 Lan Jiang
Lan Jiang Yu Chen3,4
Yu Chen3,4 Yunpeng Cao
Yunpeng Cao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 06 December 2021
Sec. Ecophysiology
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.801490
WRKY transcription factors participate in various regulation processes at different developmental stages in higher plants. Here, 98 WRKY I genes were identified in seven Rosaceae species. The WRKY I genes are highly enriched in some subgroups and are selectively expanded in Chinese pear [Pyrus bretschneideri (P. bretschneideri)] and apple [Malus domestica (M. domestica)]. By searching for intra-species gene microsynteny, we found the majority of chromosomal segments for WRKY I-containing segments in both P. bretschneideri and M. domestica genomes, while paired segments were hardly identified in the other five genomes. Furthermore, we analyzed the environmental selection pressure of duplicated WRKY I gene pairs, which indicated that the strong purifying selection for WRKY domains may contribute to the stability of its structure and function. The expression patterns of duplication PbWRKY genes revealed that functional redundancy for some of these genes was derived from common ancestry and neo-functionalization or sub-functionalization for some of them. This study traces the evolution of WRKY I genes in Rosaceae genomes and lays the foundation for functional studies of these genes in the future. Our results also show that the rates of gene loss and gain in different Rosaceae genomes are far from equilibrium.
Rosaceae species, such as black raspberry [Rubus occidentalis (R. occidentalis)], strawberry [Fragaria vesca (F. vesca)], peach [Prunus persica (P. persica)], sweet cherry [Prunus avium (P. avium)], Chinese plum [Prunus mume (P. mume)], Chinese pear [Pyrus bretschneideri (P. bretschneideri)], and apple [Malus domestica (M. domestica)], constitute the most important resources for commercial fruits. The botanical family Rosaceae can be divided into 91 genera and includes approximately 4,828 species distributed throughout the world (Christenhusz and Byng, 2016). According to previous studies, the genomes of F. vesca (Shulaev et al., 2011) and R. occidentalis (Vanburen et al., 2016; Jibran et al., 2018) (X = 7), P. persica (Verde et al., 2013), P. avium (Shirasawa et al., 2017), and P. mume (Zhang et al., 2012) (X = 8), and P. bretschneideri (Wu et al., 2013) and M. domestica (Velasco et al., 2010) (X = 17) are evolved from a common ancestor, which contained nine pairs of chromosomes (X = 9) (Illa et al., 2011). The molecular marker was used to study the evolutionary mechanism involved in the speciation process, and later more detailed studies were carried out by sequencing the genomes (Vilanova et al., 2008). These studies revealed that the Rosaceae genomes have undergone a series of chromosome translocations, fusions, and inversions during evolution (Velasco et al., 2010; Shulaev et al., 2011; Wu et al., 2013; Cao et al., 2016c). Additionally, high collinearity between Prunus and Fragaria, and high microsynteny between Malus and Pyrus were described (Vilanova et al., 2008). The above studies suggested that the Rosaceae genomes have a complex evolutionary history (Velasco et al., 2010; Shulaev et al., 2011; Wu et al., 2013; Cao et al., 2016c). Although there is knowledge of these genera of genome evolution, a paralogous or orthologous comparison is excluded. Therefore, a subgroup of the WRKY gene family was selected to analyze specific evolutionary relationships of Rosaceae-related species.
WRKY is one of the largest family of transcription factors in higher plants and has been associated with plant growth and development, abiotic and biotic stress, and others. The WRKY gene family was divided into three subgroups (i.e., types I, II, and III) based on both the number of WRKY domains and the type of zinc finger structure present in the proteins (Eulgem et al., 2000). The type III WRKY proteins have a WRKY domain (WRKYGQK) and C2HC zinc finger motif, the type II proteins contain a WRKY domain and C2H2 zinc finger motif, while type I proteins (i.e., the WRKY I subgroup) possess two WRKY domains (Eulgem et al., 2000; Wu et al., 2005). Since the cloning of the first WRKY gene, SPF1 from Ipomoea batatas (Ishiguro and Nakamura, 1994), a large number of WRKY genes have been experimentally identified from several plant species, such as sweet kumquat [Fortunella crassifolia (F. crassifolia)] (Gong et al., 2015), soybean (Glycine max) (Zhou et al., 2008), rice [Oryza sativa (O. sativa)] (Qiu et al., 2004), poplar [Populus trichocarpa (P. trichocarpa)] (Duan et al., 2015), and Arabidopsis [Arabidopsis thaliana (A. thaliana)] (Dong et al., 2003). Additionally, large-scale systematic analyses of WRKY gene family have been studied for A. thaliana (Wang et al., 2011), O. sativa (Ross et al., 2007), P. bretschneideri (Qiao et al., 2015), P. trichocarpa (He et al., 2012), and Cucumis sativus (Ling et al., 2011).
The previous studies have shown that the members of WRKY subgroup I appear to be functionally different from each other (Ishiguro and Nakamura, 1994). According to comparative genomics, evolutionary analysis of the WRKY subgroup has been reported in some model plants (Coulthart and Singh, 1988), but the specific evolutionary relationships of the WRKY subgroup are still unknown in Rosaceae genomes. To improve our understanding of the WRKY subgroup I in the sequenced genomes of Rosaceae family—F. vesca, R. occidentalis, P. persica, P. avium, P. mume, P. bretschneideri, and M. domestica—we describe the gene duplication and evolution of WRKY I genes in these seven species. We also investigate the putative orthologous or paralogous gene models. In the current study, 12 orthologous clusters and extensive synteny remains in the homologous regions were identified in Rosaceae genomes. Remarkably, we found that most WRKY genes could be traced to ancient whole genome duplication (WGD) or recent WGD, which is shared by both P. bretschneideri and M. domestica genomes. However, the gene loss and duplication rates of the WRKY gene family differ greatly among the separate lineages of Rosaceae. The current study provides the first systematic analysis and may help to push the function of the WRKY gene from one lineage to another.
The protein, cDNA, and genome sequences of seven Rosaceae were obtained from the respective genome sequence sites as follows: both P. avium and R. occidentalis from Genome Database for Rosaceae1, F. vesca from Phytozome database2, P. persica from Joint Genome Institute3, P. mume from GigaDB Dataset4, P. bretschneideri from Nanjing Agricultural University5, and M. domestica from Plant Genome Duplication Database6. The Hidden Markov Model (HMM) profiles of the WRKY-domain (PF03106) were extracted from Pfam HMM library and were used to query to search these seven genomes by HMMER 3.0 software (e-value < 0.001) (Mistry et al., 2013). The overlapping genes were manually removed, and then the Pfam database (Finn et al., 2013), InterPro database (Zdobnov and Apweiler, 2001). and SMART software (Letunic et al., 2012) were used to verify the proteins that have two WRKY domains (Figure 1), namely, WRKY I proteins.
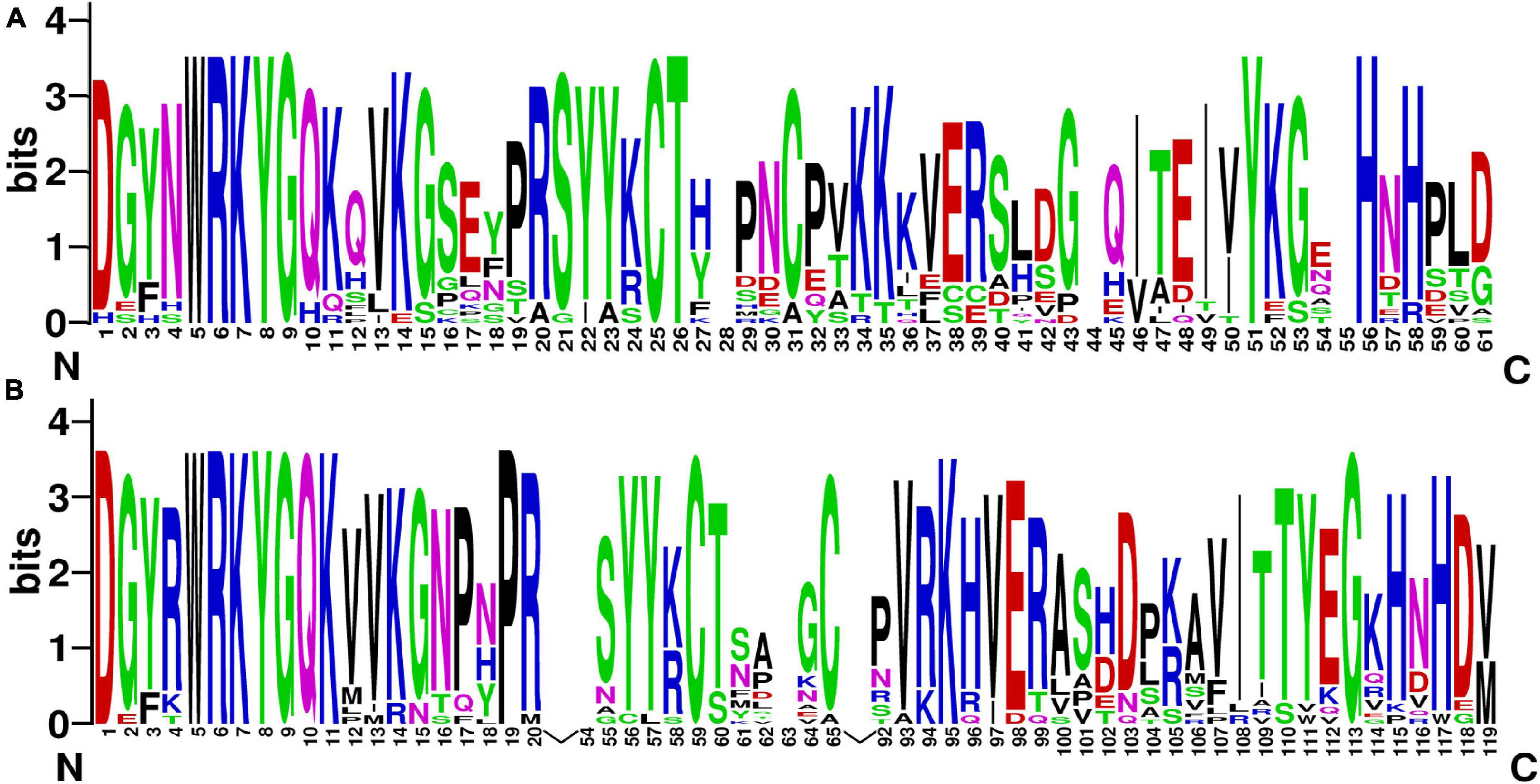
Figure 1. The sequence logos of the first WRKY domain (A) and the second WRKY domain (B). These logos were based on multiple alignments of all WRKY proteins. The bit score indicates the information content for each position in the sequence.
The gene structures of WRKY I genes were determined by the alignment of its genomic DNA sequence with the coding sequence. The conserved motif structures of WRKY I proteins were generated by Local MEME software (Bailey et al., 2015) with the following parameters: -protein, -nmotifs 20, -minw 5, and –maxw 200. The TBtools software7 was used to visualize the conserved motifs and gene structures. The WebLogo8 was used to generate the sequence logos of the first WRKY domain (a) and the second WRKY domain.
To further understand their evolutionary history, all 98 WRKY I genes from these seven Rosaceae species were used in phylogenetic analysis. The MAFFT software was used to perform the WRKY I gene alignments with default parameters (Katoh and Standley, 2013). The IQ-TREE software was used to build the maximum-likelihood (ML) phylogenetic tree with a bootstrap test for 1,000 replicates, an Shimodaira Hasegawa-approximate likelihood ratio test (SH-aLRT) for 1,000 random addition replicates (Nguyen et al., 2014), and the best nucleotide substitution model (HKY + F + I + G4) was also chosen by this software independently. The FigTree software and iTOL online were used to display the phylogenetic trees (Letunic and Bork, 2006). The Rosaceae species tree was built according to the phylogeny relationships from the NCBI taxonomy database (Federhen, 2012). To estimate the number of WRKY I genes in the ancestral species, the Notung software was used to calculate gene losses and gene gains during evolution (Chen et al., 2000).
To classify and expand WRKY I gene family in F. vesca, R. occidentalis, P. persica, P. avium, P. mume, P. bretschneideri, and M. domestica, we examined the chromosome locations of all members of this family. Subsequently, a strategy similar to that of the previously published manuscripts was executed to detect large-scale duplication events (Maher et al., 2006; Cao et al., 2016c). If two WRKY I genes were duplicated by large-scale duplication events (e.g., WGD), the two regions containing these two WRKY I genes were expected to retain some other gene duplicates, which show relatively high sequence similarity at the amino acid level (Maher et al., 2006; Jing et al., 2016). First, all WRKY I genes from F. vesca, R. occidentalis, P. persica, P. avium, P. mume, P. bretschneideri, and M. domestica were employed as the original anchor points. Next, to detect duplicated genes between two independent regions, we compared all protein-coding sequences 100-kb upstream and downstream of each anchor point by pairwise the Basic Local Alignment Search Tool (BLASTP) analysis, based on the previously published articles (Deleu et al., 2007; Cao et al., 2016c). Finally, the total number of protein-coding genes flanking the anchor point (i.e., WRKY I gene) that contained the best non-self-match (cut-off e-value = 10–10) was counted (Sato et al., 2008). According to previously published articles (Coulthart and Singh, 1988; Cao et al., 2016c), such two regions, which contained four or more gene pairs with syntenic relationships, were considered to have evolved from large-scale duplication events.
The orthologous relationships were detected in the proteomes of seven Rosaceae species using the OrthoMCL software (Li et al., 2003) with e-value < 1e–10. The ratio between non-synonymous (Ka) and synonymous (Ks) was calculated using the DnaSP software (Librado and Rozas, 2009) on protein-coding genes to detect selective pressure acting on WRKY I genes. The ratios of Ka/Ks > 1, < 1, and = 1 indicate the positive selection, negative selection, and neutral selection, respectively. To further understand the particular amino acid sites under positive selection, we carried out a sliding window analysis of Ka/Ks ratios with parameters: step size, 9 bp; window size, 150 bp (Cao et al., 2016a).
To detect the expression patterns of WRKY I genes in seven Rosaceae species, we retrieved the RNA-seq data from Sequence Read Archive (SRA) and European Bioinformatics Institute (EBI) database, and accession numbers for these RNA-seq data are presented in the “Data Availability Statement” section. The SRA toolkit software was used to decompress raw data into the FASTQ format (Sherry, 2012). The HISAT2 software was used to map each dataset to its corresponding template genome with default parameters (Pertea et al., 2016; Cao et al., 2019). The Cufflinks v2.2.1 software was used to calculate the fragments per kilobase of exon per million reads (FPKM) with the default parameters (Trapnell et al., 2012), which were then log2-transformed. The TBtools software (see text footnote 7) was used to produce the heat map.
This experiment was performed in 2017 on pear trees (P. bretschneideri cv. Dangshan Su’) planted in the experimental orchard of the farm in Dangshan, Anhui, China. Three biological replicates of fruit samples were collected from spring to summer 2017 during P. bretschneideri fruit developmental stages. The first sampling was performed on 18 April (15 days). Subsequently, remaining samples were taken on 13 May (39 days after flowering, DAF), 20 May (47 DAF), 29 May (55 DAF), 5 June (63 DAF), 21 June (79 DAF), 14 July (102 DAF), and 28 August (145 DAF) in 2017 after flowering (DAF). We used the guanidine thiocyanate extraction method to extract the total RNAs by RNA Plus (Takara, Dalian). The first-strand cDNAs were synthesized from DNaseI-treated total RNA using reverse transcriptase (Takara, Dalian) based on the instructions of the manufacturer. The CFX96 Touch™Real-Time PCR Detection System (Bio-Rad) was used to conduct the quantitative real-time PCR (qRT-PCR) experiment. We designed the primers of PbWRKY genes for qRT-PCR using Primer Premier 6.0 software (Applied Biosystems, Takara, Dalian). The relative expression level was calculated as a 2–ΔΔCt method and normalized against the P. bretschneideri Actin gene (NCBI ID AF386514) as described previously (Livak and Schmittgen, 2001; Cao et al., 2016a). For each sample, we conducted three technical replicates in our study. The SPSS v24 was used to detect the analysis of statistical significance.
The experiments did not involve endangered or protected species. No specific permits were required for these locations/activities because the P. bretschneideri genes used in this study were obtained from the tissue culture room of Anhui Agricultural University.
To identify all full-length WRKY I proteins in seven Rosaceae species, HMM searches were carried out against the annotated genomes of F. vesca, R. occidentalis, P. persica, P. avium, P. mume, P. bretschneideri, and M. domestica. A total of 6, 9, 10, 11, and 13 WRKY I genes were detected in P. avium, P. persica, P. mume, R. occidentalis, and F. vesca, respectively, while 20 and 28 WRKY I genes were identified in P. bretschneideri and M. domestica, respectively (Table 1). Subsequently, we detected the exact chromosomal locations of all WRKY I genes through a blast search of the genome sequences using TBtools software. The distribution of WRKY I genes among the chromosomes in these seven Rosaceae genomes was random and unbalanced (Supplementary Figure 1). For example, in the M. domestica genome, WRKY I genes are mainly distributed in chromosome 12, followed by chromosomes 3 (4) and 6 (4), and the others are located among chromosomes 2, 4, 11, 13, 14, 15, 16, and 17. However, in the P. bretschneideri genome, the WRKY I genes were scattered across chromosomes 2, 3, 5, 6, 8, 9, 11, 12, 13, 14, and 17.
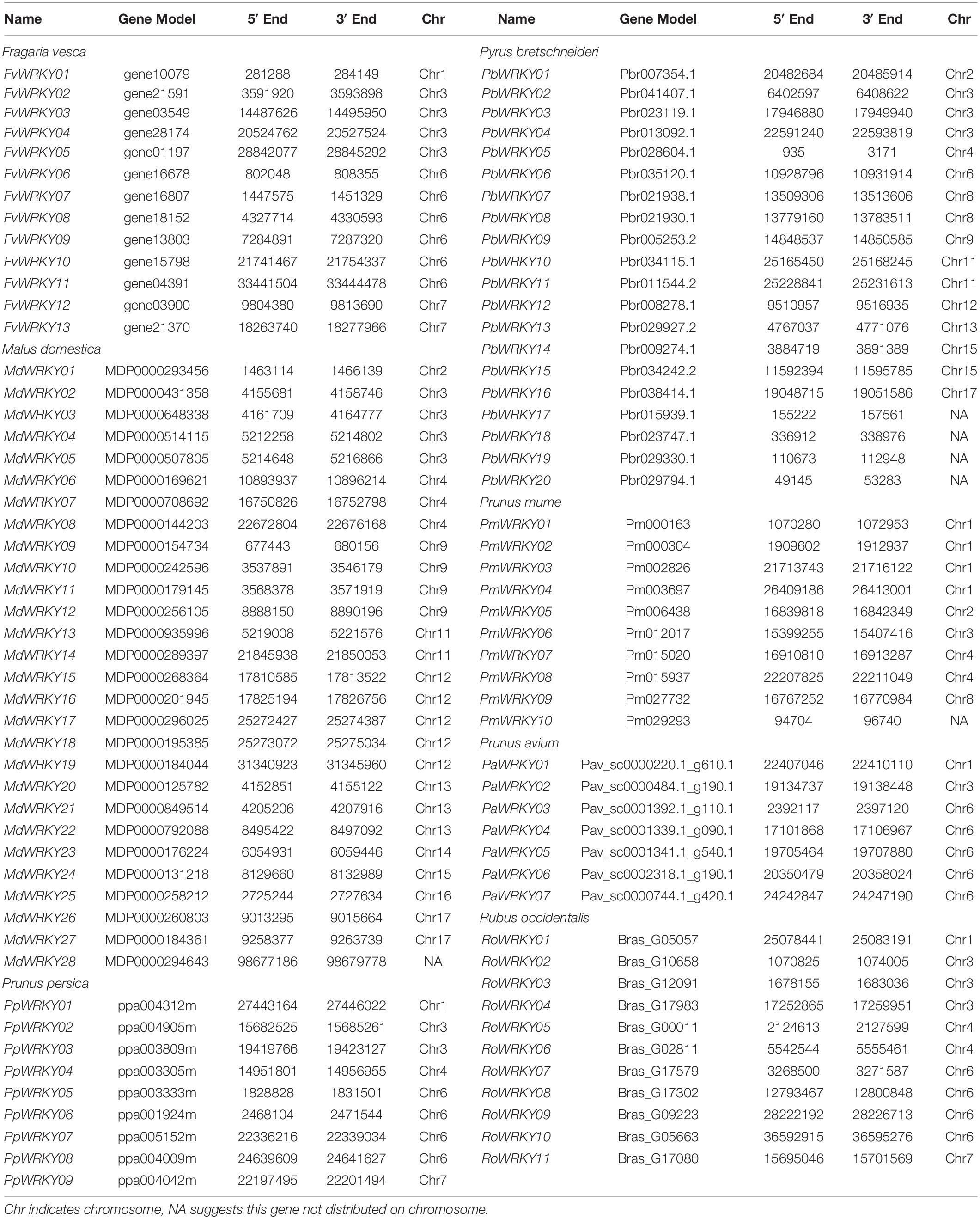
Table 1. The detailed information of seven Rosaceae species WRKY I family members.
To investigate the WRKY I genes from seven Rosaceae species, we constructed a phylogenetic tree based on the nucleotide sequence alignment (Figure 2). This tree was built according to comparisons of the full-length nucleotide sequence between the Rosaceae species by the ML method with 1,000 bootstrap replicates. The ML tree was divided into 12 subgroups (A–M), which belong to four groups (i.e., class I–IV) with higher bootstrap values. Subgroup J contained the fewest WRKY I gene members (3), but the subgroup I had the most gene members (11), followed by subgroup K (10) and subgroup M (10).
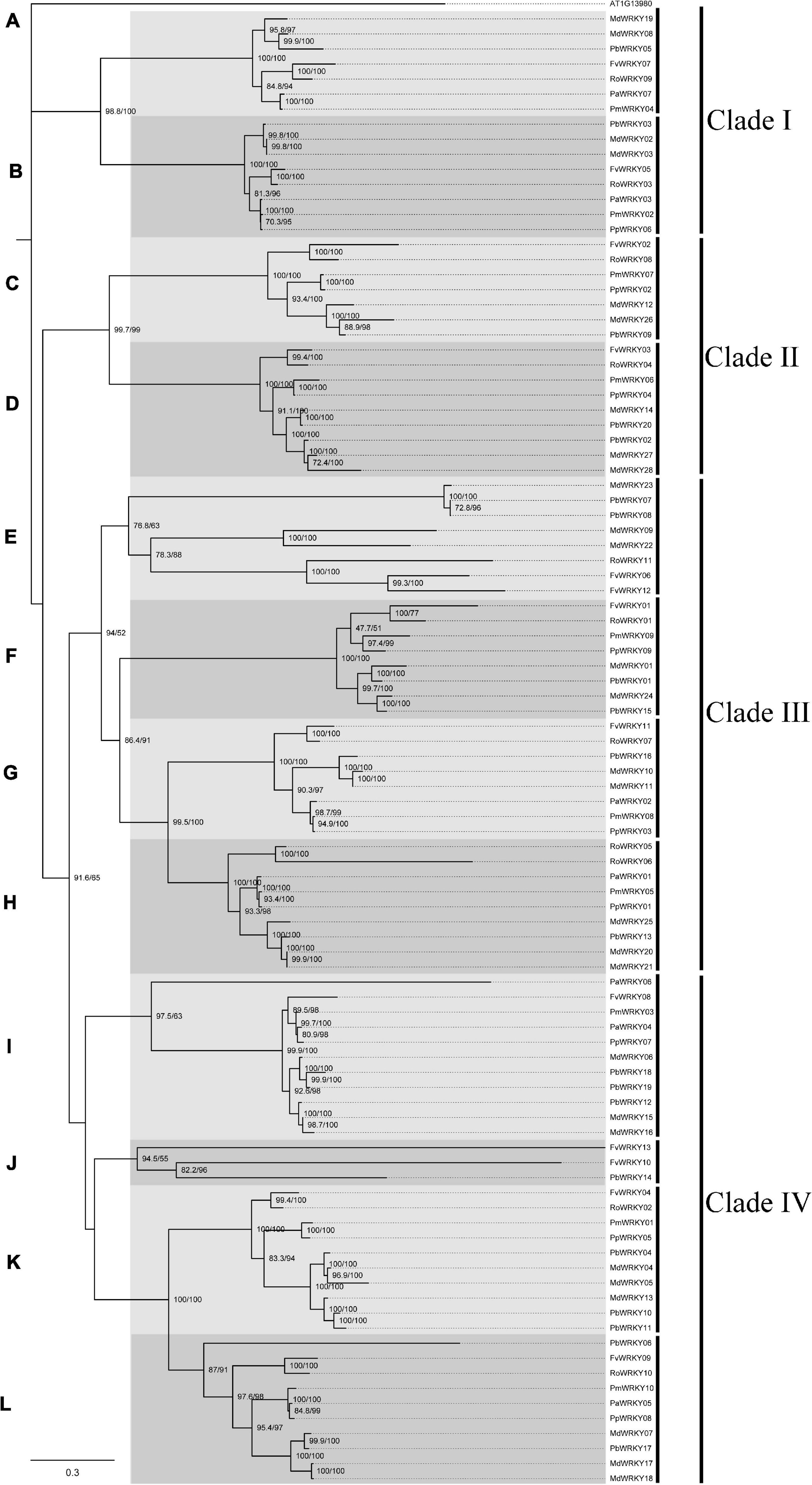
Figure 2. Maximum-Likelihood tree based on sequences of WRKY I genes from P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri, and M. domestica. The IQ-tree software was used to perform maximum likelihood (ML) phylogenetic analysis with a bootstrap test for 1,000 replicates and an SH-aLRT test for 1,000 random addition replicates. Based on the evolutionary distances and bootstrap support, this tree was divided into 12 subgroups. The tree is rooted to the A. thaliana WRKY I gene (AT1G13980).
The short branch lengths at the tips of the clades confirmed the strong conservation of nucleotide or amino acid sequence, indicating that the evolutionary relationship between these gene members was close. In each subgroup, branches with the lack of WRKY I gene in some species might be due to gene losses, whereas more than one WRKY I genes from the same species were likely to have experienced gene duplication events. In most subgroups, WRKY I genes from the P. bretschneideri and M. domestica were more abundant than WRKY I genes from the other Rosaceae species (Figure 2). Remarkably, we found that in almost every subgroup, at least one extra copy of the WRKY I genes from P. bretschneideri and M. domestica was present. In addition, the WRKY I gene members of different species in each subgroup might be due to evolving from a common ancestral gene by the divergence of the lineage. According to the ML tree, the WRKY I genes from P. bretschneideri and M. domestica have shown close pairwise relationships; the genes of F. vesca and R. occidentalis, and the genes of P. persica, P. avium and P. mume were also the most similar based on genetic distance, which consistent with the species tree of the seven Rosaceae plants (Figure 3C).
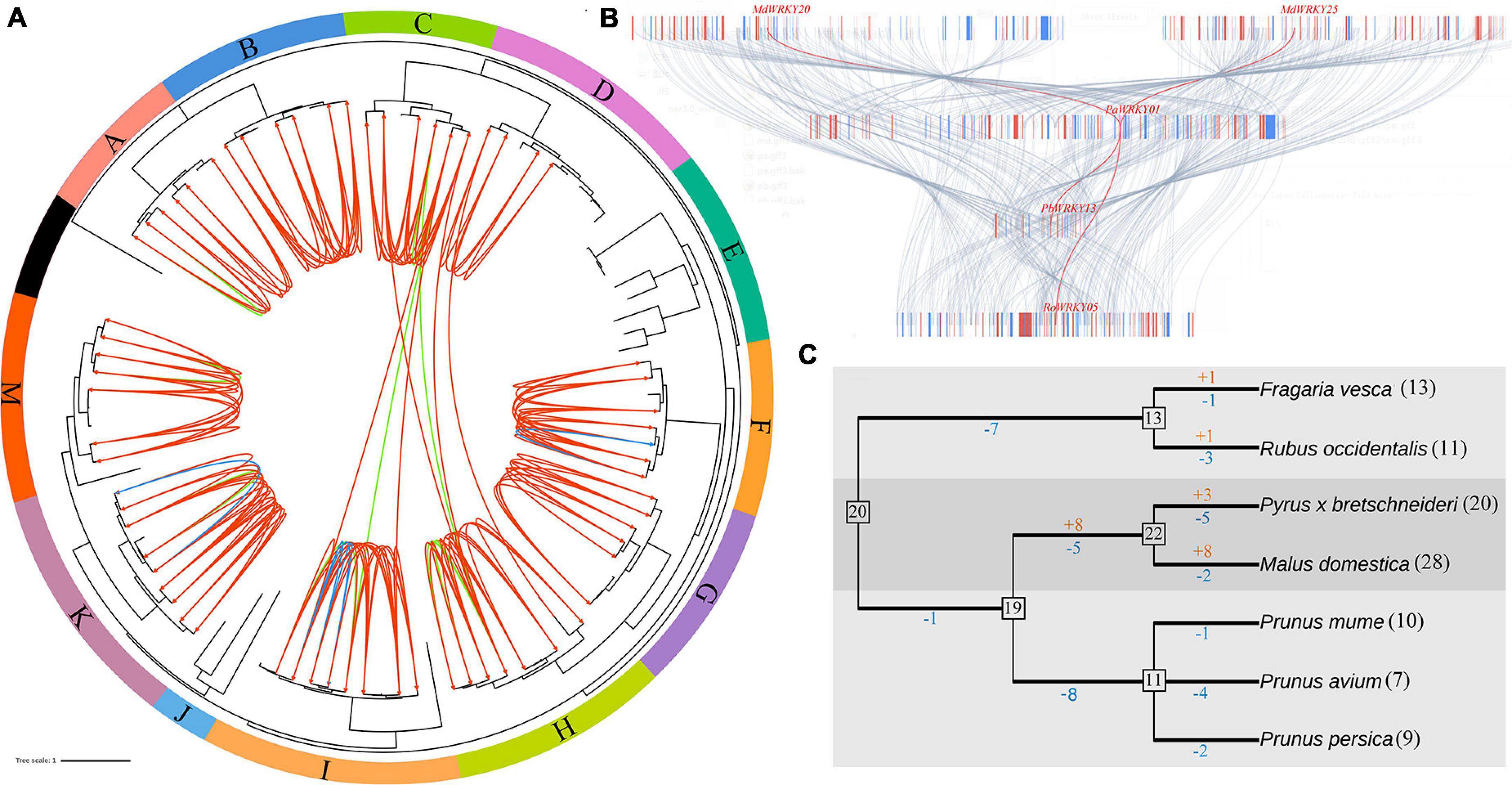
Figure 3. The expansion mechanism analysis of WRKY I genes in seven Rosaceae genomes. (A) Orthologous groups were identified among F. vesca, R. occidentalis, P. persica, P. avium, P. mume, P. bretschneideri, and M. domestica. (B) Microsynteny maps of WRKY I genes across the selected Rosaceae genomes. (C) Estimated gene numbers in ancestral species and gains/losses of genes during WRKY I genes evolution in seven Rosaceae species.
To further identify the orthologous relationships and evolutionary origins in the WRKY I gene family of Rosaceae, we carried our interspecies microsynteny analysis (Figure 3). These data presented that high-level microsynteny was maintained among these Rosaceae genomes. The PaWRKY01 was used as an example to present the high-level microsynteny of different species (Figure 3B). Subsequently, a total of 67 WRKY I genes (nine from F. vesca, 10 R. occidentalis, nine P. persica, one P. avium, four P. mume, 14 P. bretschneideri, and 20 M. domestica) were shown in the 10 orthologous groups (Supplementary Table 1). As shown in Figure 3, the orthologous groups also allowed us to confirm the 12 gene lineages inferred from the above phylogenetic analysis. There was a one-to-one correspondence between gene lineages and syntenic orthologous groups, except for the subgroups E and J (Figure 3A). These results indicated that these WRKY I genes from the E and J subgroups may be species-specific genes.
Gene families can evolve either by gene gains or by gene losses. To gain insight into the evolutionary mechanism of the WRKY I gene family in Rosaceae genomes, we compared a species tree and a bootstrap condensed gene tree based on previously published manuscripts (Nam and Nei, 2005). Subsequently, the modified reconciled-tree method was used to estimate the member of WRKY I genes in the most recent common ancestor (MRCA) of the seven plants and gene gains and losses during the evolution of the Rosaceae genomes. As shown in Figure 3C, there are 20 WRKY I genes in the MRCA, and then the Rosoideae WRKY I genes have undergone gene loss events (seven genes lost), reducing its number to 13. Subsequently, F. vesca and R. occidentalis have gained one and one, and lost one and three genes, respectively, since their divergence. In contrast, Maloideae experienced only one gene loss during the long evolutionary period resulting in the current 19 WRKY I genes in the process of evolution (Figure 3C). The number of WRKY I genes was increased to 22, with eight genes gained and five lost since the MRCA of P. bretschneideri and M. domestica. Finally, P. bretschneideri and M. domestica have gained three and eight and lost five and two genes, respectively, since their divergence. The number of WRKY I genes was reduced to 11, with one gene lost, four genes lost, and two genes lost since the MRCA of P. persica, P. avium, and P. mume. Subsequently, P. persica, P. avium, and P. mume have lost 1, 4, and two genes, respectively, since their divergence. Clearly, the WRKY I gene family of P. persica, P. avium, and P. mume has only experienced the gene loss events. Remarkably, the members of genes gained by P. bretschneideri and M. domestica were higher than those lost, resulting in abundant WRKY I gene in these genomes compared to the other Rosaceae genomes (Figure 3C).
The mechanism of multigene family evolution may be the diversity of gene structure. At the same time, exon gain or loss can be a key step in producing structural complexity and diversity. To better understand the structural diversity and functional evolution of WRKY I genes, exon-intron organization maps were generated from the coding sequence (CDS) with corresponding genome sequences of the WRKY I gene in seven Rosaceae species. The number of different exons, ranging from 2 (MdWRKY16) to 21 (FvWRKY13), was found in 98 WRKY I gene members. Furthermore, most WRKY I gene members contained four or five exons, and the members in the same subgroup had similar exon-intron structures, such as the members of subgroups B and C contained four exons. Additionally, in some subgroups, we found that different members showed significant structural diversity. For example, in subgroup J, the PbWRKY14 contained seven exons, FvWRKY10 had 11 exons, while FvWRKY13 contained 21 exons (Supplementary Figure 2). These results might explain the diversity of closely related WRKY I gene members, because these genes might have occurred exons losses with significantly higher or lower frequency during evolution.
In addition to the exon-intron structure described above, other conserved motifs may be important for the diverse functions of WRKY I proteins, as reported in previous articles (Wang et al., 2015; Jing et al., 2016). Therefore, the local MEME software was used to capture the conserved motifs, and this map is displayed in Supplementary Figures 2, 3. As expected, the motifs 1, 2, and 3 indicate WRKY domain and distribute in most all WRKY I proteins, affirm its major functional role. We also detect other well-conserved motifs outside the WRKY domain. Noteworthy, several conserved motifs were present in almost WRKY I proteins, such as motifs 7, 8, 15, and 16 (Supplementary Figure 2). The remaining motifs were scanned to be specific in the different subgroups of the WRKY I ML tree. The members of WRKY I protein in subgroup B share motif 10, in subgroup share motif 12, and in subgroup I share motif 6, indicating that these conserved motifs might have specific functions in these subgroups. Additionally, in some subgroups, we found that the motifs present a certain degree of diversity, such as subgroup J. Most of the WRKY I members from the seven Rosaceae species that clustered together with orthologous and/or paralogous gene pairs in the same subgroup contain more than one motif outside the WRKY I domain (Supplementary Figure 2). In the current study, we considered the new motifs, which identified subgroup-specific motifs, were novel. Because these new motifs have no significant similarity to any possible function assignments or known motifs by searching Pfam and SMART databases.
To survey the relationship between the expansion patterns in each Rosaceae WRKY I gene family and the corresponding genetic divergences, the gene duplication events were investigated in these Rosaceae WRKY I gene families. Rosaceae genomes have been confirmed to have undergone one or two WGD events. Therefore, we believe that the large-scale duplication events might contribute to the expansion of the WRKY I gene family. To detect this possibility, the gene similarity of the WRKY I flanking regions was searched. These two WRKY I genes were considered to be conserved and evolved from large-scale duplication events, when at least three of 100 kb downstream and upstream genes flanking two WRKY I genes achieved the best non-self-match by the Blast program. To avoid the possibility that the WRKY I gene pairs were located within more divergent blocks, we also defined a set of relaxed criteria for gene gathering based on the flanking regions of a WRKY I gene pair containing two conserved genes (Cao et al., 2016c, 2017b).
In these seven Rosaceae genomes, the gene duplication events of the WRKY I gene family were only identified in both P. bretschneideri and M. domestica, indicating gene duplication events might contribute to the expansion of the WRKY I gene family. The M. domestica genome had 28 WRKY I gene family members, 24 of which (account for 85.7%) were identified in the duplication region of the genome (Supplementary Table 2). Among them, four gene pairs were identified as tandem duplication events, such as MdW RKY04/MdWRKY05, MdWRKY02/MdWRKY03, MdWRKY15/MdWRKY16, and MdWRKY17/MdWRKY18, (Supplementary Table 2). Twelve gene pairs were found to be located in the duplicated segments of chromosomes, and these pairs were identified to be evolved from segmental duplication or WGD events (Figure 4A). As these genes, (MdWRKY25/MdWRKY10, MdWRKY21/MdWRKY25, MdW RKY20/MdWRKY25, MdWRKY20/MdWRKY10, MdWRKY 13/MdWRKY04, MdWRKY24/MdWRKY01, MdWRKY 19/MdWRKY08, MdWRKY16/MdWRKY06, MdWRKY17/Md WRKY07, MdWRKY26/MdWRKY12, and MdWR KY27/MdWRKY12), were distributed on the high synteny regions, suggesting that these gene pairs might have evolved from large-scale duplication events. Twenty WRKY I gene family members were included in the P. bretschneideri genome, and 10 of which (account for 50%) were found in the duplicated segments of chromosomes. The synteny of seven gene pairs (PbWRKY15/PbWRKY01, PbWRKY11/PbWRKY17, PbWRKY11/PbWRKY04, PbWRKY12/PbWRKY18, PbWRKY12/PbWRKY19, PbWRKY18/PbWRKY19, and PbWRKY13/PbWRKY16) was significant in the duplicated region of the genome and speculated to evolve from large-scale duplication events (Figure 4B). The flanking sequences of the gene pair PbWRKY04/PbWRKY17 contained weak synteny, with only two conserved genes (Figure 4B).

Figure 4. Microsynteny maps of WRKY I genes in M. domestica (A) and P. bretschneideri (B). The relative positions of all flanking protein-coding genes were defined by the anchored WRKY I genes, highlighted in orange. The flanking genes and their transcriptional orientations were indicated by arrows.
As we know, both P. bretschneideri and M. domestica genomes have been expanded by two WGD events. To further insight into the evolutionary constraints acting on the WRKY I gene family, the ratios of Ka/Ks were estimated for all the duplicated gene pairs of this gene family in P. bretschneideri and M. domestica. These results presented that all duplicated pairs contain the ratios of Ka/Ks less than 1, except for MdWRKY20/MdWRKY10, indicating that this gene family was slowly evolving at the protein level and had mainly undergone purifying selection (Figure 5 and Supplementary Table 3). In view of the role of two WGD events in the evolution process of P. bretschneideri and M. domestica gene family, the importance of selection intensity with evolutionary time was also emphasized, and the ratios of Ka/Ks were divided into two sets based on the WRKY I duplicated gene pairs from either the ancient (Ks ∼1.5–1.8; ∼140 MYA) or recent WGD (Ks ∼0.15–0.3; 30–45 MYA) (Supplementary Table 3). The average Ka/Ks ratio for ancient WGD WRKY I gene pairs (0.24) was lower than that of recent WGD WRKY I gene pairs (0.38), however, there was no significant difference between these ratios (t-test, P > 0.05). These results suggested that the older and the younger proteins in the WRKY I gene family had similarly stable evolutionary constraints and further supported the notion that the WRKY I genes play key roles during plant growth and development and the regulation of cellular processes in plants.
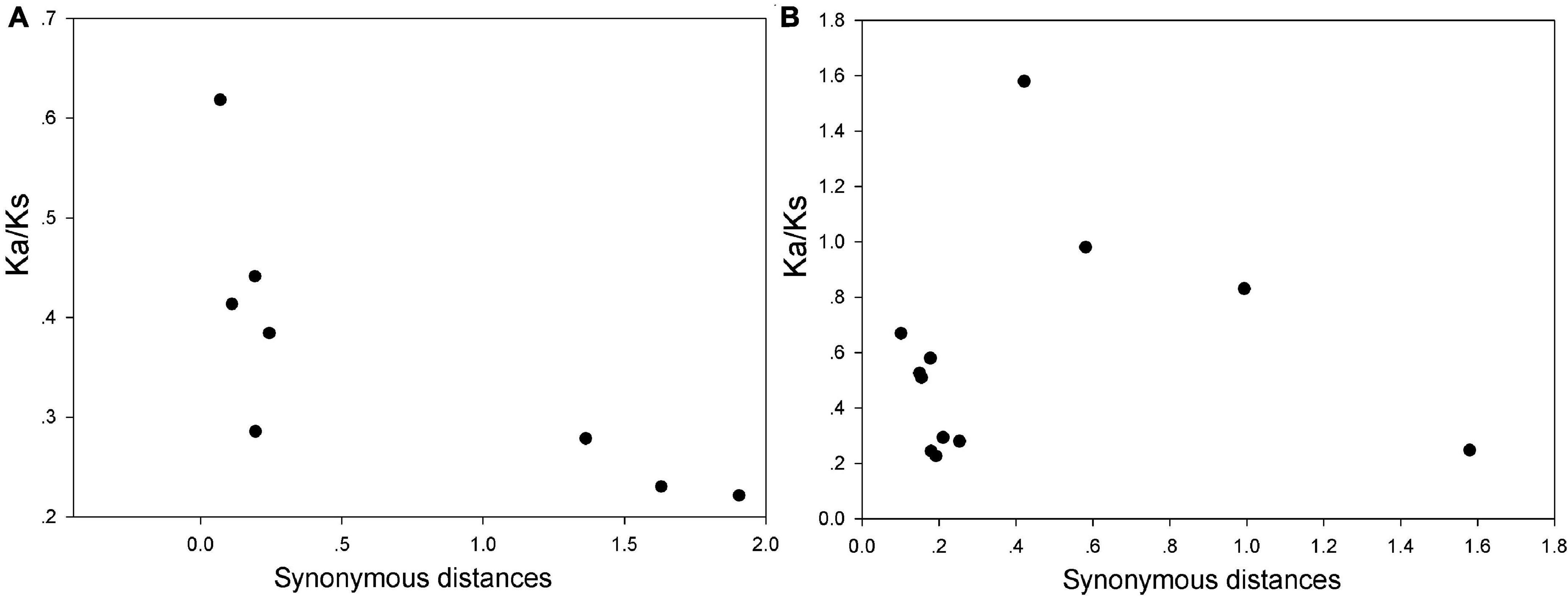
Figure 5. Estimates of Ks and Ka/Ks ratios of duplicated WRKY I genes in P. bretschneideri (A) and M. domestica (B). The x- and y-axes denote the synonymous distance and Ka/Ks ratio for each pair, respectively. Ka, non-synonymous; Ks, synonymous.
The previously published manuscripts suggested that the overall strong purifying selection could mask positive selection at a few individual codon sites (Wang et al., 2015). Therefore, we carried out a sliding-window analysis of Ka/Ks ratios between each pair of WRKY I duplicated gene pairs in P. bretschneideri (Supplementary Table 3). In the present study, we found that numerous sites/regions were under neutral to strong purifying or negative selection, as expected from the basic Ka/Ks analysis (Figure 6). The Ka/Ks ratios of the conserved WRKY domains (i.e., the first WRKY domain and the second WRKY domain) were < 1, suggesting these regions were evolving by strong purifying selection, and these sites were subjected to strong functional constraints. Additionally, the second WRKY domain of PbWRKY04/-11 contained slightly higher Ka/Ks ratios (i.e., Ka/Ks ratios > 1), implying this region was subjected to positive selection, and indicating PbWRKY04/-11 underwent somewhat different selective pressure, which reveals that this domain either displaying a higher evolutionary rate or hidden in the mean of the Ka/Ks ratio (Figure 6). At the same time, we also found that positive selection might help increase Ka/Ks ratio, but it does not guarantee that the average Ka/Ks ratio of the gene exceeds 1. Our data suggested that the WRKY I gene family members were highly conserved and have evolved by purifying selection during evolution.
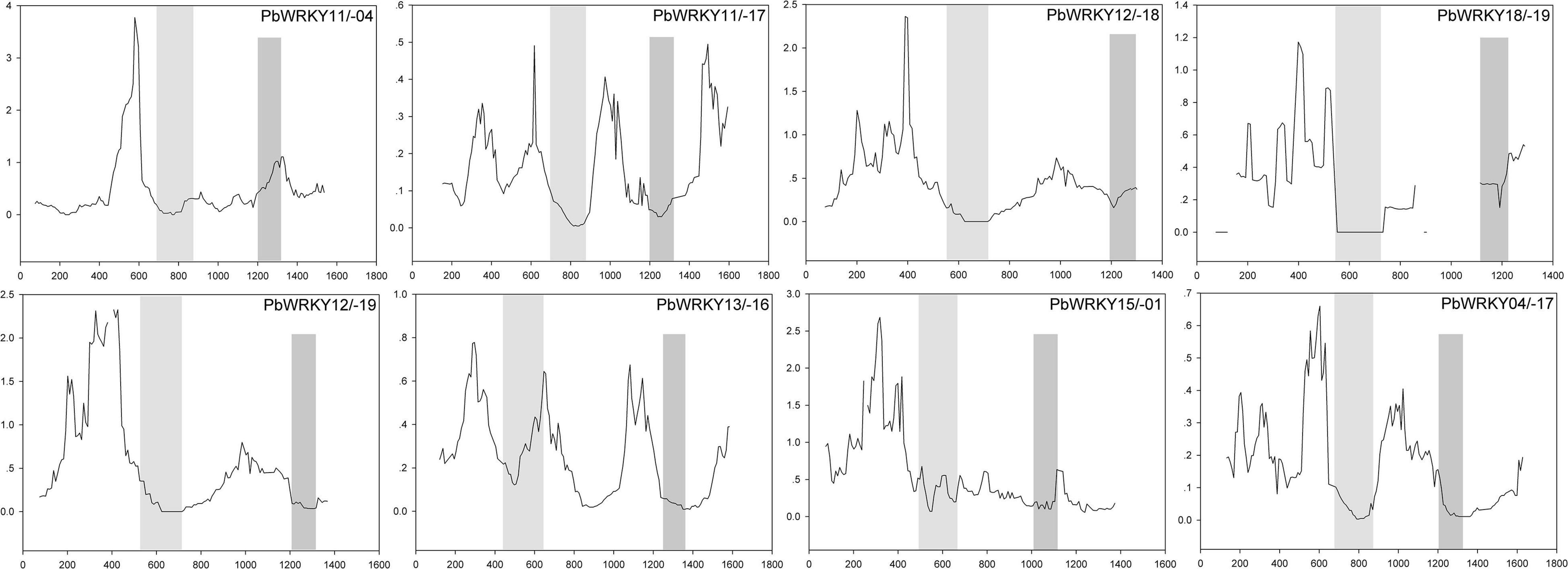
Figure 6. Sliding window analysis of duplicated WRKY I genes in P. bretschneideri. As shown in the key, the gray blocks, from light to dark, represent the positions of the first WRKY domain, and the second WRKY domain, respectively. The step size is 9 bp, and the window size is 150 bp.
To further understand the possible functions of WRKY I genes, we examined the expression patterns of all WRKY I genes using publicly available transcriptome data from the SRA database, such as tissue-specific expression, pooled organs, fruit development, and developmental biology. In P. persica, we examined the expression of WRKY I genes in four different tissues (Supplementary Figure 4). In the data, we found that most of the WRKY I genes exhibited tissue-specific expression patterns, with one, three, and four genes were highly expressed in embryos, fruits, and root, respectively, indicating that these genes might play essential roles in the process of plant development and growth. In P. avium, the expression of WRKY I genes was detected in five growth stages of floral buds: January floral buds, February floral buds, March floral buds, June floral buds, and December floral buds (Supplementary Figure 5). From these results, it was apparent that a few WRKY I genes were relatively higher expressed in P. avium floral buds, such as PaWRKY07 was highly expressed in January floral buds, and PaWRKY04 was highly expressed in March floral buds. In P. mume, we obtained the public RNA-seq data for 10 different tissues (Pollen_grains, Pollen, bud, leaf, root, stem, fruit, Cross_pollinated_pistils, Self_pollinated_pistils, and Unpollinated_pistils) of P. mume (Supplementary Figure 6). Among these WRKY I genes, only one gene (PmWRKY09) exhibited tissue-specific expression patterns, while the remaining WRKY I genes showed express at least two tissues, indicating these genes essential roles in these P. mume tissues. In R. occidentalis, RoWRKY09 and RoWRKY11 exhibited tissue-specific expression patterns, while the remaining WRKY I genes showed express at least two tissues (Supplementary Figure 7). In M. domestica, we analyzed the abundance of WRKY I gene transcripts in leaf, fruit_flesh, root_tip, growing_apex, stem, seed, flower, and during fruit development (Supplementary Figure 8). Our data indicate that 15 of the detected MdWRKYs were expressed differentially in all sampled organs. In F. vesca, we also created a heat map to characterize the expression patterns of WRKY I genes in different representative tissues (Supplementary Figure 9). Based on the heat map, we found that many FvWRKY genes (such as FvWRKY05, FvWRKY12, and FvWRKY08) were expressed and showed high expression levels in most analyzed tissues, implying a possible role in constitutive regulate processes of WRKY I genes throughout the F. vesca plant.
In P. bretschneideri, all WRKY I genes showed express at least two tissues, indicating these genes have essential roles in these P. bretschneideri tissues (Supplementary Figure 10). To further understand the possible functions of WRKY I genes during P. bretschneideri fruit development, we examined the expression patterns of all P. bretschneideri WRKY I genes using a qRT-PCR experiment. The specific primers of PbWRKY genes are presented in Supplementary Table 4. Out of 20 PbWRKY genes, the expression for PbWRKY07 and PbWRKY20 were excluded because the CT values of these two genes were more than 36. The 18 remaining PbWRKY genes were expressed in all P. bretschneideri fruit development stages investigated, but these genes presented differential patterns in terms of both expression level and specificity. Based on their expression patterns, some PbWRKY genes contain expression in particular developmental stages of fruit (Figure 7), such as PbWRKY01, PbWRKY04, and PbWRKY16, were highly expressed in 47 DAF, PbWRKY08, PbWRKY09, PbWRKY12, PbWRKY13, and PbWRKY16 had high expression in 39 DAF, and PbWRKY17 and PbWRKY03 were highly expressed in 145 DAF, suggesting that these genes might play important roles in the specific development of P. bretschneideri fruit. Generally, Pearson’s correlation coefficient (r) was used to assess the similarity between the expression patterns of duplicated gene pairs. According to the previously published manuscripts, 0.3 < r < 0.5, r > 0.5, and r < 0.3 were represented ongoing divergent, non-divergent and divergent, respectively (Blanc and Wolfe, 2004; Yim et al., 2009). To further understand the degree of expression diversity between duplicate WRKY I genes during P. bretschneideri fruit development, we estimated their expression correlations (Table 2). Finally, three duplicate gene pairs, such as PbWRKY15-PbWRKY01, PbWRKY18-PbWRKY19, and PbWRKY13-PbWRKY16, were found to be non-divergent; and five duplicate gene pairs (PbWRKY11-PbWRKY04, PbWRKY11-PbWRKY17, PbWRKY04-PbWRKY17, PbWRKY12-PbWRKY18, and PbWRKY12-PbWRKY19) were divergent (Table 2). Remarkably, we did not find any duplicate PbWRKY gene pairs that were going divergent. In addition, we found that the younger duplication pairs were non-divergent (i.e., Ks value was relatively low); however, most duplication genes were old and divergent (i.e., Ks value was relatively high). Although the structures of WRKY in plants were very conserved, there were many mutations during long evolutionary history. In summary, we observed significant expression divergence in P. bretschneideri WRKY duplication genes.
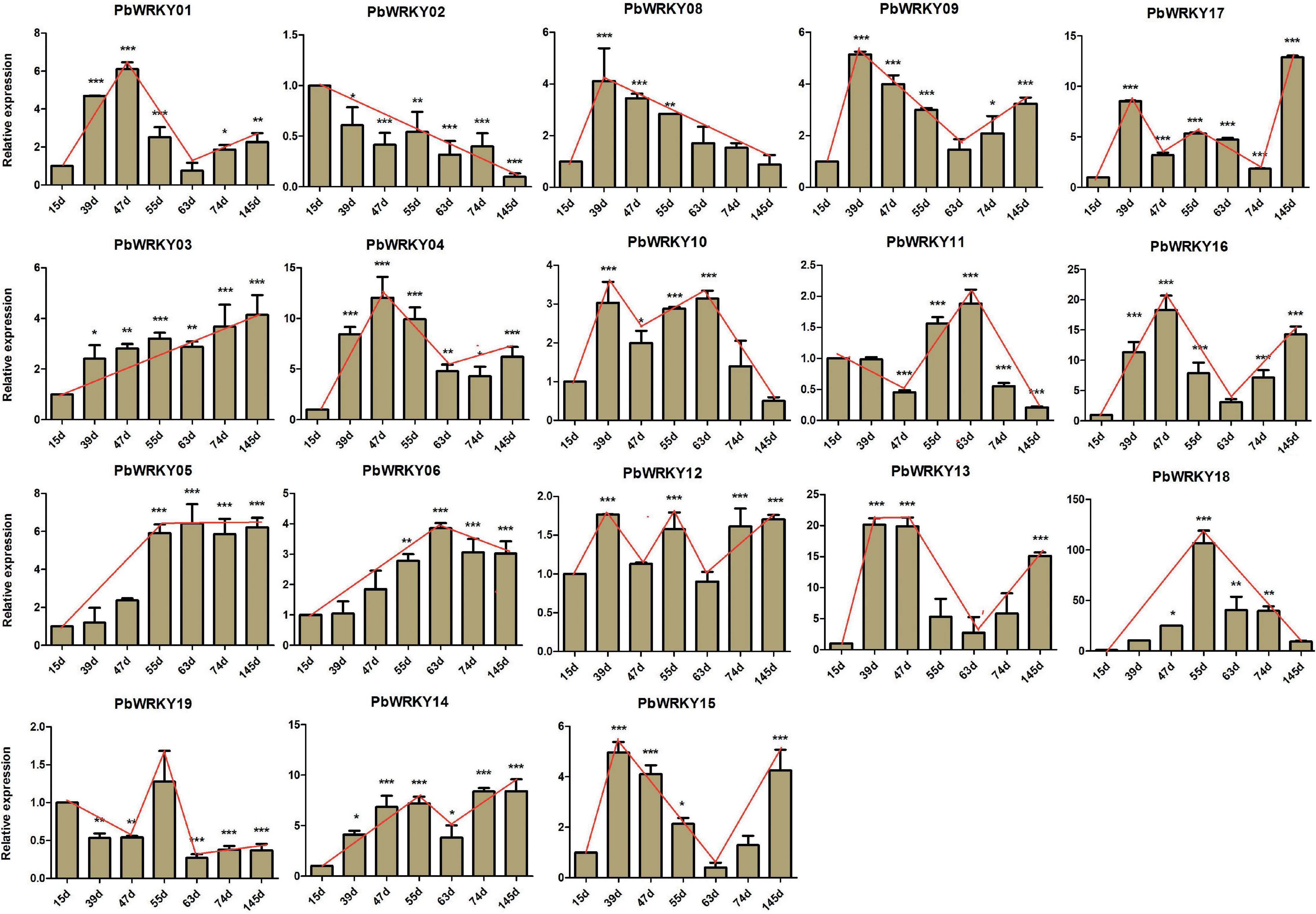
Figure 7. Expression patterns of PbWRKY genes during eight developmental stages of P. bretschneideri fruit at 15, 39, 47, 55, 63, 79, and 145 days after flowering (DAF). *Significant difference compared with the expression level at 15 DAF (p < 0.05).
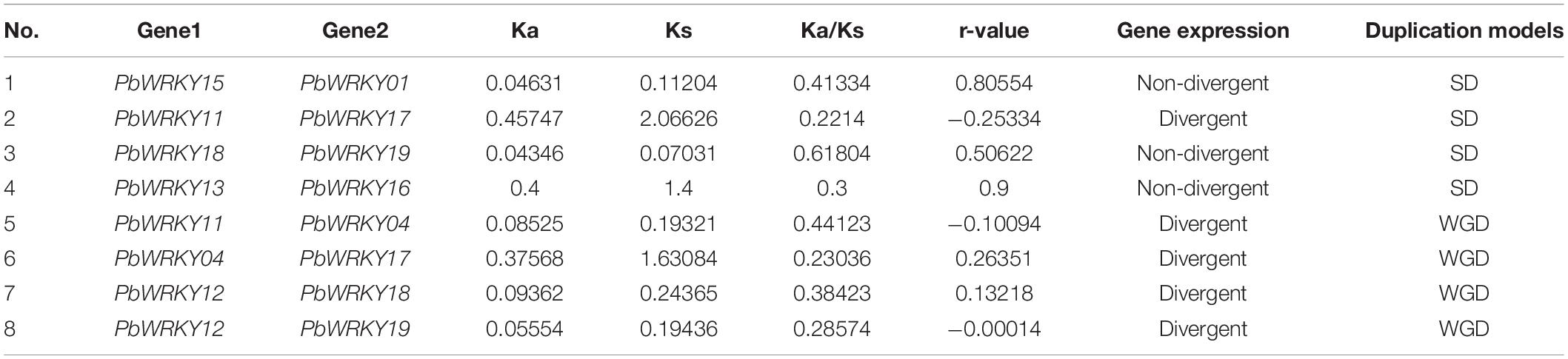
Table 2. The divergence between duplication PbWRKY gene pairs in P. bretschneideri.
WRKY I genes participate in various biological processes, such as regulating plant growth and development, responding to environmental conditions, and plant resistance to abiotic and biotic stresses (Dong et al., 2003; Ross et al., 2007; Wang et al., 2011; Yin et al., 2013). By searching the local genome database, we identified 6, 9, 10, 11, 13, 20, and 28 WRKY I genes in P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri, and M. domestica, respectively. The ML tree could divide the WRKY I genes into four clades (i.e., clades I, II, III, and VI) and noted that orthologous gene pairs of P. bretschneideri and M. domestica WRKY I genes were more widespread, suggesting some ancestor WRKY I genes have appeared before the divergence of P. bretschneideri and M. domestica. By comparing a species tree and a bootstrap condensed gene tree, we analyzed the gene gains and gene losses. We found that the gene gains and gene losses were shown by a large fraction of variability in the clades. For example, compared with the P. avium, P. persica, P. mume, R. occidentalis, and F. vesca, WRKY I genes from P. bretschneideri and M. domestica were present in at least an extra copy in almost every subgroup. At the same time, we also noted that the extra copy was very close to its potential paralogous. Our data were consistent with the proven results that both P. bretschneideri and M. domestica have experienced an extra WGD which not shared with by the other five Rosaceae genomes (Velasco et al., 2010; Wu et al., 2013). Remarkably, the number of WRKY I genes in both P. bretschneideri and M. domestica was not simply two twice compared to other Rosaceae WRKY I genes, indicating that differential gene gain or/and gene loss events might have occurred in different species. For instance, compared with the number of the most recent common ancestor genes, the number of WRKY I genes was nearly halved in P. avium, P. persica, P. mume, R. occidentalis, and F. vesca, but increased approximately 1.4-fold in M. domestica. These data largely reflect the complex evolutionary history of the WRKY I family in the Rosaceae genomes.
Gene duplication events, such as large-scale duplication and tandem duplication, are the main driving forces to generate novel genes. The previous studies have confirmed that these two duplication events are the main driving force for the expansion of gene families in plants, such as the growth-regulating factor (GRF) gene family (Cao et al., 2016c), peroxidase (PRX) gene family (Cao et al., 2016b), B-BOX gene family (Cao et al., 2017a), and MYB gene family in pear (Cao et al., 2016a), and WRKY I gene family (Jing et al., 2016) and CHS gene family in maize (Han et al., 2016). In both P. bretschneideri and M. domestica, most WRKY I genes could be distributed to the duplicated segments, which indicate that large-scale duplications (i.e., segmental duplication or WGD) might help the expansion of the WRKY I gene family in these two species. After sharing the ancient WGD event and following divergence from the other Rosaceae genomes, the lineage leading to present-day both P. bretschneideri and M. domestica are known to have experienced the recent WGD about 30–45 million years ago (MYA). This duplication did not occur in P. avium, P. persica, P. mume, R. occidentalis, F. vesca, or/and other Rosaceae genomes (Velasco et al., 2010; Zhang et al., 2012; Wu et al., 2013). Two WGDs in the ancestor of both P. bretschneideri and M. domestica lead to the expectation of up to four duplicated genes in these two genomes. As shown in Figure 2, WRKY I genes doubled twice in both P. bretschneideri and M. domestica, and formed two duplicated gene pairs, accordingly (Figure 4). Remarkably, we also noted that these four homologous WRKY I blocks were retained, such as MdWRKY25, MdWRKY20, MdWRKY10, and MdWRKY21 were distributed in four homologous blocks, respectively. Compared to the duplicated WRKY I-containing blocks derived from the ancient WGD, these gene pairs contained more conserved flanking protein-coding genes, which derived from the recent WGD. These results suggested that there are high levels of sequence conservation between the recent WGD blocks in both P. bretschneideri and M. domestica, which is consistent with the previous observations (Velasco et al., 2010; Wu et al., 2013).
The intensity and type of selection can be speculated by comparing the ratio of synonymous (silent; Ks) and non-synonymous (amino-acid altering; Ka) substitution rates (Yang, 2007). Sites with Ka/Ks more than one are regarded as undergoing positive selection with adaptive evolution. Sites showing Ka/Ks < 1 are indicative of purifying selection, suggesting that they may play key roles in structure and function (Yang, 2007). In the current study, the Ka/Ks of both P. bretschneideri and M. domestica WRKY I duplicated gene pairs was < 1, except for MdWRKY20/MdWRKY10, indicating that this gene family underwent slow evolutionary non-diversification following duplication. Because all the characteristic WRKY proteins have clear binding preferences for the same DNA motif, the WRKY domain has been assumed to be the only conservative structural feature to constitute the DNA-binding domain. These genes continue to reveal DNA-binding factors that are involved in the transcriptional regulation of key developmental processes. The WRKY I protein sequences contain two type WRKY domains, each of which has a C2-H2-motif (Figure 1), which is known as a zinc finger structure (Rushton et al., 1995). This sequence motif can be specifically bound to W box [(T)(T)TGAC(C/T)] (De Pater et al., 1996; Hara et al., 2000). These data show that the WRKY domain is very conservative. By a sliding-window analysis, we found that the WRKY domains from WRKY I proteins have experienced strong purifying or negative selection, which may provide evidence for the stability of the WRKY domain during evolution.
Previous manuscripts have confirmed that gene duplication may affect their expression patterns (Cao et al., 2017a, 2018). The fate of the gene pairs may change after the duplication event. Retention of duplicates might also be due to sub-functionalization (Force et al., 1999; Lynch and Conery, 2000), and a half or more of duplicated genes exhibited differential expression in rice, poplar, A. thaliana, and soybean. In the present study, we performed the correlation analysis of PbWRKY duplication and their expression to reveal the relationship between gene duplication and expression divergence. The expression levels of duplication PbWRKY gene pairs suggested that the majority of these gene pairs were differentially expressed during fruit development in P. bretschneideri. These results indicated that the significant functional divergence was detected in duplication PbWRKY genes, and further revealed sub-functionalization or neo-functionalization for their derived from a duplication event.
In the present study, a total of 97 WRKY I genes were detected in seven Rosaceae genomes, such as P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri, and M. domestica. Subsequently, we carried out an integrative analysis of WRKY I genes in Rosaceae, which lay the foundation for functional studies of these genes in the future. This study also shows that the rates of gene loss and gain in different Rosaceae genomes are far from equilibrium.
The protein, cDNA, and genome sequences of seven Rosaceae were obtained from the respective genome sequence sites as follows: both P. avium and R. occidentalis from Genome Database for Rosaceae (http://www.rosaceae.org/), F. vesca from Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html), P. persica from Joint Genome Institute (http://www.jgi.doe.gov/), P. mume from GigaDB Dataset (ftp://climb.genomics.cn/), P. bretschneideri from Nanjing Agricultural University (http://peargenome.njau.edu.cn/), and M. domestica from Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication/). RNA-seq data for F. vesca used in this study were available in Strawberry Genomic Resources (http://bioinformatics.towson.edu/strawberry/Default.aspx). RNA-seq data for P. bretschneideri used in this study were available in the SRA database with accession numbers SRR8119898, SRR8119899, SRR8119905, SRR8119889, SRR8119902, SRR8119903, SRR8119904, SRR8119891, SRR8119895, SRR8119890, SRR8119892, SRR8119907, SRR8119893, SRR8119894, SRR8119906, SRR8119900, SRR8119901, SRR8119896, and SRR8119897. RNA-seq data for M. domestica used in this study were available in SRA database with accession numbers SRR767660, SRR767668 to SRR767674, and SRR768127 to SRR768137. RNA-seq data for P. mume used in this study were available in the SRA database with accession numbers DRR002283, DRR002284, DRR013975 to DRR013977, and SRR542478 to SRR542482. RNA-seq data for P. persica used in this study were available in the SRA database with accession numbers SRR1556451, SRR1559275, SRR1561576, and SRR531862 to SRR531865. RNA-seq data for P. avium used in this study were available in the SRA database with accession numbers of SRR531862, SRR531863, SRR531864 and SRR531865. RNA-seq data for R. occidentalis used in this study were available in the SRA database with accession numbers SRR7274864, SRR7274865, SRR7274866, SRR7274867, SRR7274868, SRR7274869, SRR7274870, and SRR7274871.
The experiments did not involve endangered or protected species. No specific permits were required for these locations/activities because the P. bretschneideri used in this study were obtained from the tissue culture room of Anhui Agricultural University.
YPC conceived and supervised the research and wrote the manuscript. YPC and LJ designed the experiments. YPC, LJ, DB, JT, and YC performed the experiments and analyzed the results. All authors have read and approved the final manuscript.
This study was supported by the Natural Science Fund Youth Project of Hunan Province (grant no. 2021JJ41067), the Outstanding Youth of the Education Department of Hunan Province (grant no. 20B624), the Talent Scientific Research Start-up Foundation of Yijishan Hospital, Wannan Medical College (grant no. YR202001), the Opening Foundation of Key Laboratory of Non-coding RNA Transformation Research of Anhui Higher Education Institution (grant no. RNA202004), and the Key Projects of Natural Science Research of Universities in Anhui Province (grant no. KJ2020A0622). The Funding bodies were not involved in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
YC was employed by the company Anhui Zhifei Longcom Biopharmaceutical Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the reviewers and editors for their careful reading and helpful comments on this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.801490/full#supplementary-material
Supplementary Figure 1 | Chromosomal location of WRKY I genes among P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri, and M. domestica. The distribution of WRKY I genes among the chromosomes in each species is diverse. The chromosome number is indicated at the top of each chromosome.
Supplementary Figure 2 | Phylogenetic relationship, the exon-intron structure, and the motif analysis of WRKY I genes from P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri, and M. domestica. The exons and introns are indicated by green rectangles and thin lines, respectively.
Supplementary Figure 3 | The logos of the twenty motif sequences among P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri, and M. domestica. The bit score indicates the information content for each position in the sequence.
Supplementary Figure 4 | Expression profiles of P. persica WRKY I genes in different tissues. Blue and red colors correspond to downregulation and upregulation, respectively.
Supplementary Figure 5 | Expression profiles of P. avium WRKY I genes in different tissues. Blue and red colors correspond to downregulation and upregulation, respectively.
Supplementary Figure 6 | Expression profiles of P. mume WRKY I genes in different tissues. Blue and red colors correspond to down-regulation and up-regulation, respectively.
Supplementary Figure 7 | Expression profiles of R. occidentalis WRKY I genes in different tissues. Blue and red colors correspond to downregulation and upregulation, respectively.
Supplementary Figure 8 | Expression profiles of M. domestica WRKY I genes in different tissues. Blue and red colors correspond to downregulation and upregulation, respectively.
Supplementary Figure 9 | Expression profiles of F. vesca WRKY I genes in different tissues. Blue and red colors correspond to downregulation and upregulation, respectively.
Supplementary Figure 10 | Expression profiles of P. bretschneideri WRKY I genes in different tissues. Blue and red colors correspond to downregulation and upregulation, respectively.
Supplementary Table 1 | Synteny data in P. avium, P. persica, P. mume, R. occidentalis, F. vesca, P. bretschneideri and M. domestica.
Supplementary Table 2 | Duplicated WRKY I gene pairs in P. bretschneideri and M. domestica.
Supplementary Table 3 | The divergence between duplicated WRKY I gene pairs in P. bretschneideri and M. domestica.
Supplementary Table 4 | Primers in this study.
qRT-PCR, real-time PCR; Ka, non-synonymous; Ks, synonymous, NJ, neighbor joining; WGD, whole genome duplication.
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME Suite. Nucleic Acids Res. 43, W39–W49.
Blanc, G., and Wolfe, K. H. (2004). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16, 1667–1678. doi: 10.1105/tpc.021345
Cao, Y., Han, Y., Li, D., Lin, Y., and Cai, Y. (2016a). MYB transcription factors in chinese pear (Pyrus bretschneideri Rehd.): genome-wide identification, classification, and expression profiling during fruit development. Front. Plant Sci. 7:577. doi: 10.3389/fpls.2016.00577
Cao, Y., Han, Y., Meng, D., Li, D., Jin, Q., Lin, Y., et al. (2016b). Structural, Evolutionary, and Functional Analysis of the Class III Peroxidase Gene Family in Chinese Pear (Pyrus bretschneideri). Front. Plant Sci. 7:1874. doi: 10.3389/fpls.2016.01874
Cao, Y. P., Han, Y., Jin, Q., Lin, Y., and Cai, Y. (2016c). Comparative genomic analysis of the GRF genes in Chinese pear (Pyrus bretschneideri Rehd), poplar (Populous), grape (Vitis vinifera) Arabidopsis and rice (Oryza sativa). Front. Plant Sci. 7:1750. doi: 10.3389/fpls.2016.01750
Cao, Y., Han, Y., Meng, D., Abdullah, M., Yu, J., Li, D., et al. (2018). Expansion and evolutionary patterns of GDSL-type esterases/lipases in Rosaceae genomes. Funct. Integr. Genom. 18, 673–684. doi: 10.1007/s10142-018-0620-1
Cao, Y., Han, Y., Meng, D., Li, D., Jiao, C., Jin, Q., et al. (2017a). B-BOX genes: genome-wide identification, evolution and their contribution to pollen growth in pear (Pyrus bretschneideri Rehd.). BMC Plant Biol. 17:156. doi: 10.1186/s12870-017-1105-4
Cao, Y., Han, Y., Meng, D., Li, D., Jin, Q., Lin, Y., et al. (2017b). Genome-wide analysis suggests high level of microsynteny and purifying selection affect the evolution ofEIN3/EILfamily in Rosaceae. PeerJ 5:e3400.
Cao, Y., Liu, W., Zhao, Q., Long, H., Li, Z., Liu, M., et al. (2019). Integrative analysis reveals evolutionary patterns and potential functions of SWEET transporters in Euphorbiaceae. Int. J. Biol. Macromole. 139, 1–11.
Chen, K., Durand, D., and Farachcolton, M. (2000). NOTUNG: A Program for Dating Gene Duplications and Optimizing Gene Family Trees. Res. Computat. Mole. Biol. 7, 429–447. doi: 10.1089/106652700750050871
Christenhusz, M. J., and Byng, J. W. (2016). The number of known plants species in the world and its annual increase. Phytotaxa 261, 201–217. doi: 10.11646/phytotaxa.261.3.1
Coulthart, M., and Singh, R. S. (1988). High level of divergence of male-reproductive-tract proteins, between Drosophila melanogaster and its sibling species, D. simulans. Mole. Biol. Evolut. 5, 182–191.
De Pater, S., Greco, V., Pham, K., Memelink, J., and Kijne, J. W. (1996). Characterization of a Zinc-Dependent Transcriptional Activator from Arabidopsis. Nucleic Acids Res. 24, 4624–4631. doi: 10.1093/nar/24.23.4624
Deleu, W., González, V., Monfort, A., Bendahmane, A., Puigdomènech, P., Arús, P., et al. (2007). Structure of two melon regions reveals high microsynteny with sequenced plant species. Mole. Genet. Genomics Mgg 278, 611–622. doi: 10.1007/s00438-007-0277-2
Dong, J., Chen, C., and Chen, Z. (2003). Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mole. Biol. 51, 21–37.
Duan, Y., Jiang, Y., Ye, S., Karim, A., Ling, Z., He, Y., et al. (2015). PtrWRKY73, a salicylic acid-inducible poplar WRKY transcription factor, is involved in disease resistance in Arabidopsis thaliana. Plant Cell Rep. 34, 831–841. doi: 10.1007/s00299-015-1745-5
Eulgem, T., Rushton, P. J., Robatzek, S., and Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206. doi: 10.1016/s1360-1385(00)01600-9
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2013). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230.
Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y.-L., and Postlethwait, J. (1999). Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545. doi: 10.1093/genetics/151.4.1531
Gong, X., Zhang, J., Hu, J., Wang, W., Wu, H., Zhang, Q., et al. (2015). FcWRKY70, a WRKY protein of Fortunella crassifolia, functions in drought tolerance and modulates putrescine synthesis by regulating arginine decarboxylase gene. Plant Cell Environ. 38, 2248–2262. doi: 10.1111/pce.12539
Han, Y., Ding, T., Su, B., and Jiang, H. (2016). Genome-Wide Identification, Characterization and Expression Analysis of the Chalcone Synthase Family in Maize. Int. J. Mole. Sci. 17:161. doi: 10.3390/ijms17020161
Hara, K., Yagi, M., Kusano, T., and Sano, H. (2000). Rapid systemic accumulation of transcripts encoding a tobacco WRKY transcription factor upon wounding. Mole. Gener. Genet. Mgg 263, 30–37. doi: 10.1007/pl00008673
He, H., Dong, Q., Shao, Y., Jiang, H., Zhu, S., Cheng, B., et al. (2012). Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 31, 1199–1217. doi: 10.1007/s00299-012-1241-0
Illa, E., Sargent, D. J., Girona, E. L., Bushakra, J., Cestaro, A., Crowhurst, R., et al. (2011). Comparative analysis of rosaceous genomes and the reconstruction of a putative ancestral genome for the family. BMC Evolut. Biol. 11:9. doi: 10.1186/1471-2148-11-9
Ishiguro, S., and Nakamura, K. (1994). Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and β-amylase from sweet potato. Mole. Gener. Genet. MGG 244, 563–571. doi: 10.1007/BF00282746
Jibran, R., Dzierzon, H., Bassil, N., Bushakra, J. M., Edger, P. P., Sullivan, S., et al. (2018). Chromosome-scale scaffolding of the black raspberry (Rubus occidentalis L.) genome based on chromatin interaction data. Horticult. Res. 5:8. doi: 10.1038/s41438-017-0013-y
Jing, J., Kong, J., Qiu, J., Zhu, H., Peng, Y., and Jiang, H. (2016). High level of microsynteny and purifying selection affect the evolution of WRKY family in Gramineae. Devel. Genes Evolut. 226, 15–25. doi: 10.1007/s00427-015-0523-2
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mole. Biol. Evolut. 30, 772–780. doi: 10.1093/molbev/mst010
Letunic, I., and Bork, P. (2006). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128.
Letunic, I., Doerks, T., and Bork, P. (2012). SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305.
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189.
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Ling, J., Jiang, W., Zhang, Y., Yu, H., Mao, Z., Gu, X., et al. (2011). Genome-wide analysis of WRKY gene family in Cucumis sativus. BMC Genomics 12:471. doi: 10.1186/1471-2164-12-471
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2- ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155.
Maher, C., Stein, L., and Ware, D. (2006). Evolution of Arabidopsis microRNA families through duplication events. Genome Res. 16, 510–519.
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121–e121. doi: 10.1093/nar/gkt263
Nam, J., and Nei, M. (2005). Evolutionary Change of the Numbers of Homeobox Genes in Bilateral Animals. Mole. Biol. Evolut. 22, 2386–2394. doi: 10.1093/molbev/msi229
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2014). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mole. Biol. Evolut. 32, 268–274. doi: 10.1093/molbev/msu300
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11:1650. doi: 10.1038/nprot.2016.095
Qiao, X., Li, M., Li, L., Yin, H., Wu, J., and Zhang, S. (2015). Genome-wide identification and comparative analysis of the heat shock transcription factor family in Chinese white pear (Pyrus bretschneideri) and five other Rosaceae species. BMC Plant Biol. 15:12. doi: 10.1186/s12870-014-0401-5
Qiu, Y., Jing, S., Fu, J., Li, L., and Yu, D. (2004). Cloning and analysis of expression profile of 13WRKY genes in rice. Chin. Sci. Bull. 49, 2159–2168.
Ross, C. A., Liu, Y., and Shen, Q. J. (2007). The WRKY gene family in rice (Oryza sativa). J. Integr. Plant Biol. 49, 827–842.
Rushton, P. J., Macdonald, H., Huttly, A. K., Lazarus, C. M., and Hooley, R. (1995). Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of α-Amy2 genes. Plant Mole. Biol. 29, 691–702. doi: 10.1007/BF00041160
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., Kato, T., Nakao, M., et al. (2008). Genome structure of the legume, Lotus japonicus. DNA Res. 15, 227–239.
Sherry, S. (2012). “NCBI SRA toolkit technology for next generation sequence data,” in Plant and Animal Genome XX Conference (January 14-18, 2012), (San Diego: Plant and Animal Genome).
Shirasawa, K., Isuzugawa, K., Ikenaga, M., Saito, Y., Yamamoto, T., Hirakawa, H., et al. (2017). The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 24, 499–508. doi: 10.1093/dnares/dsx020
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116.
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7:562. doi: 10.1038/nprot.2012.016
Vanburen, R., Bryant, D., Bushakra, J. M., Vining, K. J., Edger, P. P., Rowley, E. R., et al. (2016). The genome of black raspberry (Rubus occidentalis). Plant J. Cell Mole. Biol. 87, 535–547.
Velasco, R., Zharkikh, A., Affourtit, J., Dhingra, A., Cestaro, A., Kalyanaraman, A., et al. (2010). The genome of the domesticated apple (Malus [times] domestica Borkh.). Nat. Genet. 42, 833–839.
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genetics 45, 487–494. doi: 10.1038/ng.2586
Vilanova, S., Sargent, D. J., Arús, P., and Monfort, A. (2008). Synteny conservation between two distantly-related Rosaceae genomes: Prunus (the stone fruits) and Fragaria (the strawberry). BMC Plant Biol. 8:67. doi: 10.1186/1471-2229-8-67
Wang, Q., Wang, M., Zhang, X., Hao, B., Kaushik, S., and Pan, Y. (2011). WRKY gene family evolution in Arabidopsis thaliana. Genetica 139:973.
Wang, Y., Feng, L., Zhu, Y., Li, Y., Yan, H., and Xiang, Y. (2015). Comparative genomic analysis of the WRKY III gene family in populus, grape, arabidopsis and rice. Biol. Direct 10:48. doi: 10.1186/s13062-015-0076-3
Wu, J., Wang, Z., Shi, Z., Zhang, S., Ming, R., Zhu, S., et al. (2013). The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396–408.
Wu, K., Guo, Z., Wang, H., and Li, J. (2005). The WRKY Family of Transcription Factors in Rice and Arabidopsis and Their Origins. DNA Res. 12, 9–26.
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mole. Biol. Evolut. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yim, W. C., Lee, B.-M., and Jang, C. S. (2009). Expression diversity and evolutionary dynamics of rice duplicate genes. Mole. Genetics Genomics 281, 483–493. doi: 10.1007/s00438-009-0425-y
Yin, G., Xu, H., Xiao, S., Qin, Y., Li, Y., Yan, Y., et al. (2013). The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 13:1–19. doi: 10.1177/1176934319857720
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhang, Q., Chen, W., Sun, L., Zhao, F., Huang, B., Yang, W., et al. (2012). The genome ofPrunus mume. Nat. Commun. 3:1318.
Zhou, Q. Y., Tian, A. G., Zou, H. F., Xie, Z. M., Lei, G., Huang, J., et al. (2008). Soybean WRKY−type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol. J. 6, 486–503. doi: 10.1111/j.1467-7652.2008.00336.x
Keywords: microsynteny, molecular evolution, WRKY, Rosaceae, WGD
Citation: Jiang L, Chen Y, Bi D, Cao Y and Tong J (2021) Deciphering Evolutionary Dynamics of WRKY I Genes in Rosaceae Species. Front. Ecol. Evol. 9:801490. doi: 10.3389/fevo.2021.801490
Received: 25 October 2021; Accepted: 08 November 2021;
Published: 06 December 2021.
Edited by:
Lin Zhang, Hubei University of Chinese Medicine, ChinaReviewed by:
Wei Hu, Chinese Academy of Tropical Agricultural Sciences, ChinaCopyright © 2021 Jiang, Chen, Bi, Cao and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunpeng Cao, eGZjeXBlbmdAMTI2LmNvbQ==; Jiucui Tong, dG9uZ2ppdWN1aUB5anN5eS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.