 Xiong Qing
Xiong Qing Kasem Kulkeaw
Kasem Kulkeaw Sirichit Wongkamchai
Sirichit Wongkamchai Stephen Kwok-Wing Tsui
Stephen Kwok-Wing Tsui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 02 February 2021
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.637805
Lymphatic filariasis is a neglected parasitic disease that is a leading cause of long-term disability. Information obtained from genome sequencing of filarial worm can help us identify systems in the worm that are likely to be useful for novel drug design. Brugia (B.) malayi is still the only lymphatic-dwelling filarial parasite with a nearly complete, fully annotated, and published genome. However, most previous studies were based on the FR3 strain of B. malayi, which originally was isolated from a human patient, and was adapted to the rodent model, then maintained in laboratories for more than 60 years. It is uncertain whether genetic variation exists, thus, sequencing of clinical isolates of lymphatic dwelling filarial parasites is a high priority. Here, we report for the first time the complete mitochondrial genome of B. malayi microfilariae from clinical isolate. Complete mitochondrial (mt) genome of the microfilariae isolated from a blood sample taken from a Thai subject living in Narathiwat Province, which is an endemic area of brugian filariasis, was assembled with sequencing reads obtained by Illumina sequencing. Gene annotation, phylogenetic analysis and single nucleotide polymorphism (SNP) were deployed. A complete 13,658-bp mt genome of B. malayi microfilaria was obtained, and it shows 68x coverage. Based on gene annotation, the mt genome consists of 12 protein-coding, two rRNA, and 23 tRNA genes. Phylogenetic analysis using all protein sequences of DNA sequences of mt genome or cytochrome c oxidase subunit I (COX1) revealed a close relationship among three lymphatic filariae (i.e., B. timori, zoonotic B. pahangi, and Wuchereria spp.). The SNPs in the COX1 gene can differentiate microfilariae of B. malayi in human from those found in canine. Furthermore, the number, order and transcription, and direction of B. malayi microfilariae mitochondrial genes were the same as those found in the FR3 strain of B. malayi. The comparison on mitochondrial genome of B. malayi could have important implications on the development of a new intervention or vaccine to treat or prevent this disease in endemic areas/regions around the world.
Lymphatic filariasis is an often-neglected tropical disease that adversely affects society and economics due to associated high morbidity, social stigmatization, and inability to work. In 2000, the World Health Organization launched the Global Programme to Eliminate Lymphatic Filariasis with the aim to eliminate lymphatic filariasis by 2020 (Turner et al., 2016). The primary pillar of this program is mass drug administration of diethylcarbamazine, albendazole, and ivermectin. However, antifilarial drug resistance has emerged (Schwab et al., 2005; Cobo, 2016), which presents a threat to the success of this treatment and elimination program. Genetic mutation of the parasite due to mass drug administration for several years is hypothesized to be an underlying cause of drug resistance (Schwab et al., 2005). To cope with drug resistance and to pave the way for the development of more effective drugs, data specific to the genetic variability of this parasite among different geographic areas are needed.
Mitochondrion, which is an intracellular organelle, functions in adenosine 5′-triphosphate (ATP) production for cellular respiration via oxidative phosphorylation and the citric acid cycle. In metazoans, including the Onchocercidae family, the mitochondria have their own genome, which is a small, circular, double-stranded DNA sequence with an average length of 15–20 kb (Boore, 1999). Metazoan mitochondrial DNA (mtDNA) typically encodes 37 genes, including 13 protein subunits of electron transport in oxidative phosphorylation, two rRNAs, and 22 tRNAs, which are for translation of proteins encoded by mtDNA. Recent complete mitochondrial genome sequencing of the filarial nematode Wuchereria bancrofti from isolates from three different geographic regions yielded evidence of a complex demographic history. Complete mt genome of W. bancrofti was performed via analysis of isolates from Papua New Guinea, India, and West Africa (Ramesh et al., 2012). A mitochondrial gene of the heme-dependent respiratory chain was reported to be a potential target for the development of a new anti-filarial drug (Strubing et al., 2010). For these reasons, mtDNA is useful for identifying drug targets, and for studying genetic variability in the same parasite from different regions.
Brugia malayi is still the only lymphatic-dwelling filarial parasite with a nearly complete, fully annotated, and published genome. Whole genome and mitochondrial genome of B. malayi are available; however, the database is derived from a laboratory strain of adult B. malayi FR3 (Ghedin et al., 2007). Since the FR3 strain has been maintained in Mongolian gerbil for many years, it is uncertain whether genetic variation exists. The sequencing of clinical isolates of lymphatic-dwelling filarial parasites is a high priority (McNulty et al., 2013). The present study reports for the first time the complete mitochondrial genome of microfilarial stage of B. malayi directly isolated from clinical blood sample of human subject living in an endemic area of nocturnally subperiodic B. malayi in Southern Thailand.
Microfilariae of B. malayi were isolated from blood samples from three Thai subjects (named BM2, BM3, and BM4) living in the Sugnai-Padee District of Narathiwat Province in Thailand, which is an endemic area of nocturnal subperiodic B. malayi, using a semi-automated microfluidic device developed by Phuakrod et al. (2019). DNA was extracted from microfilariae using a High Pure PCR Template Preparation Kit (Roche Diagnostics GmbH, Penzberg, Germany) according to the manufacturer's instructions. DNA concentration was determined using a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), after which the DNA was used as the template for sequencing.
Genomic DNA sample was sequenced to totally obtain 44.1 Gb data of Illumina short reads in three samples using Novaseq PE150 technology in 2 × 150 paired-end library with 500-bp insertion (Supplementary Table 2). After mapping to the host Homo sapiens genome (GRCh38.85) and removed by Bowtie2 v2.3.4.3 and Samtools v1.9 (Supplementary Table 2), 1.06-Gb remaining data from the BM2 sample were de novo assembled by SPAdes v3.11.0. The mitochondrial genome was de novo assembled into a single contig (named BM2 mitochondrion) and annotated by the online tool GeSeq, which combines Blat, Hmmer, Aragorn v1.2.38, Arwen v1.2.3, and tRNAscan-SE v2.0.5, and which is visualized by OGDRAW v1.3.1. Secondary structure of tRNA was predicted using ARWEN v1.2 (Laslett and Canback, 2008). Pairwise dot plot was created with the mitochondrial genome of B. malayi FR3 strain (accession no. AF538716) by Gepard 1.40. According to the dot plot, originally assembled BM2 mitochondrial genome sequence was adjusted into the final version as the mitochondrial genome of B. malayi FR3 strain.
All mitochondrial protein sequences and cytochrome c oxidase subunit I (COX1) gene sequences were downloaded from NCBI database. The mitochondrial protein sequences of B. malayi, B. timori, B. pahangi, W. bancrofti, Loa loa, Onchocera volvulus, Dirofilaria repens, D. immitis, O. ochengi, O. flexuosa, and Acanthocheilonema viteae were obtained. For the whole mitochondrial phylogeny (Figure 2), protein sequences of all 12 annotated mitochondrial genes were collected and aligned independently, and then multiple alignment results of 12 annotated mitochondrial proteins were merged for phylogenetic analysis. For the COX1-based phylogeny (Figure 3), DNA sequences of COX1 genes of B. malayi, B. timori, B. pahangi, and Gongylonema pulchrum were collected and aligned. MEGA v7.0.26 was used for multiple sequence alignment with Clustal W and phylogenetic analysis in Maximum Likelihood method with 90% site coverage and 100 bootstrap replicates, as well as for identification of single nucleotide polymorphism.
The complete mitochondrial sequence of B. malayi is available upon request to the corresponding author and was deposited in GenBank (accession no. MT149211). All sequencing data (removed human reads) are available in NCBI (BioProject ID: PRJNA613552).
The study's research protocol for the use of blood samples from human subjects was approved by the Human Research Protection Unit, Faculty of Medicine Siriraj Hospital, Mahidol University (COA no. Si459/2019).
De novo assembled circular mitochondrial genome of B. malayi has a 13,658-bp sequence and shows 68x coverage with no heterozygosity observed in the sample BM2 (Figure 1A) (hereafter referred to as BM2 mt). Regarding type of nucleotide composition, the BM2 mt genome has 24.48% GC content. BLASTn of de novo assembled BM2 mt showed 99.72% identity to that of B. malayi FR3 strain (accession no. AF538716). Genome annotation revealed 12 protein-coding, two rRNA, and 23 tRNA genes (Supplementary Table 1), and they are summarized in Figure 1B. Arrangement of the mitochondrial genes of BM2 mt was similar to that of B. malayi FR3 strain (Figures 1C,E). The extra and duplicated lysine (K) tRNA was annotated in BM2 mt (Figure 1F), while the DNA sequence of this lysine (K) tRNA also identically exists in the mitochondrial sequence of B. malayi FR3 strain, but did not annotated. Cyclization of the assembled mitochondrial sequences was confirmed in dot plot with the mitochondrion of FR3 strain (Figure 1D) and the final mitochondrial sequences were adjusted with the complementary strand and the same start site as the mitochondrion of FR3 strain. Among the annotated 23 tRNA genes, two leucine-tRNA (trnL) and two lysine-tRNA (trnK) genes were identified. There were two sets of trnL, including trnL-UAG and trnL-UAA. However, in contrast to the FR3 isolate, two sets of tRNA-lysine (trnK-CUU and trnK-UUU) were also observed in the BM2 mt. The trnK-UUU was predicted at position 4,561–4,615 of the BM2 mt genome which is a region that overlaps with cytochrome b (COB) (4,578–5,664), and the sequence of this trnK-UUU also identically existed in the mitochondrial genome of FR3 but not annotated. The 5′-end of COB gene overlapped with the 3′-end of trnK (Figure 1E; Supplementary Table 1). According to ARWEN, a tool for detection of tRNA in metazoan mitochondrial DNA, a D-loop structure of trnK-UUU was predicted (Figure 1F). Moreover, mitochondria-specific ATP synthase 6 was identified. According to the arrangement of genes in the mitochondrial genome, all annotated genes were predictably transcribed in the same direction of plus strand (Figure 1C). The region between COX3 and trnA-UGC is the AT-rich control region for replication initiation.
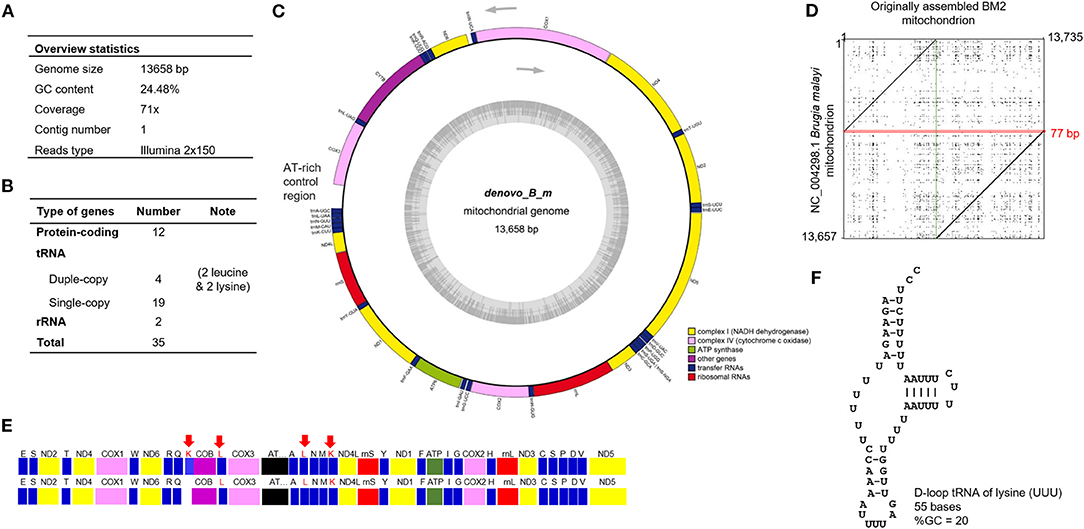
Figure 1. Whole mitochondrial genome of B. malayi isolated from clinical blood samples. (A) Overview of mitochondrial genome of B. malayi. (B) Summary of gene types annotated from mitochondrial genome of B. malayi. (C) A circular layout of mt genome visualized using GeSeq. (D) Dot plot with the sequences of originally assembled BM2 mitochondrion and the mitochondrion of FR3 strain (accession no. AF538716). Overlapped 77-bp sequences on the originally assembled BM2 mitochondrion were marked in the red square, while the junction site without any overlap of two sequences was marked with the vertical green line. (E) Illustration of genes arranged in mt genome of de novo assembled BM2 (upper) and AF538716 (lower). Each type of mt gene is illustrated as a colored box: tRNA (blue), cytochrome (violet), NADH dehydrogenase (yellow), cytochrome c oxidase (pink), ribosomal subunit RNA (red), and ATP synthase (green). Arrows indicate double copy of annotated leucine- and lysine-tRNA genes. (F) Secondary structure of predicted trnK-UUU using ARWEN.
Lymphatic filariasis that results from B. malayi, B. timori, and W. bancrofti causes high morbidity, followed by loasis and onchocerciasis (Zoure et al., 2011; Hotez et al., 2014). Therefore, we decided to compare the protein sequences of all 12 annotated mitochondrial genome of B. malayi with those of B. timori, B. pahangi, W. bancrofti, Loa loa, and Onchocera volvulus, as well as the zoonotic filarial nematodes B. pahangi, Dirofilaria repens, and D. immitis. To identify genetic variability within the Onchocercidae family, protein sequences of all 12 annotated mtDNA of O. ochengi (causative agent of cutaneous onchocerciasis in cattle), O. flexuosa (causative agent of onchocerciasis in deer), and Acanthocheilonema viteae (causative agent of rodent filariasis) were phylogenetically analyzed (Figure 2). The mtDNA of Gongylonema pulchrum, a parasitic nematode, was used as outgroup control.
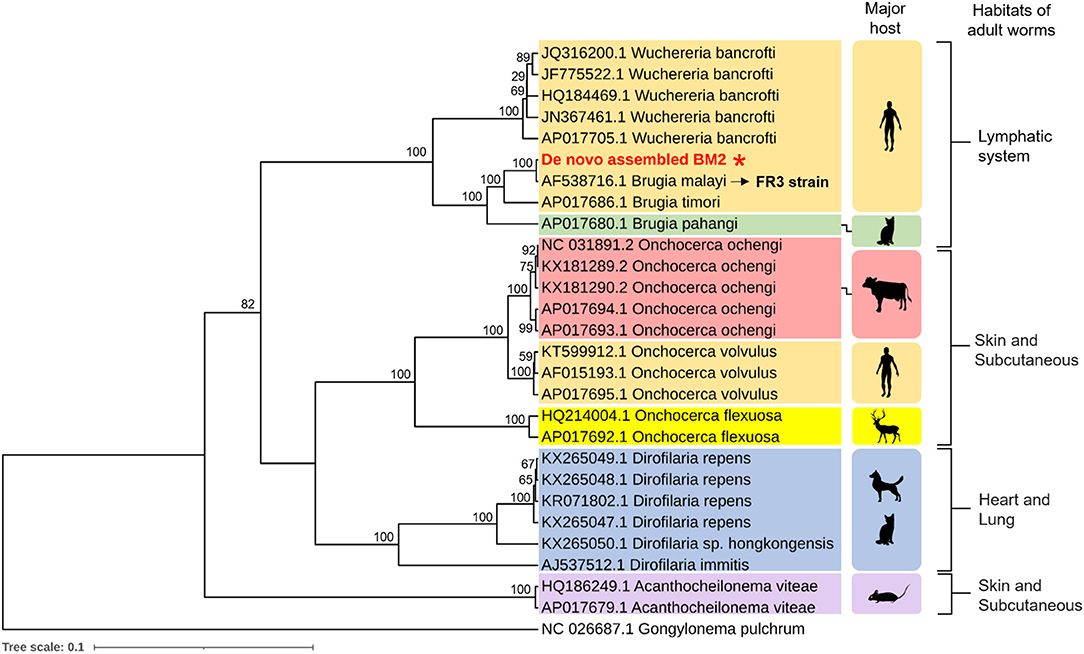
Figure 2. Phylogenetic analysis of mt genome. The protein sequences of all 12 annotated mitochondrial genome of causative agents of human filariasis (B. malayi, B. timori, B. pahangi, W. bancrofti, Loa loa, and Onchocera volvulus), zoonotic filariasis (B. pahangi, Dirofilaria repens, and D. immitis), and cutaneous onchocerciasis in cattle (O. ochengi), onchocerciasis in deer (O. flexuosa), and rodent filariasis (Acanthocheilonema viteae). Major hosts and their habitats of adult worms within the host are indicated on right hand side. The protein sequences of all 12 annotated mt genome of de novo assembled BM2 was compared to that of B. malayi FR3 strain (accession no. AF538716). The mt genome of Gongylonema pulchrum, a free-living nematode, was used as outgroup control.
Mitochondrial genome-based phylogenetic analysis categorized the aforementioned parasite species in a biological niche-specific manner. Depending on their habitat of adult worm within the host, there were three major groups, as follows: (1) lymphatic system, (2) skin and subcutaneous, and (3) heart and lung. Phylogenetic tree of the BM2 mt genome was in a clade of lymphatic-dwelling parasites, and was found to be most closely related to the B. malayi FR3 strain (accession no. AF538716). Moreover, next to B. timori, B. pahangi, which is a filarial nematode that is naturally found in cats and it causes human lymphatic filariasis, is proximate to B. malayi of human isolate in this study (Figure 2).
Using phylogenetic analysis to identify a potential source of human Brugian filariasis, we set forth to compare the mt genome sequences of Brugia isolates in Thailand. However, given the unavailability of the mt genome sequence of Thai isolates, we used the COX1 gene for phylogenetic analysis. As shown in Figure 3, COX1-based phylogenetic analysis showed a similar result, in which BM2 isolate is closely related to the FR3 laboratory strain. Interestingly, several isolates of B. malayi that were collected from domestic dogs in Bangkok, Thailand were found to be close to the human isolate (BM2) identified in the blood of a man living in Narathiwat Province, Thailand, which suggests a potential reservoir host.
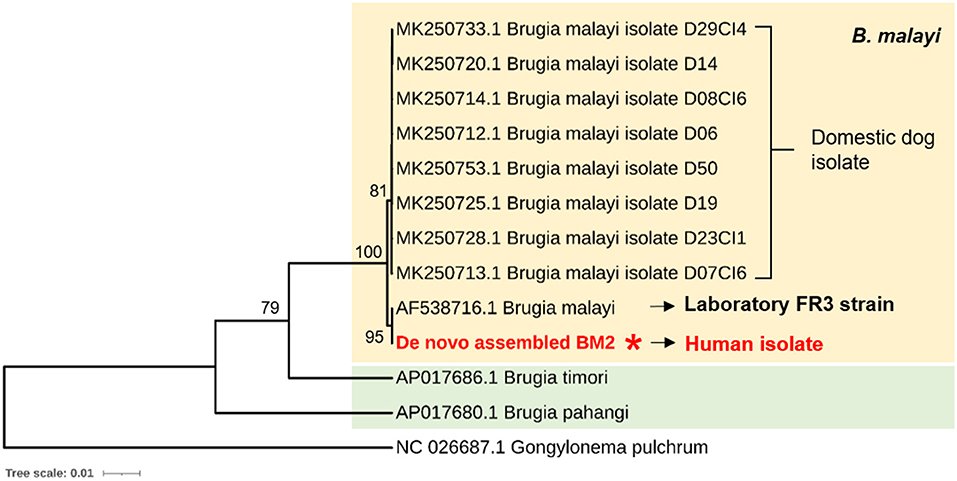
Figure 3. Cytochrome c oxidase subunit I (COX1)-based phylogenetic analysis to identify potential sources of human brugian filariasis. COX1 genes were obtained from two human isolates; one from Thailand (de novo assembled BM2 mt) and another from FR3 strain, and eight isolates of B. malayi collected from domestic dogs in Bangkok, Thailand. The COX1 gene of Gongylonema pulchrum, a free-living nematode, was used as outgroup control.
To examine type and codon alteration of the SNPs identified in the assembled COX1 gene, we compared the DNA sequence of the COX1 gene in this study with that of the FR3. There were six SNPs altered in at least two isolates and identified, as follow: C2511T, C2514T, T2637C, T2799C, G3000A, and C3130T (Figure 4A). None of SNPs had altered codons of isoleucine, glycine, phenylalanine, arginine, or leucine. Since COX1 gene sequences of canine B. malayi microfilaria were available, and all were isolated from domestic dogs in Thailand, we aligned the DNA sequences of the COX1 genes of human isolates in this study with those of the FR3 strain and canine B. malayi microfilaria using Clustal Omega. As shown in Figure 4B, we observed with following three SNPs, which are specific to human isolates: C2511T, C2514T, and C3130T.
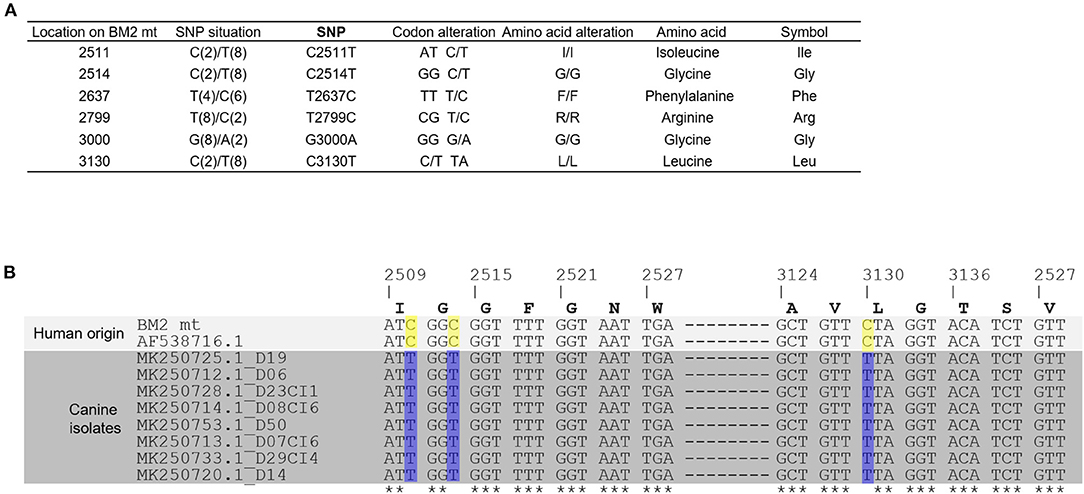
Figure 4. Single nucleotide polymorphism of COX1. COX1 genes of B. malayi were obtained from FR3 strain and from those isolated in dogs (D19, D06, D23CI1, D08CI6, D50, D07CI6, D50, D07CI6, D29CI4, and D14). (A) Types and locations of SNPs in the assembled mt DNA sequence. Six SNP sites were presented in which each nucleotide type was supported by at least two sequences, while other SNP sites caused by only one sequences were listed in the Supplementary Table 3. No SNP alterations caused a change in the type of amino acid. (B) COX1 genes of BM2 mt and other nine isolates were subjected to multiple sequence alignment using CLUSTAL OMEGA. The locations of the C/T SNPs located at 2,511, 2,514, and 3,130 were able to separate 10 B. malayi isolates into two types: human or canine origin.
Totally, 38 nucleotides are not identical between mitochondrial genomes of de novo assembled BM2 mt and the FR3 strain and cause amino acid changes in nine proteins. The amino acid sequences of COX1 of BM2 mt and AAN17806 derived from the FR3 strain (AF538716) were subjected to Phyre2 for modeling protein structure. The C1M56G, a cytochrome c oxidase of the gram-negative facultative photosynthetic bacterium Rhodobacter sphaeroides, was matched, and it ranked number 1 with both the BM2 mt and AAN17806 sequences. The matches were ranked based on the number of aligned residues and the quality of alignment (similarity of residue probability distributions for each position), secondary structure similarity, and the presence or absence of insertions and deletions. Overall, the 513 residues (93% of the sequence) of COX1 of BM2 mt were modeled with 100.0% confidence by the single highest scoring template. Thus, the secondary structure of C1M56G was used as a template for protein structure prediction. Despite a serial of amino acid changes (Supplementary File 1a, boxed), secondary structure of the protein remains unchanged (Supplementary File 1b). Given amino acid changes detected in other mitochondrial proteins (NDL4, COB, ND1, ND5, ND4, COX3, ND2, and ATP6), none of them predictively altered the secondary structure of protein (Supplementary File 2). Although there were amino acid changes in ND6 of BM2 mt; however, there were only fragments of the secondary structures of the deposited database matched with ND6, prohibiting prediction of alteration of ND6 protein structure (Supplementary File 3).
To our knowledge, this study reports the first complete mitochondrial genome of microfilarial stage of B. malayi isolated from a clinical isolate obtained from a Thai subject that lives in Southern Thailand. Major hurdle in genome sequencing of human-isolated B. malayi is an insufficient number of microfilariae circulating in blood of human host. Moreover, in an endemic area microfilaria nocturnally circulate in blood in a subperiodic manner, making the isolation of microfilaria-containing blood a difficult task. With many efforts and the recent advance in next generation sequencing, the complete mitochondrial genome of a B. malayi strain in the human host were successfully obtained at a high accuracy.
Mitochondrial genome sequences are widely used as molecular markers for phylogenetic studies. Mitochondrial genome sequencing is often preferred to whole genome sequencing. Given the rapid evolution of mitochondrial genome in animals compared to nuclear genome, the mitochondrial genome is useful for study of genetic variability and phylogenetic relation (Joy et al., 2006; Hassanin et al., 2013; Tyagi and Das, 2015; Yilmaz et al., 2016, 2019). Second, the shorter length of the mitochondrial genome makes the sequencing process easier. Third, since the adult worm habitat is in the lymphatic system of the human host, collection of adult worms is difficult and requires an invasive biopsy of tissue. Fourth, phylogenetic analysis in this study showed that B. timori, a filarial nematode causing human lymphatic filariasis, is proximate to B. malayi from human isolate. Thus, the use of mt genome for phylogenetic analysis is comparable to that of whole genome. Moreover, a mitochondrial gene of heme-dependent respiratory chain was reported to be a potential target for the development of new drugs (Strubing et al., 2010). Thus, anti-microfilaricidal drug targeting mitochondrion remains an effective therapy.
Among three DNA samples, we could obtain most reads from one sample (BM2). The main reasons for failure include: (1) the number of microfilariae in the blood vary depending on host and time of blood collection; and, (2) the integrity and purity of DNA samples. Although the success rate was low, the accuracy of the mtDNA sequence was higher than that of W. bancrofti microfilaria (Ramesh et al., 2012), as indicated by 68x coverage. The high accuracy of the mtDNA allowed us to construct a map of the mitochondrial genome and identify gene duplication and a unique arrangement of mitochondrial genes, which is information that was lacking in the study of W. bancrofti microfilariae.
Like nuclear genome-based phylogenetic analysis, mt genome of BM2 B. malayi is closely related to that of B. timori and zoonotic B. pahangi (Lau et al., 2015). Given that the genome of O. volvulus is available, phylogenetic tree of mt genome of B. malayi resembled that of O. volvulus (Cotton et al., 2016). In agreement with the previous analysis of Lefoulon et al., our mt genome-based phylogenetic tree was supported for the clades of: (1) Wucheriria spp. and Brugia spp., (2) Onchocerca spp., and (3) Dirofilaria spp. (Lefoulon et al., 2015). Moreover, similar to other animal mitochondrial genome, mitochondria-specific ATP synthase 6 gene was identified in this study. Collectively, our analyses of the mt genome are valid and they yielded a similar result to those of the mt genomes of B. timori and zoonotic B. pahangi.
The nucleotide sequences of the mitochondrial DNA (mtDNA) molecules of two nematodes, Caenorhabditis elegans (13,794 bp) and Ascaris suum (14,284 bp), were previously reported. However, ATPase subunit 8, which is common to other metazoan mtDNA, has not been identified in the mtDNA of either of these nematodes (Okimoto et al., 1992). By using Translation Table 5 (The invertebrate mitochondrial code), B. malayi mt-genetic codes are similar to C. elegans and A. suum mt-genetic codes. The O. volvulus mitochondrial genome lacks an open reading frame encoding ATPase subunit 8 (Keddie et al., 1998), a similar finding to our study. Moreover, overlap of trnK gene with adjacent COB genes and non-overlapped NAD1 and trnF gene were in agreement with annotation of W. bancrofti mt genome. By contrast, the NAD1 and trnF gene were predicted to overlap in the mt genomes of D. immitis, O. volvulus, and S. digitata (Ramesh et al., 2012).
The first complete mitochondrial genome sequence of B. malayi is available in 2007 (Ghedin et al., 2007) and the Brugia nematode was a human-derived FR3 strain that maintained in a Mongolian gerbil (Meriones unguiculatus). To our knowledge, this is the first report of complete mitochondrial genome of microfilarial B. malayi isolated from peripheral blood of human subject in endemic area. Phylogenetic tree of the BM2 mt genome sequence was closely related to the B. malayi FR3 strain (accession no. AF538716), implying accuracy of sequencing. Although, the FR3 strain has been maintained in animals since first collection from human in Kuala Lumpur, Malaysia in 1950s/early 1960s (Michalski et al., 2011), mt genome seem to be conserved and has no significant genetic variation.
Apart from vector control strategies, the infection control of reservoir hosts is necessary for more effective filariasis control. Thus, identification of the parasite reservoir host is importance. Ambily et al. firstly reported the B. malayi infection in dogs using Giemsa and histochemical staining, PCR and DNA sequencing. The authors emphasized the possible role of dogs in transmission of human filariasis (Ambily et al., 2011). In contrary, Ravindra et al., used 5.8S-ITS2-28S of rDNA gene to confirm suspected B. malayi-like parasite in the dog blood, and found that the parasite was genetically closer to B. pahangi (dogs) than to B. malayi (human). Thus, they did not find the potential of dogs as reservoirs of B. malayi (Ravindran et al., 2014). Recently, Satjawongvanit et al., using cytochrome c oxidase subunit I (COI) and internal transcribed spacer 1 (ITSI) gene-based PCR to detect filarial nematode DNA in blood collected from domestic dogs from the Bangkok Metropolitan Region in Thailand. Of the 57 blood samples, the COI and ITSI genes of 50 (87.72%) samples were amplified. Based on the partial nucleotide sequences of the COI gene (~690 bp), they classified filarial nematodes detected in the domestic dogs into three species: D. immitis (57.89%), B. pahangi (22.81%), and B. malayi (7.02%). The study also suggests that COI gene is a useful marker for distinguishing between D. immitis, B. pahangi, and B. malayi, whereas use of the ITSI gene was able to detect only D. immitis and B. pahangi. Therefore, the COI-based PCR is suitable tool for the detection of filarial nematode infections in dog (Satjawongvanit et al., 2019).
Given the unavailability of the mt genome sequence of Brugia isolates in Thailand, we set forth to compare cytochrome c oxidase subunit I or COX1 gene using phylogenetic analysis in order to identify a potential source of human brugian filariasis (Figure 3). Interestingly, several isolates of B. malayi collected from domestic dogs in Bangkok, Thailand (Satjawongvanit et al., 2019) were found to be close to the human isolate (BM2) identified in the blood of a man living in Narathiwat Province, Thailand, implying a potential reservoir host of domestic dogs. For lymphatic filariasis, the presence of B. malayi in dogs has been suspected for a long time; however, rare reports of the B. malayi infection in dogs were available. Our data is an evidence of the potential of a dog as B. malayi reservoir host. Nevertheless, further field study in human and domestic dogs is required prior to conclude the reservoir ability of dogs in endemic area.
In conclusion, the complete mitochondrial genome of microfilariae from clinical isolates was obtained in this study. To our knowledge, this report is the first to show complete mitochondrial genome of microfilariae isolated from clinical isolates. The number, order and transcription, and direction of B. malayi microfilariae mitochondrial genes were the same as those in the FR3 strain, which is B. malayi that has been adapted to the rodent model and maintaining laboratories for a long period of time.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, MT149211; https://www.ncbi.nlm.nih.gov/, PRJNA613552.
The studies involving human participants were reviewed and approved by Human Research Protection Unit, Faculty of Medicine Siriraj Hospital, Mahidol University (COA no. Si459/2019). The patients/participants provided their written informed consent to participate in this study.
XQ and KK preformed the experiments, analyzed and interpreted the data, and wrote the manuscript. SW and ST designed the study, analyzed and interpreted the data, and wrote the manuscript. All authors read and approved the final manuscript.
This work was supported by a grant from the Siriraj Research Fund, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand (Grant number R01593600), and by a Health and Medical Research Fund from the Food and Health Bureau in Hong Kong (Project number 06171016) and a General Research Fund from the Research Grant Council (Project number 14119219) in Hong Kong. The Research Career Development Grant from Thailand Science Research and Innovation financially supports KK (Funding number RSA6280102).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge the corresponding colleagues in the Mahidol University for the collection of blood samples from the patients. We thank Miss Sumart Loymek and the staff of Phikhunthong Royal Project, Narathiwat Province and Miss Achinya Phuakrod and Miss Bungoen Sermsart, Department of Parasitology, Faculty of Medicine Siriraj Hospital, Mahidol University for their assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.637805/full#supplementary-material
Supplementary File 1. Comparison of amino acid sequences of COX1 of the de novo assembled BM2 and the FR3 strain. (a) Alignment of the predicted amino acid sequences of COX1 obtained from FR3 strain (AAN17806) and the de novo assembled BM2 mt. Amino acid changes are in the blue boxes. (b) Prediction of secondary structure was performed using Phyre2. The models constructed for COX1 of two isolates were based on the C1M56G template. The position of the amino acids and the predicted secondary structure are indicated on the first and second lines, respectively. Amino acid residues are colored according to the biochemical property of the side chain of the amino acid, as follows: A,S,T,G,P - small/polar (orange); M,I,L,V - hydrophobic (green); K,R,E,N,D,H,Q - charged (red); and, W,Y,F,C - aromatic + cysteine (blue). The predicted α-helix is in green color. Changes of amino acids are indicated in the red boxes.
Supplementary File 2. Multiple sequence alignment (a) and predicted protein structure (b) of NDL4, COB, ND1, ND5, ND4, COX3, ND2, and ATP6 using CLUSTAL 2.1 and Phyre2, respectively. In Phyre2, the predicted amino acid sequences of the de novo BM2 mt are subjected to matching with the others amino acid sequences, which their protein structures are known. The matches were ranked according to the number of aligned residues and the quality of alignment (similarity of residue probability distributions for each position), secondary structure similarity, and the presence or absence of insertions and deletions. Those at the top rank are selected for examining any altered secondary structure of protein.
Supplementary File 3. Predicted protein structure of ND6 using Phyre2. Only fragments of the secondary structures of the deposited database are matched with ND6; thus, prediction of alteration of ND6 protein structure is not possible.
Ambily, V. R., Pillai, U. N., Arun, R., Pramod, S., and Jayakumar, K. M. (2011). Detection of human filarial parasite Brugia malayi in dogs by histochemical staining and molecular techniques. Vet. Parasitol. 181, 210–214. doi: 10.1016/j.vetpar.2011.04.041
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Cobo, F. (2016). Determinants of parasite drug resistance in human lymphatic filariasis. Rev. Esp. Quimioter 29, 288–295.
Cotton, J. A., Bennuru, S., Grote, A., Harsha, B., Tracey, A., Beech, R., et al. (2016). The genome of Onchocerca volvulus, agent of river blindness. Nat. Microbiol. 2:16216. doi: 10.1038/nmicrobiol.2016.216
Ghedin, E., Wang, S., Spiro, D., Caler, E., Zhao, Q., Crabtree, J., et al. (2007). Draft genome of the filarial nematode parasite Brugia malayi. Science 317, 1756–1760. doi: 10.1126/science.1145406
Hassanin, A., An, J., Ropiquet, A., Nguyen, T. T., and Couloux, A. (2013). Combining multiple autosomal introns for studying shallow phylogeny and taxonomy of Laurasiatherian mammals: Application to the tribe Bovini (Cetartiodactyla, Bovidae). Mol. Phylogenet Evol. 66, 766–775. doi: 10.1016/j.ympev.2012.11.003
Hotez, P. J., Alvarado, M., Basanez, M. G., Bolliger, I., Bourne, R., Boussinesq, M., et al. (2014). The global burden of disease study 2010: interpretation and implications for the neglected tropical diseases. PLoS Negl. Trop. Dis. 8:e2865. doi: 10.1371/journal.pntd.0002865
Joy, D. A., Mu, J., Jiang, H., and Su, X. (2006). Genetic diversity and population history of Plasmodium falciparum and Plasmodium vivax. Parasitol. 48, 561–566.
Keddie, E. M., Higazi, T., and Unnasch, T. R. (1998). The mitochondrial genome of Onchocerca volvulus: sequence, structure and phylogenetic analysis. Mol. Biochem. Parasitol. 95, 111–127. doi: 10.1016/S0166-6851(98)00102-9
Laslett, D., and Canback, B. (2008). ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. doi: 10.1093/bioinformatics/btm573
Lau, Y. L., Lee, W. C., Xia, J., Zhang, G., Razali, R., Anwar, A., et al. (2015). Draft genome of Brugia pahangi: high similarity between B. pahangi and B. malayi. Parasit. Vectors 8:451. doi: 10.1186/s13071-015-1064-2
Lefoulon, E., Bain, O., Bourret, J., Junker, K., Guerrero, R., Canizales, I., et al. (2015). Shaking the tree: multi-locus sequence typing usurps current onchocercid (filarial nematode) phylogeny. PLoS Negl. Trop. Dis. 9:e0004233. doi: 10.1371/journal.pntd.0004233
McNulty, S. N., Mitreva, M., Weil, G. J., and Fischer, P. U. (2013). Inter and intra-specific diversity of parasites that cause lymphatic filariasis. Infect. Genet. Evol. 14, 137–146. doi: 10.1016/j.meegid.2012.11.002
Michalski, M. L., Griffiths, K. G., Williams, S. A., Kaplan, R. M., and Moorhead, A. R. (2011). The NIH-NIAID Filariasis Research Reagent Resource Center. PLoS Negl. Trop. Dis. 5:e1261. doi: 10.1371/journal.pntd.0001261
Okimoto, R., Macfarlane, J. L., Clary, D. O., and Wolstenholme, D. R. (1992). The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics 130, 471–498. doi: 10.1093/genetics/130.3.471
Phuakrod, A., Sripumkhai, W., Jeamsaksiri, W., Pattamang, P., Juntasaro, E., Thienthong, T., et al. (2019). Diagnosis of feline filariasis assisted by a novel semi-automated microfluidic device in combination with high resolution melting real-time PCR. Parasit Vectors 12:159. doi: 10.1186/s13071-019-3421-z
Ramesh, A., Small, S. T., Kloos, Z. A., Kazura, J. W., Nutman, T. B., Serre, D., et al. (2012). The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol. Biochem. Parasitol. 183, 32–41. doi: 10.1016/j.molbiopara.2012.01.004
Ravindran, R., Varghese, S., Nair, S. N., Balan, V. M., Lakshmanan, B., Ashruf, R. M., et al. (2014). Canine filarial infections in a human Brugia malayi endemic area of India. Biomed. Res. Int. 2014:630160. doi: 10.1155/2014/630160
Satjawongvanit, H., Phumee, A., Tiawsirisup, S., Sungpradit, S., Brownell, N., Siriyasatien, P., et al. (2019). Molecular analysis of canine filaria and its wolbachia endosymbionts in domestic dogs collected from two animal university hospitals in Bangkok metropolitan region, Thailand. Pathogens 8:114. doi: 10.3390/pathogens8030114
Schwab, A. E., Boakye, D. A., Kyelem, D., and Prichard, R. K. (2005). Detection of benzimidazole resistance-associated mutations in the filarial nematode Wuchereria bancrofti and evidence for selection by albendazole and ivermectin combination treatment. Am. J. Trop. Med. Hyg. 73, 234–238. doi: 10.4269/ajtmh.2005.73.234
Strubing, U., Lucius, R., Hoerauf, A., and Pfarr, K. M. (2010). Mitochondrial genes for heme-dependent respiratory chain complexes are up-regulated after depletion of Wolbachia from filarial nematodes. Int. J. Parasitol. 40, 1193–1202. doi: 10.1016/j.ijpara.2010.03.004
Turner, H. C., Bettis, A. A., Chu, B. K., McFarland, D. A., Hooper, P. J., Ottesen, E. A., et al. (2016). The health and economic benefits of the global programme to eliminate lymphatic filariasis (2000-2014). Infect. Dis. Poverty 5:54. doi: 10.1186/s40249-016-0147-4
Tyagi, S., and Das, A. (2015). Mitochondrial population genomic analyses reveal population structure and demography of Indian Plasmodium falciparum. Mitochondrion 24, 9–21. doi: 10.1016/j.mito.2015.06.003
Yilmaz, E., Fritzenwanker, M., Pantchev, N., Lendner, M., Wongkamchai, S., Otranto, D., et al. (2016). The mitochondrial genomes of the zoonotic canine filarial parasites dirofilaria (Nochtiella) repens and candidatus dirofilaria (Nochtiella) honkongensis provide evidence for presence of cryptic species. PLoS Negl. Trop. Dis. 10:e0005028. doi: 10.1371/journal.pntd.0005028
Yilmaz, E., Wongkamchai, S., Ramunke, S., Koutsovoulos, G. D., Blaxter, M. L., Poppert, S., et al. (2019). High genetic diversity in the Dirofilaria repens species complex revealed by mitochondrial genomes of feline microfilaria samples from Narathiwat, Thailand. Transbound. Emerg. Dis. 66, 389–399. doi: 10.1111/tbed.13033
Zoure, H. G., Wanji, S., Noma, M., Amazigo, U. V., Diggle, P. J., Tekle, A. H., et al. (2011). The geographic distribution of Loa loa in Africa: results of large-scale implementation of the Rapid Assessment Procedure for Loiasis (RAPLOA). PLoS Negl. Trop. Dis. 5:e1210. doi: 10.1371/journal.pntd.0001210
Keywords: Brugia malayi, microfilaria, mitochondrial genome (mtDNA), phylogeny, filariasis
Citation: Qing X, Kulkeaw K, Wongkamchai S and Tsui SK-W (2021) Mitochondrial Genome of Brugia malayi Microfilariae Isolated From a Clinical Sample. Front. Ecol. Evol. 9:637805. doi: 10.3389/fevo.2021.637805
Received: 04 December 2020; Accepted: 12 January 2021;
Published: 02 February 2021.
Edited by:
Joong-Ki Park, Ewha Womans University, South KoreaReviewed by:
Steve Nadler, UC Davis Department of Entomology and Nematology, United StatesCopyright © 2021 Qing, Kulkeaw, Wongkamchai and Tsui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen Kwok-Wing Tsui, a3d0c3VpQGN1aGsuZWR1Lmhr; Sirichit Wongkamchai, c2lyaWNoaXQud29uQG1haGlkb2wuYWMudGg=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.