 Michael Tessler
Michael Tessler Spencer C. Galen4
Spencer C. Galen4 Rob DeSalle
Rob DeSalle Bernd Schierwater
Bernd Schierwater
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Ecol. Evol. , 08 December 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.1016412
This article is part of the Research Topic Insights in Phylogenetics, Phylogenomics, and Systematics: 2022 View all 6 articles
Many known evolutionary lineages have yet to be described formally due to a lack of traditional morphological characters. This is true for genetically distinctive groups within the amoeboid Placozoa animals, the protists in ponds, and the bacteria that cover nearly everything. These taxonomic tabula rasae, or blank slates, are problematic; without names, communication is hampered and other scientific progress is slowed. We suggest that the morphology of molecules be used to help alleviate this issue. Molecules, such as proteins, have structure. Proteins are even visualizable with X-ray crystallography, albeit more easily detected by and easier to work with using genomic sequencing. Given their structured nature, we believe they should not be considered as anything less than traditional morphology. Protein-coding gene content (presence/absence) can also be used easily with genomic sequences, and is a convenient binary character set. With molecular morphology, we believe that each taxonomic tabula rasa can be solved.
It is exceedingly hard for humans, whether laypeople or scientists, to communicate even modestly well about Earth’s biodiversity without commonly agreed upon names for species, groups of species, or sub-lineages within a species (Yoon, 2010). While humanity has yet to discover a perfect way to name these bins of organisms, biologists have been surprisingly consistent with their chosen mode of biodiversity classification. Linnaean taxonomy, currently spanning from Domain through binomials (genus and species), has proved durable (Schuh, 2003). The system does have some issues, such as the fact that exact cut offs between taxonomic ranks within this system are often arbitrary; one taxonomist’s genus, for example, is another’s family. Not shockingly, this has (for better or for worse) led to numerous name changes over time (Vences et al., 2013). Alternative systems such as PhyloCode (de Queiroz and Cantino, 2020) seek to improve upon Linnaeus’s system, but these alternative approaches are not broadly accepted. This is likely because these systems have their own complexities and imperfections (Nixon et al., 2003). Therefore, a system birthed in the 1700s persists as the standardized backbone for naming newly discovered species, as well as the more inclusive ranks generally called higher taxa.
Despite a consistent framework for naming groups of organisms, many diverse and distinctive evolutionary lineages have never made their way into a formalized taxonomy, largely due to a lack of distinct visualizable morphological features. We, the authors of this paper, refer to these lineages as taxonomic tabula rasae or blank slates—and we consider such lineages to present a big problem. Again, as human scientists, we can only efficiently study, talk about, or conserve those lineages that we apply a name onto for easy discourse. If one mentions reptiles or Reptilia, English speakers or scientists, respectively, will know what is being discussed. Fewer people know what Placozoa are, but many invertebrate systematists can tell you it is phylum of very simply structured animals that lack several traits that are found across Metazoa (Eitel et al., 2013; Schierwater et al., 2021). Some might even tell you it is a highly genetically-diverse and ancient lineage that has few named species or higher taxonomic ranks. Over the last decade, this group’s organization has largely relied on provisional monikers with the word “clade” in their name. These monikers were based on the phylogeny of lineages within Placozoa. However, when we mention the group called Clade VI within Placozoa, only a handful of people on the planet would even know where to look up what that meant. While we do not expect most people to become experts on Placozoa, we believe it should become a group that can be easily communicated about so that research and understanding within this phylum may more properly flourish. We believe it is time to change this lack of easily communicated ideas, not just for Placozoa, but for all taxonomic tabula rasae: groups of organisms that, while often diverse, are poorly classified.
Almost always, taxonomic tabula rasae lack names due to a paucity of easily human-identified features. For example, placozoans basically look like miniscule, shape shifting disks—essentially they are the pancake of the animals. For a taxonomist looking through a microscope, even a powerful one, there are not many characters to compare and differentiate the genetically known lineages within this phylum (Eitel et al., 2013).
The same is true for bacteria—as most look like rods, cones, or spirals—but again this does not even scratch the surface of the genetic or functional diversity in this multi-billion-year-old domain. Currently there are over 20,000 named bacterial and archaeal species and over 3,400 named genera; not too comprehensive though when one considers the total number of species of Archaea and Bacteria is estimated to be two to four orders of magnitude larger than this (Oren, 2004; Pallen et al., 2021). For most microbes, we would not even know 20%–90% of the diversity had it not been for the recent advent of environmental DNA sequencing (Tedersoo et al., 2014; Thompson et al., 2017; Lloyd et al., 2018). Accordingly, many microbial groups have spotty taxonomy outside of the lineages that are associated with or impact humans or human habitats. All of this also holds for protists. For instance, while the phylum Apicomplexa contains many well-studied pathogens of humans, such as the malaria parasites, it has been estimated that this group alone may contain tens of millions of species (Larsen et al., 2017)—this undoubtedly represents numerous unnamed yet biologically important lineages. Even within malaria parasites, which are among the best known lineages of protists, there is still need for major taxonomic revision (Galen et al., 2018). Similarly, database coverage is lower for bacteria in more remote habitats, making naming even more challenging in these areas (Tessler et al., 2017b). Bacteria, Archaea, Fungi, protists, nematodes, or anything that is microscopic (even tiny animals) or reduced (e.g., many parasites) tend to have far from complete taxonomies. To be clear, some parts of the taxonomy within each of these lineages are well worked out, but many parts are not, and we believe these fit our “blank slate” designation.
How have scientists dealt with the problem of taxonomy for taxa that lack morphological distinction? Many take the simplest solution: ignore the problem. Microbial ecologists are excellent biodiversity-oriented researchers, and they frequently skirt the issue with great success. While they often name broad groups (e.g., bacterial phyla), they do not usually give formal names to species (Tessler et al., 2017a); instead they opt for dataset driven groups, such as operational taxonomic units or amplicon sequence variants (Prodan et al., 2020). In these cases, each binned lineage is only specific to the dataset and the methods used to analyze it. It works remarkably well for the purposes of most microbial ecologists. It is a massively successful solution to a problem (lack of easily namable organisms) that could have easily stifled this flourishing field. But this lack of taxonomic resolution is not a perfect long-term solution even for microbial ecology, as it makes it much harder to realize when a species or an unnamed broader lineage keeps popping up as important across independent datasets and studies. We believe systematists need to apply taxonomic knowhow, phylogenetic mastery, and any other skills they might have to help speed up the rate of naming the diversity of visually unknowable lineages.
Of course, we are not the first systematists to care about naming lineages within morphologically plain groups of organisms. On and off, a variety of traits have been used to help break through the difficulties of naming these groups. Non-visual characters have led to rapid advances in some groups, such as the chemical differences being used as characters for lichen taxa (Lumbsch, 1998). Secondary metabolites in plants (e.g., non-protein amino acids) have also been considered as potential characters for taxonomy (Wink, 2003). Ecological preferences, while being somewhat malleable, have also been suggested as characters for certain taxonomic problems (Padial et al., 2010). Cytotaxonomy, typically the use of karyotype with taxonomy, has been used numerous times for plants and animals (Guerra, 2012; Kongim et al., 2015), and, more recently, genome architecture has been proposed as a significant evolutionary feature that can be used roughly like traditional morphology. For instance, nuclear and mitochondrial genome architecture were used to more confidently name the second and third formally classified species in Placozoa (Eitel et al., 2018; Osigus et al., 2019).
Unfortunately, progress has been slow—piecemeal at best. With so many under-classified tabula rasa groups, any solution must speed the pace if we wish to make significant progress.
We suggest that morphology is still the key to taxonomic descriptions. However, we believe there is a general fallacy in most taxonomists’ understanding of morphology, with an undue split between it and molecules (Hillis, 1987), which has resulted in only a subset of characters being frequently considered for taxonomic purposes.
Morphology is, in essence, the study of organisms’ structures (Zangerl, 1948). Yet, due to human sensory biases, taxonomists tend to circumscribe “structures” to the set of features in an organism that they can easily perceive without much mental abstraction. The number of vertebrae in fishes, for example, can clearly and easily be used as a countable morphological division (Ward and Brainerd, 2007). Taxonomists can harness this type of character with ease. Even gestalt has its place in morphological identifications (think bird watching) and is an influence on taxonomists when they produce classifications, although it is typically not mentioned as a principal character. However, characters already go far beyond this, including auditory (Angulo and Reichle, 2008) and chemical (Lumbsch, 1998) realms, to name a few. Many times characters like these may only be detectable with specialized equipment or assays; gross morphology itself often requires tools from basic light microscopy to electron micrographs or even computed tomography (Tessler et al., 2016; De Carle et al., 2022), all which go much further than basic human perception. So, we ask, why do taxonomists stop here?
Here, we propose that morphology should extend far more into molecular structures (Figure 1). As mentioned above, one compelling way to describe species within tabula rasae taxa is to look for genome architecture. This has been true for our aforementioned placozoans (Eitel et al., 2018). Genome architecture may well set a gold standard for describing the previously indescribable. Still, it is cumbersome and expensive for whole genomes (less so for mitochondrial genomes), both in terms of sequencing cost and bioinformatic resources. There can also be methodological difficulty of obtaining entire genomes from microbes that live within an organism, such as the previously mentioned malaria blood parasites (Videvall, 2019). We therefore believe molecular morphology should at times harness, but ultimately not be beholden to this high bar.
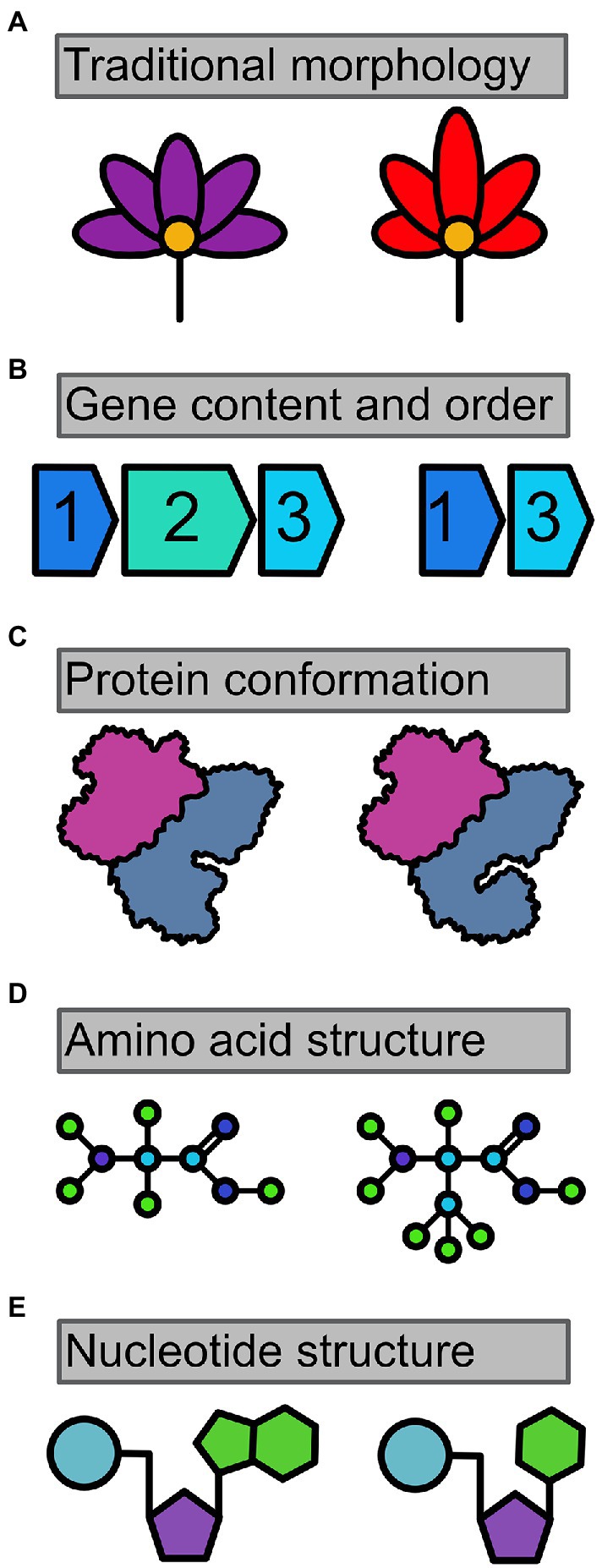
Figure 1. Molecules have morphology too. Traditionally, morphology has been limited to structures that are easily visualizable by eye or microscopy, such as the differences in (A) flower color and petal dimensions. However, morphology can be found at a number of levels when examining molecular data. (B) Gene content, gene order, and synteny are strong characters, as are karyotypes. (C) Protein conformation is three-dimensional, and easily modeled for comparisons. Lastly, even (D) amino acids and (E) nucleotides have clear differences in physical structure, much like traditional morphology.
At their core, all molecular features are structural—after all, structure is how biochemists have isolated and identified these components. Adenine and guanine may both be considered purine nucleobases, but they certainly would not be confused for one another structurally. The same is true for each amino acid, many of which also have impacts on protein structure due to features such as varying degrees of hydrophobicity (Rudinger, 1976). The presence of most functioning genes also leads to a rather clear product: protein. A number of such proteins have even been characterized since 1934, when X-ray crystallography began to make it possible to visualize these macromolecules (Giegé, 2013). Combine traditional X-ray work with protein modeling, and suddenly the shape, structures, and binding sites can become clear visually for a broad swath of proteins (Waterhouse et al., 2018). Even more proteins have been assayed for their functionality. Protein functionality is often as distinctive as morphology, such as for leech anticoagulants that are often highly distinctive for species or lineages (Iwama et al., 2021, 2022).
Even single nucleotides can be used for species delineation (DeSalle et al., 2005), although this remains controversial among systematists. However, using more nucleotides, at least one hundred (and likely many more) species have been named based principally on DNA (Renner, 2016). At least one higher taxon (a family of mosses) has also been named using DNA only (Shaw et al., 2010). Still, the argument for nucleotides as characters has not focused on morphology, which we again point out that it is. Instead the argument has focused more on genotype as being sufficient and perfectly biologically relevant to be used as a type of character for classification purposes, so long as it is sufficiently consistent within a hypothesized lineage.
So, as a practical measure, at what level should this molecular morphology be harnessed for creating enduring taxonomies for tabula rasae lineages? If all molecules are structural and thus within the purview of morphology, then the answer is easy: nucleotides. While we believe this to be true at some level, we also know that one of the special and beneficial features of morphology is that it takes genotype and processes it out as phenotype. Furthermore, morphological phenotype is often (but far from exclusively) the aggregate of multiple alleles; a much higher bar than a single genotypic character. Accordingly, molecular morphology might be best to be at minimum one step removed from the simple genetics of an organism. Amino acids, again structural, are indeed one step removed; they exist as the simple output of the instructions given by codons, triplicates of nucleotides within a genetic reading frame. We believe this again to be a fair position, but that it is unlikely to be accepted by the general community as it can be too highly malleable within some genes for most taxonomists’ tastes, varying too often among individuals.
Protein can likely provide a more stable molecular morphology standard. Proteins are separated by a few levels from base genetics, and they can be well characterized physically when needed (again, think X-ray crystallography and modeling). Much like DNA, protein structures can vary or be conserved across species (Undheim et al., 2016), and we certainly suggest this be used within molecular morphology. However, we also suggest that gene content —the presence or absence of protein coding genes—be used as a relatively easy (as far as genomics work goes) way to efficiently sort out taxonomies. Yes, it requires serious sequencing efforts, but for many groups like bacteria genomic sequences are readily available.
Gene content and protein variability can still be challenging. Orthology has various definitions and real complexity (Gabaldón and Koonin, 2013), and, like species concepts (Wheeler and Meier, 2000), providing a simple definition is difficult. While we do not think this will be solved anytime soon, we do believe that using the gene content of multiple protein coding genes will help to remove the biases dealt through orthology philosophy and algorithms. (We would similarly advocate for multiple morphological characters, whenever possible, for any data type.) Furthermore, we point out that homology was one of the most fought over topics in systematics prior to the emerging genomic era (Wagner, 1989); that is to say, the issue we face for gene orthology still remains problematic for morphological data.
There are other advantages outside of classification for molecular morphology. Some of these characters could be considered for adding to phylogenomic matrices. As we have shown, even a few dozen morphological characters can be of importance for important recalcitrant nodes, such as determining the sister to all other Metazoa (Neumann et al., 2021).
Molecular morphology can also be well integrated into a philosophy that is commonly employed by systematists when focusing on taxonomy: multiple lines of evidence. For instance, using molecules in their raw form, such as nucleotides, along with morphology or geography has been a popular tactic (DeSalle et al., 2005). Substituting traditional morphology with molecular morphology could also work as long as the focus was on something like gene content, while the raw molecular data focused on stretches of nucleotides shared across species.
Here, we will give a brief walk through of how molecular morphology might be incorporated into making a taxonomy for a tabula rasa lineage. Most importantly, we are indeed 100% stating that molecular morphology should be used in the diagnosis for species descriptions. Of course, other data would be wonderful, as they can add color and are more practical for field practitioners. Still, this extra data for visually similar taxa could be something like geography or host, which we do not suggest should be used as principal characters but can be very useful for working with given lineages when coupled with molecular morphology.
So, let us take an example of Placozoa: a phylum that persisted with only one described species for a century. While two more species have subsequently been described (Eitel et al., 2018; Osigus et al., 2019), the progress has been slow and costly. Furthermore, one of them principally relied on genome architecture (Eitel et al., 2018), likely the highest quality form of molecular morphology. Still, it has been estimated that there are potentially hundreds of species, with many lineages already clearly identified with 16S data (Eitel and Schierwater, 2010). When assessing genome assemblies for many of these lineages, we find that lineages often vary by hundreds of orthologs that are uniquely present or absent. Tens, let alone hundreds, of characters seems like more than enough to define higher taxa. Given this, members of our group published a backbone taxonomy for Placozoa, naming classes through a new species using uniquely present genes (Tessler et al., 2022). As an example of this, we present Figure 2, which highlights the 600 orthologous genes that are uniquely present for the main clade of Placozoa as well as the 76 that are uniquely present for the sister lineage. The large quantity of genes present in one clade, but not the other, gave confidence to name these as different classes (despite there being only one obvious morphological difference). Furthermore, functional groups of genes also varied, such as the GPCRs that are highlighted. The decision was also congruent with molecularly-calibrated divergence times. Within the morphologically invariant main clade of Placozoa (blue in the figure), we were able to similarly use the quantity of uniquely present and absent orthologs to name a new species, genus, families, and orders.
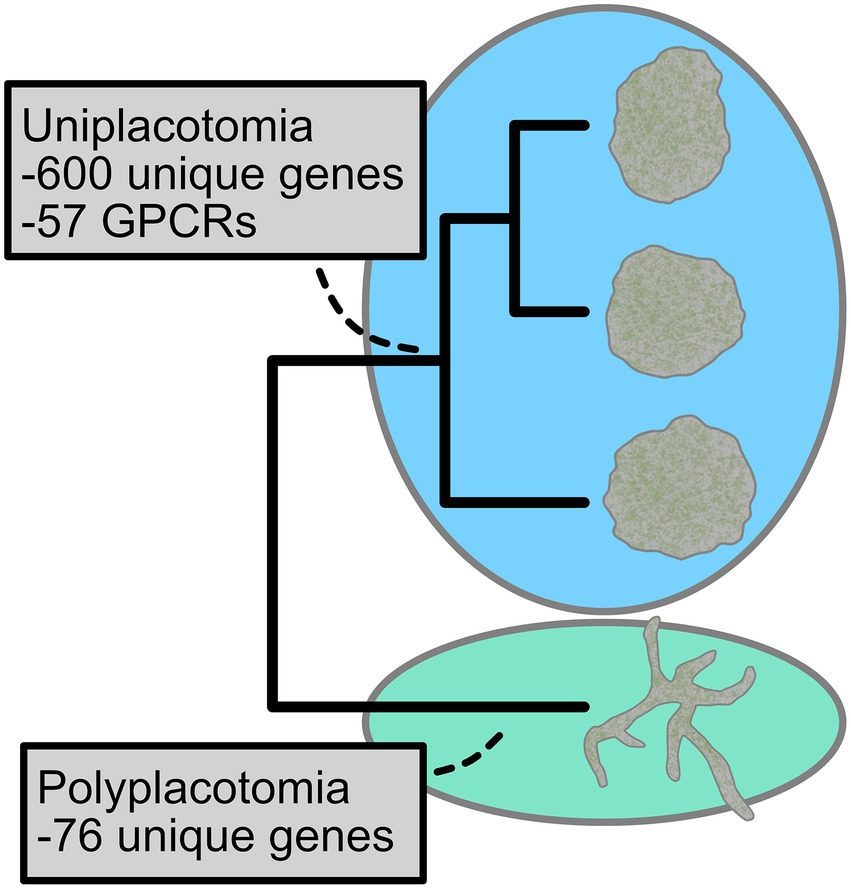
Figure 2. An example of molecular morphology in practice. Nearly all lineages of Placozoa look roughly like miniature pancakes (blue circle). Polyplacotoma mediterranea (green circle) is the only species within the phylum that is clearly morphologically distinct. Based on this single morphological trait difference, it would be easy to mistake Placozoa as a non-diverse group. However, this is not the case. While we arbitrarily depict three lineages in blue, there are at least two dozen cryptic species (with many more presumed to exist) and they are highly divergent genetically. To make a backbone higher taxonomy for Placozoa, our recent study used molecular morphology (via gene content) to clarify the size of the difference between lineages (Tessler et al., 2022). As 600 uniquely present genes were found for the main clade of Placozoa and 76 uniquely present genes were found for P. mediterranea, the two lineages were classified at the class level (which was also supported by a time-calibrated phylogeny). The uniquely present genes included functionally relevant genes, such as the 57 GPRs labeled above. Uniquely present, in this case, means orthologs that were only found in those clades.
Other molecular morphological characters like protein structure could just as easily come into play for naming species or higher taxa. For instance, molecular morphology (using protein modeling and morphometrics) has recently been used to define synapomorphies for molecularly identified clades; this is likely, in the author’s words, “the first report of a synapomorphy defined from a molecule’s shape in the study of arthropod phylogenetics” (Santibáñez-López et al., 2018). Hopefully this is just the beginning of this use of protein models. Again, adding in ecology or geography can help where applicable, but many not always be of use. For instance, placozoans function similarly (at least in the lab where most observations occur) and often have broad, overlapping ranges. Still there are some differences that can be added to help tease taxa apart.
This approach will require a large degree of expertise, as is always the case for taxonomy. However, we envision phylogenomicists helping with this role, moving from principally reconstructing relationships towards applying their data.
While interesting ideas in taxonomic automation are also being put forth for microbes (Pallen et al., 2021), for now, we suggest caution on that front. Automation has also been proposed based on DNA barcodes over the last few decades, but it failed to catch on in a broad way and has been argued against (Hubert and Hanner, 2015). Furthermore, automated approaches can be prone to the foibles of the methods and models used in the automated analysis, making it a major problem if thousands or a million taxa are named at once. For now, traditional taxonomy with expert interpretations continues to remain the most enduring system, so we suggest it remains the focus for taxonomic tabula rasae.
Overall, we believe the time to act on molecular morphology is now, as sequencing technologies and bioinformatics have matured. Whatever the exact level of molecular morphology taxonomists use to help alleviate the taxonomic tabula rasae problem is not of major concern to us. Taxonomists—as they have for centuries—will be the ones to decide when and where a cutoff is most appropriate. It will probably vary by lineage, or taxonomist, as is the case for most commonly used morphological features.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
We thank those taxonomists that have toiled and those that continue to toil with the classification of evolutionarily complex yet morphologically simple biological lineages. We also thank those, such as Jairo Arroyave, who have kindly listened to us about molecular morphology over the years.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Angulo, A., and Reichle, S. (2008). Acoustic signals, species diagnosis, and species concepts: the case of a new cryptic species of Leptodactylus (Amphibia, Anura, Leptodactylidae) from the Chapare region, Bolivia. Zool. J. Linnean Soc. 152, 59–77. doi: 10.1111/j.1096-3642.2007.00338.x
de Carle, D. B., Gajda, Ł., Bielecki, A., Cios, S., Cichocka, J. M., Golden, H. E., et al. (2022). Recent evolution of ancient Arctic leech relatives: systematics of Acanthobdellida. Zool. J. Linnean Soc. 196, 149–168. doi: 10.1093/zoolinnean/zlac006
de Queiroz, K., and Cantino, P. D. (2020). International Code of Phylogenetic Nomenclature (PhyloCode). Boca Raton, FL: CRC Press.
DeSalle, R., Egan, M. G., and Siddall, M. (2005). The unholy trinity: taxonomy, species delimitation and DNA barcoding. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 1905–1916. doi: 10.1098/rstb.2005.1722
Eitel, M., Francis, W. R., Varoqueaux, F., Daraspe, J., Osigus, H.-J., Krebs, S., et al. (2018). Comparative genomics and the nature of placozoan species. PLoS Biol. 16:e2005359. doi: 10.1371/journal.pbio.2005359
Eitel, M., Osigus, H.-J., DeSalle, R., and Schierwater, B. (2013). Global diversity of the Placozoa. PLoS One 8:e57131. doi: 10.1371/journal.pone.0057131
Eitel, M., and Schierwater, B. (2010). The phylogeography of the Placozoa suggests a taxon-rich phylum in tropical and subtropical waters. Mol. Ecol. 19, 2315–2327. doi: 10.1111/j.1365-294X.2010.04617.x
Gabaldón, T., and Koonin, E. V. (2013). Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 14, 360–366. doi: 10.1038/nrg3456
Galen, S. C., Borner, J., Martinsen, E. S., Schaer, J., Austin, C. C., West, C. J., et al. (2018). The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. R. Soc. Open Sci. 5:171780. doi: 10.1098/rsos.171780
Giegé, R. (2013). A historical perspective on protein crystallization from 1840 to the present day. FEBS J. 280, 6456–6497. doi: 10.1111/febs.12580
Guerra, M. (2012). Cytotaxonomy: the end of childhood. Plant Biosyst. 146, 703–710. doi: 10.1080/11263504.2012.717973
Hillis, D. M. (1987). Molecular versus morphological approaches to systematics. Annu. Rev. Ecol. Syst. 18, 23–42. doi: 10.1146/annurev.es.18.110187.000323
Hubert, N., and Hanner, R. (2015). DNA barcoding, species delineation and taxonomy: a historical perspective. DNA Barcodes 3, 44–58. doi: 10.1515/dna-2015-0006
Iwama, R. E., Tessler, M., and Kvist, S. (2022). Leech anticoagulants are ancestral and likely to be multifunctional. Zool. J. Linnean Soc. 196, 137–148. doi: 10.1093/zoolinnean/zlab126
Iwama, R. E., Tessler, M., Siddall, M. E., and Kvist, S. (2021). The origin and evolution of antistasin-like proteins in leeches (Hirudinida, Clitellata). Genome Biol. Evol. 13:evaa242. doi: 10.1093/gbe/evaa242
Kongim, B., Sutcharit, C., and Panha, S. (2015). Cytotaxonomy of unionid freshwater mussels (Unionoida, Unionidae) from northeastern Thailand with description of a new species. Zookeys 514, 93–110. doi: 10.3897/zookeys.514.8977
Larsen, B. B., Miller, E. C., Rhodes, M. K., and Wiens, J. J. (2017). Inordinate fondness multiplied and redistributed: the number of species on earth and the new pie of life. Q. Rev. Biol. 92, 229–265. doi: 10.1086/693564
Lloyd, K. G., Steen, A. D., Ladau, J., Yin, J., and Crosby, L. (2018). Phylogenetically novel uncultured microbial cells dominate earth microbiomes. mSystems 3:e00055-18. doi: 10.1128/mSystems.00055-18
Lumbsch, H. T. (1998). The use of metabolic data in lichenology at the species and subspecific levels. Lichenologist 30, 357–367. doi: 10.1006/lich.1998.0147
Neumann, J. S., Desalle, R., Narechania, A., Schierwater, B., and Tessler, M. (2021). Morphological characters can strongly influence early animal relationships inferred from phylogenomic data sets. Syst. Biol. 70, 360–375. doi: 10.1093/sysbio/syaa038
Nixon, K. C., Carpenter, J. M., and Stevenson, D. W. (2003). The PhyloCode is fatally flawed, and the “Linnaean” system can easily be fixed. Bot. Rev. 69, 111–120. doi: 10.1663/0006-8101(2003)069[0111:TPIFFA]2.0.CO;2
Oren, A. (2004). Prokaryote diversity and taxonomy: current status and future challenges. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 359, 623–638. doi: 10.1098/rstb.2003.1458
Osigus, H.-J., Rolfes, S., Herzog, R., Kamm, K., and Schierwater, B. (2019). Polyplacotoma mediterranea is a new ramified placozoan species. Curr. Biol. 29, R148–R149. doi: 10.1016/j.cub.2019.01.068
Padial, J. M., Miralles, A., De la Riva, I., and Vences, M. (2010). The integrative future of taxonomy. Front. Zool. 7:16. doi: 10.1186/1742-9994-7-16
Pallen, M. J., Telatin, A., and Oren, A. (2021). The next million names for archaea and bacteria. Trends Microbiol. 29, 289–298. doi: 10.1016/j.tim.2020.10.009
Prodan, A., Tremaroli, V., Brolin, H., Zwinderman, A. H., Nieuwdorp, M., and Levin, E. (2020). Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One 15:e0227434. doi: 10.1371/journal.pone.0227434
Renner, S. S. (2016). A return to Linnaeus’s focus on diagnosis, not description: the use of DNA characters in the formal naming of species. Syst. Biol. 65, 1085–1095. doi: 10.1093/sysbio/syw032
Rudinger, J. (1976). “Characteristics of the amino acids as components of a peptide hormone sequence,” in Peptide Hormones. ed. A. Parson (London, United Kingdom: Biological Council, Palgrave).
Santibáñez-López, C. E., Kriebel, R., Ballesteros, J. A., Rush, N., Witter, Z., Williams, J., et al. (2018). Integration of phylogenomics and molecular modeling reveals lineage-specific diversification of toxins in scorpions. PeerJ 6:e5902. doi: 10.7717/peerj.5902
Schierwater, B., Osigus, H.-J., Bergmann, T., Blackstone, N. W., Hadrys, H., Hauslage, J., et al. (2021). The enigmatic Placozoa part 1: exploring evolutionary controversies and poor ecological knowledge. BioEssays 43:e2100080. doi: 10.1002/bies.202100080
Schuh, R. T. (2003). The Linnaean system and its 250-year persistence. Bot. Rev. 69, 59–78. doi: 10.1663/0006-8101(2003)069[0059,tlsaiy]2.0.co;2
Shaw, A. J., Cox, C. J., Buck, W. R., Devos, N., Buchanan, A. M., Cave, L., et al. (2010). Newly resolved relationships in an early land plant lineage: Bryophyta class Sphagnopsida (peat mosses). Am. J. Bot. 97, 1511–1531. doi: 10.3732/ajb.1000055
Tedersoo, L., Bahram, M., Põlme, S., Kõljalg, U., Yorou, N. S., Wijesundera, R., et al. (2014). Fungal biogeography. Global diversity and geography of soil fungi. Science 346:1256688. doi: 10.1126/science.1256688
Tessler, M., Barrio, A., and Borda, E. (2016). Description of a soft-bodied invertebrate with microcomputed tomography and revision of the genus Chtonobdella (Hirudinea: Haemadipsidae). Zoologica 45, 552–565. doi: 10.1111/zsc.12165
Tessler, M., Brugler, M. R., DeSalle, R., Hersch, R., Velho, L. F. M., Segovia, B. T., et al. (2017a). A global eDNA comparison of freshwater bacterioplankton assemblages focusing on large-river floodplain lakes of Brazil. Microb. Ecol. 73, 61–74. doi: 10.1007/s00248-016-0834-5
Tessler, M., Neumann, J. S., Afshinnekoo, E., Pineda, M., Hersch, R., Velho, L. F. M., et al. (2017b). Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 7:6589. doi: 10.1038/s41598-017-06665-3
Tessler, M., Neumann, J. S., Kamm, K., Osigus, H.-J., Eshel, G., Narechania, A., et al. (2022). Phylogenomics and the first higher taxonomy of Placozoa, an ancient and enigmatic animal phylum. Front. Ecol. Evol. doi: 10.3389/fevo.2022.1016357
Thompson, L. R., Sanders, J. G., McDonald, D., Amir, A., Ladau, J., Locey, K. J., et al. (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463. doi: 10.1038/nature24621
Undheim, E. A. B., Mobli, M., and King, G. F. (2016). Toxin structures as evolutionary tools: using conserved 3D folds to study the evolution of rapidly evolving peptides. BioEssays 38, 539–548. doi: 10.1002/bies.201500165
Vences, M., Guayasamin, J. M., Miralles, A., and de La Riva, I. (2013). To name or not to name: criteria to promote economy of change in Linnaean classification schemes. Zootaxa 3636:201. doi: 10.11646/zootaxa.3636.2.1
Videvall, E. (2019). Genomic advances in avian malaria research. Trends Parasitol. 35, 254–266. doi: 10.1016/j.pt.2018.12.005
Wagner, G. P. (1989). The origin of morphological characters and the biological basis of homology. Evolution 43, 1157–1171. doi: 10.1111/j.1558-5646.1989.tb02566.x
Ward, A. B., and Brainerd, E. L. (2007). Evolution of axial patterning in elongate fishes. Biol. J. Linn. Soc. Lond. 90, 97–116. doi: 10.1111/j.1095-8312.2007.00714.x
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi: 10.1093/nar/gky427
Wheeler, Q. D., and Meier, R. (2000). Species Concepts and Phylogenetic Theory: A Debate. New York, NY: Columbia University Press.
Wink, M. (2003). Evolution of secondary metabolites from an ecological and molecular phylogenetic perspective. Phytochemistry 64, 3–19. doi: 10.1016/S0031-9422(03)00300-5
Yoon, C. K. (2010). Naming Nature: The Clash Between Instinct and Science. New York, NY: W. W. Norton & Company.
Keywords: molecular morphology, systematics, tabula rasa, taxonomy, biodiversity
Citation: Tessler M, Galen SC, DeSalle R and Schierwater B (2022) Let’s end taxonomic blank slates with molecular morphology. Front. Ecol. Evol. 10:1016412. doi: 10.3389/fevo.2022.1016412
Edited by:
Rodney L. Honeycutt, Pepperdine University, United StatesReviewed by:
Amy Baird, University of Houston-Downtown, United StatesCopyright © 2022 Tessler, Galen, DeSalle and Schierwater. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Tessler, bXRlc3NsZXJAc2ZjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.