
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 09 January 2023
Sec. Evolutionary Ecology of Social Behaviour
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.1055382
 Michel Diouf1*†
Michel Diouf1*† Vincent Hervé2†
Vincent Hervé2† Sophie Fréchault1
Sophie Fréchault1 Josie Lambourdière3,4Abdoulaye Baïla Ndiaye5
Josie Lambourdière3,4Abdoulaye Baïla Ndiaye5 Edouard Miambi1
Edouard Miambi1 Amélia Bourceret3
Amélia Bourceret3 My Dung Jusselme1
My Dung Jusselme1 Marc-André Selosse3,6†Corinne Rouland-Lefèvre1
Marc-André Selosse3,6†Corinne Rouland-Lefèvre1Termites have co-evolved with a complex gut microbiota consisting mostly of exclusive resident taxa, but key forces sustaining this exclusive partnership are still poorly understood. The potential for primary reproductives to vertically transmit their gut microbiota (mycobiome and bacteriome) to offspring was investigated using colony foundations from field-derived swarming alates of Macrotermes subhyalinus. Metabarcoding based on the fungal internal transcribed spacer (ITS) region and the bacterial 16S rRNA gene was used to characterize the reproductives mycobiome and bacteriome over the colony foundation time. The mycobiome of swarming alates differed from that of workers of Macrotermitinae and changed randomly within and between sampling time points, highlighting no close link with the gut habitat. The fungal ectosymbiont Termitomyces was lost early from the gut of reproductives, confirming the absence of vertical transmission to offspring. Unlike fungi, the bacteriome of alates mirrored that of workers of Macroterminae. Key genera and core OTUs inherited from the mother colony mostly persisted in the gut of reproductive until the emergence of workers, enabling their vertical transmission and explaining why they were found in offspring workers. These findings demonstrate that the parental transmission may greatly contribute to the maintenance of the bacteriome and its co-evolution with termite hosts at short time scales.
Termites have established very close mutualistic interactions with microorganisms. The gut microbiota of termites mostly consists of bacteria and archaea for the large family Termitidae (also known as “higher termites”), and with the same microorganisms plus protozoa for the paraphyletic “lower termites.” These symbionts crucially contribute to the exceptional ability of termites to digest lignocellulose compounds and exploit diverse resources, from wood to soil organic matter (Brune, 2014). The rich literature about termite symbiosis (Bignell, 2016) has demonstrated, in particular, the uniqueness of the gut microbiota and its co-evolution with hosts, likely facilitated by the social organization of termites, as in social bees (Kwong et al. 2017; Su et al. 2021).
Previous studies have considered how micro-environmental gut conditions and diet shape the gut microbiota (Mikaelyan et al., 2015, 2017). Meanwhile, other studies have emphasized the importance of vertical inheritance, thus linking microbiota and host phylogenies (Hongoh et al., 2005; Noda et al., 2007; Abdul Rahman et al., 2015; Diouf et al., 2015; Michaud et al., 2020; Arora et al., 2022). The most comprehensive study in terms of host taxa revealed that termite microbiota is primarily governed by a mixed mode of transmission that combines vertical transmission from parents to offspring and frequent colony-to-colony horizontal transfers (Bourguignon et al., 2018). The importance given to horizontal transfers, which are one-time events, is justified by their cumulative effect over the long evolutionary history of termites (around 150 million years; Engel et al., 2009; Bourguignon et al., 2014; Bucek et al., 2019). Therefore, assessing the actual transfers of symbionts at each generation should precise their significance and the microevolution of termite microbiota at shorter time scales (da Costa and Poulsen, 2018).
The symbiosis in Macrotermitinae, a sub-family of Termitidae also known as fungus-growing termites, has been widely studied, but the transmission of the gut symbionts (at the individual level) and fungal ectosymbionts (at the colony level) remains poorly documented. Macrotermitinae have an ectosymbiotic relationship with plant-degrading fungi of the genus Termitomyces (Agaricomycotina; Rouland-Lefèvre, 2000). Each generation of offspring workers often acquires fungi from the environment, resulting in frequent host switchings (Aanen et al., 2002; Diouf and Rouland-Lefevre, 2018). From an evolutionary viewpoint, some analogies could be drawn between the theorized mixed mode of transmission of the gut symbionts (Bourguignon et al., 2018) and that of the fungi in Macrotermitinae. However, while the spores of Termitomyces can survive in natura for long periods, the survival of bacterial symbionts, including many anaerobic lineages outside the digestive tract, may limit horizontal transfers.
Understanding the establishment of the gut microbiota in fungus-growing termites during the foundation of incipient colonies at each generation may provide valuable information on the colony-to-offspring transmission and, thus, the persistence of symbionts over the microevolution of these termites. The foundation of new colonies starts with the swarming flight of alates, which are mature reproductives with complete wings (Figure 1). After forming couples, alates shed their wings (becoming dealates), find a suitable substratum, and begin mating after digging out a subterranean initial colony chamber called copularium where they are sheltered with the brood. The first batch of eggs is laid between 4 and 15 days after colony initiation, and the first offspring larvae and workers emerge between 21 and 46 days and between 52 and 80 days, respectively (Grassé and Noirot, 1955; Okot-Kotber, 1981; Harit et al., 2016; Mitchell, 2020). Previous studies on reproductives of Termitidae found that alates swarmed from the mother colonies with a complete set of the host microbiota (Hongoh et al., 2006; Diouf et al., 2018). However, it is still unclear whether the gut-specific lineages persist and overlap enough with offspring development to be reliably transferred. First, the long lag time between the initiation of colony foundation and the emergence of offspring may be a potential barrier to this effective transfer. Second, the feeding behavior of reproductives during this long period is unfavorable. Indeed, unlike reproductives of lower termites, which continue to feed during colony foundation, those of Termitidae cease feeding (Nutting, 1969; Han and Bordereau, 1982; Grassé, 1984) and draw the required energy from the resorption of their fat body and of their useless alar muscles. Given the crucial role of the diet on the microbiota (Mikaelyan et al., 2015), fasting may result in the loss of the initial microbiota.
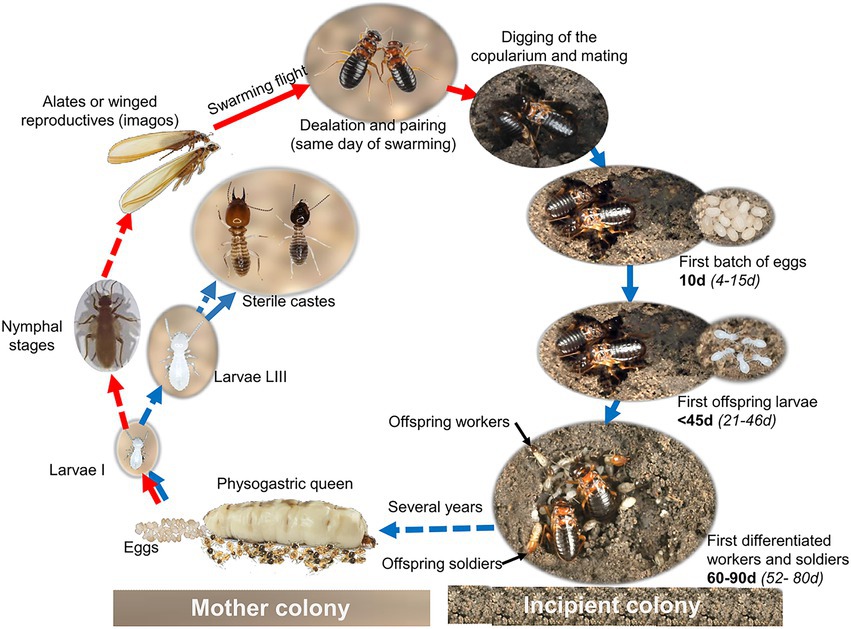
Figure 1. Simplified developmental cycle of Macrotermes subhyalinus. Red arrows indicate the developmental pathway for the reproductive caste. The chronology of the emergence of the offspring stages is indicated in bold from our own observations and in italic for Macrotermes sp. from literature.
Apart from alates, the microbiota of the reproductive caste of Termitidae has only been addressed in aged kings and queens (Poulsen et al., 2014; Otani et al., 2019). At this late stage, the microbiota is marked by the dominance of a single or very few major bacterial lineages, preventing any extrapolation of the role of reproductives in the transmission and the persistence of symbionts. In contrast to the gut microbiota, the transmission of the fungal ectosymbiont of Macrotermitinae has received more attention. One approach was to check if the fungal asexual spores (conidia) ingested from the fungus comb of the mother colony by one sex of reproductive persisted in their gut and allowed effective inoculation of primordial fungus combs in lab-founded colonies (Sands, 1960; Johnson, 1981; Johnson et al., 1981; Sieber, 1983). In several Macrotermes species, the initiation of the primordial combs occurs around 90–95 days (Grassé and Noirot, 1955; Collins, 1977; Lepage and Darlington, 2000), once the first differentiated workers can gather plant matter, which is long enough to jeopardize the viability of fungal propagules. Recent studies have instead addressed the issue indirectly through host-fungus co-cladogenesis (review by Diouf and Rouland-Lefevre, 2018). Finally, except for a few species mainly from the genus Microtermes and the species Macrotermes bellicosus, the fungal symbionts are considered acquired horizontally from the surrounding environment (Aanen et al., 2009).
The objective of our study was to assess the potential for reproductives to ensure the actual transfer of their microbiota to offspring. In this respect, we focused on the succession of reproductives microbiota (mycobiome and bacteriome) during the foundation of incipient colonies while paying attention to their gut morphology as a proxy for feeding behavior. The fungus-growing termite Macrotermes subhyalinus was used as a model to study the succession of the digestive bacteriome and mycobiome, with a special focus on Termitomyces which has been reported to be transmitted horizontally in this species (Johnson et al., 1981; Vesala et al., 2017). Deep amplicon sequencing and taxon-specific PCR for Termitomyces were used to monitor both the mycobiome and the bacteriome in the gut of reproductives from the swarming flight to the emergence of first differentiated workers and in the copularium.
Termites were collected near Diamniadio, located 30 km East of Dakar (Senegal). Reproductives were captured during the swarming flight from two distinct colonies of Macrotermes subhyalinus (colony 1 and colony 2) and one colony of the sympatric species Microtermes sp. that swarmed at the same time. Alates of Microtermes sp., and specially females that are known to carry condidia of Termitomyces for the inoculation of the comb were considered as positive control. This sampling time, considered as time zero is referred as “alates” in this manuscript. For both wild colonies of M. subhyalinus, workers were also collected to compare their bacteriome with the bacetriome of alates. As in most of Macrotermtinae species, the worker caste of M. subhyalinus encompassed two distinct morphotypes with different tasks and feeding behaviors: major workers (larger workers that develop from male larvae) and minor workers (smaller workers that develop from female larvae) (Badertscher et al., 1983). At the laboratory (IRD-ISRA Research Center of Bel-Air, Dakar), alates were allowed to form pairs. Then they broke off their wings, and each pair was isolated and transferred into an individual LAB1 box (60x45x50 mm, VWR International, LLC) partially filled with moistened non-sterile soil collected in the research center. Individuals from the colony foundations are referred as “dealates” in the manuscript. All colony foundations were set up using reproductives from colony 1 of M. subhyalinus. Two hundred and thirty colonies were set up and kept in a tropicalized termite-rearing room. The moisture of the soil was regularly checked, and adjusted when necessary by wetting the cotton balls laid on the soil surface with sterile distilled water. No offspring sterile caste (workers or soldiers) was found in colonies withdrawn from 0 to 60 days, and no foraging trace was observed in non-processed colonies during this period. Distinct offspring castes (workers and soldiers) appeared in almost all active foundations between days 60 and 90, and the foraging activity of workers was visible through foraging tunnels. From day 90, colonies were transferred into LAB2 rearing boxes (90 × 60 × 50 mm) more appropriate for that population size and fed with non-sterilized wheat straws.
For molecular analyses of the microbiota, three distinct colonies were withdrawn at 0 (alates), 10, 30, 45, 60, and 120 days. The copularoum samples on day 120 were analyzed in parallel to ensure that digestive symbionts defecated by reproductives in their surroundings did not thrive.
For the measurements of the gut size, 9 colonies were withdrawn at each sampling time point, i.e., at 0, 45, 60, and 75 days, leading to 6–9 properly dissected guts for each sex, except for the sampling time point 60 days where fewer colonies were considered. The sex of the collected individuals was always checked based on the configuration of the terminal sternites of the abdomen (Weesner, 1969). Very few false pairings, i.e., same sex couples, occurred and were discarded from analyses.
Alates were dewinged, and then rinsed three times in sterile distilled water. The surface of dealates from colony foundations was cleaned similarly. Then, under a biosafety cabinet, the whole gut was removed aseptically using fine, sterile scissors and placed individually into a 1.5-mL sterile microtube. As indicated earlier, for each sex, three replicates from three distinct colony foundations were considered at each sampling time point for molecular analyses. For major and minor workers that were collected at the same time as swarming alates from the same colonies of M. subhyalinus, the whole gut of workers was removed aseptically using fine sterile forceps. For each morphotype, DNA extraction was performed from pools of 10 guts.
For assessing the variation in gut size over the time of colony foundation, female and male reproductives were dissected as described above and photographed using the Zoom Stereo Microscope SMZ1000 of Nikon calibrated at the same scale. The images were transferred to a connected computer via a camera (Digital Sight, DS-U3, Nikon), and treated with the software NIS-Element (Basic Research, Version 4.0) from Nikon (Nikon Instruments Inc.) to measure the length of the whole gut, the areas of the most representative gut sections (the hindgut section and the paunch sub-section). We also used the method of Shimada et al. (2013) to measure the width of the most enlarged part of the paunch (Supplementary Figure S1).
Individual termite gut for alates and dealates, or pools of 10 guts for worker morphotypes from the mother colonies were crushed with a sterile polypropylene pestle in a 1.5-mL microtube containing the lysis buffer of the DNA extraction kit (NucleoSpin® DNA Insect Kit, Macherey-Nagel GmbH & Co. KG). Then, the gut homogenate was transferred to NucleoSpin® Bead tube for an additional lysis step by bead-beating with the FastPrep®-24 homogenizer (MP Biomedicals). The subsequent DNA extraction steps were performed following the manufacturer’s instructions. The yield and purity of DNA were analyzed with a ND-1000 Spectrophotometer (NanoDrop products, Wilmington, United States). The same DNA extraction protocol was applied to the copularium samples collected at the end of the experiment (day 120). DNA-based analyses were performed on aliquots adjusted at 10 ng μL−1 for all samples.
We first compared the performance of several polymerase enzymes for PCR-amplification of the Internal Transcribed Spacer (ITS) of the ribosomal DNA with the Termitomyces-specific primer ITS1FT (Aanen et al., 2007) combined with ITS4R (White et al., 1990). The endpoint PCR reactions were run in 25 μl containing 0.5 μM for each primer, according to conditions and the thermal cycle defined by Aanen et al. (2007). The Illustra PureTaq (PCR beads, GE Healthcare) was the most sensitive, producing detectable amplicons at concentrations below 20 pg. μL−1 with DNA extracts from pure mycelium of Termitomyces. Following PCR reactions, the amplicons were checked by electrophoresis onto a TAE 0.5X buffered agarose gel (1.5% (w:v) containing the DNA staining dye GelRed (GelRed® Nucleic Acid Gel Stain, Biotium, VWR International LLC.). A UV trans-illuminator (GenoSmart, VWR) allowed the visualization of the positive PCR-bands. To check the correspondence of the amplicons with the ITS of Termitomyces, some bands were randomly excised and sequenced. The corresponding sequences are referenced in GenBank under the accession numbers MN400299-MN400306. For each sampling time point, 12 to 16 individuals of M. subhyalinus were tested for each sex. The results are displayed as a percentage of positive essays at each time point.
DNA samples from the guts of minor and major workers, and alates were readjusted to 3.5 ng/μL, and then purified by Ampure XP beads (Agencourt, Beckman Coulter, United States). PCR amplifications were performed in triplicate for each sample with the primer pair 28F/338R (Cheung et al., 2018) tagged with combinations of nucleotides specific to each sample. These primers target the V1-V2 hyper variable region of the bacterial 16S rRNA gene. PCR reactions were performed in 25 μl of mixture as following: 10 ng of DNA template, 1X incomplete buffer, 0.3% bovine serum albumin, 2 mM MgCl2, 200 mM dNTPs, 300 nM of forward and reverse primer and 2 U of DFS-Taq DNA polymerase (Bioron, Ludwigshafen, Germany). The following program was used: 95°C during 5 min, then 30 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min 30 s, and to finish 10 min at 72°C. Amplicons from triplicates were pooled, checked on agarose gels (5 μl, 2%, 100 V, 20 min), and then purified twice by Ampure XP beads. The DNA concentration in purified amplicons was determined, and an equal amount of 25 ng for each sample was pooled. This final pool was purified again and reconcentrated twice by Ampure XP beads before the last quantification by Qubit ® dsDNA HS Assay Kit (Invitrogen, United States). MetaFast library preparation and sequencing were performed on an Illumina 2 × 250 MiSeq platform by Fasteris SA (Switzerland).
Subsequently, the 16S rRNA gene data were analyzed with mothur version 1.39.5 (Schloss et al., 2009), as described in Hervé and Lopez (2020). Briefly, the 16S rRNA gene reads were denoised by removing any reads having ambiguous bases, homopolymers longer than 8 bp, and having a length inferior to 280 bp or superior to 320 bp. Then sequences were aligned to the SILVA reference database 132 (Quast et al., 2012) and preclustered (pre.cluster, diffs = 1). Chimeras were detected and removed using UCHIME (Edgar et al., 2011). Sequences were classified with a naïve Bayesian classifier (Wang et al., 2007) and the SILVA database 132. Non-bacterial and unknown sequences were excluded. Sequences were clustered into operational taxonomic units (OTUs) using the OptiClust algorithm (Westcott and Schloss, 2017) with a 97% sequence similarity threshold. Finally, singletons were excluded, and each sample was rarefied to the smallest library size (21,601 reads) by random subsampling (McKnight et al., 2019). The raw sequence reads were deposited as Sequence Read Archive (SRA) in GenBank under the Bioproject accession PRJNA347254 with the BioSample accession number SAMN30890275.
Based on the preliminary results of the ITS-based detection of Termitomyces, the sequencing of the gut mycobiome was limited to 60 days, stage at which only eggs and larvae were found in brood. However, even absent from the gut of reproductives, fungal propagules especially those of Termitomyces could be kept in their immediate environment. The copularium samples were therefore collected at the end of the experiment and analyzed to check this possibility. The whole mycobiome was assessed by PCR amplifying the ITS2 region with the primer pair ITS86/ITS4 targeting a wide range of fungal lineages (Turenne et al., 1999; Waud et al., 2014), according to the PCR cycling conditions described by Schneider-Maunoury et al. (2018). Tagged primers unique for each sample were used in a second PCR step that was performed in the same conditions. The subsequent steps of the sequencing were those described by Schneider-Maunoury et al. (2018), using the Ion Torrent sequencer (Life technology, Carlsbad, United States).
For bacteria, DNA fragments spanning the V1-V2 hypervariable region of the 16S rRNA gene were amplified using the primers 28F and 338R (Cheung et al., 2018). The following thermal conditions were applied: 94°C for 5 min, followed by 25 cycles of 94°C for 30 s, 56°C for 30 s, 72°C for 60 s, and a final elongation step of 72°C for 5 min. The subsequent library preparation and sequencing steps were the same as those for fungi.
For the processing of sequencing data, demultiplexed raw data were first converted from bam format to fastq format using BEDtools version 2.25 (Quinlan and Hall, 2010). Subsequently, ITS and 16S rRNA gene data were analyzed independently with mothur version 1.39.5 (Schloss et al., 2009), following a similar procedure as the one described above.
Bacterial 16S rRNA gene amplicons were denoised by removing any reads having ambiguous bases, homopolymers longer than 8 bp, having Phred quality score < 25, and having a length inferior to 200 bp or superior to 320 bp. Then primers were removed, and sequences were aligned to the SILVA reference database 132 (Quast et al., 2012) and preclustered (pre.cluster, diffs = 1). Chimeras were detected and removed using UCHIME (Edgar et al., 2011). Sequences were classified with a naïve Bayesian classifier (Wang et al., 2007) and the SILVA database 132. Non-bacterial and unknown sequences were excluded. Sequences were clustered into operational taxonomic units (OTUs) using the OptiClust algorithm (Westcott and Schloss, 2017) with a 97% sequence similarity threshold. Finally, singletons were excluded, and each sample was rarefied to the smallest library size (2,220 reads) by random subsampling (McKnight et al., 2019).
Fungal ITS reads were denoised using the same quality parameters described, and each sequence that passed quality filtering was truncated to a 200-bp length after removing primer sequences (Brown et al., 2013) and then preclustered (pre.cluster, diffs = 1). Chimeras were detected and removed using UCHIME (Edgar et al., 2011). Sequences were classified with a naïve Bayesian classifier (Wang et al., 2007) and the Unite database v7.1 (Nilsson et al., 2018). Non-fungal and unknown sequences were excluded. Fungal ITS sequences were pairwise aligned to generate a distance matrix that was used to compute OTUs using the OptiClust algorithm (Westcott and Schloss, 2017) with a 97% sequence similarity threshold. Finally, singletons were excluded, and each sample was rarefied to the smallest library size (1,282 reads) by random subsampling (McKnight et al., 2019).
The raw sequence reads for both microbial communities were deposited as Sequence Read Archive (SRA) in GenBank under the Bioproject accession number PRJNA557705.
A particular focus has been placed on the persistence of the most representative fungal and bacterial taxa. The 40 genera displayed were the most abundant in the entire data set. Core OTUs were defined as any OTU that persisted in the gut samples at all the sampling time points for fungi and at all or at least at 5 of the 6 sampling time points from days 0 to 120 for bacteria.
For the specific case of Termitomyces (Lyophyllaceae family), the classification of the OTUs was improved by a phylogenetic approach. This consisted in combining all the OTUs affiliated to Lyophyllaceae from our data set with all the ITS sequences of Lyophyllaceae referenced in the Unite database. ITS sequences of the close genus Rhodocybe (Entolomataceae) were also included as an outgroup of Lyophyllaceae. All these sequences were aligned with MAFFT v7.427 and the L-INS-i method (Katoh and Standley, 2013). The resulting alignment was manually curated, and Smart Model Selection (Lefort et al., 2017) was used to determine the best model of nucleic acid evolution (GTR + G). Subsequently, a maximum likelihood phylogenetic tree was built with PhyML 3.0 (Guindon et al., 2010). Branch supports were calculated using a Chi2-based parametric approximate likelihood-ratio test (Anisimova and Gascuel, 2006).
The effect of time and sex on the measurements of the gut size was evaluated by Kruskall-Wallis tests. Alpha diversity was investigated using the R package hilldiv (Alberdi and Gilbert, 2019) based on abundance-based Hill numbers (qD) that integrate commonly used alpha-diversity indices according to the values of q. Hence, q = 0 refers to OTUs richness, and q = 1 or 2 refers to the converted indices of Shannon (exponential of the Shannon index) and Gini-Simpson (the multiplicative inverse of the Gini-Simpson index), respectively (Chao et al., 2016). The differences in alpha-diversity indices were analyzed by One-way ANOVA with the Fisher’s LSD Post Hoc Test, at p < 0.05, after ascertaining the normal distribution of variables. The changes in microbial community structure were investigated by analysis of similarities (ANOSIM), implemented in mothur (Schloss, 2008), using Bray-Curtis distances and 10,000 iterations. Principal coordinate analyses (PCoA) were constructed based on Bray-Curtis distances with the phyloseq R package (McMurdie and Holmes, 2013). The effect of sampling time points on the composition of the bacteriome and mycobiome was tested by non-parametric permutational multivariate analysis of variance (PERMANOVA), as implemented in the vegan function adonis with 10,000 permutations (Oksanen et al., 2007). All the other analyses were performed with R v3.4.4, and plots were generated with ggplot2 (Wickham, 2016). To test the relationship between matrices of the bacteriome and the mycobiome, a Mantel test was performed using the ecodist package (Goslee and Urban, 2007), with Bray–Curtis dissimilarity matrices, Spearman’s rank correlation coefficient, and 10,000 random permutations.
The evaluation of the bacteriome of alates and workers directly from wild colonies aimed to assess the extent to which the composition of the bacteriomes of alates leaving the mother colony could mirror the symbiotic microbiome of the colony of origin.
The phylum-level distribution of the bacterial taxa highlighted that a considerable proportion of the identified OTUs fell within Bacteroidetes, Firmicutes, and Proteobacteria, which proportion was not different between castes (Figure 2A). The distribution of OTUs was similar between minor and major workers within and between colonies and between male and female alates. This similar distribution pattern was much more visible at the genus level, with the proportion of OTUs between the most representative genus-level taxa varying hardly between castes and sexes (Figure 2B).
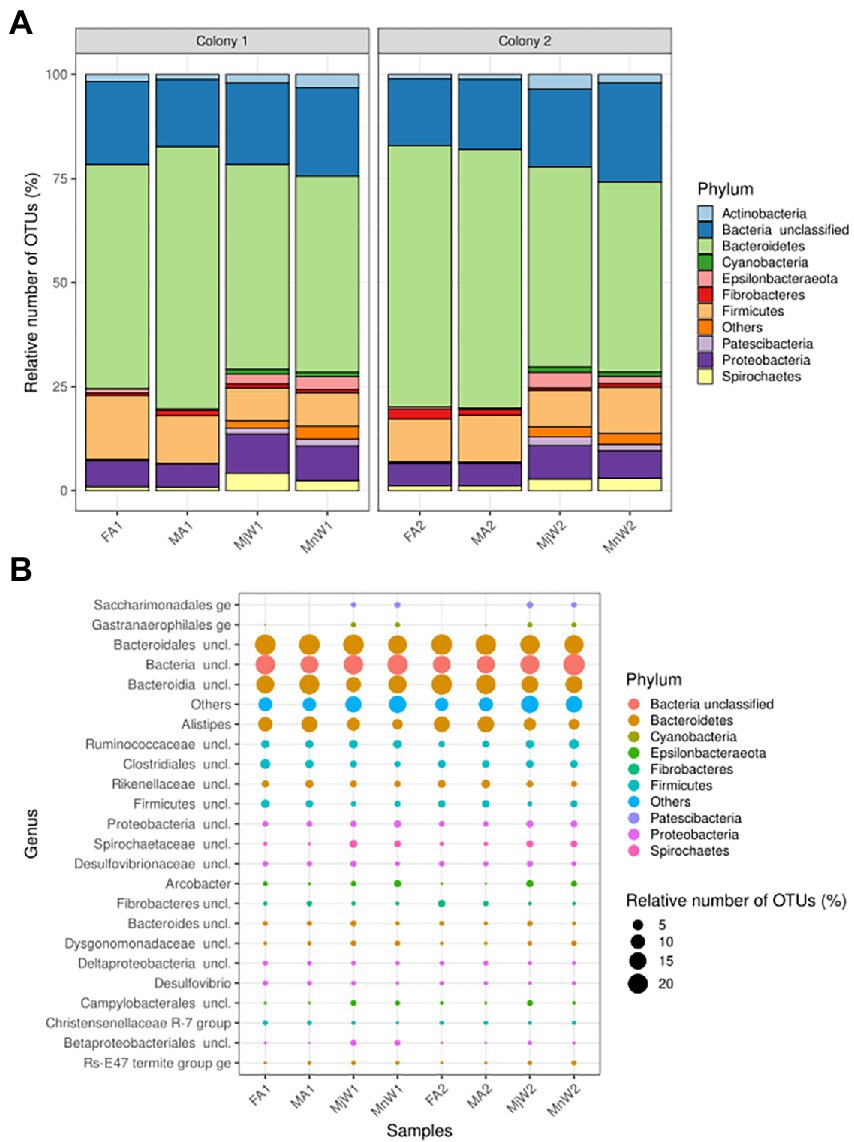
Figure 2. Phylum-level (A) and genus-level (B) distribution of bacterial OTUs in the gut of female alates (FA), male alates (MA), major workers (MjW) and minor workers (MnW) of Macrotermes subhyalinus from two distinct wild colonies. For both taxonomic levels, only taxa covering a number of OTUs >0.5% of the total number of OTUs of the dataset are displayed. The corresponding taxa covered >97% of OTUs in each library for the phylum-level and > 85% of OTUs for the genus-level. “Others” refers to the cumulative percentage of the remaining OTUs. The dot sizes are proportional to the relative number of OTUs for the corresponding taxa in each library.
Guts of reproductives of both sexes dissected over the colony foundation period starting from the swarming flight (alates) up beyond the emergence of workers were used to assess the temporal change in gut morphology and the structure of the microbiota. We first checked if the gut size and the structure of the microbiota were affected by sex of the reproductive. The various parameters of the gut morphology measured from image analyses of the dissected guts of reproductives (Supplementary Figures S1A–C1) did not significantly differ between sexes during the colony foundation period (p > 0.910 for the gut length, the hindgut area, and the paunch width; and p = 0.761 for paunch area). Likewise, there was no significant difference between sexes in the structure and taxonomic composition of the mycobiome analyzed by metabarcoding based on the fungal ITS and of the bacteriome analysed by the metabarcoding based on the 16S rRNA gene (ANOSIM, R = −0.026, p = 0.697 for the fungi; R = −0.037, p = 0.759 for the bacteria). Consequently, the results presented below were analyzed irrespective of the reproductive sexes.
The area of the hindgut significantly increased (p < 0.001) between the alates (t = 0) and dealates collected on day 45 (Figure 3). Then this area remained stable until day 75 with no significant differences between days 45, 60, and 75 (p > 0.05). The whole gut length, the paunch area and the maximum paunch width were also significantly higher in dealates at days 60 and 75 compared to the alates (p < 0.005) (Supplementary Figure S2). Beyond day 45, there were no variations of these parameters in dealates. The paunch at this time appeared distended with soil-like matter (Supplementary Figure S1D). Overall these gut morphology measures were correlated, with the hindgut area being positively correlated with the whole gut length (r = 0.734; p < 0.0001), the area of the paunch sub-section (r = 0.988; p < 0.0001) and the maximal width of the paunch (r = 0.835; p < 0.0001).
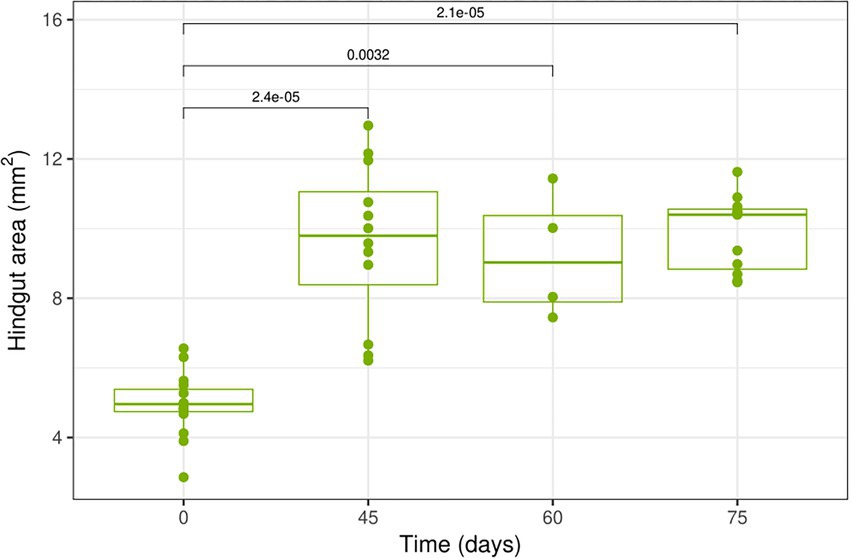
Figure 3. Boxplot representation of the change of the area of the whole hindgut of M. subhyalinus reproductives during colony foundation. For area measurements, the values in the figure represent the half of the total surface (see Supplementary Figure S1). Bars topped with p values <0.05 connect time points with statistically different measurements.
For fungi, high-quality reads of ITS clustered into 3,439 OTUs assigned to 3 phylum-level taxa from 25 classes and 165 genus-level taxa (Spreadsheet “S1_fungi,” Supplementary Table S1). Regarding the alpha diversity, both OTU richness (q = 0) and diversity (exponential of Shannon index, q = 1) remained stable between days 0 and 30 but significantly increased at days 45 and 60 (Supplementary Table S2). Similarly, these two indices were significantly higher in the copularium than in gut samples. Regarding the beta diversity the PCoA performed to assess the effect of sampling time on the structure of the mycobiome showed no clear clustering (Figure 4A). An overall significant effect of the sampling time point emerged from the PERMANOVA test (R2 = 0.275, p < 0.0001). The ANOSIM pairwise tests revealed significant differences between days 0 and 45 and between days 30 and 45 (Supplementary Table S3).
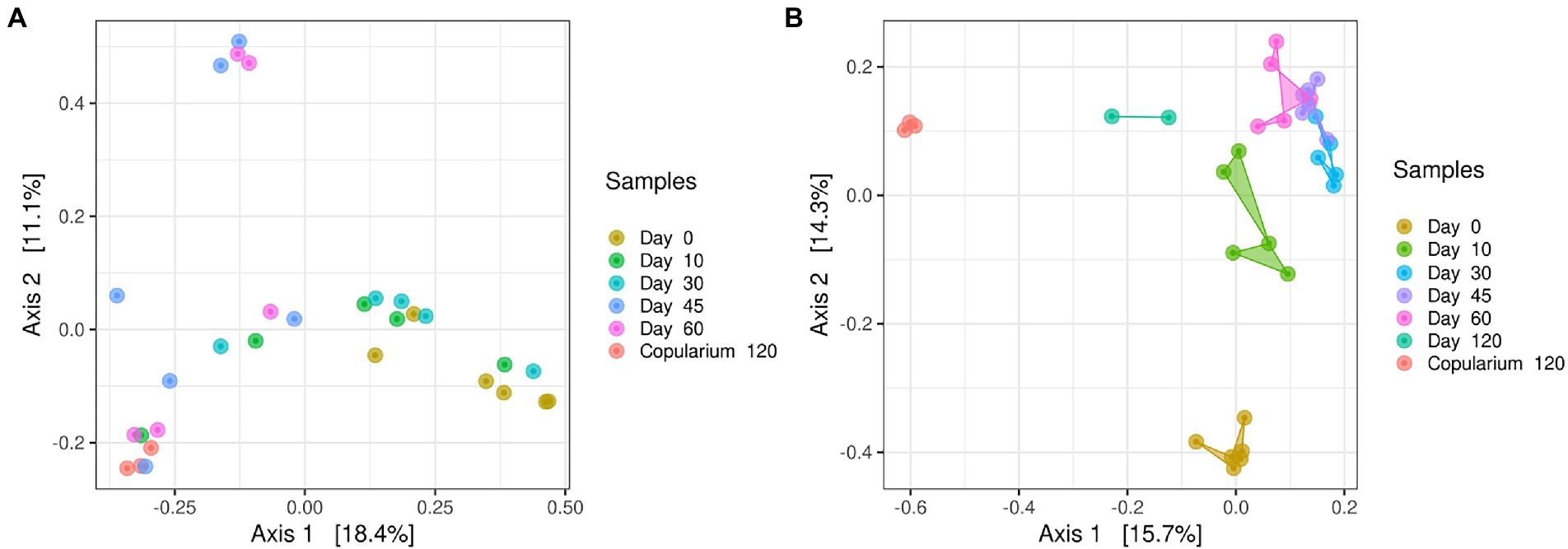
Figure 4. PCoA based on Bray-Curtis distances displaying the change in the structure of the mycobiome (A) and of the bacteriome (B) in the gut of M. subhyalinus reproductives over the colony foundation period, in comparison with the copularium at day 120. Day 0 to day 60 for fungi, and day 0 to day 120 for bacteria refer to gut samples at the corresponding time points.
For bacteria, high-quality reads of the 16S rRNA gene clustered into 8,030 distinct OTUs, assigned to 27 phylum-level taxa, 74 classes, and 479 genus-level taxa (Spreadsheet “S1_Bacteria,” Supplementary Table S1). The richness index did not vary between gut samples over the 60-first days (Supplementary Table S2). Regarding the diversity, for the exponential of Shannon index, (q = 1) and the multiplicative inverse of the Simpson index, (q = 2), there was a significant one-off decrease on days 30 to 45. The three alpha-diversity indices were higher in the copularium environment than in termite-gut samples. The structure of the bacteriomes assessed by PCoA showed clustering of gut libraries per sampling time point (Figure 4B). The bacterial communities in the copularium samples clustered separately from those of the gut samples. This clustering of the bacteriomes according to the sampling time was underpinned by the PERMANOVA test (R2 = 0.425, p < 0.0001), showing that time explained more variance for the bacteriome than for the mycobiome. Accordingly, the ANOSIM pairwise test showed significant differences between almost all time points except between days 10 and 30 and days 30 and 45 (Supplementary Table S3). Overall, there were distinct patterns between the mycobiome and bacteriome, which was further supported by the absence of significant correlation between both communities (Mantel test, rS = 0.06, p = 0.26).
The gut mycobiome of alates was co-dominated by Ascomycota and Basidiomycota (Figure 5A). From day 10, the relative abundance of Basidiomycota decreased in favor of unclassified fungi (i.e., fungal sequences that could not be taxonomically assigned below the fungal kingdom level) except at 30 days. Ascomycota accounted for about 90% of the copularium libraries. At the genus level, of the 40 major taxa of the dataset, only four clearly identified ones, from Ascomycota (Aspergillus, Retroconis, Graphium, and Mortierella), were conserved over time (Figure 5B). The other conserved lineages were unclassified groups likely from various distinct taxa. The ectosymbiotic fungus Termitomyces was present in the gut of dealates until day 30, but not beyond.
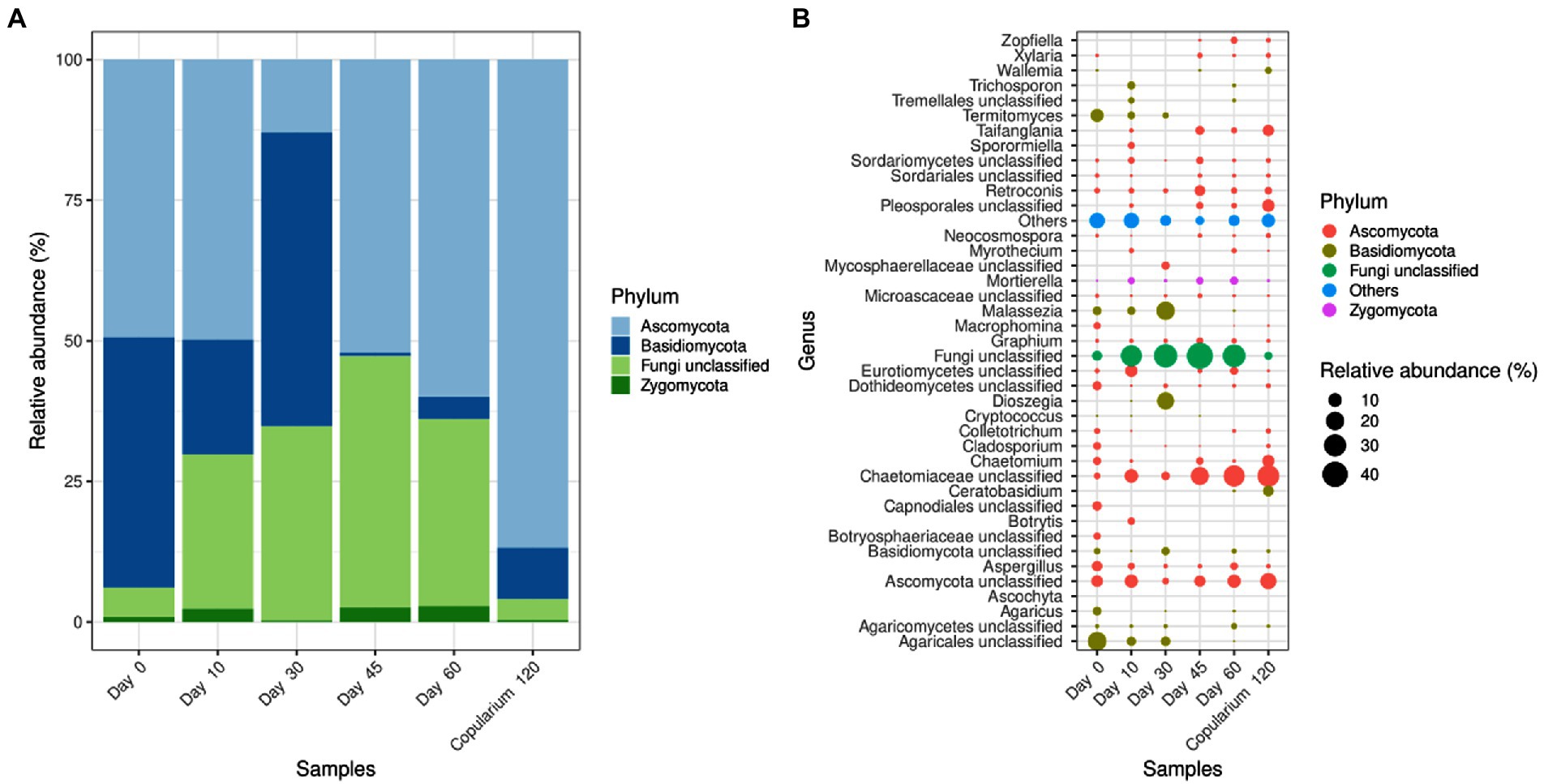
Figure 5. Phylum-level (A) and genus-level (B) distribution of fungal taxa in the gut of M. subhyalinus reproductives over the colony foundation period, in comparison with the copularium at day 120. For the genus-level, only the 40 most abundant taxa of the data set are displayed. The dot sizes are proportional to the relative abundance of the corresponding taxa in each library and the absence of dot indicates the non-detection of the corresponding taxon in a given library. « Others » refers to the sum of relative abundances of less abundant taxa.
Among the bacteria, Bacteroidetes, and to lesser extent Firmicutes were the most represented phylum-level taxa in alates gut (Figure 6A; Supplementary Table S1). Bacteroidetes remained the dominant phylum over time, but a gradual decrease was observed from day 0 to day 45. Meanwhile, the relative abundance of Epsilonbacteraeota increased. The proportion of Fibrobacteres in the gut was higher at day 120 than before. The relative abundance of Actinobacteria gradually increased over time, exceeding 7% of reads at days 60 and 120. In the copularium environment, Proteobacteria dominated, followed by Actinobacteria and Firmicutes. At the genus-level, all the major bacterial taxa of the dataset present in the gut of alates persisted in dealates over the 60 first days of the colony’s life, and only 4 were not found by the end of the experiment (Figure 6B). These major genera were dominant in the gut libraries, covering >94% of gut libraries from days 0 to 60 and 88.2% at day 120. They covered 56.2% of libraries from the copularium. These key genera primarily fell within Bacteroidia, various lineages of Proteobacteria and Clostridiales, and within well-known lineages in the gut of termites from the Campylobacterales (Epsilonbacteraeota), Chitinivibrionia (Fibrobacteres), Spirochaetaceae and Sphaerochaeta (Spirochaetes). A noticeable increase of the relative abundance of Campylobacterales, of which Arcobacter was observed from day 0 and to day 45.
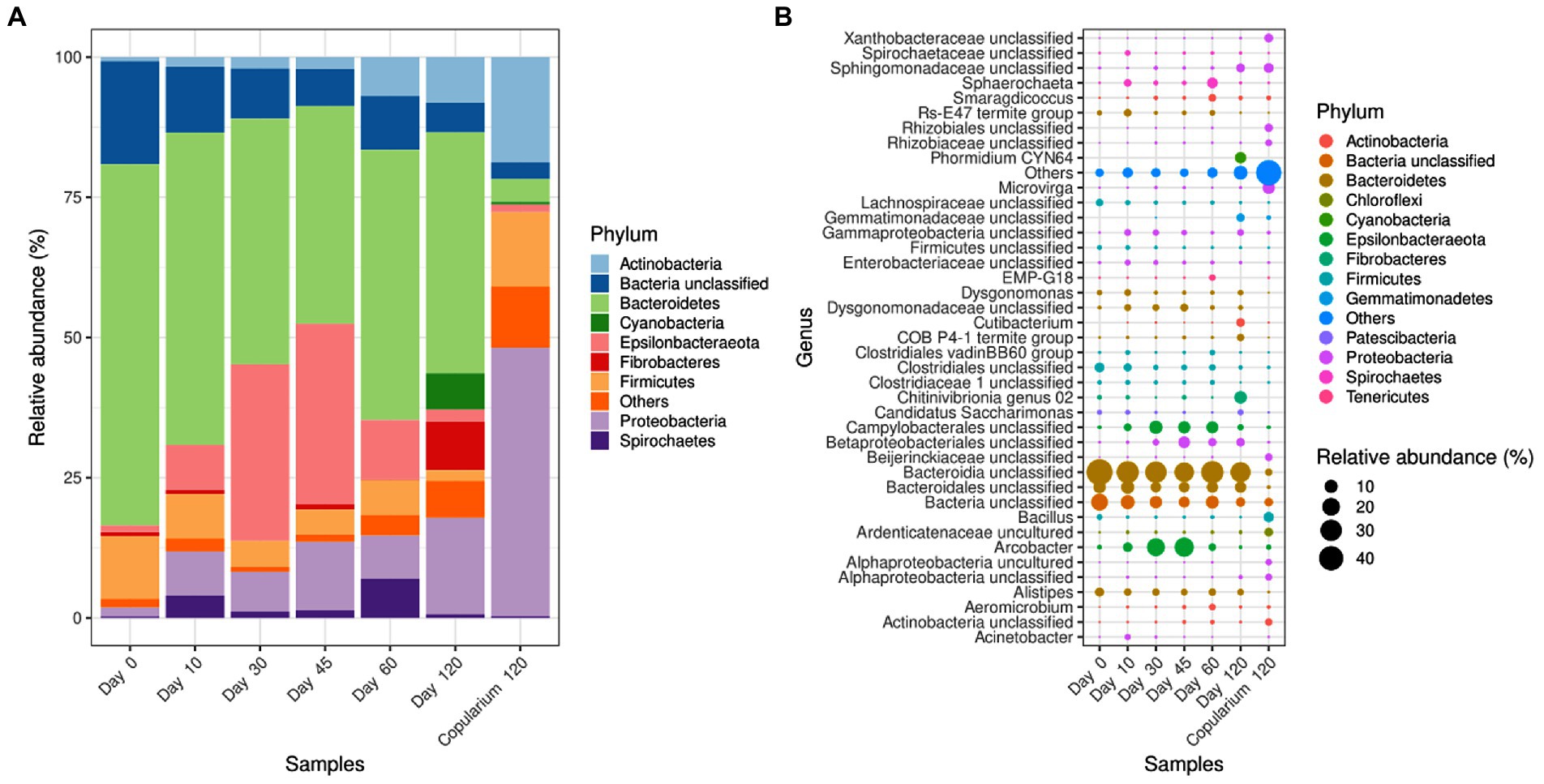
Figure 6. Phylum-level (A) and genus-level (B) distribution of bacterial taxa in the gut of M. subhyalinus reproductives over the colony foundation period, in comparison with the copularium at day 120. For the genus-level, only the 40 most abundant taxa of the data set are displayed. The dot sizes are proportional to the relative abundance of the corresponding taxa in each library and the absence of dot indicates the non-detection of the corresponding taxon in a given library. “Others” refer to the sum of relative abundances of less abundant taxa.
Core fungal OTUs were those found in the gut of reproductives at all the sampling time points from the alates up to day 60. They were only 12 OTUs (out of 3,439; Sheet “Core OTUs of fungi,” Supplementary Table S1). Together, they accounted for 5 to 10% of gut libraries from days 0 to 30 and slightly more at days 45 and 60 (≈24%). This relative increase at 45 and 60 days was primarily driven by a single OTU (unclassified Chaetomiaceae) that was the most abundant in the copularium. Core fungal OTUs were even more represented in the copularium than in gut samples.
Focusing on the ectosymbiotic fungus Termitomyces, the phylogenetic inference showed that all OTUs from the Lyophyllaceae in the dataset belonged to this genus Termitomyces (Supplementary Figure S3). In alates, their average relative abundance for both sexes was below 10%, with marked inter-individual variability (Supplementary Table S4). Their proportion sharply declined over time to zero beyond day 30. Members of Lyophyllaceae were absent from the copularium (Supplementary Table S1). With the taxon-specific PCR, 75% of tests were positive in alates of both sexes from two distinct colonies of M. subhyalinus, but no test was positive after day 10. The ectosymbionts of species from the genus Microtermes are vertically transmitted through the gut of female reproductives. Alates of a sympatric species Microtermes sp. were analysed for comparison (n = 8 for each sex), resulting in 100% positive tests (data not shown).
Considering bacteria, 419 distinct OTUs fell within core OTUs consisting of OTUs that persisted at all or at least at 5 of the 6 sampling time points from days 0 to 120 (Sheet “core OTUs of bacteria,” Supplementary Table S1). They accounted for more than 65% of OTUs in each library from days 0 to 60 and ≈ 60% at day 120 (Figure 7A). The same was true in terms of abundance, since they accounted for more than 73% of reads in all the guts libraries up to 60 days (Figure 7B) and remained highly representative at the end of experiment. Compared to gut samples, core OTUs covered only a small fraction of the number of OTUs and of the number of reads in the copularium. Their taxonomic assignment mirrors the taxonomic composition of the whole libraries. Besides unclassified bacteria (i.e., bacterial sequences that could not be taxonomically assigned below the Bacteria rank), Bacteroidetes, Firmicutes, and Proteobateria were the most represented phyla. Only a few core OTUs belonged to Epsilonibacteraeota, but these represented a substantial fraction of gut libraries, especially at days 10 to 60.
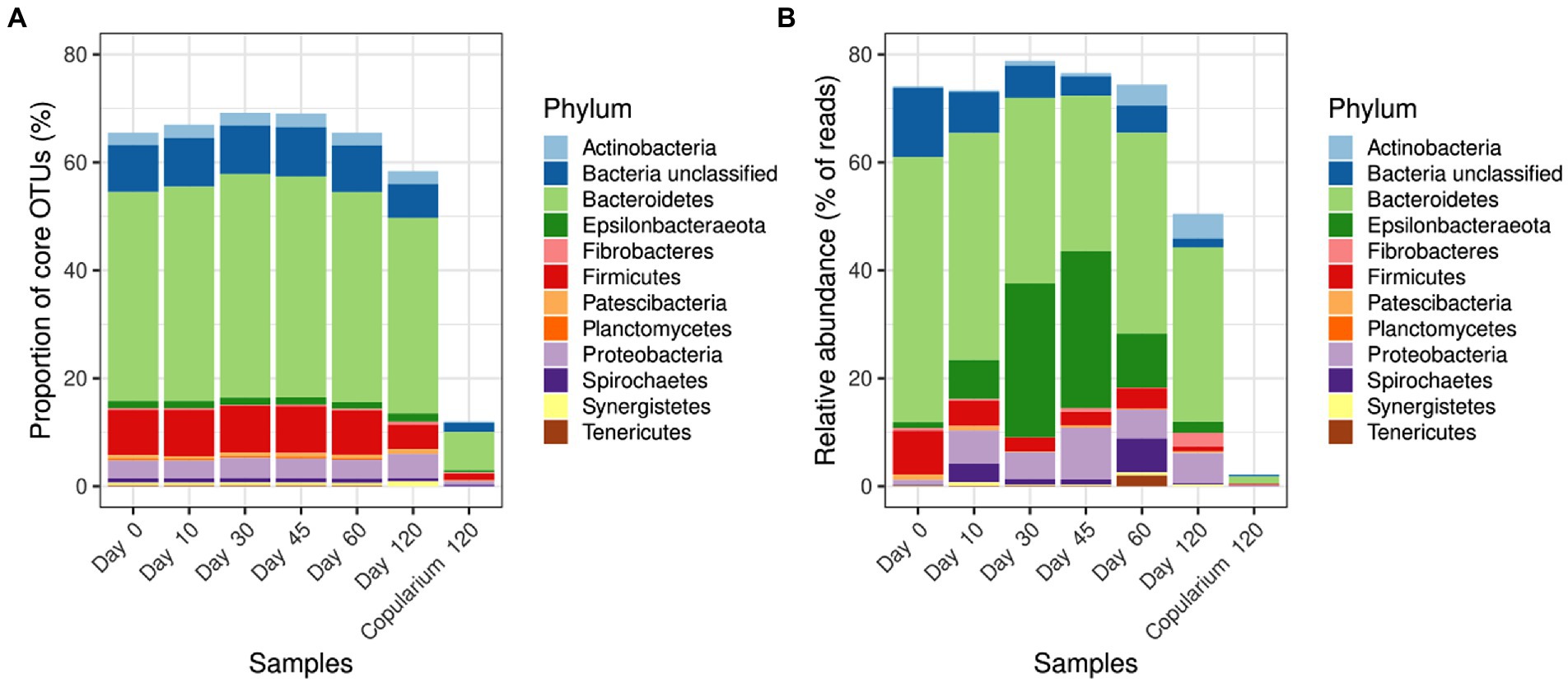
Figure 7. Taxonomic distribution of core bacterial OTUs in libraries from the gut of M. susbhyalinus reproductives over the colony foundation period, in comparison with the copularium at day 120. The significance of core OTUs in libraries is expressed either as a percentage of the number of OTUs (A) or as a percentage of the number of reads (B).
The survival of gut-resident microorganisms, and thereby their persistence in reproductives during colony foundation, was assumed to be dependent on their feeding behavior, which was approximated by the gut morphology. The total gut length and various dimensions of the hindgut did not differ between male and female reproductives, as they do in distantly related termites (Shimada et al., 2013). Similarly, the structure of the microbiota did not differ between sexes. The absence of sexual dimorphism on the microbiota was consistently observed on protozoan communities of lower termites during the first months of colony foundation (Shimada et al., 2013; Michaud et al., 2020), suggesting that both sexes are similarly involved in feeding tasks at this stage. This involvement of both parents (biparental care) was demonstrated in several studies on lower termites (Shimada et al., 2013; Benjamino and Graf, 2016; Inagaki et al., 2020; Velenovsky et al., 2021; Chouvenc, 2022) and very recently on Macrotermes natalensis (Sinotte et al., 2022) and was likely inherited from the common ancestor of termites (Nalepa, 2015).
Notably, the gut size of M. subhyalinus dealates enlarged during colony foundation, rather than shrinking as expected under the hypothesis of a fasting at this stage. As illustrated in Supplementary Figure S1, their paunch was filled with soil-like material. It is unclear whether the ingestion of soil material by dealates is natural, or rather driven by the need of particulate organic matter to compensate the unavailable wood material needed to sustain the gut microbiome. The increase in the gut size (Shimada et al., 2013) or relative weight (Nalepa et al., 2001) seems common during the first period of colony foundations in lower termites. The enlargement of the gut seems to be aligned with a gain in wet weight of reproductives during this period (Nalepa et al., 2001; Chouvenc, 2022). However, the dry body weight and the content in nutritional reserves decrease at the same time (Johnston and Wheeler, 2007; Chouvenc, 2022), likely indicating that the increase of the gut size in dealates reflects the rehydration of the gut, rather than the feeding behavior only. Albeit most of studies assessing the feeding behavior concern lower termites, the engorgement of dealates during the first weeks of colony foundation may be ubiquitous and can be a component of the observed enlargement of the gut in the current study. Further investigations taking into account weight indications and analyzing the gut content should clarify the feeding behavior of reproductives of this species during this period.
We characterized the whole gut mycobiome of Macrotermitinae reproductives along colony foundation. The gut of alates of M. subhylanus harbored dominantly Ascomycota and Basidiomycota, among which all OTUs of Lyophyllaceae clustered within Termitomyces. Despite the high frequency of detection in alates (75% of alates, regardless sex), Termitomyces surprisingly covered a low proportion of their gut mycobiome (< 10% of reads), strikingly different from the worker caste, which is dominated by Termitomyces in several Macrotermitinae species (>98% of reads; Makonde, 2017).
In contrast, the bacteriome of alates of M. subhyalinus was similar to that of workers. It predominately consists of Bacteroidetes, Firmicutes, and Proteobacteria as in workers of other Macrotermes species and some other Macrotermitinae (Hongoh et al., 2006; Dietrich et al., 2014; Otani et al., 2014; Hu et al., 2019). The similarity between alate and worker bacteriomes, which furthermore seems to be widespread (Hongoh et al., 2006; Diouf et al., 2018), indicates that the bacteriome of alates is also representative of the host species. Within colonies of Macrotermitinae, major and minor workers fulfill different tasks and have different feeding behavior (Sieber and Leuthold, 1981). Yet, there was no marked difference in the composition of the bacteriome between major and minor workers from wild colonies, which is congruent with previous reports on other species of Macrotermintinae (Otani et al., 2019; Vidkjær et al., 2021). Within-colony trophallaxis may therefore enable a suitable transfer of the main bacterial taxa between nestmates. It is worth mentioning that our study presented a fraction of reads assigned as unclassified Bacteria (Figure 6) and unclassified Fungi (Figure 5). This lack of taxonomic resolution can be explained by both the sequencing technology used here (i.e., short-read sequencing of a few hundred base pair amplicons, which limits the amount of information) and by the origin of our samples (i.e., termite guts and copularium) which are not investigated as much as human gut or soil for instance. Regarding the former point, the use of shotgun metagenomic combined with the reconstruction of metagenome-assembled genomes has been successfully used to explore at the genome level the prokaryotic diversity associated with termites (Hervé et al., 2020; Arora et al., 2022). Further studies could thus use this approach to monitor microbial transmission at the strain level (Su et al., 2021). Regarding the latter point, there are indeed clades associated with termites which has not been yet identified or characterized, and this is particularly true for fungi (Větrovský et al., 2020). This is further supported by the fact that for both fungal (Figure 5) and bacterial (Figure 6) datasets, we consistently found much less unclassified bacterial and fungal sequences in the copularium made of soil than in the termite guts.
Aside from alates, the microbiota of the reproductive caste of Termitidae has only been studied in kings and queens of Macrotermitinae from “mature” colonies several years after foundation. Their microbiota was markedly divergent and less diverse than in workers’ gut, with only a single or very few bacterial taxa undetectable in workers (Poulsen et al., 2014; Otani et al., 2019), a compositional shift likely explained by trophallactic food. This drastic divergence precluded any extrapolation on the extent of their contribution in symbionts transmission. The succession of the microbiota presented below partially fills the gap between alates and these old reproductives. However, focusing on the transitional phase where kings and queens pass from feeders (parental care) to dependency to the nutritional resources provided by offspring workers (alloparental care) should improve our understanding the process leading to this erosion of the bacteriome diversity in the gut of old kings and queens.
The succession of bacteria and fungi in the gut microbiota during colony foundation followed a distinct pattern, likely reflecting differences in the tightness of the relationship. The alpha diversity of the mycobiome, did not markedly vary up to the day 30, which may reflect the maintenance of fungal lineages imported during the flight. Beyond day 30, the richness and the exponential of Shannon indices increased together with gut dimensions, possibly due to a transition in the feeding behavior of dealates, which modified community structure. The widely variable pattern of beta diversity of the mycobiome, even between replicates, suggests that it mostly consists of transient fungi ingested from the soil rather than resident, especially the first month. The dominance of Ascomycota, which are also the main colonizers of the copularium supports an incidence of soil-borne fungi. Similarly, the majority of Sordariomycetes core OTUs are ubiquitous saprobic fungi (Zhang et al., 2006). Except for the ectosymbiont, there is little information on strict associations between Macrotermitinae and fungi. Taxon-specific PCR detected Termitomyces in 75% of male and female alates of M. subhyalinus. In comparison, 100% of positive tests were recorded for both sexes of alates of the sympatric species Microtermes sp. that inoculates its combs with conidia of Termitomyces carried in the gut of female reproductives (Johnson, 1981; Johnson et al., 1981; Nobre et al., 2011a,b). While the absence of sexual dimorphism for both species could indicate that amplified DNA included material from the food bolus, the lower score in M. subhyalinus could reflect the difference in the transmission modes. The highlighting of vertical transmission in Microtermes spp. and Macrotermes bellicosus in laboratory was based on the presence of conidia bolus of Termitomyces in the foregut of alates, and then the successful fertilization of fungus combs without inoculation by the manipulator with comb material from a mature colony (Johnson, 1981; Johnson et al., 1981). This was not the case for M. subhyalinus (Johnson et al., 1981) and though the molecular approach used here does not make it possible to distinguish the nature of the fungal material, the exhaustion of the molecular signal of Termitomyces before the emergence of offspring, even with sensitive metabarcoding, provides additional evidence of environmental acquisition of the fungal ectosymbiont in M. subhyalinus i.e., the inoculation with sexual spores harvested from the environment by the first foraging workers, as in the vast majority of Macrotermitinae. The success of the colony foundations by reproductives in this case is conditioned by the prior or simultaneous presence of Termitomyces in the dispersal area (Nobre and Aanen, 2010). This transmission mode, which is the most ancestral and the most widespread in Macrotermitinae might have contributed to their restricted geographic distribution only to Africa (the continent of origin) and to Asia (Aanen and Eggleton, 2005). The large prevalence of the horizontal transmission of Termitomyces also led to the absence of host specificity. While a lineage-specific pattern can be drawn at higher taxonomic levels, host switches are frequent at lower taxonomic levels, and no species of Termitomyces is exclusively associated to a single termite host (Aanen et al., 2002; Osiemo et al., 2010). Relevant differences were comparatively identified for bacteria. In contrast to fungi, richness indexes for bacteria remained unchanged over the first months of the study, which is compatible with the hypothesis that many bacterial taxa are not transiently ingested with the food but are rather gut residents. One original aspect of our study was to have analyzed each gut separately, preserving inter-individual variability. Despite this, gut bacteriomes clustered by age and differed from the copularium bacteriome, reinforcing the endemism of bacterial lineages in the gut habitat. The temporary decrease of Hill numbers q = 1 and q = 2 at days 30 and 45 under constant richness coincided with the change in the gut morphology. As the gut content was not analyzed, we question whether it could be attributed to a dietary transition. This period also coincides with the increase in the relative abundance of Epsilonbacteraeota, and specially of a single OTU assigned to the genus Arcobacter (21.27 to 24, 24% of reads at 30 and 45 days, respectively); which may have decreased abundance-related diversity indices. Arcobacter is a common genus of the core bacterial communities of fungus-growing termites (Otani et al., 2014, 2019). Though little is known about the metabolic functions and factors driving its abundance in termites, the increase of the relative abundance of Epsilonbacteraeota and specially of Arcobacter could be compared with the protozoan pulse in the gut lower termites during this first period of the colony foundation (Shimada et al., 2013; Benjamino and Graf, 2016; Inagaki et al., 2020; Velenovsky et al., 2021), likely resulting from the resumption of the wood-feeding activity of dealates to sustain the protozoan fauna. Alongside with the compositional analyzes performed here, quantifying bacterial density should clarify if the microbial expansion found for protozoa applied to the gut bacteriome of this higher termite.
Nevertheless, this transition did not markedly impact the core bacteriome since the most abundant genus-level taxa and core OTUs persisted for the 60 first days, indicating a high conservation of key bacterial symbionts. These same key taxa persisted until the end of the monitoring because only a few were missing from the gut samples at day 120, where they still dominated. To summarize, bacteria from the mother colony are persistent in the gut and overcome the potential diet shift associated with colony foundation, unlike fungi. As offspring appeared as larvae by day 45 and as actively foraging workers after 2 months, they co-exist with the core of bacteriome and thus enable a vertical transmission within the colony of these symbionts mostly adapted to the gut environment. Compared to the transmission of the fungal exosymbiont, the bacteriome the is likely transmitted through the same social exchanges enabling the transmission of protozoa and that were acquired in since the emergence of the ancestor of eusocial termites (Nalepa, 2015; Chouvenc et al., 2021). This vertical transmission likely explains the existence of termite-specific clusters within numerous prokaryotic lineages, indicating a coevolutionary process with termite hosts (Hongoh et al., 2005; Abdul Rahman et al., 2015; Bourguignon et al., 2018). In contrast to the gut bacteriome, the acquisition of the externally cultured fungus symbiont is a derived trait, acquired once by the ancestor of Macrotermitinae. Whether the loss of protozoa that gave rise Termitidae evolved first a soil-feeding behavior or rather an externalized digestion in combs is still discussed (Bucek et al., 2019; Chouvenc et al., 2021). Compared to the gut bacteriome, the delayed acquisition of the fungal symbiont during the colony foundation seems to support the hypothesis of postdated emergence of the ancestor Macrotermitinae relative to the loss of protozoa. As suggested by Bucek et al. (2019), fungus-growing termites may have emerged from a soil-feeding ancestor by reversion toward a wood feeding diet, fostered by the domestication of a saprobic fungus predisposed to trive in insect fecal substrate (van de Peppel et al., 2021).
The transmission of symbionts in termites has been previously addressed for Protozoa that are unique to lower termites and for fungal ectosymbionts that are unique to Macrotermitinae. To our knowledge, this is the first study addressing the transmission of the gut symbionts in Termitidae, which were assumed to have a distinct feeding behavior when compared to lower termites, with a supposed fasting during the colony foundation. To date, information on the microbiota of the reproductive caste of Termitidae has been limited to alates, which share a similar microbiota with workers, and to aged kings and queens that have a distinct microbiota. Our study describes the missing steps in the dynamics between these two extreme stages, but further investigation from the end of our study up to old kings and queens, and of the main changes taking place during the transition from biparental to alloparantal care will provide a more complete picture in the future. The persistence of gut bacteria over the first period of the colony’s life, the increase of the gut size, and the presence of soil-borne fungi demonstrated in this study did not support the existence of a fasting during the colony foundation. Our findings revealed no intimate relationship between fungi and the gut habitat and the early exhaustion of Termitomyces in the gut confirms that this symbiont is acquired from the environment. Conversely, bacteria persisted in the gut of reproductives throughout the colony foundation period, until offspring workers emerged. The presence of these taxa in offspring workers without any possible external source supports the role of both reproductives in vertical transmission of gut microbiota and, thereby, in the co-evolutionary history of bacterial symbionts and their termite hosts.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
MD, CR-L, and M-AS conceived and designed the study. CR-F and AN performed the field sampling. SF, MD, and CR-L conducted the lab rearing experiment, dissections, and morphological analyses. SF, MD, JL, AB, and M-AS conducted molecular experiments. VH, MJ, and EM conducted statistical and metabarcoding analyses. MD and VH drafted the manuscript. EM, M-AS, CR-F, AN, MJ, and AB critically revised the intellectual content of the manuscript. The final content of the manuscript was edited and approved by all authors. They declare having agreed to be accountable for all the aspects of the study. All authors contributed to the article and approved the submitted version.
This research was funded by the program Carrefour Écologie/Sciences de l’Environnement of Sorbonne Universités (CARESE-SU).
The authors would like to thank the Service de Systématique Moléculaire (UMS 2700, MNHN, CNRS) for granting access to its technical platform, the research teams of Laboratoire de Zoologie des Invertébrés Terrestres (IFAN Ch. Anta Diop) and LEMSAT (ECO&SOLS) for the allocation of human and logistic resources during the field mission. We also thank Stéphanie GIUSTI-MILLER and Maxime FERREIRA for their technical and logistical support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.1055382/full#supplementary-material
Aanen, D. K., de Fine Licht, H. H., Debets, A. J. M., Kerstes, N. A. G., Hoekstra, R. F., and Boomsma, J. J. (2009). High symbiont relatedness stabilizes mutualistic cooperation in fungus-growing termites. Science 326:1103. doi: 10.1126/science.1173462
Aanen, D. K., and Eggleton, P. (2005). Fungus-growing termites originated in African rain forest. Curr. Biol. 15, 851–855. doi: 10.1016/j.cub.2005.03.043
Aanen, D. K., Eggleton, P., Rouland-Lefèvre, C., Guldberg-Frøslev, T., Rosendahl, S., and Boomsma, J. J. (2002). The evolution of fungus-growing termites and their mutualistic fungal symbionts. Proc. Natl. Acad. Sci. U. S.A. 99, 14887–14892. doi: 10.1073/pnas.22231309
Aanen, D. K., Ros, V. I. D., de Fine Licht, H. H., Mitchell, J., de Beer, Z. W., Slippers, B., et al. (2007). Patterns of interaction specificity of fungus-growing termites and Termitomyces symbionts in South Africa. BMC Evol. Biol. 7:115. doi: 10.1186/1471-2148-7-115
Abdul Rahman, N., Parks, D. H., Willner, D. L., Engelbrektson, A. L., Goffredi, S. K., Warnecke, F., et al. (2015). A molecular survey of Australian and north American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome 3:5. doi: 10.1186/s40168-015-0067-8
Alberdi, A., and Gilbert, M. T. P. (2019). Hilldiv: an R package for the integral analysis of diversity based on hill numbers. bio Rxiv. doi: 10.1101/545665
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Arora, J., Kinjo, Y., Šobotník, J., Buček, A., Clitheroe, C., Stiblik, P., et al. (2022). The functional evolution of termite gut microbiota. Microbiome 10:78. doi: 10.1186/s40168-022-01258-3
Badertscher, S., Gerber, C., and Leuthold, R. H. (1983). Polyethism in food supply and processing in termite colonies of Macrotermes subhyalinus (Isoptera). Behav. Ecol. Sociobiol 12, 115–119. doi: 10.1007/BF00343201
Benjamino, J., and Graf, J. (2016). Characterization of the Core and caste-specific microbiota in the termite, Reticulitermes flavipes. Front. Microbiol. 7:171. doi: 10.3389/fmicb.2016.00171
Bignell, D. E. (2016). “The role of symbionts in the evolution of termites and their rise to ecological dominance in the tropics” in The Mechanistic Benefits of Microbial Symbionts. ed. C. J. Hurst (Cham: Springer International Publishing)
Bourguignon, T., Lo, N., Cameron, S. L., Šobotník, J., Hayashi, Y., Shigenobu, S., et al. (2014). The evolutionary history of termites as inferred from 66 mitochondrial genomes. Mol. Biol. Evol. 32, 406–421. doi: 10.1093/molbev/msu308
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., et al. (2018). Rampant host switching shaped the termite gut microbiome. Curr. Biol. 28, 649–654.e2. doi: 10.1016/j.cub.2018.01.035
Brown, S. P., Callaham, M. A., Oliver, A. K., and Jumpponen, A. (2013). Deep ion torrent sequencing identifies soil fungal community shifts after frequent prescribed fires in a southeastern US forest ecosystem. FEMS Microbiol. Ecol. 86, 557–566. doi: 10.1111/1574-6941.12181
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Bucek, A., Šobotník, J., He, S., Shi, M., McMahon, D. P., Holmes, E. C., et al. (2019). Evolution of termite Symbiosis informed by transcriptome-based phylogenies. Curr. Biol. 29, 3728–3734.e4. doi: 10.1016/j.cub.2019.08.076
Chao, A., Chiu, C. H., and Jost, L. (2016). “Phylogenetic diversity measures and their decomposition: a framework based on hill numbers” in Biodiversity conservation and phylogenetic systematics: Preserving our evolutionary heritage in an extinction crisis. eds. R. Pellens and P. Grandcolas, vol. 14 (Cham: Springer) doi: 10.1007/978-3-319-22461-9_8
Cheung, M. K., Wong, C. K., Chu, K. H., and Kwan, H. S. (2018). Community structure, dynamics and interactions of bacteria, archaea and fungi in subtropical coastal wetland sediments. Sci. Rep. 8:14397. doi: 10.1038/s41598-018-32529-5
Chouvenc, T. (2022). Eusociality and the transition from biparental to alloparental care in termites. Funct. Ecol. 36, 3049–3059. doi: 10.1111/1365-2435.14183
Chouvenc, T., Šobotník, J., Engel, M. S., and Bourguignon, T. (2021). Termite evolution: mutualistic associations, key innovations, and the rise of Termitidae. Cell. Mol. Life Sci. 78, 2749–2769. doi: 10.1007/s00018-020-03728-z
Collins, N. M. (1977). The population ecology and energetics of Macrotermes bellicosus Smeathman (Isoptera). Doctoral thesis London: University of London.
da Costa, R. R., and Poulsen, M. (2018). Mixed-mode transmission shapes termite gut community assemblies. Trends Microbiol. 26, 557–559. doi: 10.1016/j.tim.2018.04.005
Dietrich, C., Köhler, T., and Brune, A. (2014). The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Appl. Environ. Microbiol. 80, 2261–2269. doi: 10.1128/AEM.04206-13
Diouf, M., Hervé, V., Mora, P., Robert, A., Frechault, S., Rouland-Lefèvre,, et al. (2018). Evidence from the gut microbiota of swarming alates of a vertical transmission of the bacterial symbionts in Nasutitermes arborum (Termitidae, Nasutitermitinae). Antonie Van Leeuwenhoek 111, 573–587. doi: 10.1007/s10482-017-0978-4
Diouf, M., and Rouland-Lefevre, C. (2018). “The fungus-growing termites: biology, damage on tropical crops and specific management” in Termites and Sustainable Management: Volume 2- Economic LOSSES and Management. eds. M. A. Khan and W. Ahmad (Cham: Springer International Publishing)
Diouf, M., Roy, V., Mora, P., Frechault, S., Lefebvre, T., and Hervé, V., Cham (2015). Profiling the succession of bacterial communities throughout the life stages of a higher termite Nasutitermes arborum (Termitidae, Nasutitermitinae) using 16S rRNA gene pyrosequencing. PLoS One 10: 1–15. doi: 10.1371/journal.pone.0140014
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Engel, M. S., Grimaldi, D. A., and Krishna, K. (2009). Termites (Isoptera): their phylogeny, classification, and rise to ecological dominance. Am. Mus. Novit. 2009, 1–27. doi: 10.1206/651.1
Goslee, S., and Urban, D. (2007). The ECODIST package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 22, 1–19. doi: 10.18637/jss.v022.i07
Grassé, P.-P. (1984). “La fondation des nouvelles sociétés” in Termitologia Tome II: Fondation des sociétés et construction. ed. P.-P. Grassé (Paris: Masson)
Grassé, P.-P., and Noirot, C. (1955). La fondation de nouvelles sociétés par Bellicositermes natalensis Hav. Insect. Soc. 2, 213–220. doi: 10.1007/BF02224382
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Han, S. H., and Bordereau, C. (1982). Origin and formation of the royal fat body of the higher termite queens. J. Morphol. 173, 17–28. doi: 10.1002/jmor.1051730103
Harit, A. K., Gajalakshmi, S., and Abbasi, S. A. (2016). Studies on the development of captive termite colonies. Zoo. Ecol. 26, 301–312. doi: 10.1080/21658005.2016.1228731
Hervé, V., Liu, P., Dietrich, C., Sillam-Dussès, D., Stiblik, P., Šobotník,, et al. (2020). Phylogenomic analysis of 589 metagenome-assembled genomes encompassing all major prokaryotic lineages from the gut of higher termites. PeerJ 8:e8614. doi: 10.7717/peerj.8614
Hervé, V., and Lopez, P. J. (2020). Analysis of interdomain taxonomic patterns in urban street mats. Environ. Microbiol. 22, 1280–1293. doi: 10.1111/1462-2920.14933
Hongoh, Y., Deevong, P., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., et al. (2005). Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl. Environ. Microbiol. 71, 6590–6599. doi: 10.1128/AEM.71.11.6590-6599.2005
Hongoh, Y., Ekpornprasit, L., Inoue, T., Moriya, S., Trakulnaleamsai, S., Ohkuma, M., et al. (2006). Intracolony variation of bacterial gut microbiota among castes and ages in the fungus-growing termite Macrotermes gilvus. Mol. Ecol. 15, 505–516. doi: 10.1111/j.1365-294X.2005.02795.x
Hu, H., da Costa, R. R., Pilgaard, B., Schiøtt, M., Lange, L., and Poulsen, M. (2019). Fungiculture in termites is associated with a Mycolytic gut bacterial community. mSphere 4, e00165–e00119. doi: 10.1128/mSphere.00165-19
Inagaki, T., Yanagihara, S., Fuchikawa, T., and Matsuura, K. (2020). Gut microbial pulse provides nutrition for parental provisioning in incipient termite colonies. Behav. Ecol. Sociobiol. 74:64. doi: 10.1007/s00265-020-02843-y
Johnson, R. A. (1981). Colony development and establishment of the fungus comb in Microtermes sp. nr. Usambaricus (Sjöstedt) (Isoptera: Macrotermitinae) from Nigeria. Insect. Soc. 28, 3–12. doi: 10.1007/BF02223617
Johnson, R. A., Thomas, R. J., Wood, T. G., and Swift, M. J. (1981). The inoculation of the fungus comb in newly founded colonies of some species of the Macrotermitinae (Isoptera) from Nigeria. J. Nat. Hist. 15, 751–756. doi: 10.1080/00222938100770541
Johnston, M. L., and Wheeler, D. E. (2007). The role of storage proteins in colony-founding in termites. Insect. Soc. 54, 383–387. doi: 10.1007/s00040-007-0957-2
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kwong, W. K., Medina, L. A., Koch, H., Sing, K.-W., Soh, E. J. Y., Ascher, J. S., et al. (2017). Dynamic microbiome evolution in social bees. Sci. Adv. 3, –e1600513. doi: 10.1126/sciadv.1600513
Lefort, V., Longueville, J.-E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Lepage, M., and Darlington, J. P. E. C. (2000). “Population dynamics of termites” in Termites: Evolution, Sociality, Symbioses, Ecology. eds. T. Abe, D. E. Bignell, and M. Higashi (Dordrecht: Springer Netherlands), 333–361.
Makonde, H. (2017). Fungal diversity and community structure in gut, mound and surrounding soil of fungus-cultivating termites. Afr. J. Microbiol. Res. 11, 504–515. doi: 10.5897/AJMR2017.8484
McKnight, D. T., Huerlimann, R., Bower, D. S., Schwarzkopf, L., Alford, R. A., and Zenger, K. R. (2019). Methods for normalizing microbiome data: an ecological perspective. Methods Ecol. Evol. 10, 389–400. doi: 10.1111/2041-210X.13115
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Michaud, C., Hervé, V., Dupont, S., Dubreuil, G., Bézier, A. M., Meunier, J., et al. (2020). Efficient but occasionally imperfect vertical transmission of gut mutualistic protists in a wood-feeding termite. Mol. Ecol. 29, 308–324. doi: 10.1111/mec.15322
Mikaelyan, A., Dietrich, C., Köhler, T., Poulsen, M., Sillam-Dussès, D., and Brune, A. (2015). Diet is the primary determinant of bacterial community structure in the guts of higher termites. Mol. Ecol. 24, 5284–5295. doi: 10.1111/mec.13376
Mikaelyan, A., Meuser, K., and Brune, A. (2017). Microenvironmental heterogeneity of gut compartments drives bacterial community structure in wood- and humus-feeding higher termites. FEMS Microbiol. Ecol. 93:fiw 210. doi: 10.1093/femsec/fiw210
Mitchell, J. D. (2020). Colony foundation and the development of incipient laboratory colonies of Macrotermes natalensis (Haviland) (Termitidae: Macrotermitinae). Afr. Entomol. 28, 215–224. doi: 10.4001/003.028.0215
Nalepa, C. A. (2015). Origin of termite eusociality: trophallaxis integrates the social, nutritional, and microbial environments. Ecol. Entomol. 40, 323–335. doi: 10.1111/een.12197
Nalepa, C., Miller, L., and Lenz, M. (2001). Flight characteristics of Mastotermes darwiniensis (Isoptera, Mastotermitidae). Insect. Soc. 48, 144–148. doi: 10.1007/PL00001757
Nilsson, R. H., Larsson, K.-H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2018). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Nobre, T., and Aanen, D. K. (2010). Dispersion and colonisation by fungus-growing termites: vertical transmission of the symbiont helps, but then.? Commun. Integr. Biol. 3, 248–250. doi: 10.4161/cib.3.3.11415
Nobre, T., Koné, N. A., Konaté, S., Linsenmair, K. E., and Aanen, D. K. (2011a). Dating the fungus-growing termites’ mutualism shows a mixture between ancient codiversification and recent symbiont dispersal across divergent hosts. Mol. Ecol. 20, 2619–2627. doi: 10.1111/j.1365-294X.2011.05090.x
Nobre, T., Rouland-Lefèvre, C., and Aanen, D. K. (2011b). “Comparative biology of fungus cultivation in termites and ants” in Biology of Termites: A Modern Synthesis. eds. D. E. Bignell, Y. Roisin, and N. Lo (Dordrecht: Springer Netherlands)
Noda, S., Kitade, O., Inoue, T., Kawai, M., Kanuta, M., Hiroshima, K., et al. (2007). Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol. Ecol. 16, 1257–1266. doi: 10.1111/j.1365-294X.2006.03219.x
Nutting, W. L. (1969). “Flight and colony foundation” in Biology of Termites. eds. K. Krishna and F. Weesner (New York: Academic Press)
Okot-Kotber, B. M. (1981). Polymorphism and the development of the first progeny in incipient colonies of Macrotermes michaelseni (Isoptera, Macrotermitinae). Ins. Sci. Appl. 1, 147–150. doi: 10.1017/S174275840000031X
Oksanen, J., Kindt, R., Legendre, P., O’hara, B., Henry, M., and Maintainer, H. S. (2007). The vegan package title community ecology package. Cran R-Project Org/,Http://R-Forge R-Project Org/Projects/Vegan/
Otani, S., Mikaelyan, A., Nobre, T., Hansen, L. H., Koné, N. A., Sørensen, S. J., et al. (2014). Identifying the core microbial community in the gut of fungus-growing termites. Mol. Ecol. 23, 4631–4644. doi: 10.1111/mec.12874
Otani, S., Zhukova, M., Koné, N. A., da Costa, R. R., Mikaelyan, A., Sapountzis, P., et al. (2019). Gut microbial compositions mirror caste-specific diets in a major lineage of social insects. Environ. Microbiol. Rep. 11, 196–205. doi: 10.1111/1758-2229.12728
Osiemo, Z. B., Marten, A., Kaib, M., Gitonga, L. M., Boga, H. I., and Brandl, R. (2010). Open Relationships in the Castles of Clay: High Diversity and Low Host Specificity of Termitomyces Fungi Associated with Fungus-Growing Termites in Africa. Insect. Soc 57, 351–363. doi: 10.1007/s00040-010-0092-3
Poulsen, M., Hu, H., Li, C., Chen, Z., Xu, L., Otani, S., et al. (2014). Complementary symbiont contributions to plant decomposition in a fungus-farming termite. Proc. Natl. Acad. Sci. U. S. A. 111, 14500–14505. doi: 10.1073/pnas.1319718111
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rouland-Lefèvre, C. (2000). “Symbiosis with fungi” in Termites: Evolution, Sociality, Symbioses, Ecology. eds. T. Abe, D. E. Bignell, and M. Higashi (Dordrecht Netherlands: Springer)
Sands, W. A. (1960). The initiation of fungus comb construction in laboratory colonies of Ancistrotermes guineensis (Silvestri). Insect. Soc. 7, 251–263. doi: 10.1007/BF02224496
Schloss, P. D. (2008). Evaluating different approaches that test whether microbial communities have the same structure. ISME J. 2, 265–275. doi: 10.1038/ismej.2008.5
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schneider-Maunoury, L., Leclercq, S., Clément, C., Covès, H., Lambourdière, J., Sauve, M., et al. (2018). Is tuber melanosporum colonizing the roots of herbaceous, non-ectomycorrhizal plants? Fungal Ecol. 31, 59–68. doi: 10.1016/j.funeco.2017.10.004
Shimada, K., Lo, N., Kitade, O., Wakui, A., and Maekawa, K. (2013). Cellulolytic Protist numbers rise and fall dramatically in termite Queens and kings during Colony Foundation. Eukaryot. Cell 12, 545–550. doi: 10.1128/EC.00286-12
Sieber, R. (1983). Establishment of fungus comb in laboratory colonies of Macrotermes michaelseni and Odontotermes montanus (Isoptera, Macrotermitinae). Insect. Soc. 30, 204–209. doi: 10.1007/BF02223870
Sieber, R., and Leuthold, R. H. (1981). Behavioural elements and their meaning in incipient laboratory colonies of the fungus-growing termite Macrotermes michaelseni (Isoptera: Macrotermitinae). Insect. Soc. 28, 371–382. doi: 10.1007/BF02224194
Sinotte, V., Renelies-Hamilton, J., Andreu-Sánchez, S., Vasseur-Cognet, M., and Poulsen, M. (2022). Extensive inheritance of gut microbial communities in a superorganismal termite. doi: 10.21203/rs.3.rs-1978279/v1,
Su, Q., Wang, Q., Mu, X., Chen, H., Meng, Y., Zhang, X., et al. (2021). Strain-level analysis reveals the vertical microbial transmission during the life cycle of bumblebee. Microbiome 9:216. doi: 10.1186/s40168-021-01163-1
Turenne, C. Y., Sanche, S. E., Hoban, D. J., Karlowsky, J. A., and Kabani, A. M. (1999). Rapid identification of fungi by using the ITS2 genetic region and an automated fluorescent capillary electrophoresis system. J. Clin. Microbiol. 37, 1846–1851. doi: 10.1128/JCM.37.6.1846-1851.1999
van de Peppel, L. J. J., Nieuwenhuis, M., Auxier, B., Grum-Grzhimaylo, A. A., Cárdenas, M. E. E., de Beer, Z. W., et al. (2021). Ancestral predisposition toward a domesticated lifestyle in the termite-cultivated fungus Termitomyces. Curr. Biol. 31, 4413–4421.e5. doi: 10.1016/j.cub.2021.07.070
Velenovsky, J. F., Gile, G. H., Su, N. Y., and Chouvenc, T. (2021). Dynamic protozoan abundance of Coptotermes kings and queens during the transition from biparental to alloparental care. Insect. Soc. 68, 33–40. doi: 10.1007/s00040-021-00808-6
Vesala, R., Niskanen, T., Liimatainen, K., Boga, H., Pellikka, P., and Rikkinen, J. (2017). Diversity of fungus-growing termites (Macrotermes) and their fungal symbionts (Termitomyces) in the semiarid Tsavo ecosystem, Kenya. Biotropica 49, 402–412. doi: 10.1111/btp.12422
Větrovský, T., Soukup, P., Stiblik, P., Votýpková, K., Chakraborty, A., Odriozola, I., et al. (2020). Termites host specific fungal communities that differ from those in their ambient environments. Fungal Ecol. 48:100991. doi: 10.1016/j.funeco.2020.100991
Vidkjær, N. H., Schmidt, S., Hu, H., Bodawatta, K. H., Beemelmanns, C., and Poulsen, M. (2021). Species- and caste-specific gut metabolomes in fungus-farming termites. Meta 11:839. doi: 10.3390/metabo11120839
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Waud, M., Busschaert, P., Ruyters, S., Jacquemyn, H., and Lievens, B. (2014). Impact of primer choice on characterization of orchid mycorrhizal communities using 454 pyrosequencing. Mol. Ecol. Resour. 14, 679–699. doi: 10.1111/1755-0998.12229
Weesner, F. (1969). “External anatomy” in Biology of Termites. ed. K. Krishna (New York, USA: Academic Press)
Westcott, S. L., and Schloss, P. D. (2017). Opti Clust, an improved method for assigning amplicon-based sequence data to operational taxonomic units. mSphere 2, e00073–e00017. doi: 10.1128/mSphereDirect.00073-17
White, T., Bruns, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics” in PCR Protocols: A Guide to Methods and Applications. eds. M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (New York: Academic Press)
Wickham, H. (2016). Package ‘ggplot 2’: Elegant graphics for data analysis. Springer-Verlag New York
Keywords: termite reproductives, Macrotermitinae, gut microbiota, Termitomyces, transmission, colony foundation
Citation: Diouf M, Hervé V, Fréchault S, Lambourdière J, Ndiaye AB, Miambi E, Bourceret A, Jusselme MD, Selosse M-A and Rouland-Lefèvre C (2023) Succession of the microbiota in the gut of reproductives of Macrotermes subhyalinus (Termitidae) at colony foundation gives insights into symbionts transmission. Front. Ecol. Evol. 10:1055382. doi: 10.3389/fevo.2022.1055382
Edited by:
Aram Mikaelyan, North Carolina State University, United StatesReviewed by:
Thomas Chouvenc, University of Florida, United StatesCopyright © 2023 Diouf, Hervé, Fréchault, Lambourdière, Ndiaye, Miambi, Bourceret, Jusselme, Selosse and Rouland-Lefèvre. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Diouf, ✉ bWljaGVsLmRpb3VmQHUtcGVjLmZy
†ORCID: Michel Diouf, https://orcid.org/0000-0002-6263-8216
Vincent Hervé, https://orcid.org/0000-0002-3495-561X
Marc-André Selosse, https://orcid.org/0000-0003-3471-9067
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.