 Chun Fu†WenCong Long†ChaoBing Luo†Xiong NongXiMeng XiaoHong LiaoYuanQiu LiYing ChenJiaXin YuSiXuan Cheng
Chun Fu†WenCong Long†ChaoBing Luo†Xiong NongXiMeng XiaoHong LiaoYuanQiu LiYing ChenJiaXin YuSiXuan Cheng Saira Baloch*YaoJun Yang*
Saira Baloch*YaoJun Yang*- Bamboo Diseases and Pests Control and Resources Development Key Laboratory of Sichuan Province, Leshan Normal University, Leshan, China
Background: The most severe insect damage to bamboo shoots is the bamboo-snout beetle (Cyrtotrachelus buqueti). Bamboo is a perennial plant that has significant economic value. C. buqueti also plays a vital role in the degradation of bamboo lignocellulose and causing damage. The genome sequencing and functional gene annotation of C. buqueti are of great significance to reveal the molecular mechanism of its efficient degradation of bamboo fiber and the development of the bamboo industry.
Results: The size of C. buqueti genome was close to 600.92 Mb by building a one paired-end (PE) library and k-mer analysis. Then, we developed nine 20-kb SMRTbell libraries for genome sequencing and got a total of 51.12 Gb of the original PacBio sequel reads. Furthermore, after filtering with a coverage depth of 85.06×, clean reads with 48.71 Gb were obtained. The final size of C. buqueti genome is 633.85 Mb after being assembled and measured, and the contig N50 of C. buqueti genome is 27.93 Mb. The value of contig N50 shows that the assembly quality of C. buqueti genome exceeds that of most published insect genomes. The size of the gene sequence located on chromosomes reaches 630.86 Mb, accounting for 99.53% of the genome sequence. A 1,063 conserved genes were collected at this assembled genome, comprising 99.72% of the overall genes with 1,066 using the Benchmark Uniform Single-Copy Orthology (BUSCO). Moreover, 63.78% of the C. buqueti genome is repetitive, and 57.15% is redundant with long-term elements. A 12,569 protein-coding genes distributed on 12 chromosomes were acquired after function annotation, of which 96.18% were functional genes. The comparative genomic analysis results revealed that C. buqueti was similar to D. ponderosae. Moreover, the comparative analysis of specific genes in C. buqueti genome showed that it had 244 unique lignocellulose degradation genes and 240 genes related to energy production and conversion. At the same time, 73 P450 genes and 30 GST genes were identified, respectively, in the C. buqueti genome.
Conclusion: The high-quality C. buqueti genome has been obtained in the present study. The assembly level of this insect’s genome is higher than that of other most reported insects’ genomes. The phylogenetic analysis of P450 and GST gene family showed that C. buqueti had a vital detoxification function to plant chemical components.
Introduction
Bamboo is of great value in terms of both qualities of life and economics. Moreover, it is widely used in construction, wood-based panel, paper, bamboo tourism, and agricultural industries (Scurlock et al., 2000). It grows fast and is the most closed growing plant in the world. The bamboo forest area of China accounts for almost one-quarter of the total bamboo forest area in the world (Littlewood et al., 2013). Bamboo shoots also have rich nutritional value and health functions (Chauhan et al., 2016). However, Cyrtotrachelus buqueti (NCBI:txid1892066) is an oligotrophic bamboo forest pest that feeds exclusively on bamboo shoots. Moreover, it can destructively damage bamboo shoots, resulting in the failure of bamboo shoots to grow into bamboo, and greatly reduce the output of bamboo forest (Chen et al., 2005; Wang et al., 2005).
Cyrtotrachelus buqueti, a bamboo snout beetle, belongs to Curculionidae family of the Coleoptera. The larvae of C. buqueti grows in the soil during autumn and emerge as imagoes during the following summer (June and July). The adults of C. buqueti fly to caespitose bamboo shoot areas, feed on the bamboo shoots, and lay eggs in the bamboo shoot tips after mating (Chen et al., 2005; Wang et al., 2005; Figure 1). These beetles are mainly distributed in Fujian, Guangdong, Jiangsu, Sichuan, Guizhou, Chongqing, Guangxi and other provinces in China and Vietnam, Laos, Myanmar, and Thailand (Wen and Lu, 2006; Xiao, 2009; Li, 2010). In Sichuan Province, C. buqueti is a severe pest in caespitose bamboo forests with a risk estimate of more than 40% in 67,000 hectares of bamboo (Yang et al., 2009; Luo et al., 2018b). However, only a few studies concerning C. buqueti have been reported to date, and studies of this pest have focused on its biological features and chemical regulation. Some experiments have, for instance, been dedicated to the reproductive behavior of insects (Wang et al., 2005) and pest management of C. buqueti (Chen et al., 2005). Yang et al. (2009) investigated the effect of larval density on the number and harm of wormholes, analyzed bamboo shoot volatiles, and explored insect EAG responses (Yang et al., 2010). The semiochemical cuticular components of C. buqueti adults have also been identified and extracted (Mang et al., 2012). Previous studies’ results showed that C. buqueti played an essential role in the degradation of bamboo lignocellulose (Luo et al., 2018a, 2019a, b). However, we don’t know how many genes in C. buqueti genome are involved in the degradation of bamboo cellulose, how these genes are distributed, and which genes play an essential role in the degradation of bamboo lignocellulose. Interestingly, C. buqueti larvae only need to feed on bamboo shoots for about 20 day to complete their whole life activities. However, the molecular mechanism of energy conversion and energy storage required for a lifetime is still unclear. The beetles for which genomic sequencing has been completed include Dendroctonus ponderosae (Keeling et al., 2013), Tribolium castaneum (Tribolium Genome Sequencing Consortium et al., 2008), Anoplophora glabripennis (McKenna et al., 2016), Rhynchophorus ferrugineus (Hazzouri et al., 2020), and so on. These beetles are feeding plants and previous studies have reported specific genes for cellulose degrading in their genomes. To dissect the functional gene distribution of degrading bamboo fibers and their molecular evolutionary relationships in the genome of C. buqueti, a high-quality genome sequence was obtained by whole genome sequencing in this study.

Figure 1. Bamboo shoots and bamboo snout beetle C. buqueti.
“Based on our observation” in this study, we have achieved three goals. Firstly, the chromosome-level genome of C. buqueti has been assembled and mapped by Illumina sequencing, long-read Pacific Biosciences (PacBio) sequencing and high-throughput chromosome conformation capture (Hi-C) technology. Secondly, we’ve mined the genes Carbohydrate-Active enZymes (CAZy) involved in cellulose degradation in C. buqueti genome and other related species’ genomes. Moreover, the similarities and differences of CAZy gene family between C. buqueti and other related species were compared. Finally, we have identified that the genes were related to energy production and conversion in C. buqueti genome, and analyzed the evolutionary relationship of P450 and GST gene family in C. buqueti genome. The availability of high-quality chromosome-level genome sequence provides a theoretical basis for revealing bamboo lignocellulose degradation, energy conversion, energy storage and detoxification function of C. buqueti.
Materials and Methods
Sample Collection and Genome Sequencing
Insect Collection and Treatment
Male individuals from C. buqueti were collected from Muchuan (N103°98′, E28°96′), Sichuan, in early August 2017. After emerging bamboo shoots for 3 days, gathered all the individuals. At Sichuan Province Key Lab for Bamboo Pest Control and Resource Development, fed bamboo shoots obtained from the bamboo garden on these male individuals of C. buqueti under the following conditions in a laboratory greenhouse: 26°C, 70% humidity, and 16 h light/8 h dark. Used two male individuals fed for 1 week for genome and transcriptome sequencing, respectively.
Genome and Transcriptome Sequencing
According to the manufacturer, fresh mixed tissue samples were obtained from a male individual of C. buqueti refer to a Universal Genomic DNA Kit CW2298 (Cwbiotech Co., Ltd., Beijing, China)’s operating manual. Next, 1% agarose gel electrophoresis of the extracted genomic DNA was performed, after which the concentration was quantified using a Qubit 3 fluorometer (Thermo Fisher, Waltham, MA, United States). Until genome sequencing, long-read Pacific Biosciences (PacBio) genomic libraries (SMRTbell libraries) were built by shearing DNA into ∼20 kilobase (kb) fragments using a Covaris g-TUBE fragment (KBiosciences part No. 520079); they were then enzymatically repaired and 20-kb SMRTbell libraries were designed using a 1.0 DNA Template Prep Package (PacBio part No. 100-259-100). The size of the DNA fragments was subsequently measured using a Bioanalyzer 2100 12K DNA Chip assay (Agilent part No. 5067-1508), after which the templates were size-selected to enrich large DNA fragments (>10 kb) using BluePippin (Sage Science, Inc., Beverly, MA, United States). An Agilent Bioanalyzer 12 kb DNA Chip (Agilent Technologies, Santa Clara, CA, United States) and a Qubit fluorimeter (Invitrogen, Carlsbad, CA, United States) were used to examine the fragments’ content and measure the library. Binding Kit 2.0 (PacBio Part No. 100-862-200) was used for the design of the SMRTT.
The complex of BellPolymerase as defined in the protocols. Genome sequencing was performed using a PacBio Sequel sequencer at Biomarker Technologies Corporation (Beijing, China) (Pacific Biosciences, Menlo Park, CA, United States). Samples were mounted and sequenced onto PacBio SMRT v3.0 cells (PacBio part No. 100-171-800) of the Sequel instrument, collected in one film at 360 min per SMRT cell.
Finally, introduced MagBead loading (PacBio part No. 100-125-900) to improve the large fragments’ enrichment. As a result, a total of nine SMRT cells were programmed, and 48.71 G subread sequences with an average length of 10.53 kb were obtained (Supplementary Table 1).
For short-read sequencing using the protocols offered (San Diego, CA, United States), a single paired-end (PE) library with insert sizes of 270 bp was designed and then sequenced on an Illumina HiSeq X Ten platform as instructed by the manufacturer. The findings showed that generated generated clean sequences in total 51.12 Gb of PE (2 × 150 bp) in total (Supplementary Table 2). Only a single peak was detected in the purified sequences’ k-mer depth distribution (Figure 2), showing that the C. buqueti had low heterozygosity in the genome. Moreover, also used these Illumina sequencing data for genome size estimate, assembly adjustment, and assessment.
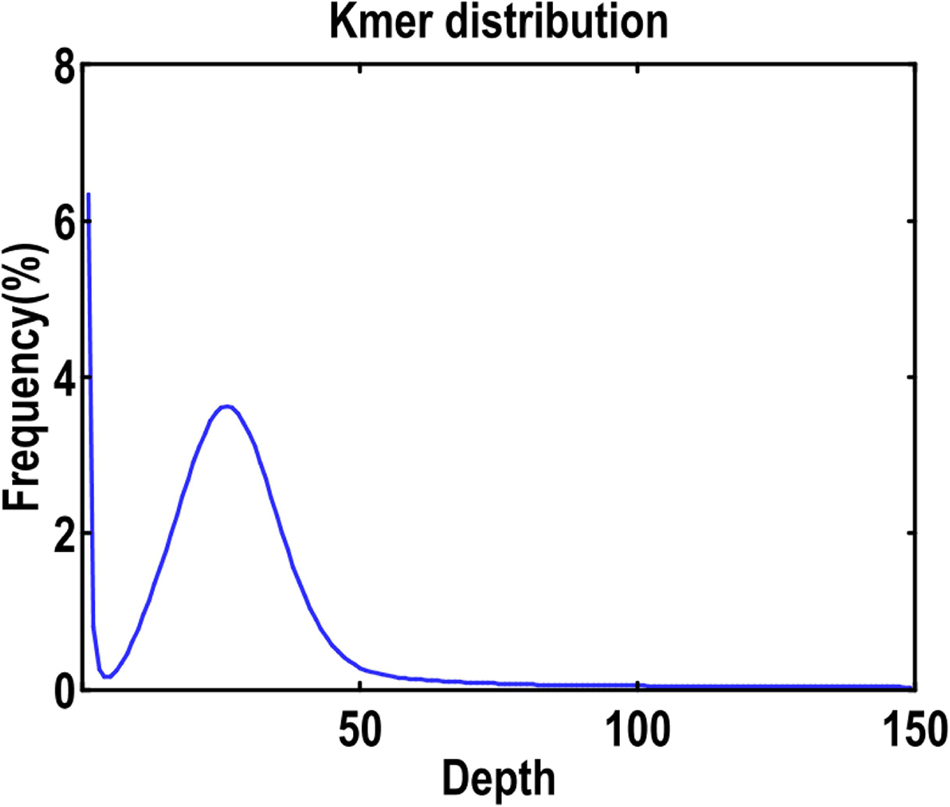
Figure 2. K-mer (K = 19) distribution of Illumina genome sequencing reads of C. buqueti. The total count of k-mers was 44976583431, and the peak k-mer depth was 64. The genome size of C. buqueti was calculated by dividing the total k-mer count by the peak depth, which was 600.92 Mb. The single peak of the k-mer distribution profile indicates that the C. buqueti genome has a low level of heterozygosity.
The total count of k-mers was 44976583431, and the peak k-mer depth was 64. The genome size of C. buqueti was calculated by dividing the total k-mer count by the peak depth, which was 600.92 Mb. The single peak of the k-mer distribution profile indicates that the C. buqueti genome has a low level of heterozygosity.
Muscle tissue from a male individual was ground in liquid nitrogen and RNA was extracted in RNA extraction reagent. RNA-seq library construction was performed using Illumina sequencing after its RNA met the requirements for transcriptome sequencing by agarose gel electrophoresis and measurement of nucleic acid instrument content. The original data were obtained by transcriptome sequencing. Clean reads obtained after quality evaluation, redundant sequences which were deleted were used for its genome sequence annotation, alignment and transcriptome analysis.
Genome Size Estimation
A PE library with 51.12 Gb clean reads from a 270 bp inserts used to estimate the C. buqueti genome size and heterozygosity using the 19-mer distribution method: G = k-mer number/average k-mer depth, with the 4^ k/genome > 200. Found the highest peak in the k-mer distribution curve at a k-mer depth of 64, and a total of 44976583431 k-mer were obtained, which resulted in 38786846512 k-mer after deleting the abnormal k-mer values (Supplementary Figure 1). According to the k-mer distribution, the C. buqueti genome size was 600.92 Mb with 52.27% repeat sequences and 0.20% heterozygosity, and the data were approximately 85 × the coverage of the genome.
High-Throughput Chromosome Conformation Capture Library Construction and Sequencing of the Cyrtotrachelus buqueti Genome
For Hi-C sequencing, chromatin isolation and library building from fresh tissues in C. buqueti was conducted using the Step Genomics animal Hi-C kit (Seattle, WA, United States). Streptavidin beads for library construction were identified as fragments containing contacts.
High-throughput chromosome conformation capture libraries were sequenced at Biomarker Technologies Corporation (Beijing, China) using the NextSeq500 platform (Illumina). The Hi-C raw reads were filtered using Trimmomatic (Bolger et al., 2014) to trim the adapter and low-quality sequences. BWA-aln (Li and Durbin, 2009) was used to align the Hi-C clean reads to the assembled contigs. A total of 55.87 Gb clean data with an 88.14 × coverage depth were obtained. Next, HiC-Pro (Burton et al., 2013) was used to screen and evaluate the Hi-C data divided into Valid Interaction Pairs and Invalid Interaction Pairs. Hi-C clean reads were aligned into genome sequences using BWA (Li and Durbin, 2009) and clustered by Phase Genomics Hi-C using LACHESIS.
Genome Assembly and Evaluation
To correct and assemble the clean data obtained from PacBio, we first used Canu assembler v1.6 (Canu, RRID:SCR 015880) (Koren et al., 2017). To complete the produced contigs, Falcon (Falcon, RRID:SCR 016089) and wtdbg1 were next used. Pilon (Pilon, RRID:SCR 014731) (Walker et al., 2014) was then carried out by aligning the transcriptome short reads to correct the sequencing errors. The assembled contigs were subsequently clustered with the default parameters by step genomics Hi-C using LACHESIS “CLUSTER_MIN_RE_SITES=22, CLUSTER_MAX_LINK_DENSITY=2, CLUSTER_ NONINFORMATIVE_RATIO=2, ORDER_MIN_N_RES_IN_ TRUN=10, ORDER_MIN_N_RES_IN_SHREDS=10.”
Genome Annotation and Comparative Genomics
LTR (long terminal repeats) and MITEs (miniature inverted transposable elements) were classified for repeat element annotation by using LTR FINDER v1.0.5 (Xu and Wang, 2007) and MITE-Hunter v1.0.0 (Bao et al., 2015), respectively. Next, PILER-DF v2.4 (Han and Wessler, 2010), which implements RepeatScout v1.0.5 findings (RepeatScout, RRID:SCR 014653) (Edgar and Myers, 2005), was used by scanning the assembly genome to produce a de novo repeat library. PASTEClassifier v1.0 (Price et al., 2005) has been described as a Transposable Elements REPET package classifier for the RepBase library repeat classification (Hoede et al., 2014). Finally, RepeatMasker v4.0.6 (RepeatMasker, RRID:SCR 012954) (Bao et al., 2015) was conducted to model repeated sequences scanning the C. buqueti genome.
Three techniques, homolog-based, RNA-sequencing (RNA-seq)-based, and initio-based gene prediction methods, were used to classify protein-coding genes. First, with Genscan, ab initio-based gene prediction was performed (Tarailo-Graovac and Chen, 2009), Augustus v2.4 (Augustus, RRID:SCR 008417) (Keilwagen et al., 2016), GlimmerHMM v3.0.4 (GlimmerHMM, RRID:SCR 002654), GeneID (v1.4) (Stanke and Waack, 2003), and SNAP v2006-07-28 (SNAP, RRID:SCR 002127) (Blanco et al., 2007), using the parameters by design. Next, using GeMoMa, homolog-based gene annotation was done (v1.3.1) (Korf, 2004), with the protein databases of Dendroctonus ponderosae (GCA 000355655.1), Tribolium castaneum (GCA 000002335.3), Drosophila melanogaster (GCA 000001215.4), Anoplophora glabripennis (GCA 000390285.2), Oryctes borbonicus (GCA 001443705.1), and from GenBank as the references. TransDecoder (V5.0)(Keilwagen et al., 2016), GeneMarkS-T v5.1 (RRID:SCR 011930) (Transdecoder, 2018), and PASA v2.0.2 (RRID:SCR 014656) (Kanehisa and Goto, 2000) were used for RNA-seq-based gene prediction, and the C. buqueti transcriptome data were assembled (NCBI bioproject accession number, PRJNA718062). Finally, to combine the three processes, EVM (v1.1.1, RRID:SCR 014659) (Tang et al., 2015) was adopted, following which the results were updated using PASA v2.0.2. Compared these predicted genes with those in the non-redundant protein sequences (NR), eukaryotic orthologous protein classes (KOG) (Campbell et al., 2006), Kyoto Encyclopedia of Genes and Genomes (KEGG) (KEGG, RRID:SCR 001120) (Burge and Karlin, 1997), Swissprot (Swissprot, RRID:SCR 002380) (Gao et al., 2018), TrEMBL (Haas et al., 2008), and Pfam (Pfam, RRID:SCR 004726) (Tatusov et al., 2001) databases for gene annotation using the Basic Local Alignment Search Tool (BLAST) with an e-value cut off of 1E-5 and hmmer V3.0 (Kanehisa and Goto, 2000). With the BLAST2GO pipeline (Boeckmann et al., 2003), Gene ontology (GO) annotation was done. GenBlastA (Mistry et al., 2013) was performed for pseudogene prediction by scanning the C. buqueti genome for homologous sequences. The stop codons or frameshift mutations were then used to recognize pseudogenes. The stop codons or frameshift mutations were subsequently used to classify pseudogenes using GeneWise (GeneWise, RRID:SCR 015054) (Zdobnov and Apweiler, 2001).
We had compared genome between C. buqueti and six other insects, D. melanogaster (GCA 000001215.4), D. ponderosae (GCA 000355655.1), T. castaneum (GCA 000002335.3), A. planipennis (GCA 000699045.2), Z. nevadensis (GCA 000696155.1), and A. glabripennis (GCA 000390285.2), as well as the giant panda (Ailuropoda melanoleuca) (GCA 000004335.1) (Birney et al., 2004; Conesa et al., 2005; Richards et al., 2008; She et al., 2009; Keeling et al., 2013; Terrapon et al., 2014; McKenna et al., 2016). OrthoMCL (Zhao et al., 2012) was used to analyze the orthologous groups of those eight species.
Manual Annotation of Specific Gene Families and Phylogenetic Analysis
Using reciprocal BLAST against NCBI nr and gene family-specific datasets, the gene families of cytochromes P450, GSTs, and CAZymes (CAZy) were identified in C. buqueti genome. Each gene model was manually annotated, and then the non-redundant translated proteins were aligned with MUSCLE (Li et al., 2003) to the corresponding proteins from several other insect species for which genomes have been sequenced. A maximum-likelihood phylogeny was created with Mega XI (Guindon et al., 2009) and drawn with iTOL2 (Ivica and Peer, 2021).
Results and Discussion
Genome Assembly and Annotation
A total of 630.86 Mb genomic sequences were located on chromosomes, accounting for 99.53% of the total sequence length, while the corresponding sequences were 105, accounting for 65.63% of the total sequences (Figure 3 and Supplementary Table 3). At last, we assembled the genome sequences of C. buqueti; this genome had an overall length of 633.85 Mb and composed of 149 contigs with an N50 length of 27.93 Mb, respectively (Supplementary Table 4). As of August 31, 2021, there are currently 2,241 insect genome assembly versions, including different assemblies of the same insect. Of all the assembled versions, 296 were at the chromosome level, 1,060 were at the contig level, 885 were at the scaffold level. After eliminating different genome assembly versions of the same species and leaving the version with the highest assembly quality, 1,525 insect genomes have been published on NCBI (Supplementary Table 5). Moreover, the maximum value of scaffold N50 in all insect genome assembly is 25.7 Mb. This value is less than the contig N50 (27.93 Mb) of C. buqueti genome. Therefore, the assembly quality of C. buqueti genome is higher than that of most published insect genomes.
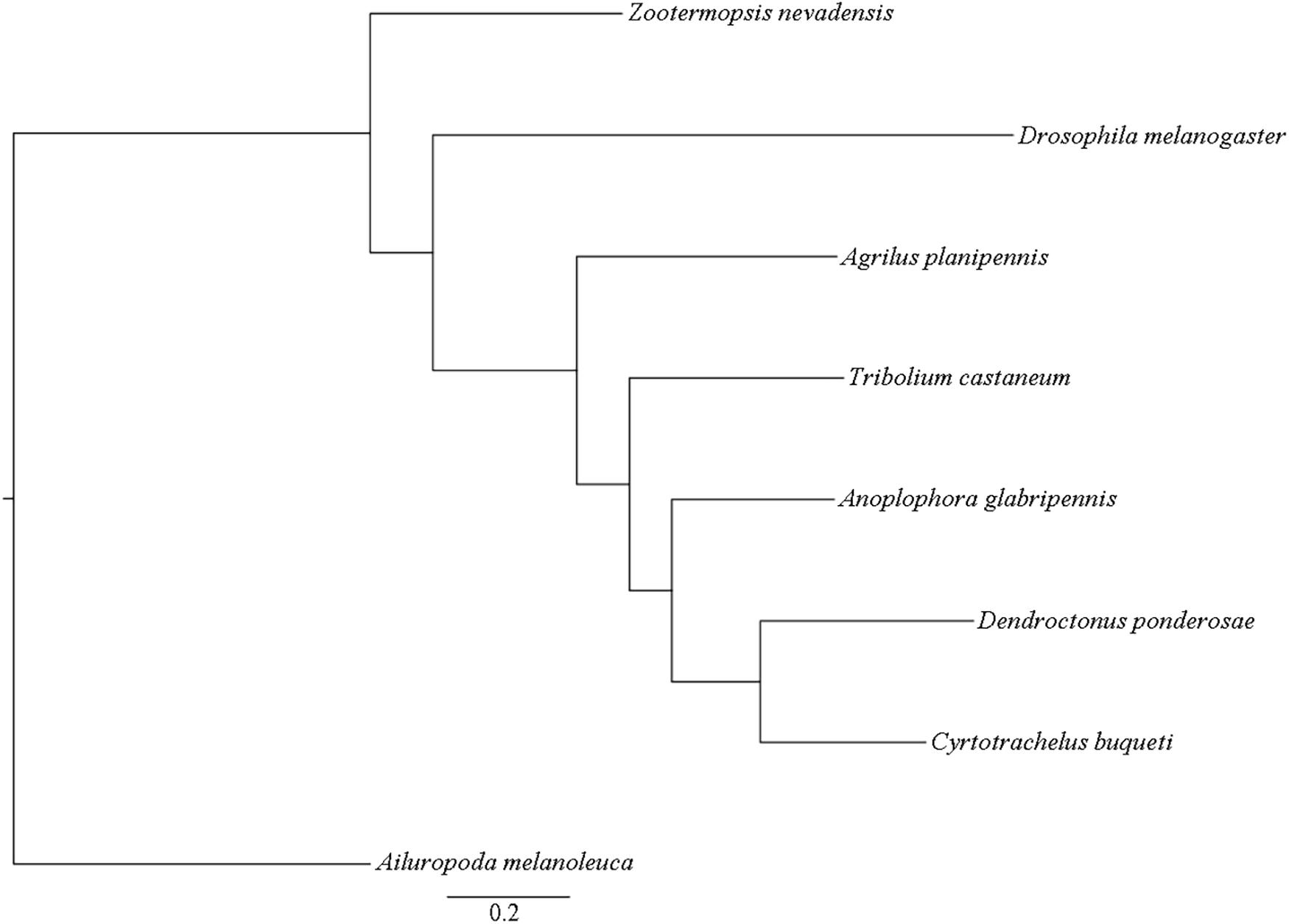
Figure 3. Phylogenetic relationships of C. buqueti and seven other species.
The 51.12 Gb transcriptome sequencing reads (NCBI bioproject number, PRJNA718062) were used for compatibility with C. buqueti to assess the genome assembly using BWA-MEM (BWA, RRID:SCR 010910) (Sudhir et al., 2018). The evaluation results showed that all Illumina reads were mapped and that 99.99% of PE reads were mapped accordingly (Supplementary Table 6). Also, the CEGMA v2.5 (CEGMA, RRID:SCR 015055) assessment revealed that there were a total of 455 CEGs in the assembly, comprising 99.34% of the database and including 243 of the 248 (97.98%) firmly retained eukaryotic core genes (CEGs) (Supplementary Table 7). Finally, the results obtained from Benchmarking Universal Single-Copy Orthologs v3.0.2 (BUSCO, RRID:SCR 015008) (Ivica and Peer, 2016) showed that partially acquired 99.71% of the CEGs and 98.87% were entirely achieved (Supplementary Table 8). With 81.41% coding regions, 7.13% introns and 11.45% intergenic regions, the GC content was 38.41% in this genome (Supplementary Table 9).
Repeat Sequence and Gene Function Annotation
There were two types, ClassI and ClassII, in the repeat sequences of C. buqueti genome. In ClassI repeat, there were 116 DIRS, 1046 LINE, 853 LTR, 124 LTR/Copia, 1336 LTR/Gypsy, 128921 PLE/LARD, 162 SINE, 13 SINE/TRIM, 2170 TRIM and 155 unknown repeats. In ClassII repeat, there were 1986 Crypton, 42679 Helitron, 2578 MITE, 164 Maverick, 160523 TIR and 1607 unknown repeats (Supplementary Table 10).
The annotation results of C. buqueti genome showed that there are 37,188 genes including protein-coding and non-protein-coding genes. The total gene length of these genes was 166,889,308 bp (Supplementary Table 11). The genome annotation results showed that there were 12,569 protein-coding genes in C. buqueti genome. Moreover, all 12,569 protein-coding genes were distributed on 12 chromosomes of C. buqueti (Table 1). A 5,912 protein-coding genes, 47.04% of all protein-coding genes in C. buqueti genome, are annotated by GO, 5,578 protein-coding genes, 44.38% of that, are annotated by KEGG, 8,388 protein-coding genes, 66.74% of that, are annotated by KOG, 9,793 protein-coding genes, 77.91% of that, are annotated by Pfam, 7,880 protein-coding genes, 62.69% of that, are annotated by Swissprot, 11,895 protein-coding genes, 94.64% of that, are annotated by TrEMBL, 11,901 protein-coding genes, 94.69% of that, are annotated by NR, 10,162 protein-coding genes, 80.85% of that, are annotated by Nt. In sum, 12,089 protein-coding genes are annotated by eight database, accounting for 96.18% of all protein-coding genes in C. buqueti genome (Table 2). The results of pseudogene prediction showed that there were 1,621 pseudogenes in C. buqueti genome (Supplementary Table 12).
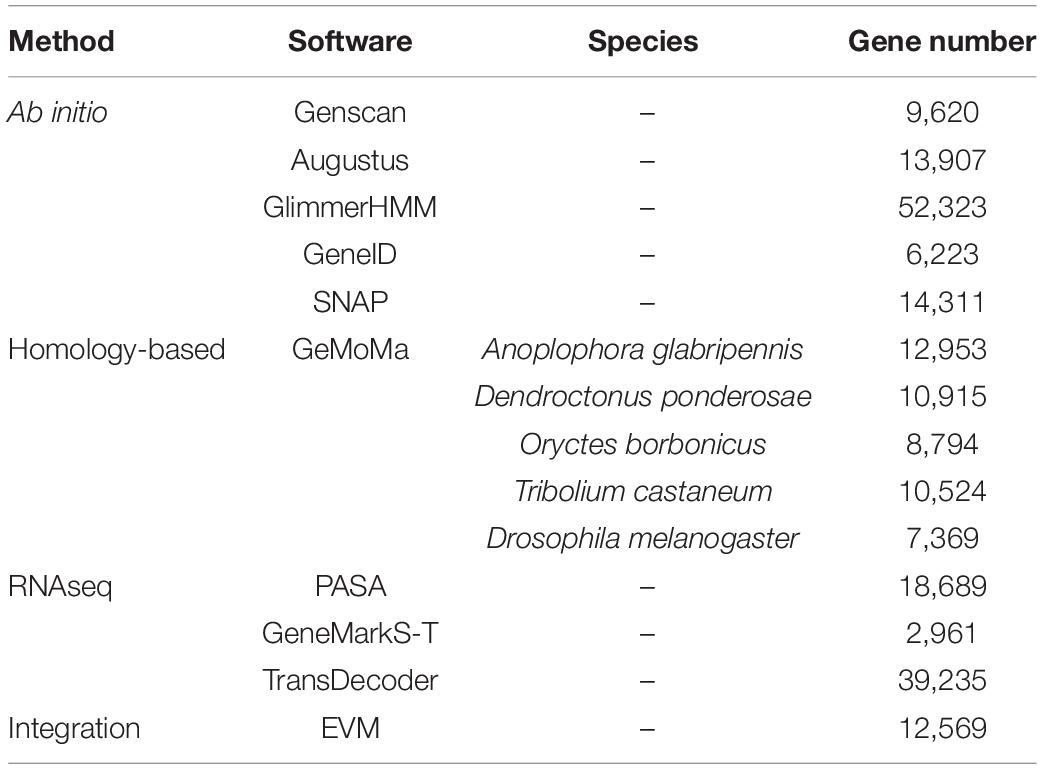
Table 1. Summary of C. buqueti genome annotation.
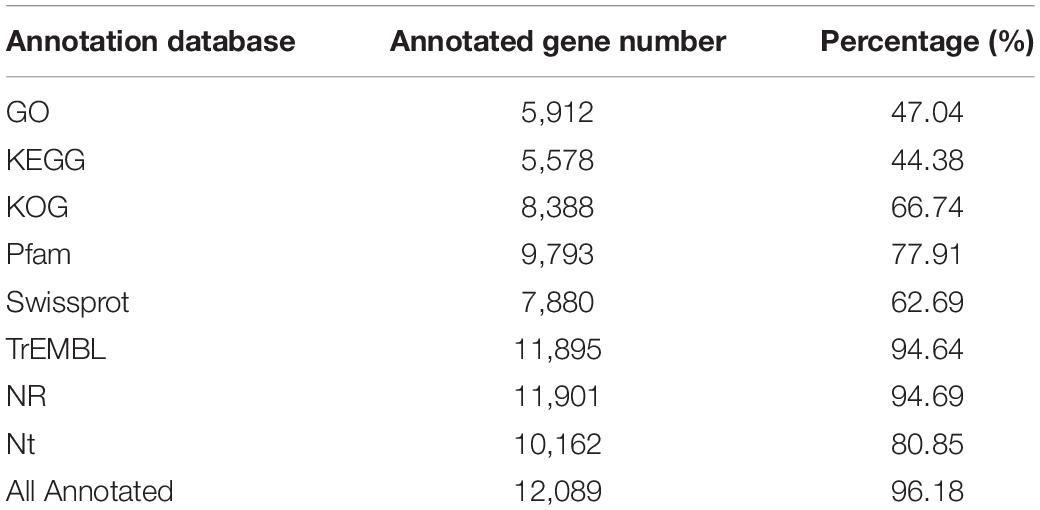
Table 2. Summary of functional annotation for the predicted genes.
Comparative Genomic Analysis
A 109,057 genes were used for comparative genomic analysis, including 12,569 genes in C. buqueti genome, 12,314 genes in Z. nevadensis genome, 12,841 genes in T. castaneum genome, 11,373 genes in A. planipennis genome, 13,886 genes in A. glabripennis genome, 12,102 genes in D. ponderosae genome, 13,886 genes in D. melanogaster genome and 19,439 genes in A. melanoleuca genome. A total of 10,956 genes in C. buqueti genome were assigned to 9,394 gene families, with 112 unique families. These results also indicate that the number of specific gene family in C. buqueti genome is only 112 as well as that in A. planipennis, which was less than that in other related genomes (Table 3). A maximum-likelihood phylogenetic tree constructed using PhyML (PhyML, RRID:SCR 014629) (Simao et al., 2015) showed that C. buqueti was closely related to D. ponderosae (Figure 3). The CodeML (Schabauer et al., 2012) module in PAML and the Branch Site model were used to analyze the selection pressure of single-copy genes. The rapid evolution single-copy genes were annotated by alignment to the KEGG and GO databases (Figure 4).
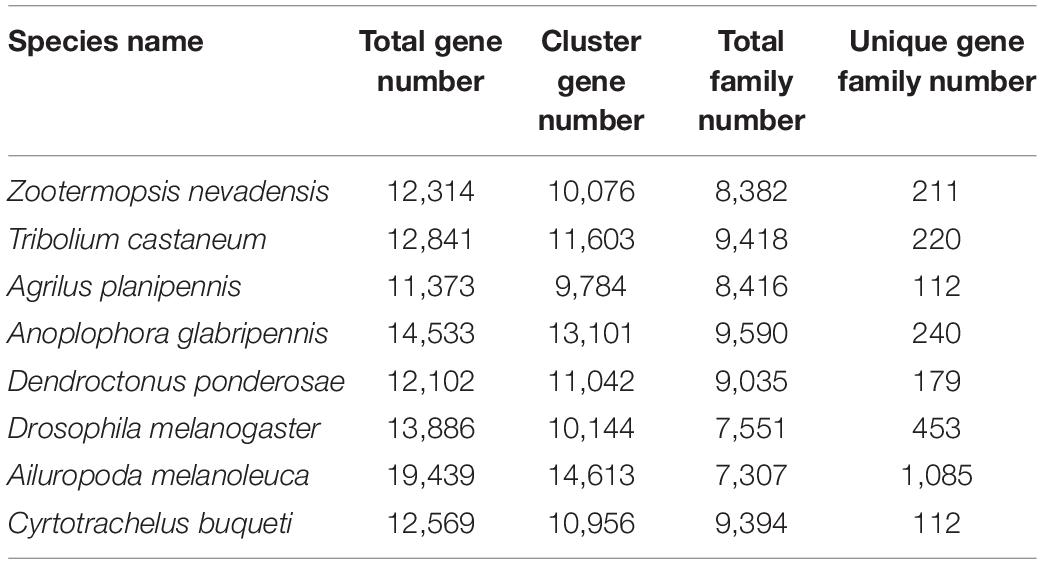
Table 3. Comparative genomic analysis of the eight species.
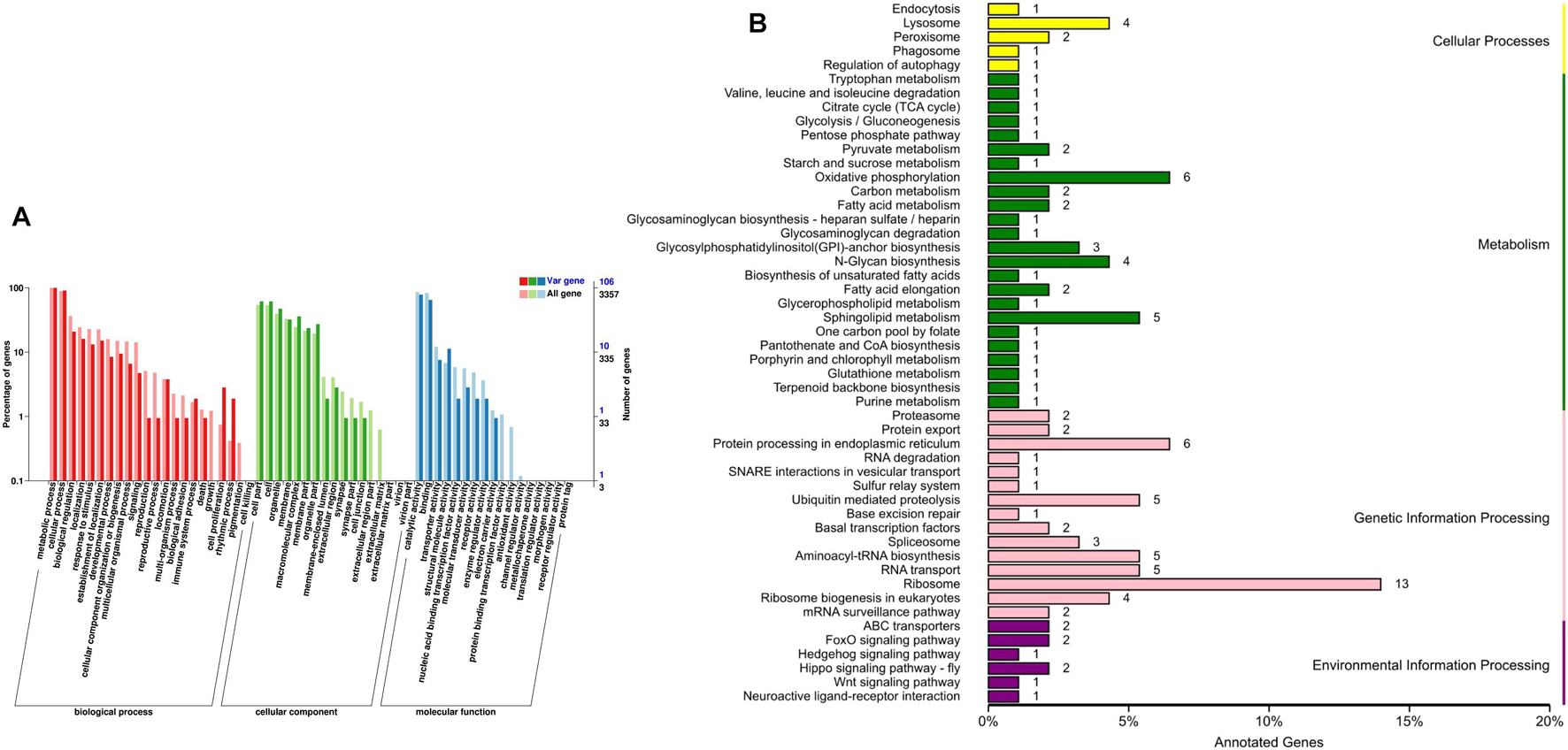
Figure 4. GO and KEGG annotation of the rapid evolution single-copy genes.
Identification and Comparative Analysis of Specific Gene Families
To complete its whole life cycle, C. buqueti a larval only has to eat on bamboo shoots for around 20 days. Furthermore, previous research has revealed that it has a high capacity to breakdown bamboo cellulose (Luo et al., 2018a, 2019a, b). However, it is unknown which gene family in the C. buqueti genome is engaged in bamboo cellulose degradation, how it is distributed, or which genes play a key role in bamboo cellulose breakdown. Therefore, its unidentified all the genes in C. buqueti genome that may be involved in bamboo cellulose degradation, but also identified those genes in other related species’ genomes. The identification results showed that the number of the CAZy genes in C. buqueti genome was only 244, account for 1.94% of total genes, which was less than that of most related species, just a little more than that of Zootermopsis nevadensis genome (236). In related species’ genomes, the number of CAZy gene family in Ailuropoda melanoleuca genome was largest, which was 701. It has been reported that Z. nevadensis has strong lignocellulose degrading enzyme activity (Pester and Brune, 2007). Moreover, A. melanoleuca is a mammal with the same food as C. buqueti as the only food. However, it has been reported that the degradation ability of A. melanoleuca to lignocellulose is not as strong as that of Z. nevadensis (Jin et al., 2021). In brief, it can be inferred that the activity of lignocellulose degrading enzyme may be inversely proportional to the number of CAZy genes in vivo. At the same time, we found that the number of AA, CBM, and CE genes in CAZy gene family in C. buqueti and Z. nevadensis genome was higher than that in A. melanoleuca genome. This finding confirms that C. buqueti has stronger ability to degrade lignocellulose than A. melanoleuca and weaker ability to degrade lignocellulose than Z. nevadensis. Moreover, this finding suggests that some members of AA, CBM, and CE gene family may play a key role in lignocellulose degradation (Table 4). Further research of which members of CAZy gene family in C. buqueti genome play a key role in bamboo cellulose degradation was needed.
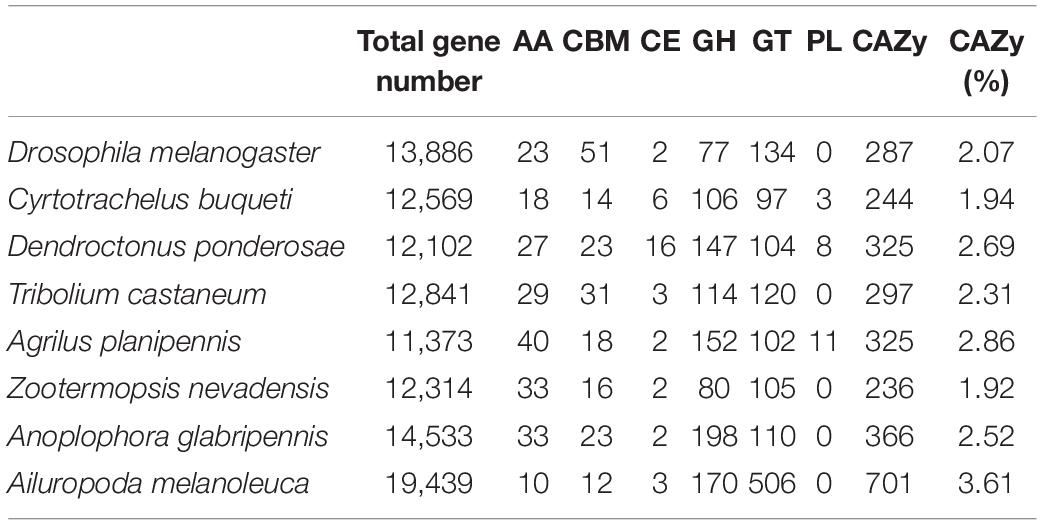
Table 4. Identification and comparative analysis of cellulose degradation enzyme gene between Cyrtotrachelus buqueti and other related species’ genomes.
Although we have identified 244 CAZy gene family genes in the C. buqueti genome involved in the rapid degradation of bamboo cellulose, which is the energy required for the rapid transformation of its life activities, its energy conversion and storage mechanism still unclear, we analyzed the genes related to energy production and conversion, energy storage in the C. buqueti genome to solve this problem. These results showed 240 genes related to energy production and transformation in C. buqueti genome, which including mitochondrial carrier protein, ATP synthase, Adenylate and so on. There are 11 genes related to fatty acid metabolism, these genes may be related to their energy storage (Table 5). Although 240 genes related to energy production and conversion have been found, it is not clear how these genes participate in the rapid energy conversion and storage of C. buqueti. Therefore, these mechanisms need to be further studied.

Table 5. Statistical table of genes related to energy production and conversion.
P450 cytochromes and glutathione S-transferases (GSTs) are commonly involved in detoxifying plant chemicals. Some members are likely to be involved in the sequential pathway of metabolizing xenobiotics by making them more polar and excretable. To reveal how C. buqueti detoxifies plant chemical components, we identified the detoxification enzyme gene, the P450 cytochromes and the glutathione S-transferases (GSTs) in its genome. At the same time, we analyzed the number and proportion of detoxification enzyme genes in its genome and other beetles. These results showed 73 P450 genes and 30 GSTs genes, accounting for 0.58 and 0.24% of the total genes, respectively, in the C. buqueti genome. Compared with other beetle genomes, the number of P450 genes in its genome is medium. However, the number of GSTs genes in its genome is relatively large, only less than T. castaneum and A. glabripennis (Table 6).
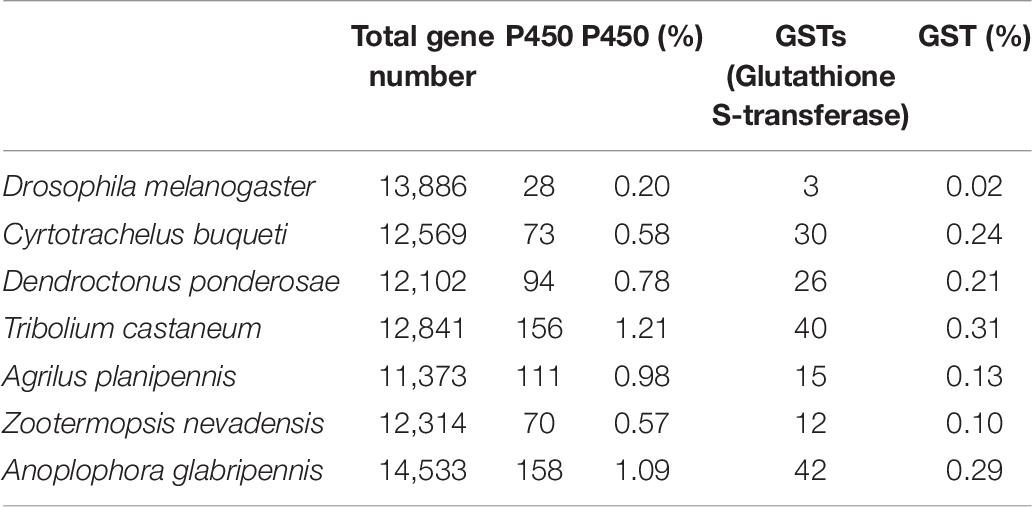
Table 6. Identification and comparative analysis of detoxification enzyme gene between Cyrtotrachelus buqueti and other insect genomes.
Tribolium castaneum is the first Coleoptera insect which be completed genome sequencing and annotation. In this study, we analyzed the evolutionary relationship of P450 and GST gene families in C. buqueti and T. castaneum genomes. All P450 family protein members were divided into 12 groups in phylogenetic tree of P450 proteins in C. buqueti and red flour beetle genome. These groups were named Group I to Group XII. In this phylogenetic tree, Group I was the fastest evolving group, and Group XII was the slowest and most conservative group. A 73 and 156 members of the P450 gene family were, respectively, found in the phylogenetic tree of C. buqueti and T. castaneum. Moreover, most P450 gene family members of C. buqueti were mainly distributed in Group II, Group VII and Group XI, most of those in T. castaneum were mainly distributed in Group I and Group IV. These results showed that most of the P450 gene family members of T. castaneum evolved faster than those of C. buqueti. Most members of the P450 gene family of C. buqueti were more conservative and probably play a more important role in detoxifying phytochemicals (Figure 5).
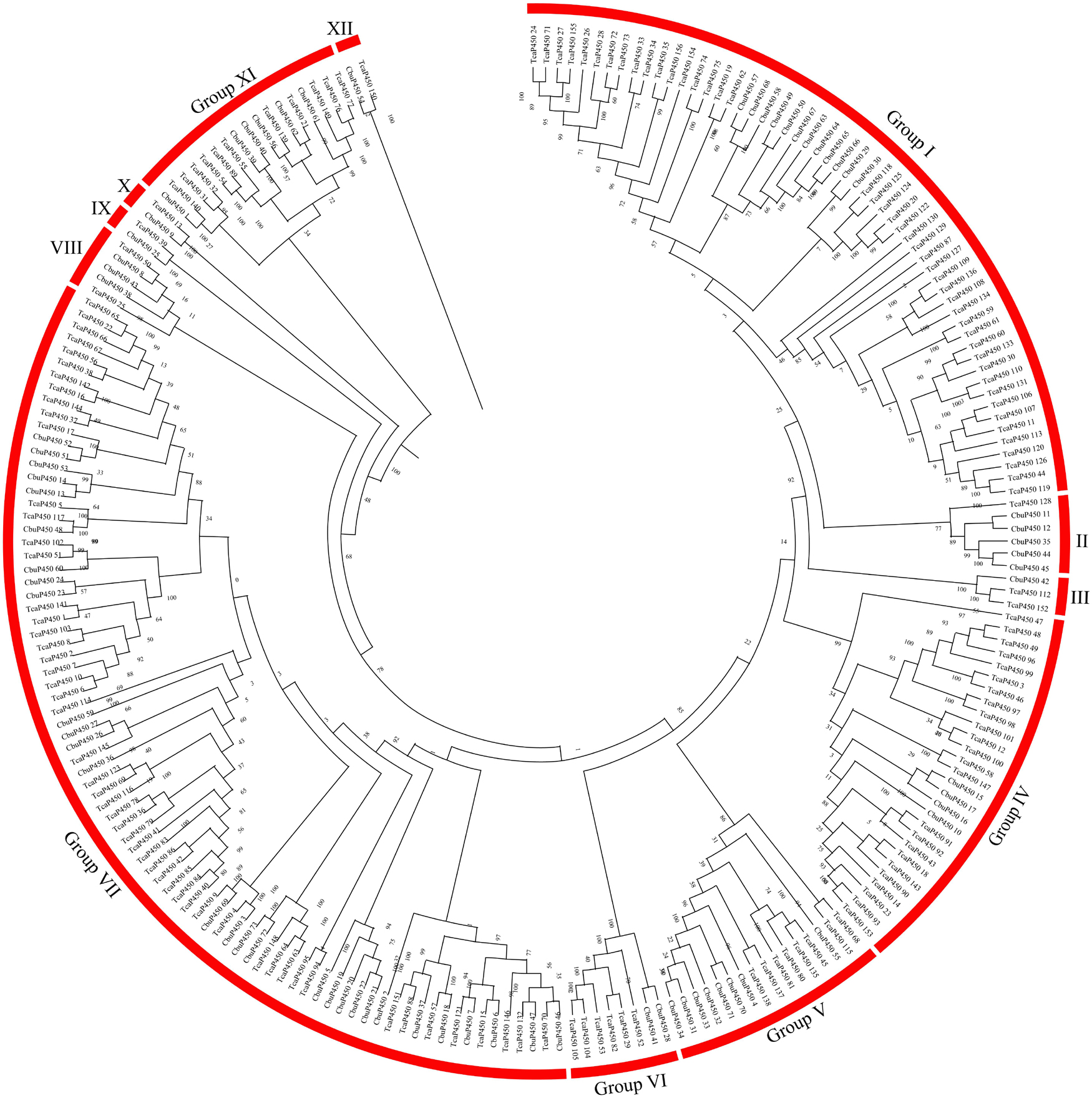
Figure 5. Phylogenetic tree of P450s in C. buqueti (C. buqueti, Cbu) and red flour beetle (T. castaneum, Tca) genome.
Glutathione S-transferase (GST) is the key enzyme in glutathione binding reaction, it exists in many forms. In the phylogenetic tree of the GSTs gene family in C. buqueti and T. castaneum genome, all the GSTs protein members were divided into 10 groups, Group I to Group X, respectively. Group I was the fastest evolving group, and Group X was the slowest and most conservative group. Interestingly, we found that most of the GST gene family members of C. buqueti were distributed in Group I, while most of the GST memory family members of T. castaneum were distributed in Group IX and Group X. These findings indicate that the evolutionary rate of GST gene family members of C. buqueti is significantly higher than that of T. castaneum (Figure 6).
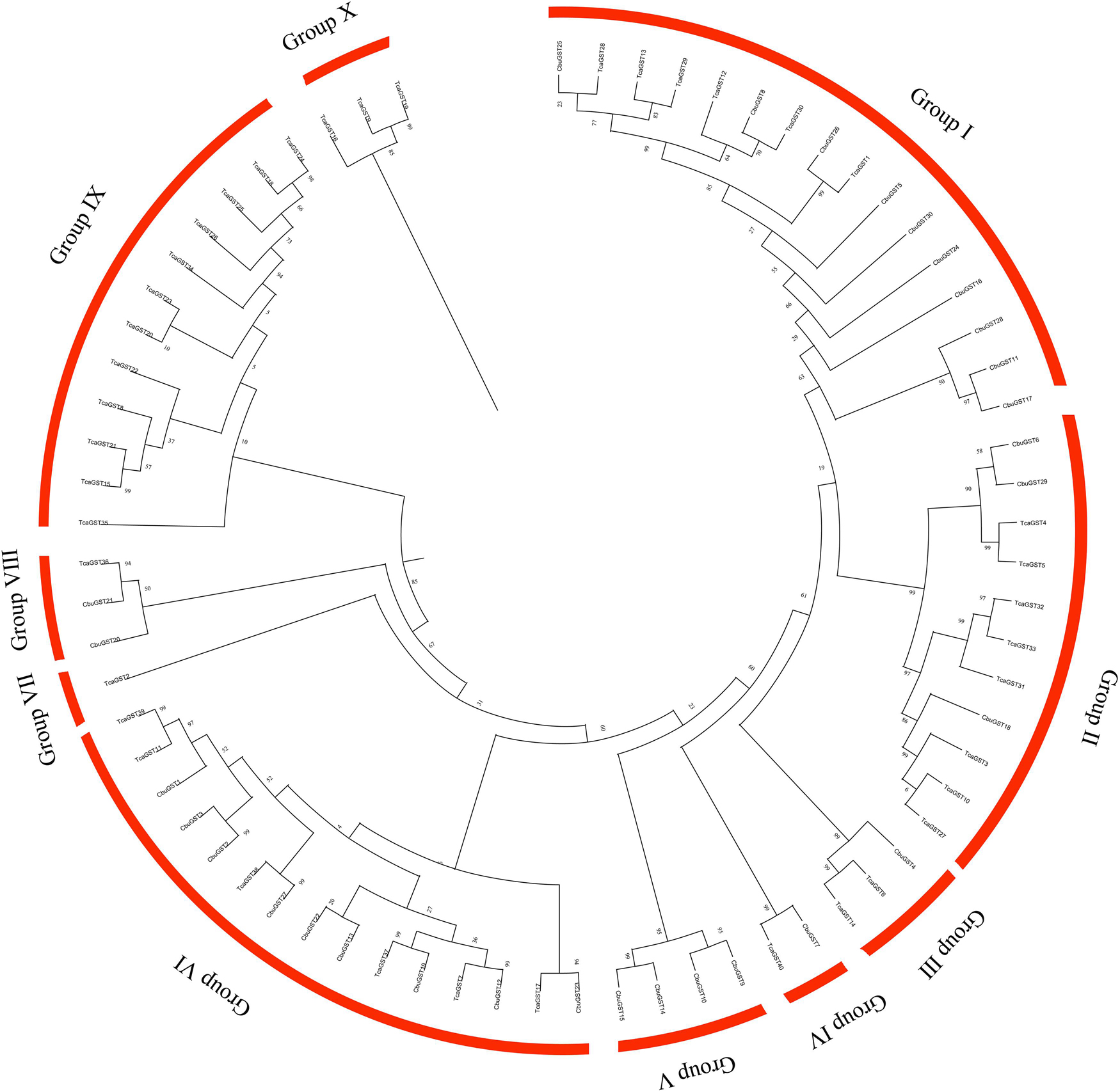
Figure 6. Phylogenetic tree of GSTs (glutathione S-transferases) in C. buqueti (C. buqueti, Cbu) and red flour beetle (T. castaneum, Tca) genome.
Conclusion
We have assembled a high-quality genome of C. buqueti, which is the pest that does the most bamboo damage. The assembly quality of this insect’s genome sequence was better than those of other published insects’ genome. In this study, we identified that 244 genes were related to bamboo cellulose degradation in C. buqueti genome. At the same time, we found that there were specific genes related to lignocellulose degradation in a genome, such as some members of AA, CBM, and CE gene families. Moreover, 240 genes related to energy production and conversion were identified in C. buqueti genome. Finally, we compared and analyzed the evolutionary relationship of the P450 and GST gene family between C. buqueti and Coleoptera model insect T. castaneum genome. The evolutionary relationship between the two gene family members showed that the detoxification function of C. buqueti was possible stronger than that of T. castaneum. Genome assembly at a chromosome level of C. buqueti, identification and comparative analysis of its specific genes laid a theoretical foundation for revealing the molecular mechanism of its bamboo degradation, energy conversion, storage and detoxification function.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accessions: PRJNA675312 and PRJNA718062).
Author Contributions
CF, WL, CL, YL, YC, XX, HL, JY, SC, XN, SB, and YY collected insect samples, extracted DNA/RNA, and performed transcriptome sequencing and gene expression analyses. CF, CL, WL, YC, XX, HL, JY, and SC performed DNA sequencing, genome assembly, gene annotation, evolution and comparative genomic analyses. CF, WL, CL, SB, and YY wrote and revised the manuscript. CF, CL, WL, XN, SB, and YY conceived strategies, designed experiments, and managed projects. All authors read and approved the manuscript.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31470655) and Leshan Normal University’s Science and Technology Program (XJR17005, LZD010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful for the support and assistance from the Bamboo Diseases and Pests Control and Resources Development Key Laboratory of Sichuan Province, Leshan Normal University.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.729100/full#supplementary-material
Abbreviations
BLAST, Basic Local Alignment Search Tool; bp, base pair; CPM, counts per million mapped read pairs; CTAB, cetyl trimethyl ammonium bromide; Gb, gigabase; Hi-C, high-throughput chromosome conformation capture; LTR, long terminal repeat; Mb, megabase; MITE, miniature inverted transposable element; NCBI, National Center for Biotechnology Information; PacBio, Pacific Biosciences; RNA-Seq, RNA sequencing; SMRT, single-molecule real-time; BUSCO, Benchmarking Universal Single-Copy Orthologs; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; KOG, eukaryotic orthologous groups of proteins; PASA, acronym for Program to Assemble Spliced Alignments; PE, paired-end; QV, quality value.
Footnotes
References
Bao, W., Kojima, K. K., and Kohany, O. (2015). Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6:11.
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Blanco, E., Parra, G., and Guigó, R. (2007). Using geneid to identify genes. Curr. Protoc. Bioinformatics 18:4.3.
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. Q., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt.2727
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M., and Buell, C. (2006). Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7:327. doi: 10.1186/1471-2164-7-327
Chauhan, O. P., Unni, L. F., Kallepalli, C., Pakalapati, S. R., and Batra, H. V. (2016). Bamboo shoots, composition, nutritional value, therapeutic role and product development for value addition. Int. J. Food Ferment. Technol. 6, 1–12. doi: 10.5958/2277-9396.2016.00021.0
Chen, F. Z., Wang, W. D., Wang, X. Q., Li, S. H., and Gong, J. W. (2005). Occurrence and control of Cyrtotrachelus buqueti Guer. Plant Prot. 31, 89–90.
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Edgar, R. C., and Myers, E. W. (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158.
Gao, F., Li, H., Xiao, Z., Wei, C., Feng, J., and Zhou, Y. (2018). De novo transcriptome analysis of Ammopiptanthus nanus and its comparative analysis with A. mongolicus. Trees 32, 287–300. doi: 10.1007/s00468-017-1631-6
Guindon, S., Delsuc, F., Dufayard, J. F., and Gascuel, O. (2009). Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 537, 113–137. doi: 10.1007/978-1-59745-251-9_6
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., and Orvis, J. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7.
Han, Y., and Wessler, S. R. (2010). MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38:e199. doi: 10.1093/nar/gkq862
Hazzouri, K. M., Sudalaimuthuasari, N., Kundu, B., Nelson, D., Al-Deeb, M. A., Le Mansour, A., et al. (2020). The genome of pest Rhynchophorus ferrugineus reveals gene families important at the plant-beetle interface. Commun. Biol. 3:323.
Hoede, C., Arnoux, S., Moisset, M., Chaumier, T., Inizan, O., Jamilloux, V., et al. (2014). PASTEC: an automatic transposable element classification tool. PLoS One 9:e91929. doi: 10.1371/journal.pone.0091929
Ivica, L., and Peer, B. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–5.
Ivica, L., and Peer, B. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Jin, L., Huang, Y., Yang, S., Wu, D., Li, C., Deng, W., et al. (2021). Diet, habitat environment and lifestyle conversion affect the gut microbiomes of giant pandas. Sci. Total Environ. 770:145316. doi: 10.1016/j.scitotenv.2021.145316
Kanehisa, M., and Goto, S. K. E. G. G. (2000). Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30.
Keeling, C. I., Yuen, M. M., Liao, N. Y., Docking, T. R., Chan, S. K., Taylor, G. A., et al. (2013). Draft genome of the mountain pine beetle, Dendroctonus ponderosae Hopkins, a major forest pest. Genome Biol. 14:R27.
Keilwagen, J., Wenk, M., Erickson, J. L., Schattat, M. H., Grau, J., and Hartung, F. (2016). Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44:e89. doi: 10.1093/nar/gkw092
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M., et al. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Li, B. Q. (2010). Present status of major forest insect pests in vietnam and countermeasures. For. Pest Dis. 29, 35–38.
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Littlewood, J., Wang, L., Turnbull, C., and Murphy, R. J. (2013). Techno-economic potential of bioethanol from bamboo in China. Biotechnol. Biofuels 6:173.
Luo, C. B., Li, Y. Q., Chen, Y., Fu, C., Long, W. C., Xiao, X. M., et al. (2019a). Bamboo lignocellulose degradation by gut symbiotic microbiota of the bamboo snout beetle Cyrtotrachelus buqueti. Biotechnol. Biofuels 12:70.
Luo, C. B., Li, Y. Q., Chen, Y., Fu, C., Nong, X., and Yang, Y. J. (2019b). Degradation of bamboo lignocellulose by bamboo snout beetle Cyrtotrachelus buqueti in vivo and vitro: efficiency and mechanism. Biotechnol. Biofuels 12:75.
Luo, C. B., Liu, A. X., Long, W. C., Liao, H., and Yang, Y. J. (2018b). Transcriptome analysis of Cyrtotrachelus buqueti in two cities in China. Gene 647, 1–12. doi: 10.1016/j.gene.2017.12.041
Luo, C. B., Li, Y. Q., Liao, H., and Yang, Y. J. (2018a). De novo transcriptome assembly of the bamboo snout beetle Cyrtotrachelus buqueti reveals ability to degrade lignocellulose of bamboo feedstock. Biotechnol. Biofuels 11:292.
Mang, D. Z., Luo, Q. H., Shu, M., and Wei, W. (2012). Extraction and identification of cuticular semiochemical components of Cyrtotrachelus buqueti Guerin-Meneville (Coleoptera: Curculionidae). Acta Entomol. Sin. 55, 291–302.
McKenna, D. D., Scully, E. D., Pauchet, Y., Hoover, K., Kirsch, R., Geib, S. M., et al. (2016). Genome of the Asian longhorned beetle (Anoplophora glabripennis), a globally significant invasive species, reveals key functional and evolutionary innovations at the beetle–plant interface. Genome Biol. 17:227.
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41:e121. doi: 10.1093/nar/gkt263
Pester, M., and Brune, A. (2007). Hydrogen is the central free intermediate during lignocellulose degradation by termite gut symbionts. ISME J. 1, 551–565. doi: 10.1038/ismej.2007.62
Price, A. L., Jones, N. C., and Pevzner, P. A. (2005). De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358.
Richards, S., Gibbs, R. A., Weinstock, G. M., Brown, S. J., Denell, R., Beeman, R. W., et al. (2008). The genome of the model beetle and pest Tribolium castaneum. Nature 452, 949–955. doi: 10.1038/nature06784
Schabauer, H., Valle, M., Pacher, C., Stockinger, H., Stamatakis, A., Robinson-Rechavi, M., et al. (2012). “SlimCodeML: An Optimized Version of CodeML for the Branch-Site Model,” in IEEE International Workshop on High Performance Computational Biology (HiCOMB’12), (Shanghai: IEEE), 706–714.
Scurlock, J. M. O., Dayton, D. C., and Hames, B. (2000). Bamboo: an overlooked biomass resource? Biomass Bioenergy 19, 229–244. doi: 10.1016/s0961-9534(00)00038-6
She, R., Chu, J. S., Wang, K., Pei, J., and Chen, N. (2009). GenBlastA: enabling BLAST to identify homologous gene sequences. Genome Res. 19, 143–149.
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–25. doi: 10.1093/bioinformatics/btg1080
Sudhir, K., Glen, S., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Tang, S., Lomsadze, A., and Borodovsky, M. (2015). Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43:e78. doi: 10.1093/nar/gkv227
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25:4.10.
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/nar/29.1.22
Terrapon, N., Li, C., Robertson, H. M., Ji, L., Meng, X., Booth, W., et al. (2014). Molecular traces of alternative social organization in a termite genome. Nat. Commun. 5:3636.
TransDecoder (2018). TransDecoder Release v5.5.0. Available online at: https://github.com/TransDecoder/TransDecoder/releases/tag/TransDecoder-v5.5.0 (accessed October 26, 2018).
Tribolium Genome Sequencing Consortium, Richards, S., Gibbs, R. A., Weinstock, G. M., Brown, S. J., Denell, R., et al. (2008). The genome of the model beetle and pest Tribolium castaneum. Nature 452, 949–955.
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, W. D., Chen, F. Z., and Wang, X. Q. (2005). Reproductive behavior of Cyrtotrachelus buqueti. Sichuan J. Zool. 24, 540–541.
Wen, L. Y., and Lu, W. X. (2006). Study on biological features of tiamlin badu bamboo’s otidognathus davidia‘s and its control. World Bamboo Rattan 4, 38–40.
Xiao, C. H. (2009). Investigation on species and damage of bamboo shoot weevil on Phyllostachys pubescens in fujian province. J. Fujian For. Sci. Technol. 36, 55–58.
Xu, Z., and Wang, H. (2007). LTR?FINDER: an efficient tool for predicting full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268.
Yang, H., Yang, M. F., Yang, W., Yang, C. P., Zhu, T. H., and Huang, Q. (2010). Behavioral and EAG responses of Cyrtotrachelus buqueti Guerin-Meneville (Coleoptera: Curculionidae) adults to host volatiles and their body extracts. Acta Entomol. Sin. 53, 286–292.
Yang, Y. J., Wang, S. F., Gong, J. W., Liu, C., Mu, C., and Qin, H. (2009). Relationships among Cyrtotrachelus buqueti larval density and wormhole number and bamboo shoot damage degree. Ying Yong Sheng Tai Xue Bao 20, 1980–5.
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan-an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Keywords: cyrtotrachelus buqueti, chromosome-level genome, genome assembly, bamboo snout beetle, functional annotation
Citation: Fu C, Long W, Luo C, Nong X, Xiao X, Liao H, Li Y, Chen Y, Yu J, Cheng S, Baloch S and Yang Y (2021) Chromosome-Level Genome Assembly of Cyrtotrachelus buqueti and Mining of Its Specific Genes. Front. Ecol. Evol. 9:729100. doi: 10.3389/fevo.2021.729100
Received: 22 June 2021; Accepted: 17 September 2021;
Published: 11 October 2021.
Edited by:
Peng Xu, Xiamen University, ChinaReviewed by:
Kecheng Zhu, Chinese Academy of Fishery Sciences (CAFS), ChinaXueyan Li, Kunming Institute of Zoology, China
Copyright © 2021 Fu, Long, Luo, Nong, Xiao, Liao, Li, Chen, Yu, Cheng, Baloch and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saira Baloch, MzA4MDczQGxzbnUuZWR1LmNu; YaoJun Yang, MzA4MDAxQGxzbnUuZWR1LmNu
†These authors have contributed equally to this work