
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 28 January 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.769565
 Mohammed Ahmed1
Mohammed Ahmed1 Nickellaus G. Roberts2Funmilola Adediran3Ashleigh B. Smythe4
Nickellaus G. Roberts2Funmilola Adediran3Ashleigh B. Smythe4 Kevin M. Kocot2
Kevin M. Kocot2 Oleksandr Holovachov1*
Oleksandr Holovachov1*Phylogenetic relationships within many lineages of the phylum Nematoda remain unresolved, despite numerous morphology-based and molecular analyses. We performed several phylogenomic analyses using 286 published genomes and transcriptomes and 19 new transcriptomes by focusing on Trichinellida, Spirurina, Rhabditina, and Tylenchina separately, and by analyzing a selection of species from the whole phylum Nematoda. The phylogeny of Trichinellida supported the division of Trichinella into encapsulated and non-encapsulated species and placed them as sister to Trichuris. The Spirurina subtree supported the clades formed by species from Ascaridomorpha and Spiruromorpha respectively, but did not support Dracunculoidea. The analysis of Tylenchina supported a clade that included all sampled species from Tylenchomorpha and placed it as sister to clades that included sampled species from Cephalobomorpha and Panagrolaimomorpha, supporting the hypothesis that postulates the single origin of the stomatostylet. The Rhabditina subtree placed a clade composed of all sampled species from Diplogastridae as sister to a lineage consisting of paraphyletic Rhabditidae, a single representative of Heterorhabditidae and a clade composed of sampled species belonging to Strongylida. It also strongly supported all suborders within Strongylida. In the phylum-wide analysis, a clade composed of all sampled species belonging to Enoplia were consistently placed as sister to Dorylaimia + Chromadoria. The topology of the Nematoda backbone was consistent with previous studies, including polyphyletic placement of sampled representatives of Monhysterida and Araeolaimida.
Nematodes are a diverse group of animals and include about 25000 species that are found in a wide range of habitats. While they are known to assume diverse lifestyles, their parasitic forms have dominated the subject of research because they directly affect humans (Sudhaus and Fitch, 2001) while the free-living forms have, to some extent, been neglected (Smythe et al., 2019), with the exception of the model genera Caenorhabditis and Pristionchus. Phylogeny and classification of nematodes has been and remains a subject of debates and disagreements between experts. It has long been acknowledged that morphology alone is not sufficient for resolving phylogenetic relationships within Nematoda (Lorenzen, 1981; Coomans, 2000) due to the paucity of informative homologous morphological characters, difficulty in distinguishing between homologous and independently evolved characters, unclear character polarity and the absence of fossil data (Blaxter et al., 1998). Excluding short-lived early classifications of the phylum (Cobb, 1919; Filipjev, 1934), the most widely accepted morphology-based classification proposed by Chitwood (1933, 1958), with the dichotomous division of Nematoda into Aphasmidia/Adenophorea and Phasmidia/Secernentea, came under scrutiny when it became evident that Adenophorea did not represent a monophyletic group (Maggenti, 1963; De Coninck, 1965). Chitwood (1933, 1958) classification continued to remain in use for quite some time (Lorenzen, 1981; Maggenti, 1981), despite strong opposition and repeated proposals to replace it with a new dichotomy or trichotomy-based framework (Goodey, 1963; Gadea, 1973; Andrássy, 1976; Drozdovsky, 1981; Malakhov, 1986; Adamson, 1987). Numerous phylum-wide molecular phylogenetic analyses utilizing the nuclear 18S ribosomal RNA gene (rDNA) have been published since 1998 (Aleshin et al., 1998; Blaxter et al., 1998; Holterman et al., 2006, 2019; Smythe et al., 2006; van Megen et al., 2009). While there was to a large extent consistency between the new molecular trees and morphological interpretation of nematode phylogeny, placements of many smaller and larger clades were far from expected (Sudhaus, 2011). As a result, a revised trichotomy-based classification system was proposed by De Ley and Blaxter (2002, 2004) that partially reflected the 18S rDNA phylogeny and partially remained morphology-based for those taxa that had not been sequenced or for which placement in the phylogeny was not unequivocal.
From the 53 taxa sampled in first phylum-wide 18S rDNA-based phylogeny of nematodes (Blaxter et al., 1998), five main clades were recognized that each comprised lineages implicated in parasitism of plants and/or animals. The first two clades equate to Dorylaimia and Enoplia, while the remaining three belong to “Secernentea.” In the same year, Aleshin et al. (1998) also analyzed the 18S rDNA of 19 nematode taxa. Both studies confirmed the paraphyly of Adenophorea as suggested by several morphological studies mentioned above, but the key limitations of both molecular phylogenies were the extremely limited taxon sampling for free-living taxa, particularly the marine nematodes. This, and the low diversity of the sampled taxa in general, meant that early branching of the nematode tree remained unresolved. To examine how broader taxon sampling and the better representation for free-living/aquatic/marine species could help address the lack of resolution at the root of Nematoda, Holterman et al. (2006) and Meldal et al. (2007), respectively assembled the 18S rDNA sequences of 339 and 212 nematode taxa for their analyses. Holterman et al. (2006) identified 12 major clades, with the earliest one to diverge from the rest of nematodes being Enoplia, thus forming a sister to the Dorylaimia + Chromadoria lineage. Clade II was formed by the subclass Dorylaimia. The remaining clades were all lineages belonging to Chromadoria. The placement of Enoplia as sister to the rest of Nematoda was not supported statistically, although morphological and embryological characters do support their position as the earliest extant branch of Nematoda (Holterman et al., 2006; Smythe et al., 2019). Meldal et al. (2007) obtained a polytomy at the base of their tree indicating that the earliest-diverging lineage could not be identified, at least not by using the 18S rDNA data. The situation did not improve even when van Megen et al. (2009) sampled more than three times the number of taxa compared to Holterman et al. (2006). Despite its popularity as a molecular marker for reconstruction of deep phylogenies, 18S rDNA alone lacks the resolving power needed to better clarify relationships at the root of the nematode tree (Blaxter and Koutsovoulos, 2015). It has also become apparent that improved taxon sampling combined with better alignments and more realistic phylogenetic algorithms are not sufficient to resolve the relationships at the root of the Nematoda tree using 18S rDNA alone. This is demonstrated in the most recent 18S rDNA-based analysis involving approximately 2,700 sequences, the Dorylaimia + Chromadoria lineage was also not well supported (Holterman et al., 2019). Another shortcomings of phylum-wide 18S rDNA phylogenetic hypotheses of nematodes published to date is a lack of support for the “backbone” of the phylogenetic tree of the group, and complete lack of resolution (phylogenetic signal) in many lineages within the phylum (Ahmed and Holovachov, 2021).
Whole-genome or transcriptome-based phylogenetic analyses are increasingly popular due to increased accessibility to high-throughput sequencing (e.g., declining costs). The integration of whole genomes in phylogenetic studies has been shown to produce a better understanding of relationships at deeper taxonomic levels (Daubin et al., 2002). By providing additional data, whole-genome analyses can potentially increase the overall resolution of phylogenetic relationships and make it possible to filter the noise out of true phylogenetic signals (Eisen and Fraser, 2003). Despite the potential benefits of genome-wide phylogenetic approaches, their application to studying nematode phylogenies is yet to have the expected impact on our understanding of relationships within the phylum. This is because the few studies that have attempted multi-gene or genome-wide approaches (Parkinson et al., 2004; Wasmuth et al., 2008; Blaxter et al., 2012, 2016; Blaxter and Koutsovoulos, 2015; Koutsovoulos, 2015) often have used very limited taxon sampling, with the taxa mostly dominated by model and parasitic species (Smythe et al., 2019). Most of these analyses had outcomes that were generally consistent with earlier 18S rDNA-based phylogenies. Smythe et al. (2019) collated the most taxonomically comprehensive dataset for Nematoda to-date, including new transcriptomes for one freshwater and seven marine species that expanded the representation of Clade I (= Enoplia) beyond a single taxon (as in Blaxter and Koutsovoulos, 2015; Koutsovoulos, 2015) to eight taxa. A key finding of Smythe et al. (2019) was support for the placement of Enoplia as sister to all other Nematoda. This result was similar to the topology of Koutsovoulos (2015) but was based on greater taxon sampling of Enoplia. However, Smythe et al. (2019) found that support for Enoplia as closest to the root of the nematode tree was weaker when orthology was inferred using a more stringent method based on HaMStR (Ebersberger et al., 2009) and PhyloTreePruner (Kocot et al., 2013).
Of the six orders within Chromadoria, three, namely Monhysterida, Desmodorida and Desmoscolecida, had no representation in the analysis of Smythe et al. (2019) and the sampled taxa were, as in previous phylogenomic analyses, massively dominated by species of the order Rhabditida. A significant number of species of these missing orders of Chromadoria are marine and their inclusion could help balance the taxonomic distribution of the sampled taxa and offer a better avenue for the comparison of genome-wide analyses with 18S rDNA-based ones. Moreover, for some of the clades included in Smythe et al. (2019), there was insufficient representation to allow for assessment of their monophyly. In the current study, we sought to address these issues by sampling an even larger set of genomes and transcriptomes from a diverse group of nematodes. We mined hundreds of genome and transcriptome datasets from public databases, including datasets published since 2019 or omitted by Smythe et al. (2019). We supplemented these data with 19 newly generated transcriptomes from distantly related marine and terrestrial nematodes. We opted to sequence transcriptomes because they are less costly and relatively easier to assemble and annotate than genomes, while still providing sufficient amount of information for phylogenetic studies.
The taxa that mostly dominate the genome and transcriptome datasets in public databases are those belonging to Rhabditina, Tylenchina, and Spirurina, all of which belong to a single order, Rhabditida. Within Dorylaimia, Trichinellida is a group with a good number of genomes in public databases. Our goal was to include most of the available genomic and transcriptomic datasets, excluding only overrepresented species or tissue-specific transcriptomes. However, since this assembly of species is still heavily biased toward parasitic and model species, we created subsets of genomes/transcriptomes for Trichinellida, Spirurina, Tylenchina and Rhabditina and analyzed them separately from the main Nematoda phylogeny. And in the main Nematoda phylogeny, we included only few taxa for overrepresented lineages. By sampling more species, our secondary goal was also to test for monophyly of higher taxa where enough data is available, especially if such were unresolved in previous phylogenomic analyses or 18S rDNA phylogenies.
Data analyzed in this study included 19 newly generated transcriptomes (Table 1, Ahmed et al., 2021), as well as 225 of publicly available genomes and 61 publicly available transcriptomes selected to represent the most complete phylogenomic dataset of the phylum Nematoda to date, albeit still heavily biased toward parasitic and model species (referred to in individual sections below, see also Supplementary Information File). Seven genomes and 49 transcriptomes were re-assembled and annotated from raw data, due to original assemblies being publicly unavailable, while 34 genomes and 12 transcriptomes were re-annotated for the same reason (annotations not available to the public).
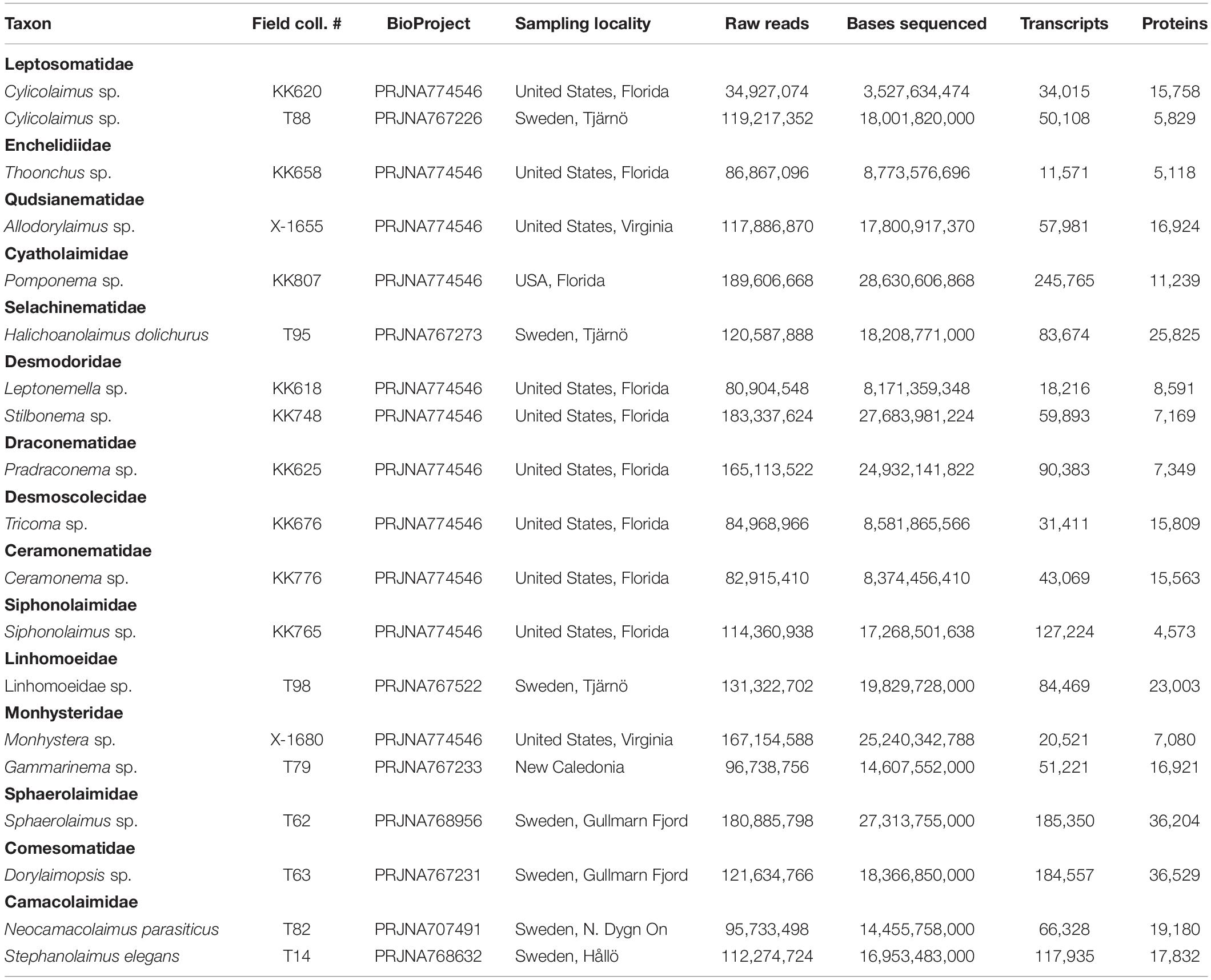
Table 1. Classification, names, accession numbers (BioProject/BioSample), sequencing and assembly statistics for newly generated transcriptomes.
Some species are represented by multiple genomes and transcriptomes. However, in the main Nematoda phylogeny, the number of conspecific datasets was kept to one for each species (with the exception of Plectus sambesi and Plectus murrayi represented each by two datasets), in order to avoid unnecessary computational burden. Several publicly available datasets were not included due to them being misidentified (see Smythe et al., 2019), contaminated (e.g., Pratylenchus brachyurus PRJNA551772 may be contaminated with Cephalobidae), of low quality or difficult to assemble. Polyploid hybrids from the genus Meloidogyne (e.g., M. arenaria PRJNA438575, Sato et al., 2018; M. arenaria PRJEB8714, M. floridensis PRJEB8714, M. incognita PRJEB8714, and M. javanica PRJEB8714, Blanc-Mathieu et al., 2017) were also excluded due to problems identifying orthologs. The classification used here and in the Supplementary Files is based on De Ley and Blaxter (2004) unless specified otherwise.
Nematodes (Figure 1) were collected and isolated from marine sediments or soil following standard protocols (Higgins and Thiel, 1988; van Bezooijen, 2006). Preservation and subsequent processing of samples varied. Cylicolaimus sp. from Sweden, Allodorylaimus sp. Halichoanolaimus dolichurus, Leptonemella sp., Stilbonema sp., Linhomoeidae sp., Monhystera sp., Gammarinema sp., Sphaerolaimus sp., Dorylaimopsis sp., Neocamacolaimus parasiticus, and Stephanolaimus elegans were preserved in RNAlater solution, while Cylicolaimus sp. from United States, Thoonchus sp., Paradraconema sp. and Tricoma sp. were frozen in RNAqueous lysis buffer for RNA extraction, and Pomponema sp., Ceramonema sp., and Siphonolaimus sp. were frozen in Takara SMART-Seq lysis buffer for reverse transcription directly from lysed cells. Subsequently, samples were stored at –80°C (samples in lysis buffer) or –20°C (samples in RNAlater).

Figure 1. Select images of target species for newly generated transcriptomes: (A,B) Tricoma sp., female, United States, Florida, anterior end (A) and tail (B); (C) Thoonchus sp. female, United States, Florida, head; (D) Allodorylaimus sp. male, United States, Virginia, tail; (E,F) Stilbonema sp., female, United States, Florida, head (E) and tail (F) showing symbiotic bacterial coat; (G) Pomponema sp. female, United States, Florida, mid-body showing cuticle and pigment spots; (H,I) Siphonolaimus sp. female, United States, Florida, multifocal composite image of head (H) and mid-body showing symbionts (I); (J) Cylicolaimus sp. male, United States, Florida, tail; (K,L) Ceramonema sp. female, United States, Florida, head (K) and mid-body showing cuticle (L).
Total RNA was extracted using the Ambion RNAqueous Micro Kit following the manufacturer’s protocol. Subsequent library preparation and cDNA synthesis was performed using the Takara SMART-Seq HT kit following the manufacturer’s instructions. Resulting double-stranded cDNA was purified using Ampure XP magnetic beads. Final sequencing library preparation was performed using the Illumina Nextera XT kit. For specimens processed in the United States, sequencing was performed using an Illumina NovaSeq (2 × 150 bp paired-end reads), while for specimens processed in Sweden, sequencing was done using an Illumina HiSeq X (2 × 150 bp paired end reads).
For both new and published transcriptomes, Illumina reads were filtered using either AfterQC (Chen et al., 2017) or fastp (Chen et al., 2018) and assembled with Trinity (Grabherr et al., 2011) installations on a local computer, on public Galaxy (Afgan et al., 2018) servers1,2,3 or using UPPMAX4, with default parameter settings and normalization of reads. The quality of each assembled transcript was assessed using the Transrate (Smith-Unna et al., 2016) installation on UPPMAX (see footnote 4), with default settings. Roche 454 reads for published transcriptomes were assembled using the Roche Newbler/GS De Novo Assembler v. 3.0 (Margulies et al., 2005) on a local computer using the following command: runAssembly -cdna -large *RR*.fastq.
Genome assemblies were generated de novo only for those datasets for which assemblies were not publicly available. Illumina-generated reads were filtered using fastp (Chen et al., 2018) and assembled with SPAdes version 3.14.0 (Bankevich et al., 2012) run on a local computer or using SPAdes version 3.13.1 on UPPMAX (see footnote 4). Roche 454 reads were assembled using the Roche Newbler/GS De Novo Assembler v. 3.0 (Margulies et al., 2005) via the command runAssembly -large *RR*.fastq. All contigs less than 500 bp in length were discarded. Scaffolding was not done for the purpose of this project.
From transcriptome assemblies, protein sequences were translated with TransDecoder 5.5.05 using the UniProt SwissProt database and PFAM (Pfam-A.hmm) versions 32 or 33 (The UniProt Consortium, 2018). From genome assemblies, protein coding genes were predicted and protein sequences were translated with the local installation of AUGUSTUS (Stanke et al., 2008) or a WebAUGUSTUS service at http://bioinf.uni-greifswald.de/webaugustus/ (Hoff and Stanke, 2013) using existing AUGUSTUS species parameters projects (Brugia malayi, Caenorhabditis elegans, or Trichinella spiralis) as training datasets and cDNA/protein sequence data from the same or closely related species, where available, as extrinsic evidence for predicting genes.
Following Phillips et al. (2017) and Huang et al. (2019), potential contaminated proteins were eliminated from newly generated (and some public) proteomes by comparing them with a high-quality reference dataset of nematode proteins. The reference dataset includes proteins derived from reference/core/telomere-to-telomere quality genomes of Caenorhabditis elegans (PRJNA13758), Pristionchus pacificus (PRJNA12644), Brugia malayi (PRJNA10729), Trichuris muris (PRJEB126), Acrobeloides nanus (PRJEB26554), and Plectus sambesii (PRJNA390260). All proteomes were combined into a single file that was used to create a reference database for DIAMOND (Buchfink et al., 2015). All proteins from query annotations not meeting the required similarity threshold to the reference dataset (E value < 10–5, see Phillips et al., 2017; Huang et al., 2019) were removed. Furthermore, CroCo (Simion et al., 2018) was used to detect and remove cross-species laboratory contamination by comparing the expression levels of different transcripts in samples prepared and sequenced together. However, instead of using the initial assemblies, we only checked for cross-contamination in the contigs that corresponded to filtered protein-coding sequences.
BUSCO scores were calculated using BUSCO version 5.1.2 (Waterhouse et al., 2018). Both “Nematoda” and “Metazoa” reference datasets were used for comparison. Two additional procedures were done to all datasets before the phylogenomic analysis: all protein coding sequences shorter than 50 amino acids and all duplicate sequences were removed using seqkit (Shen et al., 2016).
The entire dataset was not analyzed at once due to the large number of taxa. Instead, it was split and analyzed in stages. First, sub-trees were generated for the clades classified under names Trichinellida, Spirurina, Tylenchina, and Rhabditina (De Ley and Blaxter, 2004) and including all available species. From these subtrees, representative species were chosen to infer phylum-wide phylogenetic hypotheses based on the completeness of each individual proteome, relative branch length and position in the cladogram. These were combined with the remaining datasets not included in the “Trichinellida,” “Spirurina,” “Tylenchina,” and “Rhabditina” sub-trees to create a “Nematoda” tree.
Orthogroups were inferred for each of the five individual datasets (“Trichinellida,” “Spirurina,” “Tylenchina,” “Rhabditina,” and “Nematoda”) by conducting an all-versus-all comparison with Proteinortho (Lechner et al., 2014) using DIAMOND (Buchfink et al., 2015), either on UPPMAX (footnote 4) or using Galaxy Version 6.0.14 installed on usegalaxy.eu (Afgan et al., 2018). All orthogroups represented by 50% or less of the sampled species were discarded. Each candidate orthogroup was aligned with MAFFT (Katoh, 2002) using the automatic alignment strategy with a “maxiterate” value of 1000. Alignments were filtered with Aliscore and trimmed with Alicut (Kück et al., 2010) to remove ambiguously aligned regions following the strategy used by Smythe et al. (2019). To screen candidate core ortholog groups for evidence of paralogy or exogenous contamination, a maximum-likelihood (ML) tree was inferred for each alignment using IQ-TREE multicore version 2.0.6 (Minh et al., 2020) with the substitution model automatically selected by the ModelFinder (Kalyaanamoorthy et al., 2017) for each alignment, followed by 1000 bootstrap pseudoreplicates. PhyloPyPruner (latest version6, Thalén and Kocot, unpublished) was then employed to use a tree-based approach to screen each candidate ortholog for evidence of paralogy with the following settings “–min-len 50 –trim-lb 5 –min-support 0.75 –prune MI –mask pdist.” Finally, in order to eliminate orthogroups with poor taxon sampling, all groups sampled for fewer than 50% of the input taxa were discarded. PhyloPyPruner also outputs a supermatrix of all the individual orthogroup alignments concatenated into one.
For the final phylogeny reconstruction analysis, we employed two strategies. The first involved using the supermatrix alignment for ML analysis while the second involved coalescent-based inference of a species tree from individual orthogroup trees. For the supermatrix approach, ML analyses were conducted on the supermatrix alignment using RAxML 8.2.8 (Stamatakis, 2014) and IQ-TREE multicore version 2.0.6 (Minh et al., 2020). Because of the very large number of taxa in our matrices, for the RAxML analyses, data matrices were partitioned by gene but the PROTGAMMALG model was specified for all partitions rather than empirically inferring the best-fitting model for each partition. IQ-TREE analyses were performed using the site heterogeneous PMSF model (Wang et al., 2018) (-m LG + C20 + G + F) with the RAxML bipartitions tree (generated as described above) provided as the required guide tree (–ft). Nodal support was assessed with 1000 rapid bootstraps (–bb 1000). Coalescent-based phylogenies were inferred from the ML orthogroup trees using ASTRAL (a method for estimating species trees from unrooted gene trees) version 5.7.3 (Zhang et al., 2018). In order to improve accuracy of the species tree, poorly supported branches (bootstrap support < 75) were first collapsed in each orthogroup tree using newick utilities version 1.6 (Junier and Zdobnov, 2010) prior to the ASTRAL run.
Trichinellida is a part of the Clade I from Blaxter et al. (1998) or Clade 2 from Holterman et al. (2006). This subset includes only 13 species from two genera and two families (Supplementary Table 1) of the clade with nearly 400 known species distributed among over 30 genera and six families, all of which are parasites of vertebrates (Anderson, 2000). Completely missing from our analysis is the most speciose group, the family Capillariidae with over 390 known species parasitising a wide range of vertebrate hosts.
The phylogenies were constructed from a dataset of 469 orthologs for 24 ingroup taxa and Longidorus elongatus used as an outgroup (Supplementary Table 1). Concatenated alignment was 211,925 bases long. Both the IQ-TREE ML supermatrix tree (Figure 2) and ASTRAL coalescent-based species tree (Supplementary Figure 1) resulted in similar topologies, albeit with different branch length, clearly separating the two sampled genera, Trichuris and Trichinella, into two lineages. Within the Trichuris clade, there was strong support for all nodes, with Trichuris muris branching off first, while Trichuris trichiura and Trichuris suis being sister taxa, in full agreement with previous phylogenetic analyses based on COX1 (Callejón et al., 2015) and 18S rRNA (van Megen et al., 2009) genes. Within the Trichinella clade, two subclades were established representing the so-called encapsulated (T. spiralis, T. nelsoni, T. patagoniensis, T. britovi, T. murrelli, and T. nativa) and non-encapsulated (T. papuae, T. zimbabwensis, and T. pseudospiralis) species. All branches received moderate to strong bootstrap support/local posterior probabilities in both analyses (51–100%/0.64–1.0). Consistent with the analysis by Zarlenga et al. (2006) and Korhonen et al. (2016), the clade formed by the encapsulated taxa placed the lineage T. nelsoni + T. spiralis as sister to all other encapsulated taxa (T. britovi, T. murelli, T. nativa, T. patagoniensis and unidentified species T6, T8, and T9). Relationships among the non-encapsulated taxa was also identical to what has been previously reported, with the T. zimbabwensis + T. papuae lineage forming a sister group to the clade encompassing several populations of T. pseudospiralis.
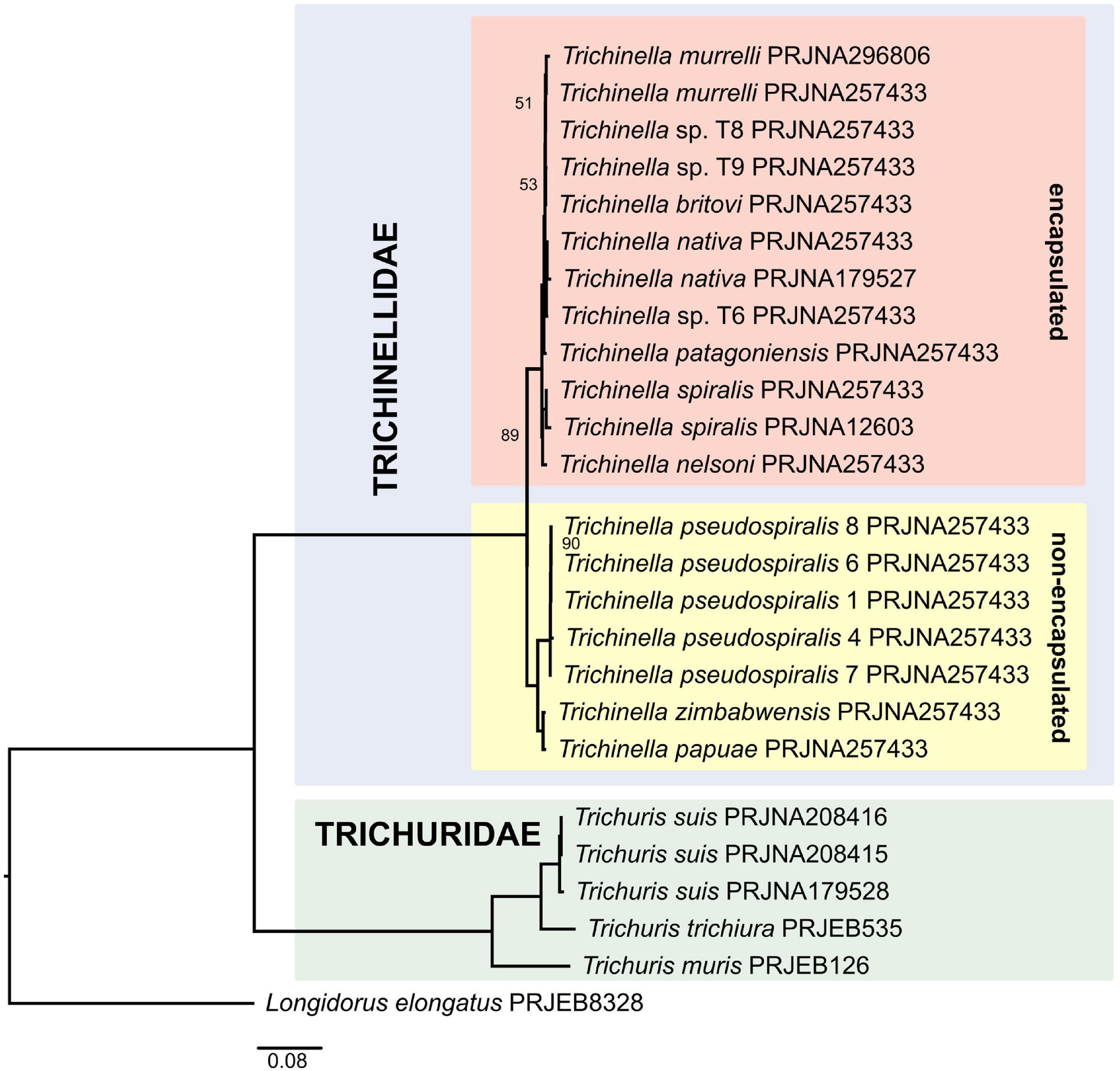
Figure 2. Phylogeny of Trichinellida inferred by the IQ-TREE maximum likelihood analysis (only bootstrap support values < 100% are shown).
Within the suborder Spirurina, De Ley and Blaxter (2004) recognized 58 families representing five infraorders. While our analysis covers four of the five infraorders (Rhigonematomorpha is not included), less than a fifth (11 families) of the 58 families are represented, including the families Dracunculidae and Aguillicolidae not classified in any infraorder (Supplementary Table 2). Moreover, a large majority of families are represented by one or two species, except for Ascarididae and Onchocercidae, with 5 and 14 species respectively.
The IQ-TREE ML supermatrix and the ASTRAL coalescent-based analyses produced trees with identical topologies (Figure 3 and Supplementary Figure 2) where all families represented in this analysis by two or more species formed well-supported clades. The clade representing Anguillicola crassus + Gnathostoma spinigerum was the earliest diverging lineage forming a sister to all remaining taxa. Although united in a well-supported clade in both analyses, Anguillicola crassus and Gnathostoma spinigerum appear to be fast evolving lineages and their phylogenetic affinities must be re-assessed with the addition of more taxa. Moreover, separate placement of Anguillicola crassus from Dracunculus medinensis (which is placed further along the phylogeny) makes the superfamily Dracunculoidea sensu De Ley and Blaxter (2004) polyphyletic.
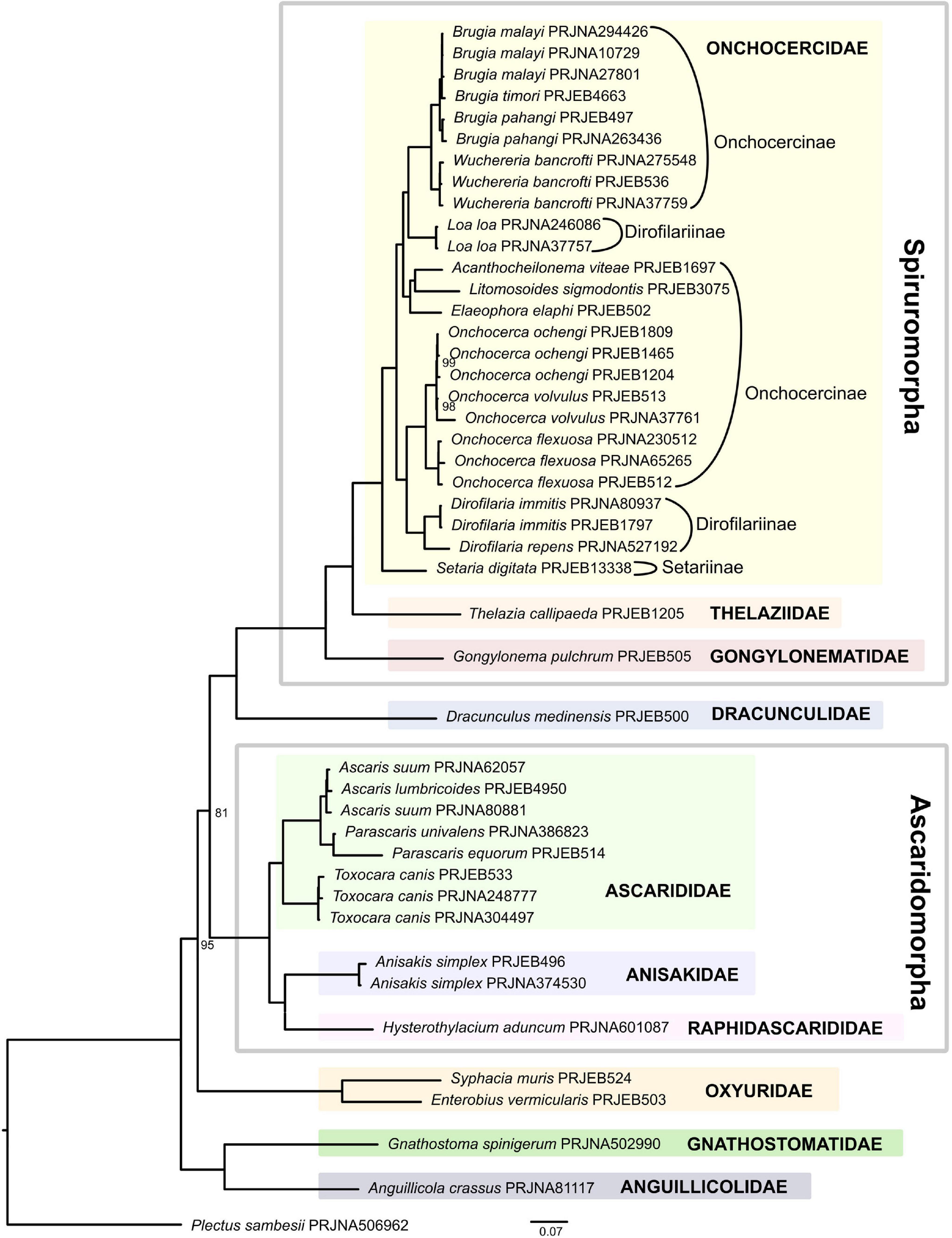
Figure 3. Phylogeny of Spirurina inferred by the IQ-TREE maximum likelihood analysis (only bootstrap support values < 100% are shown).
The next dichotomy leads to a well-supported clade represented by Syphacia muris and Enterobius vermicularis, the only two representatives of the suborder Oxyuridomorpha. Subsequent division separates Ascaridomorpha from Dracunculidae + Spiruromorpha. Within Ascaridomorpha, the family Ascarididae forms a well-supported clade, sister to a lineage uniting Anisakidae (represented by two datasets for Anisakis simplex) and Raphidascaridae (represented by Hysterothylacium aduncum), similar to the results obtained by Nadler and Hudspeth (2000). Within Ascarididae, Toxocara canis is a sister taxon to a clade uniting the genera Ascaris and Parascaris.
Within Spiruromorpha, subsequent branching events separate species representing the families Gongylonematidae (with single species Gongylonema pulchrum) and Thelazidae (Thelazia callipaeda) from the most speciose family in this analysis, Onchocercidae. Onchocercidae, represented here by three subfamilies Setariinae, Onchocercinae, and Dirofilariinae, was well supported, consistent with previous analyses based on mitochondrial genomes (Park et al., 2011; Liu et al., 2015). Thelaziidae formed a sister group to Onchocercidae with Gongylonematidae placed at their base, which is in disagreement with Liu et al. (2015), whose analysis placed Onchocercidae (=Filarioidea in Liu et al., 2015) and a clade including Gongylonematidae as sister taxa, and found Thelaziidae immediately basal to them. Within Onchocercidae, the clade composed of Dirofilaria and Onchocerca is a sister lineage to a clade representing the remaining genera of Onchocercidae. Here a small clade with the three species Elaephora elaphi, Acanthocheilonema viteae, and Litomosoides sigmodontis parasitising rodents and deer is a sister lineage to the clade that includes the genera Loa, Wuchereria, and Brugia known to cause infections in humans, domestic cats, dogs and other animals. As a result, subfamilies Onchocercinae and Dirofilariinae, as defined in Bain et al. (2014) are polyphyletic in the current analysis (Figure 3).
Four infraorders constitute the suborder Tylenchina as defined by De Ley and Blaxter (2004). These include Panagrolaimomorpha, Cephalobomorpha, Tylenchomorpha, and Drilonematomorpha, all of which except Cephalobomorpha comprise members that are parasites of animals and/or plants (Bert et al., 2008). Represented in the current phylogenetic analysis are 11 families that span three out of the four above-mentioned infraorders (Figure 4 and Supplementary Table 3). Drilonematomorpha is the only group with no representation.
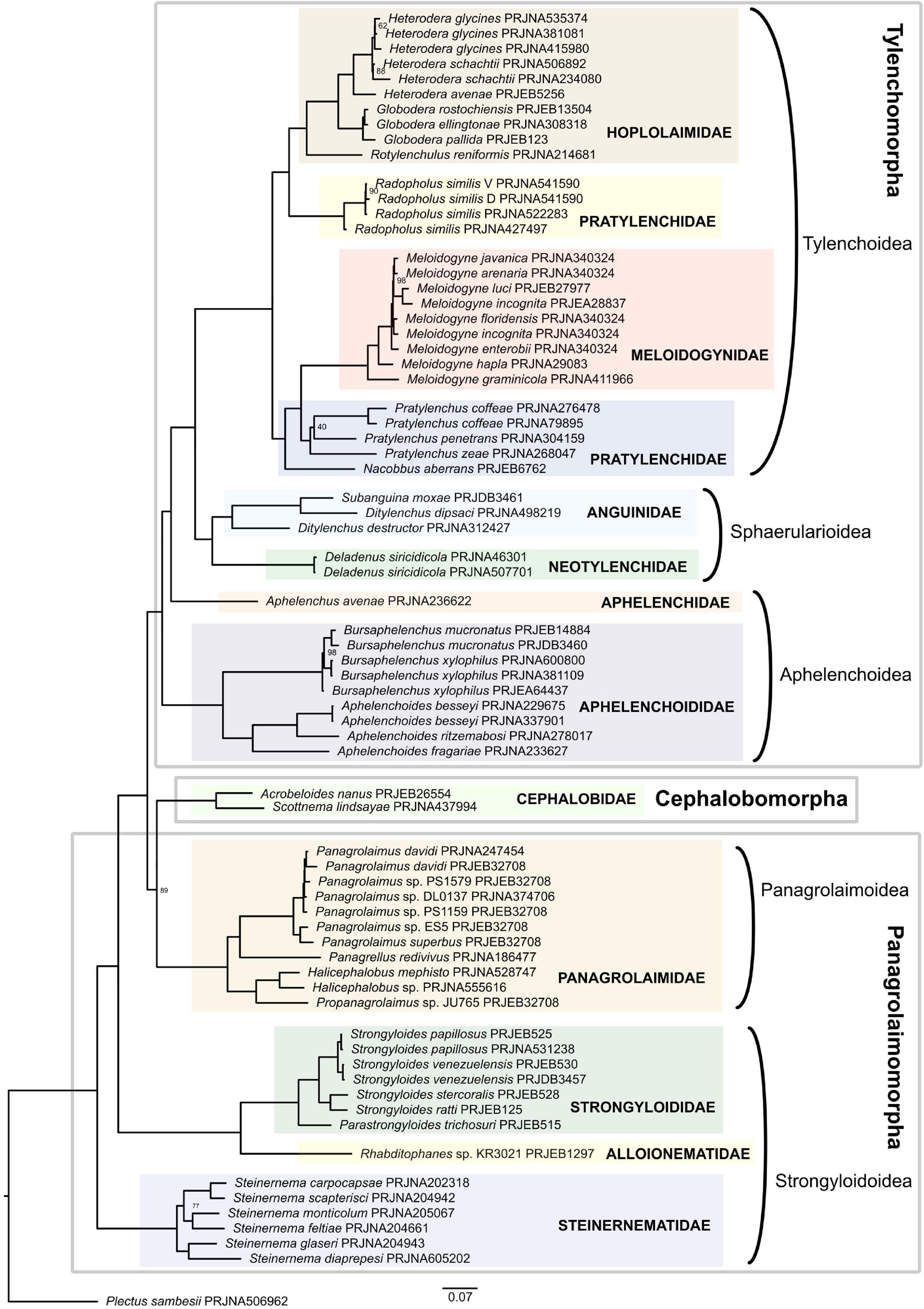
Figure 4. Phylogeny of Tylenchina inferred by the IQ-TREE maximum likelihood analysis (only bootstrap support values < 100% are shown).
Both the ML supermatrix tree and the coalescent-based tree showed the Steinernema lineage as the earliest branching clade of all Tylenchina sampled in this analysis. The next lineage splitting recovered Alloionematidae (Rhabditophanes) as sister to Strongyloididae (Strongyloides + Parastrongyloides). The next dichotomy is where lies the difference in the topologies of the two analyses. While the ML supermatrix tree placed the Cephalobomorpha + Panagrolaimomorpha as sister to all Tylenchomorpha with strong support, the placement of Cephalobomorpha in relation to the two other infraorders appears to be unresolved in the coalescent-based tree (Supplementary Figure 3). This topology according to the ML supermatrix tree is in concert with Grynberg et al. (2020) whose analysis also placed Acrobeloides nanus, the only representative of Cephalobomorpha, in a clade with Panagrolaimidae. It also contradicts previous 18S rDNA-based analyses where a close relationship between Tylenchomorpha and Cephalobomorpha was found to be moderately to well supported (De Ley and Blaxter, 2004; Holterman et al., 2006; Bert et al., 2008) and phylogenies based on mitochondrial genomes where Tylenchina is polyphyletic (Kim et al., 2020). The first lineage to diverge from within Tylenchomorpha was Aphelenchoididae. This dichotomy was followed by the splitting of Aphelenchus avenae (Aphelenchidae) from Tylenchoidea + Sphaerularioidea. Tylenchoidea + Sphaerularioidea formed a well-supported monophyletic clade in both analyses. Within this clade, a lineage consisting of Anguinidae and Neotylenchidae (=Sphaerularioidea) formed a sister clade to all other tylenchs (Tylenchoidea), agreeing with Smythe et al. (2019) but contradicting 18S rDNA-based studies (van Megen et al., 2009; Holterman et al., 2019).
Within Tylenchoidea, representatives of the family Pratylenchidae were divided into two clades, where Radopholus is a sister group to Hoplolaimidae, while Nacobbus and Pratylenchus are a sister to Meloidogyne (Meloidogynidae), both well-supported. This is in conformity with previous studies where Pratylenchidae have been found to be closely related to Meloidogynidae. There is also congruence with other studies with respect to the polyphyly of Pratylenchidae (Bert et al., 2008; van Megen et al., 2009).
All three infraorders of Rhabditina were represented in our analysis, although Bunonematomorpha was represented by just a single species. Pristionchus and Caenorhabditis constituted over half of the taxa that were sampled for this analysis (Supplementary Table 4). Contrary to Fürst von Lieven (2002) there was no support in either tree for the sister relationship between Bunonematomorpha and Diplogasteromorpha. However, this could be due to long branch attraction caused by insufficient data for the representatives of the former in this analysis. Both trees (Figure 5 and Supplementary Figure 4) placed Bunonema as sister to both Diplogasteromorpha (equal to Diplogastridae on Figure 5) and Rhabditomorpha (equal to Rhabditidae + Heterorhabditidae + Strongylida on Figure 5). In both ML supermatrix and coalescent-based trees, subsequent dichotomies resulted in the establishment of four major clades. Poikilolaimus (Rhabditidae incertae sedis) was positioned at the base of these clades, branching from the remaining Rhabditina earlier than Diplogasteromorpha [=Diplogastridae according to Sudhaus and Fürst von Lieven (2003)]. This early divergence of Poikilolaimus among rhabditids (Rhabditina) is consistent with previous analysis based on 18S rDNA (Sudhaus and Fitch, 2001; Sudhaus, 2011). Interestingly, other 18S rDNA based analyses placed the genus as an earlier branching group than even Bunonematomorpha (van Megen et al., 2009; Holterman et al., 2019). The family Diplogastridae, represented here by seven genera made up the sister clade to the remaining Rhabditina included in the analysis. There was strong support for this clade and for the monophyly of all its genera for which monophyly was testable, namely Pristionchus, Parapristionchus, and Micoletzkya. Relationships within this clade as depicted by both ML supermatrix and coalescent-based trees is incongruent with Fürst von Lieven (2002) and Sudhaus and Fürst von Lieven (2003) with regards to the time of divergence of Diplogasteroides relative to all other sampled diplogasterids. Notwithstanding this, the number and diversity of diplogastrid genera and species included in the current study remains very limited.
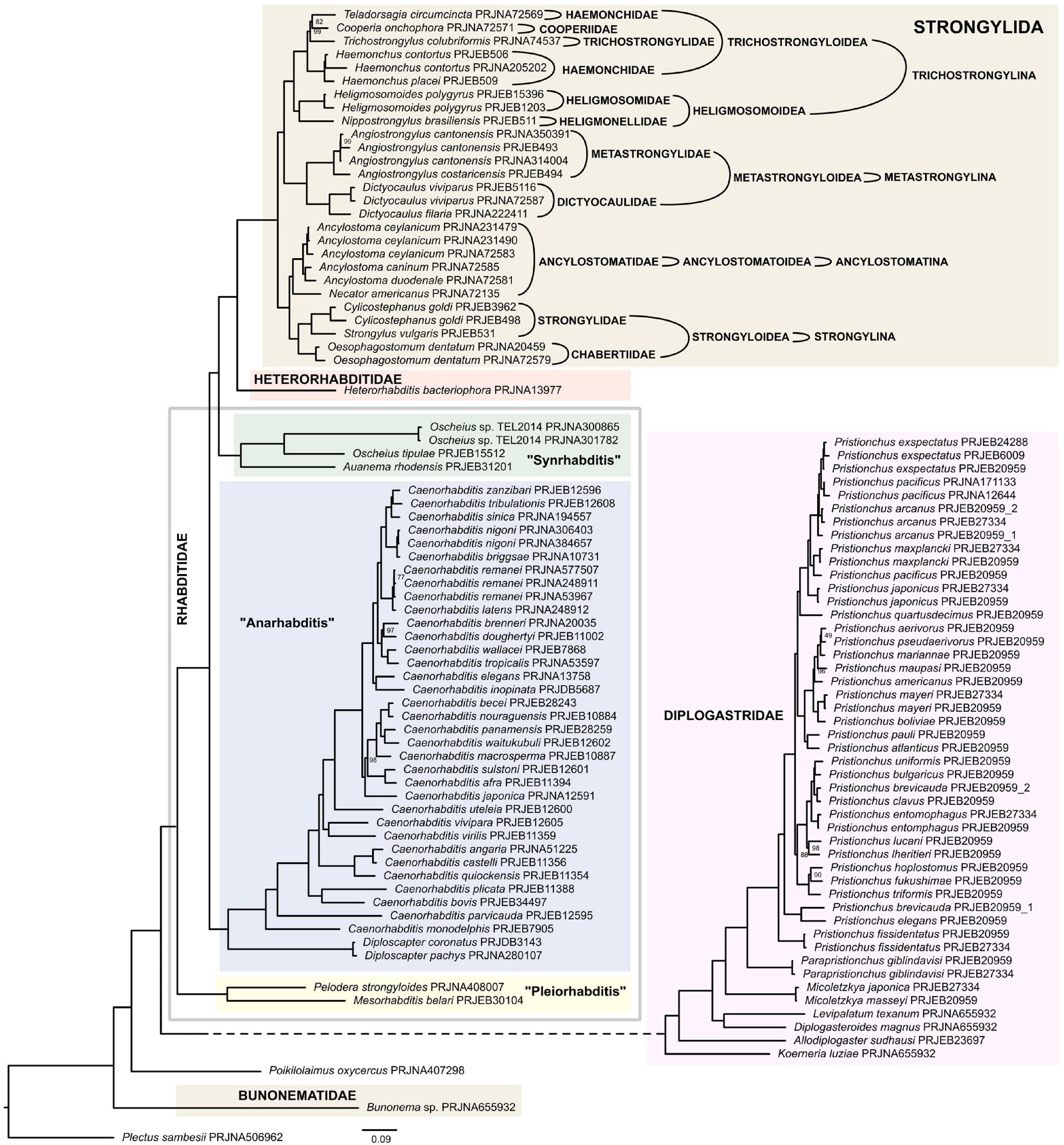
Figure 5. Phylogeny of Rhabditina inferred by the IQ-TREE maximum likelihood analysis (only bootstrap support values < 100% are shown). Clade names follow Sudhaus and Fürst von Lieven (2003) and Sudhaus (2011).
The next major bifurcation resulted in Mesorhabditioidea sensu De Ley and Blaxter (2004) or Pleiorhabditis sensu Sudhaus (2011), represented here by Mesorhabditis and Pelodera, forming the second clade. Occupying the next clade is Anarhabditis sensu Sudhaus (2011), represented here by several species of Caenorhabditis and two species of Diploscapter, both of which are supported as monophyletic. The general tree topologies from both the ML supermatrix tree and the coalescent-based tree are comparable with previous 18S rDNA-based analyses (Holterman et al., 2006; van Megen et al., 2009). The final main clade consisted of vertebrate parasites, entomopathogenic and free-living nematodes. A lineage comprising Oscheius and Auanema were the first to diverge from the rest of the members of this clade to form Synrhabditis sensu Sudhaus (2011), a sister to Strongyloidea (Heterorhabditidae and several animal parasitic families). As a result, and consistent with some previous molecular and morphology-based analyses, Rhabditidae is not a monophyletic entity if one does not include Diplogastridae, Heterorhabditidae and Strongylidae (=Strongylida sensu Beveridge et al., 2014) in it.
The families Ancylostomatidae, Metastrongylidae, and Strongylidae (sensu De Ley and Blaxter, 2004) were all recovered as monophyletic with strong branch supports but there was no support for the monophyly of Trichostrongylidae because the family Heligmosomidae, represented by Heligmosomoides, was deeply embedded within Trichostrongylidae. Comparing our phylogeny with a more conservative and detailed classification by Beveridge et al. (2014) and illustrated in the Figure 4, this clade assumes the rank of the order Strongylida. Phylogenetic relationships between species of this order closely correspond to the classification maintained by Beveridge et al. (2014), with only one exception. All four suborders identified in their classification are represented in this analysis, and all of them were well-supported as monophyletic. Trichostrongyloidea (suborder Trichostrongylina), the super-family with the most diverse taxa in this analysis, was well-supported as well. The other superfamily of Trichostrongylina, Heligmosomoidea was also well-supported. With the exception of Haemonchidae, all families consisting of more than one taxon were resolved as monophyletic. Our analysis with respect to Strongylida contradicts that of Grynberg et al. (2020) whose analysis placed Trichostrongylina and Ancylostomatina in a clade that is sister to Metrostrongylina, thus indicating Trichostrongylina as being more closely related to Ancylostomatina than it is to Metastrongylina. Similarly, phylogenies based on mitochondrial genomes propose a somewhat different topology, where Metastrongylina is a sister clade to remaining Strongylida, and Trichostrongylina is a sister clade to Anchylostomatina + Strongylina (Kim et al., 2020).
Most of the major nematode lineages (orders) are represented in our dataset by at least one species (Supplementary Table 5). The only groups missing from our dataset are the small orders Isolaimiida and Muspiceida. Benthimermithida, Marimermithida, and Rhaptothyreida previously considered as individual orders (De Ley and Blaxter, 2004) but now classified within Plectida (former) and Enoplida (latter two), are also absent from the current dataset.
After proteinortho orthology inference, 802 orthogroups were identified, almost half of which were discarded following a rather stringent screening for paralogy using PhyloPyPruner. The final supermatrix consisted of 416 putative orthologous groups with 187,750 amino acid positions and 90 OTUs. Both ML supermatrix analysis (Figure 6 and Supplementary Figure 5) and coalescent-based (Supplementary Figure 6) species trees moderately supported (94/0.92) the placement of tardigrades as sister to Nematoda, which corroborates previous analyses based on both 18S rDNA (Meldal et al., 2007) and whole genome/transcriptome data (Smythe et al., 2019). Also consistent with our analysis with respect to the Nematoda – Tardigrada sister relationship is Rota-Stabelli et al. (2011) whose analysis placed Nematoda as sister to the Panarthropoda clade which included the tardigrades. Their analysis did not include nematomorphs so could not resolve which of Tardigrada or Nematomorpha is closer to Nematoda. Campbell et al. (2011) also supported the Panarthropoda although their analysis put Nematoda and Nematomorpha together in a clade sister to the Panarthropoda which included Tardigrada. Nematoda was strongly supported, with the first major split resulting in a well-supported Enoplia clade forming a sister group to Dorylaimia and Chromadoria. Within the Enoplia clade, Triplonchida (represented by single Tobrilus) was an ingroup of Enoplida, sister to Bathylaimus, making Enoplida paraphyletic, in both ML supermatrix analyses (Figure 6 and Supplementary Figure 5), while in the coalescent-based tree (Supplementary Figure 6) Triplonchida and Enoplida were sister taxa. Such behavior can probably be explained by insufficient taxon sampling in Triplonchida and low matrix occupancy in Bathylaimus.
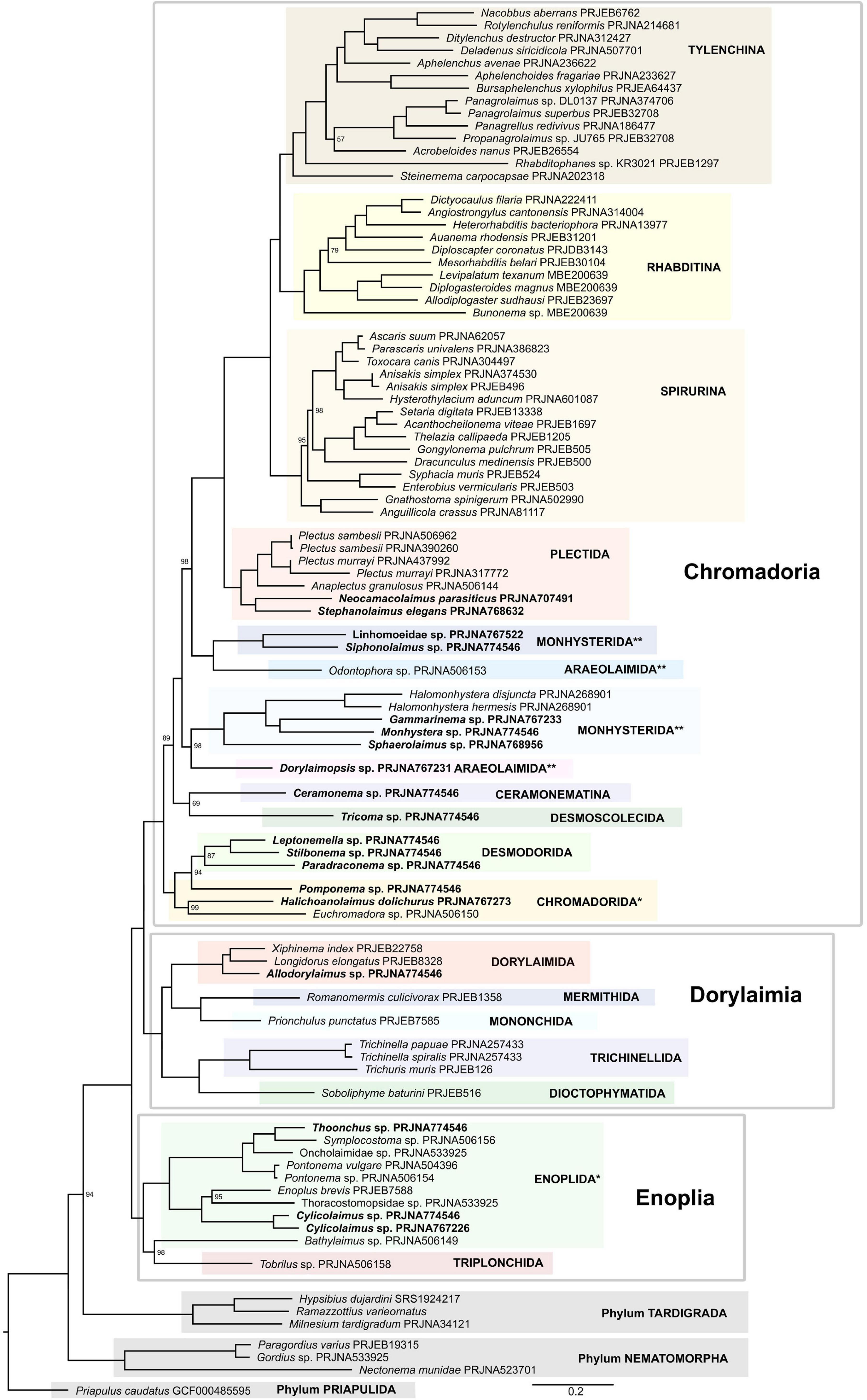
Figure 6. Phylogeny of Nematoda inferred by the IQ-TREE maximum likelihood analysis using the PMSF model (LG + C20 + G + F). Only bootstrap support values < 100% are shown. Paraphyletic orders are marked with *, polyphyletic orders are marked with **, species sequenced in this study are marked in bold.
Dorylaimia formed a well-supported clade and there was strong support for the Dorylaimia and Chromadoria sister relationship as well. Within Dorylaimia, Dioctophymatida and Trichinellida were recovered as sister to Dorylaimida and Mononchida. While Dorylaimida and Trichinellida were both recovered as monophyletic as established by Smythe et al. (2019), the monophyly of Mermithida, Mononchida and Dioctophymatida, once again, could not be assessed because they were represented each by just one species. The subsequent split resulted in a clade consisting of Chromadorida and Desmodorida (with Chromadorida being paraphyletic in respect to monophyletic Desmodorida), and together forming a sister group to the rest of Chromadoria. At the base of the remaining nematodes was a weakly supported clade consisting of Ceramonema and Tricoma. Subsequently, two well-supported clades each consisting of representatives of Monhysterida and Araeolaimida were recovered. In one, Dorylaimopsis (Araeolaimida) was recovered as a sister to Xyalidae + Monhysteridae (Monhysterida) and in the other clade Odontophora (Araeolaimida) clusters together with Linhomoeidae and Siphonolaimus (Monhysterida). Similar topology was reported in multiple 18S rDNA-based analyses (Meldal et al., 2007; Holterman et al., 2019; Ahmed and Holovachov, 2020). Plectida formed a well-supported monophyletic clade, sister to a well-supported Rhabditida clade. Two subclades were recovered within Plectida in which a lineage comprising Neocamacolaimus and Stephanolaimus (both classified in Camacolaimidae by Holovachov, 2014) formed a sister group to Plectidae represented by Plectus sambesii, Plectus murrayi, and Anaplectus granulosus.
Rhabditida was made up of three subclades equal to Spirurina, Rhabditina, and Tylenchina, all of which were strongly supported (and analyzed in details above). Within Spirurina, the families Ascarididae, Raphidascarididae, and Anisakidae clustered together in a well-supported clade, thus supporting the monophyly of the superfamily Ascaroidea. Oxyuridae, represented by Syphacia muris and Enterobius vermicularis, was also monophyletic. All three infra-orders of Rhabditina were represented in this analysis, and the topology of the tree closely corresponds to the more detailed analysis described above. Similarly, the topology of Tylenchina matches perfectly the more detailed analysis described above in this manuscript.
The placement of Enoplia as sister to the rest of Nematoda in our phylogeny corroborates similar results obtained by Smythe et al. (2019). This relationship at the root of the tree was consistently strongly supported in both the ML analysis using the PMFS model and the coalescent-based phylogeny using ASTRAL. In Smythe et al. (2019) however, Enoplia’s position as sister to all other nematodes received strong support only from one of the two analysis methods they used – the one involving the use of SCaFoS instead of PhyloTreePruner (Kocot et al., 2013) for refining the orthologs. As the authors pointed out, the algorithm employed in PhyloTreePruner can potentially cause a large number of sequences to be unnecessarily removed from the dataset. SCaFoS, on the other hand, offered a less stringent alternative and so resulted in a more complete dataset. PhyloPyPruner, which was used in this analysis, like PhyloTreePruner, uses a tree-based approach for screening putative paralogs. Because PhyloPyPruner is capable of differentiating contamination-like sequences from paralogs, there is a reduced risk of erroneously inferring paralogy and unnecessarily removing these sequences as it happens with other tree-based approaches such as PhyloTreePruner.
Backbone relationships between the major groups of Nematoda were generally similar with previously published phylogenomic analyses (Blaxter and Koutsovoulos, 2015; Koutsovoulos, 2015; Smythe et al., 2019). Aside from the taxa being represented for the first time in a phylogenomic study, no conflicting relationship can be observed between the current study and the most representative analyses to date. Compared to 18S rDNA-based phylogenies (Holterman et al., 2006, 2019; Meldal et al., 2007; van Megen et al., 2009), the same inconsistencies that were identified by Smythe et al. (2019) were also recovered here. For example, results from the current study concurred with Smythe et al. (2019) in the placement of Trichinellida and Dioctophymatida as sister to a lineage consisting of Mononchida, Mermithida and Dorylaimida, but it disagrees with placement of a clade of Dorylaimida, Trichinellida and Dioctophymatida as sister to Mononchida + Mermithida in Meldal et al. (2007) or the placement of Dorylaimida as sister to the rest of Dorylaimia in van Megen et al. (2009). The branching pattern in Holterman et al. (2006), somewhat resembles that of the current study in that, Dorylaimida + Mononchida + Mermithida formed a sister clade to Trichinellida, with the exception being the absence of Dioctophymatida in their analysis. Additionally, while the relationship between Chromadorida and Desmodorida was unresolved in van Megen et al. (2009) and Holterman et al. (2019), Chromadorida was recovered as paraphyletic in our analysis with Desmodorida being an ingroup of it, and the two were placed in a well supported clade. Relationships between the three suborders of Rhabditida have also been shown to vary, with one genome-scale analysis placing Tylenchina as sister to Spirurina + Rhabditina (International Helminth Genomes Consortium, 2018) and 18S rDNA-based analysis placing Rhabditina as sister to Tylenchina + Spirurina (Meldal et al., 2007). Consistent with our analysis, however, is that Spirurina is positioned as sister to Tylenchina + Rhabditina in Holterman et al. (2006, 2019), van Megen et al. (2009), and Smythe et al. (2019). Mitogenome-based phylogenies, on the other hand, do not support the monophyly of either Tylenchina or Spirurina (Kim et al., 2020).
Seven species of Monhysterida included in this analysis occupied two separate lineages. Altogether, the three superfamilies of the order had at least one representative. The superfamily Siphonolaimoidea was strongly supported, and within it, Siphonolaimidae and Linhomoeidae were recovered as sister taxa. This is in concordance with previous analysis based on the 18S rDNA (van Megen et al., 2009; Holterman et al., 2019). Also consistent with previous studies is the placement of this superfamily as sister to the araeolaimid family Axonolaimidae, represented in our analysis by Odontophora (Figure 6). The only other family of Araeolaimida included here is Comesomatidae and it formed a sister group to the other two superfamilies of Monhysterida, Sphaerolaimoidea and Monhysteroidea, in a well supported clade (Figure 6). This is corroborated by previous 18S rDNA-based studies where Comesomatidae is consistently recovered among Monhysterida (except Siphonolaimoidea) (van Megen et al., 2009; Holterman et al., 2019; Ahmed and Holovachov, 2020). One of the defining characters used to support the monophyly of the order Monhysterida is the presence of outstretched gonads (Fonseca and Bezerra, 2014a). Unlike Monhysterida, the order Araeolaimida comprises taxa both with reflexed and outstretched female gonads (Fonseca and Bezerra, 2014b). However, both clades in our analysis that combine representatives of Monhysterida and Araeolaimida (Figure 6, marked with **) consist of taxa with outstretched female gonads that are either not-paired (monodelphic) or paired (didelphic) – the shared apomorphic character used to establish Monhysterida in the first place. Lorenzen (1981, 1994) actually grouped all the families of Araeolaimida under the superfamily name Axonolaimoidea within Monhysterida based on this synapomorphy, but De Ley and Blaxter (2004) reinstated Araeolamida on the basis of 18S rDNA data available at that time. Morphologically, however, there is no apomorphic character to support the monophyly of Araeolamida. Moreover, recent 18S rDNA-based analyses as well as our results do not support their monophyly (Holterman et al., 2019; Ahmed and Holovachov, 2020). Not included in this analysis but frequently recovered between the two Monhysterida + Araeolaimida lineages in 18S rDNA-based phylogenies is a clade consisting of the order Isolaimida and family Aulolaimidae whose placements in the classification and phylogeny remain debatable (van Megen et al., 2009; Holterman et al., 2019). This placement may be simply a result of phylogenetic artifact which would explain why it is poorly supported. Further, having a clade (Isolaimida + Aulolaimida) whose taxa have reflexed ovaries placed between two lineages whose shared character is outstretched ovaries makes this position even more doubtful. Future phylogenomic analyses involving representatives of both Isolaimida and Aulolaimidae or at least one may finally help resolve their position with respect to Monhysterida and Areaolaimida.
Taxa that are phylogenetically close to plant-parasitic lineages have long been of interest in the quest to elucidate the origin of plant parasitism (Bert et al., 2008; Quist et al., 2015; Holterman et al., 2017). Analysis of the Tylenchina dataset gave some insight into the relationships between several important groups of plant-parasitic tylenchids as well as their close free-living relatives. Our results were generally in concurrence with previous 18S rDNA analyses. The first notable difference is with respect to the relationship between Tylenchomorpha, Cephalobomorpha, and Panagrolaimomorpha, where most 18S rDNA-based phylogenies have placed the last one as sister to the first two (Holterman et al., 2006; van Megen et al., 2009). Even though this was never strongly supported, it meant that Cephalobomorpha could belong to the same lineage as most plant-parasitic tylenchids, separate from Panagrolaimomorpha. Our analysis instead placed Tylenchomorpha as sister to Cephalobomorpha and Panagrolaimomorpha, consistent with Grynberg et al. (2020). The two species of Cephalobidae in our analysis occupied the same clade with Panagrolaimidae and this clade formed a sister group with all Tylenchomorpha, which suggests that Cephalobomorpha may not be any closer to Tylenchomorpha than Panagrolaimomorpha are (Figure 4). Another important difference is in regards to the strong support for the monophyly of Tylenchomorpha. The position of the Aphelenchoidea with respect to Tylenchoidea according to 18S rDNA has persistently deviated from morphological prediction which unites all taxa equipped with the stomatostylet (the tylenchid and aphelenchid feeding apparatus) within Tylenchomorpha. These above-mentioned analyses have Aphelenchoididae and Parasitaphelenchidae placed as sister to Panagrolaimidae, although often in a poorly supported clade (van Megen et al., 2009; Holterman et al., 2019). This lack of support for the monophyly of the superfamily Aphelenchoidea has led to speculation that the stomatostylet could have arisen multiple times during the course of evolution within Tylenchina (Quist et al., 2015). However, although our analysis does not support Aphelenchoidea as a monophyletic group, it rejects the possible multiple origins of the stomatostylet. The more substantial incongruences between our analyses and mitogenome-based phylogenies in regards to the evolution of Tylenchina are discussed in detail in section “Impact of Data Quality on the Results of Current Analyses.”
Amongst the many animal-parasitic lineages to have arisen from within Rhabditomorpha is the exclusively tetrapod vertebrate parasitic group Strongylida (Sudhaus, 2011). The group has been defined primarily on the basis of the presence of caudal bursa in males (Beveridge et al., 2014). In addition to the support given this group based on this shared presence of male bursa, molecular data have also strongly supported the group as monophyletic and our analysis is not an exception. The observed support for Heterorhabditidae as sister to Strongylida is confirmed by other 18S rDNA data as well (van Megen et al., 2009). As with the phylum itself, various classification systems have been proposed for the order Strongylida. And with these, the relationships between the suborders and superfamilies remained unclear. Most notable of these systems was the one proposed by Anderson et al. (1974) who divided the order into five superfamilies, Strongyloidea, Trichostrongyloidea, Ancylostomatoidea, Metastrongyloidea, and Diaphanocephaloidea based on morphology. Later modification to this system by Durette-Desset and Chabaud (1993) resulted in the elevation of four of these superfamilies to the ranks of suborder, with Diaphanocephaloidea being moved to Ancylostomatina. Although efforts have been made using molecular data to analyze the relationships between members of this group, very few exist that have tested the monophyly of the suborders (de Bellocq et al., 2001). The most important one in this respect was the phylogeny presented by Chilton et al. (2006) even though it was only able to establish support for Metastrongylina. All suborders in our analysis were well supported and there was a clear separation between lineages with prominent head capsule (Ancylostomatina and Strongylina) and ones without (Metastrongylina and Trichostrongylina). Detailed morphology-based cladistic interpretation of this division was presented by de Bellocq et al. (2001) and their analysis seems to suggest the presence of buccal capsule in this group as an ancestral feature. It is not clear, however, which of these character states (presence or absence of head capsule) is ancestral. The use of combined sequences of the 18S and 28S rDNA regions strongly supported Metastrongylina and placed them as sister to the other three suborders. Similar topology was obtained based on mitochondrial genomes, although all three suborders included in the analysis formed well supported distinct clades (Jex et al., 2010). Subsequent mitogenome-based phylogeny supported the monophyly of Metastrongylina, Trichostrongyina, and Ancylostomatina, while Strongylina was recovered as paraphyletic (Kim et al., 2020). According to Beveridge et al. (2014), aside from Metastrongylina being associated with the pulmonary system and the others not being associated with it, there are no other morphological or life cycle characteristics to support their separation from the remaining suborders. So far, our analysis presents a topology more consistent with morphology-based classifications than any other previous study. It will be interesting in the future to see how even more expanded taxon sampling influences the phylogeny of the group.
For nematodes, mitogenome phylogenies often produce results that are consistent with both ribosomal DNA phylogenies and genome- or transcriptome- phylogenies (Kim et al., 2017; Kern et al., 2020). For some clades, however, mitogenome phylogenies have also been shown to yield some significant topological discordances with these approaches as well. A comparison of the current analyses with some published mitochondrial genome phylogenies demonstrates this clearly. In the most updated phylum-wide mitogenome phylogeny (Kim et al., 2020), Chromadoria and Dorylaimia are both supported; and within the latter, Mermithida + Dorylaimida form a sister clade to Trichinellida, similar to the topology obtained in this study. Comparisons involving Enoplia cannot be made at the moment, since the group is yet to be represented in any mitogenome phylogeny. Therefore, the only topological differences between mitogenome phylogenies and 18S rDNA- or genome-based that can be commented on are the ones found within the Chromadoria subclass, and more specifically within Rhabditida. The order Rhabditida itself is well supported by all mitogenome trees (Sultana et al., 2013; Kim et al., 2015, 2017, 2020) except in Ivanova et al. (2021) where the presence of Plectida nested within the same polytomous clade as the other Rhabditida rendered the latter paraphyletic. All three suborders of Rhabditida have been supported by phylogenomic analysis thus far. For mitogenome-based phylogenies, only Rhabditina are recovered as monophyletic (Sultana et al., 2013; Kim et al., 2015, 2017, 2020) with the other two being polyphyletic–again the exception here being Ivanova et al. (2021) whose analysis recovered Rhabditina as polyphyletic. Within Rhabditina however, both the current analysis and mitogenome phylogenies support Diplogasteromorpha and Rhabditomorpha as monophyletic. The support for Spirurina in the current analysis is not consistent with mitogenome phylogenies involving this group. According to mitogenome trees, the infraorders of Spirurina are placed in three distinct clades, Spiruromorpha and Oxyuridomorpha occupy the first clade, immediately following this is the one that consists only of the superfamily Dracunculoidea. The third clade, which consists of Ascardiomorpha, Gnathostomatomorpha, and Rhigonematomorpha is embedded within a larger clade where it forms a sister to Rhabditina (Rhabditomorpha and Diplogasteromorpha). Between this larger clade and the Dracunculoidea lineage are Tylenchomorpha (specifically Aphelenchoidea) and Panagrolaimomorpha lineages. Within Tylenchina, the support for the infraorder Tylenchomorpha in the current analysis is also in contrast with mitogenome phylogenies which placed Tylenchoidea and Aphenchoidea in two distinct clades, with the former nested between Spirurina and Cephalobomorpha and the latter placed between Dracunculoidea and Panagrolaimomorpha. This lack of support for Tylenchomorpha therefore shows that mitogenome trees (Sultana et al., 2013; Kim et al., 2015, 2017, 2020), like some 18S rDNA trees (Quist et al., 2015), reject the single origin of the stomatostylet.
For a group as diverse and ubiquitous as nematodes, broad and balanced taxon sampling is imperative for better understanding of relationships among its members (Holterman et al., 2006). This was achieved by analyzing the overrepresented suborders separately as subsets of the Nematoda tree, thus allowing only selected sets of taxa for these groups to be sampled for the main analysis, while at the same time generating more detailed analyses for them. Nineteen transcriptomes were newly sequenced as part of the current study to further maximize representation at the order level, particularly for the hitherto excluded orders of predominantly marine nematodes, Desmodorida, Monhysterida, and Desmoscolecida. Also, our use of expanded taxon sampling for Araeolaimida and Chromadorida permits us to use our results to test the monophyly of these groups.
Data quality and completeness varied greatly between the sampled taxa, with completeness of sites reaching as low as 2.5% for Pomponema sp. and as high as 85.2% for Anisakis simplex. And for the four subset datasets, completeness was as low as 1.8% for Onchocerca volvulus (Spirurina), 0.5% for Heterodera schachtii (Tylenchina), and 2.0% for Oscheius sp. (Rhabditina). How deleterious the inclusion of patchy and incomplete data sets is on phylogenetic inference is still unclear, although most empirical evidence suggest that on its own, incomplete data is not problematic (Hejnol et al., 2009; Rubin et al., 2012; Roure et al., 2013). This was evident in the clades where the majority of species have moderate to high completeness (Onchocercidae, Hoplolaimidae, and Rhabditidae). For example, the placements of Oncholaimidae gen. sp., Bathylaimus sp., Pomponema sp., Monhystera sp., Onchocerca volvulus, Heterodera schachtii, and Oscheius sp. were all consistent with general taxonomy, morphology-based and molecular phylogenies based on the 18S rDNA (van Megen et al., 2009; Holterman et al., 2019) despite these data sets having site completion scores below 5%.
The ML supermatrix trees and the coalescent-based trees were generally similar in topology. The few conflicting nodes, particularly in the topologies of the coalescent-based trees, had low support values. The first contradiction concerned the phylogenetic affinity of Tobrilus, a member of the order Triplonchida with which Enoplida shares the subclass Enoplia. Being the only triplonchid, its placement together with Bathylaimus as observed in the ML supermatrix tree, while unexpected, could have been a result of insufficient taxon sampling for Triplonchida. The fact that the relationships between Tobrilus, Bathylaimus, and the other enoplids appeared unresolved in the coalescent-based tree supports this argument. Tobrilus belongs to one of the families of Triplonchida, namely Tobrilidae, that together with Tripylidae, Triodontolaimidae, Rhabdodemaniidae, and Pandolaimidae, was placed by Lorenzen (1981, 1994) in the Tripyloidina, the suborder to which family Tripyloididae and genus Bathylaimus belongs. The presence of metanemes was used as the synapomorphic character. These families were later moved to Triplonchida by De Ley and Blaxter (2004) based on 18S rDNA phylogenies. However, Bathylaimus together with the other genera of Tripyloididae are known to share similar stoma morphology with some triplonchid families including Tobrilidae. Some species of the family Tripyloididae have also been described as having their spicular protractor muscle modified into circular muscles similar to the capsule of suspensor muscles seen among Triplonchida (Smol et al., 2014). More taxa from these families will be necessary for comprehensive understanding of their relationships with regards to the two orders within Enoplia.
Supermatrix methods (IQ-TREE) and coalescent-based gene tree summarization methods (ASTRAL) differ in using individual DNA or amino acid characters and gene trees as input data, respectively (reviewed by Bininda-Emonds, 2004; de Queiroz and Gatesy, 2007). The question of which of the two approaches, supermatrix trees or coalescent-based trees, gives the most accurate inference of phylogeny has always been contentious (Bininda-Emonds, 2004; Gatesy and Springer, 2004; Springer and Gatesy, 2014). Bininda-Emonds (2010) proposed that rather than pushing for which approach is more accurate, the two methods can best be viewed as simply being different, in a manner similar to how statistical methods of inferring phylogenies such as ML, Bayesian inference or maximum parsimony are considered different. Thus, we can view the topological consistency in the two tree inference methods used here as only enhancing our confidence in the resulting topology and the inconsistencies as representing nodes that need further investigation. ASTRAL is a statistically consistent coalescent-based method which has been shown to be more accurate and run faster than other coalescent-based methods such as MP-EST (Liu et al., 2010) and BUCKy (Larget et al., 2010). It also performs well against concatenation methods, especially in datasets with fair amounts of incomplete lineage sorting (Mirarab et al., 2014; Mirarab and Warnow, 2015). As far as our dataset is concerned however, future analyses with better taxon sampling for Triplonchida (particularly families outside of the ones previously classified under Tripylina), Desmoscolecida and Ceramonematina can result in even more similar topologies between the two.
We sampled taxa from across the phylum Nematoda, and obtained a dataset representing almost all the major groups of the phylum for the current analysis. This is the most comprehensive phylogenomic analysis to date. Generating whole transcriptome data is becoming easier due to the increasing number of sequencing protocols that can handle small input volumes of genetic material and the continuous decrease in the cost of sequencing produced with high-throughput sequencing platforms. Nematode phylogeny is currently at a point where analysis based on single genes such as the 18S rDNA are not producing the expected resolution due to the limited amount of phylogenetic information that can be derived from the region. The most up-to-date single gene analysis of phylogeny of the entire phylum consisted of over 2,700 sequences (Holterman et al., 2019), almost a thousand more than van Megen et al. (2009). The higher taxon sampling did not appear to have resulted in increased resolution along the backbone of the nematode tree, or within many individual clades (Ahmed and Holovachov, 2021). Resolution at the root of Nematoda observed in the current and two of the previous phylogenomic analyses signifies the kind of improvement in Nematode phylogeny possible with genome-scale analysis involving transcriptomic and genomic data. Our analysis confirms the polyphyly of Monhysterida and Araeolamida and seems to suggest that Monhysterida sensu Lorenzen (1994) is paraphyletic.
The inclusion of the subtrees of Trichinellida, Spirurina, Tylenchina, and Rhabditina allowed us to carry out a more comprehensive analysis of these important groups while keeping a fairly balanced taxon sampling for the phylum-wide analysis. We found that our phylogenomic analysis supported some of the “old” morphology-based phylogenetic hypotheses which were challenged by 18S rDNA- and mitogenome- based trees. For example, within the Tylenchina, there was a lineage that gave rise to Cephalobomorpha and Panagrolaimomorpha and this lineage was placed as sister to Tylenchomorpha. Tylenchomorpha (species bearing a piercing stomatostylet) received strong support with Aphelenchoididae uniting with the other members of the infraorder in a well supported clade, thus rejecting the notion that the stomatostylet may have arisen independently within Tylenchina. The nematode stylet-like feeding apparatus in general has for a long time been known to have occurred in multiple lineages. This was already apparent from the distinct morphologies of these feeding structures. However, the stomatostylet itself has been known only amongst herbivorous/fungivorous nematodes and has been the defining feature (apomorphy) of all Tylenchomorpha. And while there may be some morphological differences between the different lineages of this infraorder, the general structure and its composition remains consistent across the group – they all consist of the distal cornus, on top of the shaft which consists of three knobs at its base. There was strong support for Strongylida as well as its division into four suborders. The presence or absence of a buccal capsule as basis for separating the lineage of Ancylostomatina + Strongylina from Metastrongylina + Trichostrongylina was also supported.
While the current study presents a significant improvement over previous phylogenomic analyses in terms of taxon sampling, topological relationships inferred herein agreed well with previous studies. Compared to studies of 18S rDNA alone, which lacks resolving power needed to clarify phylogenetic relationships across the entire nematode tree, we obtained better supported relationships in our results based on phylotranscriptomics, suggesting that whole-genome and transcriptomic approaches represent the future of phylogenetics, in general, and for Nematoda, in particular. What is left now is for there to be an extensive effort to obtain reliable transcriptomic and genomic data of as many nematode species as possible, especially those morphologically unique lineages and taxa whose placement has remained uncertain across various 18S rDNA-based phylogenetic analyses to date. The discrepancies between the evolutionary history of nuclear and mitochondrial genomes in nematodes remain to be explained as well.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GenBank, PRJNA707491, PRJNA767226, PRJNA767231, PRJNA767233, PRJNA767273, PRJNA767522, PRJNA768632, PRJNA768956, and PRJNA774546. The data generated and analyzed during the current study, including all protein datasets used for input, are available via FigShare: https://figshare.com/s/946b2bc6aef7ce4a9e6a.
MA, AS, KK, and OH collected the specimens and contributed to the writing of the manuscript. AS and OH identified the specimens. MA, FA, KK, NR, and OH performed the RNA extraction and library preparation, transcriptome assembly and protein prediction. MA, FA, and OH performed the data analysis. MA and OH conceived a study design. All authors approved the final version prior to submission.
This research was in part supported by the grant from the Swedish Taxonomy Initiative: “Systematics of poorly known marine nematodes of the class Chromadorea from Sweden” to MA and OH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Computational resources used in this project include: (1) the Galaxy Europe (usegalaxy.eu/) which is in part funded by Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and German Federal Ministry of Education and Research (BMBF grants 031 A538A/A538C RBC, 031L0101B/031L0101C de.NBI-epi, and 031L0106 de.STAIR); (2) the UseGalaxy.org server (usegalaxy.org/) at the Center for Comparative Genomics and Bioinformatics at Penn State, the Department of Biology and at Johns Hopkins University and the Computational Biology Program at Oregon Health & Science University; (3) the Trinity installation (galaxy.ncgas-trinity.indiana.edu) at the National Center for Genome Analysis Support, Pervasive Technology Institute at Indiana University (no longer available); (4) the CIPRES Science Gateway (www.phylo.org/) at the San Diego Supercomputer Center of UC San Diego, supported by NSF and NIH awards; (5) the Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX, www.uppmax.uu.se/) under Projects SNIC 2020/15-99, SNIC 2020/16-124 and SNIC 2021/22-345 partially funded by the Swedish Research Council through grant agreement no. 2018-05973; (6) the University of Alabama High-Performance Computing cluster (UAHPC). Sampling in Skagerrak (Sweden) was conducted using vessels (“Skagerak,” “Nereus,” and “Oscar von Sydow”) and facilities of the Kristineberg Marine Research Station and Tjärnö Marine Laboratory. Sampling in the United States was conducted using the vessel “Sunburst” and was supported by the Smithsonian Marine Station in Fort Pierce, FL, United States. We thank Sherry Reed, Woody Lee, David Branson, Michael Boyle, and Valerie Paul for assistance and support with field and laboratory sampling. Specimens of Gammarinema sp. used for sequencing were collected by O. Holovachov during the New Caledonia Hydrobiological expedition 2016–2018 (PI: Philippe Bouchet) which is a part of a cluster of expeditions under the “Our Planet Reviewed/La Planéte revisitée” programme, implemented by the Muséum National d’Histoire Naturelle (Pascale Joannot, Head of expeditions programme) in partnership with the Conservatoire d’Espaces naturels, with funding from the Gouvernement de la Nouvelle-Calédonie, Province Sud, Province Nord, Office des Postes et Télécommunications, Maison de la Nouvelle-Calédonie and the French Ministry for the Overseas. The expedition operated under the permits issued by the Province Sud and the Province Nord, and the organizers thank, respectively, Emmanuel Coutures and Isabelle Jurquet (Province Sud) and Jean-Jérôme Cassan and Yannick Monlouis (Province Nord) for issuing of the permits. For logistics before, during, and after the field work, we thank Sébastien Faninoz and Alice Leblond. Our Planet Reviewed/La Planéte revisitée is a global initiative founded in 2007 by the Muséum National d’histoire Naturelle and Pro-Natura International. The senior author is grateful to Nicolas Charpin, Kaj Hnauane and Christine Pöllabauer for their invaluable assistance during 2017 and 2018 sampling trips.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.769565/full#supplementary-material
Adamson, M. L. (1987). Phylogenetic analysis of the higher classification of the Nematoda. Can. J. Zool. 65, 1478–1482. doi: 10.1139/z87-230
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Čech, M., et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Ahmed, M., Adedidran, F., and Holovachov, O. (2021). A draft transcriptome of a parasite Neocamacolaimus parasiticus (Camacolaimidae, Plectida). J. Nematol. 53, e2021–e2040. doi: 10.21307/jofnem-2021-040
Ahmed, M., and Holovachov, O. (2020). Description of a new marine predatory nematode Latronema dyngi sp. nov. (Nematoda, Chromadorida, Selachinematidae) from the west coast of Sweden and an updated phylogeny of Chromadoria. Mar. Biodivers. 50:113. doi: 10.1007/s12526-020-01129-w
Ahmed, M., and Holovachov, O. (2021). Twenty years after De Ley & Blaxter – how far did we progress in understanding the phylogeny of the phylum Nematoda? Animals 11:3479. doi: 10.3390/ani11123479
Aleshin, V. V., Kedrova, O. S., Milyutina, I. A., Vladychenskaya, N. S., and Petrov, N. B. (1998). Relationships among nematodes based on the analysis of 18S rRNA gene sequences: molecular evidence for monophyly of chromadorian and secernentian nematodes. Russ. J. Nematol. 6, 175–184. doi: 10.1006/jipa.1997.4657
Anderson, R. C. (2000). Nematode Parasites Of Vertebrates: Their Development And Transmission. Wallingford: CABI Publishing.
Anderson, R. C., Chabaud, A. G., and Wilmott, S. (1974). “General introduction. Glossary of terms. Keys to subclasses, orders and superfamilies,” in CIH Keys to Nematode Parasites of Vertebrates, Vol. 1, eds R. C. Anderson, A. G. Chabaud, and S. Willmott (Farnham Royal: Commonwealth Institute of Helminthology), 1–17. doi: 10.1079/9781845935726.0001
Andrássy, I. (1976). Evolution As A Basis For The Systematization Of Nematodes. London: Pitman Publishing Ltd.
Bain, O., Mutafchiev, Y., and Junker, K. (2014). “7.21 Order Spirurida,” in Handbook of Zoology. Gastrotricha, Cycloneuralia and Gnathifera. Vol. 2: Nematoda, ed. A. Schmidt-Rhaesa (Berlin: De Gruyter), 661–732.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455-477. doi: 10.1089/cmb.2012.0021
Bert, W., Leliaert, F., Vierstraete, A. R., Vanfleteren, J. R., and Borgonie, G. (2008). Molecular phylogeny of the Tylenchina and evolution of the female gonoduct (Nematoda: Rhabditida). Mol. Phylogenet. Evol. 48, 728–744. doi: 10.1016/j.ympev.2008.04.011
Beveridge, I., Spratt, D. M., and Durette-Desset, M.-C. (2014). “7.18 Order Strongylida (Railliet & Henry, 1913,” in Handbook of Zoology. Gastrotricha, Cycloneuralia and Gnathifera. Vol. 2: Nematoda, ed. A. Schmidt-Rhaesa (Berlin: De Gruyter), 557–612.
Bininda-Emonds, O. R. P. (2004). Trees versus characters and the supertree/supermatrix “paradox”. Syst. Biol. 53, 356–359. doi: 10.1080/10635150490440396
Bininda-Emonds, O. R. P. (2010). The future of supertrees: bridging the gap with supermatrices. Palaeodiversity 3 Suppl, 99–106.
Blanc-Mathieu, R., Perfus-Barbeoch, L., Aury, J.-M., Da Rocha, M., Gouzy, J., Sallet, E., et al. (2017). Hybridization and polyploidy enable genomic plasticity without sex in the most devastating plant-parasitic nematodes. PLoS Genet. 13:e1006777. doi: 10.1371/journal.pgen.1006777
Blaxter, M. L., De Ley, P., Garey, J. R., Liu, L. X., Scheldeman, P., Vierstraete, A., et al. (1998). A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75. doi: 10.1038/32160
Blaxter, M., and Koutsovoulos, G. (2015). The evolution of parasitism in Nematoda. Parasitology 142, S26–S39.
Blaxter, M., Koutsovoulos, G., Jones, M., Kumar, S., and Elsworth, B. (2016). “Phylogenomics of nematoda,” in Next-Generation Systematics, eds J. A. Cotton, J. Hughes, and P. D. Olson (Cambridge: Cambridge University Press), 62–83.
Blaxter, M., Kumar, S., Kaur, G., Koutsovoulos, G., and Elsworth, B. (2012). Genomics and transcriptomics across the diversity of the Nematoda. Parasite Immunol. 34, 108–120. doi: 10.1111/j.1365-3024.2011.01342.x
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Callejón, R., Cutillas, C., and Nadler, S. A. (2015). Nuclear and mitochondrial genes for inferring Trichuris phylogeny. Parasitol. Res. 114, 4591–4599. doi: 10.1007/s00436-015-4705-7
Campbell, L. I., Rota-Stabelli, O., Edgecombe, G. D., Marchioro, T., Longhorn, S. J., Telford, M. J., et al. (2011). MicroRNAs and phylogenomics resolve the relationships of Tardigrada and suggest that velvet worms are the sister group of Arthropoda. Proc. Natl. Acad. Sci. U.S.A. 108, 15920–15924. doi: 10.1073/pnas.1105499108
Chen, S., Huang, T., Zhou, Y., Han, Y., Xu, M., and Gu, J. (2017). AfterQC: automatic filtering, trimming, error removing and quality control for fastq data. BMC Bioinformatics 18:80. doi: 10.1186/s12859-017-1469-3
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884-i890. doi: 10.1093/bioinformatics/bty560
Chilton, N. B., Huby-Chilton, F., Gasser, R. B., and Beveridge, I. (2006). The evolutionary origins of nematodes within the order Strongylida are related to predilection sites within hosts. Mol. Phyl. Evol. 40, 118–128. doi: 10.1016/j.ympev.2006.01.003
Chitwood, B. G. (1958). The designation of official names for higher taxa of invertebrates. Bull. Zool. Nomencl. 15, 860–895. doi: 10.5962/bhl.part.19410
Daubin, V., Gouy, M., and Perrière, G. (2002). A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 12, 1080–1090. doi: 10.1101/gr.187002
de Bellocq, J. G., Ferté, H., Depaquit, J., Justine, J.-L., Tillier, A., and Durette-Desset, M.-C. (2001). Phylogeny of the Trichostrongylina (Nematoda) Inferred from 28S rDNA Sequences. Mol. Phyl. Evol. 19, 430–442. doi: 10.1006/mpev.2001.0925
De Coninck, L. (1965). “Systématique des nématodes,” in Traité de Zoologie. Vol. 4. Némathelminthes (Nématodes), ed. P. P. Grassé (Paris: Masson & Cie), 586–668.
De Ley, P., and Blaxter, M. L. (2002). “Systematic position and phylogeny,” in The Biology of Nematodes, ed. D. Lee (London: Taylor & Francis), 1–30. doi: 10.1201/b12614-2
De Ley, P., and Blaxter, M. L. (2004). A new system for nematoda: combining morphological characters with molecular trees, and translating clades into ranks and taxa. Nematol. Monogr. Perspect. 2, 633–653. doi: 10.1163/9789004475236_061
de Queiroz, A., and Gatesy, J. (2007). The supermatrix approach to systematics. Trends Ecol. Evol. 22, 34–41. doi: 10.1016/j.tree.2006.10.002
Drozdovsky, E. M. (1981). “On the position of chromadorids in system of nematodes,” in Evolution, Taxonomy, Morphology And Ecology Of Free-Living Nematodes, eds T. A. Platonova and S. Y. Tsalolikhin (Moscow: Academy of Sciences of the USSR), 32–37.
Durette-Desset, M.-C., and Chabaud, A. G. (1993). Nomenclature des strongylida au-dessus du groupe famille. Ann. Parasitol. Hum. Comparée 68, 111–112. doi: 10.1051/parasite/1993682111
Ebersberger, I., Strauss, S., and von Haeseler, A. (2009). HaMStR: profile hidden markov model-based search for orthologs in ESTs. BMC Evol. Biol. 9:157. doi: 10.1186/1471-2148-9-157
Eisen, J. A., and Fraser, C. M. (2003). Phylogenomics: intersection of evolution and genomics. Science 300, 1706–1707. doi: 10.1126/science.1086292
Filipjev, I. N. (1934). The Classification Of Freeliving Nematodes And Their Relation To The Parasitic Nematodes. Washington, DC: Smithsonian Institution.
Fonseca, G., and Bezerra, T. N. (2014a). “7.14 Order Monhysterida Filipjev, 1929,” in Handbook of Zoology. Gastrotricha, Cycloneuralia and Gnathifera: Nematoda, Vol. 2, ed. A. Schmidt-Rhaesa (Berlin: De Gruyter), 433–465.
Fonseca, G., and Bezerra, T. N. (2014b). “7.15 Order Araeolaimida De Coninck & Schuurmans Stekhoven, 1933,” in Handbook of Zoology. Gastrotricha, Cycloneuralia and Gnathifera: Nematoda, Vol. 2, ed. A. Schmidt-Rhaesa (Berlin: De Gruyter), 467–486. doi: 10.1515/9783110274257.467
Fürst von Lieven, A. (2002). The sister group of the Diplogastrina (Nematoda). Russian J. Nematol. 10, 127–137.
Gadea, E. (1973). Sobre la filogenia interna de los Nematodos. Publicationes Inst. Biol. Apll. (Barcelona) 54, 87–92.
Gatesy, J., and Springer, M. S. (2004). “A critique of matrix representation with parsimony supertrees,” in Phylogenetic Supertrees: Combining Information To Reveal The Tree Of Life, ed. O. R. P. Bininda-Emonds (Dordrecht: Springer), 369–388. doi: 10.1007/978-1-4020-2330-9_18
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644-652. doi: 10.1038/nbt.1883
Grynberg, P., Coiti Togawa, R., Dias de Freitas, L., Antonino, J. D., Rancurel, C., Mota do Carmo Costa, M., et al. (2020). Comparative genomics reveals novel target genes towards specific control of plant-parasitic nematodes. Genes 11:1347. doi: 10.3390/genes11111347
Hejnol, A., Obst, M., Stamatakis, A., Ott, M., Rouse, G. W., Edgecombe, G. D., et al. (2009). Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. Biol. Sci. 276, 4261–4270. doi: 10.1098/rspb.2009.0896
Higgins, R. P., and Thiel, H. (1988). Introduction To The Study Of Meiofauna. Washington, DC: Smithsonian Institution Press.
Hoff, K. J., and Stanke, M. (2013). WebAUGUSTUS – a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res. 41, W123-W128. doi: 10.1093/nar/gkt418
Holovachov, O. (2014). “7.16 Order Plectida Gadea, 1973,” in Handbook of Zoology. Gastrotricha, Cycloneuralia and Gnathifera: Nematoda, Vol. 2, ed. A. Schmidt-Rhaesa (Berlin: De Gruyter), 487–535.
Holterman, M., Karegar, A., Mooijman, P. van Megen, H. van den Elsen, S., Vervoort, M. T. W., et al. (2017). Disparate gain and loss of parasitic abilities among nematode lineages. PLoS ONE 12:e0185445. doi: 10.1371/journal.pone.0185445
Holterman, M., Schratzberger, M., and Helder, J. (2019). Nematodes as evolutionary commuters between marine, freshwater and terrestrial habitats. Biol. J. Linn. Soc. 128, 756–767. doi: 10.1093/biolinnean/blz107
Holterman, M., van de Wurff, A., van den Elsen, S., van Megen, H., Bongers, T., Holovachov, O., et al. (2006). Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution towards crown clades. Mol. Biol. Evol. 23, 1792–1800. doi: 10.1093/molbev/msl044
Huang, X., Xu, C.-L., Yang, S.-H., Li, J.-Y., Wang, H.-L., Zhang, Z.-X., et al. (2019). Life-stage specific transcriptomes of a migratory endoparasitic plant nematode, Radopholus similis elucidate a different parasitic and life strategy of plant parasitic nematodes. Sci. Rep. 9:6277. doi: 10.1038/s41598-019-42724-7
International Helminth Genomes Consortium (2018). Comparative genomics of the major parasitic worms. Nat. Genet. 51, 163–174. doi: 10.1038/s41588-018-0262-1
Ivanova, E. S., Efeykin, B. D., and Spiridonov, S. E. (2021). The re-description of Synoecnema hirsutum Timm, 1959 (Synoecneminae, Ungellidae, Drilonematoidea) from a pheretimoid earthworm in Vietnam with the analysis of its phylogenetic relationships. ZooKeys 1076, 135–150. doi: 10.3897/zookeys.1076.75932
Jex, A. R., Hall, R. S., Littlewood, D. T. J., and Gasser, R. B. (2010). An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 38, 522–533. doi: 10.1093/nar/gkp883
Junier, T., and Zdobnov, E. M. (2010). The newick utilities: high-throughput phylogenetic tree processing in the UNIX shell. Bioinformatics 26, 1669–1670. doi: 10.1093/bioinformatics/btq243
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kern, E. M. A., Kim, T., and Park, J.-K. (2020). The mitochondrial genome in nematode phylogenetics. Front. Ecol. Evol. 8:250. doi: 10.3389/fevo.2020.00250
Kim, J., Kern, E., Kim, T., Sim, M., Kim, J., Kim, Y., et al. (2017). Phylogenetic analysis of two Plectus mitochondrial genomes (Nematoda: Plectida) supports a sister group relationship between Plectida and Rhabditida within chromadorea. Mol. Phylogenet. Evol. 107, 90–102. doi: 10.1016/j.ympev.2016.10.010
Kim, J., Lee, S., Gazi, M., Kim, T., Jung, D., Chun, J. Y., et al. (2015). Mitochondrial genomes advance phylogenetic hypotheses for Tylenchina (Nematoda: Chromadorea). Zool. Scr. 44, 446–462. doi: 10.1111/zsc.12112
Kim, T., Lee, Y., Kil, H.-J., and Park, J.-K. (2020). The mitochondrial genome of Acrobeloides varius (Cephalobomorpha) confirms non-monophyly of Tylenchina (Nematoda). PeerJ 8:e9108. doi: 10.7717/peerj.9108/table-2
Kocot, K. M., Citarella, M. R., Moroz, L. L., and Halanych, K. M. (2013). PhyloTreePruner: a phylogenetic tree-based approach for selection of orthologous sequences for phylogenomics. Evol. Bioinformatics Online 9, 429–435. doi: 10.4137/EBO.S12813
Korhonen, P. K., Pozio, E., La Rosa, G., Chang, B. C. H., Koehler, A. V., Hoberg, E. P., et al. (2016). Phylogenomic and biogeographic reconstruction of the Trichinella complex. Nat. Comm. 7:10513. doi: 10.1038/ncomms10513
Koutsovoulos, G. (2015). Reconstructing The Phylogenetic Relationships Of Nematodes Using Draft Genomes And Transcriptomes. Ph.D. dissertation. Edinburgh: University of Edinburgh.
Kück, P., Meusemann, K., Damback, J., Thormann, B., von Reumont, B. M., Wägele, J. W., et al. (2010). Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front. Zool. 7:10. doi: 10.1186/1742-9994-7-10
Larget, B. R., Kotha, S. K., Dewey, C. N., and Ané, C. (2010). BUCKy: gene tree/species tree reconciliation with the Bayesian concordance analysis. Bioinformatics 26, 2910–2911. doi: 10.1093/bioinformatics/btq539
Lechner, M., Hernandez-Rosales, M., Doerr, D., Wieseke, N., Thévenin, A., Stoye, J., et al. (2014). Orthology detection combining clustering and synteny for very large datasets. PLoS One 9:e105015. doi: 10.1371/journal.pone.0105015
Liu, G. H., Jia, Y. Q., Wang, Y. N., Zhao, G. H., and Zhu, X. Q. (2015). The complete mitochondrial genome of the gullet worm Gongylonema pulchrum: gene content, arrangement, composition and phylogenetic implications. Parasites Vectors 8:100. doi: 10.1186/s13071-015-0697-5
Liu, L., Yu, L., and Edwards, S. V. (2010). A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 10:302. doi: 10.1186/1471-2148-10-302
Lorenzen, S. (1981). Entwurf eines phylogenetischen Systems der freilebenden Nematoden. Veröff. Inst. Meeresforsch. Bremerh Suppl. 7, 1–472.
Maggenti, A. (1963). “Comparative morphology in nemic phylogeny,” in The Lower Metazoa, Comparative Biology And Phylogeny, ed. E. C. Dougherty (Berkeley, CA: University of California Press), 273–282.
Malakhov, V. V. (1986). Nematodes: Structure, Development, Classification And Phylogeny. Moscow: Mauka.
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., et al. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380. doi: 10.1038/nature03959
Meldal, B. H. M., Debenham, N. J., De Ley, P., Tandingan De Ley, I., Vanfleteren, J. R., Vierstraete, A. R., et al. (2007). An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol. Phyl. Evol. 42, 622–636. doi: 10.1016/j.ympev.2006.08.025
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mirarab, S., and Warnow, T. (2015). ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31, i44–i52. doi: 10.1093/bioinformatics/btv234
Mirarab, S., Reaz, R., Bayzid, M. D. S., Zimmermann, T., Swenson, M. S., and Warnow, T. (2014). ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, i541–i548. doi: 10.1093/bioinformatics/btu462
Nadler, S. A., and Hudspeth, D. S. (2000). Phylogeny of the Ascaridoidea (Nematoda: Ascaridida) based on three genes and morphology: hypotheses of structural and sequence evolution. J. Parasitol. 86, 380–393. doi: 10.1645/0022-3395(2000)086[0380:POTANA]2.0.CO;2
Park, J.-K., Sultana, T., Lee, S.-H., Kand, S., Kim, H. K., Min, G.-S., et al. (2011). Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics 12:392. doi: 10.1186/1471-2164-12-392
Parkinson, J., Mitreva, M., Whitton, C., Thomson, M., Daub, J., Martin, J., et al. (2004). A transcriptomic analysis of the phylum Nematoda. Nat. Genet. 36, 1259–1267. doi: 10.1038/ng1472
Phillips, W. S., Howe, D. K., Brown, A. M. V., Eves-van den Akker, S., Dettwyler, L., Peetz, A. B., et al. (2017). The draft genome of Globodera ellingtonae J. Nematol. 49, 127–128. doi: 10.21307/jofnem-2017-054
Quist, C. W., Smant, G., and Helder, J. (2015). Evolution of plant parasitism in the phylum nematoda. Ann. Rev. Phytopathol. 53, 289–310. doi: 10.1146/annurev-phyto-080614-120057
Rota-Stabelli, O., Campbell, L., Brinkmann, H., Edgecombe, G. D., Longhorn, S. J., Peterson, K. J., et al. (2011). A congruent solution to arthropod phylogeny: phylogenomics, microRNAs and morphology support monophyletic Mandibulata. Proc. Royal Soc. B 278, 298–306. doi: 10.1098/rspb.2010.0590
Roure, B., Baurain, D., and Philippe, H. (2013). Impact of missing data on phylogenies inferred from empirical phylogenomic data sets. Mol. Biol. Evol. 30, 197–214. doi: 10.1093/molbev/mss208
Rubin, B. E., Ree, R. H., and Moreau, C. S. (2012). Inferring phylogenies from RAD sequence data. PLoS One 7:e33394. doi: 10.1371/journal.pone.0033394
Sato, K., Kadota, Y., Gan, P., Bino, T., Uehara, T., Yamaguchi, K., et al. (2018). High-quality genome sequence of the Root-Knot nematode Meloidogyne arenaria genotype A2-O. Genome Announc. 6:26. doi: 10.1128/genomeA.00519-18
Shen, W., Le, S., Li, Y., and Hu, F. (2016). SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11:e0163962. doi: 10.1371/journal.pone.0163962
Simion, P., Belkhir, K., Francois, C., Veyssier, J., Rink, J. C., Manuel, M., et al. (2018). A software tool ‘CroCo’ detects pervasive cross-species contamination in next generation sequencing data. BMC Biol. 16:28. doi: 10.1186/s12915-018-0486-7
Smith-Unna, R., Boursnell, C., Patro, R., Hibberd, J. M., and Kelly, S. (2016). TransRate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 26, 1134–1144. doi: 10.1101/gr.196469.115
Smol, N., Muthumbi, A., and Sharma, J. (2014). “7.3 Order Enoplida,” in Handbook of Zoology. Gastrotricha, Cycloneuralia and Gnathifera: Nematoda, Vol. 2, ed. A. Schmidt-Rhaesa (Berlin: De Gruyter), 192–249.
Smythe, A. B., Sanderson, M. J., and Nadler, S. A. (2006). Nematode small subunit phylogeny correlates with alignment parameters. Syst. Biol. 55, 972–992. doi: 10.1080/10635150601089001
Smythe, A., Holovachov, O., and Kocot, K. (2019). Improved phylogenomic sampling of free-living nematodes enhances resolution of higher-level nematode phylogeny. BMC Evol. Biol. 19:121. doi: 10.1186/s12862-019-1444-x
Springer, M. S., and Gatesy, J. (2014). Land plant origins and coalescence confusion. Trends Plant Sci. 19, 267–269. doi: 10.1016/j.tplants.2014.02.012
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Sudhaus, W. (2011). Phylogenetic systematisation and catalogue of paraphyletic “Rhabditidae” (Secernentea, Nematoda). J. Nematode Morphol. Syst. 14, 113–178.
Sudhaus, W., and Fitch, D. (2001). Comparative studies on the phylogeny and systematics of the Rhabditidae (Nematoda). J. Nematol. 33, 1–70.
Sudhaus, W., and Fürst von Lieven, A. (2003). A phylogenetic classification and catalogue of the Diplogastridae (Secernentea, Nematoda). J. Nematode Morph. Syst. 6, 43–90.
Sultana, T., Kim, J., Lee, S.-H., Han, H., Kim, S., Min, G.-S., et al. (2013). Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol. Biol. 13:12. doi: 10.1186/1471-2148-13-12
The UniProt Consortium (2018). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. doi: 10.1093/nar/gky1049
van Megen, H., van den Elsen, S., Holterman, M., Karssen, S., Mooyman, P., Bongers, T., et al. (2009). A phylogenetic tree of nematodes based on about 1,200 full length small subunit ribosomal DNA sequences. Nematology 11, 927–950. doi: 10.1163/156854109x456862
Wang, H. C., Minh, B. Q., Susko, S., and Roger, A. J. (2018). Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst. Biol. 67, 216–235. doi: 10.1093/sysbio/syx068
Wasmuth, J., Schmid, R., Hedley, A., and Blaxter, M. (2008). On the extent and origins of genic novelty in the phylum Nematoda. PLoS Negl. Trop. Dis. 2:e258. doi: 10.1371/journal.pntd.0000258
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Zarlenga, D. S., Rosenthal, B. M., La Rosa, G., Pozio, E., and Hoberg, E. P. (2006). Post-Miocene expansion, colonization, and host switching drove speciation among extant nematodes of the archaic genus Trichinella. Proc. Natl. Acad. Sci. U.S.A. 103, 7354–7359. doi: 10.1073/pnas.0602466103
Keywords: roundworms, evolution, transcriptome, genome, parasitism, classification
Citation: Ahmed M, Roberts NG, Adediran F, Smythe AB, Kocot KM and Holovachov O (2022) Phylogenomic Analysis of the Phylum Nematoda: Conflicts and Congruences With Morphology, 18S rRNA, and Mitogenomes. Front. Ecol. Evol. 9:769565. doi: 10.3389/fevo.2021.769565
Received: 02 September 2021; Accepted: 13 December 2021;
Published: 28 January 2022.
Edited by:
Joong-Ki Park, Ewha Womans University, South KoreaReviewed by:
Justin C. Bagley, Jacksonville State University, United StatesCopyright © 2022 Ahmed, Roberts, Adediran, Smythe, Kocot and Holovachov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oleksandr Holovachov, b2xla3NhbmRyLmhvbG92YWNob3ZAbnJtLnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.