 Euki Yazaki
Euki Yazaki Akinori Yabuki
Akinori Yabuki Yuki Nishimura
Yuki Nishimura Takashi Shiratori4
Takashi Shiratori4 Tetsuo Hashimoto
Tetsuo Hashimoto Yuji Inagaki
Yuji Inagaki- 1Interdisciplinary Theoretical and Mathematical Sciences (iTHEMS), RIKEN, Wako, Saitama, Japan
- 2Japan Agency for Marine-Earth Science and Technology, Yokosuka, Kanagawa, Japan
- 3Department of Integrated Biosciences, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwa, Chiba, Japan
- 4Faculty of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan
- 5Center for Computational Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan
The mitochondrial genomes are very diverse, but their evolutionary history is unclear due to the lack of efforts to sequence those of protists (unicellular eukaryotes), which cover a major part of the eukaryotic tree. Cryptista comprises cryptophytes, goniomonads, kathablepharids, and Palpitomonas bilix, and their mitochondrial genomes (mt-genomes) are characterized by various gene contents, particularly the presence/absence of an ancestral (bacterial) system for the cytochrome c maturation system. To shed light on mt-genome evolution in Cryptista, we report the complete mt-genome of Microheliella maris, which was recently revealed to branch at the root of Cryptista. The M. maris mt-genome was reconstructed as a circular mapping chromosome of 61.2 kbp with a pair of inverted repeats (12.9 kbp) and appeared to be the most gene-rich among the mt-genomes of the members of Diaphoretickes (a mega-scale eukaryotic assembly including Archaeplastida, Cryptista, Haptista, and SAR) studied so far, carrying 53 protein-coding genes. With this newly sequenced mt-genome, we inferred and discussed the evolution of the mt-genome in Cryptista and Diaphoretickes.
Introduction
Mitochondria, which originated from an endosymbiotic prokaryote, are fundamental and unique eukaryotic organelles. All eukaryotes possess mitochondria or organelles derived from mitochondria, with a single exception, the amitochondriate eukaryote Monocercomonoides exilis (Karnkowska et al., 2016), and their cellular functions have been studied extensively (Atteia et al., 2009; Panigrahi et al., 2009; Calvo and Mootha, 2010; Lee et al., 2013; Gawryluk et al., 2014). Nevertheless, it is still controversial as to when and how the first mitochondrion was established in eukaryotic evolution (Gray et al., 2001; Gray, 2015; Roger et al., 2017; Zachar and Boza, 2020). The diversity of the mitochondrial genomes of unicellular eukaryotes (protists) is very high with respect to both gene content and genomic structure (Smith and Keeling, 2015). To gain deeper insights into the diversity of mitochondrial genomes among protists, we need to sample data from phylogenetically diverse protists, as the mitochondrial genomes of the members of a well-defined taxonomic group are similar to one another (Smith, 2015). In other words, novel and/or poorly studied protists may shed light on previously overlooked aspects of mitochondrial genome evolution. In particular, early branching species of the taxonomic group of interest, or even those of eukaryotes as a whole, are anticipated to retain the mitochondrial genomes with some ancestral features. Indeed, Ancoracysta twista, which was described recently and was shown to belong to diaphoretickes but no clear phylogenetic affinity to any known diaphoretickes lineages, was shown to possess a large number of genes in its mitochondrial genomes (Janouškovec et al., 2017). Besides A. twista, jakobids, which belong to Discoba, a taxonomic group excluded from Diaphoretickes, have the most gene-rich mitochondrial genomes among the eukaryotes studied to date (e.g., 67 protein-coding genes in the mitochondrial genome of Andalucia godoyi; Burger et al., 2013; Smith and Keeling, 2015; Janouškovec et al., 2017; Roger et al., 2017).
The evolution of the mitochondrial genome of Cryptista, which is composed of cryptophytes, goniomonads, kathablepharids, and Palpitomonas bilix (Adl et al., 2019), remains to be understood. To our knowledge, the complete mitochondrial genomes are available for multiple cryptophytes and goniomonads, two kathablepharids, and P. bilix (Hauth et al., 2005; Nishimura et al., 2016, 2020; Cenci et al., 2018; Kim et al., 2018; Suo et al., 2018; Wideman et al., 2019). The cryptist mitochondrial genomes analyzed so far are circular, except for P. bilix, the deepest branching member of Cryptista. The P. bilix mitochondrial genome is a linear molecule that bears a pair of repeat sequences at both ends of the genome (~30 kb in length), which have been found in three distantly related protists, namely the green alga Polytomella parva, alveolate Acavomonas peruviana, and stramenopile Proteromonas lacertae (Fan and Lee, 2002; Pérez-Brocal et al., 2010; Janouškovec et al., 2013). Comparative studies of cryptist mitochondrial genomes have suggested the complex evolution of the cytochrome c maturation system in this protist assemblage (Nishimura et al., 2016, 2020). The genes encoding the components of the “bacterial” cytochrome c maturation system (System I) have been found in the mitochondrial genomes of P. bilix and the goniomonad Hemiarma marina, although other cryptists use nucleus-encoded, eukaryote-specific enzymes for cytochrome c maturation (System III). Although the evolution of the proteins involved in Systems I and III could not be elucidated with confidence, the two evolutionarily distinct systems for cytochrome c maturation were exclusively distributed in the Cryptista clade. Thus, the shift from System I to System III (or that from System III to System I) needs to be invoked multiple times during Cryptista evolution (Nishimura et al., 2016, 2020). To precisely understand the distribution of System I/III in Cryptista, we need the mitochondrial genomes of more diverse cryptists as well as other eukaryotes, including Microheliella maris, which showed an intimate affinity to Cryptista in a recent phylogenomic study (Yazaki et al., 2022).
Microheliella maris is a protist that was originally classified into the phylum Heliozoa (centroheliozoans) based on several characteristics, such as the radiating axopodia with tiny granules and the centroplast, which are similar to those of centroheliozoans (Cavalier-Smith and von der Heyden, 2007; Yabuki et al., 2012; Cavalier-Smith et al., 2015). In the phylogenetic analyses based on alignments comprising 319 genes, M. maris branched at the base of Cryptista, and the clade of M. maris plus Cryptista was further united with Archaeplastida (Yazaki et al., 2022). The clade of M. maris plus Cryptista was proposed to be called Pancryptista, and the union of Pancryptista and Archaeplastida was named the CAM clade (Yazaki et al., 2022). In the present study, we sequenced the mitochondrial genome of M. maris. The mitochondrial genome of M. maris was found to be a circular form with a pair of inverted repeats, and there were 53 protein-coding genes, which were greater than those of any other Diaphoretickes, including A. twista. In light of the M. maris mitochondrial genome (Mm mt-genome), we discuss the evolution of the mitochondrial genomes of Pancryptista and the CAM clade, as well as Diaphoretickes, and that of cytochrome c maturation system in Pancryptista.
Materials and methods
Sequencing of Microheliella maris mitochondrial DNA
A culture of Microheliella maris, which was studied by Yabuki et al. (2012) and kept in our laboratory, was used in this study. The same strain is available in the Culture Collection of Algae & Protozoa (CCAP).1 Approximately 200 ml of mid-exponential phase cell culture was subjected to DNA extraction. The cells were then centrifuged at 2,400 × g for 5 min. Total DNA was extracted from the cell pellet using a DNeasy Plant Mini Kit (Qiagen) following the manufacturer’s protocol. About 4.1 μg of total DNA was sent to Macrogen Japan Corp. (Kyoto). The sequence library was reconstructed and run on the NovaSeq 6000 system. A total of 1.4 × 108 pair reads, each up to 150 bp in length, were generated and subsequently subjected to contig assembly using SPAdes 3.13 (Bankevich et al., 2012). From the contig sequences, three possible mitochondrial genomic fragments were detected by BLASTN using the mitochondrial genome sequence of Ophirina amphinema (GenBank accession number: LC369600.1) as a query sequence. A circular mitochondrial genome (61,620 bp in length) was reconstructed from three fragments by PCR and Bowtie2 (Langmead and Salzberg, 2012) analyses (see Supplementary Information). The mitochondrial genome sequence was annotated using MFannot.2 Each open reading frame (ORF) was annotated based on the result of MFannot, but the position of the start codon was determined following the results of Artemis analysis (Carver et al., 2012). Infernal 1.1.2 (Eddy and Durbin, 1994) was also used to identify transfer-messenger RNA (tmRNA) using the covariance model built by Hafez et al. (2013).
Phylogenetic analyses of cox11, tufA, ccmA, ccmC, and ccmF
We prepared cox11 and tufA alignments, of which taxon samplings refer to the cox15 alignments analyzed in He et al. (2016). The cox11 and tufA sequences of M. maris, Marophrys sp. SRT127, several eukaryotes, and several alphaproteobacteria were also added. The phylogenetic alignments of ccmA, ccmC, and ccmF analyzed by Nishimura et al. (2020) were updated by adding the M. maris sequences retrieved from the genome data. The sequences were aligned using MAFFT with the L-INS-i option (Katoh and Standley, 2013). Ambiguously aligned positions were manually removed from each alignment. The details of each final alignment for phylogenetic analyses are described in Supplementary Table S3. The final alignments were subjected to maximum likelihood (ML) phylogenetic analyses using IQ-TREE 2.2.0 (Minh et al., 2020), coupled with a 100-replicate standard bootstrap analysis. The substitution models applied in each alignment selected by ModelFinder, based on Bayesian information criteria, are summarized in Supplementary Table S3. Bayesian analysis of each alignment was conducted under the CAT + GTR model using PHYLOBAYES-mpi v. 1.8a (Lartillot and Philippe, 2004; Lartillot et al., 2013). Two chains of Markov chain Monte Carlo (MCMC) were run until maxDiff was less than 0.1. The first quarter of the trees sampled during MCMC was discarded as burn-in, and the consensus tree with branch lengths and Bayesian posterior probabilities (BPPs) was calculated from the remaining three-quarters of the trees. The cycle numbers of MCMC runs and the calculated maxDiff values are summarized in Supplementary Table S3.
Results
Overview of the Microheliella maris mitochondrial genome and mitochondrial genes of interest
The circular-mapping of the mitochondrial genome of Microheliella maris (Mm mt-genome) was found to be 61,620 bp in length (Figure 1) with G + C contents of 28.7% (Table 1). A total of 79 genes were annotated in this genome, namely three ribosomal RNAs (rnl, rns, and rrn5), one RNase-P RNA (rnpB), one tmRNA (ssrA), 53 protein-coding genes, one functionally unassignable ORF, and 21 tRNAs. trnA and trnT were not detected in the Mm mt-genome. The Mm mt-genome possesses rnpB and ssrA, which are very rare genes in the mitochondrial genomes studied to date. rnpB has been found only in some jakobids and restricted species in Fungi, and ssrA is found only in some jakobids and Palpitomonas bilix (Aguileta et al., 2014; Nishimura et al., 2016; Zardoya, 2020). No introns were detected in any of the ORFs. The Mm mt-genome possesses a pair of inverted repeats, 12,850 bp in length, and contains 22 genes (three ribosomal RNAs, seven tRNAs, and 12 protein-coding genes; Figure 1). We could not detect nad10 and ccmB in the Mm mt-genome, which were found in the mitochondrial genome of P. bilix (Pb mt-genome), but detected nad10 transcripts in the RNA-seq data generated by Yazaki et al. (2022). The protein sequence of M. maris nad10 showed an extension at the N-terminus compared to that of P. bilix encoded in its mt-genome. MitoFate (Fukasawa et al., 2015) predicted the N-terminus of M. maris nad10 sequence as a mitochondrial-targeting signal, suggesting that this particular gene is encoded in the nucleus and the gene product is imported post-translationally to the mitochondrion in M. maris.
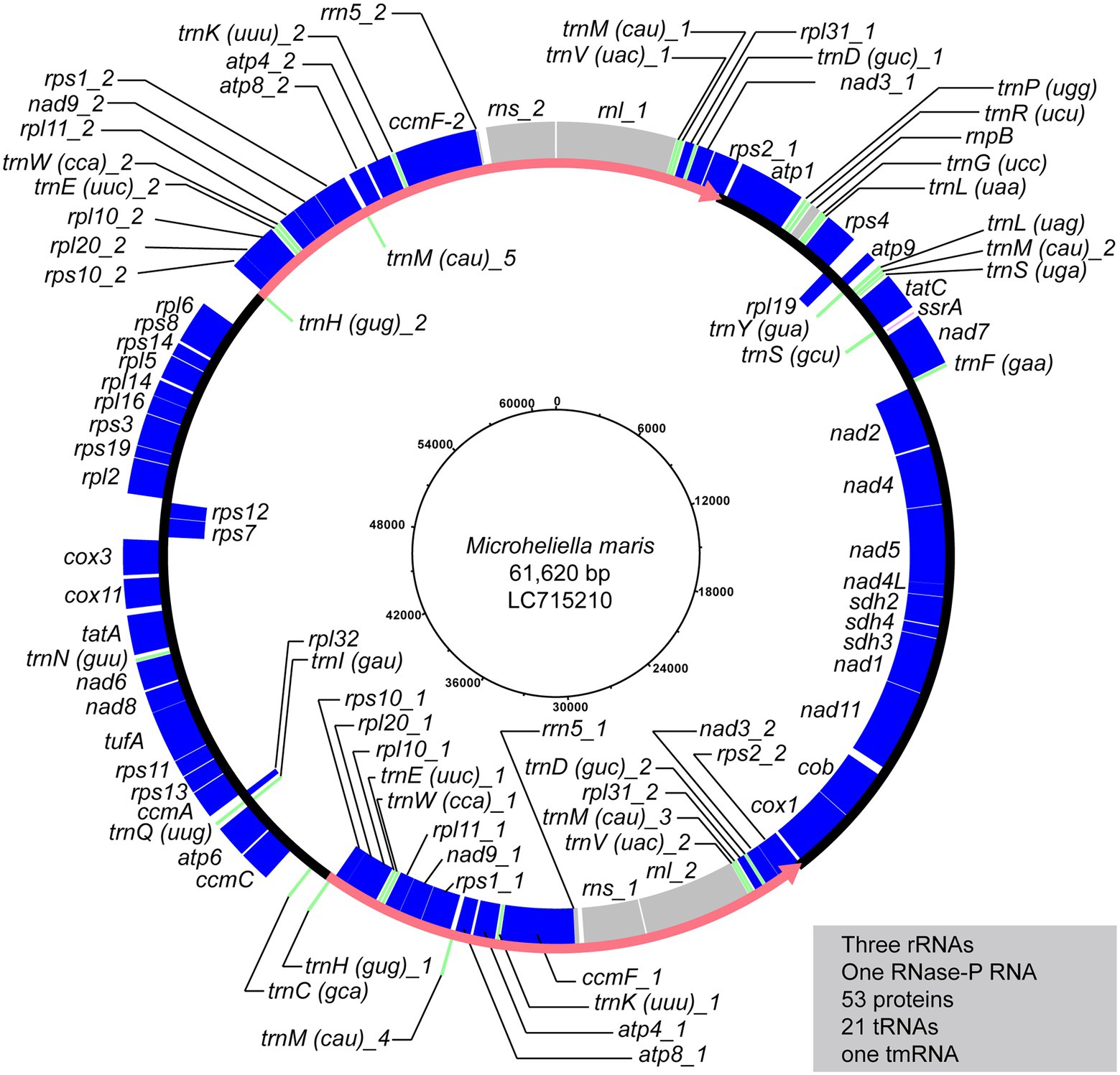
Figure 1. The mitochondrial genome map of Microheliella maris. Protein-coding regions are shown in blue. Transfer RNA genes and other structural RNA genes (i.e., rns, rnl, rrn5, rnpB, and ssrA) are shown in green and gray, respectively. Pink arrows indicate a pair of inverted repeats.
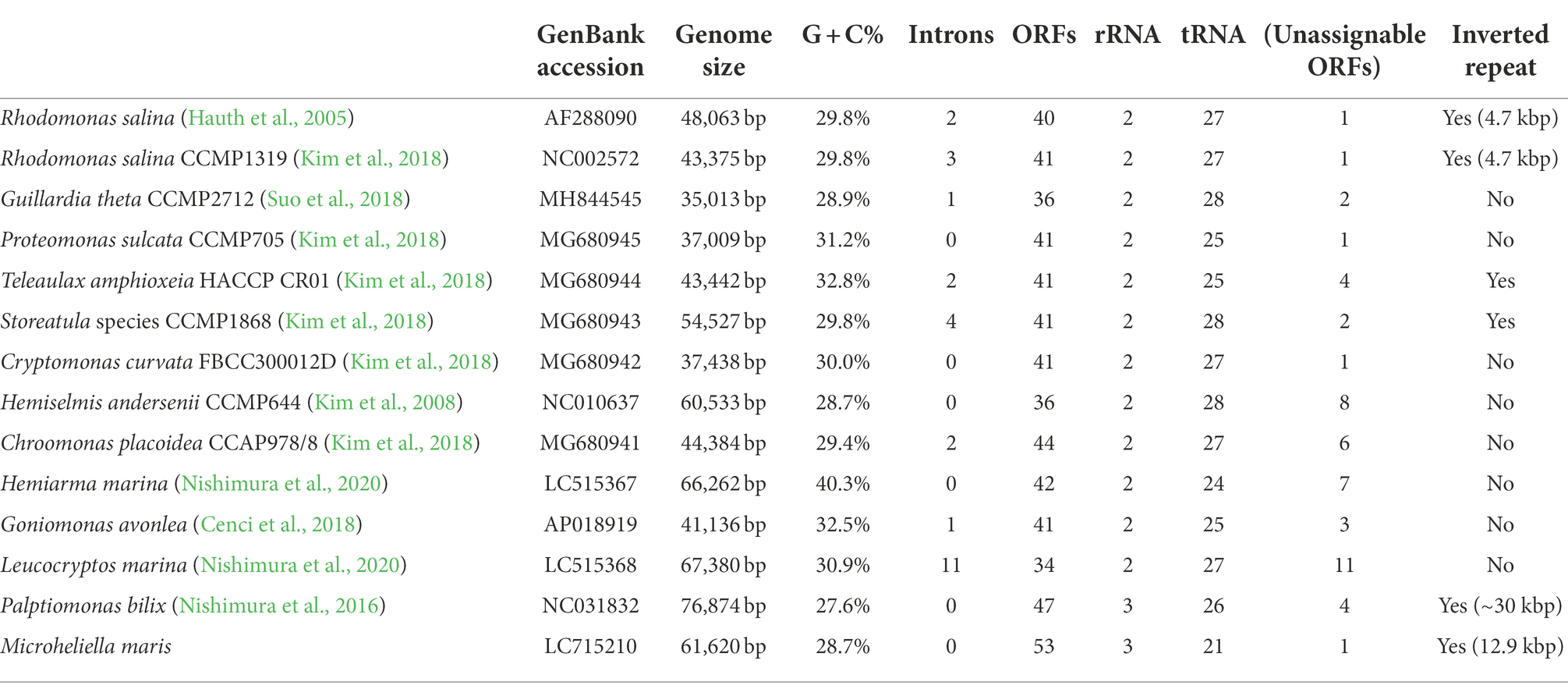
Table 1. Characteristics of the Pancryptista mitochondrial genome.
Prior to this study, the mitochondrial genome-encoded cox11 and tufA were only detected in members of Discoba (jakobids and Tsukubamonas globosa), a centrohelid Marophrys sp. SRT127, and Diphylleia rotans, a member of the CRuMs (only cox11 was detected in the D. rotans mt-genome; Kamikawa et al., 2016; Janouškovec et al., 2017; Nishimura et al., 2019). Hence, this is the second case in Diaphoretickes, following Marophrys, in which the two genes were detected in the mt-genome. As neither cox11 nor tufA have been reported from any other member of the CAM clade, it was unclear whether they were vertically inherited from ancestral eukaryotes or independently acquired from a phylogenetically distinct organism via lateral gene transfer. Therefore, we addressed their origins using phylogenetic analysis. In the cox11 phylogenetic tree, all eukaryotic sequences, including cox11 of M. maris (Mm cox11), formed a single clade. Mm cox11 was monophyletic with the mitochondrion-encoded cox11 of a jakobid, Ophirina amphinema. Unfortunately, owing to the lack of phylogenetic resolution in the cox11 phylogeny, the precise position of Mm cox11 in the eukaryotic clade was unclear (Supplementary Figure S1A). In the tufA phylogenetic tree, the monophyly of eukaryotes was also recovered, and this clade split into nucleus-encoded and mitochondrial genome-encoded sequences, but the monophyly of each subclade was not well supported. The mitochondrial genome-encoded tufA sequences, including tufA of M. maris (Mm tufA), grouped together and the Mm tufA branched with the sequences of two jakobids (Reclinomonas americana and Jakoba libera), although neither of the two relationships mentioned above received high statistical support (Supplementary Figure S1B).
The origin of the mitochondrial genome-encoded cytochrome c maturation system in Cryptista is still under debate (Nishimura et al., 2016, 2020). Therefore, the information on M. maris, which represents the lineage most closely related to Cryptista known to date, is important. We detected three cytochrome c maturation genes (ccmA, ccmC, and ccmF) in the Mm mt-genome; however, we did not detect ccmB or holocytochrome c synthase (HCCS) in either the Mm mt-genome or RNAseq data generated by Yazaki et al. (2022). In the ccmA, ccmC, and ccmF phylogenies, the M. maris sequences were included in the major eukaryotic clades (Supplementary Figures S2A–C). In the ccmA phylogenic tree (Supplementary Figure S2A), the M. maris sequence is monophyletic with the Ancoracysta twista sequences. The ccmC and ccmF phylogenies commonly connected the M. maris sequence with the Hemiarma marina sequence. Nevertheless, none of the three analyses resolved the relationship between M. maris and the Ancoracysta/Hemiarma sequences with high statistical support.
Discussion
The evolution of the mitochondrial genome in Pancryptista
Protein-coding genes
We successfully determined the mitochondrial genome of Microheliella maris (Mm mt-genome), which represents the most basal lineage of Pancryptista (Yazaki et al., 2022). The size of the Mm mt-genome (61.6 kbp) is similar to those of the Palpitomonas bilix mt-genome (Pb mt-genome, 76.9 kbp) and the kathablepharid Lecocryptos marina mt-genome (67.3 kbp), whereas those of goniomonads and cryptophytes ranges from 35.0 to 66.2 kbp (Table 1). Overall, the Mm mt-genome and Pb mt-genome appeared to contain 53 and 47 ORFs, respectively, which were greater than the ORF numbers found in other cyrptists (ranging from 34 to 44; Table 1). If no acquisition of ORF (except those associated with mobile elements) is assumed during the evolution of the pancryptist mt-genomes, the ORF content of the mt-genome of the common ancestor of Pancryptsita should be the union set of those of the mt-genomes of all members of Pancryptista known to date. The putative ORF content in the ancestral pancryptist mt-genome likely resembles that in the Mm mt-genome, which carries eight extra genes compared to the putative ancestral cryptist mt-genome, namely five ribosomal protein genes, sdh2, cox11, and tufA (Figure 2B). On the other hand, nad10 and ccmB are present in the Pb mt-genome (P. bilix is the putative most basal cryptist) but absent in the Mm mt-genome. The M. maris transcriptome data contains transcripts encoding nad10, the N-terminus of which was predicted to function as a mitochondrial-targeting signal. Therefore, nad10 in M. maris is thought to have migrated from the mt-genome to the nucleus. nad10 was not found in the mt-genome of Leucocryptos marina but no evidence for the nucleus-encoded version has not been found in the transcriptome data. We currently consider that nad10 gene was mitochondrion-encoded in the common ancestor of pancryptists but translocated to the nuclear genome on the separate branches leading to M. maris and L. marina after the divergence of pancryptists. As ccmB transcripts were not found in the transcriptome data of M. maris, it is still unclear whether ccmB is (i) truly absent in M. maris or (ii) present but missing from the transcriptome data. If the M. maris ccmB sequence was highly divergent, the homology search may have overlooked the ccmB sequence in the transcriptome data.
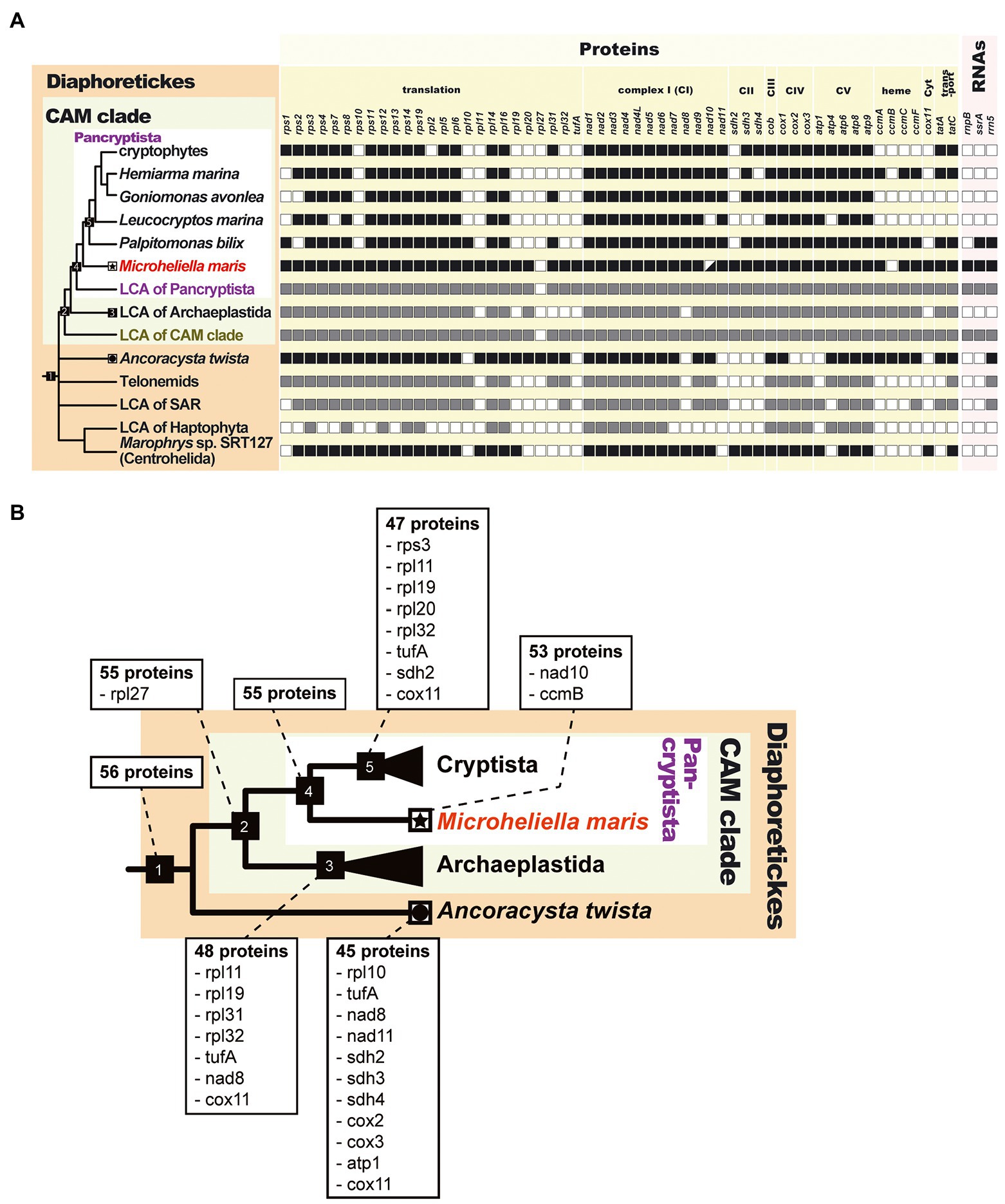
Figure 2. Evolution of mitochondrial genomes in Diaphoretickes. (A) Comparison of the gene contents of the mitochondrial genomes in Diaphoretickes. We only considered the genes containing functionally assignable open reading frames in this comparison. The presence and absence of genes in a mitochondrial genome are shown by closed and open squares, respectively. nad10 was absent in the mitochondrial genome but the corresponding transcript was detected in Microheliella maris (highlighted by a square with lower right half painted in black). The putative presence and absence of genes in the last common ancestors (LCAs) of Pancryptista, Archaeplastida, SAR, and Haptophyta are shown by gray and open squares, respectively. (B) Putative gene loss events during the mitochondrial genome evolution in Diaphoretickes. Only members of Pancryptista, Archaeplastida, and Ancoracysta twista with the second gene-rich mitochondrial genome among the member of Diaphoretickes, are depicted in this figure. For each node of interest, the putative number of genes and the gene/genes lost comparing to the previous node are indicated. Node 1, last common ancestor (LCA) of Diaphoretickes; node 2, LCA of CAM clade; node 3, LCA of Archaeplastida; node 4, LCA of Pancryptista; node 5, LCA of Cryptista. The terminal node for M. maris is labeled by a star. The 11 genes lost on the branch from the LCA of Diaphoretickes to A. twista are also shown in a shaded box.
RNA genes
Here, we discuss putative changes in the content of RNA genes in the pancryptist mt-genomes. As seen in other mt-genomes, the Mm and cryptist mt-genomes possess both small and large subunit rRNA genes (rns and rnl). Intriguingly, Mm and Pb mt-genomes appeared to possess a unique set of RNA genes, namely the 5S rRNA gene (rrn5), which is also present in the mt-genomes of cryptophytes, and tmRNA genes (ssrA). Thus, we assumed that the ancestral pancryptist mt-genome carried rns, rnl, rrn5, and ssrA. After the separation of P. bilix and the common ancestor of kathablephalids, goniomonads, and cryptophytes, the mt-genome of the latter lineage secondarily lost rrn5 and ssrA (Table 1; Figure 2). The mitochondrion-encoded RNAase P RNA gene (rnpB) should have been lost from the ancestral cryptist mt-genome, as rnpB was detected only in the Mm mt-genome, but not in any cryptist mt-genomes. In contrast, the number of tRNA species in the Mm mt-genome is 21, which is smaller than that identified in the cryptist mt genomes. The Mm mt-genome discarded alaninyl and threoninyl tRNAs. We assume that over 28 tRNA species were present in the ancestral pancryptist mt-genome, although a reduction in the tRNA repertory occurred in the Mm mt-genome. Regarding tRNAs, the cryptist mt-genomes typically encodes approximately 27 tRNAs (24–28), whereas the Mm mt-genome encodes only 21 tRNAs (Table 1). Despite no experimental validation, the tRNA, of which genes were not found in the mitochondrial genome, are likely imported from the cytosol, as anticipated for other mitochondria (Gray et al., 1998; Tan et al., 2002; Kolesnikova et al., 2004; Schneider, 2011; Salinas et al., 2012; Lang, 2018). The Mm mt-genome has more protein-coding genes but less tRNA genes than the cryptist mt-genomes, suggesting that protein-coding genes and tRNA genes have been under different evolutionary constraints.
Inverted repeats
Gene synteny is not conserved among the cryptist mt-genomes, and likewise, the Mm mt-genome does not share gene synteny with any of the cryptist mt-genomes. The Mm mt-genome has a pair of inverted repeats, which contain multiple genes, as seen in Pb and several cryptophyte mt-genomes, but not in goniomonad mt-genomes [Table 1; note that short repeats in mt-genomes (e.g., less than 50 bp containing no gene) are not regarded as “inverted repeats” in this study]. However, the inverted repeats found in the pancryptist mt-genomes are not consistent in length or gene content, suggesting that they are not of the same origin. The inverted repeats in the Mm mt-genome are longer than those found in other protists except P. bilix (Pérez-Brocal et al., 2010; Janouškovec et al., 2013; Tikhonenkov et al., 2014; Nishimura et al., 2016). The features of the inverted repeats in the pancryptist mt-genomes suggest that they occurred independently during the evolution of Pancryptista.
Cytochrome c maturation
Previous studies assessing the cryptist mt-genomes have focused on the evolution of the cytochrome c maturation system (Nishimura et al., 2016, 2020). Two types of cytochrome c maturation systems, System I with ccmA-H and System III with holocytochrome c synthase (HCCS), were found in Cryptista, but each cryptist possesses either of these and co-existing cases in a single cryptist has not been reported. Whether each system was vertically inherited in Cryptista or acquired horizontally is not clear, and the evolution of cytochrome c maturation in Cryptista was assumed in several scenarios because of the insufficiency of the phylogenetic signal of the genes associated with cytochrome c maturation. Although the presence/absence of ccmB remains uncertain, ccmA, ccmC, and ccmF were detected in the Mm mt-genome (Figure 2), and HCCS could not be detected in the transcriptome data mentioned above. Thus, in M. maris, cytochrome c maturation is likely to occur via System I. Molecular phylogenetic analyses of ccmA, ccmC, and ccmF, including the M. maris sequences, showed that all M. maris sequences were included within the eukaryotic clade. Unfortunately, the relationship among the eukaryotic sequences in each gene tree was not well resolved, and we could not propose the scenario of the transition of the cytochrome c maturation system in Pancryptista with confidence (Nishimura et al., 2016, 2020).
New insights into mitochondrial genome evolution in Diaphoretickes
A recent phylogenomic analysis suggested that Pancryptista and Archaeplastida are sisters to each other in Diaphoretickes (CAM clade; Yazaki et al., 2022). Combining the gene contents of the previously determined mt-genomes and the Mm mt-genomes, we reconstructed the putative gene contents of the mt-genomes in the common ancestors of the CAM clade and Diaphoretickes. Significantly, the Mm mt-genome appeared to possess six genes that are absent in the mt-genome of Ancoracysta twista, and thus are the most gene-rich among the mt-genomes of Diaphoretickes determined prior to this study. The putative mt-genomes of the common ancestors of the CAM clade and Diaphoretickes were predicted to possess two extra genes (i.e., ccmB and nad10) and three extra genes (i.e., rpl27, ccmB, and nad10), respectively, compared to the Mm mt-genome (Figure 2B). The “gene-richness” of the Mm mt-genome seemingly corresponds to the deep-branching position of M. maris in the tree of Diaphoretickes inferred from a phylogenomic alignment (Yazaki et al., 2022). In future studies, it will be important to sequence the mitochondrial genomes of phylogenetically diverged (particularly deep-branching) members of Diaphoretickes to infer the early evolution of mt-genomes in this assemblage more accurately. For instance, the mt-genomes of Hemimastigophora are intriguing for predicting the gene content of the common ancestor of Diaphoretickes. A phylogenomic analysis revealed no obvious affinity between Hemimastigophora and any of the major lineages in Diaphoretickes, suggesting that Hemimastigophora represents an early branch of Diaphoretickes (Lax et al., 2018). It is certain that a number of diaphoretickes remain unstudied and some of them are of early branching in the tree of Diaphoretickes. Thus, Hemimastigophora, as well as the as-yet undescribed early branching diaphoretickes, potentially possess more gene-rich mt-genomes than M. maris.
After we obtain more mt-genome data from early branching diaphoretickes (see the above discussion), we can achieve a better understanding of the evolution of cox11 and tufA in Diaphoretickes. The mitochondrion-encoded cox11 was found only in Marophrys sp. SRT127 and M. maris among diaphoretickes. Likewise, no mitochondrial genomes of diaphoretickes, except for maris, retain tufA. As both the cox11 and tufA phylogenies were essentially unresolved (Supplementary Figures S1A,B), we cannot discard one of the two scenarios for the punctuated distributions of cox11 and tufA in Diaphoretickes. First, the current distribution of cox11/tufA in Diaphoretickes may have been shaped by the strict vertical inheritance of cox11/tufA from the common ancestor of Diaphoretickes, followed by a (potentially large) number of secondary losses. Alternatively, one can incorporate the lateral transfer of cox11/tufA to explain the punctuated distribution of the two genes of interest in Diaphoretickes. If mitochondrion-encoded cox11 and tufA are identified in additional early branching Diaphoretickes in the future, it will be clear whether these genes were transferred vertically or horizontally.
Data availability statement
The datasets presented in this study can be found in online repositories, Dryad, and accession number(s) can be https://doi.org/10.5061/dryad.8pk0p2nr3.
Author contributions
EY and AY participated in the design of the study, carried out the molecular lab work and data analyses including determination of the mitochondrial genome and phylogenetic analyses, and drafted and revised the manuscript. YN prepared the phylogenetic alignments and drafted the manuscript. TS prepared and assembled the genome-seq data. TH prepared the phylogenetic alignments. YI prepared the phylogenetic alignments and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Japan Society for the Promotion of Science awarded to YI (18KK0203 and 19H03280) and AY (11J04684 and 17K19434).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.1030570/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Phylogenetic trees of cox11 and tufA sequences. Maximum likelihood (ML) trees inferred from the cox11 and tufA alignments are shown in (A,B), respectively. The eukaryotic sequences (and branches) are shown in blue. Archaeal and bacterial branches are colored orange and gray, respectively. The pink shade indicates nuclear-coded sequences and the right blue shade indicates mitochondrial-coded sequences. The sequences identified in the mitochondrial genome of Microheliella maris are shown in red. Only ML bootstrap values/Bayesian posterior probabilities equal to or > 50% / 0.90 are shown.
SUPPLEMENTARY FIGURE S2 | Phylogenetic trees of ccmA, ccmC, and ccmF sequences. Maximum likelihood (ML) trees inferred from ccmA, ccmC, and ccmF alignments are shown in (A-C) respectively. The eukaryotic sequences (and branches) are shown in blue. Archaeal and bacterial branches are colored orange and gray, respectively. Sequences identified in the mitochondrial genomes of Microheliella maris, Palpitomonas bilix, and Hemiarma marina are shown in red. Only ML bootstrap values/Bayesian posterior probabilities equal to or > 50% / 0.90 are shown.
Footnotes
1. ^https://www.ccap.ac.uk/catalogue/strain-1945-1
2. ^http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl
References
Adl, S. M., Bass, D., Lane, C. E., Lukeš, J., Schoch, C. L., Smirnov, A., et al. (2019). Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 66, 4–119. doi: 10.1111/jeu.12691
Aguileta, G., De Vienne, D. M., Ross, O. N., Hood, M. E., Giraud, T., Petit, E., et al. (2014). High variability of mitochondrial gene order among fungi. Genome Biol. Evol. 6, 451–465. doi: 10.1093/GBE/EVU028
Atteia, A., Adrait, A., Brugire, S., Tardif, M., Van Lis, R., Deusch, O., et al. (2009). A proteomic survey of Chlamydomonas reinhardtii mitochondria sheds new light on the metabolic plasticity of the organelle and on the nature of the α-proteobacterial mitochondrial ancestor. Mol. Biol. Evol. 26, 1533–1548. doi: 10.1093/molbev/msp068
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Burger, G., Gray, M. W., Forget, L., and Lang, B. F. (2013). Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol. Evol. 5, 418–438. doi: 10.1093/gbe/evt008
Calvo, S. E., and Mootha, V. K. (2010). The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44. doi: 10.1146/annurev-genom-082509-141720
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/BIOINFORMATICS/BTR703
Cavalier-Smith, T., Chao, E. E., and Lewis, R. (2015). Multiple origins of Heliozoa from flagellate ancestors: new cryptist subphylum Corbihelia, superclass Corbistoma, and monophyly of Haptista, Cryptista, Hacrobia and Chromista. Mol. Phylogenet. Evol. 93, 331–362. doi: 10.1016/j.ympev.2015.07.004
Cavalier-Smith, T., and von der Heyden, S. (2007). Molecular phylogeny, scale evolution and taxonomy of centrohelid heliozoa. Mol. Phylogenet. Evol. 44, 1186–1203. doi: 10.1016/j.ympev.2007.04.019
Cenci, U., Sibbald, S. J., Curtis, B. A., Kamikawa, R., Eme, L., Moog, D., et al. (2018). Nuclear genome sequence of the plastid-lacking cryptomonad Goniomonas avonlea provides insights into the evolution of secondary plastids. BMC Biol. 16:137. doi: 10.1186/s12915-018-0593-5
Eddy, S. R., and Durbin, R. (1994). RNA sequence analysis using covariance models. Nucleic Acids Res. 22, 2079–2088. doi: 10.1093/NAR/22.11.2079
Fan, J., and Lee, R. W. (2002). Mitochondrial genome of the colorless green alga Polytomella parva: two linear DNA molecules with homologous inverted repeat termini. Mol. Biol. Evol. 19, 999–1007. doi: 10.1093/oxfordjournals.molbev.a004180
Fukasawa, Y., Tsuji, J., Fu, S. C., Tomii, K., Horton, P., and Imai, K. (2015). MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol. Cell. Proteomics 14, 1113–1126. doi: 10.1074/mcp.M114.043083
Gawryluk, R. M. R., Chisholm, K. A., Pinto, D. M., and Gray, M. W. (2014). Compositional complexity of the mitochondrial proteome of a unicellular eukaryote (Acanthamoeba castellanii, supergroup Amoebozoa) rivals that of animals, fungi, and plants. J. Proteomics 109, 400–416. doi: 10.1016/j.jprot.2014.07.005
Gray, M. W. (2015). Mosaic nature of the mitochondrial proteome: implications for the origin and evolution of mitochondria. Proc. Natl. Acad. Sci. U. S. A. 112, 10133–10138. doi: 10.1073/pnas.1421379112
Gray, M. W., Burger, G., and Lang, B. F. (2001). The origin and early evolution of mitochondria. Genome Biol. 2, reviews1018.1–reviews1018.5. doi: 10.1186/gb-2001-2-6-reviews1018
Gray, M. W., Lang, B. F., Cedergren, R., Golding, G. B., Lemieux, C., Sankoff, D., et al. (1998). Genome structure and gene content in protist mitochondrial DNAs. Nucleic Acids Res. 26, 865–878. doi: 10.1093/NAR/26.4.865
Hafez, M., Burger, G., Steinberg, S. V., and Lang, B. F. (2013). A second eukaryotic group with mitochondrion-encoded tmRNA: in silico identification and experimental confirmation. RNA Biol. 10, 1117–1124. doi: 10.4161/RNA.25376
Hauth, A. M., Maier, U. G., Lang, B. F., and Burger, G. (2005). The Rhodomonas salina mitochondrial genome: bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res. 33, 4433–4442. doi: 10.1093/NAR/GKI757
He, D., Fu, C.-J., and Baldauf, S. L. (2016). Multiple origins of eukaryotic cox15 suggest horizontal gene transfer from bacteria to jakobid mitochondrial DNA. Mol. Biol. Evol. 33, 122–133. doi: 10.1093/molbev/msv201
Janouškovec, J., Tikhonenkov, D. V., Burki, F., Howe, A. T., Rohwer, F. L., Mylnikov, A. P., et al. (2017). A new lineage of eukaryotes illuminates early mitochondrial genome reduction. Curr. Biol. 27, 3717–3724.e5. doi: 10.1016/j.cub.2017.10.051
Janouškovec, J., Tikhonenkov, D. V., Mikhailov, K. V., Simdyanov, T. G., Aleoshin, V. V., Mylnikov, A. P., et al. (2013). Colponemids represent multiple ancient alveolate lineages. Curr. Biol. 23, 2546–2552. doi: 10.1016/j.cub.2013.10.062
Kamikawa, R., Shiratori, T., Ishida, K. I., Miyashita, H., and Roger, A. J. (2016). Group II intron-mediated trans-splicing in the gene-rich mitochondrial genome of an enigmatic eukaryote, Diphylleia rotans. Genome Biol. Evol. 8, 458–466. doi: 10.1093/GBE/EVW011
Karnkowska, A., Vacek, V., Zubáčová, Z., Treitli, S. C., Petrželková, R., Eme, L., et al. (2016). A eukaryote without a mitochondrial organelle. Curr. Biol. 26, 1274–1284. doi: 10.1016/j.cub.2016.03.053
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, E., Lane, C. E., Curtis, B. A., Kozera, C., Bowman, S., and Archibald, J. M. (2008). Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Gnomics 9:215. doi: 10.1186/1471-2164-9-215
Kim, J. I., Yoon, H. S., Yi, G., Shin, W., and Archibald, J. M. (2018). Comparative mitochondrial genomics of cryptophyte algae: gene shuffling and dynamic mobile genetic elements. BMC Genomics 19, 1–14. doi: 10.1186/S12864-018-4626-9/FIGURES/5
Kolesnikova, O. A., Entelis, N. S., Jacquin-Becker, C., Goltzene, F., Chrzanowska-Lightowlers, Z. M., Lightowlers, R. N., et al. (2004). Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum. Mol. Genet. 13, 2519–2534. doi: 10.1093/HMG/DDH267
Lang, B. F. (2018). “Mitochondrial genomes in fungi,” in Molecular Life Sciences. eds. R. D. Wells, J. S. Bond, J. Klinman, B. S. S. Masters (New York: Springer), 722–728.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lartillot, N., and Philippe, H. (2004). A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21, 1095–1109. doi: 10.1093/molbev/msh112
Lartillot, N., Rodrigue, N., Stubbs, D., and Richer, J. (2013). PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62, 611–615. doi: 10.1093/sysbio/syt022
Lax, G., Eglit, Y., Eme, L., Bertrand, E. M., Roger, A. J., and Simpson, A. G. B. (2018). Hemimastigophora is a novel supra-kingdom-level lineage of eukaryotes. Nature 564, 410–414. doi: 10.1038/s41586-018-0708-8
Lee, C. P., Taylor, N. L., and Millar, A. H. (2013). Recent advances in the composition and heterogeneity of the Arabidopsis mitochondrial proteome. Front. Plant Sci. 4:4. doi: 10.3389/fpls.2013.00004
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/MOLBEV/MSAA015
Nishimura, Y., Kume, K., Sonehara, K., Tanifuji, G., Shiratori, T., Ishida, K., et al. (2020). Mitochondrial genomes of Hemiarma marina and Leucocryptos marina revised the evolution of cytochrome c maturation in Cryptista. Front. Ecol. Evol. 8:140. doi: 10.3389/FEVO.2020.00140
Nishimura, Y., Shiratori, T., Ishida, K., Hashimoto, T., Ohkuma, M., and Inagaki, Y. (2019). Horizontally-acquired genetic elements in the mitochondrial genome of a centrohelid Marophrys sp. SRT127. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-41238-6
Nishimura, Y., Tanifuji, G., Kamikawa, R., Yabuki, A., Hashimoto, T., and Inagaki, Y. (2016). Mitochondrial genome of Palpitomonas bilix: derived genome structure and ancestral system for cytochrome c maturation. Genome Biol. Evol. 8, 3090–3098. doi: 10.1093/gbe/evw217
Panigrahi, A. K., Ogata, Y., Zíková, A., Anupama, A., Dalley, R. A., Acestor, N., et al. (2009). A comprehensive analysis of Trypanosoma brucei mitochondrial proteome. Proteomics 9, 434–450. doi: 10.1002/pmic.200800477
Pérez-Brocal, V., Shahar-Golan, R., and Clark, C. G. (2010). A linear molecule with two large inverted repeats: the mitochondrial genome of the stramenopile Proteromonas lacertae. Genome Biol. Evol. 2, 257–266. doi: 10.1093/GBE/EVQ015
Roger, A. J., Muñoz-Gómez, S. A., and Kamikawa, R. (2017). The origin and diversification of mitochondria. Curr. Biol. 27, R1177–R1192. doi: 10.1016/j.cub.2017.09.015
Salinas, T., Duby, F., Larosa, V., Coosemans, N., Bonnefoy, N., Motte, P., et al. (2012). Co-evolution of mitochondrial tRNA import and codon usage determines translational efficiency in the green alga Chlamydomonas. PLoS Genet. 8:e1002946. doi: 10.1371/JOURNAL.PGEN.1002946
Schneider, A. (2011). Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu. Rev. Biochem. 80, 1033–1053. doi: 10.1146/ANNUREV-BIOCHEM-060109-092838
Smith, D. R. (2015). The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief. Funct. Genomics 15, 47–54. doi: 10.1093/bfgp/elv027
Smith, D. R., and Keeling, P. J. (2015). Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc. Natl. Acad. Sci. 112, 10177–10184. doi: 10.1073/pnas.1422049112
Suo, F., Ma, Y., Manzilamu, Z., and Huang, L. (2018). The complete mitochondrial genome of a transitional form in secondary endosymbiotic Cryptophyte algae Guillardia theta. Strain CCMP2712. Mitochondrial DNA B Resour. 3, 1304–1305. doi: 10.1080/23802359.2018.1535866
Tan, T. H. P., Pach, R., Crausaz, A., Ivens, A., and Schneider, A. (2002). tRNAs in Trypanosoma brucei: genomic organization, expression, and mitochondrial import. Mol. Cell. Biol. 22, 3707–3717. doi: 10.1128/MCB.22.11.3707-3716.2002
Tikhonenkov, D. V., Janouškovec, J., Mylnikov, A. P., Mikhailov, K. V., Simdyanov, T. G., Aleoshin, V. V., et al. (2014). Description of Colponema vietnamica sp. n. and Acavomonas peruviana n. gen. n. sp., two new alveolate phyla (Colponemidia nom. nov. and Acavomonidia nom. nov.) and their contributions to reconstructing the ancestral state of alveolates and eukaryotes. PLoS One 9:e95467. doi: 10.1371/journal.pone.0095467
Wideman, J. G., Monier, A., Rodríguez-Martínez, R., Leonard, G., Cook, E., Poirier, C., et al. (2019). Unexpected mitochondrial genome diversity revealed by targeted single-cell genomics of heterotrophic flagellated protists. Nat. Microbiol. 5, 154–165. doi: 10.1038/s41564-019-0605-4
Yabuki, A., Chao, E. E., Ishida, K.-I., and Cavalier-Smith, T. (2012). Microheliella maris (Microhelida Ord. N.), an ultrastructurally highly distinctive new axopodial protist species and genus, and the unity of phylum Heliozoa. Protist 163, 356–388. doi: 10.1016/j.protis.2011.10.001
Yazaki, E., Yabuki, A., Imaizumi, A., Kume, K., Hashimoto, T., and Inagaki, Y. (2022). The closest lineage of Archaeplastida is revealed by phylogenomics analyses that include Microheliella maris. Open Biol. 12:210376. doi: 10.1098/RSOB.210376
Zachar, I., and Boza, G. (2020). Endosymbiosis before eukaryotes: mitochondrial establishment in protoeukaryotes. Cell. Mol. Life Sci. 77, 3503–3523. doi: 10.1007/s00018-020-03462-6
Keywords: Microheliella maris, Pancryptista, Diaphoretickes, mitochondrial genome, cox11, tufA
Citation: Yazaki E, Yabuki A, Nishimura Y, Shiratori T, Hashimoto T and Inagaki Y (2022) Microheliella maris possesses the most gene-rich mitochondrial genome in Diaphoretickes. Front. Ecol. Evol. 10:1030570. doi: 10.3389/fevo.2022.1030570
Edited by:
Xiaofan Zhou, South China Agricultural University, ChinaReviewed by:
Shinichiro Maruyama, Tohoku University, JapanDing He, Technical University of Denmark, Denmark
Copyright © 2022 Yazaki, Yabuki, Nishimura, Shiratori, Hashimoto and Inagaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Euki Yazaki, ZXVraTg3QGdtYWlsLmNvbQ==; Akinori Yabuki, eWFidWtpYUBqYW1zdGVjLmdvLmpw
†These authors have contributed equally to this work