 Heather R. Kates1*
Heather R. Kates1* Fernando López Anido2
Fernando López Anido2 Guillermo Sánchez-de la Vega3
Guillermo Sánchez-de la Vega3 Luis E. Eguiarte3
Luis E. Eguiarte3 Pamela S. Soltis1
Pamela S. Soltis1 Douglas E. Soltis1,4
Douglas E. Soltis1,4- 1Florida Museum of Natural History, University of Florida, Gainesville, FL, United States
- 2Facultad de Ciencias Agrarias, Universidad Nacional de Rosario-IICAR CONICET, Rosario, Argentina
- 3Departamento de Ecología Evolutiva, Instituto de Ecología, Universidad Nacional Autónoma de México, Mexico City, Mexico
- 4Department of Biology, University of Florida, Gainesville, FL, United States
Studies of domestication genetics enrich our understanding of how domestication shapes genetic and morphological diversity. We characterized patterns of genetic variation in two independently domesticated pumpkins and their wild progenitors to assess and compare genetic consequences of domestication. To compare genetic diversity pre- and post-domestication and to identify genes targeted by selection during domestication, we analyzed ∼15,000 SNPs of 48 unrelated accessions, including wild, landrace, and improved lines for each of two pumpkin species, Cucurbita argyrosperma and Cucurbita maxima. Genetic diversity relative to its wild progenitor was reduced in only one domesticated subspecies, C. argyrosperma ssp. argyrosperma. The two species have different patterns of genetic structure across domestication status. Only 1.5% of the domestication features identified for both species were shared between species. These findings suggest that ancestral genetic diversity, wild-crop gene flow, and domestication practices shaped the genetic diversity of two similar Cucurbita crops in different ways, adding to our understanding of how genetic diversity changes during the processes of domestication and how trait improvement impacts the breeding potential of modern crops.
Introduction
Plant domestication has produced hundreds of crop species that differ dramatically from their wild ancestors, both genetically and phenotypically (Meyer and Purugganan, 2013). The differences between wild and domesticated plants result from broad evolutionary changes that include selection associated with crops’ coevolution with human domesticators and the demographic processes that accompany domestication, including population bottlenecks, genetic drift, and introgression with wild relatives. Because of the recent and severe genetic bottleneck that often accompanies domestication, domesticated plants typically possess only a subset of the genetic diversity present in their wild ancestors (e.g., Meyer and Purugganan, 2013). Genetic diversity in the initial domesticate is often further reduced by modern breeding (Meyer and Purugganan, 2013). Unlike modern improved lines produced by modern breeding (hereafter referred to as “improved”), landrace varieties (sometimes referred to as “folk” or “primitive” varieties, but hereafter referred to as “landrace”) are local varieties typically developed by small-scale farmers in traditional agricultural systems over hundreds of years (Villa et al., 2005). Landraces are often highly variable as they continue to evolve within a defined ecogeographical area under the influence of local human culture (Casañas et al., 2017). The loss of a crop’s genetic diversity through modern breeding is increasingly alarming as breeders cannot access genetic diversity underlying traits such as disease resistance and drought tolerance needed to respond to pressures of climate change and human population growth (Esquinas-Alcázar, 2005).
Despite the importance of genetic diversity for crop improvement, much of what we know about how the domestication process shapes the genetic diversity of crops comes from studies of just one branch of the plant tree of life, cereals (e.g., rice: Zhu et al., 2007; corn: Hufford et al., 2012; and wheat: Haudry et al., 2007), representing the grass family (Poaceae). Cereals include the most economically important domesticated species, but the life-history and domestication traits that these annual species grown for their shared fruit-type (caryopsis) have in common may bias our understanding of how domestication and breeding shape crop diversity in general. Recent work to reconstruct the domestication processes in a greater diversity of crops [e.g., apple (Malus, Rosaceae): Cornille et al., 2012; olive (Olea, Oleaceae): Diez et al., 2015; carrot (Daucus, Apiaceae): Iorizzo et al., 2013; peach (Prunus, Rosaceae): Cao et al., 2014; and soybean (Glycine, Fabaceae): Guo et al., 2010] has led to broader characterizations of domestication as an evolutionary process. Population genomic studies in apple (Cornille et al., 2012) and carrot (Iorizzo et al., 2013) revealed that these crops did not experience the severe domestication bottlenecks that accompanied the domestications of rice (Oryza, Poaceae), corn (Zea, Poaceae), and wheat (Triticum, Poaceae) and highlight the need to characterize domestication in crops that better represent the diversity of wild plant species.
Establishing a single demographic model of crop domestication is not possible, because of the diversity of crop wild ancestors and domestication and diversification processes (Meyer and Purugganan, 2013). Comparisons of the domestications of diverse crop species are therefore vital to an accurate understanding of how domestication and breeding affect genetic diversity, but such comparisons are limited by the differences in traits that diverse domesticated species possess. Here we characterize and compare domestication in two pumpkin species (Cucurbita, Cucurbitaceae) that were independently domesticated from closely related wild species.
Of the six independently domesticated pumpkin (2n = 40) species, only Cucurbita argyrosperma ssp. argyrosperma (“cushaw,” “calabaza pipiana”) and Cucurbita maxima ssp. maxima (“buttercup squash,” “zapallo”) have a clearly supported sister relationship to an extant wild species that is likely the crop wild ancestor (Kates et al., 2017). The domestication syndromes of buttercup squash and cushaw are similar, based on shared initial domestication traits and overlapping modern breeding objectives. Both species were domesticated from monecious, outcrossing, bee-pollinated, herbaceous annual vine species that bear round fruits, 3.5–8.0 cm in diameter, with a green exocarp that may be striped or unstriped and may be yellow or green at maturity (Nee, 1990). The rinds of the wild relatives are hard and lignified (Robinson and Decker-Walters, 1997), and the flesh contains cucurbitacins that render the flesh inedible unless repeatedly boiled (Nabhan and Felger, 1985). Wild plants were likely initially selected by semi-nomadic humans for their edible, nutritious seeds and use of durable rinds as containers (Small, 2013; Ranere et al., 2009, Sánchez-de la Vega et al., 2018). Discovery of rare, non-bitter or less-bitter Cucurbita fruits led to the eventual non-bitter Cucurbita crops we know today.
The traits that define the domestication syndrome of C. argyrosperma and C. maxima include more uniform germination, a bush habit, a reduction in size and abundance of trichomes that interfere with harvesting, an increase in the size of fruits and seeds, and a reduction in the bitter taste of the flesh (Lira-Saade and Montes Hernández, 1994). There are also striking differences in the fruit phenotypes of C. maxima ssp. maxima and C. argyrosperma ssp. argyrosperma and in the economic importance, geographic extent, and ecological diversity of their cultivars (Table 1).

Table 1. Summary information about each domesticated subspecies.
Domesticated C. maxima ssp. maxima is among the most economically important and widely cultivated Cucurbita species and is also the most morphologically diverse Cucurbita crop species (Chigmura Ngwerume and Grubben, 2004). C. maxima ssp. maxima was domesticated in South America ∼4,000 years ago from C. maxima ssp. andreana, a taxon that occurs in warm, temperate areas of Argentina and Uruguay (Decker-Walters and Walters, 2000) and as far north as Bolivia (Figure 1). C. maxima ssp. maxima was brought to the Old World during the Columbian exchange (Decker-Walters and Walters, 2000) and is now cultivated all over the world, including in a secondary center of crop diversity in India and Southeast Asia (Zeven and Zhukovskii, 1975), where extensive breeding and improvement of new varieties have occurred. In contrast, C. argyrosperma ssp. argyrosperma is the only domesticated Cucurbita that is still primarily cultivated within the area where it was domesticated (Robinson and Decker-Walters, 1997). C. argyrosperma ssp. argyrosperma was likely domesticated over 8,000 years ago (Smith, 2006; Ranere et al., 2009) from C. argyrosperma ssp. sororia, a wild taxon that today occurs mostly along the Pacific coast of Mexico and more rarely in semi-arid areas of northwestern Mexico and northern Central America (Lira-Saade and Montes Hernández, 1994; Figure 1).
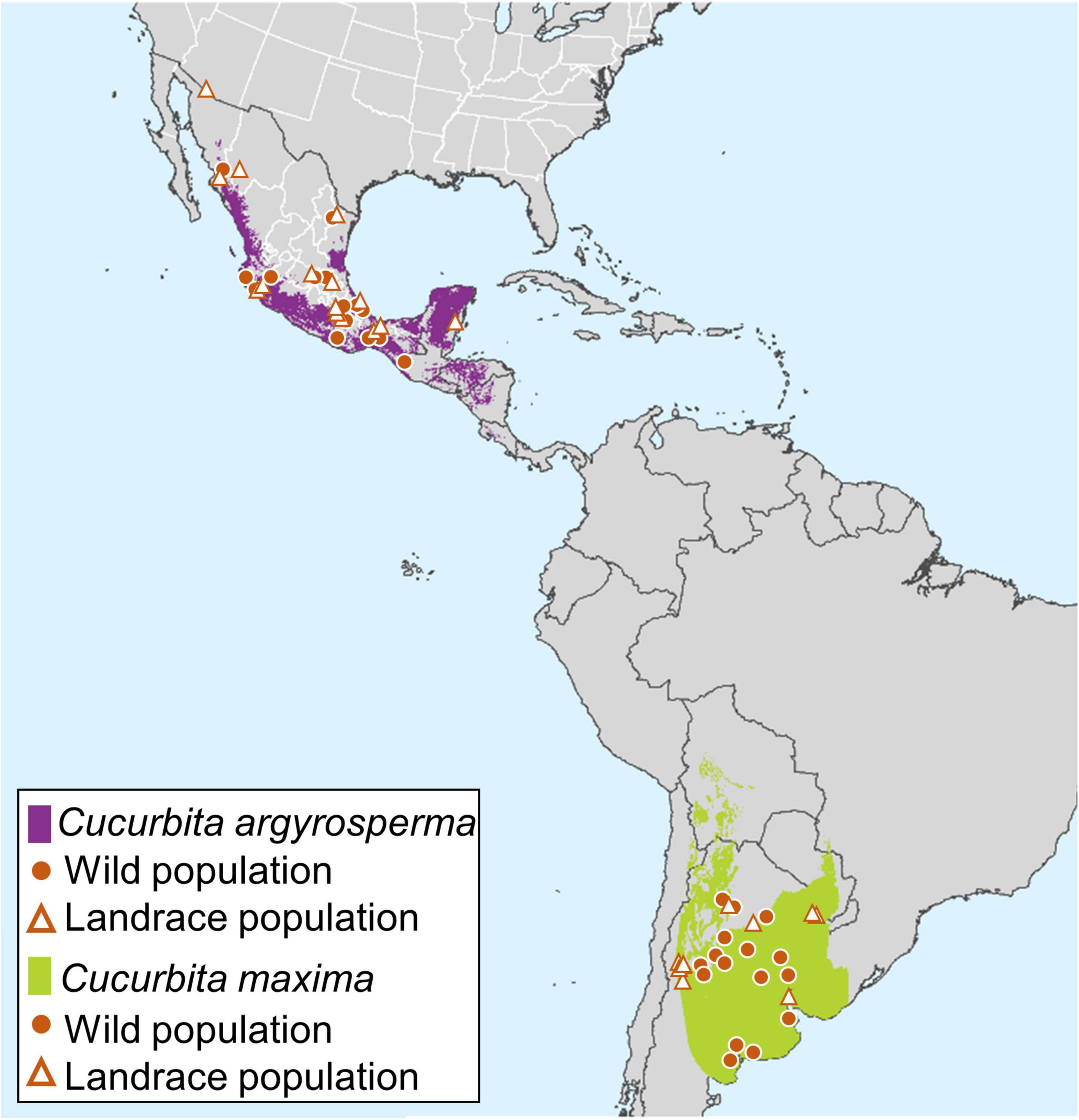
Figure 1. Map of putative range of wild and landrace C. maxima and C. argyrosperma with sampling localities (if known) for landrace and wild samples included in this study. For Cucurbita argyrosperma (purple), distribution is based on 620 occurrence points from GBIF (gbif.org). For C. maxima (green), distribution is based on 58 occurrence points from GBIF (gbif.org), Specieslink (splink.cria.org.br/), Discover life (www.discoverlife.org), and CONABIO (conabio.gob.mx). Landrace samples are indicated by triangles and wild samples are indicated by solid circles.
The incredible diversity of fruit morphology in C. maxima ssp. maxima, which includes a variety that produces the largest fruit on Earth (over one metric ton and over five meters in circumference; Decker-Walters and Walters, 2000), is not seen in C. argyrosperma ssp. argyrosperma. Along with perhaps Cucurbita ficifolia (figleaf gourd, “chilacayote”), C. argyrosperma ssp. argyrosperma is the least diverse domesticated Cucurbita species in terms of developed varieties and fruit morphology (Lira-Saade and Montes Hernández, 1994), although cultivars grown in the United States and Canada do show differences in fruit and seed size, shape, and color (OECD, 2016). The fruits of C. argyrosperma ssp. argyrosperma resemble larger versions of the wild type with or without a crookneck (Lira-Saade and Montes Hernández, 1994), and C. argyrosperma ssp. argyrosperma is mostly cultivated for seed rather than fruit (Lira-Saade and Montes Hernández, 1994).
We performed population genomic analyses using diverse wild, landrace, and improved accessions of each species and over 15,000 single nucleotide polymorphisms (SNPs) evenly distributed across the Cucurbita genome to investigate the following: (1) Is there evidence for population subdivision within each species from which we can infer more specific geographic origins of domestication?; (2) How large is the contribution of wild species to the genome of the domesticates?; and (3) What consequences have domestication and subsequent crop improvement had for the genetic variation in each domesticated pumpkin species, and how do patterns of genetic variation relate to the variable morphological and ecological diversity and economic importance of the modern crops? Our general overarching hypothesis related to all three questions is that the apparently less intense improvement and comparatively lower diversity of fruit morphology of C. argyrosperma compared to C. maxima would be evidenced by its lower population subdivision in C. argyrosperma, a larger contribution of wild C. argyrosperma to its domesticate, and a less drastic reduction of its genetic diversity in C. argyrosperma relative to C. maxima.
Materials and Methods
Plant Material and DNA Extraction
For each species, we selected a panel of 48 wild and domesticated lines to maximize coverage of the wild and landrace geographic ranges and include 12 unique cultivars (Supplementary Table 1). Identification of domesticated lines as landrace or improved can be inexact. We designated germplasm of cultivated material collected from the geographic region of domestication as “landrace” and material from outside of this region as “improved.” We assumed that forms cultivated in the region of domestication are more representative of the initial domesticates and those cultivated outside of this area are more likely products of modern breeding programs. We also used varietal and cultivar information for the accessions to inform these designations when available. Limitations of this approach are addressed in the discussion. Germplasm for all samples was obtained from the collections of various institutes [United States Department of Agriculture-Agricultural Research Service (USDA-ARS), Universidad Nacional de Rosario (UNR), Instituto Nacional de Tecnología Agropecuaria (INTA)] and the Institute of Ecology at Universidad Nacional Autónoma de México (UNAM) for C. argyrosperma and at UNR for C. maxima. Leaf samples for DNA extraction were obtained from seedlings grown at the University of Florida, Gainesville, FL, United States.
DNA was extracted from fresh leaf tissue using a modified 2× CTAB method (Doyle and Doyle, 1987; Kates et al., 2017) yielding ∼50–120 ng/μl of DNA per sample. DNA was extracted from fresh leaf tissue using a modified 2× CTAB method (Doyle and Doyle, 1987; Kates et al., 2017) yielding ∼50–120 ng/μl of DNA per sample. DNA quantity and quality were analyzed using the Agilent 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, United States). For 42 accessions of C. maxima, DNA was extracted directly from non-viable germplasm using the following modifications: seeds were dissected, and the endosperm and seed coat removed from the embryonic tissue using a sterile razor blade; DNA was extracted from the embryo, and an additional phenol-phenol purification following the first addition of chloroform-isoamyl alcohol allowed for stronger protein dissolution and separation from aqueous DNA.
Targeted Enrichment and DNA Sequencing
Genomic library building, probe design, and targeted enrichment were performed by Rapid Genomics LLC (Gainesville, FL, United States). Between 250 ng and 1 μg of genomic DNA of each sample was fragmented to an average size of 400 bp. DNA libraries were constructed by end-repairing the sheared DNA, A-tailing and adapter ligation, bar-coding, and PCR amplification. Targeted enrichment of Illumina libraries using biotinylated RNA baits (Gnirke et al., 2009) was used to reduce genomic complexity prior to sequencing to increase the number of samples that could be multiplexed on a single sequencing lane. A custom RNA probe kit was developed and synthesized by Rapid Genomics LLC that included 10,000 150-mer probes targeting 9,175 genomic sequences, including 7,922 previously identified SNPs and 1,253 putatively single-copy genes. The probe sequences and the targeted SNPs and single-copy genes are available on Zenodo (10.5281/zenodo.4773140).
Single nucleotide polymorphism loci targets were based on SNPs in C. maxima ssp. maxima (Zhang et al., 2015), with 500-bp flanking sequences, mined from the C. maxima ssp. maxima genome (Sun et al., 2017). Single-copy genes were identified using a custom all-by-all BLAST plus single-linkage-clustering pipeline described in Kates et al. (2017) where the BLAST database included four C. argyrosperma transcriptomes and three C. maxima transcriptomes sequenced and assembled by the COMAV Cucurbits Breeding Group and Bioinformatics at Universidad Politécnica de Valencia (Huang et al., 2019).
Putatively single-copy genes ranged in size from 350 to 900 bp. Whole plastome sequences of C. maxima and C. argyrosperma obtained from GenBank were used to identify and remove potential probes that would hybridize with high-copy plastid genes. We designed 7,922 probes to target SNPs, and probes were centered on the SNP region. A total of 2,081 probes were designed to capture the putatively single-copy genes. A single probe was used to target exons that were less than 350 bp, two probes for those that were more than 350–500 bp, and three probes for exons >500. The probes were placed to capture both intron and exon sequence. The probes were hybridized to the libraries and enriched for the targets specified. Samples were then pooled equimolar, and 61,778,473 reads were generated on an Illumina HiSeq 3000 PE100 (Illumina, San Diego, CA, United States).
Read Filtering, Mapping, and SNP Calling
Sequencing reads were split by barcode, filtered, and trimmed by quality using the FASTX toolkit1. Filtered reads were aligned to the C. maxima ssp. maxima reference genome (Sun et al., 2017) using MOSAIK 2.3.2 (Lee W.-P. et al., 2014) with a mismatch threshold (option -mmp) of 0.05. SNPs were identified in the nuclear genome using FREEBAYES 0.9.15 (Garrison and Marth, 2012). Sites with less than 8× coverage (–min-coverage 8) and alleles with a base quality of less than 20 (–min-base-quality 20) were excluded from the analysis, and indels and multi-nucleotide polymorphisms were ignored (–no-indels, –no-mnps). SNPs were quality-filtered using VCFtools (Danecek et al., 2011) to include only biallelic sites with quality values greater than 10 and fewer than 50 missing genotypes, mean depth value between 3 and 750, a minor allele frequency greater than or equal to 0.01, and minor allele count greater than or equal to 1. All analyses below were performed for C. maxima and C. argyrosperma independently using sets of within-species SNPs parsed from the full dataset using VCFtools (Danecek et al., 2011), which resulted in datasets of 15,236 SNPs for C. argyrosperma and 17,235 SNPs for C. maxima.
Population Structure and Genetic Diversity
Population structure within each species was estimated using STRUCTURE 2.3.4 (Pritchard et al., 2000). We also separately estimated population structure within domesticated C. maxima. For all STRUCTURE analyses, 10 independent runs with a burn-in length of 50,000 and a run length of 100,000 were performed for each K value from 1 to 10 with the admixture model and correlated allele frequencies between populations. A priori population information (i.e., wild, landrace, improved) was not used. The most likely K value was determined following Evanno et al. (2005). STRUCTURE results were visualized using the R package Pophelper 2.1.0 (Francis, 2017). Principal component analysis (PCA) and FST analysis were performed using the R package SNPRelate (Zheng et al., 2012). To assess variation among different sample sets, FST was calculated using two population definitions: (1) two STRUCTURE-defined clusters (K = 2) based on majority proportion of inferred ancestry (q) for C. maxima and C. argyrosperma (except for one wild C. maxima sample that did not belong to the wild cluster based on the inferred ancestry coefficient and was excluded from subsequent analyses) (Supplementary Table 2), and (2) three a priori population designations: wild, landrace, and improved. For all PCA and FST analyses, pruning based on linkage disequilibrium (LD) was performed with an LD threshold of 0.20, that resulted in a set of 1,926 SNPs for C. maxima and 2,024 SNPs for C. argyrosperma.
Phylogenetic trees were built using SNPhylo (Lee T.-H. et al., 2014) with 100 bootstrap replicates and were rooted with four outgroups (i.e., four C. argyrosperma accessions were included in the phylogenetic analysis of C. maxima accessions, and four C. maxima accessions were included in the phylogenetic analysis of C. argyrosperma accessions). Although population genetic datasets comprising SNP data for closely related individuals violate assumptions of the evolutionary models underlying phylogenetic analysis [including that implemented in DNAML (Felsenstein, 1981) used in SNPhylo], phylogenetic trees can provide additional information about sample groupings along with population genetic analyses when some characteristics of the data are taken into account. SNPhylo extracts representative SNPs from the original dataset to reduce SNP bias due to high levels of LD (Lee T.-H. et al., 2014); this results in a set of aligned sites (SNPs) less in violation of the model’s assumption that each site evolved independently (Felsenstein, 1981). For phylogeny reconstructions, we used an LD threshold of 0.40, resulting in a phylogenetic dataset of 4,285 SNPs for C. maxima and 2,988 SNPs for C. argyrosperma.
Genetic diversity calculations were performed using the total number of SNPs described in section “Read Filtering, Mapping, and SNP Calling.” Expected heterozygosity (HE) and observed heterozygosity (HO) were calculated with the 4P software (Benazzo et al., 2015) as locus-by-locus and population mean estimates. Watterson’s (1975) estimator of nucleotide diversity θW and Nei and Li’s (1979) nucleotide diversity π were calculated by gene across each chromosome using the PopGenome package for R (Pfeifer et al., 2014). Chromosome positions correspond to the C. maxima ssp. maxima loci (Zhang et al., 2015) used to target the SNP loci (described above). To identify regions that may have been subject to selection during domestication, we scanned for loci that had both the highest differences in genetic diversity [π log-ratio, ln(πwild)-ln(πdomesticated)] and extreme divergence in allele frequency between wild and domesticated sample sets (FST). We compared (1) wild and improved samples of C. maxima and (2) wild samples of C. argyrosperma to landrace and improved samples separately. We identified SNPs with outlier FST values and loci with outlier π log-ratios using Z-tests (P < 0.05). We used AmiGo 22 to identify orthologous gene products and perform gene ontology enrichment analysis.
A GFF file created using the chromosome position information in the BED file that accompanied the SNP data was used to map chromosome-wide positions for each SNP in PopGenome. For C. maxima, populations were defined as the two STRUCTURE-defined clusters that corresponded to improved and wild accessions. C. maxima landrace accessions were excluded from the genetic diversity analyses because population structure analyses failed to identify these samples as distinct from improved samples (Figures 2–4) and because assigning them to the improved population a posteriori may have led to issues with uneven sampling between populations. For C. argyrosperma, populations were defined as the three a priori designations–wild, landrace, or improved–as these were supported by STRUCTURE analysis and PCA.
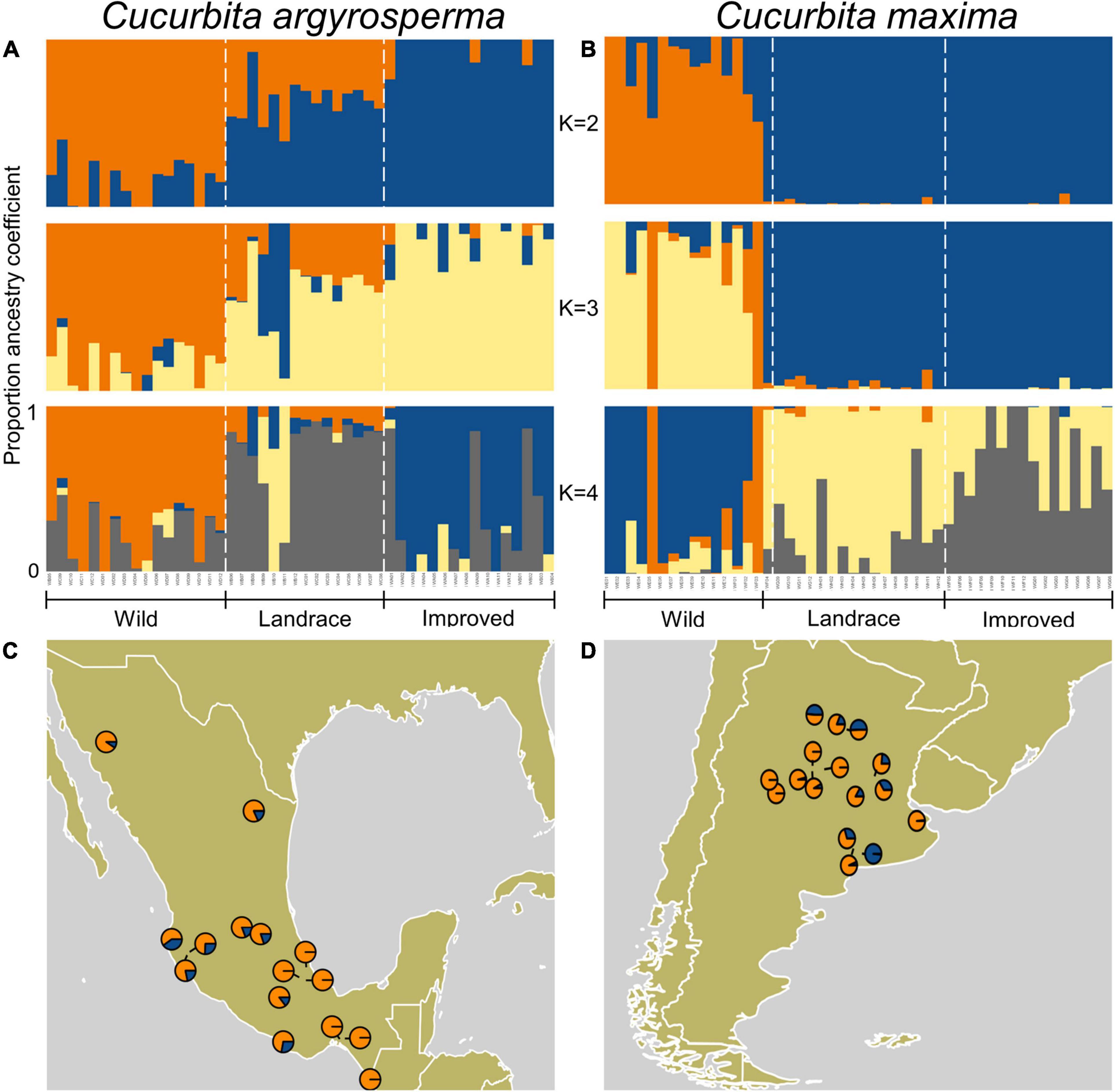
Figure 2. Bayesian clustering (STRUCTURE) results for C. maxima and C. argyrosperma and ancestry coefficients mapped onto sampling localities. (A,B) Bayesian clustering (STRUCTURE, K = 2–4) of (A) C. argyrosperma accessions and (B) C. maxima accessions. (C,D) Ancestry coefficients mapped onto sampling localities for wild accessions of (C) C. argyrosperma and (D) C. maxima. Colors correspond to ancestry-coefficient colors in panels (A,B).
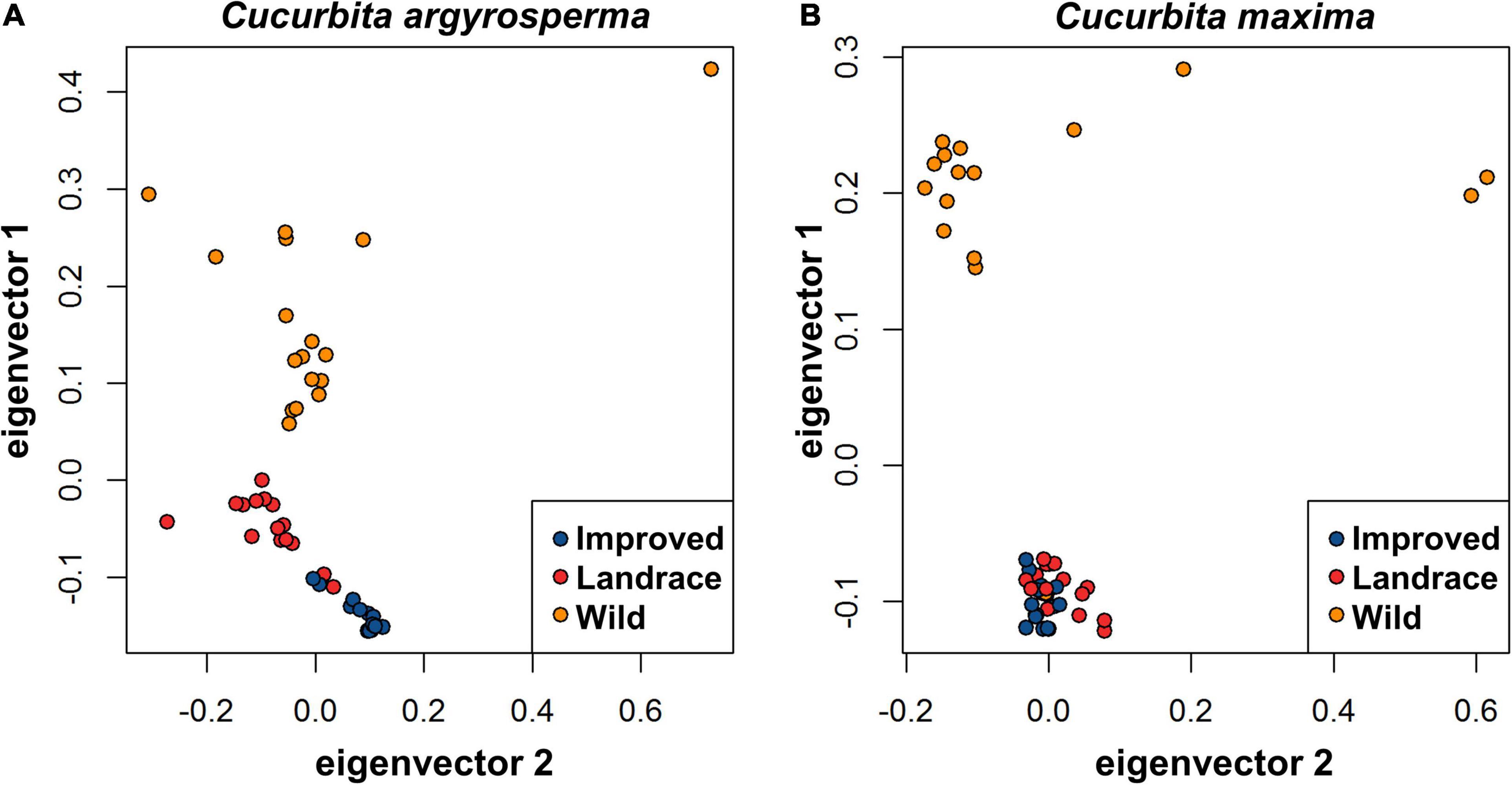
Figure 3. Principal component analysis of improved (blue), landrace (red), and wild (orange) (A) C. argyrosperma accessions and (B) C. maxima accessions based on 2,024 SNPs for C. argyrosperma and 1,926 SNPs for C. maxima. For C. argyrosperma, the percent of variation accounted for by eigenvector 1 and eigenvector 2 is 9.0 and 5.2%, respectively. For C. maxima, the percent of variation accounted for by eigenvector 1 and eigenvector 2 is 8.8 and 6.7%, respectively.
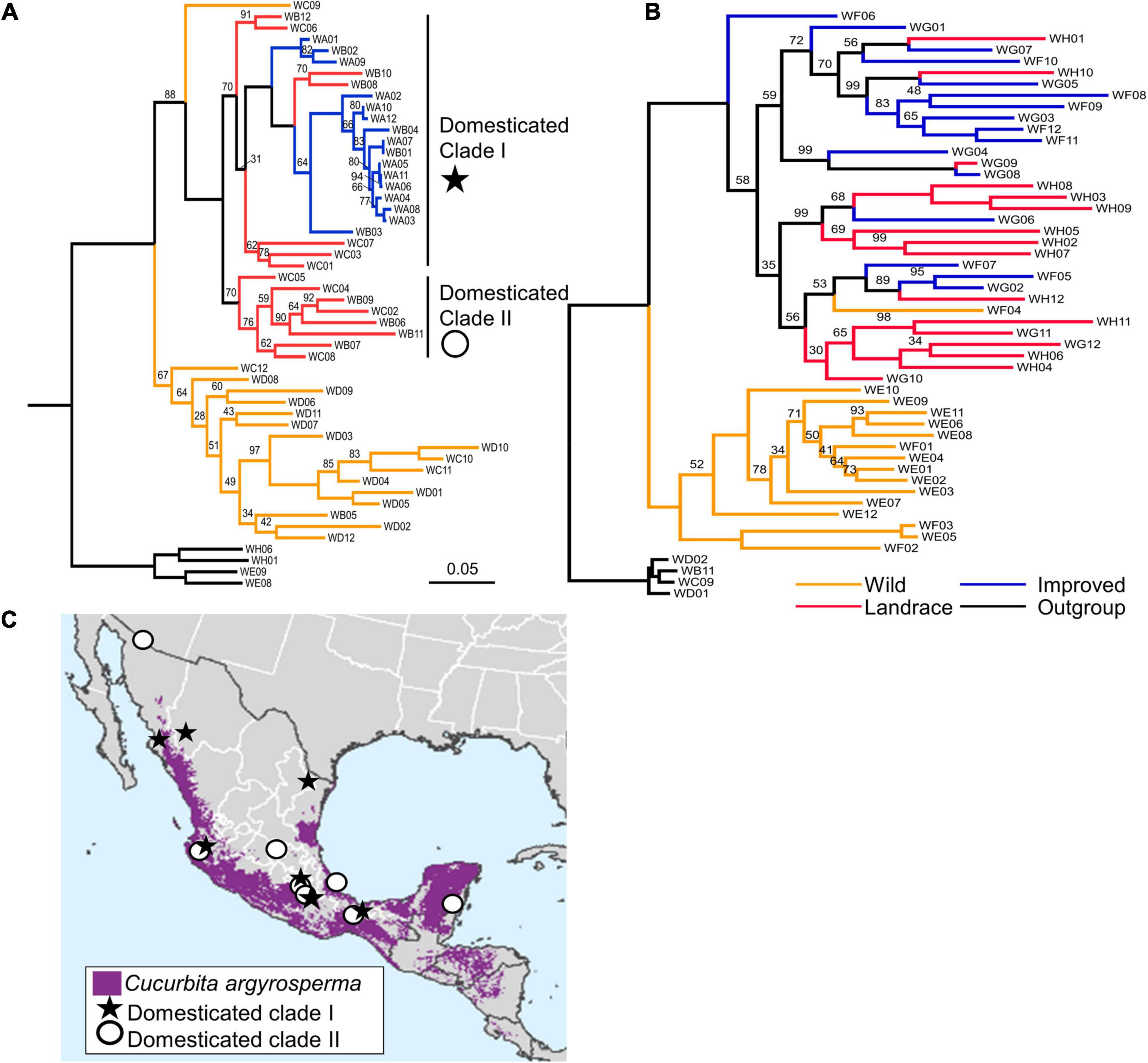
Figure 4. Results of phylogenetic analysis of (A) Cucurbita argyrosperma accessions and (B) Cucurbita maxima accessions. Branches are colored by improvement status. Bootstrap values of 100 are not shown. Two clades of domesticated C. argyrosperma are indicated by Domesticated Clade I and Domesticated Clade II (C) Map of sampling localities of C. argyrosperma landraces with samples’ membership in Domesticated Clade I or Domesticated Clade II indicated by * or ∘, respectively. Scale bar indicates the proportion of site changes along each branch.
Results
SNP Data
An initial set of 67,362 inter-species SNPs was identified from 61.8 million merged paired-end reads for 96 samples mapped to the C. maxima ssp. maxima genome. After filtering SNPs and separating by species, 15,236 SNPs for C. argyrosperma and 17,235 SNPs for C. maxima were used for the analyses.
Population Structure and Pairwise Population Differentiation
The STRUCTURE analysis using the method of Evanno et al. (2005) suggested two clusters (K = 2) as optimal for both C. argyrosperma and C. maxima (Supplementary Figure 1). For K = 2 in C. argyrosperma, the wild and improved samples are differentiated as distinct clusters (ancestry coefficient >0.7), and the landrace samples are a mixture of individuals from both ancestral populations (Figure 2A). For K = 2 in C. maxima, the wild and improved samples are clearly differentiated, but in contrast to the structuring of C. argyrosperma, the landrace samples are not admixed, and instead all belong to the same ancestral population as the improved samples (Figure 2B). We observed less admixture in landrace and in improved samples of C. maxima than for landrace and improved samples of C. argyrosperma. Because there are limitations to considering clustering results for only one value of K (Meirmans, 2015), we present the clustering results for K = 3 and K = 4 as well as K = 2. As values of K change from 2 to 4, subpopulation structure appeared in the domesticated C. maxima accessions; subpopulation structure as K increased from K = 2 was less pronounced for wild accessions and domesticated C. argyrosperma.
Mapping of admixed wild samples onto their sampling locations can provide information about likely sites of domestication by highlighting geographic regions, where admixed wild samples that contain a relatively high proportion of alleles common in the domesticated samples occur, although gene flow between wild and domesticated populations can obscure this pattern. In C. argyrosperma, such samples occur mostly in western and central Mexico and not in the easternmost or southernmost part of the distribution of the wild taxon (Figure 2C). In C. maxima, the region identified is the northernmost part of the distribution of the wild taxon (Figure 2D).
For both C. maxima and C. argyrosperma, the results of PCA were congruent with the clusters differentiated by STRUCTURE (Figures 3A,B). For both species, pairwise FST was higher in the comparison of wild samples and domesticated samples (both landrace and improved) than in analyses of the two domesticated subclasses (landrace and improved) compared to each other or to the wild samples (Table 2). Pairwise FST between wild and domesticated samples was nearly equal in C. maxima and C. argyrosperma. In C. argyrosperma, pairwise FST between the two STRUCTURE-defined populations was 0.30; values were nearly equal for wild vs. landrace and landrace vs. improved (0.23 and 0.22, respectively), but considerably higher (0.32) for wild vs. domesticated (landrace and improved). In C. maxima, pairwise FST was 0.33 between the two STRUCTURE-defined populations, 0.30 for both wild vs. domesticated and wild vs. landrace, and only 0.07 between landrace and improved.
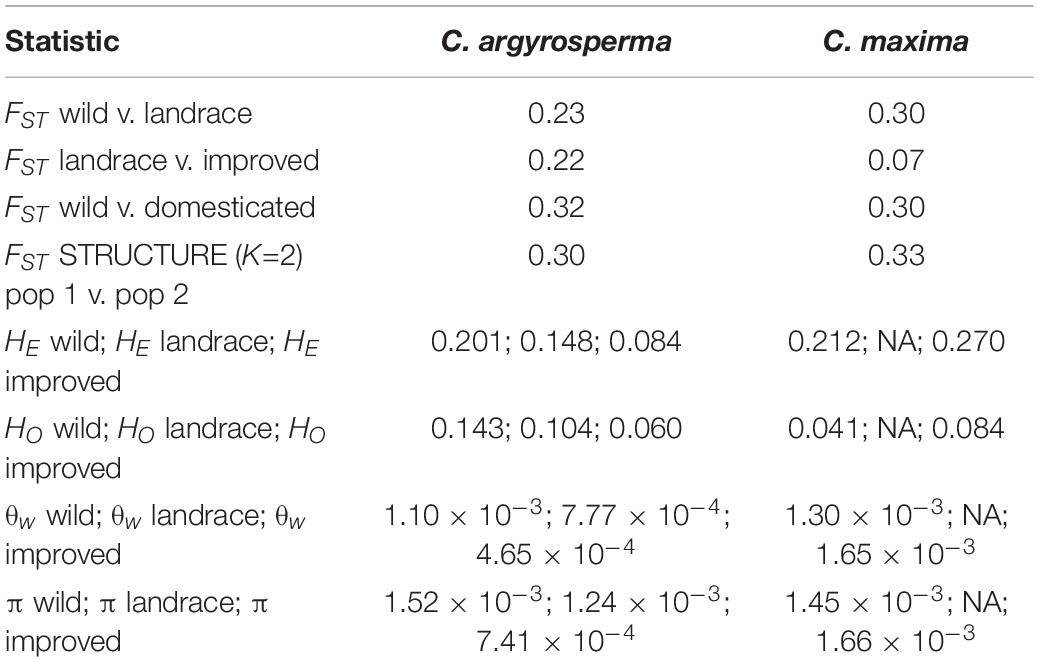
Table 2. Summary of genetic diversity statistics.
Phylogenetic Analysis
Cucurbita argyrosperma–Wild and domesticated samples form separate clades (BS 67 and 88%, respectively) (Figure 4A). Domesticated samples occur in two subclades: clade I (BS 70%) includes landrace and improved samples, and clade II is composed entirely of landrace samples (BS 70%). There is not a clear geographic pattern to the landrace samples in the two separate clades (Figure 4C). One sample designated as “wild” (WC09) was sister to the rest of the domesticated samples. However, this sample was originally submitted to the USDA Plant Genetic Resources Conservation Unit as “Cucurbita cf. palmeri” and was later re-named as C. argyrosperma ssp. sororia. Cucurbita cf. palmeri is thought to be a feral escape from cultivation (Merrick, 1990) and is very difficult to distinguish from the wild subspecies and was thus excluded. Based on our phylogenetic results, this sample is likely a feral escape from cultivation rather than a true wild sample.
Cucurbita maxima–The wild and domesticated samples each form well-defined clades (BS 100%) (Figure 4B). The domesticated clade includes the wild sample (WF04) that was also assigned to the domesticated genetic cluster by STRUCTURE based on its ancestry coefficient and was subsequently removed from genetic diversity analyses. One improved sample (WF06), “Nan kwa,” is sister to the rest of the domesticated samples and is the only cultivar included in this study that is likely of Chinese origin.
Genetic Diversity
The mean values of expected heterozygosity (HE) and observed heterozygosity (HO) in both species are summarized in Table 2. Observed heterozygosity was more than three times higher in wild C. argyrosperma than in wild C. maxima. Both wild and improved C. maxima exhibited much higher expected heterozygosity than observed heterozygosity. In C. argyrosperma, mean HE and HO were 0.201 and 0.143, respectively, in the wild samples, 0.148 and 0.104 in the landrace samples, and 0.084 and 0.060 in the improved samples. In C. maxima, HE and HO were 0.212 and 0.041 in the wild samples and 0.270 and 0.084 in the improved samples, respectively.
The by-locus values of within-population nucleotide diversity (θw and π) across chromosomes are illustrated in Figures 5, 6, and the mean θw and π for each population are summarized in Table 2. Mean θw was similar for wild C. argyrosperma and C. maxima, but highest in improved C. maxima. A similar pattern was observed for mean π. In C. argyrosperma, mean θw was 1.10 × 10–3 in the wild samples, 7.77 × 10–4 in the landrace samples, and 4.65 × 10–4 in the improved samples. Mean π was 1.52 × 10–3 in the wild samples, 1.24 × 10–3 in the landrace samples, and 7.41 × 10–4 in the improved samples. In C. maxima, mean θw was 1.30 × 10–3 in the wild samples and 1.65 × 10–3 in the improved samples, and mean π was 1.45 × 10–3 in the wild samples and 1.66 × 10–3 in the improved samples. Although the overall trend across loci is lower genetic diversity in improved C. argyrosperma relative to domesticated C. argyrosperma (Figures 5A, 6A), and lower genetic diversity in wild C. maxima relative to improved C. maxima (Figures 5B, 6B), nevertheless there are multiple loci for each species that exhibit the reverse (i.e., a locus with higher genetic diversity for improved C. argyrosperma than for wild C. argyrosperma) (Figures 5, 6).
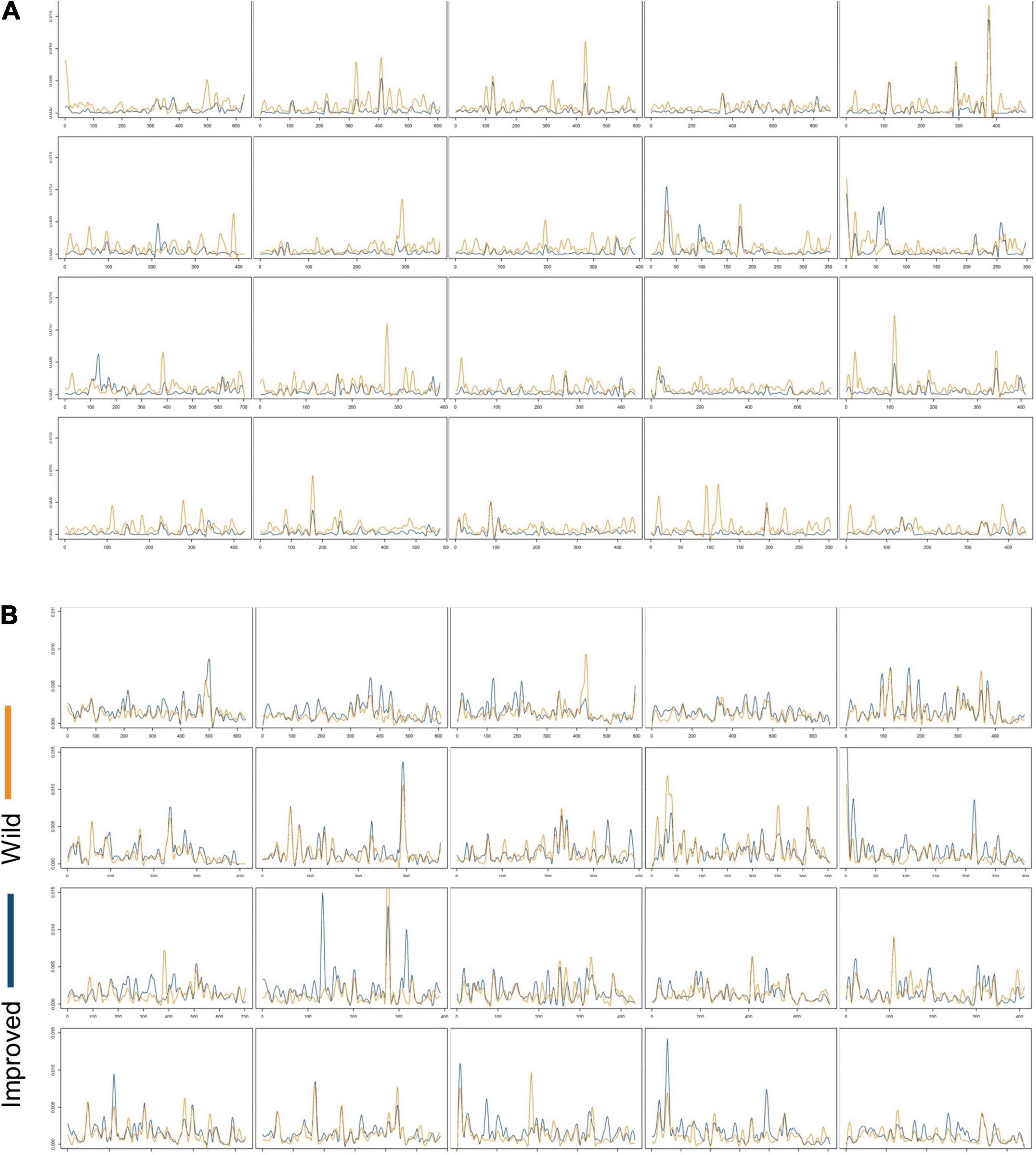
Figure 5. By-locus values of within-population nucleotide diversity (θw) for improved (blue) and wild (orange) “populations” across chromosomes 1–20 in (A) Cucurbita argyrosperma and (B) Cucurbita maxima.
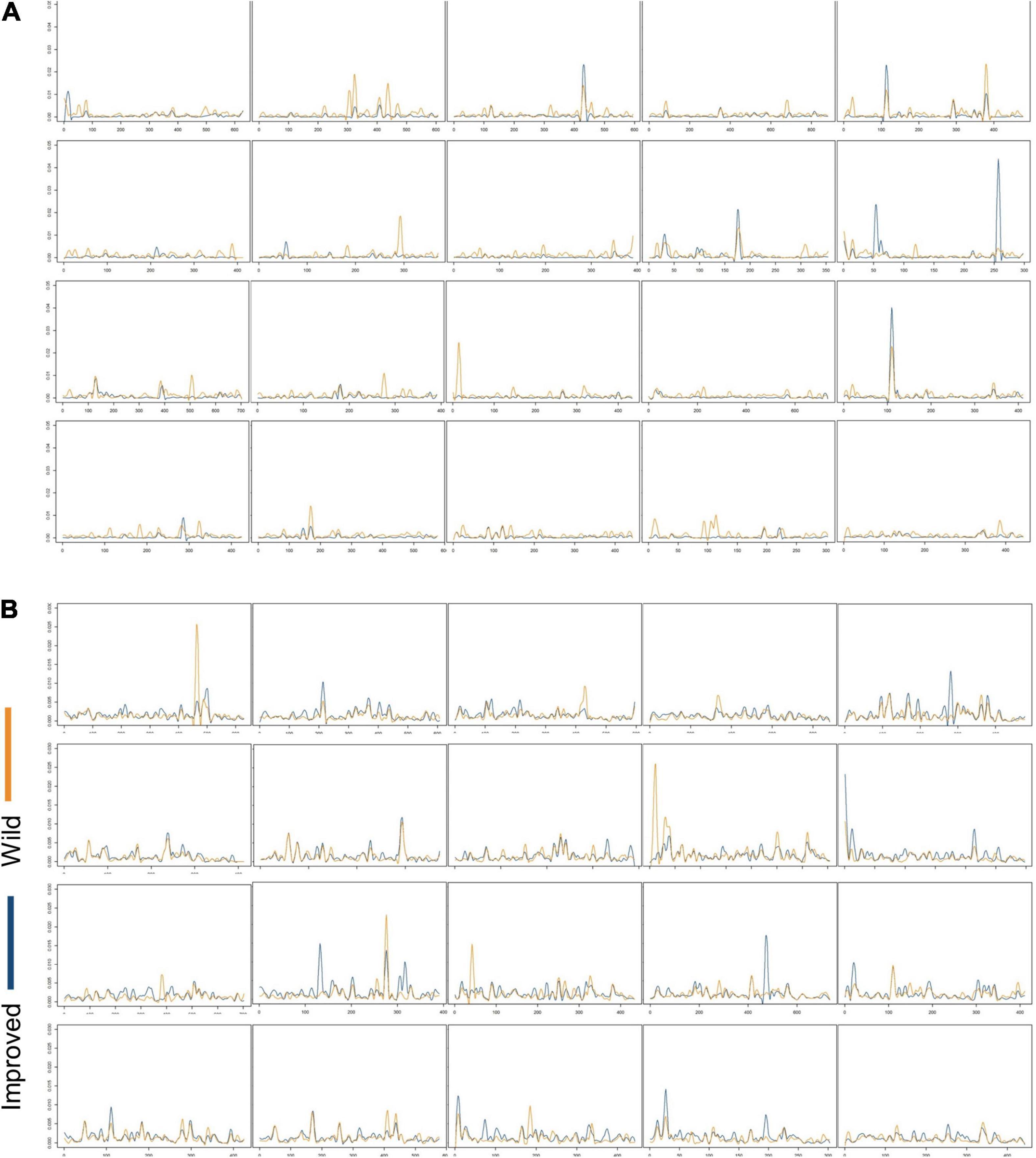
Figure 6. By-locus values of within-population nucleotide diversity (π) for improved (blue) and wild (orange) “populations” across chromosomes 1–20 in (A) Cucurbita argyrosperma and (B) Cucurbita maxima.
We detected genomic regions that may have been subject to selection as inferred from high wild/domestic π log-ratios and an extreme population differentiation between wild and domesticated samples. Using this test, we identified four domestication features in improved C. argyrosperma, 17 in landrace C. argyrosperma, and 20 in improved C. maxima (Supplementary Table 3). Domestication features comprise a locus or multiple loci mapped to a single predicted gene product with FST and π log-ratio above thresholds determined by our outlier tests: C. argyrosperma (wild vs. improved/wild vs. landrace) π log-ratio > 2.84/2.09 and FST > 0.80/0.54; C. maxima (wild vs. improved) π log-ratio > 1.34 and FST > 0.77. We annotated domestication features by similarity to Arabidopsis gene models. There was no overlap between species in these two sets of putative domestication genes or associated gene models. A less stringent test for selection based on high wild/domestic π log-ratio alone recovered 115 and 171 domestication features in C. argyrosperma (landrace and improved) and C. maxima, respectively, four of which were identified in both species. We found seven shared GO terms from the 13 and 76 significantly enriched full GO terms identified for C. maxima and C. argyrosperma, respectively (Supplementary Table 4). To consider gene ontology more broadly, we assessed overlap in each species’ top 5 GO slim terms and found 2 overlapping GO slim terms (Supplementary Table 4).
Discussion
Origins of Domestication
We provide the first molecular investigation of the site and number of origins of buttercup squash and cushaw domestication. Our results support a single origin each of buttercup squash and cushaw (Figures 4A,B) and reveal geographic structuring of the wild ancestors of both crops (Figures 2A,B). The site of buttercup squash domestication has been sought in the accepted range of C. maxima ssp. andreana in Argentina (Nee, 1990), although several authors have referenced its occurrence north of this region, in Peru and Bolivia (Rosas et al., 2004; Cutler and Whitaker, 1961), where a large number of cultivars of C. maxima are grown (Cutler and Whitaker, 1961). We could not obtain records or collections from these populations, but our finding that populations from the northern part of our sampling range share the highest proportion of alleles with the domesticated subspecies (Figure 2D) suggests a more northern origin and highlights the importance of including wild populations from Peru and Bolivia in future efforts to pinpoint the ancestry of buttercup squash.
These northern populations occur in regions where there may be ongoing gene-flow between wild and domesticated C. maxima. Evidence for gene-flow is based on the discovery of fruits in the Jesús María region in northern Cordoba province that appear to be hybrid forms between C. maxima ssp. maxima and C. maxima ssp. andreana (Millán, 1945; Lira-Saade, 1995). Our sampling was limited by what was readily available, and additional genetic analyses of targeted sampling from this area may allow for differentiation between admixture due to ongoing gene flow between wild and domesticated accessions and admixture that is evidence of an origin of domestication.
Although archeological remains suggest that southern Mexico is the site of initial domestication for cushaw (Piperno et al., 2009), we found that populations of C. argyrosperma ssp. sororia from the southern part of its range share fewer alleles with domesticated cushaw than populations from the Pacific coast and central Mexico (Figure 2C). The cluster of populations of wild C. argyrosperma ssp. sororia on the Pacific coast that share more alleles with the domesticate may represent a more specific geographic origin for the initial domestication of cushaw in Mexico. This result is congruent with recent findings by Sánchez-de la Vega et al. (2018) and in particular Barrera-Redondo et al. (2021) that used coalescent models of domestication and genetic differentiation tests (FST) to show that C. argyrosperma ssp. sororia populations from Jalisco, Mexico, are most closely related to domesticated C. argyrosperma ssp. argyrosperma accessions included in those studies, suggesting domestication may have occurred in western Mexico.
However, it is complicated to point to patterns of shared alleles between wild and domesticated C. argyrosperma as evidence for an origin of domestication within Mexico because wild-crop gene-flow likely occurs throughout the region (Montes-Hernandez and Eguiarte, 2002; Decker-Walters et al., 1990). Wild C. argyrosperma ssp. sororia is frequently found growing in or near cultivated fields of C. argyrosperma ssp. argyrosperma where hybridization between the two taxa has been documented (Montes-Hernandez and Eguiarte, 2002; Decker-Walters et al., 1990). Additional genetic analyses of increased sampling from this region may allow for differentiation between admixture due to ancient and current gene flow, but this was beyond the scope of our study.
Ancient Crop-Wild Gene Flow May Be Central to Contrasting Domestication Bottlenecks in C. maxima and C. argyrosperma
Some domesticated plants exhibit decreased genetic diversity relative to their wild ancestors (e.g., maize: Hufford et al., 2012; African rice: Li et al., 2011; and wheat: Haudry et al., 2007), but others do not (e.g., apple: Cornille et al., 2012; carrot: Iorizzo et al., 2013). Recent archaeogenomic evidence also calls into question histories of early domestication bottlenecks in extant crops that have reduced genetic diversity relative to their wild ancestors (Allaby et al., 2019; Brown, 2019). Our results did not support our hypothesis that the domestication and improvement history of C. argyrosperma would yield lower population subdivision, a larger contribution of wild genetic diversity to its domesticated forms, and a less drastic reduction of its genetic diversity relative to C. maxima. To the contrary, we found no evidence for a domestication bottleneck in C. maxima nor did we find differentiation between landraces and improved lines. This finding is contrasted by our results that show C. argyrosperma experienced reductions in genome-wide diversity consistent with both a domestication bottleneck and a subsequent improvement bottleneck. Erroneous treatment of weedy populations secondarily derived from a domesticate as wild populations could obscure patterns of reduced genetic diversity in a crop compared with its wild relative and explain the lack of evidence for a domestication bottleneck in C. maxima. However, secondarily derived weedy populations are themselves the product of a genetic bottleneck and can be identified by reduced genetic diversity relative to the crop (Qiu et al., 2017), and we did not observe this in the wild populations of C. maxima.
Biological explanations for the differences in domestication footprints among crops include (1) outcrossing vs. inbred breeding strategies; (2) annual vs. perennial growth; (3) time since domestication; and (4) domestication traits. The difference in the footprint of domestication for these two crops with similar domestication syndromes derived from closely related wild species suggests broad variability in domestication practices and response to domestication.
Examples of domesticated species that retain or increase morphological diversity relative to wild ancestors, potentially via crop-wild gene flow, are well documented [e.g., common bean (Phaseolus): Singh et al., 1991; brassicas (Brassica): Liu et al., 2014]. This contrasts with the domestication process as commonly described based on maize, wheat, or rice, because the domestications of these crops involved a radical shift in morphology (e.g., loss of shattering) and/or polyploidization that resulted in a loss of sexual compatibility with wild progenitors (Li, 2006; Haudry et al., 2007; Hufford et al., 2012). The most radical phenotypic shift in the domestication of Cucurbita was the loss of bitter cucurbitacins, but unlike the loss of the hard kernel covering in maize, cucurbitacins in wild and hybrid Cucurbita do not preclude human use. Cucurbita fruits were likely first selected for the consumption of seeds that do not contain cucurbitacins present in the fruit flesh (Small, 2014), and boiling can remove cucurbitacins from the fruit’s flesh (Nabhan and Felger, 1985).
Archeological evidence supports a broad domestication of C. maxima characterized by ongoing gene flow between wild and cultivated populations. Wild, intermediate, and domesticated C. maxima morphotypes were identified among archeological remains from the Pampa Grande archeological site in northern Argentina (1720 ± 50 bp) (Lema, 2011). The domesticated types were morphologically diverse and included both thin-rind types adapted for use as food and others with thick, lignified rinds suited for use as containers (Lema, 2011). “Intermediate” remains likely resulted from hybridization between wild C. maxima ssp. andreana and domesticated C. maxima ssp. maxima (Lema, 2011). The diversity of morphotypes suggests that intentional or unintentional early cultivation practices allowed frequent crosses between sympatric wild and domesticated populations (Lema, 2011; Martínez et al., 2018) that introduced novel traits. This practice is still common in modern rural agriculture in Mexico (Altieri et al., 1987).
Gene flow is also well documented between wild and domesticated populations of C. argyrosperma (Sánchez-de la Vega et al., 2018; Montes-Hernandez and Eguiarte, 2002), but we do find evidence for a domestication bottleneck in this species, in contrast to a recent study based on nine microsatellite loci that found similar levels of polymorphism in wild and domesticated C. argyrosperma (Sánchez-de la Vega et al., 2018). Gene flow between wild and domesticated C. argyrosperma has not produced the same phenotypic diversity that characterizes domesticated C. maxima (Lira-Saade and Montes Hernández, 1994); little diversity in fruit morphology is present in domesticated C. argyrosperma, and this crop does not tolerate a wide variety of growth conditions (Lira-Saade and Montes Hernández, 1994). In contrast, there are over 52 cultivars of C. maxima with wide variation in morphological traits (e.g., flesh and fruit color, shape, rind texture, fruit size, and lobing) and agronomic traits (e.g., annual cycle, yield, and adaptive plasticity) (Lema, 2009). The relationship between gene flow, phenotypic diversity, and evidence for a domestication bottleneck may be affected by how readily human-mediated crosses produce novel genotypes. We found similar expected heterozygosity in wild populations of both C. argyrosperma and C. maxima, but C. maxima ssp. andreana had much lower observed heterozygosity, suggesting that C. maxima is predisposed to developing novel diversity through crosses of highly diverged individuals that are more likely to yield novel allelic combinations. The likelihood of novel diversity arising in C. maxima also raises the possibility that a domestication bottleneck did occur in this species, and that our failure to detect this was due to diversity that originated after domestication, rather than retained ancestral diversity. The lack of admixture in any domesticated C. maxima samples (Figure 2B) also provides support for this scenario, and additional studies of population structure that include broader sampling of C. maxima landraces are needed to investigate this hypothesis more carefully.
Considering the domestication and cultivation of other Cucurbita species provides additional context to evaluate the differences in cultivation practices and genetic diversity in C. maxima and C. argyrosperma. C. argyrosperma was most likely domesticated and is commonly grown in Mexico, where at least one other Cucurbita species (Cucurbita pepo) was also domesticated (Nee, 1990). It is possible that humans may not have selected as many different forms of C. argyrosperma, which is primarily grown for its seed (Lira-Saade and Montes Hernández, 1994), if multiple fruit morphotypes were readily available in C. pepo (Lira-Saade et al., 1995). On the other hand, C. maxima is the only Cucurbita species domesticated in southern South America (Lira-Saade and Montes Hernández, 1994), and domestication and management of this species likely sought to achieve a greater diversity of uses than in C. argyrosperma (Ferriol et al., 2004).
Improvement
A shift to modern breeding techniques often yields modern lines that can be differentiated from landraces (e.g., carrot: Iorizzo et al., 2013). Landraces are expected to be more representative of the initial domesticate and older breeding practices (Zeven, 1998), and modern-improved cultivars may represent a subset of the genetic diversity present in landraces (e.g., peach: Cao et al., 2014), but this is not always the case (e.g., pigeon pea: Kassa et al., 2012). Our results indicate a lack of genetic differentiation between landrace and improved accessions of C. maxima ssp. maxima and no evidence of a secondary domestication bottleneck associated with improvement. Although C. maxima ssp. maxima and other Cucurbita crops are naturally outcrossing, breeding F1 hybrid squash through self-pollination has been the industry standard for developing new, more uniform varieties of C. maxima ssp. maxima since the 1960s (Whitaker and Robinson, 1986; Della Vecchia et al., 1993; Robinson and Decker-Walters, 1997), due to the unusual ability of Cucurbita to withstand inbreeding (Allard, 1960; Whitaker and Robinson, 1986; Robinson, 1999). This approach is distinct from previous outcrossing breeding practices that produced Cucurbita cultivars characterized by high genetic variability (Bisognin, 2002). Although some breeders have made efforts to create new varieties of C. maxima ssp. maxima that maintain maximum heterozygosity (see Dallman and Dallman, 2009), this practice is rare (Kates, 2019), and is not likely to have affected our results.
Patterns of genetic differentiation between landrace and improved C. argyrosperma are more typical of the consequences of initial domestication and subsequent improvement. Landrace accessions are mostly admixed between wild and domesticated genotypes at K = 2, and they form a separate population at K = 4. We also observed lower genetic diversity in the landrace samples than in the wild samples, consistent with a loss of genetic diversity following initial domestication and a subsequent loss of genetic diversity or secondary bottleneck following modern improvement. This pattern has been observed in many other crops (e.g., maize: Hufford et al., 2012; soybean: Wen et al., 2015; and cotton: Fang et al., 2017) and occurs due to a founder effect when a small subset of the initial domesticate is introduced to new areas and/or modern breeding practices severely reduce heterozygosity (Zeven, 1998). A strong secondary bottleneck in C. argyrosperma suggests that although there are few commercial cultivars of C. argyrosperma (Merrick, 1990), they may be more uniform than commonly thought.
Domestication Features
The different domestication features identified in C. maxima and C. argyrosperma suggest that non-parallel genetic changes underlie the shared domestication syndromes of the two species, and that selection under improvement is stronger in C. maxima. Genome scans for selection are more appropriate for identifying targets of selection under domestication than improvement (Baute et al., 2015), so comparisons of overall genes involved in domestication and/or improvement in C. maxima vs. C. argyrosperma may be limited by our analyses’ exclusion of C. maxima landrace samples because these samples were not identified as a distinct population from improved samples. Additional sampling of C. maxima landrace accessions that better represent early C. maxima domesticates could improve the scope of this analysis. Evidence for convergent selection on the same genes has been found in independently domesticated Brassica (Cheng et al., 2016) and in much more distantly related domesticated grasses (Glémin and Bataillon, 2009). An almost complete lack of convergence in domestication loci and orthologous gene products in two closely related crops is therefore surprising, but a study of two domestication events in common bean found a similar result (Schmutz et al., 2014). The only evidence we found for convergence in the domestication process between the two pumpkin species is that over half of the GO terms significantly enriched in the domestication genes identified for C. argyrosperma were also significantly enriched in C. maxima. Functional relevance of these enriched GO terms should be considered with caution, as our SNP data were generated by targeted sequencing and we cannot know whether the domestication loci we identified are themselves the targets of selection. Furthermore, even genome scans performed with large resequencing panels and whole genome sequencing are not suited to directly identify domestication genes. Identifying adaptation at the genetic level will require experimental tests of selection on the genes underlying phenotypic traits and would be best investigated by incorporating genomics into field experiments to characterize the fitness effects of individual mutations (Barret and Hoekstra, 2011).
This first genomic investigation into the domestication of squashes and pumpkins offers a rare comparison of the genetic consequences of domestication in two crops that were independently domesticated from closely related species. As such, our results provide novel insights into how multiple factors influence the effect of domestication on the genetic diversity of crops. We found that two species that share many characteristics bear contrasting signatures of selection, and we identify broad morphological diversity of cultivars as a possible indicator of high genetic diversity in a crop. In addition, modern breeding practices do not always reduce crop genetic diversity, and breeders may discover untapped genetic diversity underlying traits of interest in improved germplasm.
Data Availability Statement
The sequencing data generated for this study can be found in the NCBI Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) under BioProject ID PRJNA699883.
Author Contributions
HK, PS, and DS designed the project. FA, GS, and LE conducted fieldwork and collected samples. HK generated and analyzed the data, produced the figures, and drafted the manuscript. All authors critically reviewed the manuscript, contributed to writing, and read and approved the submitted version.
Funding
This work was supported by NSF grant DEB-1406960 (Doctoral Dissertation Improvement Grant to DS, PS, and HK). Sampling of Mexican accessions was supported by grants CONABIO KE 004 and PE001 to LE and Rafel Lira-Saade (FES-Iztacala. UNAM). Fieldwork by FA was supported by PICT 2012-0709 (ANPCyT) and P-UE 0043 CONICET.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AC declared a shared affiliation, though no collaboration, with two of the authors GS and LE, to the handling editor.
Acknowledgments
We thank D. Barrera for help with lab work and T. Fields, K. R. Reitsma, and S. Tennies for facilitating access to seed material from USDA ARS repositories. We thank C. Khoury for preparing the maps.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.618380/full#supplementary-material
Supplementary Figure 1 | Delta K plots from the Evanno method for detecting the appropriate number of population clusters from STRUCTURE results for C. maxima (A) and C. argyrosperma (B) for values of K = 1 through 6.
Supplementary Table 1 | Accession information for samples included in this study.
Supplementary Table 2 | STRUCTURE ancestry coefficients per accession for values of K = 2, 4, and 5. These values correspond to the bar graph in Figures 2A,B. Note that the population number is assigned arbitrarily by STRUCTURE and independently between species-level analyses. The column headers denote which population corresponds to which color in Figure 2.
Supplementary Table 3 | Domestication features in landrace and improved C. maxima ssp. maxima and C. argyrosperma ssp. argyrosperma based on loci with FST and π log-ratio above thresholds determined by our outlier tests.
Supplementary Table 4 | Significantly enriched full GO terms and GO slim terms identified for C. maxima and C. argyrosperma.
Footnotes
References
Allaby, R. G., Ware, R. L., and Kistler, L. (2019). A re-evaluation of the domestication bottleneck from archaeogenomic evidence. Evol. Appl. 12, 29–37. doi: 10.1111/eva.12680
Altieri, M. A., Anderson, M. K., and Merrick, L. C. (1987). Peasant agriculture and the conservation of crop and wild plant resources. Conserv. Biol. 1, 49–58. doi: 10.1111/j.1523-1739.1987.tb00008.x
Barrera-Redondo, J., Sánchez de la Vega, G., Aguirre-Ligouri, J. A., Castellanos-Morales, G., and Gutierrez-Guerrero, Y. T. (2021). The domestication of cucurbita argyrosperma as revealed by the genome of its wild relative. Horticult. Res. 8:109. doi: 10.1038/s41438-021-00544-9
Barret, R. D. H., and Hoekstra, H. E. (2011). Molecular spandrels: tests of adaptation at the genetic level. Nat. Rev. Genet. 12, 767–780. doi: 10.1038/nrg3015
Baute, G. J., Kane, N. C., Grassa, C. J., Lai, Z., and Rieseberg, L. H. (2015). Genome scans reveal candidate domestication and improvement genes in cultivated sunflower, as well as post-domestication introgression with wild relatives. New Phytol. 206, 830–838. doi: 10.1111/nph.13255
Benazzo, A., Panziera, A., and Bertorelle, G. (2015). 4P: fast computing of population genetics statistics from large DNA polymorphism panels. Ecol. Evol. 5, 172–175. doi: 10.1002/ece3.1261
Bisognin, D. A. (2002). Origin and evolution of cultivated cucurbits. Ciência Rural 32, 715–723. doi: 10.1590/s0103-84782002000400028
Brown, T. A. (2019). Is the domestication bottleneck a myth? Nat. Plants 5, 337–338. doi: 10.1038/s41477-019-0404-1
Cao, K., Zheng, Z., Wang, L., Liu, X., Zhu, G., Fang, W., et al. (2014). Comparative population genomics reveals the domestication history of the peach, prunus persica, and human influences on perennial fruit crops. Genome Biol. 15:415.
Casañas, F., Simó, J., Casals, J., and Prohens, J. (2017). Toward an evolved concept of landrace. Front. Plant Sci. 8:145. doi: 10.3389/fpls.2017.00145
Cheng, F., Wu, J., Cai, C., Fu, L., Liang, J., Borm, T., et al. (2016). Genome resequencing and comparative variome analysis in a Brassica rapa and Brassica oleracea collection. Sci. Data 3:160119.
Chigmura Ngwerume, F., and Grubben, G. J. H. (2004). “Cucurbita,” in Vegetables, ed. G. J. H. Grubben (New York, NY: PROTA).
Cornille, A., Gladieux, P., Smulders, M. J. M., Roldán-Ruiz, I., Laurens, F., Le Cam, B., et al. (2012). New insight into the history of domesticated apple: secondary contribution of the european wild apple to the genome of cultivated varieties. PLoS Genet. 8:e1002703. doi: 10.1371/journal.pgen.1002703
Cutler, H. C., and Whitaker, T. W. (1961). History and distribution of the cultivated cucurbits in the Americas. Am. Antiquity 26, 469–485. doi: 10.2307/278735
Dallman, N., and Dallman, M. (2009). Breeding classic Cucurbita maxima Buttercup Squash for increased genetic diversity. Cucurbit Genet. Cooperative Rep. 31, 17–18.
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Decker-Walters, D. S., and Walters, T. W. (2000). “Squash,” in The Cambridge World History of Food, eds K. C. Ornelas and K. F. Kiple (Cambridge: Cambridge University Press).
Decker-Walters, D. S., Walters, T. W., Posluszny, U., and Kevan, P. G. (1990). Genealogy and gene flow among annual domesticated species of cucurbita. Can. J. Bot. 68, 782–789. doi: 10.1139/b90-104
Della Vecchia, P. T., Terenciano Sobrinho, P., and Terenciano, A. (1993). Breeding Bush Types of C. moschata with Field Resistance to PRSV-w. Cucurbit Genet. Cooperative Rep. 16, 70–72.
Diez, C. M., Trujillo, I., Martinez-Urdiroz, N., Barranco, D., Rallo, L., Marfil, P., et al. (2015). Olive domestication and diversification in the mediterranean basin. New Phytol. 206, 436–447. doi: 10.1111/nph.13181
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 3, 11–15.
Esquinas-Alcázar, J. (2005). Protecting crop genetic diversity for food security: political, ethical and technical challenges. Nat. Rev. Genet. 6, 946–953. doi: 10.1038/nrg1729
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294x.2005.02553.x
Fang, L., Wang, Q., Hu, Y., Jia, Y. H., Chen, J. D., Liu, B. L., et al. (2017). Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 49:1089. doi: 10.1038/ng.3887
Felsenstein, J. (1981). Evolutionary trees from gene frequencies and quantitative characters: finding maximum likelihood estimates. Evol. Int. J. Organ. Evol. 35, 1229–1242. doi: 10.2307/2408134
Ferriol, M., Picó, B., and Nuez, F. (2004). Morphological and molecular diversity of a collection of Cucurbita maxima landraces. J. Am. Soc. Horticult. Sci. 129, 60–69. doi: 10.21273/jashs.129.1.0060
Francis, R. M. (2017). pophelper: an R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 17, 27–32. doi: 10.1111/1755-0998.12509
Garrison, E. P., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv Genom. [Preprint]. arXiv: 1207.3907.
Glémin, S., and Bataillon, T. (2009). A comparative view of the evolution of grasses under domestication. New Phytol. 183, 273–290. doi: 10.1111/j.1469-8137.2009.02884.x
Gnirke, A., Melnikov, A., Maguire, J., Rogov, P., LeProust, E. M., Brockman, W., et al. (2009). Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189. doi: 10.1038/nbt.1523
Guo, J., Wang, Y., Song, C., Zhou, J., Qiu, L., Huang, H., et al. (2010). A single origin and moderate bottleneck during domestication of soybean (Glycine max): implications from microsatellites and nucleotide sequences. Ann. Bot. 106, 505–514. doi: 10.1093/aob/mcq125
Haudry, A., Cenci, A., Ravel, C., Bataillon, T., Brunel, D., Poncet, C., et al. (2007). Grinding up wheat: a massive loss of nucleotide diversity since domestication. Mol. Biol. Evol. 24, 1506–1517. doi: 10.1093/molbev/msm077
Huang, H.-X., Yu, T., Li, J.-X., Qu, S.-P., Wang, M.-M., Wu, T.-Q., et al. (2019). Characterization of cucurbita maxima fruit metabolomic profiling and transcriptome to reveal fruit quality and ripening gene expression patterns. J. Plant Biol. 62, 203–216. doi: 10.1007/s12374-019-0015-4
Hufford, M. B., Xun, X., van Heerwaarden, J., Pyhäjärvi, T., Jer-Ming, C., Cartwright, R. A., et al. (2012). Comparative population genomics of maize domestication and improvement. Nat. Genet. 44, 808–811.
Iorizzo, M., Senalik, D. A., Ellison, S. L., Grzebelus, D., Cavagnaro, P. F., Allender, C., et al. (2013). Genetic structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am. J. Bot. 100, 930–938. doi: 10.3732/ajb.1300055
Kassa, M. T., Penmetsa, R. V., Carrasquilla-Garcia, N., Sarma, B. K., Datta, S., Upadhyaya, H. D., et al. (2012). Genetic patterns of domestication in pigeonpea (Cajanus cajan (L.) Millsp.) and wild cajanus relatives. PLoS One 7:e39563. doi: 10.1371/journal.pone.0039563
Kates, H. R. (2019). “Pumpkins, squashes, and gourds (Cucurbita L.) of North America,” in North American Crop Wild Relatives, eds S. Greene, K. Williams, C. Khoury, M. Kantar, and L. Marek (Cham: Springer).
Kates, H. R., Soltis, P. S., and Soltis, D. E. (2017). Evolutionary and domestication history of cucurbita (pumpkin and squash) species inferred from 44 nuclear loci. Mol. Phylogenet. Evol. 111, 98–109. doi: 10.1016/j.ympev.2017.03.002
Lee, T.-H., Guo, H., Wang, X., Kim, C., and Paterson, A. H. (2014). SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15:162. doi: 10.1186/1471-2164-15-162
Lee, W.-P., Stromberg, M. P., Ward, A., Stewart, C., Garrison, E. P., and Marth, G. T. (2014). MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS One 9:e90581. doi: 10.1371/journal.pone.0090581
Lema, V. S. (2009). Domesticación Vegetal y Grados de Dependencia Ser Humano-Planta en el Desarrollo Cultural Prehispánico del Noroeste Argentino. Tesis Doctoral. Argentina: Facultad de Ciencias Naturales y Museo, Universidad Nacional de La Plata.
Lema, V. S. (2011). The possible influence of post-harvest objectives on cucurbita maxima subspecies maxima and subspecies andreana evolution under cultivation at the argentinean northwest: an archaeological example. Archaeol. Anthropol. Sci. 3, 113–139. doi: 10.1007/s12520-011-0057-0
Li, C. (2006). Rice domestication by reducing shattering. Science 311, 1936–1939. doi: 10.1126/science.1123604
Li, Z.-M., Zheng, X.-M., and Ge, S. (2011). Genetic diversity and domestication history of African rice (Oryza glaberrima) as inferred from multiple gene sequences. Theor. Appl. Genet. 123, 21–31.
Lira-Saade, R. (1995). Estudios Taxonómicos y Ecogeográficos de las Cucurbitácea Latinoamericanas de Importancia Económica. Systematic and Ecogeographic Studies on Crop Genepools 9. Rome: International Plant Genetic Resources Institute.
Lira-Saade, R., Andres, T. C., and Nee, M. (1995). “Cucurbita L,” in Systematic and Ecogeographic Studies on Crop Genepools, ed. J. M. Edmonds (Roma: International Plant Genetic Resources Institute), 1–115. doi: 10.1007/978-3-319-23534-9_1
Lira-Saade, R., and Montes Hernández, M. (1994). “Cucurbits (Cucurbita spp.),” in Neglected Crops : 1492 from a Different Perspective, eds J. Hernando Bermejo and J. Leon (Rome: FAO), 63–77.
Liu, S., Liu, Y., Yang, X., Tong, C., David, E., Parkin, I. A. P., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5:3930.
Martínez, A., Lema, V., Capparelli, A., Bartoli, C., Anido, F. L., and Pérez, I. (2018). Multidisciplinary studies in Cucurbita maxima (squash). Veg. Hist. Archaeobot. 27, 207–217. doi: 10.1007/s00334-017-0637-8
Meirmans, P. G. (2015). Seven common mistakes in population genetics and how to avoid them. Mol. Ecol. 24, 3223–3231. doi: 10.1111/mec.13243
Merrick, L. C. (1990). “Systematics and evolution of a domesticated squash, Cucurbita argyrosperma, and its wild and weedy relatives,” in Biology and utilization of the Cucurbitaceae, eds D. M. Bates, R. W. Robinson, and C. Jeffrey (New York, NY: Cornell University Press), 77–95. doi: 10.7591/9781501745447-009
Meyer, R. S., and Purugganan, M. D. (2013). Evolution of crop species: genetics of domestication and diversification. Nat. Rev. Genet. 14, 840–852. doi: 10.1038/nrg3605
Millán, R. (1945). Variaciones del Zapallito Amargo Cucurbita andreana y el Origen de Cucurbita maxima. Rev. Argentina Agron. 12, 86–93.
Montes-Hernandez, S., and Eguiarte, L. E. (2002). Genetic structure and indirect estimates of gene flow in three taxa of Cucurbita (Cucurbitaceae) in Western Mexico. Am. J. Bot. 89, 1156–1163. doi: 10.3732/ajb.89.7.1156
Nabhan, G. P., and Felger, R. S. (1985). “Wild desert relatives of crops: their direct uses as food,” in Plants for Arid Lands, eds G. E. Wickens, J. R. Goodin, and D. V. Field (Dordrecht: Springer).
Nee, M. (1990). The domestication of cucurbita (Cucurbitaceae). Econ. Bot. 44:56. doi: 10.1007/bf02860475
Nei, M., and Li, W. H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E., and Martin, J. L. (2014). PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. doi: 10.1093/molbev/msu136
Piperno, D. R., Ranere, A. J., Holst, I., Iriarte, J., and Dickau, R. (2009). Starch grain and phytolith evidence for early ninth millennium B.P. maize from the Central Balsas River Valley, Mexico. Proc. Natl. Acad. Sci. U.S.A. 106, 5019–5024.
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Qiu, J., Zhou, Y., Mao, L., Ye, C., Wang, W., Zhang, J., et al. (2017). Genomic variation associated with local adaptation of weedy rice during de-domestication. Nat. Commun. 8:15323.
Ranere, A. J., Piperno, D. R., Holst, I., Dickau, R., and Iriarte, J. (2009). The cultural and chronological context of early holocene maize and squash domestication in the Central Balsas River Valley, Mexico. Proc. Natl. Acad. Sci. U.S.A. 106, 5014–5018. doi: 10.1073/pnas.0812590106
Robinson, R. W. (1999). Rationale and methods for producing hybrid cucurbit seed. J. New Seeds 1, 1–47. doi: 10.1300/j153v01n03_01
Rosas, R. V., Andres, T., and Nee, M. (2004). The Goldman Cucurbit Collecting Expedition in Peru, 2004. Available online at: http://www.cucurbit.org/Peru/PeruReport.html (accessed August 08, 2017).
Sánchez-de la Vega, G., Castellanos-Morales, G., Gámez, N., Hernández-Rosales, H. S., Vázquez-Lobo, A., Aguirre-Planter, E., et al. (2018). Genetic resources in the ‘calabaza pipiana’ squash (Cucurbita argyrosperma) in Mexico: genetic diversity, genetic differentiation and distribution models. Front. Plant Sci. 9:400. doi: 10.3389/fpls.2018.00400
Schmutz, J., McClean, P. E., Mamidi, S., Wu, G. A., Cannon, S. B., Grimwood, J., et al. (2014). A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713.
Singh, S. P., Gepts, P., and Debouck, D. G. (1991). Races of common bean (Phaseolus vulgaris, Fabaceae). Econ. Bot. 45, 379–396. doi: 10.1007/bf02887079
Small, E. (2013). North American Cornucopia: Top 100 Indigenous Food Plants. Boca Raton, FL: CRC Press.
Small, E. (ed.). (2014). “Squash (Cucurbita pepo squash),” in North American Cornucopia: Top 100 Indigenous Food Plants, 1st Edn, (Boca Raton, FL: CRC Press).
Smith, B. D. (2006). Eastern North America as an independent center of plant domestication. Proc. Natl. Acad. Sci. U.S.A. 103, 12223–12228. doi: 10.1073/pnas.0604335103
Sun, H., Wu, S., Zhang, G., Jiao, C., Guo, S., Ren, Y., et al. (2017). Karyotype stability and unbiased fractionation in the paleo-allotetraploid cucurbita genomes. Mol. Plant 10, 1293–1306. doi: 10.1016/j.molp.2017.09.003
Villa, T., Maxted, N., Scholten, M., and Ford-Lloyd, B. (2005). Defining and identifying crop landraces. Plant Genet. Resourc. 3, 373–384. doi: 10.1079/PGR200591
Watterson, G. A. (1975). On the number of segregating sites in genetical models without recombination. Theor. Population Biol. 7, 256–276. doi: 10.1016/0040-5809(75)90020-9
Wen, Z. X., Boyse, J. F., Song, Q. J., Cregan, P. B., and Wang, D. C. (2015). Genomic consequences of selection and genome-wide association mapping in soybean. BMC Genomics 16:671. doi: 10.1186/s12864-015-1872-y
Whitaker, T. W., and Robinson, R. W. (1986). “Squash breeding,” in Breeding Vegetable Crops, ed. M. J. Bassett (Westport, CT: AVI Publishing Company).
Zeven, A. C. (1998). Landraces: a review of definitions and classifications. Euphytica 104, 127–139. doi: 10.1023/a:1018375009640
Zeven, A. C., and Zhukovskii, P. M. (1975). Dictionary of Cultivated Plants and Their Centres of Diversity: Excluding Ornamentals, Forest Trees and Lower Plants. Wallingford: CABI.
Zhang, G., Ren, Y., Sun, H., Guo, S., Zhang, F., Zhang, J., et al. (2015). A high-density genetic map for anchoring genome sequences and identifying QTLs associated with dwarf vine in pumpkin (Cucurbita maxima Duch.). BMC Genomics 16:1101. doi: 10.1186/s12864-015-2312-8
Zheng, X., Levine, D., Shen, J., Gogarten, S. M., Laurie, C., and Weir, B. S. (2012). A high-performance computing toolset for relatedness and principal component analysis of SNP Data. Bioinformatics 28, 3326–3328. doi: 10.1093/bioinformatics/bts606
Keywords: domestication, ancestral gene flow, population genomics, Cucurbita, targeted sequencing
Citation: Kates HR, Anido FL, Sánchez-de la Vega G, Eguiarte LE, Soltis PS and Soltis DE (2021) Targeted Sequencing Suggests Wild-Crop Gene Flow Is Central to Different Genetic Consequences of Two Independent Pumpkin Domestications. Front. Ecol. Evol. 9:618380. doi: 10.3389/fevo.2021.618380
Received: 16 October 2020; Accepted: 31 May 2021;
Published: 12 July 2021.
Edited by:
Raymond L. Tremblay, University of Puerto Rico, Puerto RicoReviewed by:
Alejandro Casas, Universidad Nacional Autónoma de México, MexicoAlessandro Alves-Pereira, State University of Campinas, Brazil
Copyright © 2021 Kates, Anido, Sánchez-de la Vega, Eguiarte, Soltis and Soltis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heather R. Kates, aGthdGVzQHVmbC5lZHU=