 Qian Li
Qian Li Guang-Mei Li
Guang-Mei Li Yong-Li Zheng
Yong-Li Zheng Shu-Jun Wei
Shu-Jun Wei- 1College of Life Sciences, China Jiliang University, Hangzhou, China
- 2Zhejiang Agricultural Products Quality and Safety Center, Hangzhou, China
- 3Institute of Plant and Environmental Protection, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China
The striped flea beetle (SFB) Phyllotreta striolata (Fabricius) (Coleoptera: Chrysomelidae) is a major pest of cruciferous vegetables in southern China. The population diversity and genetic structure of SFB are unknown. Here, we assembled a draft genome for the SFB and characterized the distribution of microsatellites. Then, we developed 12 novel microsatellite markers across the genome. We used a segment of the cox1 gene and newly developed microsatellite markers to genotype the genetic diversity of SFB across southern China. There were 44 mitochondrial haplotypes in the SFB populations, with haplotype 2 as the most widespread. The population genetic differentiation was very low, indicated by FST-values (<0.05 except for Guangxi population with other populations based on cox1), high gene flow (4.10 and 44.88 of cox1 and microsatellite, respectively) and Principal Coordinate Analysis across all populations. Mantel test showed genetic distance in SFB was significantly associated with geographic distance based on microsatellites (R2 = 0.2373, P = 0.014) while result based on cox1 (R2 = 0.0365, P = 0.155) showed no significant difference. The phylogenetic analysis did not find any geographically related clades among all haplotypes. Analyses based on microsatellites showed a lack of population genetic structure among all populations. Our study provides a foundation for the future understanding of the ecology and evolution of SFB and its management.
Introduction
The striped flea beetle (SFB) Phyllotreta striolata (Fabricius) (Coleoptera: Chrysomelidae) is a pest of cruciferous crops (Brassicaceae) (Soroka et al., 2018; Cao et al., 2020; Atirach et al., 2021). The adults chew on leaves and larvae feed on fiber. The field control of this pest heavily relys on insecticides (Andersen et al., 2006), leading to insecticide resistance (Feng et al., 2000; James et al., 2019).
The SFB is mainly distributed in Asia, Europe, and North America (Soroka et al., 2018; Cao et al., 2020; Atirach et al., 2021). Native to Eurasia, SFB was introduced to North America in 1663 from Europe (Rousseau and Lesage, 2016). In China, the SFB is one of the most important cruciferous crops pests in southern areas. When the adult density of SFB is 20∼50 individuals every 100 Brassica pekinensis, the damage rate will be 50∼76%; if the adult density more than 50, the damage rate will be 100% in Shenzhen (Zhang et al., 2000). There are six to nine generations of SFB throughout the year without overwintering in southern China (Sun et al., 2010), compared to one to two generations per year in North America (Olfert et al., 2017). Recently, damage of SFB has increased. Its geographic range expanded both in China and North America (Chai, 2010; Sun, 2010; Lee et al., 2011; Kielen, 2012). Current studies mainly focus on this pest’s biology and control methods (Gao et al., 2000; Chai, 2010; Lee et al., 2011; Soroka et al., 2018; Yan et al., 2018; Atirach et al., 2021); however, no studies that we are aware of on the ecology and evolution of the SFB.
In this study, we examined the population genetic diversity and genetic structure of the SFB among populations across its main distribution range in China. Due to the lack of genetic markers, we developed a novel set of microsatellite markers from first-time assembled random genomic sequences of SFB using Illumina sequencing. We used both microsatellites and a segment of the mitochondrial cytochrome oxidase subunit I (cox1) gene to determine the genetic diversity and differentiation among eight representative populations across southern China. We tested the hypothesis that levels of genetic diversity and population differentiation for the SFB are low due to high levels of gene flow among populations. These results provide a basis to understand the ecology and evolution of this pest and its management.
Materials and Methods
Sample Collection and DNA Extraction
Adults of SFB were collected across southern China from cruciferous vegetables (Table 1 and Figure 1). To avoid collecting siblings, we collected specimens from about 20 sites at least 10 m apart at each sampling location. In total, eight populations were collected. The species were first identified by morphology (He et al., 2012) and then validated by molecular identification using the mitochondrial cox1 gene (see below). All samples were kept in 100% ethanol before DNA extraction.
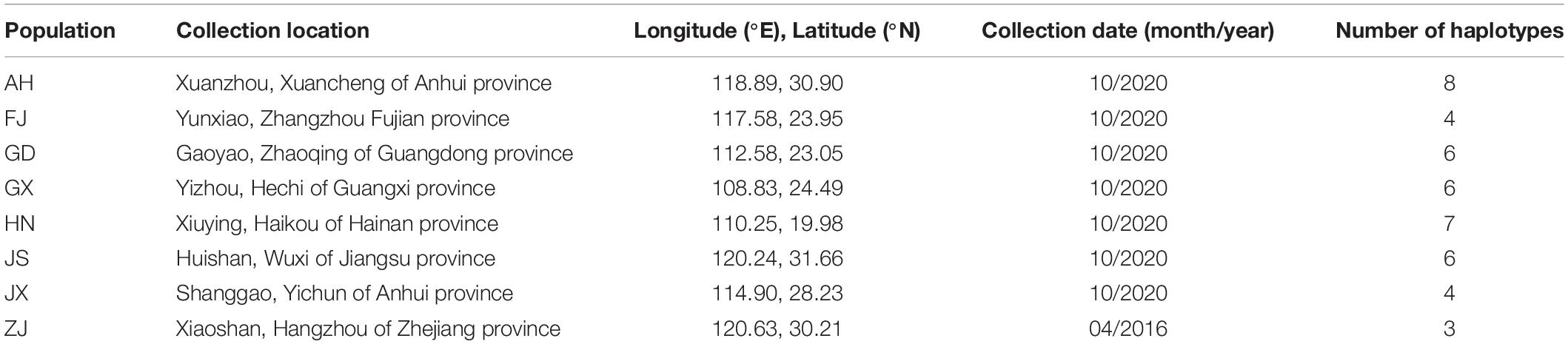
Table 1. Population information of Phyllotreta striolata used in this study.
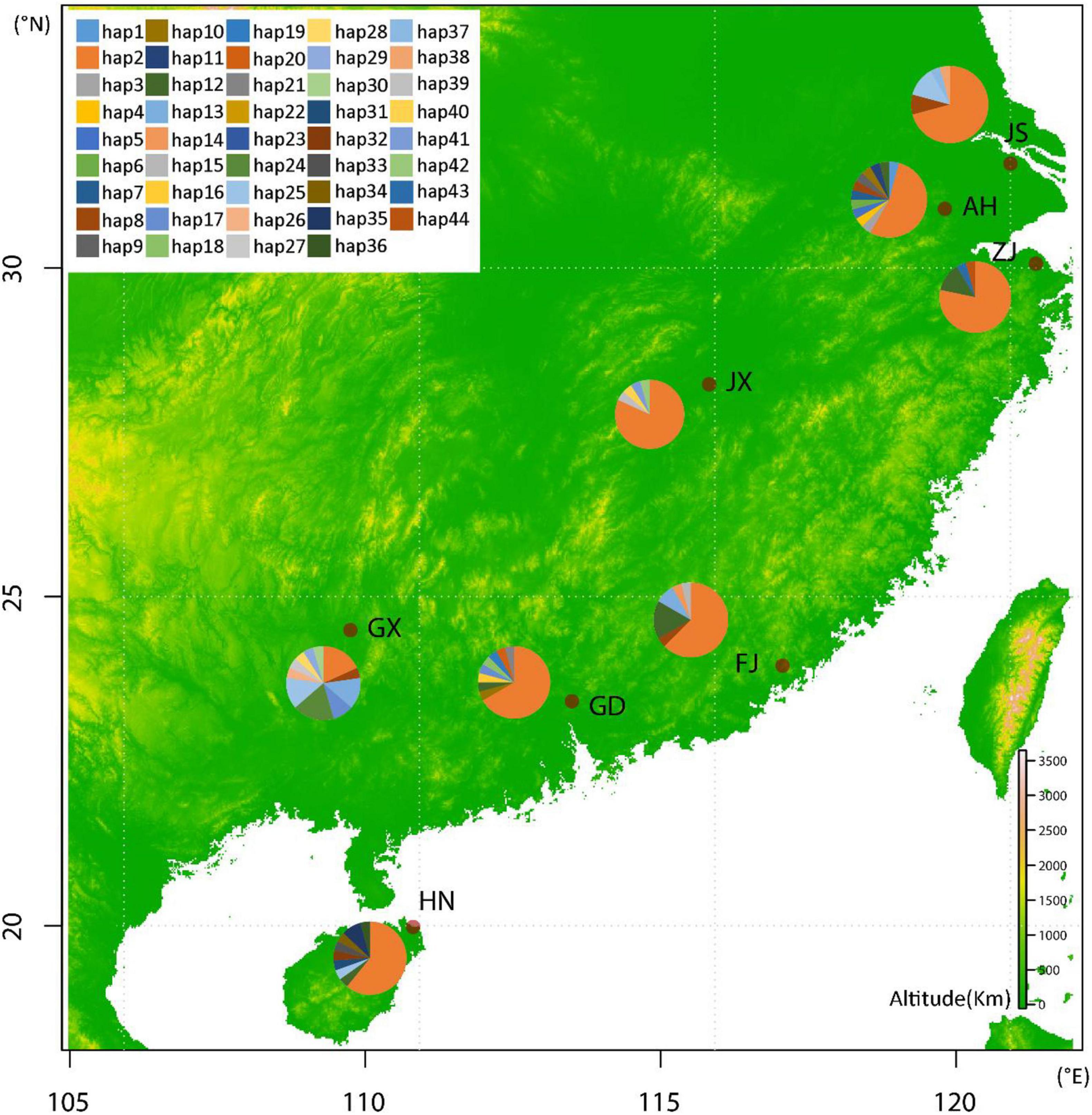
Figure 1. Collection sites and geographical distribution of the mitochondrial cox1 haplotypes of Phyllotreta striolata. The haplotype frequency for each population is represented by colors in the pie charts. See Table 1 for population codes.
To construct a high-throughput sequencing library for microsatellites development, we extracted genomic DNA from 50 adults collected in Xiaoshan, Zhejiang province, using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Then, we used 192 individuals for population-level genotyping, with 24 individuals from each population. Total genomic DNA was extracted from individual whole adult using the DNeasy Blood & Tissue Kit. The voucher specimens were stored at –80°C in the Integrated Pest Management Laboratory of the Beijing Academy of Agriculture and Forestry Sciences.
Library Construction, Genome Sequencing, and Assembly
The high-throughput sequencing library with 500-bp insert size was prepared using the Illumina TruSeq DNA PCR-Free HT Library Prep Kit (Illumina, San Diego, CA, United States). The prepared library was sequenced on an Illumina Hiseq4000 Sequencer using the Hiseq Reagent Kit v3 (Illumina, San Diego, CA, United States) by Beijing BerryGenomics Co., Ltd. The paired-end 150 bp raw data were trimmed by removing the low-quality reads using Trimmomatic 0.36 (Bolger et al., 2014), and then the sequences were evaluated by FastQC v 0.11.5 (Andrews, 2004). The genome size of P. striolata was estimated by JELLYFISH v2.2.6 software with a K-mer method (Kingsford, 2011). IDBA_UD v1.1.1 was used to assemble the generated genomic sequences with K-mer from 20 to 140 (Peng et al., 2010).
Development of Universal Microsatellite Markers
The Microsatellite Search and Building Database (MSDB)1 was used to identify potential microsatellite sites from the whole genome sequences of P. striolata. A minimum of 12, 5, 5, 5, 5, and 5 repeats was used to distinguish mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide motifs, respectively (Lianming et al., 2013). The QDD3 program (Emese et al., 2010) was then used to extract microsatellites and their flanking sequences (300 bp each) and design the primers. The criteria and parameters for the primer design for the isolated microsatellite markers followed Cao et al. (2016). The primer design was set according to the following parameters: annealing temperature (Tm): 54°C < Tm < 60°C; the difference in Tm between paired upstream and downstream primers was < 4°C; only one pair of microsatellite primers is kept on each locus; the repeat motif of the amplified product was ≥ 3. The microsatellite loci were further filtered under stringent criteria as described in previous studies (Song et al., 2018): (i) the microsatellites had to be pure and specific, (ii) the design strategy of “A” was used, and (iii) the minimum distance between the 3′ end of a primer pair and its target region had to be no shorter than 10 bp.
We selected 70 pairs of primers for initial validation using one individual from each representative population (AH, FJ, GD, GX, HN, JS, JX, ZJ). We used three fluorescences (FAM, ROX, HEX) to label the amplified products of each locus (Schuelke, 2000; Blacket et al., 2012). The fluorescences were independently added to a primer C tail (PC-tail) (5′ CAGGACCAGGCTACCGTG 3′). The sequence of the PC-tail was added to the 5′ end of each upstream primer (Blacket et al., 2012). The reaction system was set to 10 μL, including 0.5 μL of template DNA (30–160 ng/μL), 5 μL of Master Mix (Promega, Madison, WI, United States), 0.08 μL of the PC tail modified forward primer (10 mM), 0.16 μL of reverse primer (10 mM), 0.32 μL of fluorescence-labeled PC-tail (10 mM) and 3.94 μL of ddH2O. The following PCR amplification program was used to amplify the microsatellites: 3 min at 94°C; 35 cycles of 15 s at 94°C, 30 s at 56°C and 1 min at 72°C, followed by a final 10 min extension at 72°C. Primer pairs fitting the following criteria were retained in subsequent analysis: (i) primers with PCR amplification rate equal or higher than 75%; (ii) at least two alleles present in tested individuals; (iii) without non-specific amplification.
Length of PCR products was analyzed on an ABI 3730xl DNA Analyzer (Applied Biosystems, United States) using the GeneScan™ 500 LIZ™ dye Size Standard (Applied Biosystems, United States) for sizing DNA fragments. Genotypes were determined using GENEMAPPER v4.0 (Applied Biosystems, United States) (Chatterji and Pachter, 2006). The remaining primer pairs were used for population-level genotyping.
Mitochondrial Gene Amplification, Sequencing, and Homologous Gene Downloading
To characterize the mitochondrial variation and validate correct identification of the specimens, a fragment of the cox1 gene involved the DNA barcoding region of insects was sequenced using the universal primers LCO1490 (5′ GGTCAACAAATCATAAAGATATTGG 3′) and HCO2198 (5′ TAAACTTCAGGGTGACCAAAAAATCA 3′) (Folmer et al., 1994). Polymerase chain reactions (PCR) were conducted using the Mastercycler Pro system (Eppendorf, Germany) in a 15 μL volume consisting of 1 μL of template DNA (30–160 ng/μL), 7.5 μL of Master Mix (Promega, Madison, WI, United States), 0.6 μL of forward primer (10 mM), 0.6 μL of reverse primer (10 mM) and 5.3 μL of ddH2O. The thermal profiles for DNA amplification were as follows: 3 min at 94°C; 35 cycles of 15 s at 94°C, 20 s at 54°C, and 40 s at 72°C, followed by a final 10-min extension at 72°C. Amplified products were purified and sequenced on an ABI 3730xl DNA Analyzer by Tsingke Biotechnology Co. Ltd. (Beijing, China).
Apart from sequences obtained in this study, we searched orthologous cox1 gene sequences from the NCBI nucleotide database and included those with sampling locations in our phylogenetic analysis (Pentinsaari et al., 2014; Hendrich et al., 2015; Nie et al., 2017; Coral şahin et al., 2018; Lalrinfeli et al., 2019) (Supplementary Table 1). Sequences with obvious errors were deleted, and others were used for follow-up analysis.
Population Genetic Diversity Analysis
For mitochondrial DNA, sequencing results from both strands were assembled using SeqMan in the LASERGENE version 7.1.2 (DNASTAR, Inc., United States). Sequences of the cox1 were aligned with MUSCLE (Edgar, 2004) implemented in MEGA version X (Kumar et al., 2018). The number of polymorphic sites (S), number of haplotypes (H), haplotype diversity (Hd), nucleotide diversity (Pi), Tajima’s D, Fu’s Fs and gene flow (Nm) were analyzed in DnaSP version 6 (Rozas et al., 2017). Pairwise mean population differentiation (FST) was estimated using Arlequin 3.5 (Excoffier and Lischer, 2010).
For microsatellites, the number of alleles and the observed heterozygosity (Ho) were analyzed using the macros Microsatellite Tools (Park, 2001). The null allele frequencies and FST with excluding of null alleles (ENA) were estimated using the software FreeNA (Chapuis and Estoup, 2007). Deviation from Hardy-Weinberg equilibrium (HWE) for each locus/population combination, FST and inbreeding coefficients (FIS) were estimated in GENEPOP v4.0.11 (Rousset, 2008). Principal Coordinate Analysis (PCoA) and genetic differentiation coefficient (GST) were performed with the GenAlex program ver. 6.502 (Peakall and Smouse, 2012). The gene flow was calculated using the formula Nm = 0.5 (1- GST)/GST. HP-RARE v1.1 (Kalinowski, 2010) was used to test allelic richness (AR) and allelic richness of private alleles (PAR) of each site. GENCLONE v2.0 (Arnaud-Haond and Belkhir, 2006) was used to estimate the total number of alleles (AT) and the unbiased expected heterozygosity (HE). We compared the number of alleles (AS) among samples with different sample sizes in GENCLONE. Mantel test (Mantel, 1967), used to estimate correlation between genetic and geographic distances of populations to test isolation by distance, was performed with 1,000 permutations based on Genepop.2
Phylogenetic and Population Genetic Structure Analysis
A Bayesian-inference phylogenetic tree was constructed for mitochondrial DNA using Mrbayes v3.2.2 (Ronquist et al., 2012) to examine phylogenetic relationships among cox1 haplotypes of SFB. Four independent Markov chains for 50 million MCMC generations were run with tree sampling every 5,000 generations and a burn-in of 2,500 trees.
For microsatellite data, the population genetic structure was first investigated using the STRUCTURE v2.3.4 program (Pritchard et al., 2000). We used 30 replicates of each K-value from 1 to 8, with 200,000 Markov chain Monte Carlo iterations and a burn-in of 100,000 iterations. The results were uploaded to the online software Structure Harvester v0.6.94 (Earl and Vonholdt, 2012) to determine the optimal K-value by a Delta K method (last accessed on 28, Dec. 2020).3 The K-value with the highest ΔK represents the number of potential genetic clusters in the population. Membership coefficient matrices (Q-matrices) associated with the optimal K were processed using CLUMPP v1.12 (Jakobsson and Rosenberg, 2007) and then visualized using the DISTRUCT v1.1 (Taubert et al., 2019). Finally, we used discriminant analysis of principal component (DAPC) to analyze population genetic structure under default settings to complement the STRUCTURE analysis. This analysis was run using an R-package adegenet v1.4-2 (Jombart et al., 2008).
Results
Genome Assembly and Characterization of Microsatellites Across the Genome
A total of 47.88 Gb paired-end (PE) sequences (159,598,840 reads each with a length of 150 bp) were obtained. Trimmed reads were assembled into 595,192 scaffolds with a total length of 342.147 MB ranging from 200 bp to 118.198 KB with an N50 of 600 bp. These contigs were used for microsatellite discovery.
In total, 28,512 microsatellites were isolated from the randomly sequenced genome sequences of SFB with 8,635 mononucleotide repeat sites (30.29%), 6,735 (23.62%) dinucleotide repeat (DNR) sites, 8,115 (28.46%) trinucleotide repeat (TNR) sites, 3,550 (12.4%) tetranucleotide repeat (TTNR) sites, 905 (3.17%) pentanucleotide repeats (PNR) sites and 572 (2.00%) hexanucleotide repeat (HNR) sites. In decreased order, A, AC, AT, and AAT were the most frequent repeat motifs. The microsatellites with the three most frequently repeated motifs occupied 51.9% of all microsatellites. The average length of the repeat region ranged from 14.4 to 49.44 bp.
Development of Microsatellite Markers
The QDD3 program designed 2,585 primer pairs for 1,994 din-, 359 tri-, 70 tetra-, 29 penta-, and 21 hexa-nucleotide microsatellites. Following our stringent filter, 70 primer pairs flanking perfect microsatellites were retained. The number of primer pairs flanking tri-, tetra-and penta-nucleotide microsatellites was 67, 1, and 2, respectively. In our initial test of these 70 primer pairs, 16 pairs generated polymorphic genotypes, 12 pairs failed to amplify in any samples, and 46 pairs failed to amplify in more than two individuals. In the final test, 12 microsatellite markers were retained for population-level genotyping (Table 2).

Table 2. Twelve microsatellite markers developed for Phyllotreta striolata in this study.
Population Genetic Diversity
Based on the mitochondrial cox1 genes which were 647 bp long, we found 44 haplotypes from the collected SFB populations. Haplotype #2 was the most common (61.17% of individuals). Thirty-four haplotypes appeared in only one population. The GX population had the most haplotypes with thirteen, while the ZJ population had the least number of haplotypes (n = 4). The haplotype diversity ranged from 0.338 to 0.931, and the nucleotide diversity ranged from 0.00059 to 0.00497. There were 11 polymorphic sites in the HN population which represented the highest number across the most in all populations (Table 3). The data of Tajima’s D and Fu’s Fs of each population were all negative except for FS of GX (FS = 0.57 without statistical significance). For all populations Tajima’s D and Fu’s Fs were –2.53 and –6.01, respectively, with statistical significance, which showed the SFB populations followed the neutral evolution model and there was a population expansion in recent history. The Nei‘s Nm of haplotypes and all sequences was 4.10 and 3.37, respectively.
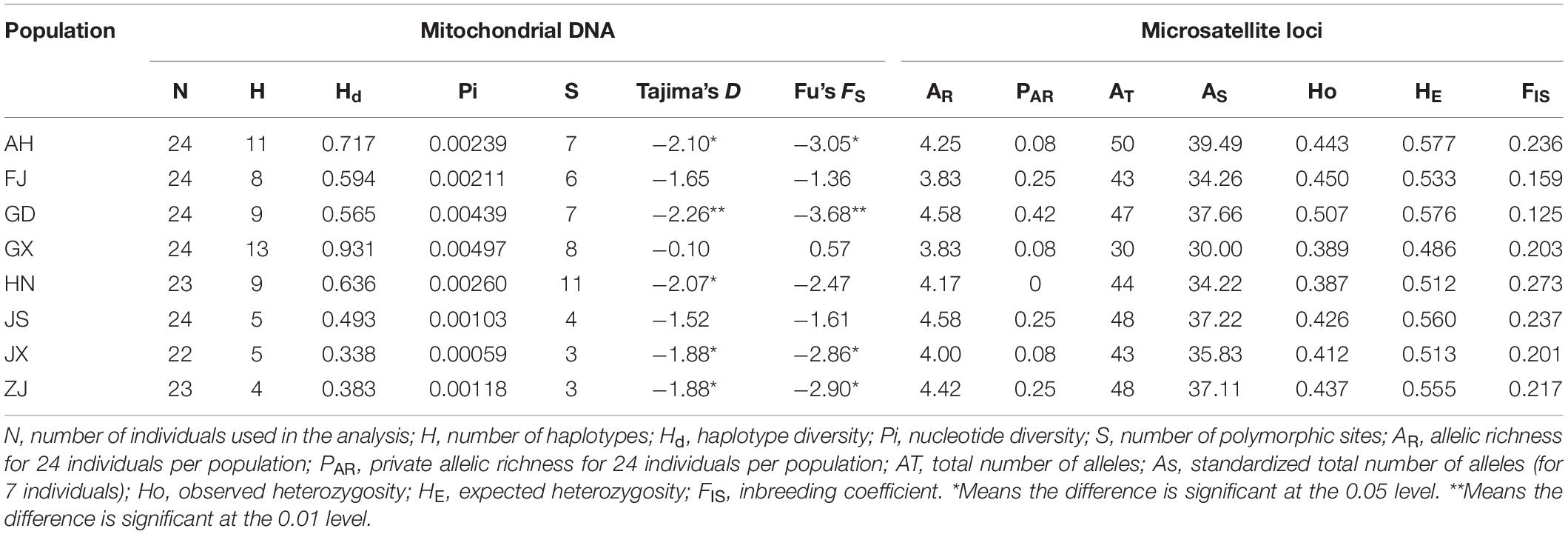
Table 3. Population genetic diversity of Phyllotreta striolata collected from southern China.
Using all cox1 sequences from the public database, haplotypes were reanalyzed and all populations were divided into thirty-four haplotypes. Haplotypes #30–34 were unique to populations outside China. Haplotype #2 was shared by populations in and out of China. The other twenty-eight haplotypes were unique to the populations of China. The pairwise distance among all haplotypes ranged from 0.0018 to 0.0143 (Table 4). We also calculated the pairwise population genetic differentiation among eight populations and FST-values ranged from –0.0187 (FJ-ZJ) to 0.1374 (JX-GX). A moderate genetic differentiation was observed between GX and other populations based on the FST-values ranging from 0.0367 to 0.1374 (Table 5).
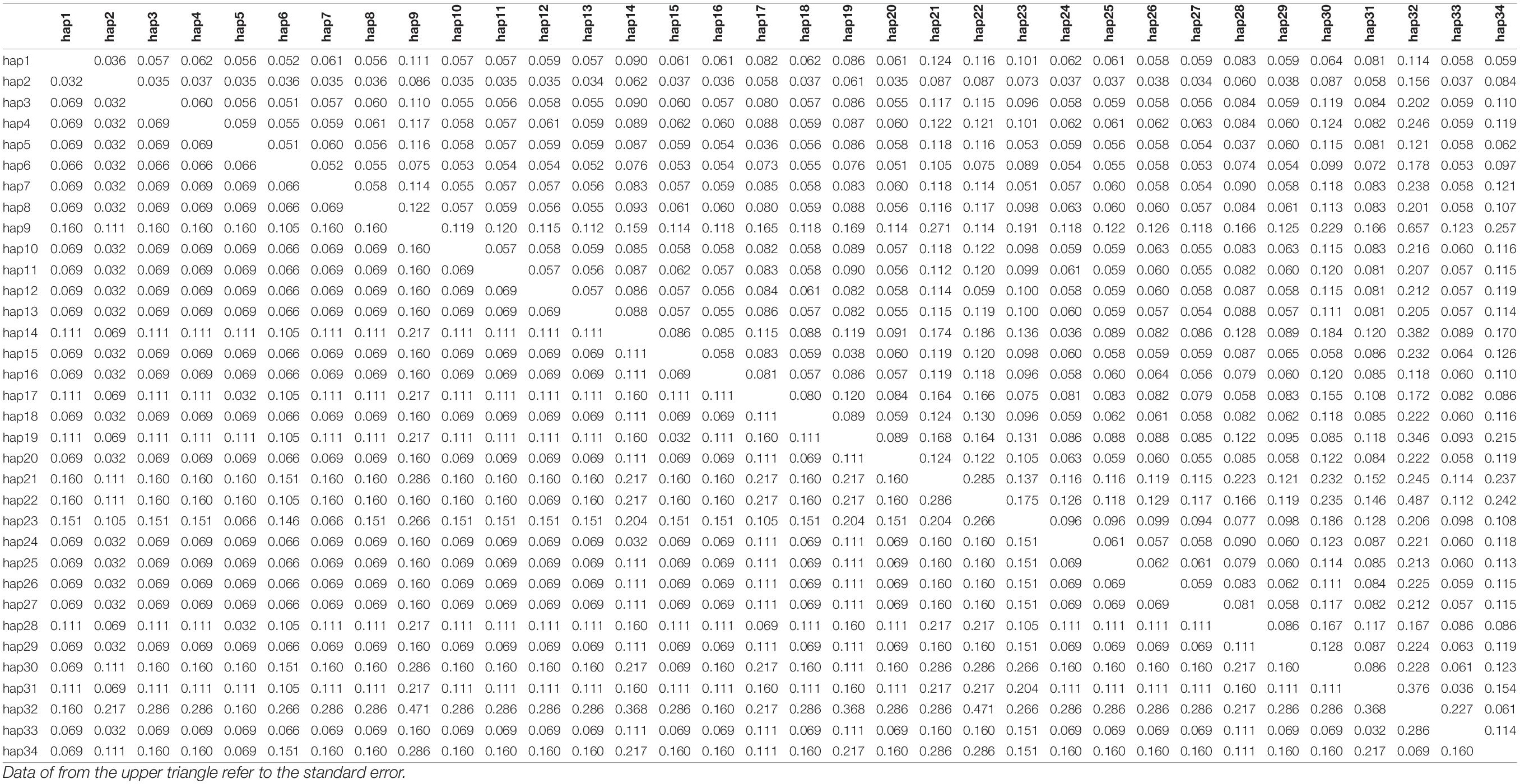
Table 4. Pairwise genetic distance among 34 mitochondrial cox1 haplotypes.
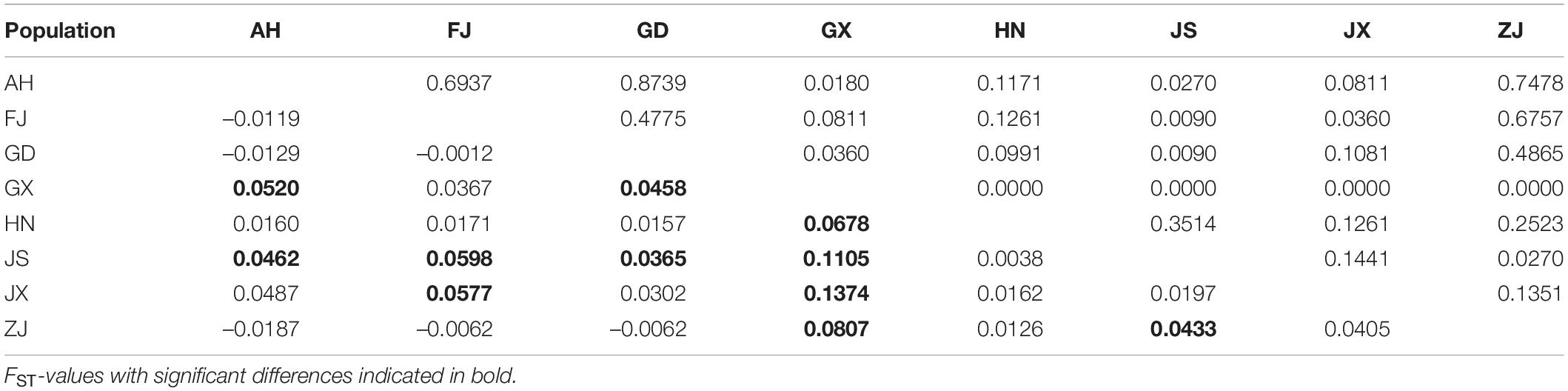
Table 5. Pairwise population genetic differentiation (FST, lower triangle) and P-values (upper triangle) among eight populations of Phyllotreta striolata based on haplotypes of the cox1 gene.
Based on the 12 developed microsatellite markers, we evaluated the genetic diversity in eight populations of SFB. Significant departures from HWE (p < 0.05) were detected in 53 of the 96 population-locus combinations after sequential Bonferroni correction. The population-locus pairs colonized by primer PS-22 all showed significant departures from HWE. Thirty-five of the 528 locus-locus pairs showed linkage disequilibrium in at least one population (p < 0.05), whereas five (S08-S21, S20-S13, S09-S22, S22-S33, and S07-S33 pair) of 66 locus pairs showed linkage disequilibrium across all populations.
Genetic diversity parameters varied among populations. The observed (Ho) and expected (HE) heterozygosity values ranged from 0.387 to 0.507 and from 0.486 to 0.577, respectively. The total number of alleles was the highest in population AH, with a value of 50. In calculating the standardized total number of alleles, we removed locus S06 because of the failure of amplification in the population JX. The inbreeding coefficient of all populations ranged from 0.125 to 0.273 (Table 3). FST-values ranged from –0.0125 (JX-FJ) to 0.0458 (JS-GX). FST-values of populations pairs JX-FJ, JX-GD, and JX-GX are below 0 (Table 6). FST-values had significant differences among 15 populations (AH-FJ, AH-GD, AH-GX, AH-HN, AH-JX, FJ-GD, FJ-GX, FJ-HN, FJ-JS, FJ-ZJ, GD-JS, GD-JX, GX-JS, HN-JS, and JS-JX). The GST of total alleles is 0.011 with a Nm of 44.88, indicating high geneflow among SFB populations. We also calculated the FST-values with excluding of ENA but the results are similar to that without ENA (Table 6). Populations GX and JS have the highest geographical distance and genetic distance. However, the JX and FJ have the nearest geographical distance but not the least genetic distance. The Principal Coordinate Analysis (PCoA) showed no obvious geographic structure (Figure 2). The first and second principal coordinates explained 21.78 and 16.65% of the total variation, respectively.
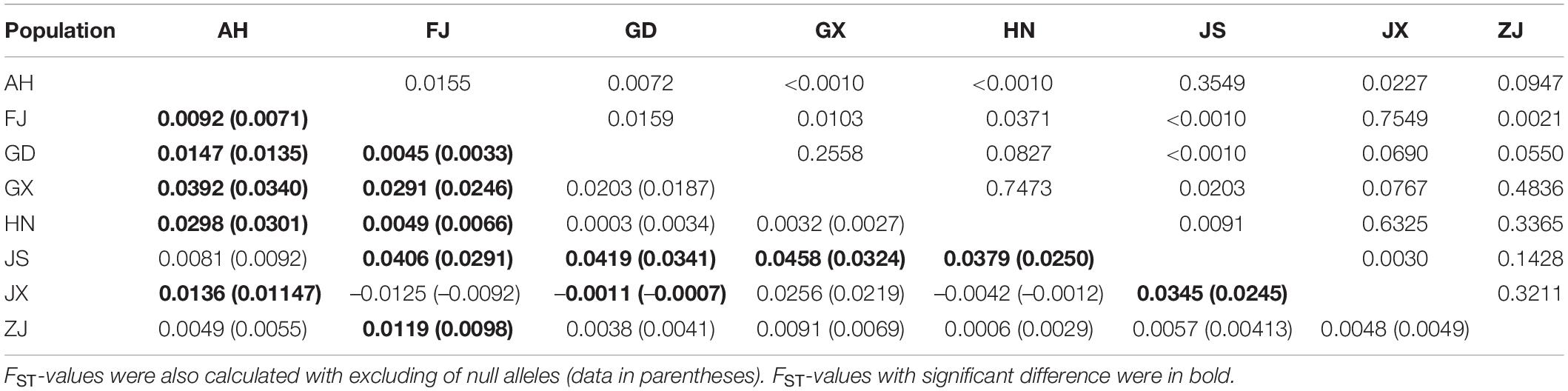
Table 6. Pairwise population genetic differentiation (FST, lower triangle) and P-values (upper triangle) among eight populations of Phyllotreta striolata based on microsatellites.
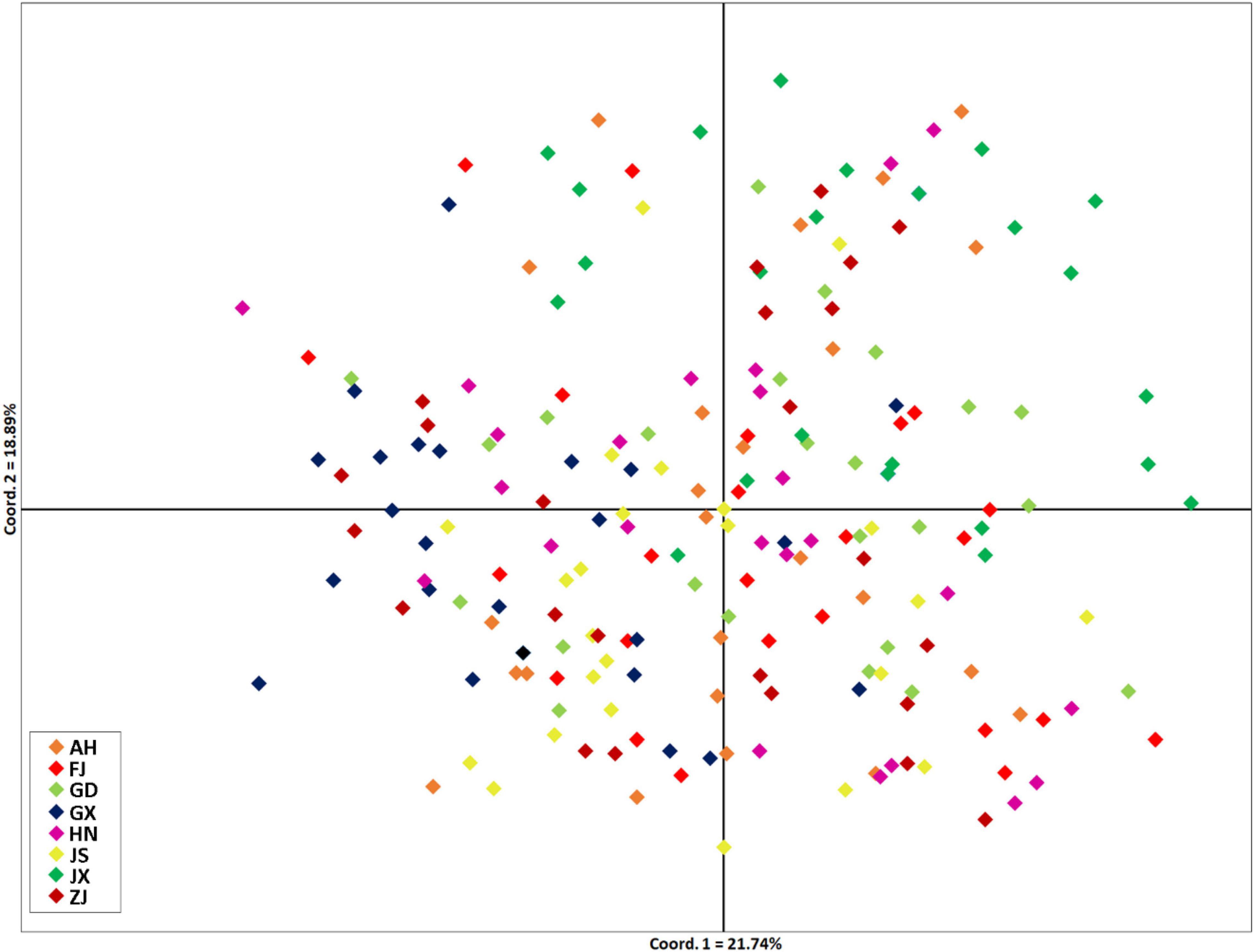
Figure 2. PCoA based on pairwise Nei’s genetic distance estimates for the populations.
To determine whether the genetic distance in SFB is significantly associated with geographic distance, we conducted Mantel tests between pairwise genetic differentiation [FST/(1 – FST)] and geographical distance matrix based on haplotypes and microsatellites (Figure 3). The relationships of genetic distance and geographic distance were not identical based on microsatellites (R2 = 0.2373, P = 0.014) and mtDNA (R2 = 0.0365, P = 0.155).
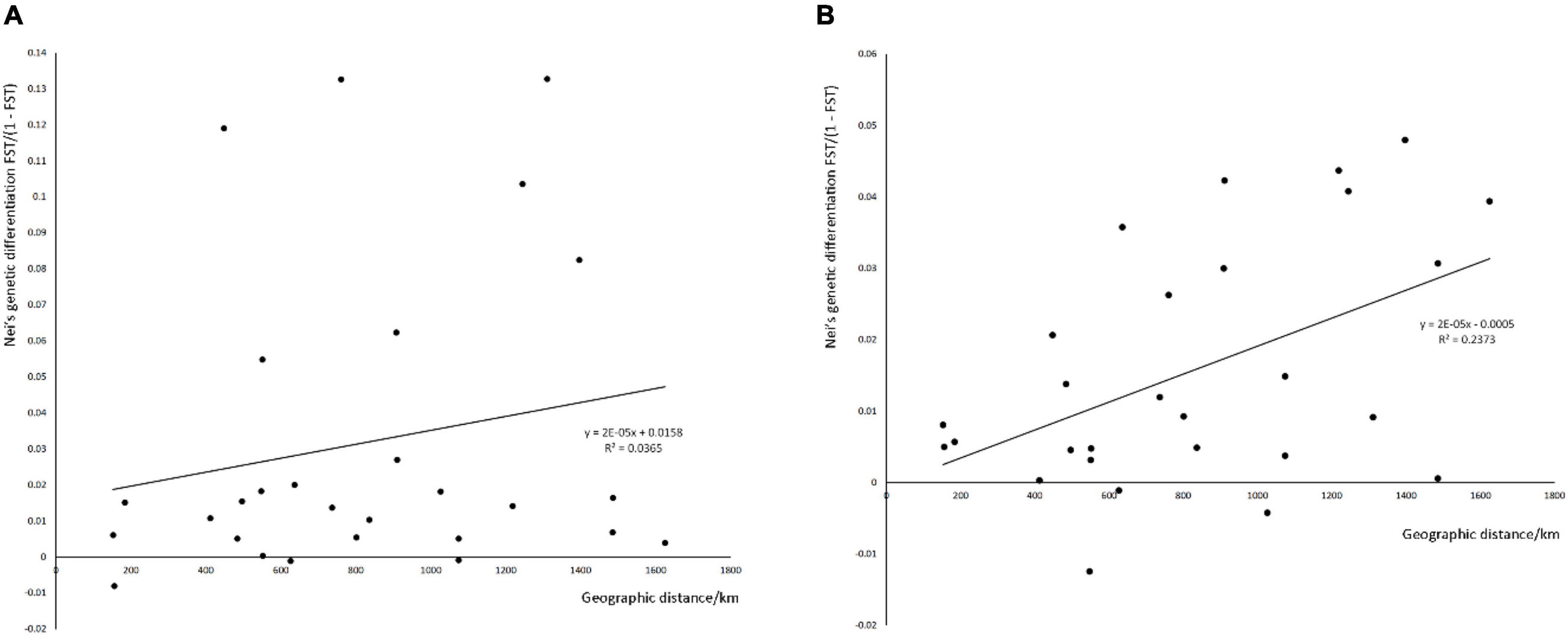
Figure 3. Mantel test of the geographic distance and Nei’s genetic distance based on haplotypes of cox1 gene (A) and microsatellites (B).
Population Genetic Structure and Phylogenetic Relationship Analysis
We reconstructed the phylogenetic tree based on thirty-two haplotypes (Figure 4). Haplotypes from different geographical regions were mixed in the phylogenetic tree, which was consistent with PCoA analysis. We did not find any geographically related clades with low supporting data, except for one clade composed of two haplotypes (30 and 32) unique to populations outside of China, and another clade composed of three haplotypes (28, 29, and 31) unique to populations outside of China.
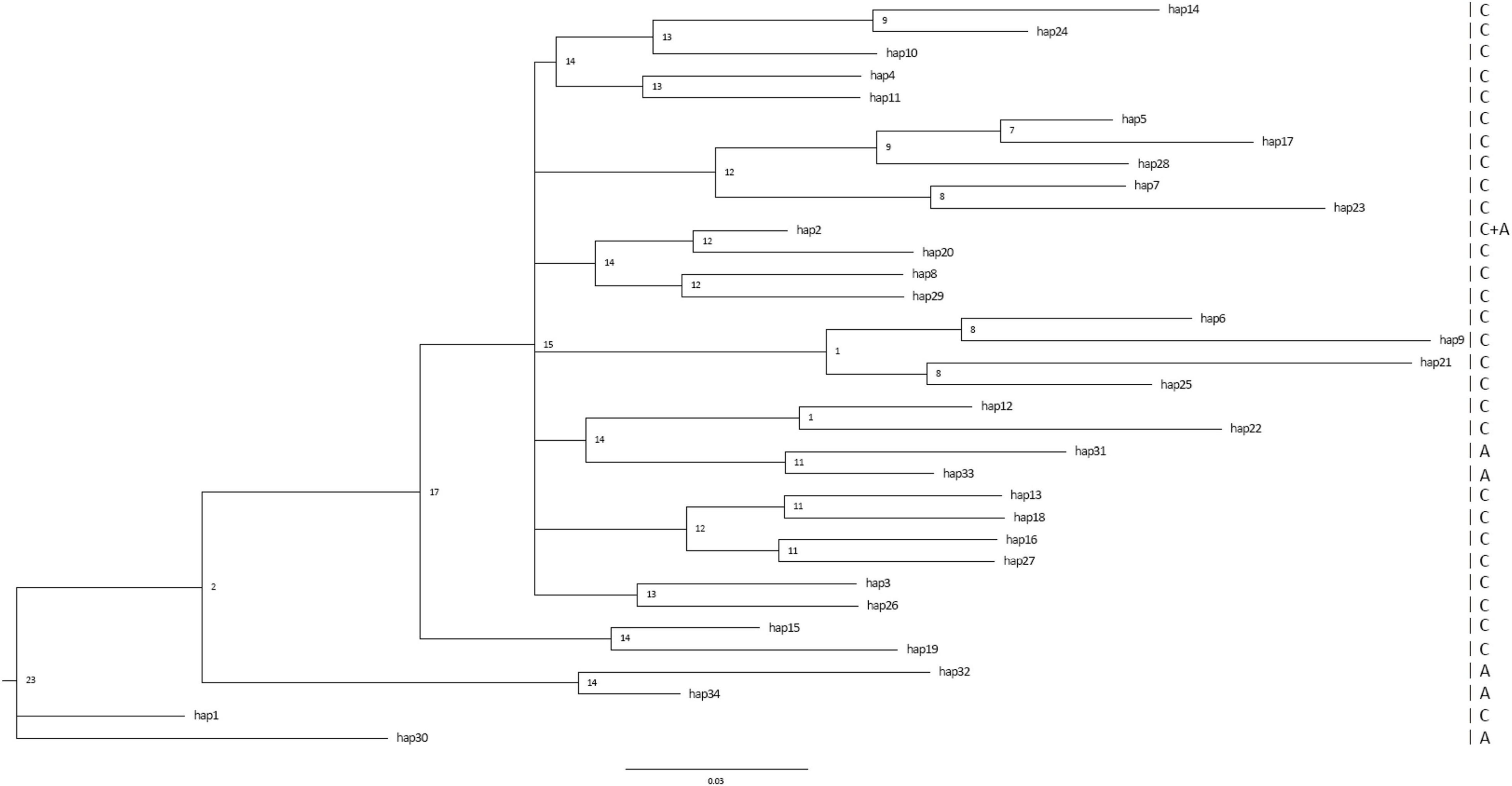
Figure 4. Bayesian-inference phylogenetic relationships among mitochondrial cox1 haplotypes of Phyllotreta striolata worldwide. C, haplotypes present in China. A, haplotypes present outside China.
The STRUCTURE analysis indicated that the optimal K-value was two. There was a lack of population structure when K increased from 2 to 3 (Figure 5A). DAPC analysis revealed similar patterns to those obtained from the STRUCTURE clustering analyses with all populations clumping together (Figure 5B).
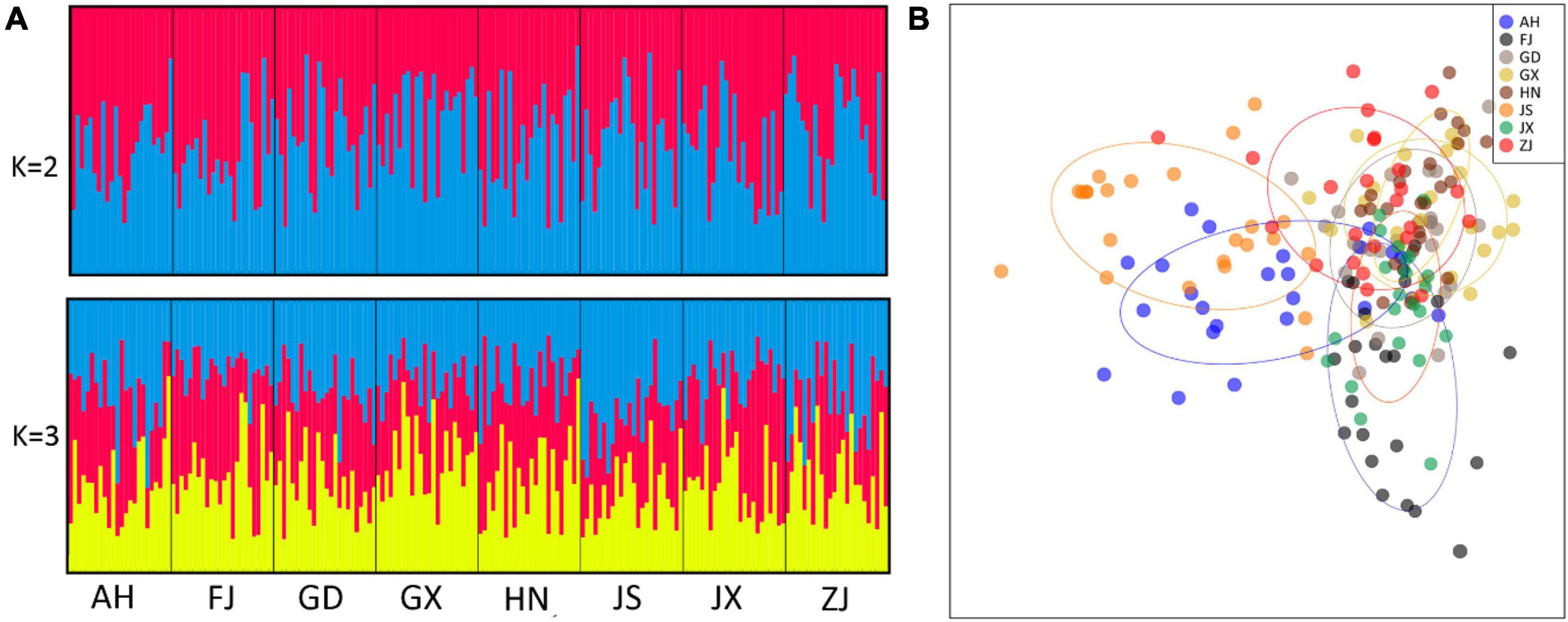
Figure 5. Genetic structure of Phyllotreta striolata populations from southern China inferred from STRUCTURE (A) and DAPC (B) analyses based on 12 microsatellite loci. The optimal number of clusters for the STRUCTURE analysis was three; K = 2 and 3 are presented (A) arranged by population. DAPC analysis shows all populations are clumping together (B).
Discussion
This study developed universal microsatellite markers of SFB based on a de novo assembled draft genome. Both mitochondrial DNA and microsatellite markers point to the low population genetic diversity of SFB in southern China. The haplotype analyses using partial mitochondrial cox1 genes also confirms the low genetic diversity in Europe and Asia since only two haplotypes were identified from the public databases. A lack of population structure was supported by population genetic structure analysis results using STRUCTURE and DAPC.
The mononucleotide repeat sites included four types as (A)n, (T)n, (C)n, and (G)n. The number of (A)n and (T)n far exceeded that of (C)n and (G)n in SFB, which is similar to other eukaryotic species (Katti et al., 2001). (A)n, (AT)n, and (AAT)n were the most frequent motifs in SFB, as in other beetle species (Song et al., 2020). The frequency and density of microsatellite markers varied among species (Selkoe and Toonen, 2010; Xu et al., 2017; Liu et al., 2019). The dominant repeats in SFB are dinucleotides except for an occasional mononucleotide. Interestingly, this differs from other beetle species that have been sequenced in the Family Chrysomelidae, in which the dominant repeats are trinucleotide or tetranucleotide repeats in Leptinotarsa decemlineata (Coleoptera, Chrysomelidae) and Diabrotica virgifera (Coleoptera, Chrysomelidae), respectively (Kim et al., 2008; Liu et al., 2018).
In general, the results based on microsatellites and haplotypes of the cox1 gene support the notion that the sampled populations of SFB have low genetic variation and lack population genetic structure in southern China, as suggested by multiple lines of analyses such as pairwise F-statistics, PCoA, DAPC, STRUCTURE and phylogenetic reconstruction. The lack of population genetic structure was found in some species, such as recently introduced species Obolodiplosis robiniae and Frankliniella occidentalis (Shang et al., 2015; Cao et al., 2017) or migratory species like Plutella xylostella, whose Sichuan populations are sources populations for northern immigrants, and southern China and Yunnan populations are sources populations for central-eastern populations (Chen et al., 2020). In order to escape suboptimal environmental or weather conditions, some insects have annual migrations (Zhan et al., 2011). In Coleoptera, some species, like Leptinotarsa decemlineata and Pachysternum capense can migrate (Ferro et al., 1991; Greń and Górz, 2020). We did not find a records of migration events for the of SFB and studied have shown that it has been established in Eurasia for several centuries (Rousseau and Lesage, 2016). Our hypothesis, that the low levels of genetic diversity and population differentiation for the SLB are due to high levels of gene flow among populations, needs to be further tested.
Consistently low variation among groups is indicative of high levels of gene flow across the geographic range as seen microsatellite data. Pairwise comparisons of FST-values from mtDNA data are also consistent with high gene flow across the range, with generally low FST-values between populations. Haplotypes (n = 2) widely exist in all populations also suggest a high-level of gene flow among 8 populations. Cruciferous vegetables are a kind of common vegetable in China and is the preferential host of SFB. The high gene flow maybe due to the transportation of fresh vegetable from south to north in China, such as from Anhui Province to Zhejiang Province. The passive transportation accelerates gene flow among different populations (Martinez-Hernandez et al., 2021). The high geneflow, however, may be the evidence of its migration (Seymour et al., 2016; Yang et al., 2020).
But there are also some divergences between these two types of molecular markers. A moderate genetic differentiation was observed between GX and other populations based on genetic distances of mtDNA, which were not observed based on microsatellites. Because mtDNA is sensitive to founder effects and small population size, the probable loss or gain of a mtDNA haplotype will be greater for small populations (Roderick, 1996). Furthermore, the Mantel test of microsatellites suggests evidence of a positive relationship in genetic distances (FST) between genetic and geographic distances, while the result based on mtDNA had no significant difference. This may be caused by the use of only a single marker of mtDNA vs. 12 markers of microsatellite. Different parts of mtDNA evolve at different rates, thus different parts of mitochondrial DNA should be considered for future studies to find higher-level population differentiation (Avise et al., 1992). The significant Mantel test suggests that while genetic differences between populations are rather small, they tend to accumulate as geographic distances increase, supporting the non-negligible population structure.
We found that the population collected from Guangxi province of southwest China had the highest number of unique haplotypes. Many species in East Asia were found to have originated from southwestern China and expanded their distribution range northward during the interglacial periods in the Quaternary (Dong et al., 2013; Wei et al., 2015; Yang et al., 2020). Our study did not include samples collected from southwestern areas such as Yunnan and Sichuan. The inclusion of Guangxi populations and the relatively higher level of genetic diversity in this population may indicate that the SFB may also have originated from southwestern China and subsequently dispersed to other areas.
Conclusion
In conclusion, our study provides a draft genome and a set of microsatellites developed from the genome. Based on these microsatellites and a segment of mitochondrial gene, we investigated the population genetic structure of the SFB in southern China, where it is a common pest. We found a lack of population differentiation of SFB among populations in southern China, which might be caused by high frequencies of gene flow among populations. Either passive transportation or migration events will accelerate the gene flow among different populations. Species collected from Guangxi Province showed more genetic divergence than others based on mitochondrial gene which indicates that the SFB may originate from southwestern China. In consideration of the limited collection range, the hypothesized movements need further investigation.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA761897. Data are also available on the Barcode of Life Datasystems (BOLD) database under Project–SFB “Barcoding of Phyllotreta striolata in southern China” (doi: 10.5883/DS-188PSLQ).
Author Contributions
QL analyzed the data and wrote the article. QL and G-ML collected the data. Y-LZ and S-JW revised the article. All authors contributed significantly to the drafts and gave final approval for publication.
Funding
This research was funded by the China Agriculture Research System of MOF and MARA (CARS-23-C05), and “Three rural issues and six participants” of Scientific and Technological Cooperation Projects of Zhejiang Province-9#. The integration, demonstration and promotion of green technology of prevention and control of Phyllotreta striolata (CTZB-F180706LWZ-SNY1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Lina Sun and Lijun Cao for their help in experiments and data analyses. We are also very grateful to all collectors for collecting specimens.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.775414/full#supplementary-material
Footnotes
- ^ http://msdb.biosv.com/
- ^ https://genepop.curtin.edu.au/
- ^ http://taylor0.biology.ucla.edu/structureHarvester/
References
Andersen, C. L., Hazzard, R., VanDriesche, R., and Mangan, F. X. (2006). Alternative management tactics for control of Phyllotreta cruciferae and Phyllotreta striolata (Coleoptera: Chrysomelidae) on Brassica rapa in Massachusetts. J. Econ. Entomol. 99, 803–810. doi: 10.1603/0022-0493-99.3.803
Andrews, S. (2004). FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Arnaud-Haond, S., and Belkhir, K. (2006). GENCLONE: a computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol. Ecol. Notes 7, 15–17.
Atirach, N., Sirirut, M., and Lewis, E. E. (2021). Biological control potential of entomopathogenic nematodes against the striped flea beetle, Phyllotreta sinuata Stephens (Coleoptera: Chrysomelidae). Crop Prot. 141:105448.
Avise, J. C., Alisauskas, R. T., Nelson, W. S., and Ankney, C. D. (1992). Matriarchal population genetic structure in an avian species with female natal philopatry. Evolution 46, 1084–1096. doi: 10.1111/j.1558-5646.1992.tb00621.x
Blacket, M. J., Robin, C., Good, R. T., Lee, S. F., and Miller, A. D. (2012). Universal primers for fluorescent labelling of PCR fragments–an efficient and cost-effective approach to genotyping by fluorescence. Mol. Ecol. Resour. 12, 456–463. doi: 10.1111/j.1755-0998.2011.03104.x
Bolger, A. M., Marc, L., and Bjoern, U. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cao, C., Huang, D., Yao, J., Zhu, Z., Zheng, J., Zhou, R., et al. (2020). Field application techniques for control of Phyllotreta striolata with microbial insecticides on radish. Chin. J. Biol. Control. 36, 987–991.
Cao, L. J., Li, Z. M., Wang, Z. H., Zhu, L., Gong, Y. J., Chen, M., et al. (2016). Bulk development and stringent selection of microsatellite markers in the western flower thrips Frankliniella occidentalis. Sci. Rep. 6:26512. doi: 10.1038/srep26512
Cao, L. J., Wang, Z. H., Gong, Y. J., Zhu, L., and Wei, S. J. (2017). Low genetic diversity but strong population structure reflects multiple introductions of western flower thrips(Thysanoptera: Thripidae) into China followed by human-mediated spread. Evol. Appl. 10, 391–401. doi: 10.1111/eva.12461
Chai, W. (2010). The habit and control method of Phyllotreta striolata (Fabricius) on Brassica napus in Hexizoulang in Gan su Provence, China. China Plant Prot. 30, 23–24.
Chapuis, M. P., and Estoup, A. (2007). Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631. doi: 10.1093/molbev/msl191
Chatterji, S., and Pachter, L. (2006). Reference based annotation with GeneMapper. Genome Biol. 7:R29. doi: 10.1186/gb-2006-7-4-r29
Chen, M. Z., Cao, L., Li, B., Chen, J., and Wei, S. (2020). Migration trajectories of the diamondback moth Plutella xylostella in China inferred from population genomic variation. Pest Manag. Sci. 77, 1683–1693. doi: 10.1002/ps.6188
Coral şahin, D., Magoga, G., Özdikmen, H., and Montagna, M. (2018). DNA barcoding as useful tool to identify crop pest flea beetles of Turkey. J. Appl. Entomol. 143, 105–117. doi: 10.1111/jen.12566
Dong, L., Heckel, G., Liang, W., and Zhang, Y. (2013). Phylogeography of Silver pheasant (Lophura nycthemera L.) across China: aggregate effects of refugia, introgression and riverine barriers. Mol. Ecol. 22, 3376–3390. doi: 10.1111/mec.12315
Earl, D. A., and Vonholdt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emese, M., Caroline, C., Vincent, D., André, G., and Thibaut, M. (2010). QDD: a user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics 26, 403–404. doi: 10.1093/bioinformatics/btp670
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Feng, H. T., Huang, Y. J., and Hsu, J. C. (2000). Insecticide susceptibility of cabbage flea beetle (Phyllotreta striolata (Fab.)) in Taiwan. Plant Prot. Bull. 42, 67–72.
Ferro, D. N., Tuttle, A. F., and Weber, D. C. (1991). Ovipositional and flight behavior of overwintered Colorado potato beetle (Coleoptera: Chrysomelidae). Environ. Entomol. 20, 1309–1314. doi: 10.1093/ee/20.5.1309
Folmer, O., Black, M., Wr, H., Lutz, R., and Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome C oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Gao, Z. Z., Wu, W. J., and Cui, Z. X. (2000). Study the host range of Phyllotreta striolata (Fabricius). Ecol. Sci. 19, 70–72.
Greń, C., and Górz, A. (2020). Coprophagous hydrophilid beetles (Coleoptera, Hydrophilidae, Sphaeridiinae) distribution in the Polish Carpathians. Insects 11:355. doi: 10.3390/insects11060355
He, H. L., Bin, S. Y., and Lin, J. T. (2012). The research progress of biology, ecology characters and occurrence reasons of Phyllotreta striolata. J. Anhui Agric. Sci. 40, 10683–10686.
Hendrich, L., Morinière, J., Haszprunar, G., Hebert, P. D. N., Hausmann, A., Köhler, F., et al. (2015). A comprehensive DNA barcode database for Central European beetles with a focus on Germany: adding more than 3500 identified species to BOLD. Mol. Ecol. Resour. 15, 795–818. doi: 10.1111/1755-0998.12354
Jakobsson, M., and Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
James, M., Adam, M. A., and Thomas, P. K. (2019). Flea beetle (Coleoptera: Chrysomelidae) populations, effects of feeding injury, and efficacy of insecticide treatments on eggplant and cabbage in southwest Virginia. J. Econ. Entomol. 113, 887–895. doi: 10.1093/jee/toz355
Jombart, T., Devillard, S., Dufour, A. B., and Pontier, D. (2008). Revealing cryptic spatial patterns in genetic variability by a new multivariate method. Heredity 101, 92–103. doi: 10.1038/hdy.2008.34
Kalinowski, S. T. (2010). Hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 5, 187–189. doi: 10.1111/j.1471-8286.2004.00845.x
Katti, M. V., Ranjekar, P. K., and Gupta, V. S. (2001). Differential distribution of simple sequence repeats in eukaryotic genome sequences. Mol. Biol. Evol. 18, 1161–1167. doi: 10.1093/oxfordjournals.molbev.a003903
Kim, K. S., Ratcliffe, S. T., French, B. W., Liu, L., and Sappington, T. W. (2008). Utility of EST-derived SSRs as population genetics markers in a beetle. J. Heredity 99, 112–124. doi: 10.1093/jhered/esm104
Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Kumar, S., Steched, G., Li, M., Knyaz, C., Tamura, K., and Notes, A. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lalrinfeli, R., Behere, G., Firake, D., Sharma, B., Banerjee, A., and Rajesh, T. (2019). Development of DNA barcodes for major insect pest of cole crops in Mid-Hills of Meghalaya. Int. J. Curr. Microbiol. Appl. Sci. 8, 789–799.
Lee, C. F., Chang, H. Y., Wang, C. L., and Chen, W. S. (2011). A review of Phyllotreta Chevrolat in Taiwan (Coleoptera: Chrysomelidae: Galerucinae: Alticini). Zool. Stud. 50, 525–533.
Lianming, D., Yuzhi, L., Xiuyue, Z., and Bisong, Y. (2013). MSDB: a user-friendly program for reporting distribution and building databases of microsatellites from genome sequences. J. Heredity 104, 154–157.
Liu, M., Wang, X., Ma, L., Cao, L., Liu, H., Pu, D., et al. (2019). Genome-wide developed microsatellites reveal a weak population differentiation in the hoverfly Eupeodes corollae (Diptera: Syrphidae) across China. PLoS One 14:e0215888. doi: 10.1371/journal.pone.0215888
Liu, Y., Fu, K., He, J., and Guo, W. (2018). Verfication SSR primers by datamining genome SSR loci in Leptinotarsa decemlineata. J. Environ. Entomol. 40, 633–644.
Martinez-Hernandez, F., Villalobos, G., and Martínez-Ibarra, J. A. (2021). Population structure and genetic diversity of Triatoma longipennis (Usinger, 1939) (Heteroptera: Reduviidae: Triatominae) in Mexico. Infect. Genet. Evol. 89:104718.
Mantel, N. A. (1967). The detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220.
Nie, R. E., Breeschoten, T., Timmermans, M. J. T. N., Nadein, K., Xue, H. J., Bai, M., et al. (2017). The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics 33, 1–18. doi: 10.1111/cla.12196
Olfert, O., Weiss, R. M., Soroka, J. J., and Elliott, R. H. (2017). Bioclimatic approach to assessing factors influencing shifts in geographic distribution and relative abundance of two flea beetle species (Coleoptera: Chrysomelidae) in North America. Can. Entomol. 150, 1–14.
Park, S. D. E. (2001). Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Ph.D. thesis. Dublin: University of Dublin.
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2010). “IDBA – a practical iterative de Bruijn graph de novo assembler,” in Proceedings of the 14th Annual international conference on Research in Computational Molecular Biology (Berlin: Springer).
Pentinsaari, M., Hebert, P. D. N., Mutanen, M., and Fontaneto, D. (2014). Barcoding beetles: a regional survey of 1872 species reveals high identification success and unusually deep interspecific divergences. PLoS One 9:e108651. doi: 10.1371/journal.pone.0108651
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Roderick, G. K. (1996). Geographic structure of insect populations: gene flow, phylogeography, and their uses. Annu. Rev. Entomol. 41, 325–352. doi: 10.1146/annurev.en.41.010196.001545
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rousseau, M., and Lesage, L. (2016). Earliest North American occurrence of Phyllotreta striolata (Coleoptera: Chrysomelidae) from Québec, Canada. Can. Entomol. 148, 476–478.
Rousset, F. (2008). Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 8, 103–106. doi: 10.1111/j.1471-8286.2007.01931.x
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J., Guirao-Rico, S., Librado, P., Ramos-Onsins, S., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Schuelke, M. (2000). An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 18, 233–234. doi: 10.1038/72708
Selkoe, K. A., and Toonen, R. J. (2010). Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol. Lett. 9, 615–629. doi: 10.1111/j.1461-0248.2006.00889.x
Seymour, M., Perera, O. P., Fescemyer, H. W., Jackson, R. E., Fleischer, S. J., and Abel, C. A. (2016). Peripheral genetic structure of Helicoverpa zea indicates asymmetrical panmixia. Ecol. Evol. 6, 3198–3207. doi: 10.1002/ece3.2106
Shang, X., Yao, Y., Huai, W., and Zhao, W. (2015). Population genetic differentiation of the black locust gall midge Obolodiplosis robiniae (Haldeman) (Diptera: Cecidomyiidae): a North American pest invading Asia. Bull. Entomol. Res. 105, 736–742. doi: 10.1017/S000748531500070X
Song, W., Cao, L., Li, B., Gong, Y., Hoffmann, A. A., and Wei, S. (2018). Multiple refugia from penultimate glaciations in East Asia demonstrated by phylogeography and ecological modelling of an insect pest. BMC Evol. Biol. 18:152. doi: 10.1186/s12862-018-1269-z
Song, X., Yang, T., Yan, X., Zheng, F., Xu, X., and Zhou, C. (2020). Comparison of microsatellite distribution patterns in twenty-nine beetle genomes. Gene 757:144919. doi: 10.1016/j.gene.2020.144919
Soroka, J., Grenkow, L., Otani, J., Gavloski, J., and Olfert, O. (2018). Flea beetle (Coleoptera: Chrysomelidae) species in canola (Brassicaceae) on the northern Great Plains of North America. Can. Entomol. 150, 100–115. doi: 10.1603/0022-0493(2008)101[159:dipcap]2.0.co;2
Sun, J., Xian, Z., Liu, X., and Liu, Y. (2010). The cause of serious occurrence of Phyllotreta striolata in Guangxi Nanning and its control measures. Guangxi Agric. Sci. 41, 21–23.
Sun, Z. (2010). Occurrence regularity and control measures of Phyllotreta striolata in Chaidamu area. Qinghai Agro Technol. Ext. 4, 38–39.
Taubert, O., Reinartz, I., Meyerhenke, H., and Schug, A. (2019). diSTruct v1.0: generating biomolecular structures from distance constraints. Bioinformatics 35, 5337–5338. doi: 10.1093/bioinformatics/btz578
Wei, S., Cao, L., Gong, Y., Shi, B., Wang, S., Zhang, F., et al. (2015). Population genetic structure and approximate Bayesian computation analyses reveal the southern origin and northward dispersal of the oriental fruit moth Grapholita molesta (Lepidoptera: Tortricidae) in its native range. Mol. Ecol. 24, 4094–4111. doi: 10.1111/mec.13300
Xu, Y., Li, W., Hu, Z., Zeng, T., Shen, Y., Liu, S., et al. (2017). Genome-wide mining of perfect microsatellites and tetranucleotide orthologous microsatellites estimates in six primate species. Gene 643, 124–132. doi: 10.1016/j.gene.2017.12.008
Yan, X., Lin, Y., Huang, Z., and Han, R. (2018). Characterisation of biological and biocontrol traits of entomopathogenic nematodes promising for control of striped flea beetle (Phyllotreta striolata). Nematology 20, 503–518.
Yang, N., Dong, Z., Chen, A. D., Yin, Y. Q., Li, X. Y., and Chu, D. (2020). Migration of Sogatella furcifera between the Greater Mekong Subregion and northern China revealed by mtDNA and SNP. BMC Ecol. Evol. 20:154. doi: 10.1186/s12862-020-01722-4
Zhan, S., Merlin, C., Boore, J. L., and Reppert, S. M. (2011). The monarch butterfly genome yields insights into long-distance migration. Cell 147, 1171–1185. doi: 10.1016/j.cell.2011.09.052
Keywords: Phyllotreta striolata, southern China, population genetic structure, microsatellite, mitochondrial gene
Citation: Li Q, Li G-M, Zheng Y-L and Wei S-J (2022) Lack of Genetic Structure Among Populations of Striped Flea Beetle Phyllotreta striolata (Coleoptera: Chrysomelidae) Across Southern China. Front. Ecol. Evol. 9:775414. doi: 10.3389/fevo.2021.775414
Received: 14 September 2021; Accepted: 29 December 2021;
Published: 03 February 2022.
Edited by:
Charles K. Lee, University of Waikato, New ZealandReviewed by:
Ruth Freire Alvarez, Heinrich Heine University of Düsseldorf, GermanyIan D. Hogg, Polar Knowledge Canada (POLAR), Canada
Copyright © 2022 Li, Li, Zheng and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Li Zheng, eW9uZ2xpemhlbmdAeWVhaC5uZXQ=; Shu-Jun Wei, c2h1anVuMjY4QDE2My5jb20=