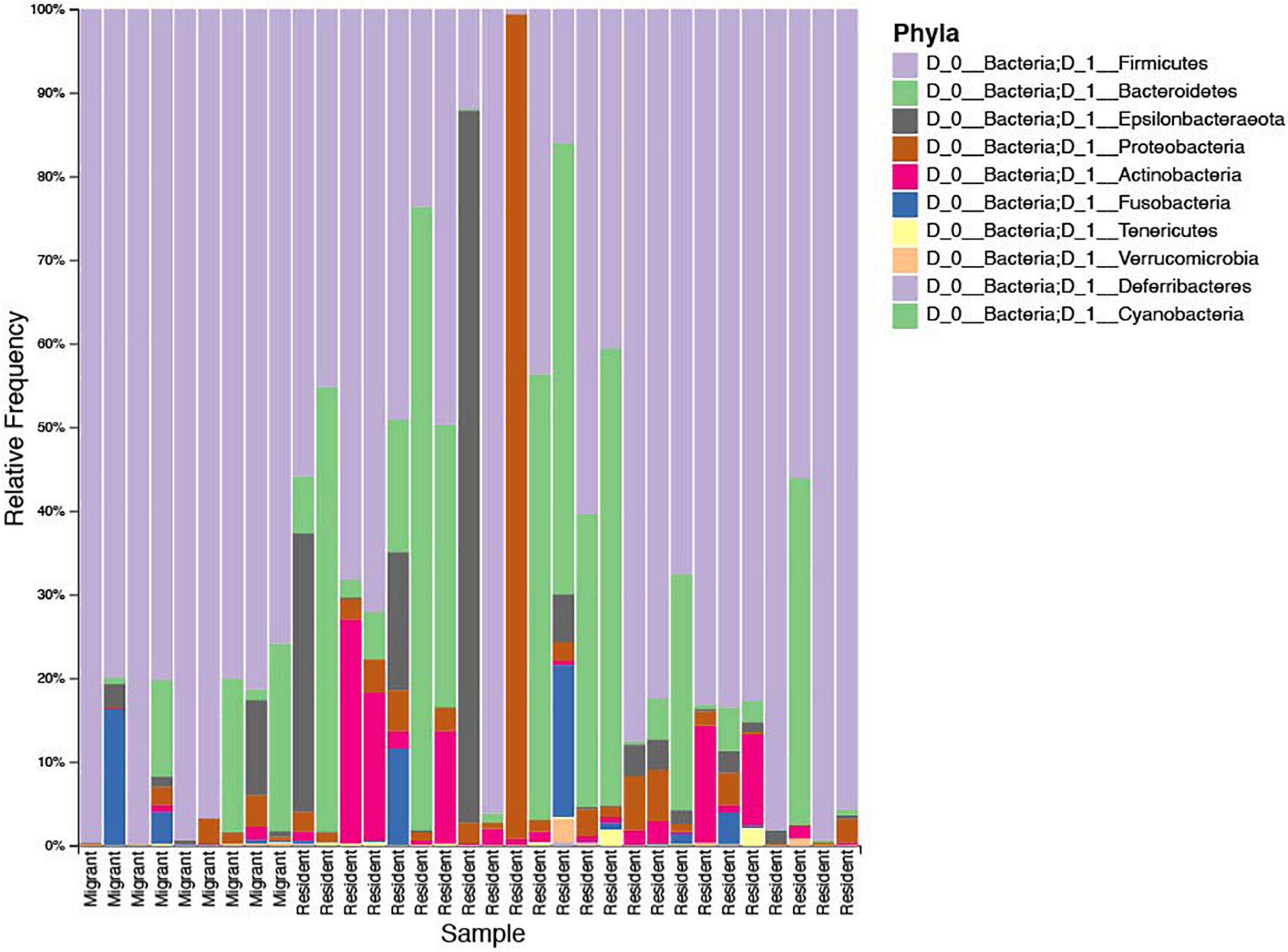
Figure 6. Bar plot showing the relative abundance of microbial phyla of each sample, represented as the percent frequency of sequences. The samples from individual geese are ordered along the x axis by migrant status.
Discussion
In this study, we tested the hypothesis that year-long urban resident Canada geese have a distinct microbiome compared to migrants. While our overall hypothesis was supported, contrary to our predictions, we found that migrant geese had less overall diversity and less within-group variation compared to residents. However, in support of our predictions, migrants had a higher relative abundance of Firmicutes, a bacterial phylum that is believed to be important for digestion and metabolism in wild birds (Grond et al., 2018) and has been associated with starch and cellulose breakdown in humans (Flint et al., 2012).
Given the wide range of ecological and physiological factors that differ between migrant and year-long urban resident individuals of partially migratory species, it is perhaps not surprising that we detected a difference in the gut microbiome composition of migrant and resident Canada geese in the Chicago area. Interestingly, however, the differences in overall composition were only detectable using an unweighted UniFrac method. This pattern suggests that the presence/absence of rarer microbial taxa differ more between groups than the relative abundances of the more abundant taxa and that migrant and resident geese share a substantial portion of their microbiome. This finding is similar to that reported for migrating migratory swan geese sampled before and after migration (Wu et al., 2018) and signals the potential presence of a core microbiome that is resilient to environmental change.
While the lower microbial diversity in migrants contradicted our original prediction, there are at least two non-exclusive potential explanations for this pattern. First, we expected migrant Canada geese to exhibit higher microbial diversity than residents because they are exposed to multiple habitats with potentially distinct dietary and environmental microbial inputs during migration. The rapid digestive rates and frequent foraging of geese (i.e., retention rate of approximately 2 h for other goose species; Prop and Vulink, 1992) would facilitate goose uptake of environmental microbes (Drovetski et al., 2018). However, it is possible that this uptake does not occur. If geese incorporate environmental microbiomes readily into their own we would expect the microbiome of migrant geese to converge with those of resident geese by the time we sampled them in an urban environment up to a week after arrival, but migrant microbiome composition was significantly different than residents. Similar patterns of reduced microbial diversity in migrant red-necked stints (Calidris ruficollis) (Risely et al., 2017) and curlew sandpipers (Calidris ferruginea) (Risely et al., 2018) compared to resident conspecifics have also been reported. It is possible that migrant microbiome resistance to invasion by environmental bacteria could be an adaptive mechanism that prevents pathogenic bacteria from being incorporated into the microbiome as migrants are exposed to so many new environments in a short time period (Risely et al., 2017).
Additionally, the resident geese in this study lived in urban areas year-round, which likely affects goose microbiome diversity and composition. Multiple studies have shown that increased urbanization alters the bird gut microbiome (Teyssier et al., 2018; Murray et al., 2020). While there are a variety of environmental factors that differ between urban and rural habitats, it is possible that diet may play an important role in driving observed patterns. Canada geese are known to actively seek out and anticipate food handouts from humans in urban areas (Conover, 1991). Resident urban geese may be able to exploit these artificial resources better than migrants, as they spend more time in the same place and have more time to learn where handouts occur. Geese that seek out human food sources are known to also eat plant matter (Conover, 1991), and so geese that consume anthropogenic food may have a more diverse diet than geese that purely rely on plants, and this greater diversity in diet could lead to greater diversity in gut microbes. Whether resident urban Canada geese actually receive more food from humans (or have a more diverse diet) than their migratory counterparts is unknown. However, similar to our results, polar bears (Ursus maritimus) that forage on land, and potentially include anthropogenic food waste in their diets, exhibit higher gut microbiome alpha diversity and a higher abundance of Firmicutes bacteria than bears foraging on sea ice (Watson et al., 2019). Baboons that feed in closer proximity to humans also have higher gut microbiome diversity (Barelli et al., 2020). In coyotes (Canis latrans), urbanization is correlated with greater anthropogenic food consumption, higher gut microbiome diversity, and unhealthy body conditions (Sugden et al., 2020). To better assess these relationships in geese, future studies should incorporate a quantitative assessment of diet and other environmental factors in urban and rural habitats.
With respect to variation in specific microbial taxa, the relative abundance of bacteria in the phylum Firmicutes was higher in migrants than residents. Firmicutes is a dominant bacterial phylum in the Canada geese gut microbiome in our sample and other studies (Drovetski et al., 2018). However, Firmicutes is a large and diverse phylum, so examining which genera and ASVs were higher in migrants can be more useful for elucidating any possible physiological or functional shifts. Three genera within Firmicutes that were significantly more represented in the migrant group have implications for gut function: Terrisporobacter, Turicibacter, Cellulosilyticum. Bacteria in Terrisporobacter are known to produce acetate (Gerritsen et al., 2014), a compound found to be an important metabolite for skeletal muscle systems in mammals (Frampton et al., 2020). It is possible that bacteria in Terrisporobacter have a role in providing energy for avian skeletal muscle during migration. Turicibacter includes species that metabolize maltose and produce lactate (Bosshard et al., 2002). This may suggest that it aids in breaking down intermediary sugars in the gut, although more research would be needed to elucidate its role in the microbiome. Bacteria in Cellulosilyticum metabolize a variety of carbohydrates, most notably cellulose and cellobiose (Cai and Dong, 2010). As cellulose makes up 25–50% of grass biomass (Sun and Cheng, 2002), and grass is the primary food source for geese in urban environments (Conover, 1991), it is likely that Cellulosilyticum aid goose digestion. Most of the significant taxa at the ASV level were simply subsets of significant genera, although notably, three ASVs of Clostridium sensu stricto (Cluster I) were significantly higher in migrants. Clostridium sensu stricto bacteria are known to metabolize a variety of compounds including carbohydrates, amino acids, and alcohols, and therefore may play an important part in digestive function (Gupta and Gao, 2009; Alou et al., 2017). In total, the taxa that differed significantly between goose populations at the phylum, genus, and ASV levels provide preliminary evidence that migrant geese are characterized by more bacteria that may contribute to the digestion of key dietary components. Whether this pattern results in nutrition and health risks in then populations of urban resident Canada geese or simply indicates adaptive microbial shifts in response to diet remains to be seen. Even in other bird species, these relationships are not well studied. For example, resident curlew sandpipers occupying a less urban environment have a higher abundance of Firmicutes relative to migrants, potentially in response to diet differences (Risely et al., 2017). Nevertheless, diet was not well quantified, and impacts on health were unknown.
Our results highlight differences in microbiome composition and diversity among migratory and resident urban birds. However, as implied above, the precise mechanism that drives microbiome shifts in human-dominated landscapes has yet to be untangled, with diet, stress, and pollution representing the potential competing or synergistic factors (Fuirst et al., 2018; Murray et al., 2020). Further, little is known about how microbes are transmitted among flocking birds, which may lead to non-independence among birds in the same flock. We believe it is unlikely that our results (i.e., that migrants exhibited less inter-individual variation) are due to migrants flocking together because they arrived back in Chicago on different dates to different sites and were not observed together during sampling events. Some of the migrants did use the same sites as residents but if this affected our results, it would have diminished rather than exacerbated any differences in microbiome composition between residents and migrants. Further, the traits of individual birds such as body condition or age may mediate the relationship between migration and urbanization on the microbiome. Future studies that sample social groups in multiple species simultaneously at multiple time points along urban to rural gradients could reveal demographic, social, and landscape drivers of microbial diversity.
The lower microbiome diversity seen in migrants, taken without context, might suggest that the migrant microbiomes are in poor health. However, while reduced microbial diversity is often associated with negative health consequences, this is not always the case. For example, low gut microbiome diversity is associated with a healthy state in human infants (Duar et al., 2020), and higher microbiome diversity has been associated with worse health in urban coyotes (Sugden et al., 2020). Migration can induce a drastic change in the digestive physiology in birds, reducing gut size and requiring birds to metabolize fat and protein reserves (McWilliams and Karasov, 2001). It is possible that the changes in the microbiome may promote energy storage and fat deposition through endotoxins produced by gram-negative bacteria or exotoxins produced by some gram-positive bacteria that provoke an inflammatory response (Risely et al., 2017). Thus, the stresses of migration may shape the microbiome in predictable ways, leading to less inter-individual variation within migrants. The relatively high similarity in the microbiomes of migrants compared to residents suggests a more stable microbial composition and a potential convergence of microbiome composition and function indicative of adaptation. In contrast, the relatively high inter-individual variation in resident geese could signal reduced microbiome resilience, increased microbiome stochasticity, and potentially associated health risks. The “Anna Karenina Principle” (Zaneveld et al., 2017) posits that unstable microbial communities tend to be each unstable in their own unique way, resulting in increased inter-individual variation among “unhealthy” individuals. The patterns we observed in our data are consistent with these dynamics.
Future studies should more directly measure the impact of these microbial shifts on host health. For example, in house sparrows (Passer domesticus), a shift in the microbiome of urbanized populations correlated with lower metabolic gene expression in the microbiome (Teyssier et al., 2018), which could ultimately affect fitness via impacts on processes such as digestion and nutrient intake. Also, urbanization has been linked to a decrease in Firmicutes relative abundance in bird gut microbiomes (Murray et al., 2020), which could impact digestion and nutrition. Given the lower relative abundances of Firmicutes that we observed in resident birds in our study, it seems that the stresses of urbanization may be affecting resident geese more strongly than migrants, who often only winter in urban areas. Quantifying the habitat selection of migrant and resident urban birds along the urban gradient in relation to microbiome composition and host physiology could help identify the consequence of urbanization and migration for wildlife health.
In conclusion, we identified differences in microbiome composition and diversity between migrant and resident Canada geese in urban areas. Determining the relative importance of environment, diet, and physiology in driving these differences, and whether either migrants or residents are healthier, represent important avenues for future research. For example, the differences we detected in the gut microbiomes of migrant and resident Canada geese could have broader implications for the health of migratory bird populations. As urban areas expand around the world, more migratory bird populations may become urban residents (Bonnet-Lebrun et al., 2020; Soriano-Redondo et al., 2020) due to pressures from climate change or the potential benefits of cities such as reliable resources and warmer temperatures. However, the potential benefits to these “colonizing” residents could develop disordered gut microbiomes as a result of altered diets and habitat use. Because microbes affect immune function, including helping hosts resist infections (Harris et al., 2009), a greater understanding of migratory bird microbiomes could be one pathway to improve the chances of success in conservation efforts (Amato, 2013; Murray et al., 2020). Novel techniques that restore natural microbiodiversity could hold meaningful, but relatively untapped conservation potential (Amato, 2013; Teyssier et al., 2018; Wu et al., 2018; West et al., 2019). Beginning to integrate gut microbiome data into more studies of migratory bird ecology represents an important first step toward these novel approaches.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SRA, PRJNA774835.
Ethics Statement
Ethical review and approval was not required for the animal study because fecal samples were collected non-invasively from geese that had been GPS-tracked as part of a separate study by different investigators.
Author Contributions
MM designed the study and collected biological samples. SO and MS performed laboratory work. SO performed statistical analysis and wrote the original manuscript draft. KA supervised laboratory work and statistical analysis. MM, KA, and MS provided edits. All authors approved of the final version.
Funding
The Office of Undergraduate Research at Northwestern University Academic-Year Undergraduate Research Grant, the Northwestern University Libraries Open Access Fund, and the Davis Foundation provided funding for this project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Ryan Askren and Michael Ward’s lab for providing GPS data for the geese. We would also like to thank Gary Galbreath for advising on this project as an Honors Thesis in the Northwestern Biology Department and DNA Service Center at UIC for sequencing the DNA samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.742369/full#supplementary-material
References
Alou, M. T., Ndongo, S., Frégère, L., Labas, N., Andrieu, C., Richez, M., et al. (2017). Taxonogenomic description of four new Clostridium species isolated from human gut: ‘Clostridium amazonitimonense’, ‘Clostridium merdae’, ‘Clostridium massilidielmoense’ and ‘Clostridium nigeriense.’. New Microb. New Infect. 21, 128–139. doi: 10.1016/j.nmni.2017.11.003
Amato, K. (2013). Co-evolution in context: the importance of studying gut microbiomes in wild animals. Microbiome Sci. Med. 1, 10–29. doi: 10.2478/micsm-2013-0002
Askren, R. J., Dorak, B. E., Hagy, H. M., Eichholz, M. W., Washburn, B. E., and Ward, M. P. (2019). Tracking Canada geese near airports: using spatial data to better inform management. Hum. Wildlife Interact. 13, 344–355.
Balkcom, G. D. (2010). Demographic parameters of rural and urban adult resident canada Geese in Georgia. J. Wildlife Manag. 74, 120–123. doi: 10.2193/2009-007
Barelli, C., Albanese, D., Stumpf, R. M., Asangba, A., Donati, C., Rovero, F., et al. (2020). The gut microbiota communities of wild arboreal and ground-feeding tropical primates are affected differently by habitat disturbance. mSystems 5:e00061-20. doi: 10.1128/mSystems.00061-20
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonnet-Lebrun, A.-S., Manica, A., and Rodrigues, A. S. L. (2020). Effects of urbanization on bird migration. Biol. Conserv. 244:108423. doi: 10.1016/j.biocon.2020.108423
Bosshard, P. P., Zbinden, R., and Altwegg, M. (2002). Turicibacter sanguinis gen. nov., sp. nov., a novel anaerobic, gram-positive bacterium. Int. J. Syst. Evol. Microbiol. 52, 1263–1266. doi: 10.1099/00207713-52-4-1263
Cai, S., and Dong, X. (2010). Cellulosilyticum ruminicola Gen. Nov., Sp. Nov., isolated from the rumen of yak, and reclassification of Clostridium lentocellum as Cellulosilyticum lentocellum Comb. Nov. Int. J. Syst. Evol. Microbiol. 60, 845–849. doi: 10.1099/ijs.0.014712-0
Cao, J., Hu, Y., Liu, F., Wang, Y., Bi, Y., Lv, N., et al. (2020). Metagenomic analysis reveals the microbiome and resistome in migratory birds. Microbiome 8:26. doi: 10.1186/s40168-019-0781-8
Conover, M. R. (1991). Herbivory by Canada Geese: diet selection and effect on lawns. Ecol. Appl. 1, 231–236. doi: 10.2307/1941816
Drovetski, S. V., O’Mahoney, M., Ransome, E. J., Matterson, K. O., Chuan Lim, H., Chesser, R. T., et al. (2018). Spatial organization of the gastrointestinal microbiota in Urban Canada Geese. Sci. Rep. 8:3713. doi: 10.1038/s41598-018-21892-y
Duar, R. M., Henrick, B. M., Casaburi, G., and Frese, S. A. (2020). Integrating the ecosystem services framework to define dysbiosis of the breastfed infant gut: the role of b. infantis and human milk oligosaccharides. Front. Nutr. 7:33. doi: 10.3389/fnut.2020.00033
Flint, H. J., Scott, K. P., Duncan, S. H., Louis, P., and Forano, E. (2012). Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306. doi: 10.4161/gmic.19897
Fox, J., and Weisberg, S. (2019). An R Companion to Applied Regression, Third Edn. Thousand Oaks CA: Sage.
Frampton, J., Murphy, K. G., Frost, G., and Chambers, E. S. (2020). Short-chain fatty acids as potential regulators of skeletal muscle metabolism and function. Nat. Metab. 2, 840–848. doi: 10.1038/s42255-020-0188-7
Friendly, M., Monette, G., and Fox, J. (2013). Elliptical insights: understanding statistical methods through elliptical geometry. Stat. Sci. 28, 1–39. doi: 10.1214/12-STS402
Fuirst, M., Veit, R. R., Hahn, M., Dheilly, N., and Thorne, L. H. (2018). Effects of urbanization on the foraging ecology and microbiota of the generalist seabird Larus argentatus. PLoS One 13:e0209200. doi: 10.1371/journal.pone.0209200
Gerritsen, J., Fuentes, S., Grievink, W., van Niftrik, L., Tindall, B. J., Timmerman, H. M., et al. (2014). Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the Genus Clostridium into the Genera romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int. J. Syst. Evol. Microbiol. 64(Pt_5), 1600–1616. doi: 10.1099/ijs.0.059543-0
Grond, K., Lanctot, R. B., Jumpponen, A., and Sandercock, B. K. (2017). Recruitment and establishment of the gut microbiome in arctic shorebirds. FEMS Microbiol. Ecol. 93:fix142. doi: 10.1093/femsec/fix142
Grond, K., Sandercock, B. K., Jumpponen, A., and Zeglin, L. H. (2018). The avian gut microbiota: community, physiology and function in wild birds. J. Avian Biol. 49:e01788. doi: 10.1111/jav.01788
Gupta, R. S., and Gao, B. (2009). Phylogenomic analyses of Clostridia and identification of novel protein signatures that are specific to the Genus Clostridiumsensu stricto (Cluster I). Int. J. Syst. Evol. Microbiol. 59, 285–294.
Harris, R. N., Brucker, R. M., Walke, J. B., Becker, M. H., Schwantes, C. R., Flaherty, D. C., et al. (2009). Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824.
Hird, S. M., Carstens, B. C., Cardiff, S. W., Dittmann, D. L., and Brumfield, R. T. (2014). Sampling locality is more detectable than taxonomy or ecology in the gut microbiota of the brood-parasitic brown-headed cowbird (Molothrus ater). PeerJ 2:e321. doi: 10.7717/peerj.321
Liu, M., and Swanson, D. L. (2014). Stress physiology of migrant birds during stopover in natural and anthropogenic woodland habitats of the northern prairie region. Conserv. Physiol. 2:cou046. doi: 10.1093/conphys/cou046
Mallott, E. K., and Amato, K. R. (2018). The microbial reproductive ecology of white-faced capuchins (Cebus capucinus). Am. J. Primatol. 80:e22896. doi: 10.1002/ajp.22896
McLandress, M. R., and Raveling, D. G. (1981). Changes in diet and body composition of canada geese before spring migration. Auk 98, 65–79. doi: 10.1093/auk/98.1.65
McWilliams, S. R., and Karasov, W. H. (2001). Phenotypic flexibility in digestive system structure and function in migratory birds and its ecological significance. Comp. Biochem. Physiol. A Mol. Integr. Physiol. Physiol. Consequences Feed. Anim. 128, 577–591. doi: 10.1016/S1095-6433(00)00336-6
Meillère, A., Brischoux, F., Bustamante, P., Michaud, B., Parenteau, C., Marciau, C., et al. (2016). Corticosterone levels in relation to trace element contamination along an urbanization gradient in the common blackbird (Turdus merula). Sci. Total Environ. 566–567, 93–101.
Møller, A. P., Jokimäki, J., Skorka, P., and Tryjanowski, P. (2014). Loss of migration and urbanization in birds: a case study of the blackbird (Turdus merula). Oecologia 175, 1019–1027. doi: 10.1007/s00442-014-2953-3
Mueller, E. A., Wisnoski, N. I., Peralta, A. L., and Lennon, J. T. (2020). Microbial rescue effects: how microbiomes can save hosts from extinction. Funct. Ecol. 34, 2055–2064. doi: 10.1111/1365-2435.13493
Murray, M. H., Lankau, E. W., Kidd, A. D., Welch, C. N., Ellison, T., Adams, H. C., et al. (2020). Gut microbiome shifts with urbanization and potentially facilitates a zoonotic pathogen in a wading bird. PLoS One 15:e0220926. doi: 10.1371/journal.pone.0220926
Murray, M. H., Sánchez, C. A., Becker, D. J., Byers, K. A., Worsley-Tonks, K. E., and Craft, M. E. (2019). City sicker? A meta-analysis of wildlife health and urbanization. Front. Ecol. Environ. 17, 575–583.
Pérez-Tris, J., and Tellería, J. L. (2002). Migratory and sedentary blackcaps in sympatric non-breeding grounds: implications for the evolution of avian migration. J. Anim. Ecol. 71, 211–224. doi: 10.1046/j.1365-2656.2002.00590.x
Prop, J., and Vulink, T. (1992). Digestion by barnacle geese in the annual cycle: the interplay between retention time and food quality. Funct. Ecol. 6, 180–189.
Pulido, F., and Berthold, P. (2010). Current selection for lower migratory activity will drive the evolution of residency in a migratory bird population. Proc. Natl. Acad. Sci. U.S.A. 107, 7341–7346. doi: 10.1073/pnas.0910361107
R Core Team (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Risely, A., David, W. W., Ujvari, B., Hoye, B. J., and Klaassen, M. (2018). Active migration is associated with specific and consistent changes to gut microbiota in calidris shorebirds. J. Anim. Ecol. 87, 428–437. doi: 10.1111/1365-2656.12784
Risely, A., Waite, D., Ujvari, B., Klaassen, M., and Hoye, B. (2017). Gut microbiota of a long-distance migrant demonstrates resistance against environmental microbe incursions. Mol. Ecol. 26, 5842–5854. doi: 10.1111/mec.14326
Ruiz, G., Rosenmann, M., Novoa, F. F., and Sabat, P. (2002). Hematological parameters and stress index in rufous-collared sparrows dwelling in urban environments. Condor 104, 162–166. doi: 10.1093/condor/104.1.162
Runge, C. A., Watson, J. E. M., Butchart, S. H. M., Hanson, J. O., Possingham, H. P., and Fuller, R. A. (2015). Protected areas and global conservation of migratory birds. Science 350, 1255–1258. doi: 10.1126/science.aac9180
Smith, A. E., Craven, S. R., and Curtis, P. D. (1999). Managing Canada Geese in Urban Environments: A Technical Guide. Ithaca, NY: Cornell Cooperative Extension.
Soriano-Redondo, A., Gutiérrez, J. S., Hodgson, D., and Bearhop, S. (2020). Migrant birds and mammals live faster than residents. Nat. Commun. 11:5719. doi: 10.1038/s41467-020-19256-0
Strimmer, K. (2008). Fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462. doi: 10.1093/bioinformatics/btn209
Sugden, S., Sanderson, D., Ford, K., Stein, L. Y., and St Clair, C. C. (2020). An altered microbiome in urban coyotes mediates relationships between anthropogenic diet and poor health. Sci. Rep. 10:22207.
Sun, Y., and Cheng, J. (2002). Hydrolysis of Lignocellulosic materials for ethanol production: a review. Bioresour. Technol. Rev. Issue 83, 1–11. doi: 10.1016/S0960-8524(01)00212-7
Teyssier, A., Rouffaer, L. O., Hudin, N. S., Strubbe, D., Matthysen, E., Lens, L., et al. (2018). Inside the guts of the city: urban-induced alterations of the gut microbiota in a wild passerine. Sci. Tot. Environ. 612, 1276–1286. doi: 10.1016/j.scitotenv.2017.09.035
Thompson, L. R., Sanders, J. G., McDonald, D., Amir, A., Ladau, J., Locey, K. J., et al. (2017). A communal catalogue reveals earth’s multiscale microbial diversity. Nature 551, 457–463. doi: 10.1038/nature24621
Waite, D. W., and Taylor, M. W. (2015). Exploring the avian gut microbiota: current trends and future directions. Front. Microbiol. 6:673. doi: 10.3389/fmicb.2015.00673
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., et al. (2016). Improved bacterial 16S RRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1:e00009-15. doi: 10.1128/mSystems.00009-15
Watson, S. E., Hauffe, H. C., Bull, M. J., Atwood, T. C., McKinney, M. A., Pindo, M., et al. (2019). Global change-driven use of onshore habitat impacts polar bear faecal microbiota. ISME J. 13, 2916–2926. doi: 10.1038/s41396-019-0480-2
West, A. G., Waite, D. W., Deines, P., Bourne, D. G., Digby, A., McKenzie, V. J., et al. (2019). The microbiome in threatened species conservation. Biol. Conserv. 229, 85–98. doi: 10.1016/j.biocon.2018.11.016
Wickham, H., Averick, M., Bryan, J., Chang, W., McGowan, L. D., François, R., et al. (2019). Welcome to the tidyverse. J. Open Sour. Softw. 4:1686. doi: 10.21105/joss.01686
Wiener, P., and Tuljapurkar, S. (1994). Migration in variable environments: exploring life-history evolution using structured population models. J. Theor. Biol. 166, 75–90. doi: 10.1006/jtbi.1994.1006
Wu, Y., Yang, Y., Cao, L., Yin, H., Xu, M., Wang, Z., et al. (2018). Habitat environments impacted the gut microbiome of long-distance migratory swan geese but central species conserved. Sci. Rep. 8:13314. doi: 10.1038/s41598-018-31731-9
Xie, Y., Xia, P., Wang, H., Yu, H., Giesy, J. P., Zhang, Y., et al. (2016). Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 6:33350. doi: 10.1038/srep33350
Zaneveld, J. R., McMinds, R., and Thurber, R. V. (2017). Stress and stability: applying the anna karenina principle to animal microbiomes. Nat. Microbiol. 2, 1–8. doi: 10.1038/nmicrobiol.2017.121
Keywords: migration, Branta canadensis, urban ecology, Firmicutes, microbiome, microbial diversity
Citation: Obrochta S, Savo Sardaro ML, Amato KR and Murray MH (2022) Relationships Between Migration and Microbiome Composition and Diversity in Urban Canada Geese. Front. Ecol. Evol. 10:742369. doi: 10.3389/fevo.2022.742369
Received: 16 July 2021; Accepted: 20 January 2022;
Published: 16 February 2022.
Edited by:
Joël Meunier, UMR 7261 Institut de Recherche sur la Biologie de l’Insecte (IRBI), FranceReviewed by:
Heidi Christine Hauffe, Fondazione Edmund Mach, ItalyFrederick R. Adler, The University of Utah, United States
Copyright © 2022 Obrochta, Savo Sardaro, Amato and Murray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maureen H. Murray, bWF1cmVlbm11cnJheUBscHpvby5vcmc=