 Ke He
Ke He Ying Zhu
Ying Zhu Shang-Chen Yang1
Shang-Chen Yang1 Sheng-Guo Fang
Sheng-Guo Fang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 13 December 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.1078058
Background: The major histocompatibility complex (MHC) gene family, a vital immune gene family in vertebrates, helps animals defend against pathogens. The polymorphism of MHC genes is important for a species and is considered to be caused by the numerous alleles of MHC antigen-presenting genes. However, the mechanism of this process is unclear due to the lack of data on the MHC structure. The evolutionary trajectories of the tetrapod MHC are also unclear because of insufficient studies on the reptile MHC architecture. Here, we studied the Chinese alligator (Alligator sinensis), which experienced a population bottleneck, but the population increased rapidly in the past 30 years and is proposed to have a unique MHC system to face pathogenic challenges.
Results: We successfully constructed a 2 Mb MHC region using bacterial artificial chromosome (BAC) library and genome data of the Chinese alligator and checked the antigen-presenting genes using transcriptome data and the rapid amplification of cDNA ends (RACE) technique. The MHC architecture reported here uncovers adjacent Class I and Class II subregions and a unique CD1 subregion. This newly added information suggested that the Class I-II structure pattern was more ancient in tetrapods and helped reconstruct the evolution of the MHC region architecture. We also found multiple groups of MHC class I (MHC-I) (12 duplicated loci, belonging to three groups, two of which were novel) and MHC class II (MHC-II) (11 duplicated loci, belonging to two groups) inside the 2 Mb MHC region, and there were three more duplicated MHC-I loci outside it. These highly duplicated antigen-presenting loci had differences in expression, amino acid length of antigen-presenting exons, and splice signal of exon and intron, which together promoted the polymorphism of duplicated genes. Although the MHC antigen-presenting genes were identified as monomorphic or oligomorphic in our previous population study, the loci with high copy numbers and many differences can make up for this loss, presenting another mechanism for polymorphism in antigen presentation. These MHC-I and MHC-IIB loci with low polymorphism for each locus, but high numbers in all, may also contribute to MHC antigen-presenting binding variability in a population.
Conclusion: To summarize, the fine MHC region architecture of reptiles presented in this study completes the evolutionary trajectories of the MHC structure in tetrapods, and these distinctive MHC gene groups in the Chinese alligator may have helped this species to expand rapidly in the past recent years.
The major histocompatibility complex (MHC) genes are a critical component of the adaptive immune system and are considered to respond to pathogens by encoding polymorphic antigen-presenting proteins. Two types of antigen-presenting genes related to T-cells play vital roles in innate and adaptive immunity. MHC class I (MHC-I) genes mainly present endogenous peptides to CD8+ T-cells, and MHC class II (MHC-II) genes, composed of IIA and IIB, present peptides derived from extracellular pathogens to CD4+ T helper cells (Pishesha et al., 2022). MHC-related antigen-processing genes (Taps, Psmbs) are also present in this gene cluster (Belov et al., 2006; Zhou et al., 2021). The MHC gene family is one of the most studied gene regions in vertebrates; it is found in cartilaginous fish, bony fish, and tetrapods (Flajnik, 2018). Unlike unlinked Class I and Class II genes in bony fish (Sato et al., 2000), MHC genes are located in one contiguous region in most jawed vertebrates, including cartilaginous fish (Ohta et al., 2000) and tetrapods. However, the gene organization and size are dynamic and different among different lineages.
Except in fish, there are mainly two patterns of MHC architecture in studied jawed vertebrates. Firstly, as the best-illustrated MHC region, the MHC regions of humans and mice consist of syntenic clusters of Class I, Class II, and Class III regions (Shiina et al., 2017). This Class I-III-II structure is conserved among the eutherian group but with slight changes. For example, in the Class II region, ruminants have DI/DY genes instead of DP genes (Andersson et al., 1988), the felines have expanded DR genes, but no DQ genes (Yuhki et al., 2003), and pinnipeds do not have DP genes (de Sá et al., 2019); in the Class I region, the numbers of putatively functional MHC-I genes were different in the extended Class I region and κ and β duplication blocks (sub-region of Class I region) in different mammal groups (Abduriyim et al., 2019).
Another pattern of the MHC region had adjacent Class I and Class II regions, with no intervening Class III region. It was reported in monotremes [platypus, Ornithorhynchus anatinus (Zhou et al., 2021)] and marsupials [tammar wallaby, Macropus eugenii (Siddle et al., 2011); Tasmanian devil, Sarcophilus harrisii, (Cheng et al., 2012); gray short-tailed opossum, Monodelphis domestica (Belov et al., 2006)]. The MHC of birds also has a linked Class I and Class II region, e.g., the MHC of chicken is well-known as “minimal and essential,” with no more than 100 kb (Kaufman et al., 1999); this compact and simple structure was also confirmed in our previous study on golden pheasant (Chrysolophus pictus) (Ye et al., 2012) and in other Galliformes (Hosomichi et al., 2006; He et al., 2021). However, the fewer, smaller, and more densely packed MHC pattern in chickens (compared with other groups) is not typical for birds. Based on third-generation sequencing (TGS, e.g., PacBio SMRT Sequencing and Oxford Nanopore Technologies) genome data, we found the vastly different copy numbers of MHC-I and MHC-II genes in birds (He et al., 2020) and discovered that the MHC organization varies greatly, both in length and gene arrangement (He et al., 2021). The ancient MHC architecture in birds was predicted to have tightly linked Class I and Class II regions and to differ from the eutherian MHC in size and the orientation of its three MHC regions (He et al., 2021).
To date, it is unclear whether the Class I-III-II structure in eutherians or the linked Class I-II in marsupials and birds is the original MHC pattern. Furthermore, the mechanism for the organization of these distinct MHC regions remains unknown. Kaufman (2018) revealed that the typical mammalian MHC (Class I-III-II pattern) could arise from the ancestral organization of non-mammalian vertebrates (Class I-II pattern, with Class III region not far away) by an inversion; however, the key part lacks strong supporting evidence. This is mainly because the MHC region architecture is not deeply understood in non-mammalian tetrapods, except birds. Amphibians occupy a basal phylogenetic position among tetrapods. The MHC region of the western clawed frog (Xenopus laevis) has tightly linked MHC-I and MHC-II genes in L chromosomes; however, because of the allotetraploid origin of X. laevis, there also exists notable gene loss on the S subgenome (Session et al., 2016). The MHC region of urodele species (axolotl, Ambystoma mexicanum) has a conserved arrangement syntenic with humans (Schloissnig et al., 2021), whereas an analysis of axolotl mostly included showed framework genes. The MHC architecture in tetrapods still needs further resolving; furthermore, it is unclear whether the hypothetical ancient bird’s MHC is still applicable in its sister group, Crocodylia.
Knowledge of the complete MHC architecture in all tetrapod lineages can help us answer the above mentioned questions. To our knowledge, research on non-avian reptiles’ MHC regions has been conducted on saltwater crocodile (Crocodylus porosus) (Jaratlerdsiri et al., 2014a), tuatara (Sphenodon punctatus) (Miller et al., 2015), Komodo dragon (Varanus komodoensis) (Reed and Settlage, 2021), and two anoles (Anolis carolinensis and Anolis sagrei) (Card et al., 2022). However, these studies have not sufficiently explored the genomic organization of reptiles. A bacterial artificial chromosome (BAC) study of saltwater crocodile (in Crocodylia clade) revealed that MHC-I and MHC-II genes were present in separate scaffold regions and that MHC-I genes were located along with Tap and Trim39 genes (Jaratlerdsiri et al., 2014a). In tuatara (in Rhynchocephalia clade), seven MHC-I and 11 MHC-IIB genes were found through BAC screening, and the cytogenetic location of these genes indicated duplication and translocation of MHC genes outside the core region (Miller et al., 2015). Based on genome data, multiple Class Iα and Class IIβ genes were observed in Komodo dragon (in the Squamata clade), but most were alone, without any other linked genes (Reed and Settlage, 2021). The latest reported core MHC genomic region in two Anolis lizards (in the Squamata clade) provided the first detailed characterization of reptiles’ MHC, with a single core MHC region (Card et al., 2022). Although some TGS-based genomes of reptiles have been published, the complex MHC region cannot be easily illustrated, e.g., 56 MHC genes spanning 13 scaffolds were identified in tuatara’s genome (Gemmell et al., 2020), and the detail MHC gene maps of Anolis were obtained using two independent datasets and approaches (BAC screening and sequencing and BLAST-derived inference of homology from genome) (Card et al., 2022). This limited and fragmented information on Crocodylia clade’s MHC region thus far does not allow the comparison of gene gain and loss and gene rearrangement in this lineage.
Here, we focused on the Chinese alligator, which belongs to Crocodylia. This species experienced a severe population bottleneck about 30 years ago, but its population has recovered to more than 20,000 individuals in recent years, starting with 219 founder alligators (7 in Changxing Yinjiabian Chinese Alligator Nature Reserve and 212 in Anhui Research Center for Chinese Alligator Reproduction) (Zhai et al., 2017). In our previous study, the BAC library was constructed (He et al., 2012), the genome was sequenced (Wan et al., 2013), and the differential mRNA expression of several tissues of the Chinese alligator was investigated (Lin et al., 2018) to understand the immune system and genetic basis of sex determination and long-diving behavior. Furthermore, MHC genes were investigated to study MHC-associated mate choice in Chinese alligators (Han et al., 2019). Together, all these studies make it possible to tackle the unanswered questions about the MHC architecture in Crocodylia.
As a species that experienced a population bottleneck, the Chinese alligator is expected to have a limited degree of genetic polymorphism in the population, which is not beneficial to the rapid growth of the population. The genetic and phenotypic variation is key to successful establishment (Forsman, 2014), and more variable populations are less vulnerable to environmental changes, compared with less variable populations or species (Forsman and Wennersten, 2016). In this study, we aimed to explore whether there are any compensation mechanisms for the lack of genetic diversity in the immune system, which is critical for species protection and population growth. Specifically, we aimed (1) to illustrate the MHC architecture in Chinese alligator based on BAC contigs and identify the groupings and phylogenetic relationships of antigen-presenting genes, (2) to infer major evolutionary trajectories of the MHC architecture in tetrapods, and (3) to explore the potential mechanisms for antigen-presenting polymorphism in this bottleneck population.
We searched the antigen-presenting genes (MHC-I, MHC-IIA, and MHC-IIB) in the gff file of the previously published genome of Chinese alligator (GCA_000455745.1) using “class I histocompatibility” and “class II histocompatibility” as keywords, and extracted the complete gene annotation to explore the arrangement of antigen-presenting genes and MHC-located genes (genes in MHC region, but not antigen-presenting and antigen processing genes, Step 1.1 in Figure 1). Scaffold 1303 was used as the draft of the Chinese alligator’s MHC core region (details in Supplementary file 1). Observations suggested a tight linkage of MHC-IIA-IIB-BRD2-DMA-DMB and that Psmbs were close to MHC-I genes. However, the incompletely sequenced Scaffold 1303, with 82 Ns-regions (details in Supplementary file 1), was insufficient for understanding the fine MHC architecture of this species, and the gene prediction was not precise due to the multiple duplications of antigen-presenting genes. Thus, more detailed sequence information and more accurate predictions are required.
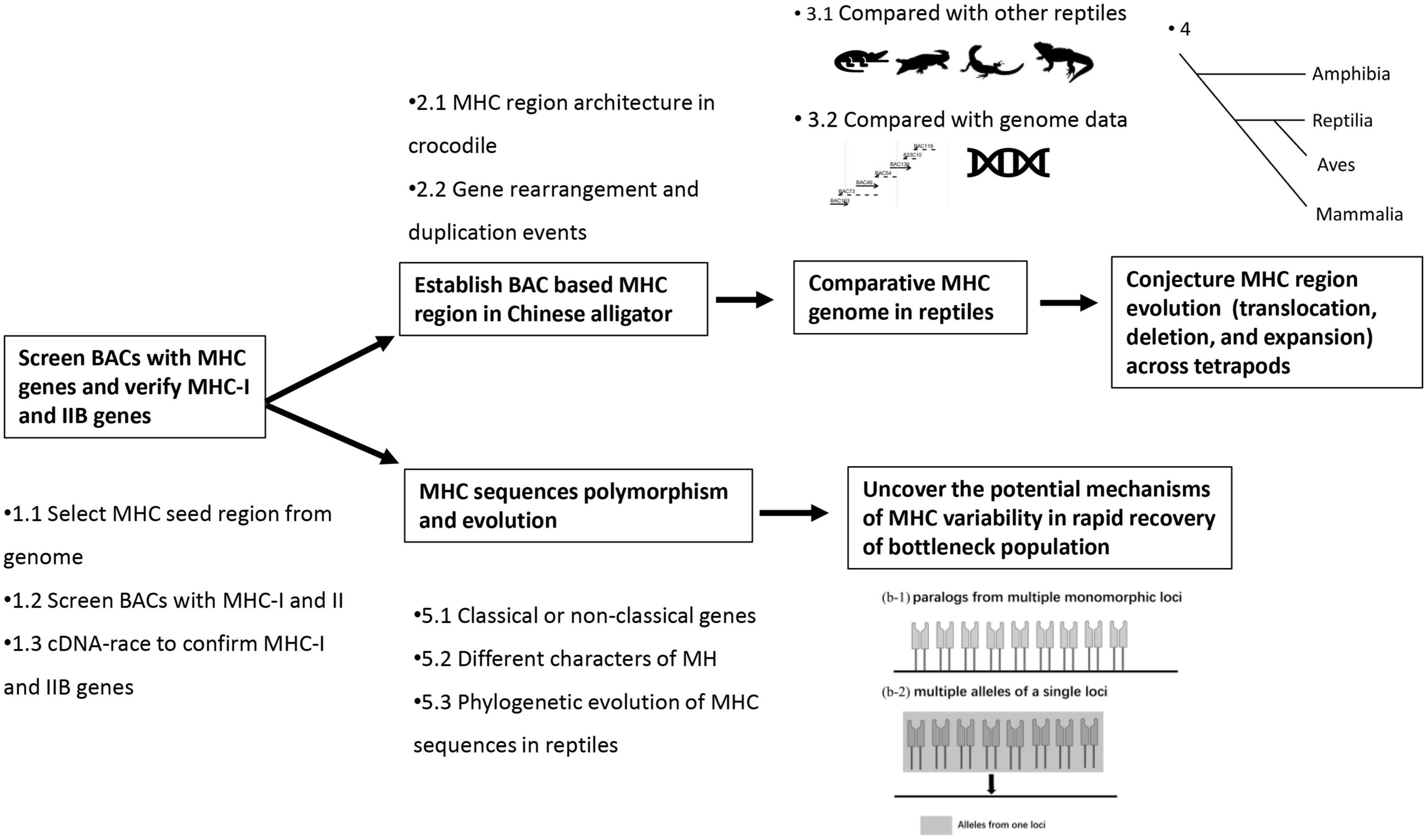
Figure 1. Key steps in this study.
Based on information from Scaffold 1303, we screened BACs containing both MHC-IIA and MHC-IIB (Step 1.2 in Figure 1). Primers were designed according to one predicted MHC-IIA (exon 2), ten predicted MHC-IIB (exon 3), and 16 predicted MHC-I (exon 3) sequences of the genome (Supplementary Table 1). The 16 predicted MHC-I sequences from the genome showed various differences (sequences similarity 36–89%), so three pairs of primers were designed to target this region. BAC screening was conducted as described in our previous study (He et al., 2012), and the process of BAC screening and information about the contigs are detailed in Supplementary files 2–4.
After constructing the Alligator sinensis (Alsi)-BAC-MHC contig, gene structure annotation was performed (Step 2.1 in Figure 1) based on two aspects. According to one aspect, antigen-presenting genes are highly polymorphic, with multiple gene duplications, so we adopted three ways to locate these homologies in our Alsi-BAC-MHC contig. First, after using the RepeatMasker program1, GeneScan (Burge and Karlin, 1997) was used to predict the coding sequence (CDS). Second, to ignore results across duplicated gene copies, we extracted regions 5 kb upstream and downstream of the PCR-positive site for analyses. Some exons of antigen-presenting genes were no longer than 100 bp, so exon 2–exon 4 was mainly used. Rapid amplification of cDNA ends (RACE) was also employed using the universal primers in groups to check gene expression and to explore if the genes are novel. Third, the results based on the predictions from the first two steps were manually adjusted using Geneious Prime v.2022.1.1 (Biomatters Ltd., Auckland, New Zealand).
Based on the other aspect, we used GeneScan to predict and compare other genes (including antigen processing genes and MHC-located genes) with homologous genes to determine the gene location. The results were plotted using gggenes,2 an extension of ggplot2 (Wickham 2016), in R 3.5.2 (R Core Team, 2018) (Step 2.1 in Figure 1).
The Alsi-BAC-MHC contig presented in this study is the first detailed MHC architecture of reptiles. We took the published MHC gene arrangement of three other reptiles to compare if the gene arrangement is conserved among all reptiles (Step 3.1 in Figure 1). The references used were saltwater crocodile (Jaratlerdsiri et al., 2014a), tuatara (Miller et al., 2015), Komodo Dragon (Reed and Settlage, 2021), and green anole (Card et al., 2022). We used the tree constructed using VertLife3 (Tonini et al., 2016) to describe the phylogenetic distribution of these four species. BAC was considered the best and precise method to study gene arrangement. To analyze whether there is a mismatch or annotation mistake in the MHC region, we compared the gene annotation of this region from second-generation sequencing (SGS) (Illumina short read sequencing, GCA_000455745.1) and TGS [which was available during our organizing this article, (Yang et al., 2022)] with our Alsi-BAC-MHC contig (Step 3.2 in Figure 1).
We also compared the representative MHC architecture from other tetrapod lineages to remodel the evolutionary trajectories of the MHC structure tetrapods (Step 4 in Figure 1). For this purpose, we selected the African clawed frog (X. laevis, representing the amphibian species, GCA_000004195.4 chromosome 8 containing the MHC core and extended subregions), chicken (as bird group, chromosome 16, the classical MHC region), gray short-tailed opossum [M. domestica, as marsupial group, BAC study (Belov et al., 2006)], and human (as eutherian group, chromosome 6, MHC Class I-III-II regions). Collinearity analyses were conducted using TBtools (Chen et al., 2020). Furthermore, we analyzed the evolutionary events by region (MHC-I related region, MHC-II related region, and some unique regions) using Rxrb-Col11A2 and C4-Cyp21-CenpA-TNXB as flanking markers, aiming to understand the story of MHC evolution in tetrapods more clearly.
The polymorphism and evolution of MHC sequences were analyzed in several ways. First, to classify the classical and non-classical genes, we evaluated the expression pattern of MHC-I, MHC-IIB, and CD1 genes using our previously published RNA-seq data (NCBI BioProjects: PRJNA556093), which were based on 12 tissues (thyroid, testicle, spleen, small intestine, ovary, lung, liver, kidney, hypothalamus, heart, brain, and adipose) of eight Chinese alligators (Step 5.1 in Figure 1). Most of the tissues had four replicates, except hypothalamus and thyroid (n = 2). We quantified the expression of each gene using Salmon v1.8.0 (Patro et al., 2017). The expression level of each MHC gene was normalized by calculating the number of transcripts mapped to the mRNA per million transcripts (TPM).
Second, after confirming the presence of multiple copies of MHC-I, MHC-IIB, and CD1 genes in the Alsi-BAC-MHC contig, it was necessary to analyze the evolutionary history of these duplicated genes. All alignments were performed using Geneious (Step 5.2 in Figure 1). The MrBayes software was used to establish the phylogenetic relationships of MHC-I, MHC-IIB, and CD1 genes using reference sequences from the NCBI database (Step 5.3 in Figure 1, species and Genbank ID are listed in Supplementary Table 2).
Finally, considering the extensive number of characters suggested by multiple loci, we proposed a hypothesis about how this extinct population faces threats from pathogens (Step 6 in Figure 1).
After sequencing, we constructed a BAC-based contig of the Chinese alligator’s MHC region, involving 19 BACs and three scaffolds of genome data (results of Step 1.1 in Figure 1, detail results in Supplementary file 3, Supplementary Figure 1, Supplementary Tables 3, 4). These BACs and selected scaffolds were approximately 2 Mb (2,089,021 bp, Supplementary Figure 1) in size, and we labeled it as the core MHC region of the Chinese alligator. Currently, this contig is the largest and most well illustrated MHC-BAC-based contig sequenced in crocodylia.
Subsequently, we performed gene prediction of the 2 Mb contig. In the core MHC region, 101 genes were identified, comprising 12 copies of MHC-I genes, 11 copies of MHC-IIB genes, five antigen-processing genes, and 68 other adjacent genes and pseudogenes (Figure 2). All the functional antigen-presenting genes (referred to as MHC genes) were distributed in two relatively concentrated regions, from Alsi-I1 to Alsi-I11 and Alsi-IIA to Alsi-IIB11, except for Alsi-I12 and Alsi-IIB1. Notably, there were five duplications of CD1 genes, which belong to innate immunity, located upstream of the MHC-I enriched region. This contig also included genes present closely next to MHC antigen-presenting genes studied in chicken and frog (defined as MHC-located genes), e.g., DMA, DMB, and BRD2, and genes in flanking regions (defined as MHC-landmark genes), e.g., RXRB, COL11A2, TNXB, and C4.
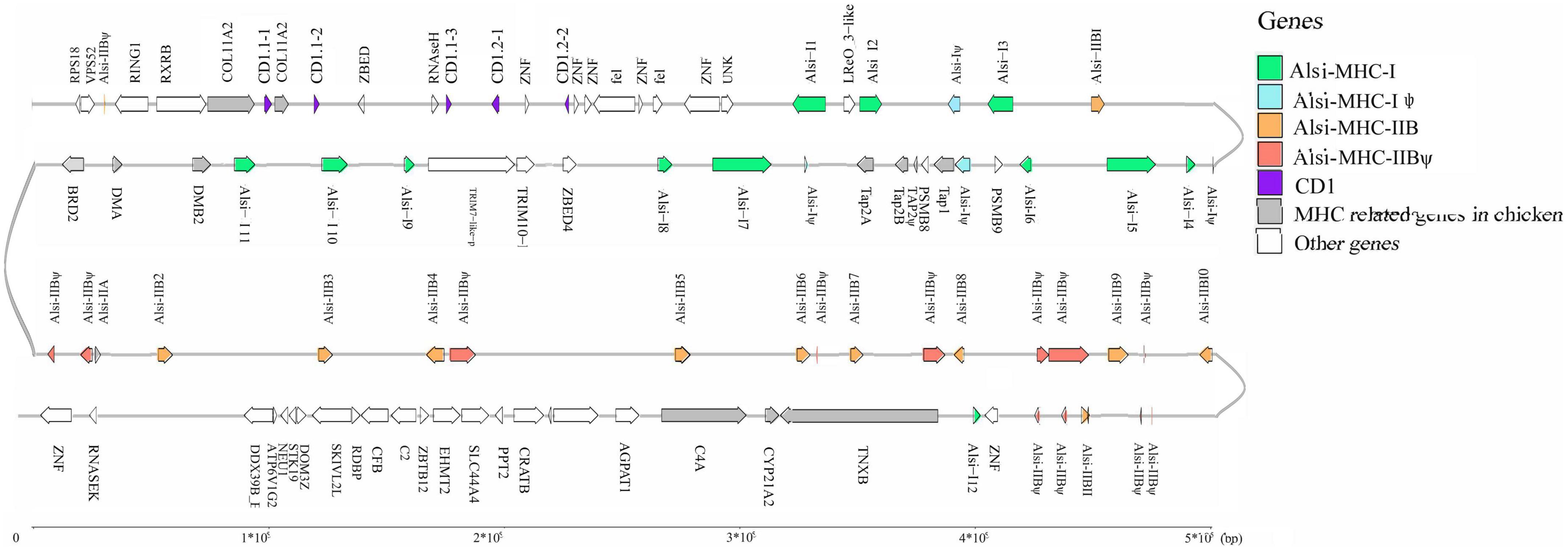
Figure 2. Gene arrangement in 2M BACs MHC region in Chinese alligator.
The Alsi-MHC-BAC region revealed the complete and delicate MHC architecture in reptiles. In most previously published studies on reptiles, the target BACs or contigs usually had single or duplicated MHC antigen genes. Some gene linkages reported in other reptiles were verified in our study, e.g., MHC-I with Tap2, Trim with MHC-I, and MHC-I with MHC-II in saltwater crocodile; MHC-I with Mr1 and Tap2 with Psmbs and Tnxb-Cyp21-Cenpa-C4 in the related region in Komodo dragon; MHC-I with MHC-II in tuatara and Anolis (Figure 3A). However, in the Chinese alligator’s MHC core region, we did not find framework genes that are present in the other reptiles, e.g., ABCB9 in the MHC-I region (Komodo dragon) and GUK1 in MHC-I genes (tuatara).
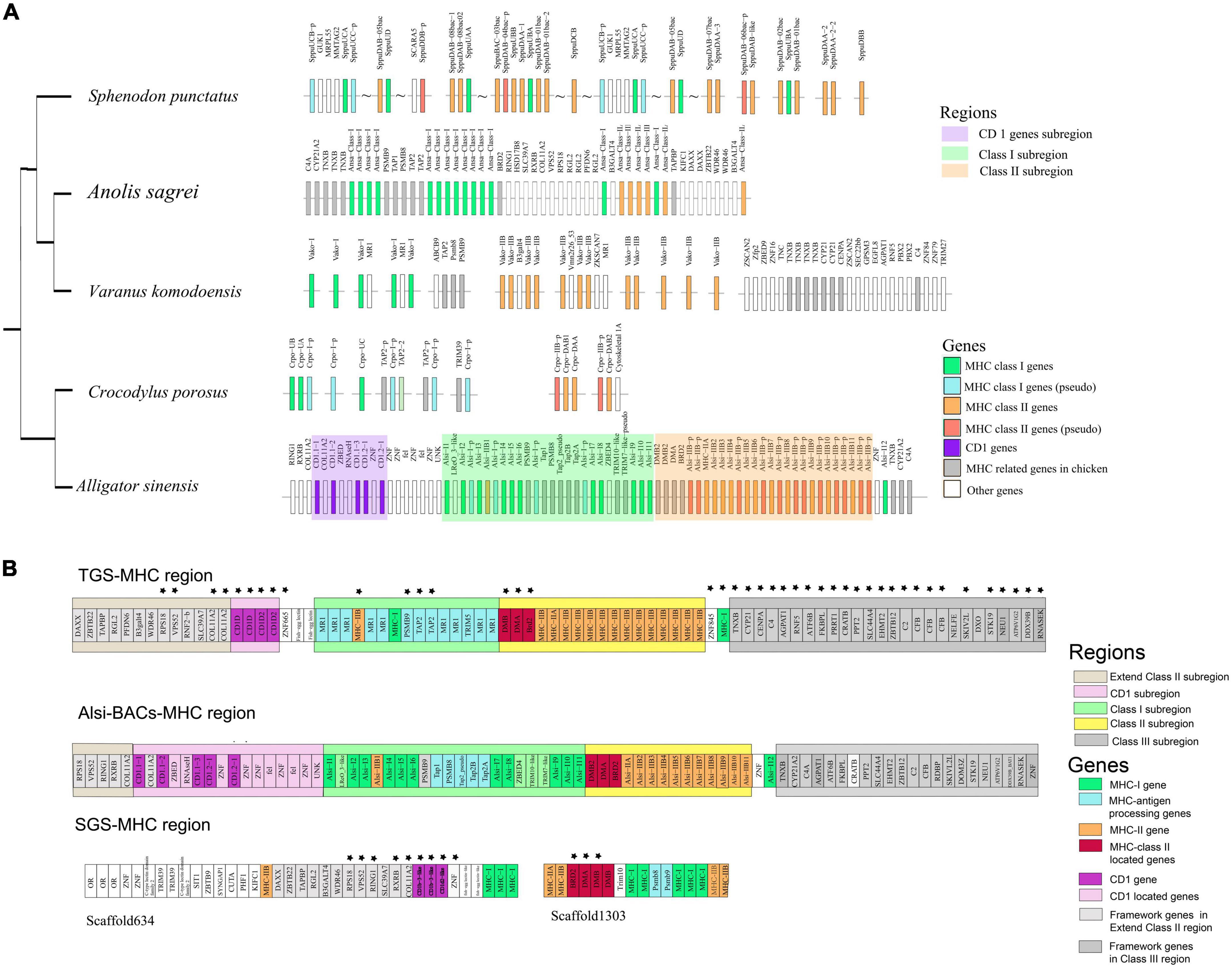
Figure 3. (A) Gene arrangement in the Chinese alligator and other published MHC regions of reptiles. In Sphenodon punctatus, wavy lines suggest existence on the same chromosome but with uncertain distance. Molecular distances between genes and subregions are not reflected on the map. (B) The comparison of gene annotation in the SGS-based genome, gene prediction in Alsi-BAC-MHC region, and gene annotation in TGS-based genome. The genes having a conserved location with the Alsi-BAC-MHC region (except MHC-I and MHC-II genes) are marked with stars.
When comparing the Alsi-MHC-BAC region with the other sources of MHC data, Alis-SGS genome and Alis-TGS genome, it is still suggested that BAC is the most effective and accurate method to deal with the MHC gene family (Table 1). The MHC region found in the SGS-based genome (SGS-MHC region) was significantly more fragmented than the Alsi-BAC-MHC region, with MHC-I and MHC-IIB genes in separate scaffolds. Only nine genes upstream of the Class I subregion and three genes in the Class II subregion in SGS based-MHC were consistent with those in the Alsi-BAC-MHC region. This situation was better in the MHC region found in the TGS-based genome (TGS-MHC region). The TGS-MHC region had a closely linked Class I subregion and Class II subregion, consistent with the Alsi-BAC-MHC region, and had gene consistency with three of five and 22 of 23 genes in the left (Extend Class II subregion) and right boundary regions (Class III subregion) of the Alsi-BAC-MHC region, respectively. The notable difference was mainly in the copy numbers of the antigen-presenting genes (Alsi-TGS vs. Alsi-BAC-MHC, MHC-I: 2 vs. 12, and MHC-IIB: 14 vs. 11). Although the copy numbers were different, the gene location was conserved, including one MHC-IIB gene in the Class I subregion and one MHC-I gene next to the right boundary of the Class II subregion. The three more MHC-IIB genes in the Alis-TGS genome might be pseudogenes with partial CDS, and the MHC-I genes in the Class I subregion were considered MR1 genes in the Alsi-TGS genome. MR1 is considered a non-classical MHC Class I molecule. The expression data and RACE results suggested that most of the predicted MHC-I (9 of 12) genes in Alsi-BAC-MHC were expressed in all tissues, so we considered them as MHC-I genes. Above all, the numbers based on BAC, RACE, and manual checking are more reliable.
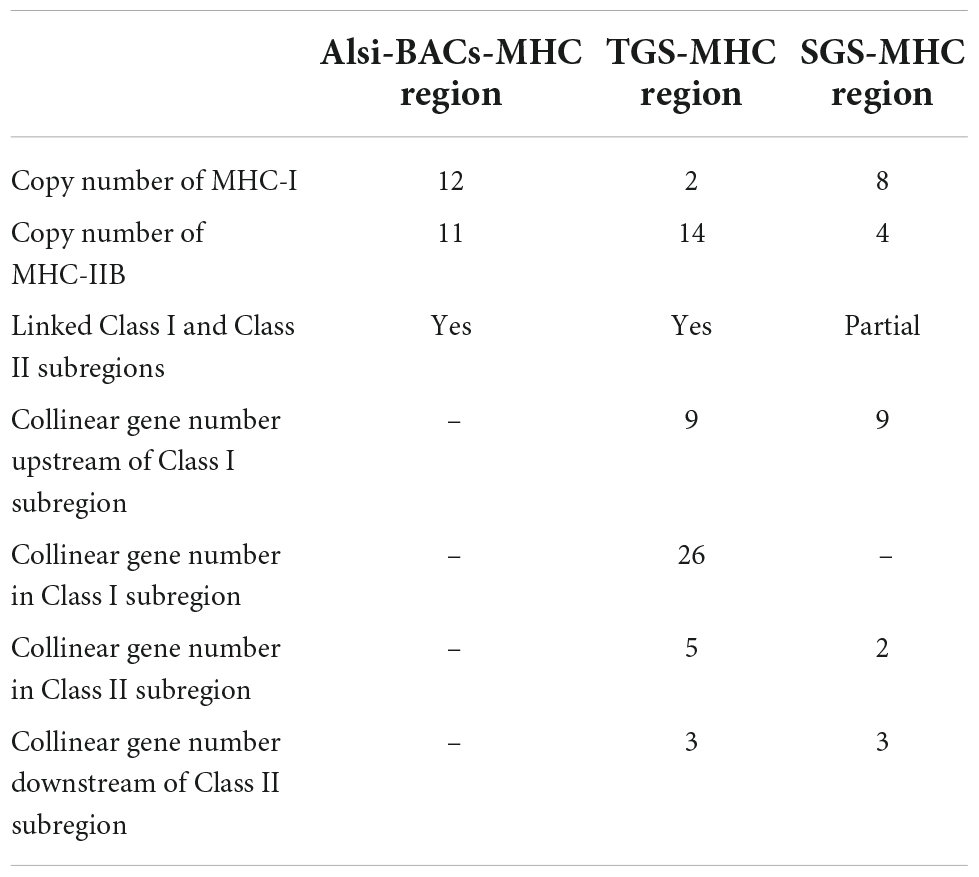
Table 1. Comparison of MHC region from three sources.
With the fine elaboration of MHC architecture in reptiles, we further compared the evolution of the MHC architecture in tetrapods. First, we redefined the MHC core region based on the collinearity analysis between amphibians, reptiles, birds, and mammals (Supplementary Figure 2). Two linked gene clusters (Rxrb-Col11a2 and C4-Cyp21-Cenpa-Tnxb) were taken as the flanking markers at both ends. In all studied species, C4, Cyp21, Cenpa, and Tnxb were conserved and were adjacent to the MHC core region (considered as the MHC-II region in mammals). At the other end, Rxrb and Col11A2 were considered as the boundary of the MHC core region, which was proved in African clawed frogs, Chinese alligators, gray short-tailed opossums, and humans, but not in chickens (Figure 4). Then, the MHC core region of four species, except humans, was all divided into Class I subregion (with MHC-I and antigen-processing genes) and Class II subregion (with MHC-II genes and some MHC-located genes, e.g., BRD2). This delimited MHC core region had adjacent Class I and II subregions. This suggested that the proximity of Class I and Class II subregions existed in the tetrapod ancestor (Figure 4).

Figure 4. Model of tetrapod MHC evolution. To distinguish the Homo-Class I subregion (with MHC-I genes) and the original location of antigen-processing genes, we labeled them with dark green (with MHC-I inside) and light green (the sub-region with antigen-processing genes).
The arrangement of Class I and Class II subregions was also variable in the MHC core region. The locations of Class I and Class II regions in the five species were distinct (Figure 4). To distinguish the MHC regions from different species, we prefixed them with the corresponding species names. The Xela-Class II region was next to Rxrb-Col11A2 in African clawed frog, but the Alsi-Class II subregion was next to C4-Cyp21-Cenpa-Tnxb in Chinese alligator. Based on the information from the other species (especially chicken), we conjectured that the Alsi-Class I and Alsi-Class II subregions inverted as a whole in Chinese alligators. This hypothesis is further supported by the Alsi-MHC-I genes between the Alsi-Class II subregion and C4-Cyp21-Cenpa-Tnxb and the Alsi-MHC-IIB genes at the beginning of the Alsi-Class I subregion. Moreover, it strongly supported that Taps and Psmbs genes are located along with MHC-I genes in the Class I subregion and the BRD2 gene is located along with MHC-II genes in the Class II subregion.
Within Class I and Class II subregions, gene deletion, duplication, and translocation has also occurred. The close linkage of BRD2 with MHC-II was consistent in all species. However, Psmb was missing from the Gaga-Class I subregion in chickens and the MHC-I genes translocated to another subregion (homo-Class I region) in humans, leaving only antigen processing genes (Psbms and Taps) among the two homo-Class II regions. In the Class II region, all species had linked IIA and IIB, in pairs or otherwise, except chicken, in which IIA translocated 5.6 cM away (Salomonsen et al., 2003). However, some birds still have IIA-IIB dryads in the Class II subregion (He et al., 2021). In mammals (both marsupial and eutherian), the Class II subregion was divided into two by the Class I subregion or Class I antigen processing genes. It is speculated that this inversion of the Class II subregion and part of the Class I subregion occurred in the ancestor of mammals.
Some order-specific characters were also observed in the MHC core region (Figure 4). A unique CD1 region was inserted between Rxrb-Col11A2 and MHC antigen genes in Chinese alligators. However, no homologous CD1 genes were found in African clawed frogs, chickens, gray short-tailed opossums, humans, and green anole, suggesting that it might be a specific character of Chinese alligators or this group. Chicken is also considered to have two specific subregions (at least in Galliformes): BG and Blec. Blec genes are also closely linked to the innate immune function of birds.
The 15 true MHC-I genes (12 in the 2 Mb contig, with five pseudogenes, Table 2 and Supplementary file 5) diversified in expression patterns and amino acid similarities. First, according to the expression data, these 15 true MHC-I genes were classified as “classical” (expressed in all tissues) and “non-classical” (with low expression in some tissues; Figure 5A). Four non-classical MHC-I genes were determined, including three in the 2 Mb BAC-based contig (Alsi-I3, Alsi-I9, and Alsi-I10) and one (Alsi-I14) in an unlinked BAC. These 15 sequences formed three groups (I0, I1, and I2) in the phylogenetic analysis (Figure 3A, marked with different colors, both with high bootstrap support) and based on the characters of translated amino acids (Supplementary Figure 3). The I0 group (blue in Figure 5A) was the most abundant, including seven loci in the 2 Mb contig and three separated ones. The I1 (pink in Figure 3A) and I2 groups (green in Figure 3A) were found only in the 2 Mb contig, with three and one copies, respectively. All the groups expressed classical loci.
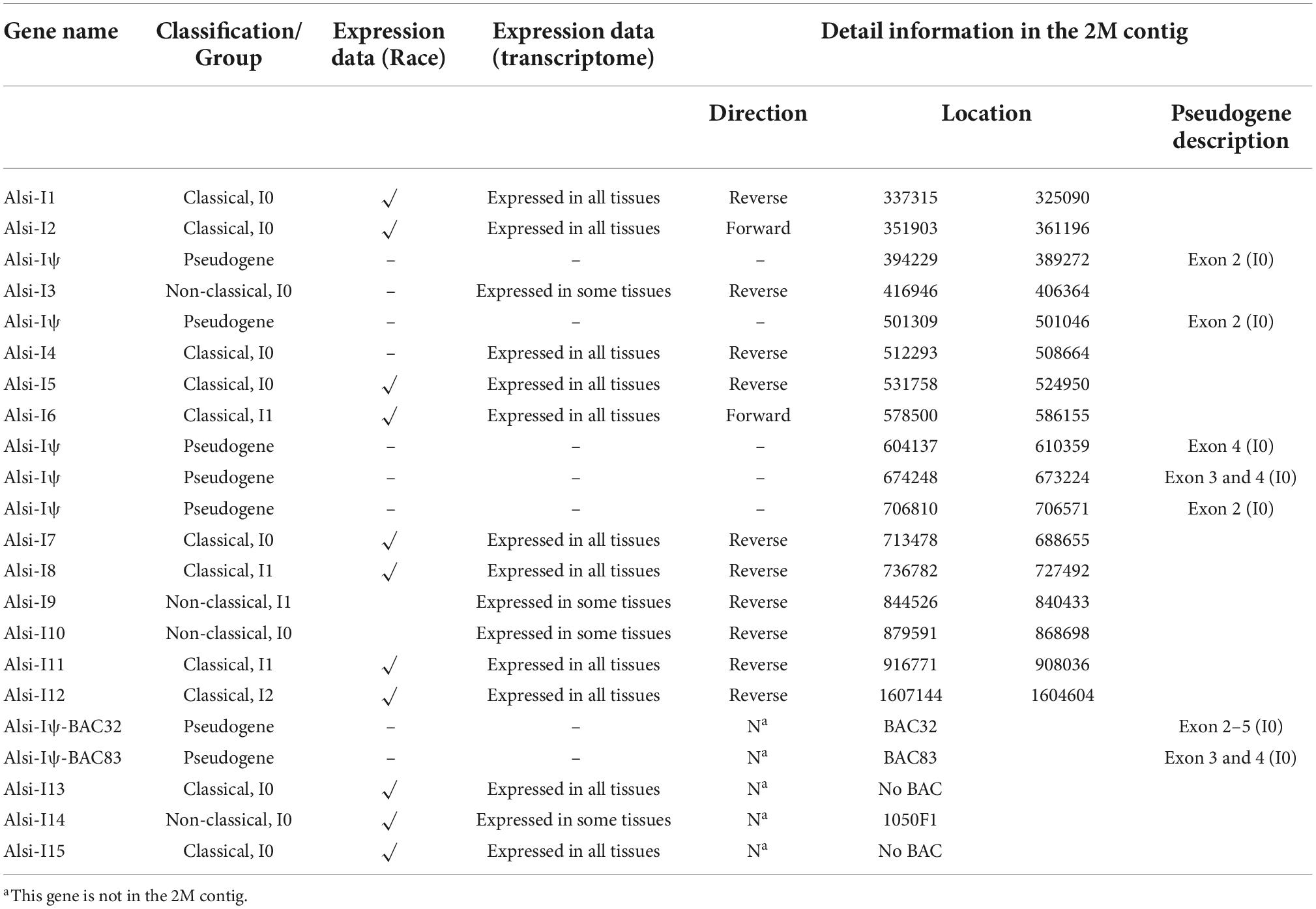
Table 2. The information about MHC-I genes in Chinese alligator.
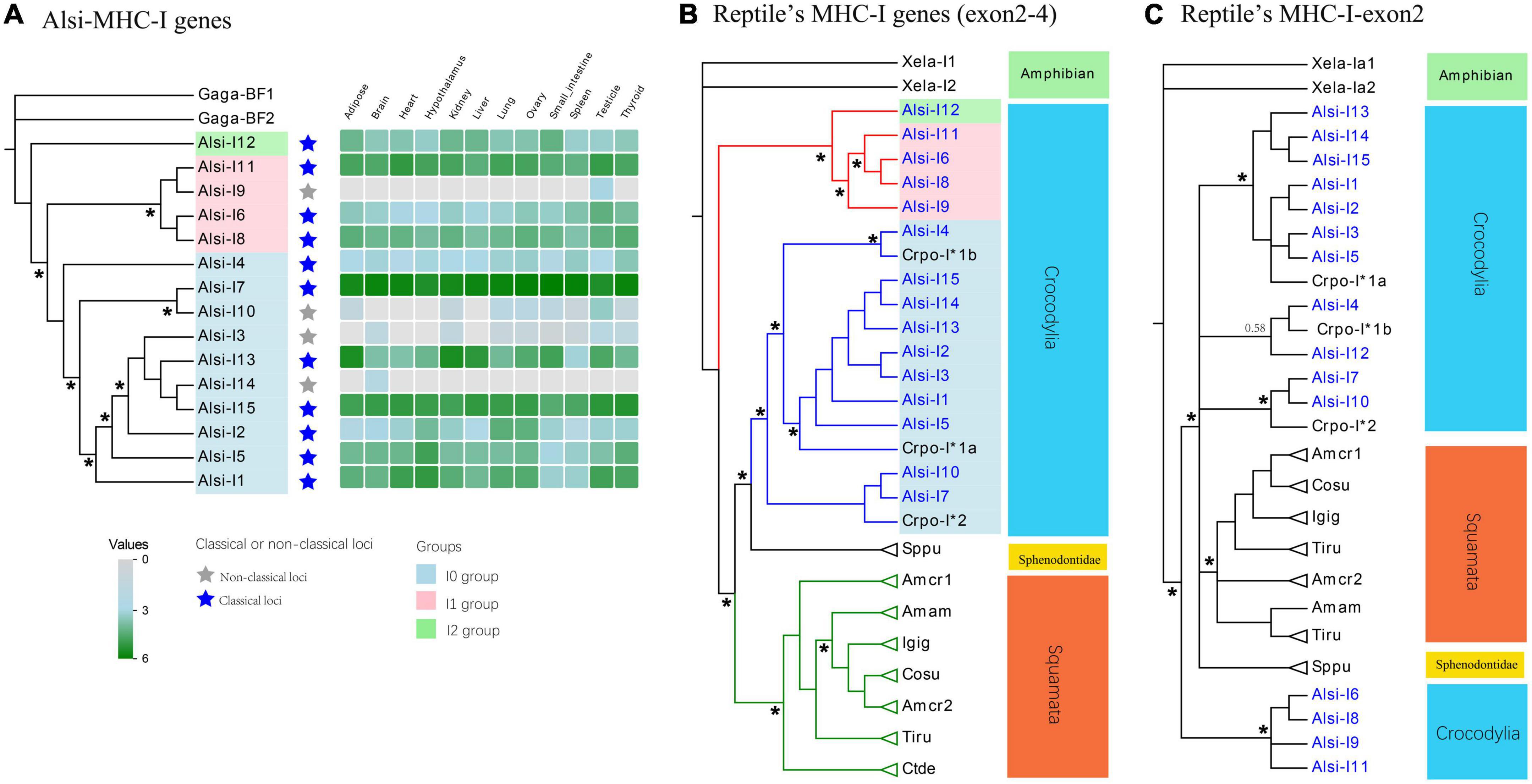
Figure 5. Phylogenetic evolution of MHC-I genes in (A) Chinese Alligator, (B) in reptiles using the main CDS (exon 2 to exon 4), and (C) in reptiles using the antigen-presenting exon 2. The expression data in panel (A) were calculated using the log10 of TPM values, and all the values under one were treated as zero. The genes extracted in our study are labeled with blue names in panels (B,C). Stars on phylogenetic trees suggest bootstrap values >0.7, and some vital bootstrap values less than 0.7 are also listed with numbers. All the corresponding sequences are listed in Supplementary Table 2. Gaga, Gallus gallus; Xela, Xenopus laevis; Amcr, Amblyrhynchus cristatus; Sppu, Sphenodon punctatus; Crpo, Crocodylus porosus; Amam, Ameiva ameiva; Cosu, Conolophus subcristatus; Ctde, Ctenophorus decresii; Igig, Iguana iguana; Tiru, Tiliqua rugosa.
Analysis with other MHC-I sequences of reptiles revealed the evolution of MHC-I genes in this order. Based on the main part of sequences (exon 2–exon 4), there were three apparent clades (clade 1: I1 and I2 groups from Chinese alligator, clade 2: I0 group with sequences from Crocodilia and Sphenodontidae, and clade 3: sequences from Squamata); all the bootstrap values were higher than 0.90 (Figure 5B). The MHC-I genes were gathered by groups rather than by species, and it was apparent that the I1 and I2 groups were novel MHC-I groups, which were detected for the first time, and all the published MHC-I data of Crocodilia belonged to the I0 group. Focusing on the antigen-presenting region (exon 2, Figure 5C; exon 3 showed similar results, Supplementary Figure 4) suggested that the I1 and I2 groups were still distinct from I0 and other reported sequences.
Based on the transcriptome data, the 11 true genes (all in the 2 Mb contig, with 15 pseudogenes, Table 3 and Supplementary file 5) were grouped as “classical” (N = 7) and “non-classical” (N = 4) (Figure 6B). Highly similar MHC-IIB genes were observed, e.g., Alsi-IIB7 and Alsi-IIB10 (identical CDS, 100% similarity, but in different non-overlapping BACs) and Alsi-IIB4 and Alsi-IIB9 (99% similarity, also in different BACs). The amino acid similarity was higher than 80% in the seven classical MHC-IIB genes, but it was 58–75% with non-classical genes (Supplementary Figure 5). However, the 11 expressed MHC-IIB genes maintained diversity in several aspects. First, nearly half of the genes (5 of 11) differed in the length of exon 2, with one amino acid deletion (Alsi-IIB1), one amino acid insertion (Alsi-IIB3), and two amino acid deletions (Alsi-IIB6, Alsi-IIB7, and Alsi-IIB10) (Figure 6A). Secondly, the splice site of exon 2 and 3 was diversified. Compared with the conserved “GU-AG” intron splice signal, we found four genes with “CA-TC” signals, and Alsi-IIB10 had some other signals as well (“CG-CC” and “CT-TC”) (Figure 6A).
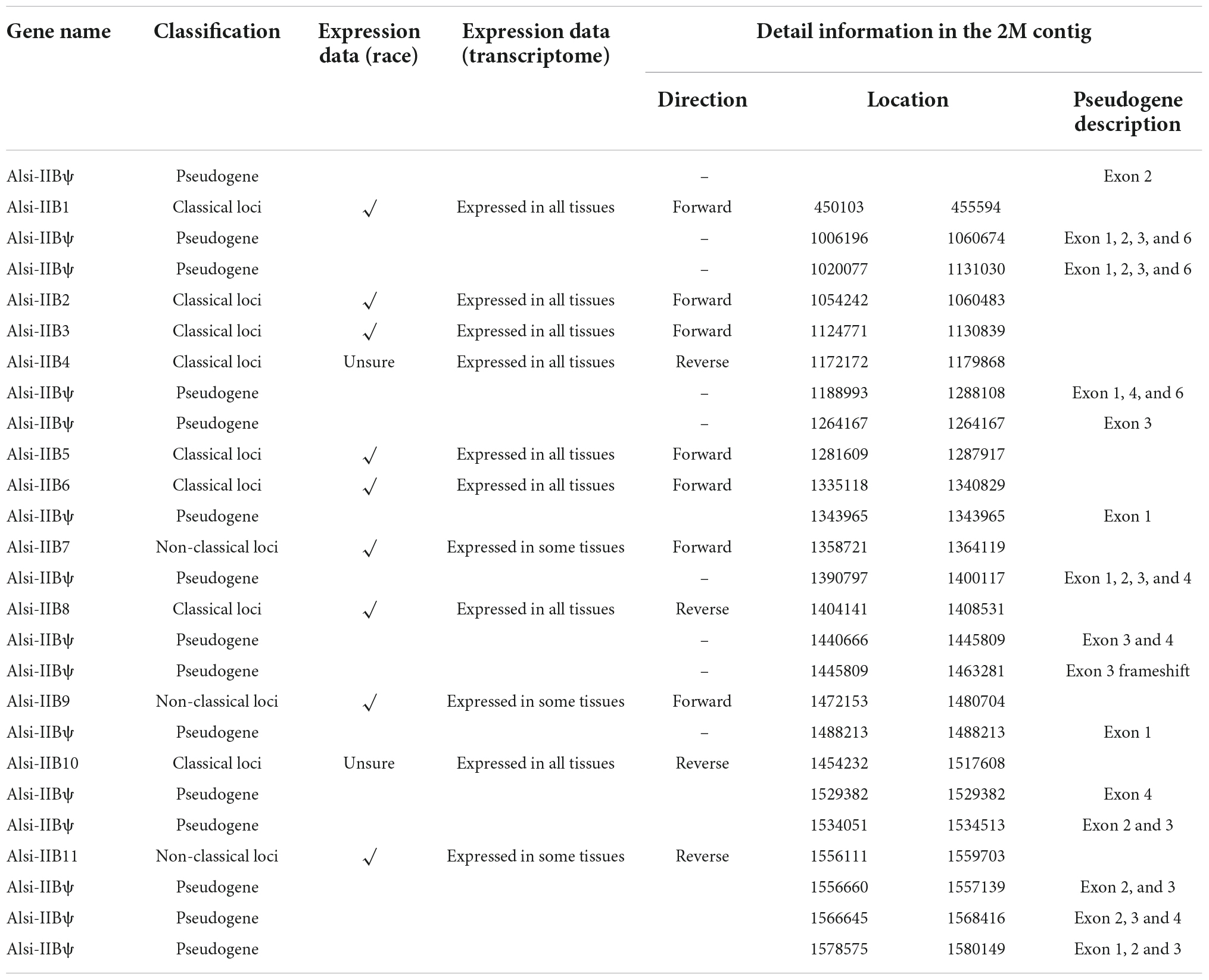
Table 3. The information about MHC-IIB genes in Chinese alligator.
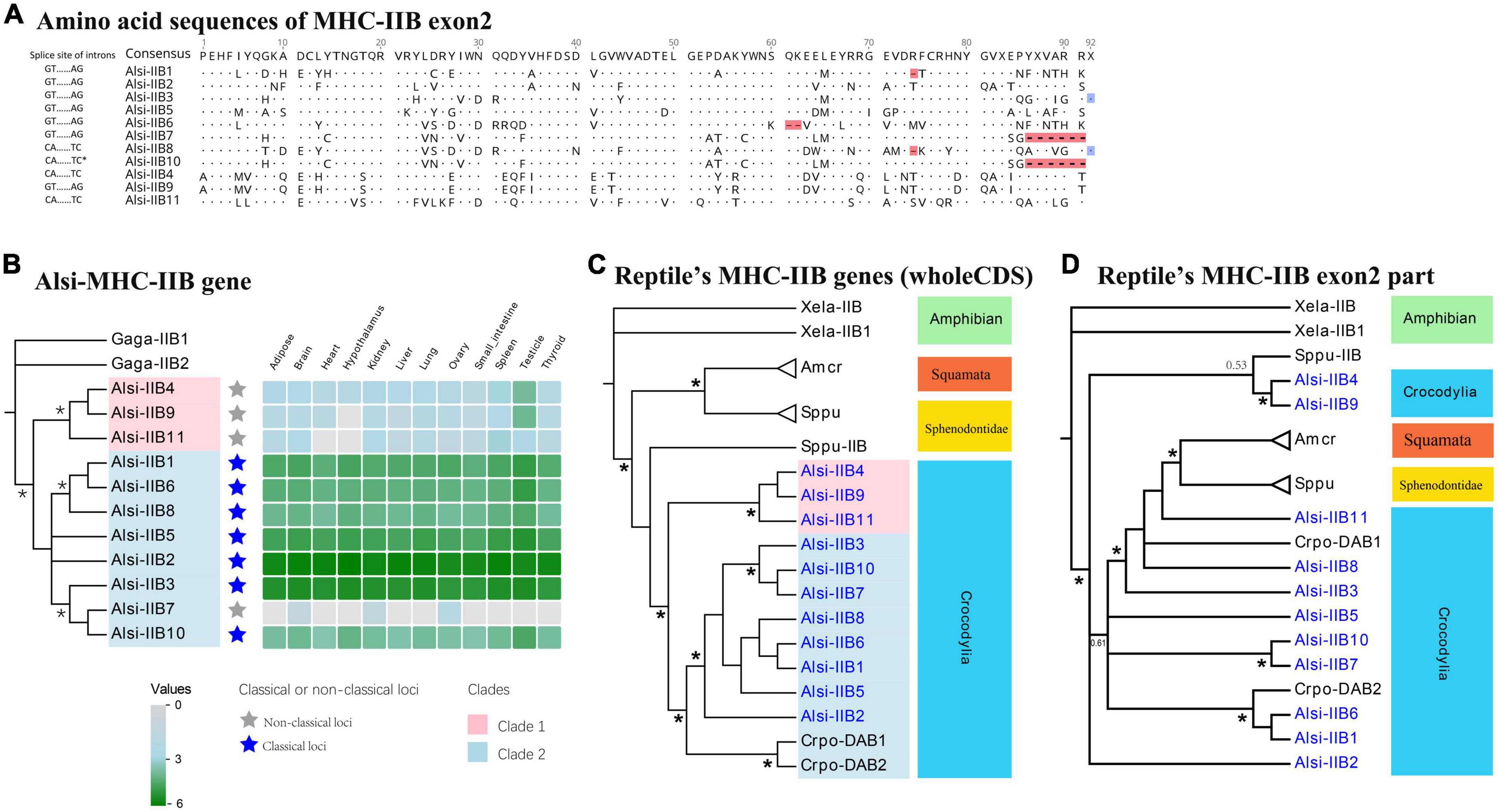
Figure 6. The phylogenic evolution and expression of MHC-IIB genes in the Chinese alligator. (A) Amino acid sequences and exon/intron splice signals in MHC-IIB exon 2. Deletions and insertions are marked with red and blue, respectively. Phylogenetic evolution of MHC-I genes in (B) Chinese Alligator, (C) in reptiles using the whole CDS, and (D) in reptiles using the antigen-presenting exon 2. Explanatory notes are the same as in Figure 5. Stars on phylogenetic trees suggest bootstrap values >0.7.
The difference between classical MHC-IIB genes and non-classical ones was also confirmed by the phylogenetic relationship (Figure 6B). The 11 expressed MHC IIB genes formed two well-supported clades using the whole CDS, and one clade included three non-classical genes (red in Figure 6B). When compared with other reptiles’ data, all the available MHC-IIB sequences of Crocodilia form a single clade, with two subclades (represented by two in Figure 6C, in blue clade). One clade in Crocodilia included non-classical loci, and the other had classical loci in this study and other sequences from previous studies. The significantly isolated clades of Squamata and Sphenodontidae suggested that non-classical MHC-IIB genes emerged after the split of Crocodilia and other reptiles. We also used the antigen-presenting region (exon 2) to study how it evolved under pathogen pressure. The phylogenetic tree showed that trans-species polymorphism (Figure 6D) might be the primary mechanism for maintaining polymorphism.
CD1 genes are considered the third family of antigen-presenting molecules. In the 2 Mb contig, we found five CD1 genes in proximity with each other, located upstream of the MHC-I region (Figure 2, Table 4). According to the phylogenetic analysis, three of five CD1 genes were defined as the CD1.1 group, and two others as the CD1.2 group (Figure 7). The duplicated CD1.1 and CD1.2 genes suggested a gene duplication event might have formed them. Based on the transcriptome data, one CD1.1 gene was found to be lowly expressed (Alsi-CD1.1-1).
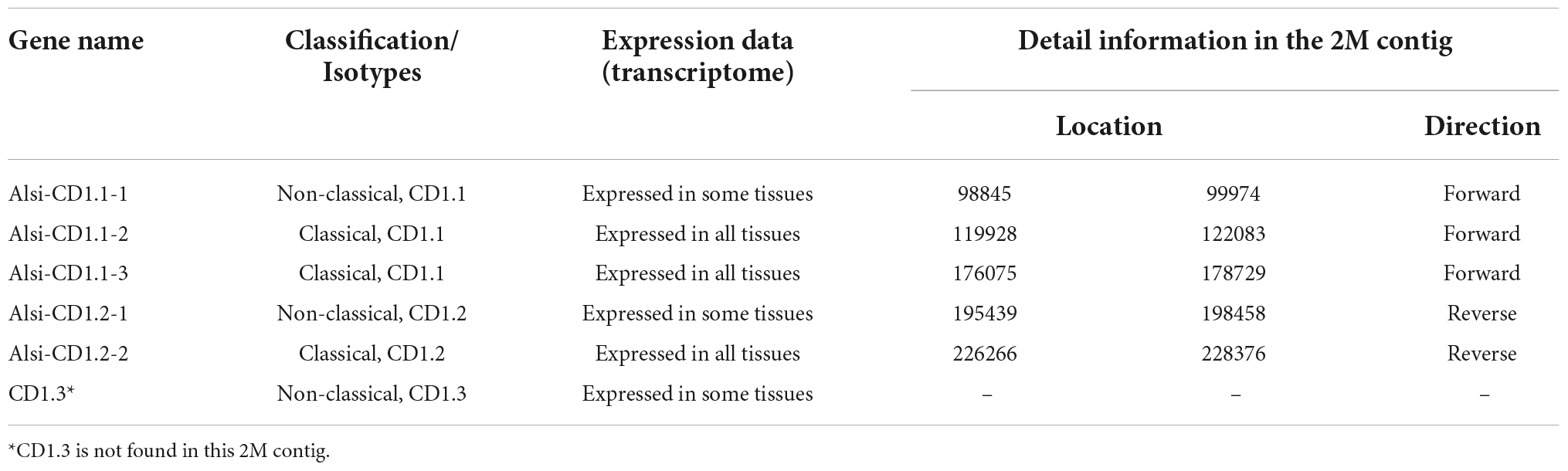
Table 4. The information about CD1 genes in Chinese alligator.
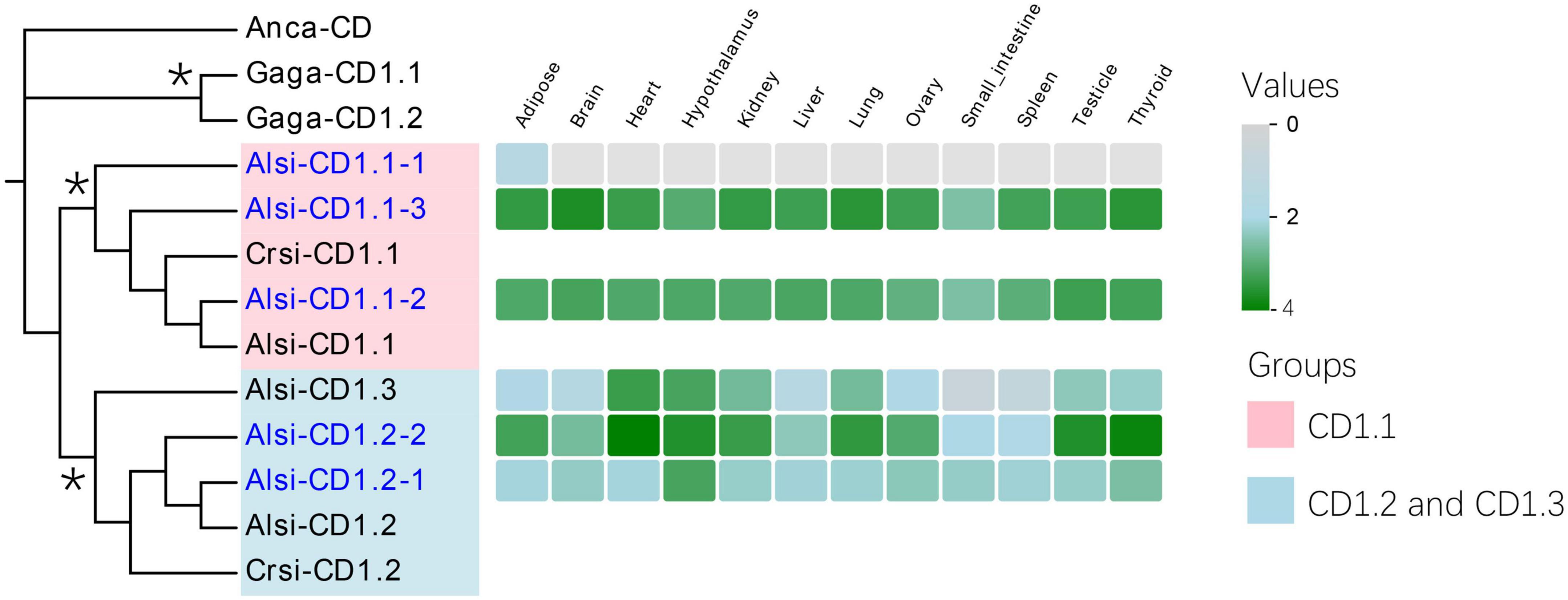
Figure 7. Phylogenic evolution and expression of CD1.1 genes in the Chinese alligator. The significant clades are marked with different colors (purple-CD1.1 and blue-CD1.2 and CD1.3). Explanatory notes are the same as in Figure 5. Stars on phylogenetic trees suggest bootstrap values >0.7.
The CD1 gene was assumed to have three groups in reptiles (Yang et al., 2015); however, only two groups were observed in our 2 Mb contig. The third group (Alsi-CD1.3, defined by Yang et al., 2015) was gathered with the CD1.2 clade in our phylogenic tree (85.91% bootstraps, Figure 7) and expressed in only some tissues. However, we found no other homologous genes in the 2 Mb contig. Based on our previous genome data, this special gene might have resulted from the transposition of Alsi-CD1.1.
In this study, we systematically characterized the genomic architecture, gene expression, gene grouping, and phylogeny of the MHC genes in the Chinese alligator. The results provide the first comprehensive physical map of MHC architecture in crocodylia. We also found that Chinese alligator MHC includes a continuous region with adjacent Alsi-Class I and Alsi-Class II subregions and a unique CD1 subregion close by. Based on this newly added information, we reconstructed the evolutionary history of MHC in tetrapods and inferred the evolutionary trajectories of this vital immune family. It provided new evidence regarding chromosomal rearrangements and translocation events that led to the diverse organization of the tetrapod MHC.
The multiple copies of antigen-presenting genes were clustered into several groups, some of which were described for the first time. We believe that the pseudogenes might also function in history, and they can provide a great repertoire for this species. All these results suggested that Chinese alligator has a robust immune system.
To date, the architecture of the MHC has been revealed in amphibians, birds, marsupials, and eutherians, but it is poorly understood in reptiles. A few studies on reptile MHC have reported polymorphisms at MHC-I or MHC-IIB genes using high-throughput sequencing or Sanger sequencing (Nie et al., 2013; Jaratlerdsiri et al., 2014b,c) but did not examine the arrangement of MHC genes on a chromosomal scale. Previous studies in reptiles only revealed a few characteristics of MHC, e.g., duplicated MHC-I and MHC-IIB genes [saltwater crocodile (Jaratlerdsiri et al., 2014a), tuatara (Miller et al., 2015), Komodo dragon (Reed and Settlage, 2021), and two anoles (Card et al., 2022)], Tap2 with MHC-I (saltwater crocodile), and MHC-I and MHC-II genes were not far away from each other (tuatara and anoles). These findings were just part of the story. Here, combining genome data and the BAC library, we completed the missing information in this group. We found that (1) the alligator’s whole MHC region was continuous, and its size (approximately 2 Mb) was comparable to that of other tetrapods, except birds; (2) the Alsi-Class I and Alsi-Class II subregions were close together, the antigen-presenting genes (Taps and Psmbs) were found in the Alsi-Class I subregion with MHC-I genes, and the MHC-landmark genes (C4, Cenpa, and Tnxb) were next to the Alsi-Class II subregion; (3) a unique CD1 subregion existed upstream of the MHC core region, next to the Alsi-Class I subregion, which has been reported for the first time in the MHC core region.
It is widely known that the difficulties of assembling the MHC region are due to extensive variation in gene sequence and haplotype composition of multigene families. Here, we took a relatively conservative method to detect the antigen-presenting genes in the BAC library and assemble the MHC region. With the help of genome annotation (Wan et al., 2013), we figured out this puzzle and renewed the BAC and alligator MHC-I and MHC-IIB repertoires using the RACE sequences. The results (adjacent genes, closely linked Class I and Class II subregions and gene arrangement) were also supported by the latest TGS-based genome of the Chinese alligator (Figure 6B; Yang et al., 2022). Combined with transcriptome data, the discrepant copy numbers of MHC-I and MHC-IIB genes were more accurate in our results, which were based on BAC, RACE, and manually checking. As MHC genes have numerous copies, which means several gene paralogs with different degrees of similarity, sequencing by BAC is still the best way to illustrate the homologous genes with multiple copies. Although TGS can cover more than 1 Mb, more detailed sequence information and more accurate predictions are required.
The MHC architecture revealed in crocodiles has refreshed our understanding of MHC evolution. Firstly, as in birds (Kaufman et al., 1999), marsupials (Zhou et al., 2021), and amphibians (Ohta et al., 2006), adjacent Class I and Class II subregions were also revealed in reptiles. This suggested that proximity between Class I and Class II subregions existed in the tetrapod ancestor (Figure 6A). This conclusion is further supported by the ancient MHC model inferred in birds (He et al., 2021) and the conserved MHC model studied in amphibians (personal communication with Wieslaw Babik). The MHC-I gene translocation event, from the original Class I subregion to the eutherian Class I subregion, might have happened after the split of eutherians, marsupials, and monotremes, about 161–185 MYA. This translocation event included the entire MHC-I genes, except antigen-presenting genes (Taps and Psmbs), so it is common to find these antigen-presenting genes, which should be part of the previous Class I region, in the Class II region in eutherians (Shiina et al., 2017).
Secondly, inversion of a region of the Class II subregion might have happened in reptiles and mammals. The differences in the order of different regions in crocodile, X. laevis, and chicken were reliable and proved using both BACs and long-reads sequencing (Kaufman et al., 1999; Ohta et al., 2006). This chromosome rearrangement resulted in Rxrb-Class I subregion-Class II subregion-TNXB in crocodiles. Such an inversion event has been found in cetaceans and ruminants, wherein the Class II subregion was separated into two subregions, one with DR and DQ loci and the other with non-classical genes (de Sá et al., 2019).
Thirdly, all the lineages exhibited distinct features in gene content and organization. Notably, we found a CD1 subregion in the MHC core region of the Chinese alligator. CD1s are considered a third family of antigen-presenting molecules and originated from MHC-I in some stage of vertebrate evolution (Yang et al., 2015). Yang et al. found CD1 gene numbers, groups, and genomic locations in reptiles. In our study, two CD1 genes were found upstream of the Class I subregion, consistent with the findings of (Yang et al., 2015) that CD1 genes are linked to the MHC region and MHC paralogs region. We also inferred that the compaction of avian MHC occurred after the crocodilian-avian split (approximately 240 MYA) (Kumar and Hedges, 1998). Each lineage also has varying degrees of duplicated antigen-presenting genes, and the MHC-I and MHC-IIB genes have undergone large-scale expansion in the Chinese alligator, resulting in more than 10 MHC-I and MHC-IIB loci. Longer intron length is one reason for gene expansion. It is suggested that the mean length of MHC-I and MHC-IIB is about five to six times and two to three times, respectively, greater in saltwater crocodiles than in the fowl species (about 2 kb and about 1.8 kb, respectively, on an average) (Jaratlerdsiri et al., 2014a). These reports, including ours, provide opportunities to investigate how the MHC function evolves in different genomic landscapes and may challenge the functional importance of conserving the MHC organization.
We also performed the first comprehensive analysis of the loci of antigen-presenting genes of this lineage. Among the predicted MHC-I and MHC-IIB loci, we proposed 15 functional MHC-I genes and 11 functional MHC-IIB genes based on transcriptome files and RACE data. This thorough understanding of MHC-I and MHC-IIB genes allowed us to investigate Crocodylia phylogenetic relationships. Multiple copies of MHC-I clustered into various clades, rather than all together by species (Figure 5B), which might have originated from different lineages of MHC-I genes. However, all the published data in reptiles could not be clustered with clade (Figure 3B), so it is unclear if I1 and I2 group MHC-I genes are species-specific or newly found. For MHC-IIB genes, all the sequences, except Sppu-IIB, were gathered by order (Figure 6B). The study in tuatara suggested two lineages of Class II genes in reptiles (Miller et al., 2015). SppuDBB (listed as Sppu-IIB in Figure 6) in turtle and crocodile were in a separate clade. However, sequences of the Chinese alligator were only detected in the SppuDBB clade, and no clues of another clade were obtained (Figure 6C). It can be inferred that one of the ancient MHC-IIB lineages was missed in crocodiles, as some lineages lost one of the two ancient avian MHC class IIB (MHCIIB) lineages in birds (Goebel et al., 2017). Furthermore, the phylogenetic clustering changed when compared with the peptide region. The two significantly different clades of MHC-I (Figure 5C) and MHC-IIB genes (Figure 6C) allow the Chinese alligator to respond to different environmental pressures or perform novel immune functions.
We took a population survey for MHC-I and MHC-IIB genes in previous studies (Zhai et al., 2017; Han et al., 2019). Unlike the various high alleles found in MHC-IIB in Chinese alligator, only I1327 (Alsi-I7), I20 (Alsi-Iψ-BAC32), and Beta1085 (Alsi-IIB11) were found all dimorphic: two alleles at each locus (Zhai et al., 2017). All the alleles had a few amino acid differences: three in Alsi-I7 exon 2, two in Alsi-Iψ-BAC32 exon 3, and zero in Alsi-IIB11 exon 2 (the single mutation did not cause any change in amino acid). These results contradicted the diversifying selection reported previously (Liu et al., 2007; Nie et al., 2013). The high polymorphism reported by Liu et al. (2007) was caused by the amplification method with conservative primers for multiple loci, which might consider sequences from different loci as alleles. The usage of universal primers could produce a lower than expected ds value, leading to an elevated dN/dS ratio and thus giving “anomalous” results (Miller and Lambert, 2004). With the prevalence of long-read sequencing, this shortage of primers might be overcome.
Realizing that multiple loci may also be the primary source of MHC polymorphism, we first proposed the hypothesis of the “MHC antigen-presenting binding variability” model (Figure 8), which included two aspects. First, the hundreds or thousands of alleles are the main reason for MHC polymorphism, and it is well-accepted in birds, especially chickens, with only one major expressed MHC-I gene (Kaufman, 2018). In another aspect, with the new findings from this study, the multiple copies of MHC genes can also enrich the diversity of antigens. In a simplistic scenario of antigen-presenting binding variability, the polymorphism might have resulted from many alleles from one locus or paralogs homologies from many duplicated loci. However, in reality, it is hard to find these extreme examples, especially in a species with multiple monomorphic loci. Our findings on the Chinese alligator suggested that it is almost an extreme case and might be related to its bottleneck event. In an individual, the co-expression of multiple monomorphic loci is supposed to have more antigen-presenting binding variability than the expression of a single locus with many alleles. This might be part of the magic key to the success of bottleneck populations. A similar phenomenon was also found in crested ibis (Lan et al., 2019) and Père David’s deer (Wan et al., 2011).
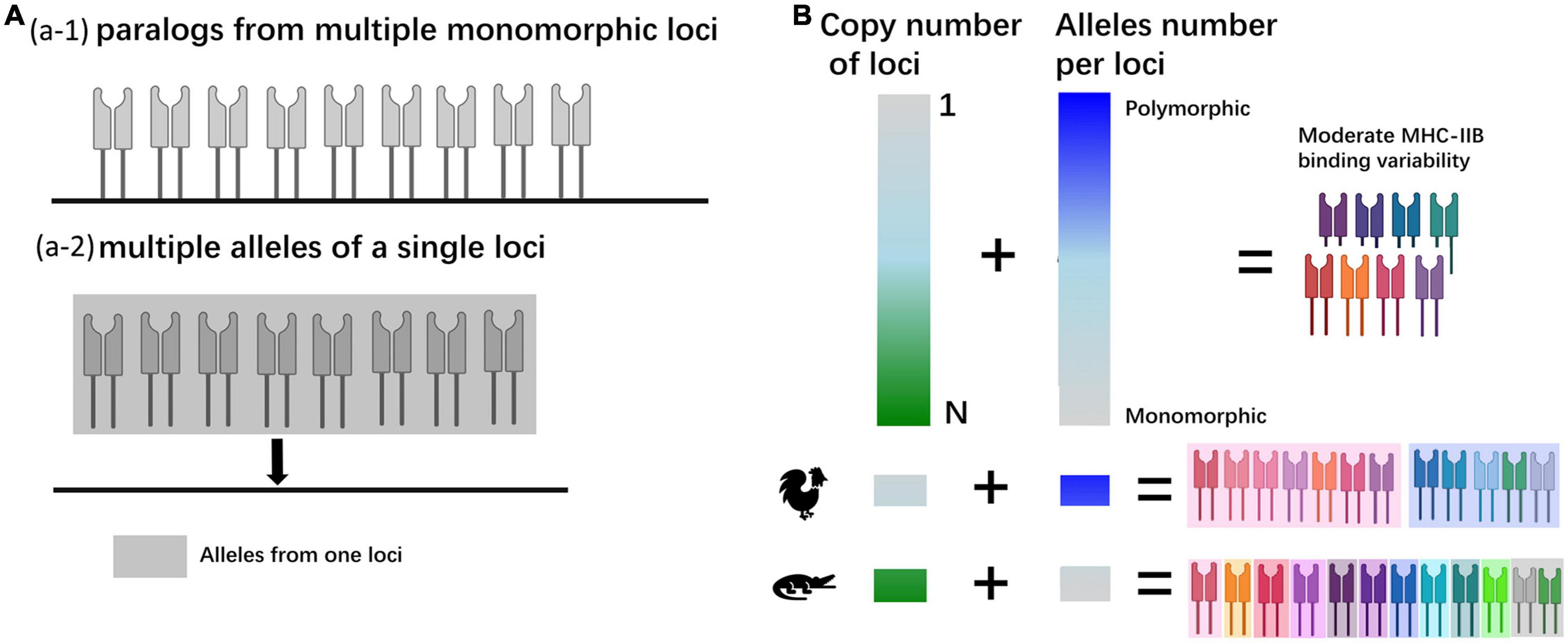
Figure 8. (A) The mechanism of polymorphism in antigen-presenting proteins. (a-1) An extreme model with paralogs from multiple loci, and each locus is monomorphic. (a-2) An extreme model with all the alleles from one locus. (B) The causes of gene polymorphism in two aspects and the speculated models for chicken and the Chinese alligator.
Taken together, our results indicate a delicate and unique MHC architecture in the Chinese alligator. It presented adjacent Class I and Class II subregions and a unique CD1 subregion. The novel MHC architecture of reptiles filled the missing parts of the MHC cluster’s evolution and inspired some constructive ideas about gene rearrangement events. The sufficient data also revealed both classical and non-classical genes, classified as three groups of MHC-I genes and two groups of MHC-IIB genes in this species. We can infer from these findings that these highly duplicated antigen-presenting loci may have contributed to MHC antigen-presenting binding variability in this extinct population and helped this species to expand rapidly in recent years.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal study was reviewed and approved by Zhejiang University (ZJU2015-154-13).
KH, YZ, QY, and Q-HW performed the experiments, analyzed the data, and wrote the manuscript. S-CY analyzed the data. Q-HW and S-GF designed and edited the manuscript. All authors reviewed the manuscript.
This work was supported by the Fundamental Research Funds for the Central Universities of China, Natural Science Foundation of Zhejiang Province, China (LQ21C030008), and the National Natural Science Foundation of China (No. 32101243).
QY was employed by Zhejiang University Press Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.1078058/full#supplementary-material
Abduriyim, S., Zou, D. H., and Zhao, H. (2019). Origin and evolution of the major histocompatibility complex class I region in eutherian mammals. Ecol. Evol. 9, 7861–7874. doi: 10.1002/ece3.5373
Andersson, L., Lundén, A., Sigurdardottir, S., Davies, C. J., and Rask, L. (1988). Linkage relationships in the bovine MHC region. High recombination frequency between class II subregions. Immunogenetics 27, 273–280. doi: 10.1007/BF00376122
Belov, K., Deakin, J. E., Papenfuss, A. T., Baker, M. L., Melman, S. D., Siddle, H. V., et al. (2006). Reconstructing an ancestral mammalian immune supercomplex from a marsupial major histocompatibility complex. PLoS Biol. 4:e46. doi: 10.1371/journal.pbio.0040046
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94.
Card, D. C., Van Camp, A. G., Santonastaso, T., Jensen-Seaman, M. I., Anthony, N. M., and Edwards, S. V. (2022). Structure and evolution of the squamate major histocompatibility complex as revealed by two Anolis lizard genomes. Front. Genet. 13:979746. doi: 10.3389/fgene.2022.979746
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Cheng, Y., Stuart, A., Morris, K., Taylor, R., Siddle, H., Deakin, J., et al. (2012). Antigen-presenting genes and genomic copy number variations in the Tasmanian devil MHC. BMC Genomics 13:87. doi: 10.1186/1471-2164-13-87
de Sá, A. L. A., Breaux, B., Burlamaqui, T. C. T., Deiss, T. C., Sena, L., Criscitiello, M. F., et al. (2019). The marine mammal class II major histocompatibility complex organization. Front. Immunol. 10:696. doi: 10.3389/fimmu.2019.00696
Forsman, A. (2014). Effects of genotypic and phenotypic variation on establishment are important for conservation, invasion, and infection biology. Proc. Natl. Acad. Sci. U.S.A. 111, 302–307. doi: 10.1073/pnas.1317745111
Forsman, A., and Wennersten, L. (2016). Inter-individual variation promotes ecological success of populations and species: Evidence from experimental and comparative studies. Ecography 39, 630–648.
Gemmell, N. J., Rutherford, K., Prost, S., Tollis, M., Winter, D., Macey, J. R., et al. (2020). The tuatara genome reveals ancient features of amniote evolution. Nature 584, 403–409.
Goebel, J., Promerová, M., Bonadonna, F., Mccoy, K. D., Serbielle, C., Strandh, M., et al. (2017). 100 million years of multigene family evolution: Origin and evolution of the avian MHC class IIB. BMC Genomics 18:460. doi: 10.1186/s12864-017-3839-7
Han, Q. H., Sun, R. N., Yang, H. Q., Wang, Z. W., Wan, Q. H., and Fang, S. G. (2019). MHC class I diversity predicts non-random mating in Chinese alligators (Alligator sinensis). Heredity 122, 809–818. doi: 10.1038/s41437-018-0177-8
He, C., Zhao, L., Xiao, L., Xu, K., Ding, J., Zhou, H., et al. (2021). Chromosome level assembly reveals a unique immune gene organization and signatures of evolution in the common pheasant. Mol. Ecol. Resour. 21, 897–911. doi: 10.1111/1755-0998.13296
He, K., Minias, P., and Dunn, P. O. (2020). Long-read genome assemblies reveal extraordinary variation in the number and structure of MHC loci in birds. Genome Biol. Evol. 13:evaa270. doi: 10.1093/gbe/evaa270
He, K., Ye, Q., Zhu, Y., Chen, H., Wan, Q. H., and Fang, S. G. (2012). A bacterial artificial chromosome library for the Chinese alligator (Alligator sinensis). Gene 507, 74–78. doi: 10.1016/j.gene.2012.06.035
Hosomichi, K., Shiina, T., Suzuki, S., Tanaka, M., Shimizu, S., Iwamoto, S., et al. (2006). The major histocompatibility complex (Mhc) class IIB region has greater genomic structural flexibility and diversity in the quail than the chicken. BMC Genomics 7:322. doi: 10.1186/1471-2164-7-322
Jaratlerdsiri, W., Deakin, J., Godinez, R. M., Shan, X., Peterson, D. G., Marthey, S., et al. (2014a). Comparative genome analyses reveal distinct structure in the saltwater crocodile MHC. PLoS One 9:e114631. doi: 10.1371/journal.pone.0114631
Jaratlerdsiri, W., Isberg, S. R., Higgins, D. P., Ho, S. Y., Salomonsen, J., Skjodt, K., et al. (2014b). Evolution of MHC class I in the order Crocodylia. Immunogenetics 66, 53–65.
Jaratlerdsiri, W., Isberg, S. R., Higgins, D. P., Miles, L. G., and Gongora, J. (2014c). Selection and trans-species polymorphism of major histocompatibility complex class II genes in the order Crocodylia. PLoS One 9:e87534. doi: 10.1371/journal.pone.0087534
Kaufman, J. (2018). Unfinished business: Evolution of the MHC and the adaptive immune system of jawed vertebrates. Annu. Rev. Immunol. 36, 383–409. doi: 10.1146/annurev-immunol-051116-052450
Kaufman, J., Milne, S., Göbel, T., Walker, B. A., and Beck, S. (1999). The chicken B locus is a minimal essential major histocompatibility complex. Nature 401, 923–925.
Kumar, S., and Hedges, S. B. (1998). A molecular timescale for vertebrate evolution. Nature 392, 917–920.
Lan, H., Zhou, T., Wan, Q. H., and Fang, S. G. (2019). Genetic diversity and differentiation at structurally varying MHC haplotypes and microsatellites in bottlenecked populations of endangered crested ibis. Cells 8:377. doi: 10.3390/cells8040377
Lin, J.-Q., Zhou, Q., Yang, H.-Q., Fang, L.-M., Tang, K.-Y., Sun, L., et al. (2018). Molecular mechanism of temperature-dependent sex determination and differentiation in Chinese alligator revealed by developmental transcriptome profiling. Chin. Sci. Bull. 63, 209–212.
Liu, H., Wu, X., Yan, P., and Jiang, Z. (2007). Polymorphism of exon 3 of MHC class II B gene in Chinese alligator (Alligator sinensis). J. Genet. Genomics 34, 918–929. doi: 10.1016/S1673-8527(07)60103-9
Miller, H. C., and Lambert, D. M. (2004). Genetic drift outweighs balancing selection in shaping post-bottleneck major histocompatibility complex variation in New Zealand robins (Petroicidae). Mol. Ecol. 13, 3709–3721. doi: 10.1111/j.1365-294X.2004.02368.x
Miller, H. C., O’meally, D., Ezaz, T., Amemiya, C., Marshall-Graves, J. A., and Edwards, S. (2015). Major histocompatibility complex genes map to two chromosomes in an evolutionarily ancient reptile, the tuatara Sphenodon punctatus. G3 5, 1439–1451. doi: 10.1534/g3.115.017467
Nie, C., Zhao, J., Li, Y., and Wu, X. (2013). Diversity and selection of MHC class IIb gene exon3 in Chinese alligator. Mol. Biol. Rep. 40, 295–301. doi: 10.1007/s11033-012-2061-6
Ohta, Y., Goetz, W., Hossain, M. Z., Nonaka, M., and Flajnik, M. F. (2006). Ancestral organization of the MHC revealed in the amphibian Xenopus. J. Immunol. 176, 3674–3685. doi: 10.4049/jimmunol.176.6.3674
Ohta, Y., Okamura, K., Mckinney, E. C., Bartl, S., Hashimoto, K., and Flajnik, M. F. (2000). Primitive synteny of vertebrate major histocompatibility complex class I and class II genes. Proc. Natl. Acad. Sci. U.S.A. 97, 4712–4717. doi: 10.1073/pnas.97.9.4712
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pishesha, N., Harmand, T. J., and Ploegh, H. L. (2022). A guide to antigen processing and presentation. Nat. Rev. Immunol. 22, 751–764.
R Core Team (2018). R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. Available online at: https://www.R-project.org
Reed, K. M., and Settlage, R. E. (2021). Major histocompatibility complex genes and locus organization in the Komodo dragon (Varanus komodoensis). Immunogenetics 73, 405–417. doi: 10.1007/s00251-021-01217-6
Salomonsen, J., Marston, D., Avila, D., Bumstead, N., Johansson, B., Juul-Madsen, H., et al. (2003). The properties of the single chicken MHC classical class II alpha chain (B-LA) gene indicate an ancient origin for the DR/E-like isotype of class II molecules. Immunogenetics 55, 605–614. doi: 10.1007/s00251-003-0620-7
Sato, A., Figueroa, F., Murray, B. W., Málaga-Trillo, E., Zaleska-Rutczynska, Z., Sültmann, H., et al. (2000). Nonlinkage of major histocompatibility complex class I and class II loci in bony fishes. Immunogenetics 51, 108–116. doi: 10.1007/s002510050019
Schloissnig, S., Kawaguchi, A., Nowoshilow, S., Falcon, F., Otsuki, L., Tardivo, P., et al. (2021). The giant axolotl genome uncovers the evolution, scaling, and transcriptional control of complex gene loci. Proc. Natl. Acad. Sci. U.S.A. 118:e2017176118. doi: 10.1073/pnas.2017176118
Session, A. M., Uno, Y., Kwon, T., Chapman, J. A., Toyoda, A., Takahashi, S., et al. (2016). Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343. doi: 10.1038/nature19840
Shiina, T., Blancher, A., Inoko, H., and Kulski, J. K. (2017). Comparative genomics of the human, macaque and mouse major histocompatibility complex. Immunology 150, 127–138.
Siddle, H. V., Deakin, J. E., Coggill, P., Whilming, L. G., Harrow, J., Kaufman, J., et al. (2011). The tammar wallaby major histocompatibility complex shows evidence of past genomic instability. BMC Genomics 12:421. doi: 10.1186/1471-2164-12-421
Tonini, J. F. R., Beard, K. H., Ferreira, R. B., Jetz, W., and Pyron, R. A. (2016). Fully-sampled phylogenies of squamates reveal evolutionary patterns in threat status. Biol. Conserv. 204, 23–31.
Wan, Q. H., Pan, S. K., Hu, L., Zhu, Y., Xu, P. W., Xia, J. Q., et al. (2013). Genome analysis and signature discovery for diving and sensory properties of the endangered Chinese alligator. Cell Res. 23, 1091–1105. doi: 10.1038/cr.2013.104
Wan, Q. H., Zhang, P., Ni, X. W., Wu, H. L., Chen, Y. Y., Kuang, Y. Y., et al. (2011). A novel HURRAH protocol reveals high numbers of monomorphic MHC class II loci and two asymmetric multi-locus haplotypes in the Père David’s deer. PLoS One 6:e14518. doi: 10.1371/journal.pone.0014518
Yang, S., Lan, T., Zhang, Y., Wang, Q., Li, H., Dussex, N., et al. (2022). Genomic investigation of the Chinese alligator reveals wild-extinct genetic diversity and genomic consequences of their continuous decline. Mol. Ecol. Resour. [Epub ahead of print]. doi: 10.1111/1755-0998.13702
Yang, Z., Wang, C., Wang, T., Bai, J., Zhao, Y., Liu, X., et al. (2015). Analysis of the reptile CD1 genes: Evolutionary implications. Immunogenetics 67, 337–346. doi: 10.1007/s00251-015-0837-2
Ye, Q., He, K., Wu, S.-Y., and Wan, Q.-H. (2012). Isolation of a 97-kb minimal essential MHC B locus from a new reverse-4D BAC library of the golden pheasant. PLoS One 7:e32154. doi: 10.1371/journal.pone.0032154
Yuhki, N., Beck, T., Stephens, R. M., Nishigaki, Y., Newmann, K., and O’brien, S. J. (2003). Comparative genome organization of human, murine, and feline MHC class II region. Genome Res. 13, 1169–1179.
Zhai, T., Yang, H. Q., Zhang, R. C., Fang, L. M., Zhong, G. H., and Fang, S. G. (2017). Effects of population bottleneck and balancing selection on the Chinese alligator are revealed by locus-specific characterization of MHC genes. Sci. Rep. 7:5549. doi: 10.1038/s41598-017-05640-2
Keywords: MHC architecture, Chinese alligator, evolution of tetrapod MHC, MHC polymorphism, MHC evolution
Citation: He K, Zhu Y, Yang S-C, Ye Q, Fang S-G and Wan Q-H (2022) Major histocompatibility complex genomic investigation of endangered Chinese alligator provides insights into the evolution of tetrapod major histocompatibility complex and survival of critically bottlenecked species. Front. Ecol. Evol. 10:1078058. doi: 10.3389/fevo.2022.1078058
Received: 24 October 2022; Accepted: 28 November 2022;
Published: 13 December 2022.
Edited by:
Alison G Nazareno, Federal University of Minas Gerais, BrazilReviewed by:
Vladimir A. Trifonov, Institute of Molecular and Cellular Biology (RAS), RussiaCopyright © 2022 He, Zhu, Yang, Ye, Fang and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiu-Hong Wan, cWl1aG9uZ3dhbkB6anUuZWR1LmNu; Sheng-Guo Fang, c2dmYW5nbGFiQHpqdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.