 Marco Gerdol1
Marco Gerdol1 Claudia La Vecchia2
Claudia La Vecchia2 Maria Strazzullo2
Maria Strazzullo2 Pasquale De Luca3Stefania Gorbi4Francesco Regoli4
Pasquale De Luca3Stefania Gorbi4Francesco Regoli4 Alberto Pallavicini1,2
Alberto Pallavicini1,2 Enrico D’Aniello2*
Enrico D’Aniello2*- 1Department of Life Sciences, University of Trieste, Trieste, Italy
- 2Department of Biology and Evolution of Marine Organisms, Stazione Zoologica Anton Dohrn, Naples, Italy
- 3Research Infrastructures for Marine Biological Resources Department, Stazione Zoologica Anton Dohrn, Naples, Italy
- 4Department of Life and Environmental Sciences, Polytechnic University of Marche, Ancona, Italy
DNA methylation is an essential epigenetic mechanism influencing gene expression in all organisms. In metazoans, the pattern of DNA methylation changes during embryogenesis and adult life. Consequently, differentiated cells develop a stable and unique DNA methylation pattern that finely regulates mRNA transcription during development and determines tissue-specific gene expression. Currently, DNA methylation remains poorly investigated in mollusks and completely unexplored in Mytilus galloprovincialis. To shed light on this process in this ecologically and economically important bivalve, we screened its genome, detecting sequences homologous to DNA methyltransferases (DNMTs), methyl-CpG-binding domain (MBD) proteins and Ten-eleven translocation methylcytosine dioxygenase (TET) previously described in other organisms. We characterized the gene architecture and protein domains of the mussel sequences and studied their phylogenetic relationships with the ortholog sequences from other bivalve species. We then comparatively investigated their expression levels across different adult tissues in mussel and other bivalves, using previously published transcriptome datasets. This study provides the first insights on DNA methylation regulators in M. galloprovincialis, which may provide fundamental information to better understand the complex role played by this mechanism in regulating genome activity in bivalves.
Introduction
Epigenetic modifications play a key role in regulating gene expression. They are involved in many cellular processes, such as cellular differentiation, development, tumorigenesis, germ cell pluripotency, and inheritable genome imprinting. Accumulating evidence established that epigenetic alterations consist of a complex regulatory transcriptional pathway (DNA methylation, histone modifications, and micro-RNA mediated gene silencing) (Ho and Burggren, 2010; Becker and Workman, 2013; Greally, 2018; Skvortsova et al., 2018). These modifications reflect the high plasticity of the genome and contribute to modify gene expression independent of DNA sequence alterations (Peaston and Whitelaw, 2006). Additionally, recent evidence shows that these modifications can be passed from generation to generation in a phenomenon known as transgenerational inheritance (Bošković and Rando, 2018). The major epigenetic modification is the methylation of cytosines in genomic DNA (5mC methylation), which is necessary to modulate gene expression both in specialized tissue-specific cell types and in pluripotent cells undergoing lineage-commitment. DNA methylation is an ancient mechanism shared by all living organisms, from prokaryotes to humans (Willbanks et al., 2016). However, the leading molecular players in the context of DNA methylation often significantly differ among taxa, highlighting partial functional conservation in the landscape of methylation patterning. Vertebrate genomes are subject to high levels of DNA methylation, with a global pattern that varies from regulatory regions, promoters, and enhancers to transposable elements and gene bodies (Keller et al., 2015). On the other hand, invertebrates DNA methylation is mostly concentrated in gene bodies (Zemach et al., 2010), with species like Caenorhabditis elegans and Drosophila melanogaster mainly being free of 5mC (Bird et al., 1995).
Mollusks represent the second-largest animal phylum and the class Bivalvia includes marine organisms with high ecological and economic value due to their impact on aquaculture. Notwithstanding the growing research interest put on DNA methylation in invertebrates over the past few years, these studies are still at their infancy. The information recently gathered from bivalves have only provided a first glimpse into the basic organization of the DNA methylation in mollusks and on its interplay with the environment (Gavery and Roberts, 2010, 2014; Díaz-Freije et al., 2014; Rivière, 2014; Sun et al., 2014; Li et al., 2015, 2019; Saint-Carlier and Riviere, 2015; Riviere et al., 2017; Song et al., 2017; Eirin-Lopez and Putnam, 2019; Suarez-Ulloa et al., 2019; Fallet et al., 2020; Lim et al., 2021). DNA methylation, catalyzed by DNA methyltransferases (DNMTs also called “writers”), consists in the addition of a methyl-group to the 5′ position of cytosine to give methylcytosine. Among the three main DNMTs identified in metazoans, those involved in DNA methylation are DNMT1, which participates in methylation maintenance during cell division, and DNMT3, functioning primarily in de novo methylation during embryonic development (Iyer et al., 2011). On the other hand, the third DNA methyltransferase, DNMT2, does not play any role in DNA methylation in mammals, being instead involved in aspartic acid tRNA methylation (Goll et al., 2006). Its possible role in DNA methylation in other organisms, where it is the only present DNA methyltransferase, is still poorly understood (Ashapkin et al., 2016). Another crucial mechanism, catalyzed by the ten-eleven translocation enzymes (TET, also called “erasers”), is responsible for active DNA demethylation (Tahiliani et al., 2009). The recent discovery of TETs can be considered as one of the hallmarks in epigenetics, due to the key role these genes play in oncogenesis, but also in development (Rasmussen and Helin, 2016). TETs have never been reported in bivalves to date. Another class of very important proteins that are believed to convert the information of methylation patterns are the methyl-CpG-binding domain proteins (MBD), also called “readers.” These proteins, associated with other factors, play critical functions in regulating transcriptional repression and/or heterochromatin formation (Fatemi and Wade, 2006). While the majority of the sequenced bilaterian genomes display the presence of at least one representative gene for each of the aforementioned classes, others, such as yeast, fruit fly, and roundworm, have entirely lost DNA methyltransferase homologs and the associated DNA methylation enzymatic activities (Bhattacharyya et al., 2020). These losses have been occasionally reported also in some protostomes, such as the gastropod mollusk Aplysia californica, which lacks DNMT3 (Dabe et al., 2015).
As of today, a comprehensive investigation of the key genes involved in the DNA methylation machinery in the marine bivalve Mytilus galloprovincialis is still missing. Here, we provide the first insights on this process in this species by identifying the DNA methylation-related genes through a genome-wide analysis, studying their phylogenetic relationships and characterizing their expression pattern in different adult tissues in comparison with other bivalves.
Materials and Methods
Retrieval of Mytilus galloprovincialis Sequences
The human sequences of DNMT1 (P26358.2), DNMT2 (O14717.1), DNMT3a (Q9Y6K1.4), DNMT3b (Q9UBC3.1), DNMT3L (Q9UJW3.3), MBD1 (Q9UIS9.2), MBD2 (Q9UBB5.1), MBD3 (O95983.1), MBD3L1 (Q8WWY6.2), MBD3L2 (Q8NHZ7.3), MBD3L3 (A6NE82.1), MBD3L5 (A6NJ08.1), MBD3L4 (A6NE82.1), MBD4 (O95243.1), MeCp2 (P51608.1), and TET1 (Q8NFU7.2) were retrieved from UniprotKB. These were used as queries for tBLASTn searches (Altschul et al., 1990) against the M. galloprovincialis genome assembly, version mg10 (Gerdol et al., 2020). Moreover, to take into account the possibility of the presence of highly divergent ortholog sequences, the same analyses were carried out using the previously reported sequences from the mollusk Lottia gigantea as queries (Albalat, 2008). Missing hits were re-checked against previous versions of the same genome assembly (mg3 and mg5, more redundant, but slightly more complete) to rule out the possibility that the genes of interest were erroneously discarded during the assembly phases aimed at removing uncollapsed heterozygous genomic regions. All analyses were initially carried out using an e-value threshold of 1E-5, and overlapping gene annotations were translated to protein sequences to verify their reliability and completeness, based on the pairwise comparison with human queries and on the evaluation of the presence of the expected conserved domains with InterProScan 5 (Jones et al., 2014). Predicted CDS were further validated with available assembled transcriptomes from the same species (Gerdol et al., 2014; Moreira et al., 2015), which allowed to detect and correct erroneous exon annotations. Exon and intron boundaries were therefore refined, whenever needed, through the alignment between genomic DNA and cDNA sequences with Splign (Kapustin et al., 2008). To provide a comparative overview on the architecture of DNA methylation-related genes, the genomes of Crassostrea gigas (Zhang et al., 2012), Pomacea canaliculata (or the congeneric species P. maculata) (Sun et al., 2019), and Lingula anatina (Luo et al., 2015) were considered as representative species for Bivalvia, non-bivalve mollusks, and non-molluscan lophotrochozoans, respectively. The accession IDs of these sequences are provided in Supplementary Table 1.
Comparative Genomics Analyses
The validated translated amino acid sequences of M. galloprovincialis were used as queries for BLASTp searches against the virtually-translated proteomes derived from all the available genomes of species belonging to the class Bivalvia. In addition, tBLASTn searches were carried out whenever no ortholog could be identified in the proteomes, to detect the presence of non-annotated partial genes. In detail, the following bivalve genomes were screened: Argopecten irradians (Liu et al., 2020), Argopecten purpuratus (Li et al., 2018), Bathymodiolus platifrons (Sun et al., 2017), C. gigas (Zhang et al., 2012), Crassostrea virginica (Gómez-Chiarri et al., 2015), Limnoperna fortunei (Uliano-Silva et al., 2018), Magallana hongkongensis (Peng et al., 2020), Mizuhopecten yessoensis (Wang et al., 2017), Modiolus philippinarum (Sun et al., 2017), Mytilus coruscus (Li et al., 2020), Pecten maximus (Kenny et al., 2020), Pinctada fucata (Du et al., 2017), Saccostrea glomerata (Powell et al., 2018), and Scapharca broughtonii (Bai et al., 2019) among Pteriomorphia; Archivesica marissinica (Ip et al., 2020), Cyclina sinensis (Wei et al., 2020), Dreissena rostriformis (Calcino et al., 2019), Lutraria rhynchaena (Thai et al., 2019), Panopea generosa (unpublished), Ruditapes philippinarum (Yan et al., 2019), and Sinonovacula constricta (Ran et al., 2019) among Euheterodonta; Megalonaias nervosa among Palaeoheterodonta (Rogers et al., 2021). Details about the taxonomic classification of the species are provided in Supplementary Table 2. In addition, the presence of DNA methylation machinery transcripts was also assessed in the basal bivalve subclade Protobranchia: in this case, due to the lack of genomic data, tBLASTn searches were carried out in the transcriptomes of three representative species, obtained from a previously published study (Smith et al., 2011): Solemya velum (order Solemiyda), Ennucula tenuis (order Nuculida), and Yoldia limatula (order Nuculanida).
The genomes of other representative non-bivalve molluscan species (i.e., P. canaliculata, Lottia gigantea, Biomphalaria glabrata, Aplysia californica, and Octopus bimaculoides) were also similarly searched to identify ortholog sequences to be used as outgroups in subsequent phylogenetic analyses (Simakov et al., 2013; Albertin et al., 2015; Adema et al., 2017; Liu et al., 2018).
The best matching sequences for each species were retrieved to build a list of ortholog sequences. The correct identification of the sequences was evaluated by the assessment of primary sequence homology the presence of the expected conserved domains based on literature data. The reliability of the hits was evaluated as described above for M. galloprovincialis. Partial sequences unusable for downstream phylogenetic analyses [i.e., those covering less than 50% of the length of the multiple sequence alignment (MSA)] were replaced with assembled transcripts recovered from the NCBI SRA database, whenever available (the accession IDs of all the sequences are provided in Supplementary Table 1). Highly fragmented sequences with unequivocal homology with the queries were marked as present, but they were not used for downstream analyses as they were not deemed to be phylogenetically informative.
Phylogenetic Inference
Five sets of ortholog sequences (i.e., DNMT1, DNMT3, TET, MBD2/3, and MBD4) identified in Bivalvia were subject to MSA with MUSCLE v. 3.8.31 (Edgar, 2004), and highly divergent, phylogenetically uninformative regions were removed with Gblocks v.0.91b (Castresana, 2000). The refined MSA files were then analyzed with ModelTest-ng (Darriba et al., 2020) to identify the best fitting molecular model of evolution, based on the corrected Akaike Information Criterion (Cavanaugh, 1997). This was found to be the Le-Gascuel model (Le and Gascuel, 2008), with a proportion of invariable sites and a gamma-shaped distribution of rate variation across sites (LG + I + G) in most cases, except MBD4 and MBD2/3, where the LG + G and JTT + G (Jones et al., 1992) models were selected, respectively. Bayesian phylogenetic inference was implemented in MrBayes v.3.2.1 (Huelsenbeck and Ronquist, 2001), with two independent Markov Chain Monte Marco (MCMC) analyses run in parallel for one million generations each. This amount of generations was sufficient to reach convergence in all cases, as assessed by the achievement of an Effective Sample Size > 200 for all the parameters of the model with Tracer (Rambaut et al., 2018). The resulting trees were represented with a 50% majority consensus topology, and nodes with low statistical support (i.e., <50% posterior probability) were collapsed. Non-bivalve molluscan sequences were used as outgroups for rooting purposes.
In silico Gene Expression Analysis
The expression levels of the genes of interest were calculated in silico in M. galloprovincialis, M. philippinarum, C. gigas, A. purpuratus, and M. yessoensis as representative species for Pteriomorphia, and A. marissinica as a representative species for Heterodonta. All these species had a fully sequenced genome and tissue-specific RNA-seq sequencing libraries available (Supplementary Table 3). In detail, raw reads, downloaded from the NCBI SRA database, were mapped to the reference genomes, with high stringency (length fraction = 0.75, similarity fraction = 0.98), using the CLC Genomics Workbench v. 20 (Qiagen, Hilden, Germany). Gene expression levels were calculated as Transcripts Per Million (TPM), to allow a more reliable comparison both within and between samples (Wagner et al., 2012), and represented as a heat map based on Log (TPM + 1) values.
Real-Time Quantitative PCR Analyses
Adult M. galloprovincialis with shells 8–10 cm in length were obtained from a commercial shellfish farm (Bacoli, Napoli, Italy). Three living adult animals were used for RNA extraction from different tissues. From each mussel, hemolymph was withdrawn from the adductor muscle of each mussel with a 0.5 mm diameter (25G) disposable needle. The hemolymph sampled at time zero was centrifuged at 4°C a 3000 g for 10 min, and the pellet containing the hemocytes was resuspended in 350 μl of RTL buffer included in the RNasy Micro Kit (Qiagen) and stored at −80°C until RNA isolation.
To be consistent with RNA extraction, tissue samples from adductor muscle, gills, digestive gland, mantle edge, and foot were isolated from the same three animals that were used to isolate hemocytes and processed with the same kit. RNA integrity was estimated on a 1.0% agarose gel. Then, 500 ng of total RNA was retrotranscribed with the QuantiTect Reverse Transcription Kit (Qiagen), following the manufacturer’s instructions. Quantitative PCR (qPCR) was carried out with Applied Biosystems (Life Technologies) ViiA7 384 wells format. The absence of genomic DNA contamination was ensured thanks to two DNase treatments carried out during the RNA extraction and during the cDNA retro-transcription phases, respectively. Real-time qPCR reactions were performed with three biological replicates, and each of them with technical triplicates. Each reaction was performed in a final volume of 9 μL containing 5 μL of Fast SYBR Green Master Mix, 1 μL diluted cDNA (1:5), and 0.7 pmol/μL specific primers (Supplementary Table 4). The primers were specifically designed either on different exons or on exon/exon junctions to prevent the amplification of contaminant genomic DNA or to detect its presence through a melting curve analysis and agarose gel electrophoresis. The amplification efficiency of each primer pair was calculated using a dilution series of cDNA. Dissociation curves were carried out to ensure that primer pairs amplified single products with no detectable primer dimers. In addition, the expected size of each amplicon was verified by 1.2% agarose gel electrophoresis with GeneRuler 100 bp ladder (ThermoFisher) as a size marker. Expression levels were standardized to EF1α as a reference gene, and all the data were analyzed using 2–ΔΔCt Livak Method. We decided to use the expression of Dnmt1 in hemocytes as a baseline of expression level, so all the ΔΔCt values were calculated using the Dnmt1 ΔCt in hemocytes as a reference for each individual. Data shown are the average of the relative expression levels in all three animals.
Results
Identification of Mytilus galloprovincialis DNA-Methylation Machinery Genes
In order to identify DNA-methylation genes in M. galloprovincialis, we employed a survey analysis by either extracting annotations from the mg10 version assembly, or by recovering full-length CDS from previous versions and/or transcriptome data (full details in Table 1 below).
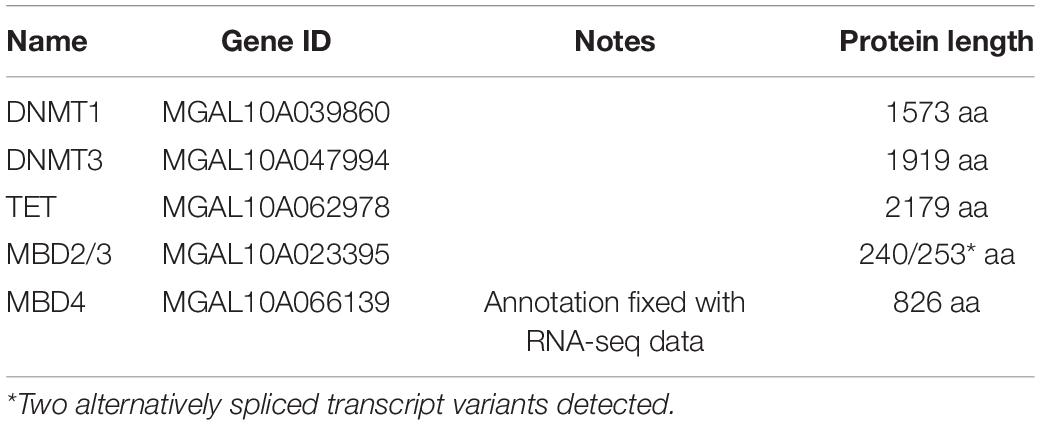
Table 1. List of genes involved in the DNA-methylation machinery in Mytilus galloprovincialis, with gene ID (genome assembly version mg10), protein length, and GenBank accession ID.
These survey analyses revealed that M. galloprovincialis, compared with the reference human sequences, has:
(i) A single DNMT1 ortholog. The identified sequence displayed an unequivocal homology with human DNMT1, with 53% pairwise amino acid sequence identity, detectable from the N-terminal to the C-terminal end, and preservation of all expected conserved domains (see below).
(ii) A single DNMT3 ortholog. The mussel sequence, in this case, showed some remarkable structural differences compared with human, as it displayed a much longer N-terminal region (see below). Nevertheless, the alignable portion of the mussel sequence nearly entirely covered the full protein sequence of both human DNMT3 paralogs, with a slightly superior homology with DNMT3B than DNMT3A (35 vs. 33% identity).
(iii) A single TET ortholog. The BLAST alignment revealed a similar degree of similarity with the three human TET paralogs, which all share a very similar domain architecture (50.91% identity with TET1, 53.19% identity with TET2, and 56.48% identity with TET3), with significant homologies detected in the conserved C-terminal domain the protein (see below).
(iv) A single ortholog to MBD2/MBD3 with two variants, originated by a single gene by alternative splicing, that we called MBD2/3a and MBD2/3b. In this case, the level of sequence homology was very similar between the two human paralogs, with 48.81% identity with MBD2 and 50.00% identity with MBD3 at the amino acid level. Although both human sequences share the typical tripartite structure of MBD proteins (see below), human MBD2 is characterized by a N-terminal glycine-rich low complexity region, which was not detected in mussel, making the M. galloprovincialis orthologs structurally slightly more similar to MBD3.
(v) A single MBD4 ortholog. In this case, just 24% of the mussel sequence could be aligned with human MBD4. However, the orthology relationship was made quite clear by the presence of two distinct regions of homology, corresponding to the MBD (57% sequence identity) and to the DNA glycosylase (71% sequence identity) domains, respectively.
No reliable ortholog sequence to the other human DNA methylation-related proteins could be identified in M. galloprovincialis. In particular, we did not find any orthologs for the MeCP2, considered the founding member of the MBD family of vertebrates. This observation was not surprising, considering that vertebrate MBD4 and MeCP2 are thought to derive from a lineage-specific gene duplication event that occurred several million years after the split between protostomes and deuterostomes and that MBD4 is thought to have maintained a structural organization closer to that of the ancestral protein. Hence, the MBD4 orthologs found in invertebrates have been often referred to as MBD4/MeCP2 (Albalat et al., 2012; de Mendoza et al., 2021).
Although our survey also allowed the identification of two DNMT2 orthologs in M. galloprovincialis (one complete plus one partial gene), these sequences have not been subject to in-depth analyses in the present study due to their involvement in a different biological process, the methylation of aspartic acid tRNA.
Mussel DNA Methylation Related Gene Structure
Next, we took advantage of the SPLIGN NCBI software,1 in order to determine the exon-intron structure of genes of interest. By an accurate and fast alignment of the cDNA sequences against their genomic counterparts, we were able to define the number of exons of each gene and their organization, as shown in Figure 1.
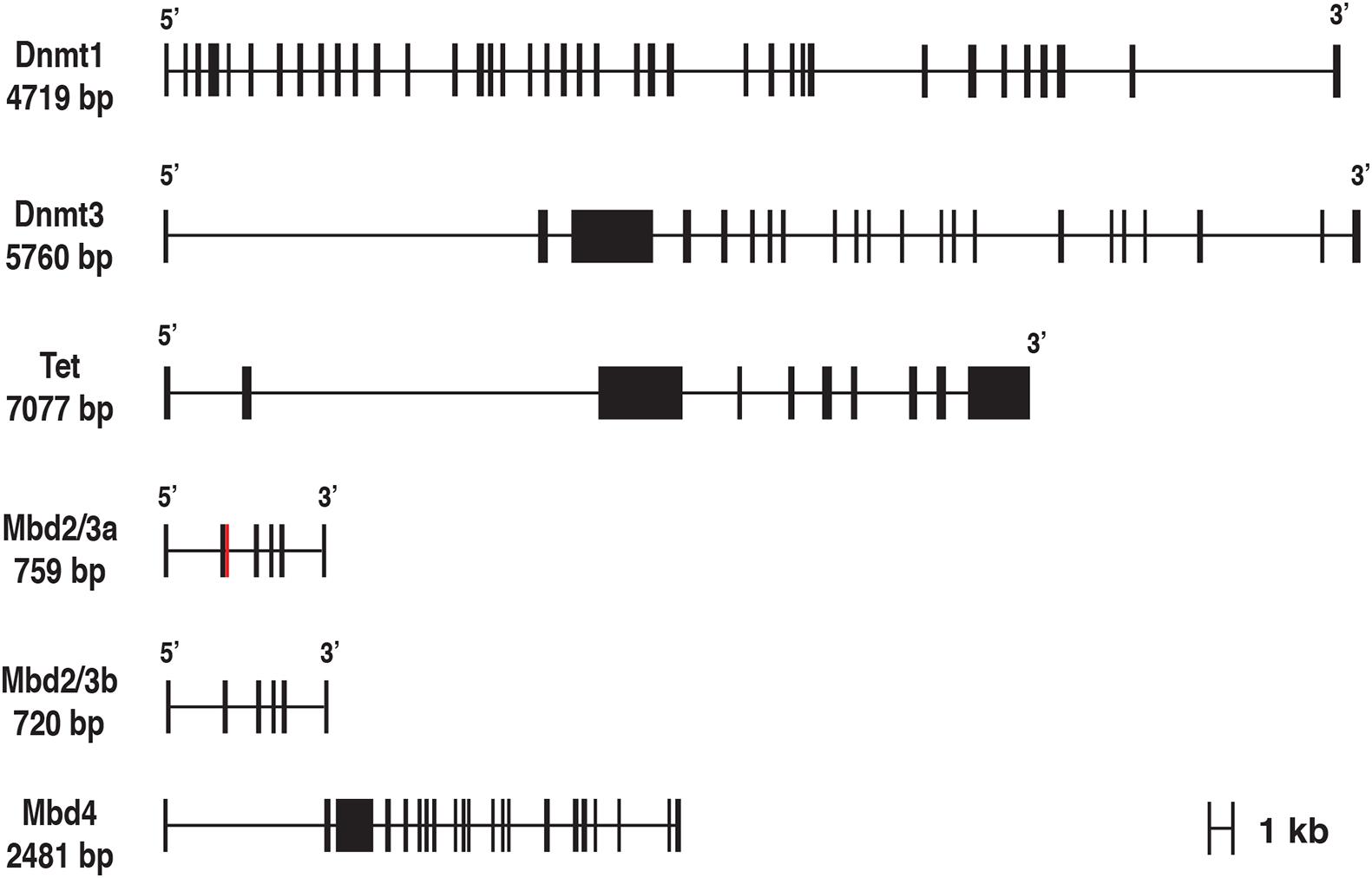
Figure 1. Exon-Intron structure of M. galloprovincialis Dnmt1, Dnmt3, Tet, Mbd 2/3, and Mbd4 genes. The red square indicates the optional 39 bp region alternatively spliced in the exon 2 of the Mbd2/3 gene. The two different lengths refer to the Mbd2/3a and Mbd2/3b variants.
The mussel Dnmt1 gene contains 38 exons, 3 less than the human ortholog (Dabe et al., 2015). The number of exons found in the Mytilus Dnmt3 gene (22) was very similar to the human Dnmt3b gene (24 exons) and much higher than Dnmt3a (9 exons) (Lees-Murdock et al., 2004). The unique M. galloprovincialis Tet gene contains a slightly lower number of exons than the three human paralogs, i.e., 10 vs. the 12-11-11 found in Tet 1-2-3, respectively (NCBI). The Mytilus MBD2/3 gene has six exons, just one less than the human MBD2 and MBD3 genes (NCBI). Finally, the M. galloprovincialis MBD4 gene was characterized by a much higher number of exons than the human ortholog, i.e., 21 vs. 8 (NCBI).
However, a pairwise comparison between the structure of mussel and human ortholog genes is complicated by the high phylogenetic divergence between protostomes and deuterostomes and by the consequent possible lineage-specific acquisition or loss of exons at the 5′ and 3′ ends of the genes. A comprehensive comparative analysis of the structural organization of DNA methylation-related genes in protostomes goes beyond the scope of this manuscript, due to the impossibility of verifying the accuracy of genome assemblies and gene annotations of all species. Nevertheless, we briefly compared the architecture of M. galloprovincialis genes with the ortholog sequences found in other lophotrochozoans (i.e., the bivalve C. gigas, gastropod mollusks belonging to the genus Pomacea and the brachiopod L. anatina). This preliminary characterization pointed out that the number of coding exons and their boundaries remained relatively well conserved during evolution within the protein regions showing the highest homology and the presence of recognizable protein domains (see the section “Domain Architecture of M. galloprovincialis DNA Methylation Toolkit Related Proteins”). On the other hand, exons of variable size, encoding highly divergent protein sequences were often found at the 5′ and 3′ ends of some target genes (most notably, DNMT3 and TET), suggesting that multiple independent gene architecture rearrangements might have happened in these regions in protostomes (Supplementary Figure 1).
Domain Architecture of M. galloprovincialis DNA Methylation Toolkit Related Proteins
The structural comparison between the mussel sequences and those from human and from representative lophotrochozoan species were then extended at the protein level. To this aim we employed the pfam and interpro databases (Figures 2, 3). As previously mentioned, the M. galloprovincialis DNMT1 contains the same highly conserved domains of the human protein (Jeltsch and Jurkowska, 2016; Ren et al., 2018), as well as of the ortholog sequences from most other Lophotrochozoans. These include the N-terminal DMAP1 domain (undetectable in L. anatina), which can bind the transcriptional co-repressor of the same name (Rountree et al., 2000). Mussel DNMT1 shows a replication foci domain (RFD) necessary for mediating the anchoring to the replication fork (Leonhardt et al., 1992), a zing finger CXXC motif able to bind unmethylated CpG (Pradhan et al., 2008) and two bromo-adjacent homology domains that may work in mediating protein–protein interaction necessary for mediating gene expression silencing. As expected, the C-terminal region contains a methylase domain (C5 cytosine methyltransferase), responsible for the catalytic activities on a cytosine residue in DNA sequence and transfer the methyl group from S-Adenosyl methionine to the 5′ position in the cytosine ring (Razin, 1989). It is interesting to note that the correct orchestration of all these domains contributes to the allosteric regulation of DNMT1 activity on maintenance methylation (Jeltsch and Jurkowska, 2016).
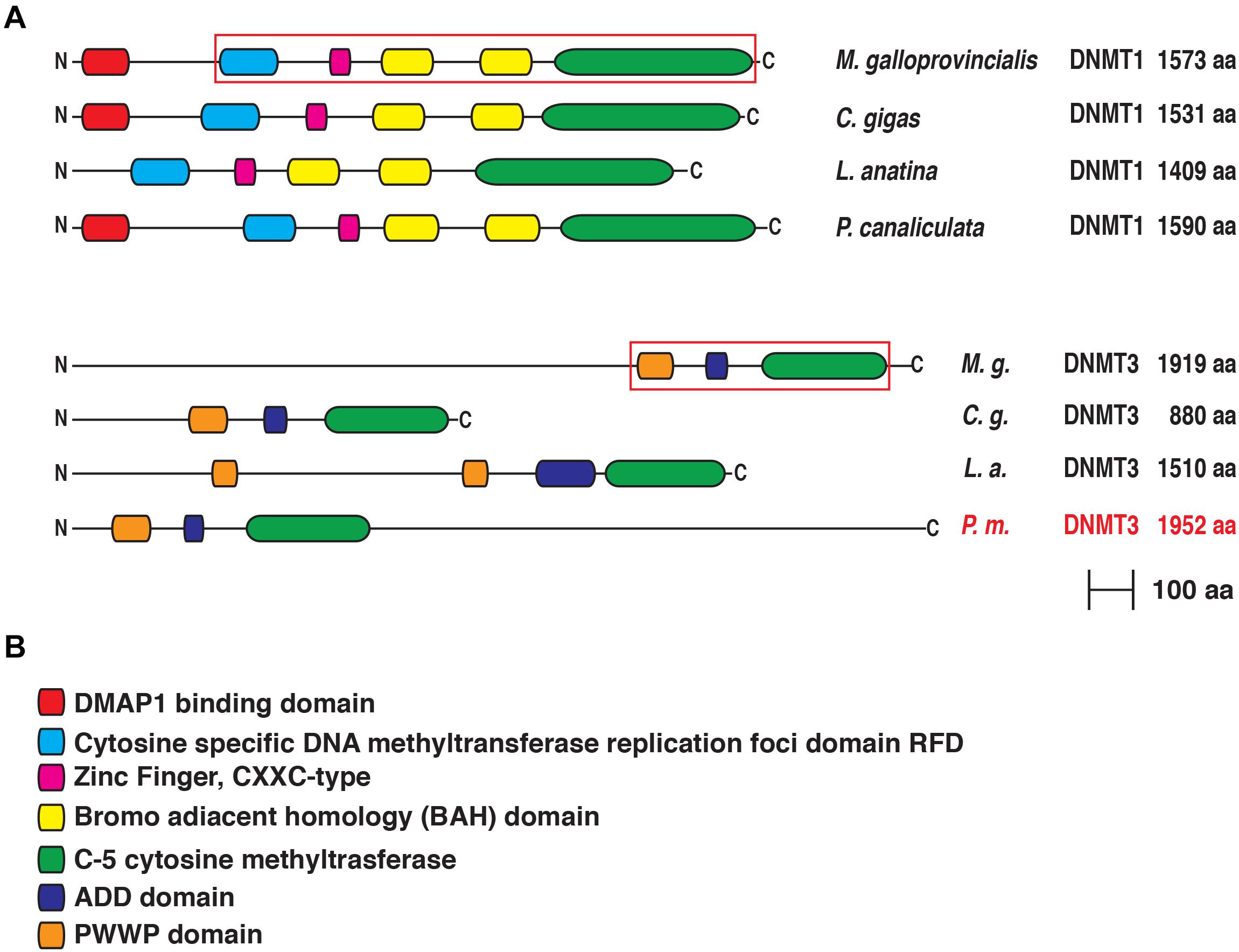
Figure 2. (A) Conserved domain organization of DNMT1 and DNMT3 of Mytilus galloprovincialis in comparison with those of Crassostrea gigas, Lingula anatina and Pomacea canaliculata. For DNMT3 we used the sequence of Pomacea maculata (in red) instead of Pomacea canaliculata. The phylogenetically informative regions used for Bayesian inference are included in a red box. (B) List of the functional domains. For each protein the N-terminus is on the left and the C-terminus is on the right.
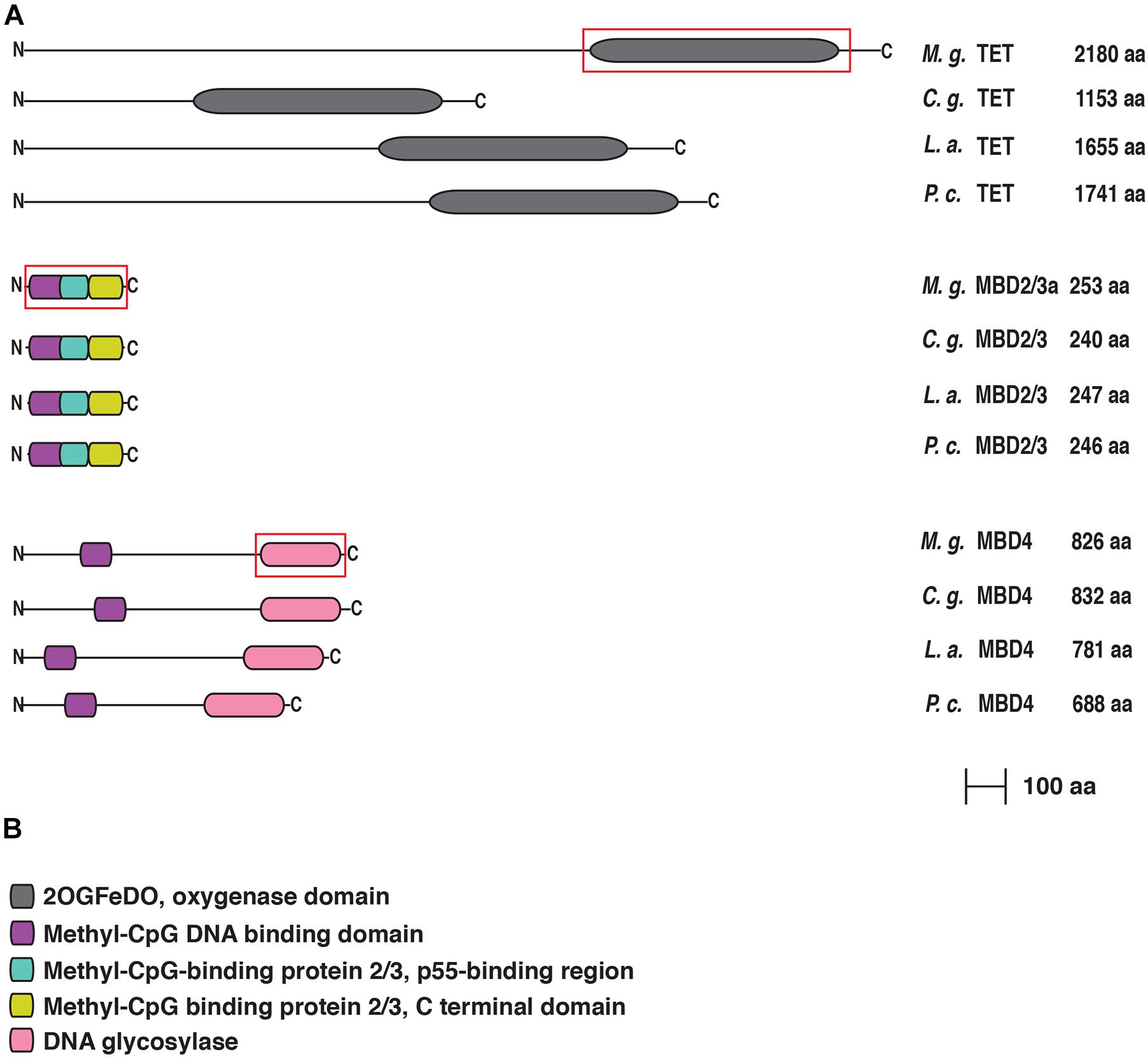
Figure 3. (A) Conserved domain organization of TET, MBD2/3, and MBD4 of Mytilus galloprovincialis in comparison with those of Crassostrea gigas, Lingula anatina, and Pomacea canaliculata. The phylogenetically informative regions used for Bayesian inference are included in a red box. (B) List of the functional domains. For each protein the N-terminus is on the left and the C-terminus is on the right.
The domain architecture of the Mytilus DNMT3 (Figure 2) is also very similar to that of human DNMT3a and DNMT3b even though this protein includes a very long N-terminal region devoid of recognizable domains, which is encoded by exons that are not conserved across Lophotrochozoa (Supplementary Figure 1). In brief, the sequence harbored three main domains. The most N-terminal domain, the Pro-Trp-Trp-Pro (PWWP) domain, is required for DNMT3 chromatin association via recognition of Histone H3 trimethylated at lysine 36. This binding is necessary for directing the de novo methylation activity at the pericentric heterochromatin (Chen et al., 2004). The central ADD domain binds to histone H3 unmethylated tails at lysine 4 (Zhang et al., 2010). Similarly to DNMT1, the C-terminal domain is the C-5 cytosine-specific DNA methylase.
The unique M. galloprovincialis TET protein has only a single well defined structural domain, i.e., the double-stranded β-helix 2-oxoglutarate and Fe(II)-dependent dioxygenase domain (Figure 3). The function of this domain is to convert 5 methylcytosine to 5-hydroxymethylcytosine (Tahiliani et al., 2009), inducing demethylation or further oxidation to 5-formylcytosine and 5-carboxylcytosine (He et al., 2011). Our analysis showed that the Mytilus TET lacks the CXXC motif and the cysteine-rich domain that characterizes two of three mammalian TET (Nakajima and Kunimoto, 2014). Like DNMT3, the mussel TET protein shows a long N-terminal region with no detectable domains, encoded by exons which are not conserved in different Lophotrochozoan species (Supplementary Figure 1).
We then analyzed the methyl CpG binding domain proteins (MBD2/3 and MBD4). In M. galloprovincialis, both alternatively spliced isoforms of MBD2/3 generate similar proteins, which only differ by a 13 residues indel, which does not impair the overall tripartite structural organization. This consists of a typical N-terminal MBD domain, shared by all MBD proteins and by MeCP2, followed by a central domain which, in vertebrate MBD2, includes the transcriptional repressor domain (TRD) responsible for the recruitment of histone deacetylases, and by a C-terminal domain containing coiled-coil regions (Hendrich and Bird, 1998; Figure 3). It is, however, important to note that the precise localization of TRD in the mussel sequence would require additional functional characterization, since the transcriptional repression function is carried out by different non-homologous regions in different MBD family members (Boeke et al., 2000). Finally, MBD4 displayed a structural architecture similar to that of other metazoan orthologs, with a slight difference in the length of the N-terminal region, showing the presence of the characterizing MBD domain followed by a larger DNA glycosylase domain at the C-terminal end (Kondo et al., 2005). The two domains are separated by a low complexity region significantly longer than human MBD4 (Figure 3).
The DNA-Methylation Machinery Is Conserved Across All Bivalves
The largely different sequencing strategies and computational approaches used to obtain the 23 bivalve genomes comparatively analyzed in this study resulted in genome assemblies with different quality and completeness, ranging from chromosome-scale (Ran et al., 2019; Peng et al., 2020; Wei et al., 2020) to highly fragmented and partial drafts (Uliano-Silva et al., 2018). Hence, the lack of detection of some of the genes involved in the DNA-methylation machinery in some of the target species does not necessarily imply the absence of a given gene, as several technical factors could explain these apparent gene losses.
In brief, we could recover evidence in support of the existence of Dnmt1, Dnmt3, Tet, Mbd2/3, and Mbd4 orthologs, either with a full CDS or with partial sequences, in all the species analyzed (Figure 4). On the other hand, Tet could not be retrieved from M. nervosa and P. generosa, two species with a relatively low assembly and annotation quality (i.e., with a high rate of fragmented and absent BUSCOs), which also lacked transcriptome data to fill-in such gaps. We can therefore hypothesize that such missed detections are unlikely to indicate the absence of Tet genes, but rather their inclusion in unassembled genomic regions. Moreover, the identification of Dnmt1, Dnmt3, Mbd2/3, Mbd4, and Tet orthologs in the transcriptomes available for three representative species belonging to the basal bivalve subclass Protobranchia further supports the high conservation of this system in bivalves. Overall, shorter sequences were more likely to be detected as complete or as nearly complete, as the consequence of their relatively simple gene structure (Figure 1) and exon computational annotation. Conversely, possibly owing to the very low expression level of Dnmt3 in adult tissues (Figures 6, 7), this particular gene was often poorly annotated, and the encoded sequences could not be included in phylogenetic inference analysis.
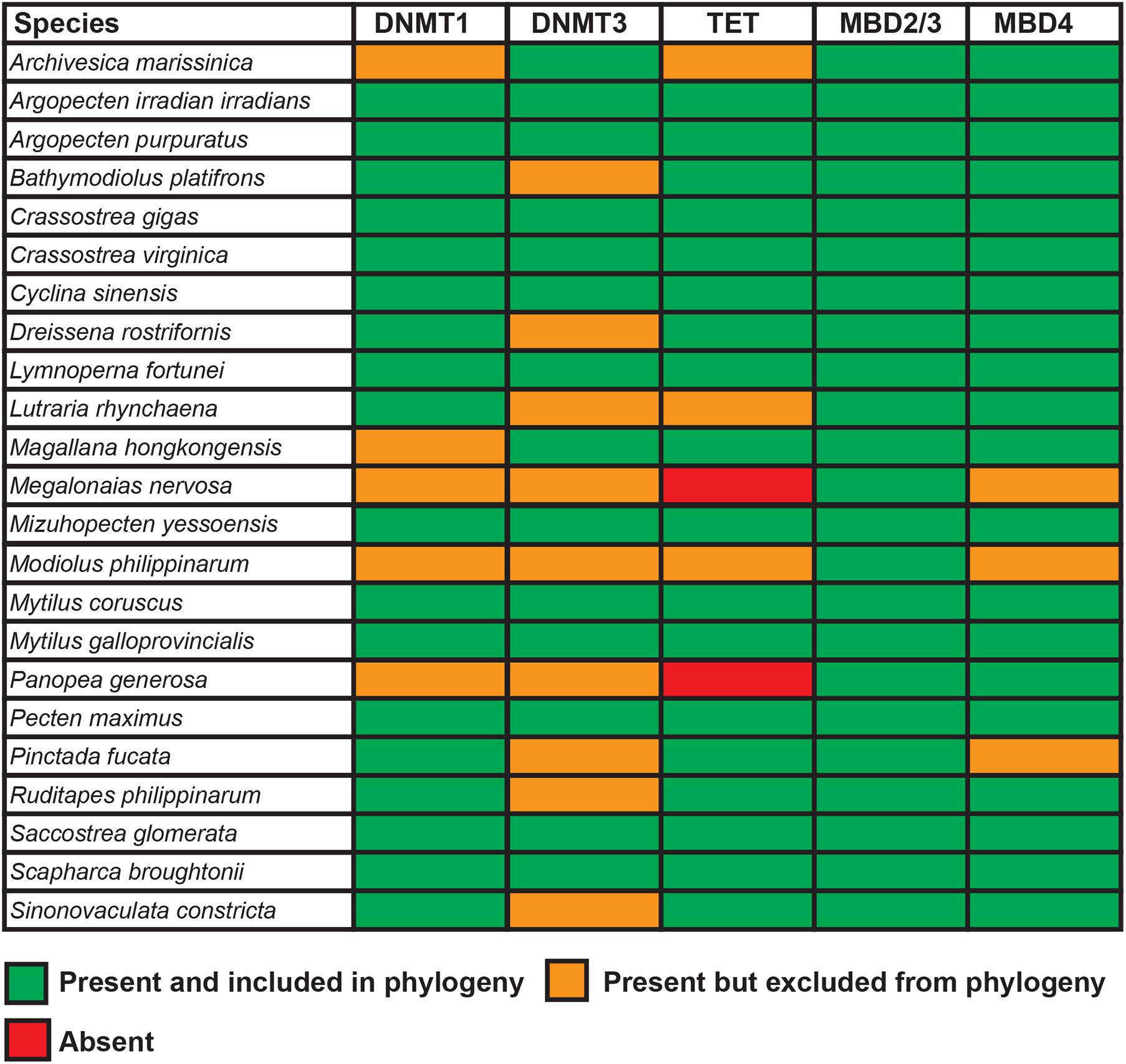
Figure 4. Summary of the genes involved in the DNA methylation machinery found in the 23 bivalve species analyzed in this study.
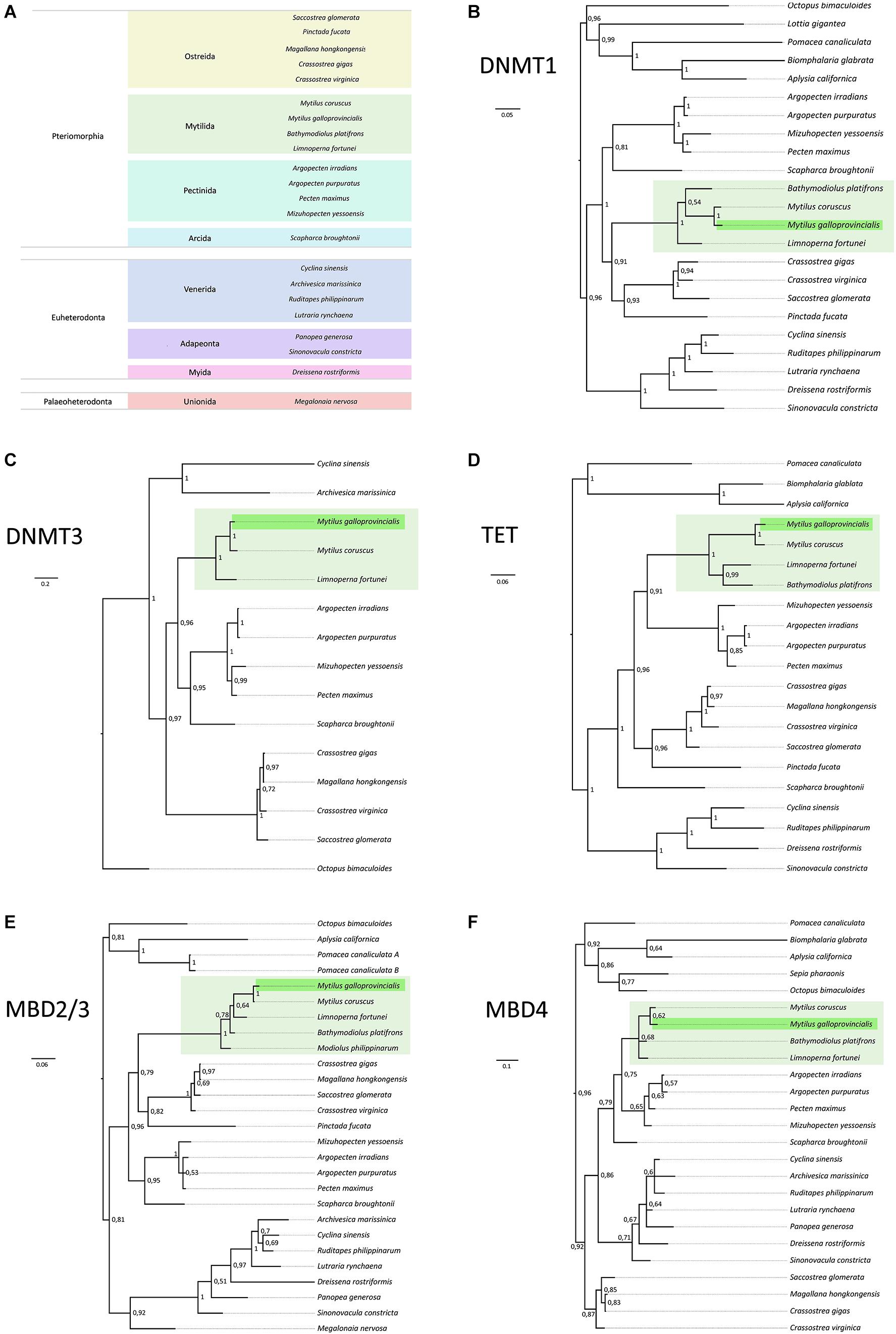
Figure 5. (A) Taxonomic classification (infraclass and order) of the bivalve species considered in this study. (B–F) Bayesian phylogeny of bivalve DNMT1, DNMT3, TET, MBD2/3, and MBD4 orthologs. Sequences from M. galloprovincialis and mytilid bivalves are highlighted with a green and light green background, respectively. Posterior probability support values are shown close to each node. Poorly supported nodes (posterior probability <0.5) have been collapsed in polytomies.
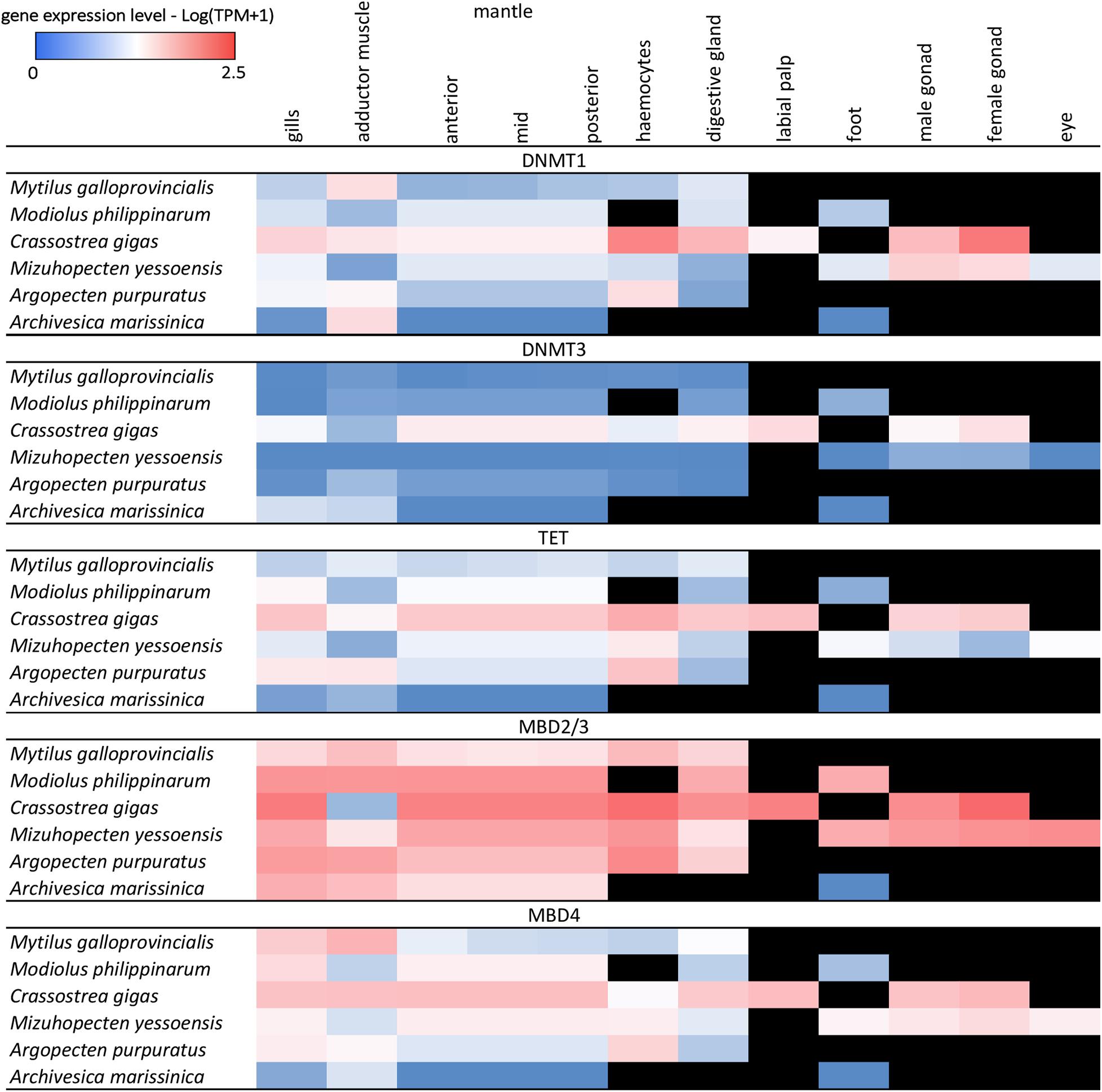
Figure 6. Heat map of DNA methylation-related genes expression in adult tissues of 6 bivalve species from transcriptome database. Black cells indicate missing data.
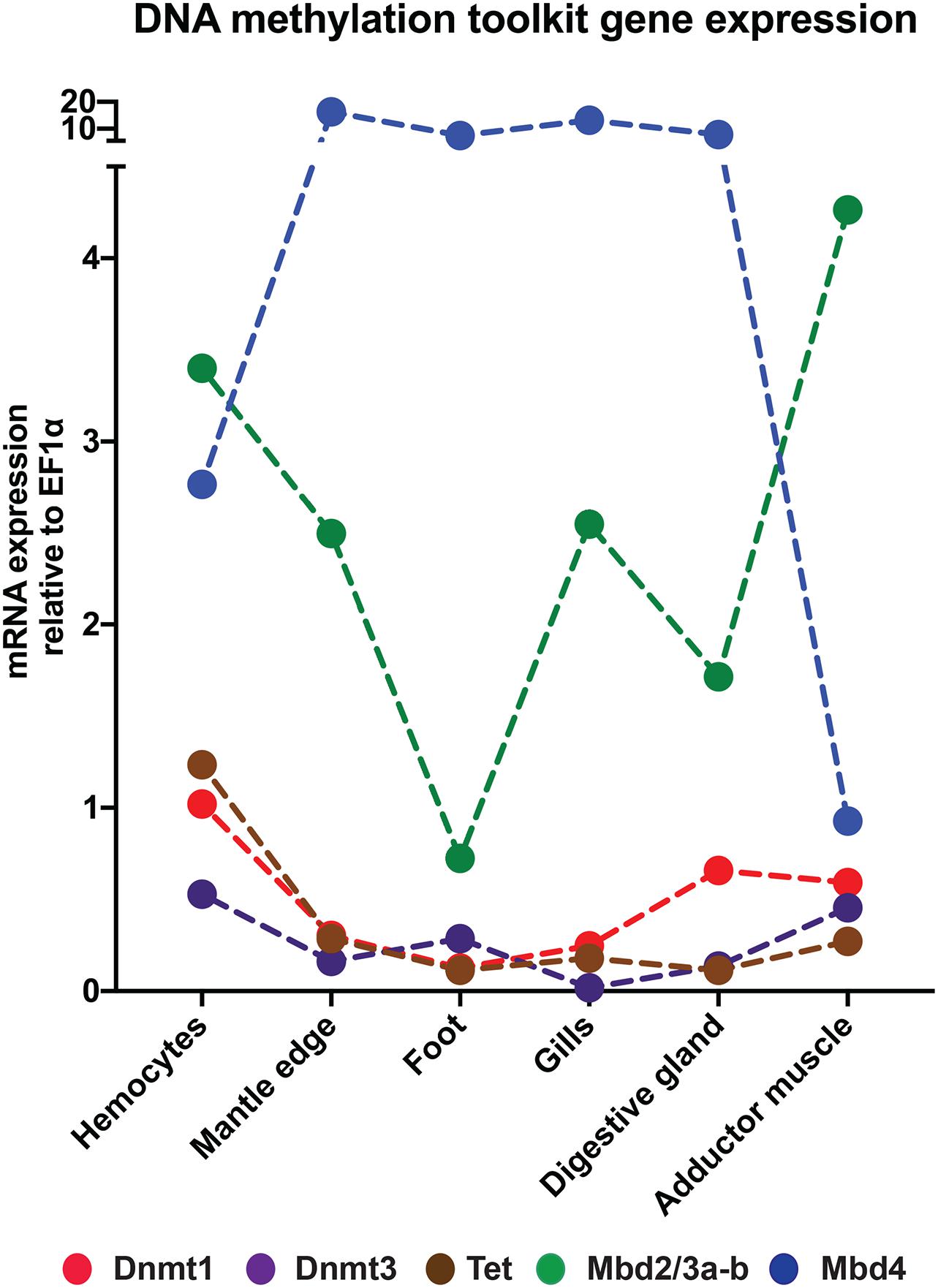
Figure 7. Expression profiles of the Dnmt1, Dnmt3, Tet, Mbd2/3a-b, and Mbd4 in different Mytilus galloprovincialis adult tissues.
In summary, while the presence of gene duplications among Dnmt and Mbd orthologs is not uncommon among Metazoa (Albalat et al., 2012), all the genes involved in the DNA methylation machinery appear to be nearly invariably present with a single copy in bivalves (Figure 4). The results we report are, for the most part, in line with the data recently reported by Planques et al. (2020) in a large-scale comparative study among metazoans. However, we here clarify that the previously reported apparent lack of Dnmt3 in a few bivalve species was most likely a technical artifact linked with mis-assemblies or incorrect gene annotation.
Evolutionary Analysis of Bivalve DNA Methylation Machinery Genes
Previous comparative studies on the genes involved in DNA methylation processes have only targeted a very few molluscan species with a fully sequenced genome, such as the owl limpet Lottia gigantea (Albalat et al., 2012). However, Mollusca is an extraordinarily large and diverse phylum, which includes over 43,000 recognized species adapted to marine, freshwater, and terrestrial life, and over 8000 bivalve species have been formally described to date (Rosenberg, 2014). Therefore, the use of a single bivalve species as a representative for the entire class may not be appropriate to fully capture the high degree of morpho-ecological diversity of these organisms. Compared with previous studies, our comparative Bayesian inference approach allowed us to investigate with more depth the evolution of DNMT, TET, and MBD genes in bivalve species with complete genome sequence available.
In line with the rare occurrence of gene duplication and loss events, most genes involved in the bivalve DNA methylation machinery displayed sequence phylogenies nearly identical to the currently accepted species phylogeny (Figure 5A and Supplementary Table 1). As of note, the relatively high degree of inter-species sequence divergence, as well as the fragmentation of some of the ortholog genes identified, often reduced the phylogenetically informative regions that could be included in the MSA to a fraction of the full-length proteins (Figures 2–4).
Not surprisingly, this structural conservation of DNMT1 (Figure 2) was mirrored by phylogenetic inference, which produced a well-supported clade for Mytilida sequences (posterior probability = 1) within an equally well-supported monophyletic Pteriomorphia clade. Overall, Bayesian phylogeny produced an unambiguous phylogeny, nearly devoid of taxonomic inconsistencies (Figure 5B).
The phylogenetic reconstruction of DNMT3 evolution was complicated by the usually high degree of fragmentation of the annotated genes, which resulted in the exclusion of several species from the analysis (Figure 5C). In any case, the topology of the tree was consistent with the expected species phylogeny, with mytilid sequences once again being placed in a monophyletic clade (posterior probability = 1).
As previously mentioned, bivalve TET proteins displayed a largely divergent N-terminal region and therefore the MSA was trimmed to include only the conserved C-terminal domain (Figure 3). Overall, phylogenetic inference produced a gene tree highly consistent with the expected species tree, which strongly supported the monophyly of mytilid sequences (Figure 5D).
Consistently with the similar length and structural organization of bivalve MBD2/3 orthologs (Figure 3) the topology of the phylogenetic tree deriving from the full alignment of the protein sequences was well-consistent with the species tree (Figure 5E).
The low levels of primary sequence homology of bivalve MBD4 led to the inclusion of just the relatively short C-terminal DNA glycosylase domain in the trimmed MSA (Figure 3). Possibly owing to the relatively short length of the MSA, the phylogeny of MBD4 was the most problematic among all those studied in this work, as the tree was characterized by some discrepancies in the relative relationships among bivalve subclasses (Figure 5F). In particular, Pteriomorphia was not identified as a monophyletic group, and the monophyly of sequences from the Mytilida clade was supported by a relatively low posterior probability (i.e., 0.68).
In summary, our phylogenetic investigations did not reveal any unexpected peculiarity in the evolution of the bivalve DNA methylation machinery. The few incongruences observed between gene trees and species trees came to no surprise due to the relatively short length of the MSAs analyzed, incomplete taxonomical sampling and the possibility of missing data linked with past gene duplication and loss events (Pamilo and Nei, 1988; Maddison, 1997; Nichols, 2001). In general, the availability of additional genomic data from other bivalves would be expected to improve the topology of the trees obtained with this approach, likely resolving most of the incongruences between gene and species phylogeny.
Expression of DNA Methylation-Related Genes in Bivalve Adult Tissues
Overall, the in silico assessment of gene expression levels of DNA methylation-related genes in bivalve adult tissues revealed a poor transcriptional activity, seldom reaching values higher than 10 TPM and most often displaying values lower than 3 TPM, which is close to the arbitrary threshold generally used to define whether a transcript is expressed at biologically significant levels (Figure 6). The only exception was Mbd2/3, which displayed higher expression in all tissues, possibly due to the involvement of this gene in other processes, which are provided in vertebrates by the three paralogous genes Mbd1, Mbd2, and Mbd3 (Figure 6). M. galloprovincialis was generally characterized by gene expression patterns in line with those of the other bivalve species analyzed for the available tissues, i.e., gills, posterior adductor muscle, anterior, mid and posterior mantle, hemocytes, and digestive gland (Figure 6). With the exception of MBD2/3 which, as mentioned above, was broadly expressed at values comprised between ∼8 TPM in mantle and ∼22 TPM in hemocytes, just a very few transcripts exceeded the threshold of 5 TPM: namely, Dnmt1 in the posterior adductor muscle (TPM = 9.34) and MBD4 in gills and posterior adductor muscle (14.15 and 25.48, respectively). Among the genes investigated, Dnmt3 emerged as the one displaying the lowest expression levels, in all adult tissues and all species, with the only notable exception represented by the foot of A. marissinica (TPM = 24.34).
Although reproductive tissues were only available for two species (C. gigas and M. yessoensis), RNA-seq data suggests that Dnmt1, Mbd2/3, Mbd4, and Tet are expressed in both male and female gonads at levels comparable to other adult tissues. On the other hand, Dnmt3 maintained a poor expression in the gonads of M. yessoensis (TPM < 1) and a moderate expression in the gonads of C. gigas (TPM ∼5). As of note, gene expression levels in bivalve gonads are expected to undergo significant seasonal variations due to the gametogenic cycle (Banni et al., 2011) and it is presently unknown whether and to which extent DNA methylation-related genes are affected by this factor.
In summary, RNA-seq data highlights that all DNA methylation related genes, except Mbd2/3, appear to be poorly expressed in the adult tissues of all bivalve species, including M. galloprovincialis.
We verified the expression of Dnmt1, Dnmt3, Tet, Mbd2/3a-b, and Mbd4 by qPCR in different M. galloprovincialis adult tissues: hemocytes, mantle edge, foot, gills, digestive gland, and posterior adductor muscle. The results obtained mainly were consistent with RNA-seq data, confirming the very low expression of Dnmt1, Dnmt3, and Tet and the moderate expression of Mbd2/3 in all adult tissues (Figure 7). As a further confirmation of RNA-seq gene expression data, the adductor muscle and hemocytes were the two tissues characterized by the highest absolute expression levels in M. galloprovincialis (Figure 7). The most significant discrepancy between the two approaches concerned Mbd4, which appeared to be strongly expressed in all tissues by qPCR, in contrast with the poor expression levels highlighted by RNA-seq in all tissues, except gills, and adductor muscle (Figures 6, 7). This inconsistency may be due to a number of different reasons, including the genetic variability between the animals that were subjected to the two types of analyses, their different age, the diverse geographical background and ecological parameters of sampling sites. Unfortunately, transcriptome data were not available for some M. galloprovincialis tissues, which prevented further discussion concerning the expression of DNA methylation-related genes in the foot and, in particular, their seasonal regulation in reproductive tissues (that we carefully excluded from qPCR analyses by dissecting the mantle edge only).
Discussion
Although DNA methylation is a widespread phenomenon in eukaryotes, most of the studies on this subject in metazoans have so far targeted vertebrates and a few invertebrate models. To expand these investigations to marine bivalves, in this study we selected the Mediterranean mussel M. galloprovincialis as a reference species, revealing for the first time a full complement of DNA methylation-related genes in its genome, comprising Dnmt1, Dnmt3, Tet, Mbd2/3, and Mbd4. We characterized the architecture of the mussel genes and the structural organization of the encoded proteins, revealing relatively good conservation of exon number and position, as well as of the key conserved domains with lophotrochozoan and human orthologs. Phylogenetic inference strongly supported the monophyletic origins of each of the five sequence groups, highlighting a high concordance between gene family trees and species taxonomy. The evolutionary analyses also revealed that no lineage-specific gene duplication events occurred in bivalves, and confirmed that only a single Mbd2/3 ortholog and a single Mbd4/MeCP2 ortholog (with high structural homology with Mbd4) are present in mollusks. Moreover, we investigated the expression profiles of all the genes involved in the DNA methylation machinery, using both RNA-seq and qPCR data, revealing that most of them (excluding Mbd2/3 and Mbd4) are expressed at poor levels in adult tissues. These results provide a framework to better understand how DNA methylation regulates several fundamental biological processes in marine bivalves, paving the way for future studies focused on the role of DNA methylation during early developmental stages. Indeed, while this process is well known to play a key role during mammalian development, its possible involvement in the reprogramming of the DNA methylome in gametes or during embryonic development in invertebrates, and mollusks in particular, is still poorly understood. Furthermore, despite the growing number of studies focused on this subject in mollusks, many aspects need to be further investigated. In oyster, as well as in other mollusks, DNA methylation seems to mostly occur with a mosaic distribution, with an alternation of hypermethylated (housekeeping genes) and hypomethylated (inducible genes) DNA regions (Fneich et al., 2013; Wang et al., 2014; Riviere et al., 2017; Venkataraman et al., 2020). Moreover, unlike vertebrates, where DNA methylation is more common in intergenic regions and regulatory elements, the distribution in mollusks appears to be predominant in the gene body (Gavery and Roberts, 2013; Keller et al., 2015). However, the mechanism by which gene body methylation can differentially regulate gene transcription remains to be elucidated. Moreover, the very recent discovery of DNA methylation occurring also in the promoters and in regions associated with transposable elements in C. virginica highlights that the role of DNA methylation in bivalves is still not fully understood (Venkataraman et al., 2020). In summary, this manuscript provides the first insights on the DNA methylation machinery genes in M. galloprovincialis, offering a unique opportunity for further in-depth investigations that should be aimed at clarifying the mechanisms underpinning DNA methylation and the functional role of this process in mollusks. Ultimately, additional studies will be necessary in the near future to better understand how the environment and emerging contaminants can influence DNA methylation patterns in bivalves, and how the induced environmental phenotypic plasticity, in which may be associated with new physiological responses and adaptation, can be inherited through generations.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
MG, AP, SG, FR, and ED’A conceived and designed the experiments. MG, AP, and ED’A performed the bioinformatics and phylogenetic analyses. CLV, MS, PD, ED’A carried out qPCR experiments and analyzed the data. MG and ED’A contributed to design figures and wrote the initial manuscript. All the authors contributed to revising the final version of the manuscript.
Funding
CLV was supported by a Ph.D. fellowship co-founded by Stazione Zoologica Anton Dohrn, Napoli, Italy and Università Politecnica delle Marche, Ancona, Italy.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.698561/full#supplementary-material
Supplementary Figure 1 | Schematic representation of Dnmt1, Dnmt3, Tet, Mbd2/3, and Mbd4 gene architecture in Mytilus galloprovincialis, compared with Crassostrea gigas, Pomacea canaliculata (replaced by Pomacea maculata for Dnmt3), and Lingula anatina. Only protein-coding exons (represented by arrows) are shown. The gene architecture of M. galloprovincialis was used as a reference. Highly divergent exons with non-detectable sequence homology with mussel have been omitted and replaced by a symbol which indicates an extension of the gene structure toward the 5′ or 3′ ends.
Supplementary Table 1 | Accession numbers of the mollusk proteins considered in this work.
Supplementary Table 2 | Taxonomical classification of the bivalve species considered in this work, according to the World Register of Marine Species.
Supplementary Table 3 | List of RNA-sequencing experiments analyzed in this study.
Supplementary Table 4 | List of primers used for qPCR. (Mbd2/3 primer amplifies both variants).
Footnotes
References
Adema, C. M., Hillier, L. W., Jones, C. S., Loker, E. S., Knight, M., Minx, P., et al. (2017). Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat. Commun. 8:15451.
Albalat, R. (2008). Evolution of DNA-methylation machinery: DNA methyltransferases and methyl-DNA binding proteins in the amphioxus Branchiostoma floridae. Dev. Genes Evol. 218, 691–701.
Albalat, R., Martí-Solans, J., and Cañestro, C. (2012). DNA methylation in amphioxus: from ancestral functions to new roles in vertebrates. Brief. Funct. Genomics 11, 142–155. doi: 10.1093/bfgp/els009
Albertin, C. B., Simakov, O., Mitros, T., Wang, Z. Y., Pungor, J. R., Edsinger-Gonzales, E., et al. (2015). The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224. doi: 10.1038/nature14668
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Ashapkin, V. V., Kutueva, L. I., and Vanyushin, B. F. (2016). Dnmt2 is the most evolutionary conserved and enigmatic cytosine DNA methyltransferase in eukaryotes. Russ. J. Genet. 52, 237–248. doi: 10.1134/s1022795416030029
Bai, C.-M., Xin, L.-S., Rosani, U., Wu, B., Wang, Q.-C., Duan, X.-K., et al. (2019). Chromosomal-level assembly of the blood clam, Scapharca (Anadara) broughtonii, using long sequence reads and Hi-C. Gigascience 8:giz067. doi: 10.1093/gigascience/giz067
Banni, M., Negri, A., Mignone, F., Boussetta, H., Viarengo, A., and Dondero, F. (2011). Gene expression rhythms in the mussel Mytilus galloprovincialis (Lam.) across an annual cycle. PLoS One 6:e18904. doi: 10.1371/journal.pone.0018904
Becker, P. B., and Workman, J. L. (2013). Nucleosome remodeling and epigenetics. Cold Spring Harb. Perspect. Biol. 5:a017905. doi: 10.1101/cshperspect.a017905
Bhattacharyya, M., De, S., and Chakrabarti, S. (2020). Origin and Evolution of DNA methyltransferases (DNMT) along the tree of life: a multi-genome survey. bioRxiv doi: 10.1101/2020.04.09.033167
Bird, A., Tate, P., Nan, X., Campoy, J., Meehan, R., Cross, S., et al. (1995). Studies of DNA methylation in animals. J. Cell Sci. Suppl. 19, 37–39. doi: 10.1242/jcs.1995.supplement_19.5
Boeke, J., Ammerpohl, O., Kegel, S., Moehren, U., and Renkawitz, R. (2000). The minimal repression domain of MBD2b overlaps with the methyl-CpG-binding domain and binds directly to Sin3A. J. Biol. Chem. 275, 34963–34967. doi: 10.1074/jbc.m005929200
Bošković, A., and Rando, O. J. (2018). Transgenerational Epigenetic Inheritance. Annu. Rev. Genet. 52, 21–41.
Calcino, A. D., de Oliveira, A. L., Simakov, O., Schwaha, T., Zieger, E., Wollesen, T., et al. (2019). The quagga mussel genome and the evolution of freshwater tolerance. DNA Res. 26, 411–422. doi: 10.1093/dnares/dsz019
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Cavanaugh, J. E. (1997). Unifying the derivations for the Akaike and corrected Akaike information criteria. Stat. Probab. Lett. 33, 201–208. doi: 10.1016/s0167-7152(96)00128-9
Chen, T., Tsujimoto, N., and Li, E. (2004). The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell Biol. 24, 9048–9058. doi: 10.1128/mcb.24.20.9048-9058.2004
Dabe, E. C., Sanford, R. S., Kohn, A. B., Bobkova, Y., and Moroz, L. L. (2015). DNA Methylation in Basal Metazoans: insights from Ctenophores. Integr. Comp. Biol. 55, 1096–1110. doi: 10.1093/icb/icv086
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
de Mendoza, A., Poppe, D., Buckberry, S., Pflueger, J., Albertin, C. B., et al. (2021). The emergence of the brain non-CpG methylation system in vertebrates. Nat. Ecol. Evol. 5, 369–378. doi: 10.1038/s41559-020-01371-2
Díaz-Freije, E., Gestal, C., Castellanos-Martínez, S., and Morán, P. (2014). The role of DNA methylation on Octopus vulgaris development and their perspectives. Front. Physiol. 5:62. doi: 10.3389/fphys.2014.00062
Du, X., Fan, G., Jiao, Y., Zhang, H., Guo, X., Huang, R., et al. (2017). The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. GigaScience 6, 1–12. doi: 10.1093/gigascience/gix059
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eirin-Lopez, J. M., and Putnam, H. M. (2019). Marine Environmental Epigenetics. Ann. Rev. Mar. Sci. 11, 335–368. doi: 10.1146/annurev-marine-010318-095114
Fallet, M., Luquet, E., David, P., and Cosseau, C. (2020). Epigenetic inheritance and intergenerational effects in mollusks. Gene 729:144166. doi: 10.1016/j.gene.2019.144166
Fatemi, M., and Wade, P. A. (2006). MBD family proteins: reading the epigenetic code. J. Cell Sci. 119, 3033–3037. doi: 10.1242/jcs.03099
Fneich, S., Dheilly, N., Adema, C., Rognon, A., Reichelt, M., Bulla, J., et al. (2013). 5-methyl-cytosine and 5-hydroxy-methyl-cytosine in the genome of Biomphalaria glabrata, a snail intermediate host of Schistosoma mansoni. Parasit. Vectors 6:167. doi: 10.1186/1756-3305-6-167
Gavery, M. R., and Roberts, S. B. (2010). DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genomics 11:483. doi: 10.1186/1471-2164-11-483
Gavery, M. R., and Roberts, S. B. (2013). Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1:e215. doi: 10.7717/peerj.215
Gavery, M. R., and Roberts, S. B. (2014). A context dependent role for DNA methylation in bivalves. Brief. Funct. Genomics 13, 217–222. doi: 10.1093/bfgp/elt054
Gerdol, M., De Moro, G., Manfrin, C., Milandri, A., Riccardi, E., Beran, A., et al. (2014). RNA sequencing and de novo assembly of the digestive gland transcriptome in Mytilus galloprovincialis fed with toxinogenic and non-toxic strains of Alexandrium minutum. BMC Res. Notes 7:722. doi: 10.1186/1756-0500-7-722
Gerdol, M., Moreira, R., Cruz, F., Gómez-Garrido, J., Vlasova, A., Rosani, U., et al. (2020). Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 21:275.
Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder, J. A., Hsieh, C.-L., Zhang, X., et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. doi: 10.1126/science.1120976
Gómez-Chiarri, M., Warren, W. C., Guo, X., and Proestou, D. (2015). Developing tools for the study of molluscan immunity: the sequencing of the genome of the eastern oyster, Crassostrea virginica. Fish Shellfish Immunol. 46, 2–4. doi: 10.1016/j.fsi.2015.05.004
Greally, J. M. (2018). A user’s guide to the ambiguous word “epigenetics.”. Nat. Rev. Mol. Cell Biol. 19, 207–208. doi: 10.1038/nrm.2017.135
He, Y.-F., Li, B.-Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Hendrich, B., and Bird, A. (1998). Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell Biol. 18, 6538–6547. doi: 10.1128/mcb.18.11.6538
Ho, D. H., and Burggren, W. W. (2010). Epigenetics and transgenerational transfer: a physiological perspective. J. Exp. Biol. 213, 3–16. doi: 10.1242/jeb.019752
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Ip, J. C.-H., Xu, T., Sun, J., Li, R., Chen, C., Lan, Y., et al. (2020). Host–Endosymbiont Genome Integration in a Deep-Sea Chemosymbiotic Clam. Mol. Biol. Evol. 38, 502–518. doi: 10.1093/molbev/msaa241
Iyer, L. M., Abhiman, S., and Aravind, L. (2011). Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 101, 25–104. doi: 10.1016/b978-0-12-387685-0.00002-0
Jeltsch, A., and Jurkowska, R. Z. (2016). Allosteric control of mammalian DNA methyltransferases – a new regulatory paradigm. Nucleic Acids Res. 44, 8556–8575. doi: 10.1093/nar/gkw723
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kapustin, Y., Souvorov, A., Tatusova, T., and Lipman, D. (2008). Splign: algorithms for computing spliced alignments with identification of paralogs. Biol. Direct 3:20. doi: 10.1186/1745-6150-3-20
Keller, T. E., Han, P., and Yi, S. V. (2015). Evolutionary Transition of Promoter and Gene Body DNA Methylation across Invertebrate–Vertebrate Boundary. Mol. Biol. Evol. 33, 1019–1028. doi: 10.1093/molbev/msv345
Kenny, N. J., McCarthy, S. A., Dudchenko, O., James, K., Betteridge, E., Corton, C., et al. (2020). The gene-rich genome of the scallop Pecten maximus. Gigascience 9:giaa037. doi: 10.1093/gigascience/giaa037
Kondo, E., Gu, Z., Horii, A., and Fukushige, S. (2005). The Thymine DNA Glycosylase MBD4 Represses Transcription and Is Associated with Methylated p16INK4a and hMLH1 Genes. Mol. Cell Biol. 25, 4388–4396. doi: 10.1128/mcb.25.11.4388-4396.2005
Le, S. Q., and Gascuel, O. (2008). An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. doi: 10.1093/molbev/msn067
Lees-Murdock, D. J., McLoughlin, G. A., McDaid, J. R., Quinn, L. M., O’Doherty, A., Hiripi, L., et al. (2004). Identification of 11 pseudogenes in the DNA methyltransferase gene family in rodents and humans and implications for the functional loci. Genomics 84, 193–204. doi: 10.1016/j.ygeno.2004.02.004
Leonhardt, H., Page, A. W., Weier, H. U., and Bestor, T. H. (1992). A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71, 865–873. doi: 10.1016/0092-8674(92)90561-p
Li, C., Liu, X., Liu, B., Ma, B., Liu, F., Liu, G., et al. (2018). Draft genome of the Peruvian scallop Argopecten purpuratus. Gigascience 7:giy031. doi: 10.1093/gigascience/giy031
Li, R., Zhang, W., Lu, J., Zhang, Z., Mu, C., Song, W., et al. (2020). The Whole-Genome Sequencing and Hybrid Assembly of Mytilus coruscus. Front. Genet. 11:440. doi: 10.3389/fgene.2020.00440
Li, Y., Zhang, L., Li, Y., Li, W., Guo, Z., Li, R., et al. (2019). Dynamics of DNA Methylation and DNMT Expression During Gametogenesis and Early Development of Scallop Patinopecten yessoensis. Mar. Biotechnol. 21, 196–205. doi: 10.1007/s10126-018-09871-w
Li, Y.-G., Guan, Y.-Y., Li, Q., and He, M.-X. (2015). Analysis of DNA methylation in tissues and development stages of pearl oyster Pinctada fucata. Genes Genomics 37, 263–270. doi: 10.1007/s13258-014-0246-1
Lim, Y.-K., Cheung, K., Dang, X., Roberts, S. B., Wang, X., and Thiyagarajan, V. (2021). DNA methylation changes in response to ocean acidification at the time of larval metamorphosis in the edible oyster, Crassostrea hongkongensis. Mar. Environ. Res. 163:105217. doi: 10.1016/j.marenvres.2020.105217
Liu, C., Zhang, Y., Ren, Y., Wang, H., Li, S., Jiang, F., et al. (2018). The genome of the golden apple snail Pomacea canaliculata provides insight into stress tolerance and invasive adaptation. GigaScience 7:giy101. doi: 10.1093/gigascience/giy101
Liu, X., Li, C., Chen, M., Liu, B., Yan, X., Ning, J., et al. (2020). Draft genomes of two Atlantic bay scallop subspecies Argopecten irradians irradians and A. i. concentricus. Sci. Data 7:99.
Luo, Y.-J., Takeuchi, T., Koyanagi, R., Yamada, L., Kanda, M., Khalturina, M., et al. (2015). The Lingula genome provides insights into brachiopod evolution and the origin of phosphate biomineralization. Nat. Commun. 6:8301.
Moreira, R., Pereiro, P., Canchaya, C., Posada, D., Figueras, A., and Novoa, B. (2015). RNA-Seq in Mytilus galloprovincialis: comparative transcriptomics and expression profiles among different tissues. BMC Genomics 16:728. doi: 10.1186/s12864-015-1817-5
Nakajima, H., and Kunimoto, H. (2014). TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 105, 1093–1099. doi: 10.1111/cas.12484
Nichols, R. (2001). Gene trees and species trees are not the same. Trends Ecol. Evol. 16, 358–364. doi: 10.1016/s0169-5347(01)02203-0
Pamilo, P., and Nei, M. (1988). Relationships between gene trees and species trees. Mol. Biol. Evol. 5, 568–583.
Peaston, A. E., and Whitelaw, E. (2006). Epigenetics and phenotypic variation in mammals. Mamm. Genome 17, 365–374. doi: 10.1007/s00335-005-0180-2
Peng, J., Li, Q., Xu, L., Wei, P., He, P., Zhang, X., et al. (2020). Chromosome-level analysis of the Crassostrea hongkongensis genome reveals extensive duplication of immune-related genes in bivalves. Mol. Ecol. Resour. 20, 980–994. doi: 10.1111/1755-0998.13157
Planques, A., Kerner, P., Ferry, L., Grunau, C., Gazave, E., and Vervoort, M. (2020). DNA methylation during development and regeneration of the annelid Platynereis dumerilii. bioRxiv doi: 10.1101/2020.11.13.381673
Powell, D., Subramanian, S., Suwansa-Ard, S., Zhao, M., O’Connor, W., Raftos, D., et al. (2018). The genome of the oyster Saccostrea offers insight into the environmental resilience of bivalves. DNA Res. 25, 655–665. doi: 10.1093/dnares/dsy032
Pradhan, M., Estève, P.-O., Chin, H. G., Samaranayke, M., Kim, G.-D., and Pradhan, S. (2008). CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry 47, 10000–10009. doi: 10.1021/bi8011725
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ran, Z., Li, Z., Yan, X., Liao, K., Kong, F., Zhang, L., et al. (2019). Chromosome-level genome assembly of the razor clam Sinonovacula constricta (Lamarck, 1818). Mol. Ecol. Resour. 19, 1647–1658. doi: 10.1111/1755-0998.13086
Rasmussen, K. D., and Helin, K. (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750. doi: 10.1101/gad.276568.115
Razin, A. (1989). “DNA Methylases,” in Genetic Engineering, Vol. 11, ed. J. K. Setlow (Boston: Springer), 1–11. doi: 10.1007/978-1-4615-7084-4_1
Ren, W., Gao, L., and Song, J. (2018). Structural Basis of DNMT1 and DNMT3A-Mediated DNA Methylation. Genes 9:620. doi: 10.3390/genes9120620
Rivière, G. (2014). Epigenetic features in the oyster Crassostrea gigas suggestive of functionally relevant promoter DNA methylation in invertebrates. Front. Physiol. 5:129. doi: 10.3389/fphys.2014.00129
Riviere, G., He, Y., Tecchio, S., Crowell, E., Gras, M., Sourdaine, P., et al. (2017). Dynamics of DNA methylomes underlie oyster development. PLoS Genet. 13:e1006807. doi: 10.1371/journal.pgen.1006807
Rogers, R. L., Grizzard, S. L., Titus-McQuillan, J. E., Bockrath, K., Patel, S., Wares, J. P., et al. (2021). Gene family amplification facilitates adaptation in freshwater unionid bivalve Megalonaias nervosa. Mol. Ecol. 30, 1155–1173. doi: 10.1111/mec.15786
Rosenberg, G. (2014). A New Critical Estimate of Named Species-Level Diversity of the Recent Mollusca. Am. Malacol. Bull. 32, 308–322. doi: 10.4003/006.032.0204
Rountree, M. R., Bachman, K. E., and Baylin, S. B. (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277. doi: 10.1038/77023
Saint-Carlier, E., and Riviere, G. (2015). Regulation of Hox orthologues in the oyster Crassostrea gigas evidences a functional role for promoter DNA methylation in an invertebrate. FEBS Lett. 589, 1459–1466.
Simakov, O., Marletaz, F., Cho, S.-J., Edsinger-Gonzales, E., Havlak, P., Hellsten, U., et al. (2013). Insights into bilaterian evolution from three spiralian genomes. Nature 493, 526–531. doi: 10.1038/nature11696
Skvortsova, K., Iovino, N., and Bogdanoviæ, O. (2018). Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 19, 774–790. doi: 10.1038/s41580-018-0074-2
Smith, S. A., Wilson, N. G., Goetz, F. E., Feehery, C., Andrade, S. C. S., Rouse, G. W., et al. (2011). Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature 480, 364–367. doi: 10.1038/nature10526
Song, K., Li, L., and Zhang, G. (2017). The association between DNA methylation and exon expression in the Pacific oyster Crassostrea gigas. PLoS One 12:e0185224. doi: 10.1371/journal.pone.0185224
Suarez-Ulloa, V., Rivera-Casas, C., Michel, M., and Eirin-Lopez, J. M. (2019). Seasonal DNA Methylation Variation in the Flat Tree Oyster Isognomon Alatus from a Mangrove Ecosystem in North Biscayne Bay, Florida. J. Shellfish Res. 38, 79–88. doi: 10.2983/035.038.0108
Sun, J., Mu, H., Ip, J. C. H., Li, R., Xu, T., Accorsi, A., et al. (2019). Signatures of Divergence, Invasiveness, and Terrestrialization Revealed by Four Apple Snail Genomes. Mol. Biol. Evol. 36, 1507–1520. doi: 10.1093/molbev/msz084
Sun, J., Zhang, Y., Xu, T., Zhang, Y., Mu, H., Zhang, Y., et al. (2017). Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 1:0121.
Sun, Y., Hou, R., Fu, X., Sun, C., Wang, S., Wang, C., et al. (2014). Genome-wide analysis of DNA methylation in five tissues of Zhikong scallop, Chlamys farreri. PLoS One 9:e86232. doi: 10.1371/journal.pone.0086232
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Thai, B. T., Lee, Y. P., Gan, H. M., Austin, C. M., Croft, L. J., Trieu, T. A., et al. (2019). Whole Genome Assembly of the Snout Otter Clam, Lutraria rhynchaena, Using Nanopore and Illumina Data, Benchmarked Against Bivalve Genome Assemblies. Front. Genet. 10:1158. doi: 10.3389/fgene.2019.01158
Uliano-Silva, M., Dondero, F., Dan Otto, T., Costa, I., Lima, N. C. B., Americo, J. A., et al. (2018). A hybrid-hierarchical genome assembly strategy to sequence the invasive golden mussel, Limnoperna fortunei. Gigascience 7:gix128. doi: 10.1093/gigascience/gix128
Venkataraman, Y. R., Downey-Wall, A. M., Ries, J., Westfield, I., White, S. J., Roberts, S. B., et al. (2020). General DNA Methylation Patterns and Environmentally-Induced Differential Methylation in the Eastern Oyster (Crassostrea virginica). Front. Mar. Sci. 7:225. doi: 10.3389/fmars.2020.00225
Wagner, G. P., Kin, K., and Lynch, V. J. (2012). Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285. doi: 10.1007/s12064-012-0162-3
Wang, S., Zhang, J., Jiao, W., Li, J., Xun, X., Sun, Y., et al. (2017). Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat. Ecol. Evol. 1:120.
Wang, X., Li, Q., Lian, J., Li, L., Jin, L., Cai, H., et al. (2014). Genome-wide and single-base resolution DNA methylomes of the Pacific oyster Crassostrea gigas provide insight into the evolution of invertebrate CpG methylation. BMC Genomics 15:1119. doi: 10.1186/1471-2164-15-1119
Wei, M., Ge, H., Shao, C., Yan, X., Nie, H., Duan, H., et al. (2020). Chromosome-Level Clam Genome Helps Elucidate the Molecular Basis of Adaptation to a Buried Lifestyle. iScience 23:101148. doi: 10.1016/j.isci.2020.101148
Willbanks, A., Leary, M., Greenshields, M., Tyminski, C., Heerboth, S., Lapinska, K., et al. (2016). The Evolution of Epigenetics: from Prokaryotes to Humans and Its Biological Consequences. Genet. Epigenet. 8, 25–36.
Yan, X., Nie, H., Huo, Z., Ding, J., Li, Z., Yan, L., et al. (2019). Clam Genome Sequence Clarifies the Molecular Basis of Its Benthic Adaptation and Extraordinary Shell Color Diversity. iScience 19, 1225–1237.
Zemach, A., McDaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zhang, G., Fang, X., Guo, X., Li, L., Luo, R., Xu, F., et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54.
Keywords: mollusk, bivalve, methylation, epigenetics, DNMT, TET, MBD
Citation: Gerdol M, La Vecchia C, Strazzullo M, De Luca P, Gorbi S, Regoli F, Pallavicini A and D’Aniello E (2021) Evolutionary History of DNA Methylation Related Genes in Bivalvia: New Insights From Mytilus galloprovincialis. Front. Ecol. Evol. 9:698561. doi: 10.3389/fevo.2021.698561
Received: 21 April 2021; Accepted: 28 May 2021;
Published: 09 July 2021.
Edited by:
Giulia Furfaro, University of Salento, ItalyReviewed by:
Celine Cosseau, Université de Perpignan Via Domitia, FranceRicard Albalat, University of Barcelona, Spain
Copyright © 2021 Gerdol, La Vecchia, Strazzullo, De Luca, Gorbi, Regoli, Pallavicini and D’Aniello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrico D’Aniello, ZW5yaWNvLmRhbmllbGxvQHN6bi5pdA==