 Jasmin Gegner
Jasmin Gegner- 1Branch for Bioresources, Fraunhofer Institute for Molecular Biology and Applied Ecology IME, Gießen, Germany
- 2Department of Entomology, Max-Planck Institute for Chemical Ecology, Jena, Germany
- 3Bioinformatics and Systems Biology, Justus-Liebig-University of Gießen, Gießen, Germany
- 4Institute for Insect Biotechnology, Department of Applied Entomology, Justus-Liebig-University of Gießen, Gießen, Germany
The transition between morphologically distinct phenotypes during complete metamorphosis in holometabolous insects is accompanied by fundamental transcriptional reprogramming. Using the tobacco hornworm (Manduca sexta), a powerful model for the analysis of insect evolution and development, we conducted a genome-wide comparative analysis of gene expression and DNA methylation in caterpillars and adults to determine whether complete metamorphosis has an epigenetic basis in this species. Bisulfite sequencing indicated a generally low level of DNA methylation with a unimodal CpGO/E distribution. Expression analysis revealed that 24 % of all known M. sexta genes (3.729) were upregulated in last-instar larvae relative to the adult moth, whereas 26 % (4.077) were downregulated. We also identified 4.946 loci and 4.960 regions showing stage-specific differential methylation. Interestingly, genes encoding histone acetyltransferases and histone deacetylases were differentially methylated in the larvae and adults, indicating there is crosstalk between different epigenetic mechanisms. The distinct sets of methylated genes in M. sexta larvae and adults suggest that complete metamorphosis involves epigenetic modifications associated with profound transcriptional reprogramming, involving approximately half of all the genes in this species.
1. Introduction
Transformation from one morphologically distinct phenotype to another during development is known as metamorphosis. The complete metamorphosis of holometabolous insects involves a pupal stage, during which larval tissues and organs are remodeled and/or replaced by the structures required in the imagoes. A fundamental question in developmental biology is how the same genome can generate morphologically and ecologically distinct phenotypes, such as the caterpillars, pupae, and imagoes in the order Lepidoptera. Many if not all developmental processes involve the orchestrated large-scale transcriptional reprogramming of genes. We therefore postulate that the metamorphosis of holometabolous insect larvae into adults involves fundamental changes in gene expression, and that epigenetic mechanisms may be involved in this process. Epigenetics can explain the phenotypic diversity of insect larvae, pupae, and adults sharing a common genome because such mechanisms globally modulate gene expression rather than altering the DNA sequence (Belles, 2017; Glastad et al., 2019).
There are three major epigenetic mechanisms, one of which is the expression of microRNAs. These short, non-coding RNAs (18–24 bp) operate at the post-transcriptional level to inhibit the translation of specific mRNAs by base-pairing with the untranslated regions or occasionally the coding region (Asgari, 2013; Hussain and Asgari, 2014). Individual microRNAs can regulate the expression of single genes or hundreds of genes. The role of microRNAs in the regulation of complete metamorphosis in holometabolous insects is well-established (Belles, 2017; Ylla et al., 2017). The first microRNAs that are differentially expressed during lepidopteran metamorphosis have been identified in the greater wax moth Galleria mellonella (Mukherjee and Vilcinskas, 2014).
The two other principal epigenetic mechanisms regulate transcriptional initiation. The first is the acetylation and deacetylation of histones by enzymes with opposing activities, thus controlling the ability of transcription factors to access chromatin and initiate gene expression. The addition of acetyl groups to histones is mediated by histone acetyltransferases (HATs), which enhance access to DNA by loosening the chromatin, whereas the removal of acetyl groups by histone deacetylases (HDACs) has the opposite effect and therefore causes gene silencing (Marks et al., 2003). We have previously shown that histone acetylation/deacetylation modulates gene expression during the complete metamorphosis of G. mellonella (Mukherjee et al., 2012). The involvement of both histone modification and microRNAs in this process provides evidence for cross-talk between different epigenetic mechanisms (Mukherjee and Vilcinskas, 2014).
The final major epigenetic mechanism is DNA methylation (Glastad et al., 2019). The addition of a methyl group to a cytosine residue in the dinucleotide sequence CpG results in the formation of 5-methylcytosine, which retains the base-pairing specificity of the unmodified nucleoside but influences its interactions with regulatory proteins. The transfer of methyl groups to DNA is mediated by evolutionarily-conserved enzymes collectively known as DNA methyltransferases (DNMTs). These can be divided further into maintenance methyltransferases (DNMT1), which complete the symmetrical methylation marks on newly-replicated DNA by recognizing the hemimethylated sequences inherited from each parent, and de novo methyltransferases (DNMT3), which establish new methylation marks on unmethylated DNA (Bestor, 2000; Klose and Bird, 2006). In insects, DNMT1 is evolutionarily conserved and widely distributed whereas DNMT3 is restricted to some species (Bewick et al., 2017). Despite the apparent loss of DNMT3 in the order Lepidoptera, these insects have functional methylation systems (Provataris et al., 2018). Indeed, the DNA of holometabolous insects is methylated, although typically only in exon sequences (Provataris et al., 2018).
Here, we tested the hypothesis that metamorphosis-related transcriptional reprogramming in the lepidopteran model species M. sexta involves DNA methylation. We carried out a genome-wide comparative analysis of gene expression and DNA methylation in caterpillars and adults, and identified correlations between stage-specific genes and methylation marks, including marks affecting genes involved in other epigenetic processes.
Our data provide insight into the potential role of epigenetic modifications of complete metamorphosis of holometabolous insects.
2. Results
2.1. Methylation in Manduca sexta
Comparing DNA methylation in different developmental stages in M. sexta, the mean mapping efficiency for Bismark mapping for paired end mapping was 45.6 % and could be increased to 57.2 % due to the following single end mapping (Supplementary Table 1). The HISAT2 mean mapping for transcriptome sequencing results was 67.6 % (Supplementary Table 1).
The overall level of DNA methylation in each sample (L1—3, A1—3) was 0.18–0.48 %, but when focusing on specific positions (found in two out of three larvae or adults) the level of DNA methylation was much lower (0.05 %), as shown in Supplementary Material (Supplementary Table 2). To gain more insight into the methylated sites in the M. sexta genome, we compared the positions based on the sequence context (CpG, CHG, or CHH) and the sample type (larvae, adults, or both). This showed that the CpG context was the most prominent (62 % of all methylated sites) compared to CHG at 6 % and CHH at 32 % (Figure 1A and Supplementary Table 3). The positions were almost equally distributed in all three samples: 28 % in larvae, 37 % in adults, and 35 % in both (Figure 1B and Supplementary Table 3). The normalized CpG content (CpGO/E) revealed a unimodal distribution (Figure 2). Given the relatively large number of methylated positions in the sequence contexts CHG and CHH (Figure 1A), we carried out a global analysis of differential methylation in M. sexta (CX) rather than focusing on CpG, CHG or CHH methylation alone. We identified 4946 differentially methylated loci (DMLs) and 4960 differentially methylated regions (DMRs) in total. These were classified according to their genomic location as belonging to the gene body (gb), regulatory region (rr), or intergenic region (ir). Interestingly, metamorphosis was accompanied by a significant shift in the location of DMLs and DMRs, resulting in an increase in the number of methylated sites in gene bodies at the expense of sites especially in the intergenic regions, whereas the number of methylated sites in regulatory regions was almost the same in both developmental stages (Figure 3 and Supplementary Table 4).
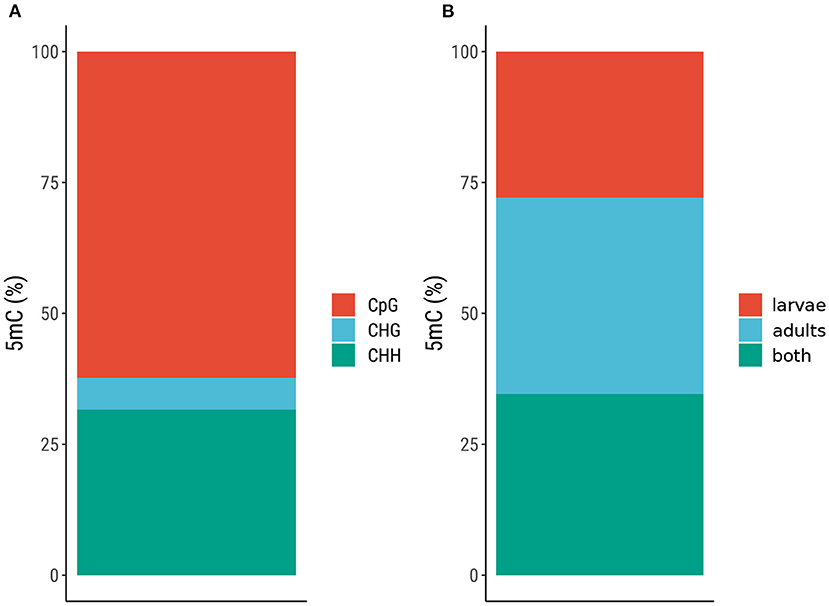
Figure 1. Distribution of 5′-methylcytosine (5mC) in the genome of Manduca sexta. (A) Classification by sequence context. (B) Distribution across different life stages (larvae and adults).
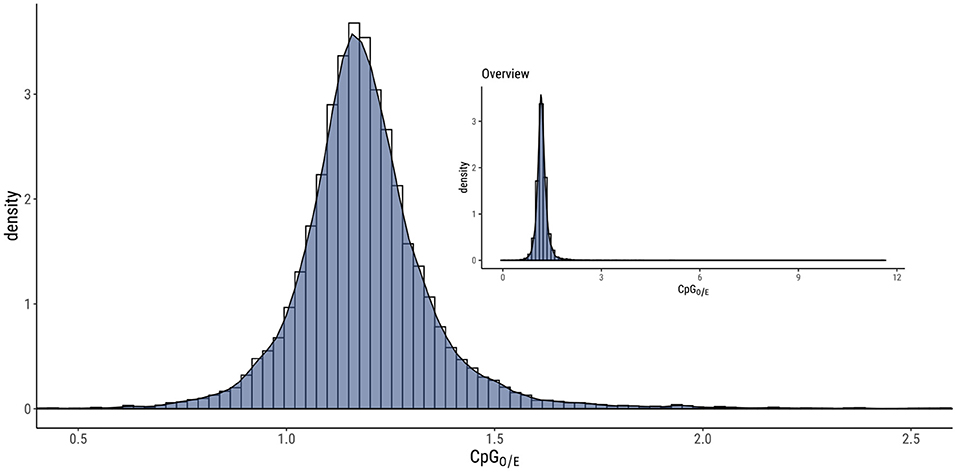
Figure 2. CpGO/E analysis of Manduca sexta showing a unimodal distribution, which represents a low rate of C to T transitions in the sequence context CpG and therefore indicates a generally low level of CpG methylation.
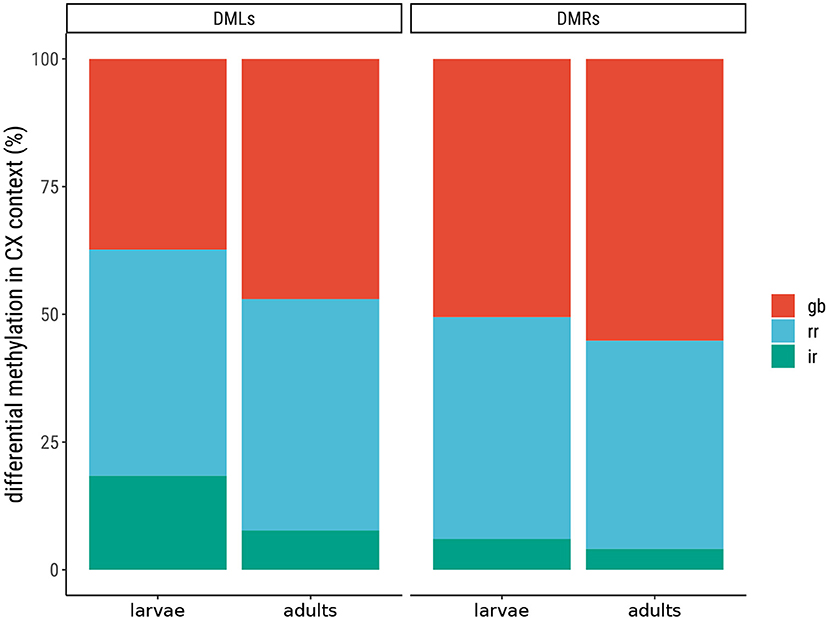
Figure 3. Differential methylation in Manduca sexta according to genomic location. The differentially methylated loci (DMLs) and differentially methylated regions (DMRs) are classified by their location in the gene body (gb), regulatory region (rr), or intergenic region (ir).
2.2. Differential Gene Expression
The annotated M. sexta genome sequence contains 15.542 genes (Kanost et al., 2016a,b,c). We compared the expression profiles of all these genes in larvae and adults, and found that about half of them were either not expressed at detectable levels during either stage (n.d., 35 %) or showed no evidence of significant differential expression (n.s., 15 %). The remaining genes were either upregulated (24 %) or downregulated (26 %) in larvae compared to imagoes (Supplementary Table 5). Gene ontology enrichment analysis revealed eight functional categories with stage-specific over-representation, seven of which were related to reproduction and one involved in sensory perception (Supplementary Table 6).
2.3. Correlation Between Differential Methylation and Gene Expression
To investigate the correlation between differential methylation and gene expression, we selected all genes expressed at detectable levels at one or both developmental stages that also contained DMLs or DMRs (without distinguishing between the type of methylated site). Given the possibility that genes might overlap with multiple DMLs or DMRs, we also distinguished between genes that were exclusively methylated in the larvae or adults (L_5mC or A_5mC), methylated more extensively but not exclusively in the larvae or imagoes (pL_5mC or pA_5mC), and those methylated to the same extent in larvae and adults (mixed). As shown in Table 1, most of the differentially expressed genes showed no evidence of methylation (74 %). After this, the most abundant categories were the exclusively differentially methylated genes (L_5mC 14 % and A_5mC 10 %). Only a small proportion of the genes were assigned to the mixed, pL_5mC, and pA_5mC categories.
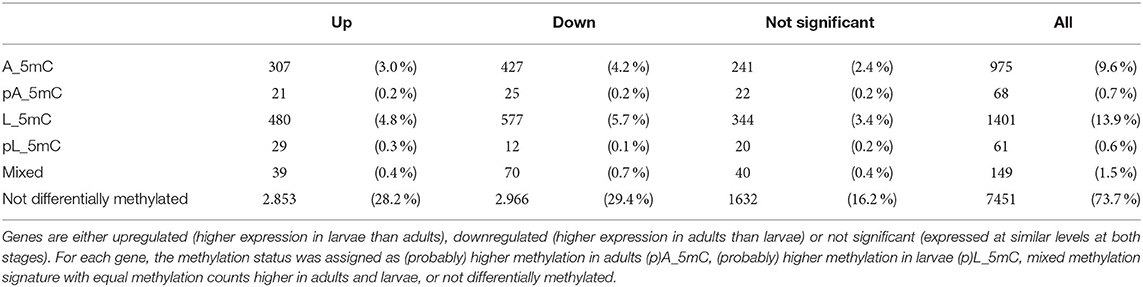
Table 1. Classification of gene expression based on differential methylation analysis.
For the investigation of a potential correlation between methylation pattern and expression status, we analyzed the log fold change for all differential expressed genes in dependency of their methylation status. With an exception of pL_5mC all medians for log fold changes are negative and indicate a down regulation of the gene expression. For pL_5mC this trend is reversed and the median of the log fold changes is positive and represents an up regulation (Figure 4).
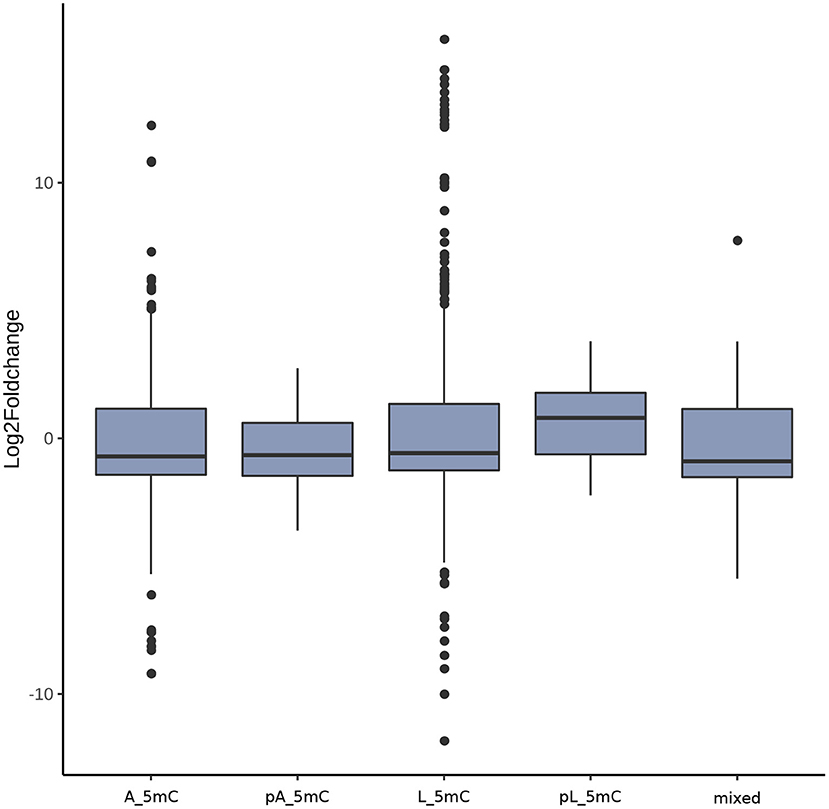
Figure 4. Correlation between gene expression (log2 fold change) and the different possible groups based on methylation status per gene. The methylation status of each gene was defined as mixed, for equal numbers of methylated loci and regions in larvae and adults, as higher methylation in larvae (L_5mC) or higher methylation in adults (A_5mC) where methylation was exclusively state-specific, or as probably higher methylation in larvae (pL_5mC) or probably higher methylation in adults (pA_5mC) for loci/regions methylated in both samples but with a preference for one stage over the other.
2.4. Genes of Interest
To understand the functional implications of the correlation between gene expression and DNA methylation during metamorphosis, we focused on 850 M. sexta genes representing eight different functional categories: chitin metabolism, detoxification, immunity, epigenetic modification, juvenile hormone/ecdysone signaling, chorion proteins, and vitellogenin (Supplementary Table 7). The genes coding for chorion proteins and vitellogenin showed no evidence of differential methylation. Where differential methylation was detected, in general there was no correlation between gene expression levels and the degree of DNA methylation at each developmental stage (Table 2 and Supplementary Table 8). For example, seven genes encoding chitin-binding proteins were higher expressed in larvae, three of which were hypermethylated in larvae and four hypermethylated in imagoes. However, one exceptional case was the immunity-related gene Atg8 (Msex2.12227), which produces two separate transcripts encoding protein isoforms involved in autophagy. Transcript Msex2.12227-RA was upregulated in larvae relative to adults, whereas transcript Msex2.12227-RB was downregulated. The genomic region for that gene contain three differentially methylated regions: two hypermethylated in adults covering both transcripts and one hypermethylated in larvae exclusive covering the transcript Msex2.12227-RA. We also identified genes with roles in histone acetylation/deacetylation that were upregulated in imagoes and generally hypermethylated in the larvae, with the exception of one HDAC gene that hypermethylated in adults.
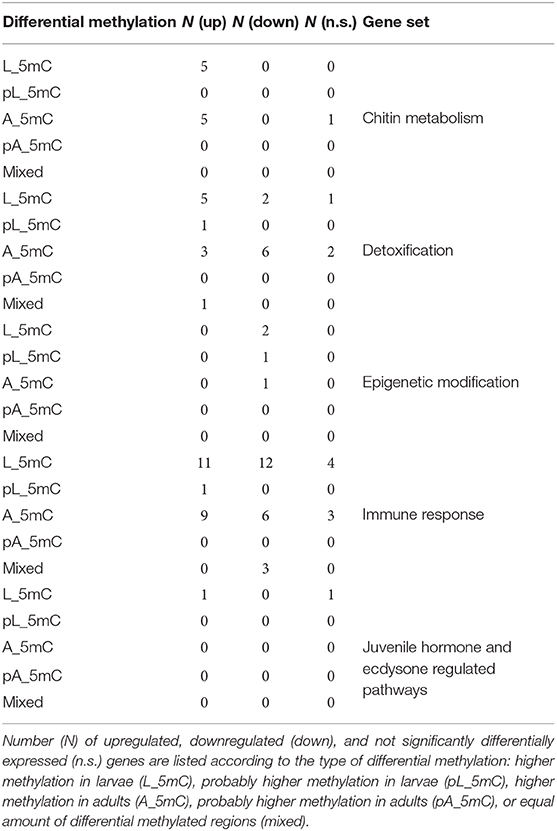
Table 2. Summary of differential gene expression and methylation patterns in genes of interest.
3. Discussion
DNA methylation is involved in the regulation of many physiological and developmental processes in insects, but its role in metamorphosis has not been investigated in detail. Previous studies have shown that the transcriptional reprogramming that accompanies metamorphosis is associated with other forms of epigenetic regulation, specifically microRNA expression and the acetylation/deacetylation of histones (Mukherjee et al., 2012; Mukherjee and Vilcinskas, 2014). Therefore, we investigated the relationship between genomic DNA methylation and developmentally-regulated gene expression during metamorphosis in the model lepidopteran species M. sexta.
Initially we measured the general prevalence of DNA methylation in the M. sexta genome and found that the overall level of methylation was low. This is in line with recent findings in other holometabolous insects, which tend to have lower levels of DNA methylation in exons in comparison to hemimetabolous insects (Provataris et al., 2018). Accordingly, CpGO/E analysis revealed a unimodal distribution which indicates a limited degree of C to T transitions over evolutionary timescales, consistent with a low level of CpG methylation (Provataris et al., 2018). Similar unimodal distributions have been reported in the silkworm Bombyx mori, the red flour beetle Tribolium castaneum, and the large milkweed bug Oncopeltus fasciatus (Bewick et al., 2017) as well as the mosquito Anopheles gambiae (Bewick et al., 2017). Two different distributions in one specimen were found in the ant species Camponotus floridanus and Harpegnathos saltator, which also show low DNA methylation levels. While CpG dinucleotides have a bimodal distribution, the CHH and CHG context showed a unimodal one (Bonasio et al., 2012). In contrast, the honeybee Apis mellifera features a bimodal CpGO/E distribution reflecting DNA methylation associated with differential gene expression in different bee castes (Elango et al., 2009). However, positive correlation between gene expression and cytosines located in CpG context in species with a unimodal CpGO/E and low methylation levels, like it was observed for T. castaneum (Song et al., 2017). Consequently, the sparse DNA methylation in M. sexta does not exclude the possibility of a role during complete metamorphosis. We therefore expanded our comparative analysis of DNA methylation in larvae and adults beyond the CpGO/E distribution to include the separate analysis of sites located in gene bodies, regulatory regions, and intergenic regions. This revealed that both stages showed preponderance for DMLs and DMRs in the gene body. Nevertheless, a significant shift during metamorphosis was detected with a greater proportion of gene body sites in the adult moths primarily at the expense of intergenic sites, which were more prevalent in the larvae. The number of sites in gene regulatory regions remained almost steady throughout development. These observations are in contrast with the report of Song et al. (2017) showing that more than 80 % of the methylated cytosine residues in T. castaneum are almost entirely restricted to intergenic regions (Song et al., 2017).
We were particularly interested to probe the relationship between differential methylation and differential gene expression during metamorphosis in M. sexta. We found that almost half of all genes were differentially expressed, the remainder either are expressed at similar levels in both larvae and adults or not expressed at detectable levels in either stage. There was little overall correlation between the methylation patterns and gene expression profiles, but there were a small number of striking correlations for highly relevant genes. One of the differentially methylated genes in M. sexta encoded chitin synthase 2. In B. mori a gene from the same class is also differentially methylated during development, with the methylation mark being erased from the promoter in the adult allowing its expression in the wings (Xu et al., 2018). We also observed a correlation between expression and methylation for two different transcripts of the atg8, which is involved in autophagy. Isoform RA was upregulated in larvae and the corresponding genomic region was potentially hypermethylated in imagoes, whereas isoform RB was downregulated in larvae and the corresponding genomic region was exclusively hypermethylated in the adults. Similarly, G. mellonella Atg8 is induced during metamorphosis, with the increasing abundance of the transcript and protein suggesting a role in midgut transformation (Khoa and Takeda, 2012). Combined with our findings, the data suggest that DNA methylation contributes to the regulation of Atg8 during metamorphosis in the Lepidoptera. Juvenile hormone and ecdysone are particularly important regulators in insect development, and the corresponding genes were recently found to be regulated by histone acetylation and deacetylation (Roy and Palli, 2018). We therefore focused on the corresponding M. sexta genes, but only two of these were differentially methylated genes and there was no relationship with the observed expression profiles. The absence of a link between DNA methylation and juvenile hormone has also been demonstrated in honeybees (Cardoso-Junior et al., 2018). The latter study also showed that DNA hypomethylation led to an increase in vitellogenin expression, which is important for reproduction. In contrast, DNA hypomethylation in the brown planthopper Nilaparvata lugens caused a loss of fecundity (Zhang et al., 2015). In M. sexta, we found no evidence that genes encoding chorion proteins or vitellogenin were differentially methylated, suggesting they are not regulated directly by this epigenetic mechanism.
The expression of differentially methylated genes related to the RNA interference pathway declined during the transition from larvae to imagoes, suggesting that RNA-dependent regulation may play a more prominent role in M. sexta larvae than adults. We identified few differentially methylated genes involved in detoxification and immunity-related functions, but interestingly we found that some genes with roles in histone acetylation/deacetylation were upregulated in adults and generally hypermethylated in the larvae, with the exception of one HDAC gene that hypermethylated in imagoes. This indicates there is crosstalk between different epigenetic mechanisms and that histone modification may work in concert with DNA methylation during metamorphosis in M. sexta. Similarly, we found that the specific inhibition of HATs in G. mellonella delays pupation whereas the inhibition of HDACs leads to precocious pupation and accelerated development (Mukherjee et al., 2012). Given that the opposing activities of HATs and HDACs are tightly regulated, we conclude that differential methylation of the corresponding genes has the potential to contribute to the epigenetic regulation of transcriptional reprogramming during metamorphosis in M. sexta and other lepidopterans. Furthermore, histone acetylation and deacetylation in T. castaneum influence the production of juvenile hormone (Roy and Palli, 2018). Thus, our findings suggest that DNA methylation could indirectly regulate the juvenile hormone system.
Metamorphosis in the oyster Crassostrea gigas is accompanied by changes in the methylation status of exons, introns, promoters, repeats and transposons, including the increased methylation of exons at the expense of introns, which contrasts with our findings in M. sexta (Riviere et al., 2017). The same study revealed that, regardless of the level of methylation, the pattern of methylation within genes is associated with transcriptional regulation. Another recent study revealed that the model cephalochordate Amphioxus lanceolatum has also low levels of CpG methylation, but this nevertheless is involved in the regulation of metamorphosis (Marletaz et al., 2018). Changes in DNA methylation patterns therefore have the capability to accompany metamorphosis in all organisms that contain methylated cytosines, but there is immense diversity in terms of the methylation targets: gene bodies, regulatory regions, and intergenic regions. Even low levels of methylation can help to regulate the transcriptional reprograming that occurs during metamorphosis in holometabolous animals, potentially in an indirect manner involving other epigenetic mechanisms. We have provided the first evidence that DNA methylation is potentially involved in the complete metamorphosis of M. sexta, a model lepidopteran.
4. Methods
4.1. Sample Preparation
Samples were taken from our in-house M. sexta stock, which was reared as previously described (Gegner et al., 2019). We selected three female L5 during their feeding stage (2–3 days after molting) and three female adults 2–3 days after their eclosion. Individual insects were frozen and ground in liquid nitrogen. RNA was isolated from 50 mg of ground tissue of one animal using the Direct-zol RNA MiniPrep Kit (ZymoResearch) including DNA digestion according to the manufacturer's recommendations. Sample quantity and purity were assessed using a NanoDrop ND-1000 UV/Vis spectrophotometer (Thermo Fisher Scientific) and the integrity was verified using an Agilent 2100 Bioanalyzer and a RNA 6000 Nano Kit (Agilent Technologies). DNA was extracted from equal amounts of tissue from the same individual using the Genomic-tip 20/G and Genomic DNA Buffer Set (Qiagen) according to the manufacturer's protocol. RNA-Seq using poly(A)+ enriched RNA fragmented to an average of 250 bp and bisulfite sequencing were carried out using the llumina 2000 HiSeq2500 platform at GATC Biotech (Eurofins Genomics) based on single samples of 2 μg RNA or 1 μg DNA, respectively. Sample preparation for Bisulfite and RNA sequencing was performed by GATC Biotech using their in house protocols.
4.2. Raw Read Processing and Methylation Analysis
Sequence data were processed using GNU parallel v20141022 (Tange, 2011). The quality of the raw and processed reads was controlled using FastQC v0.11.7 (Andrews, 2010). Sequences were trimmed using Trim Galore v0.4.5 (Krueger, 2012) based on FastQC v0.11.7 and Cutadapt v1.9.1 (Martin, 2011). The software was combined into a Docker Trim Galore image (Förster, 2018i) tagged v0.4.5.1. Trim Galore was executed for each Fastq file pair using the following parameters: –retain_unpaired –paired –fastqc_args –nogroup –noextract –outdir QC/after_trimming' –gzip –length 20 –quality 20 –fastqc.
The M. sexta draft genome sequence (Kanost et al., 2016b) was used as a reference (Kanost et al., 2016a,b,c). Reads were mapped by paired-end mapping in Bismark v0.15.0 (Krueger and Andrews, 2011) using Bowtie2 v2.3.4.1 with default settings (Langmead and Salzberg, 2012). The software was combined into a Bismark Docker image (Förster, 2018a) tagged v0.15.0. Unmapped reads were remapped separately by single-end mapping, and all mapped reads were deduplicated using the Perl script deduplicate_bismark_alignment_output.pl from Bismark software package. Positions of one biological sample with coverage > 10 and ≥ 10% methylated cytosine residues were defined as methylated (Wang et al., 2013). If positions were called methylated in two out of three biological samples per developmental stage, they were defined as methylated within that stage. Differential methylation analysis was processed using DSS v2.26.0 (Wu et al., 2013, 2015; Feng et al., 2014; Park and Wu, 2016) in R v3.4.4 (R Development Core Team, 2020) with default settings and an unspecified cytosine context (CX). The p-value threshold was set to 0.001 for differentially methylated loci and to 0.01 for differentially methylated regions. The DSS installation was combined into a DSS Docker image (Förster, 2018c) and tagged v2.28.0.
4.3. Characterization of Methylated Regions
The original genome annotation file contained only some annotations for 5′ and 3′ untranslated regions (UTRs). Therefore, the complete file was re-analyzed and annotations for 5′ and 3′ UTRs were added to the transcript annotation, if the coding region was shorter than the complete transcript. The genome was sectioned into three sequence categories: gene bodies (gb: exons, introns, 5′ and 3′ UTRs), regulatory regions (rr: sequences 2 kbp beyond the UTRs), and intergenic regions (ir). DMLs and DMRs were characterized by their location in the genome and the expression of any genes. These results were summarized per gene and the methylation status of each gene was defined as mixed, for equal numbers of methylated loci and regions in larvae and imagoes, as higher methylation in larvae (L_5mC) or higher methylation in adults (A_5mC) where methylation was exclusively state-specific, or as probably higher methylation in larvae (pL_5mC) or probably higher methylation in adults (pA_5mC) for loci/regions methylated in both samples but with a preference for one stage over the other.
4.4. RNA Raw Read Processing and Differential Expression Analysis
Quality control measures, including the filtering of high-quality reads based on fastq file scores, the removal of reads containing primer/adapter sequences, and trimming of the read length, were carried out using CLC Genomics Workbench v8.1 (Qiagen Bioinformatics, 2018). Predicted transcript sequences in the M. sexta draft genome (Kanost et al., 2016a) sequence were downloaded from (Kanost et al., 2016c) and matched to our RNA-Seq data using HISAT2 v2.1.0 (Kim et al., 2015). The software was combined into a HISAT2 Docker image (Förster, 2018d) and tagged v2.1.0. The resulting SAM files were converted into BAM files using samtools v1.9 (Li et al., 2009). The BAM files were processed by stringtie v1.3.4d (Pertea et al., 2015, 2016) to generate count information for each transcript and sample. Stringtie includes the script prepDE.py which was used to prepare the mapping counts for the differential expression analysis. Docker images are available containing samtools (Förster, 2018g) and stringtie (Förster, 2018h), tagged v1.9 and v1.3.4d, respectively.
Differential expression analysis was performed using DESeq2 v1.14.1 (Love et al., 2014) for GNU R v3.4.4, which is available as a Docker image (Förster, 2018b) tagged v1.14.1. All transcripts with normalized count sums of < 1.000 across all samples were defined as not detected (n.d.). Transcripts with adjusted p ≥ 0.05 showed no significant differential expression profiles (n.s.). Where the adjusted p-values were < 0.05, a log2 fold change < 0 reflected the upregulation of the corresponding gene in larvae compared to imagoes whereas a log2 fold change >0 reflected the downregulation of that gene in larvae compared to adults. Genes were classified as modulated when the expression of at least one transcript was upregulated or downregulated. If the same gene was simultaneously represented by upregulated and downregulated transcripts, the expression class was defined by the mRNA with the highest raw mean value.
4.5. CpGO/E Analysis
The normalized CpG dinucleotide content was used as a proxy for the presence of DNA methylation because methylated cytosine residues are prone to spontaneous deamination into thymine, leading to a gradual decline in the prevalence of CpG dinucleotides, a phenomenon known as CpG depletion (Provataris et al., 2018). Based on the published gene set, CpGO/E analysis was performed according to Bird (1980). We used the script cpgoe.pl with the gene annotation file and the draft genome scaffolds as inputs.
4.6. Gene Set (Re-)annotation
The original M. sexta gene set was re-annotated using InterproScan v5.27-66.0 (Jones et al., 2014), including separately-licensed versions of TMHMM v2.0c (Sonnhammer et al., 1998; Krogh et al., 2001), SignalP v4.1 (Petersen et al., 2011), and Phobius v1.01 (Kall et al., 2004). A Docker image (Förster, 2018e) comprising the installation of InterproScan and all the additional publicly available software is tagged v5.27.66. Separately-licensed software was added to the running Docker container as volume.
4.7. Gene Ontology Enrichment
Gene ontology terms were obtained by InterproScan reannotation, and gene enrichment analysis was carried out using ontologizer v2.1 (Bauer et al., 2008). The docker image providing the software installation (Förster, 2018f) was tagged as v2.1. The required go.obo file (Ashburner et al., 2000; The Gene Ontology, Consortium, 2019) was downloaded from (The Gene Ontology, Consortium, 2018).
4.8. Genes of Interest
We analyzed 850 genes of interest to determine their methylation status and expression profile. We selected genes involved in chitin metabolism (65), detoxification (255), immunity (411), epigenetic modification (22), and juvenile hormone/ecdysone signaling (15), as well as those encoding chorion proteins (79) or vitellogenin (3) (Palli et al., 1992; Segraves and Woldin, 1993; Fujiwara et al., 1995; Hiruma et al., 1995; Jindra et al., 1996, 1997; Zhou et al., 1998a,b; Weller et al., 2001; Stilwell et al., 2003; Dubrovskaya et al., 2004; Cao et al., 2015a,b; Tetreau et al., 2015a,b; Kanost et al., 2016a). A full list is available in Supplementary Material (Supplementary Table 7).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA517460.
Author Contributions
JG and FF contributed to analysis and interpretation of data. HV, AB, and AV designed the study. AV inspired the study and acquired funding. All authors contributed to the drafting of the manuscript and gave their final approval.
Funding
AV acknowledges generous funding by the Hessen State Ministry of Higher Education, Research and the Arts (HMWK) via the LOEWE Centers Insect Biotechnology and Bioresources and Translational Biodiversity Genomics. HV acknowledges funding by the Max Planck Gesellschaft.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Richard M. Twyman for editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.646281/full#supplementary-material
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Asgari, S. (2013). MicroRNA functions in insects. Insect Biochem. Mol. Biol. 43, 388–397. doi: 10.1016/j.ibmb.2012.10.005
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bauer, S., Grossmann, S., Vingron, M., and Robinson, P. N. (2008). Ontologizer 2.0-a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650–1651. doi: 10.1093/bioinformatics/btn250
Belles, X. (2017). MicroRNAs and the evolution of insect metamorphosis. Annu. Rev. Entomol. 62, 111–125. doi: 10.1146/annurev-ento-031616-034925
Bestor, T. H. (2000). The DNA methyltransferases of mammals. Hum. Mol. Genet. 9, 2395–2402. doi: 10.1093/hmg/9.16.2395
Bewick, A. J., Vogel, K. J., Moore, A. J., and Schmitz, R. J. (2017). Evolution of DNA methylation across insects. Mol. Biol. Evol. 34, 654–665. doi: 10.1093/molbev/msw264
Bird, A. P. (1980). DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8, 1499–1504. doi: 10.1093/nar/8.7.1499
Bonasio, R., Li, Q., Lian, J., Mutti, N. S., Jin, L., Zhao, H., et al. (2012). Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 22, 1755–1764. doi: 10.1016/j.cub.2012.07.042
Cao, X., He, Y., Hu, Y., Wang, Y., Chen, Y. R., Bryant, B., et al. (2015a). The immune signaling pathways of Manduca sexta. Insect Biochem. Mol. Biol. 62, 64–74. doi: 10.1016/j.ibmb.2015.03.006
Cao, X., He, Y., Hu, Y., Zhang, X., Wang, Y., Zou, Z., et al. (2015b). Sequence conservation, phylogenetic relationships, and expression profiles of nondigestive serine proteases and serine protease homologs in Manduca sexta. Insect Biochem. Mol. Biol. 62, 51–63. doi: 10.1016/j.ibmb.2014.10.006
Cardoso-Junior, C. A. M., Guidugli-Lazzarini, K. R., and Hartfelder, K. (2018). DNA methylation affects the lifespan of honey bee (Apis mellifera L.) workers - Evidence for a regulatory module that involves vitellogenin expression but is independent of juvenile hormone function. Insect Biochem. Mol. Biol. 92, 21–29. doi: 10.1016/j.ibmb.2017.11.005
Dubrovskaya, V. A., Berger, E. M., and Dubrovsky, E. B. (2004). Juvenile hormone regulation of the E75 nuclear receptor is conserved in Diptera and Lepidoptera. Gene 340, 171–177. doi: 10.1016/j.gene.2004.07.022
Elango, N., Hunt, B. G., Goodisman, M. A., and Yi, S. V. (2009). DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc. Natl. Acad. Sci. U.S.A. 106, 11206–11211. doi: 10.1073/pnas.0900301106
Feng, H., Conneely, K. N., and Wu, H. (2014). A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 42:e69. doi: 10.1093/nar/gku154
Förster, F. (2018a). Bismark Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_bismark
Förster, F. (2018b). DESeq2 Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_deseq2
Förster, F. (2018c). DSS Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_dss
Förster, F. (2018d). Hisat2 Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_hisat2
Förster, F. (2018e). InterProScan Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_interproscan
Förster, F. (2018f). Ontologizer Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_ontologizer
Förster, F. (2018g). Samtools Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_samtools
Förster, F. (2018h). Stringtie Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/greatfireball/ime_stringtie
Förster, F. (2018i). TrimGalore Docker Image. Dockerhub. Available online at: https://hub.docker.com/r/imetools/trimgalore
Fujiwara, H., Jindra, M., Newitt, R., Palli, S. R., Hiruma, K., and Riddiford, L. M. (1995). Cloning of an ecdysone receptor homolog from Manduca sexta and the developmental profile of its mRNA in wings. Insect Biochem. Mol. Biol. 25, 845–856. doi: 10.1016/0965-1748(95)00023-O
Gegner, J., Baudach, A., Mukherjee, K., Halitschke, R., Vogel, H., and Vilcinskas, A. (2019). Epigenetic mechanisms are involved in sex-specific trans-generational immune priming in the lepidopteran model host Manduca sexta. Front. Physiol. 10:137. doi: 10.3389/fphys.2019.00137
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. D. (2019). Epigenetics in insects: genome regulation and the generation of phenotypic diversity. Annu. Rev. Entomol. 64, 185–203. doi: 10.1146/annurev-ento-011118-111914
Hiruma, K., Carter, M. S., and Riddiford, L. M. (1995). Characterization of the dopa decarboxylase gene of Manduca sexta and its suppression by 20-hydroxyecdysone. Dev. Biol. 169, 195–209. doi: 10.1006/dbio.1995.1137
Hussain, M., and Asgari, S. (2014). MicroRNAs as mediators of insect host-pathogen interactions and immunity. J. Insect Physiol. 70, 151–158. doi: 10.1016/j.jinsphys.2014.08.003
Jindra, M., Huang, J. Y., Malone, F., Asahina, M., and Riddiford, L. M. (1997). Identification and mRNA developmental profiles of two ultraspiracle isoforms in the epidermis and wings of Manduca sexta. Insect Mol. Biol. 6, 41–53. doi: 10.1046/j.1365-2583.1997.00153.x
Jindra, M., Malone, F., Hiruma, K., and Riddiford, L. M. (1996). Developmental profiles and ecdysteroid regulation of the mRNAs for two ecdysone receptor isoforms in the epidermis and wings of the tobacco hornworm, Manduca sexta. Dev. Biol. 180, 258–272. doi: 10.1006/dbio.1996.0299
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kall, L., Krogh, A., and Sonnhammer, E. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Kanost, M. R., Arrese, E. L., Cao, X., Chen, Y. R., Chellapilla, S., Goldsmith, M. R., et al. (2016a). Multifaceted biological insights from a draft genome sequence of the tobacco hornworm moth, Manduca sexta. Insect Biochem. Mol. Biol. 76, 118–147. doi: 10.1016/j.ibmb.2016.07.005
Kanost, M. R., Arrese, E. L., Cao, X., Chen, Y. R., Chellapilla, S., Goldsmith, M. R., et al. (2016b). Manduca sexta Genome Sequence. Available online at: https://i5k.nal.usda.gov/data/Arthropoda/mansex-%28Manduca_sexta%29/Current%20Genome%20Assembly/1.Genome%20Assembly/Manduca_sexta_v1.0/Scaffolds/
Kanost, M. R., Arrese, E. L., Cao, X., Chen, Y. R., Chellapilla, S., Goldsmith, M. R., et al. (2016c). Manduca sexta Genome Annotation. Available online at: https://i5k.nal.usda.gov/data/Arthropoda/mansex-(Manduca_sexta)/Current%20Genome%20Assembly/2.Official%20or%20Primary%20Gene%20Set/OGS2.0/ms_ogs_transcripts.fa
Khoa, D. B., and Takeda, M. (2012). Expression of autophagy 8 (Atg8) and its role in the midgut and other organs of the greater wax moth, Galleria mellonella, during metamorphic remodelling and under starvation. Insect Mol. Biol. 21, 473–487. doi: 10.1111/j.1365-2583.2012.01152.x
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Klose, R. J., and Bird, A. P. (2006). Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31, 89–97. doi: 10.1016/j.tibs.2005.12.008
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Krueger, F. (2012). Trim Galore: A Wrapper Tool Around Cutadapt and FastQC. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore
Krueger, F., and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. doi: 10.1093/bioinformatics/btr167
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Marks, P. A., Miller, T., and Richon, V. M. (2003). Histone deacetylases. Curr. Opin. Pharmacol. 3, 344–351. doi: 10.1016/S1471-4892(03)00084-5
Marletaz, F., Firbas, P. N., Maeso, I., Tena, J. J., Bogdanovic, O., Perry, M., et al. (2018). Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 564, 64–70. doi: 10.1038/s41586-018-0734-6
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
Mukherjee, K., Fischer, R., and Vilcinskas, A. (2012). Histone acetylation mediates epigenetic regulation of transcriptional reprogramming in insects during metamorphosis, wounding and infection. Front. Zool. 9:25. doi: 10.1186/1742-9994-9-25
Mukherjee, K., and Vilcinskas, A. (2014). Development and immunity-related microRNAs of the lepidopteran model host Galleria mellonella. BMC Genomics 15:705. doi: 10.1186/1471-2164-15-705
Palli, S. R., Hiruma, K., and Riddiford, L. M. (1992). An ecdysteroid-inducible Manduca gene similar to the Drosophila DHR3 gene, a member of the steroid hormone receptor superfamily. Dev. Biol. 150, 306–318. doi: 10.1016/0012-1606(92)90244-B
Park, Y., and Wu, H. (2016). Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 32, 1446–1453. doi: 10.1093/bioinformatics/btw026
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Provataris, P., Meusemann, K., Niehuis, O., Grath, S., and Misof, B. (2018). Signatures of DNA methylation across insects suggest reduced DNA methylation levels in holometabola. Genome Biol. Evol. 10, 1185–1197. doi: 10.1093/gbe/evy066
Qiagen Bioinformatics (2018). CLCWorkbench. Available online at: http://www.clcbio.com
R Core Team (2020). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna. Available online at: https://www.R-project.org/
Riviere, G., He, Y., Tecchio, S., Crowell, E., Gras, M., Sourdaine, P., et al. (2017). Dynamics of DNA methylomes underlie oyster development. PLoS Genet. 13:e1006807. doi: 10.1371/journal.pgen.1006807
Roy, A., and Palli, S. R. (2018). Epigenetic modifications acetylation and deacetylation play important roles in juvenile hormone action. BMC Genomics 19:934. doi: 10.1186/s12864-018-5323-4
Segraves, W. A., and Woldin, C. (1993). The E75 gene of Manduca sexta and comparison with its Drosophila homolog. Insect Biochem Mol. Biol. 23, 91–97. doi: 10.1016/0965-1748(93)90086-8
Song, X., Huang, F., Liu, J., Li, C., Gao, S., Wu, W., et al. (2017). Genome-wide DNA methylomes from discrete developmental stages reveal the predominance of non-CpG methylation in Tribolium castaneum. DNA Res. 24, 445–457. doi: 10.1093/dnares/dsx016
Sonnhammer, E. L., von Heijne, G., and Krogh, A. (1998). A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182.
Stilwell, G. E., Nelson, C. A., Weller, J., Cui, H., Hiruma, K., Truman, J. W., et al. (2003). E74 exhibits stage-specific hormonal regulation in the epidermis of the tobacco hornworm, Manduca sexta. Dev. Biol. 258, 76–90. doi: 10.1016/S0012-1606(03)00105-2
Tange, O. (2011). GNU parallel - the command-line power tool. USENIX Magaz. 36, 42–47. doi: 10.5281/zenodo.16303
Tetreau, G., Cao, X., Chen, Y. R., Muthukrishnan, S., Jiang, H., Blissard, G. W., et al. (2015a). Overview of chitin metabolism enzymes in Manduca sexta: identification, domain organization, phylogenetic analysis and gene expression. Insect Biochem. Mol. Biol. 62, 114–126. doi: 10.1016/j.ibmb.2015.01.006
Tetreau, G., Dittmer, N. T., Cao, X., Agrawal, S., Chen, Y. R., Muthukrishnan, S., et al. (2015b). Analysis of chitin-binding proteins from Manduca sexta provides new insights into evolution of peritrophin A-type chitin-binding domains in insects. Insect Biochem. Mol. Biol. 62, 127–141. doi: 10.1016/j.ibmb.2014.12.002
The Gene Ontology Consortium (2018). Available online at: http://geneontology.org/page/download-ontology
The Gene Ontology Consortium (2019). The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. doi: 10.1093/nar/gky1055
Wang, X., Wheeler, D., Avery, A., Rago, A., Choi, J. H., Colbourne, J. K., et al. (2013). Function and evolution of DNA methylation in Nasonia vitripennis. PLoS Genet. 9:e1003872. doi: 10.1371/journal.pgen.1003872
Weller, J., Sun, G. C., Zhou, B., Lan, Q., Hiruma, K., and Riddiford, L. M. (2001). Isolation and developmental expression of two nuclear receptors, MHR4 and betaFTZ-F1, in the tobacco hornworm, Manduca sexta. Insect Biochem. Mol. Biol. 31, 827–837. doi: 10.1016/S0965-1748(00)00188-0
Wu, H., Wang, C., and Wu, Z. (2013). A new shrinkage estimator for dispersion improves differential expression detection in RNA-seq data. Biostatistics 14, 232–243. doi: 10.1093/biostatistics/kxs033
Wu, H., Xu, T., Feng, H., Chen, L., Li, B., Yao, B., et al. (2015). Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 43:e141. doi: 10.1093/nar/gkv715
Xu, G., Zhang, J., Lyu, H., Song, Q., Feng, Q., Xiang, H., et al. (2018). DNA methylation mediates BmDeaf1-regulated tissue- and stage-specific expression of BmCHSA-2b in the silkworm, Bombyx mori. Epigenet. Chromatin 11:32. doi: 10.1186/s13072-018-0202-4
Ylla, G., Piulachs, M. D., and Belles, X. (2017). Comparative analysis of miRNA expression during the development of insects of different metamorphosis modes and germ-band types. BMC Genomics 18:774. doi: 10.1186/s12864-017-4177-5
Zhang, J., Xing, Y., Li, Y., Yin, C., Ge, C., and Li, F. (2015). DNA methyltransferases have an essential role in female fecundity in brown planthopper, Nilaparvata lugens. Biochem. Biophys. Res. Commun. 464, 83–88. doi: 10.1016/j.bbrc.2015.05.114
Zhou, B., Hiruma, K., Jindra, M., Shinoda, T., Segraves, W. A., Malone, F., et al. (1998a). Regulation of the transcription factor E75 by 20-hydroxyecdysone and juvenile hormone in the epidermis of the tobacco hornworm, Manduca sexta, during larval molting and metamorphosis. Dev. Biol. 193, 127–138. doi: 10.1006/dbio.1997.8798
Keywords: Manduca sexta, metamorphosis, epigenetics, DNA-methylation, differential gene expression, development
Citation: Gegner J, Vogel H, Billion A, Förster F and Vilcinskas A (2021) Complete Metamorphosis in Manduca sexta Involves Specific Changes in DNA Methylation Patterns. Front. Ecol. Evol. 9:646281. doi: 10.3389/fevo.2021.646281
Received: 25 December 2020; Accepted: 08 February 2021;
Published: 11 March 2021.
Edited by:
Nico Posnien, University of Göttingen, GermanyReviewed by:
Michael Kanost, Kansas State University, United StatesYuichiro Suzuki, Wellesley College, United States
Copyright © 2021 Gegner, Vogel, Billion, Förster and Vilcinskas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Vilcinskas, andreas.vilcinskas@agrar.uni-giessen.de; Frank Förster, frank.foerster@computational.bio.uni-giessen.de