
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Ecol. Evol. , 19 April 2021
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.565649
This article is part of the Research Topic DNA Barcodes: Controversies, Mechanisms and Future Applications View all 13 articles
 David S. Thaler1,2*
David S. Thaler1,2*Animal and plant biodiversity is decreasing. In contrast, the global direction and the pace of change in microbial, including viral, biodiversity is unknown. Important niches for microbial diversity occur in highly specific associations with plants and animals, and these niches are lost as hosts become extinct. The taxonomic diversity of human gut bacteria is reported to be decreasing. On the other hand, SARS-CoV-2 variation is increasing. Where microbes are concerned, Darwin’s “tangled bank” of interdependent organisms may be composed mostly of other microbes. There is the likelihood that as some classes of microbes become extinct, others evolve and diversify. A better handle on all processes that affect microbial biodiversity and their net balance is needed. Lack of insight into the dynamics of evolution of microbial biodiversity is arguably the single most profound and consequential unknown with regard to human knowledge of the biosphere. If some or all parts of microbial diversity are relentlessly increasing, then survey approaches may be too slow to ever catch up. New approaches, including single-molecule or single-cell sequencing in populations, as well as focused attention on modulators and vectors of vertical and horizontal evolution may offer more direct insights into some aspects of the pace of microbial evolution.
Animal and plant biodiversity on earth is decreasing. Many important features of this decrease are unclear, including ways in which the pace, i.e., the rate of decrease, is comparable to great extinctions defined by paleontology and how the current decrease is distributed among different phylogenetic domains and ecosystems (Di Marco et al., 2019; Trisos et al., 2020). However, the overall trajectory is clearly downward (Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services, 2020). The decrease in global biodiversity of “macrobes,” i.e., eukaryotic multicellular differentiated organisms (EMDOs), commonly known as plants and animals, is a key issue of the Anthropocene.
In contrast to what we know of the world of plants and animals, we have no idea whether global microbial diversity is increasing, decreasing, or staying the same. For the purpose of this discussion microbial biodiversity includes eubacteria, archaea, protists, single-celled fungi, and viruses of all forms, bacteriophages, archaeaphages, and viruses of eukaryotes, including viruses of animals and plants. It is a blind spot– almost a scandal– that the question of the global trends of microbial biodiversity seems never to have been raised. We raise it here. Once the question is asked, are there direct ways to address it? The intent here is to review literature in search of additional ways to address pace and direction. If there are ways to short-circuit survey approaches, what might they be? Can the enormous amount of data on microbial genomes and metagenomics be examined with rate of change in mind? What new approaches might shed light on the question? The approaches proposed in this article are by no means final and are certainly not protocols to solve the problem. The purpose here is to frame the rate of change of microbial biodiversity as an interesting and important question on which consequential progress is possible.
Consider the graphs below in which the Y axis represents biological diversity and the X axis is time Figure 1). The trajectory of biodiversity at any point in time is the first derivative (defined in calculus as the tangent of the curve at that point) of total biodiversity versus time. Our goal is to directly measure the straight line that is tangent to the curve at the present time).
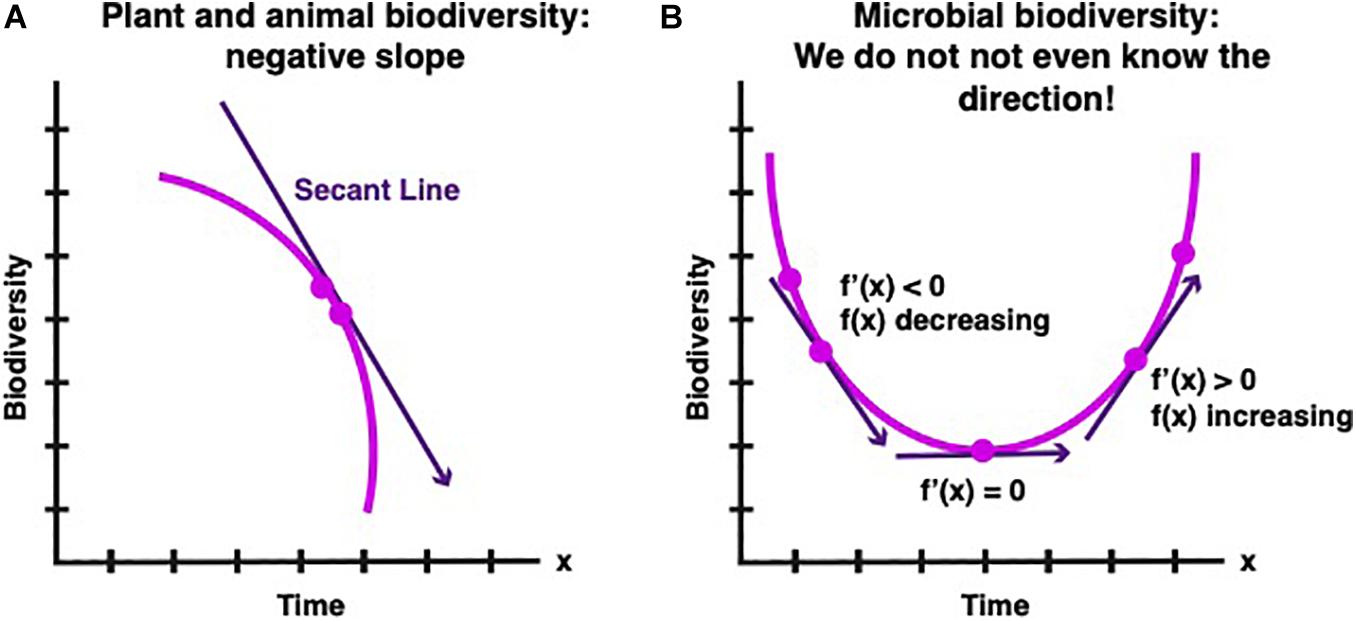
Figure 1. (A) In pre-calculus, the secant is a straight line connecting two points on a curve. The present-day approach to measure changes in biodiversity is to count the number of species at different times and determine the slope of the secant. In the case of animal and plant biodiversity the slope of the secant is negative. (B) The breakthrough of calculus allows defining and finding the slope at a single point. A breakthrough analogous to calculus would be tremendously helpful to determining the trajectory of microbial biodiversity because the secant approach is in many cases difficult or seemingly impossible to implement.
Three preliminary issues require consideration: (a) What is meant by microbial biodiversity? (b) By what metrics is biodiversity in microbial realms comparable to the biodiversity of EMDOs (i.e., animals and plants?) (c) What baseline knowledge of microbial biodiversity is necessary in order to analyze how that diversity changes over time? I propose the following point of view: (a) Microbial biodiversity is the distribution of individuals in sequence space. (b) Microbial distribution in sequence space is similar enough to EMDO distribution that meaningful comparisons are possible. (c) Global directions of microbial evolution need not depend on catalogs of species and phylogenies. In some cases, direct measurement of the derivative at a single point need not depend on knowledge of the shape of the curve or the equation for the entire line. These interrelated issues are expanded below. The answers proposed below are meant to initiate discussion of the problem, not to prescribe specific approaches.
Carl Woese pioneered the use of small subunit (SSU) RNA as a tool for species identification and phylogenetic analysis across the entirety of the living world (with the notable exception of viruses) (Woese and Fox, 1977; Woese et al., 1990). Woese’s insight of universal sequence with sufficient variation also applies to mitochondrial cytochrome oxidase I (mtCOI) DNA barcoding in the animal kingdom. The key recognized by Woese is the need for sequences similar enough across the groups of interest that they can be aligned and compared. Small subunit RNA works marvelously well to divide life into large groups. Subsequent work has moved beyond genes encoding SSU RNA, using other genes and gene families (Zhu et al., 2019; Williams et al., 2020). Whole genomes have also been used for parsing bacteria and are able to distinguish clusters of individuals (species or strains) separated by horizontal gene transfer (HGT) rather than only by point mutations in shared genes (Rodriguez et al., 2018; Murray et al., 2020). The relative roles of vertical and horizontal evolution are of great interest (Woese, 2004; Frazao et al., 2019).
DNA barcodes and their relationship to animal species invite comparison to measures of microbial biodiversity. DNA barcoding by mtCOI has been more extensively discussed elsewhere (Stoeckle and Thaler, 2018). The clustering pattern of macroscopic life was elegantly articulated by Dobzhansky in his foundational book Genetics and the Origin of Species (Dobzhansky, 1937), page 4:
If we assemble as many individuals living at a given time as we can, we notice that the observed variation does not form a single probability distribution or any other kind of continuous distribution. Instead, a multitude of separate, discrete, distributions are found. In other words, the living world is not a single array of individuals in which any two variants are connected by unbroken series of intergrades, but an array of more or less distinctly separate arrays, intermediates between which are absent or at least rare. Each array is a cluster of individuals, usually possessing some common characteristics and gravitating to a definite modal point in their variation. Therefore the biological classification is simultaneously a man-made system of pigeonholes devised for the pragmatic purpose of recording observations in a convenient manner and an acknowledgement of the fact of organic discontinuity.
DNA barcodes constitute a single metric by which the “feeling that it must be right” can be given a single quantitative meaning across the entire animal kingdom. Important findings have emerged from analysis of several million COI DNA barcodes. In groups throughout the animal kingdom, DNA barcode clusters largely correspond to what experts in each group have determined to be species. The extent of variance within clusters is similar and small (0.0% to 0.5% with most ∼0.2%) as determined through average pairwise difference (APD) within species from widely different groups including birds, mammals, fish, and insects. In most cases the APD separating nearest neighbor clusters is 1% to 2%. A key controversy regarding sequence clustering is the extent to which variance within clusters is neutral. For mitochondrial DNA barcodes most variation changes synonymous codons. The conclusion is not certain but the preponderance of evidence is that synonymous codon variation in the mitochondrial genome is neutral (Stoeckle and Thaler, 2018). If so, then synonymous variation is a passive passenger and also an indicator of population processes such that the accumulated variation is a function of population size and time (Kimura, 1986).
One takeaway is that the clustering pattern seen in macroscopic life is in a general way also a property of microscopic forms of life (Tibayrenc et al., 2015). Mechanisms necessarily differ because in many microbes (and fewer macrobes) the reproductive life cycle is not coupled to genetic exchange.
One avenue for further work will be the critical comparison of variation within SSUs (genes encoding small subunit RNA, 16S in eubacteria and archaea; 18S from the cytoplasmic ribosomes of eukaryotes) with the variance of mitochondrial COI DNA barcodes. In this way we could learn whether the amount of variation accumulated in extant species of EMDOs corresponds to that found in groups of microbes. The same APD approach should be applicable to mtCOI DNA barcodes and SSU analysis. A related area of important uncertainty in both cases (SSU RNA and mtCOI) is which variants are selectively neutral and which are subject to selection. Patterns of clustering of variation among individuals in extant populations of microbes may prove meaningfully comparable with analogous measures in plants and animals.
Microbial diversity is generated through both vertical and horizontal mechanisms. Evolution involves both the generation of diversity and selection among variants. However, these two processes are not always neatly separated (Thaler, 1994). HGT and mutation in vertical lineages are each generated through the action of enzymes encoded by genes that are themselves subject to evolution. The presence of genes involved in HGT, their activity and allelic state are indicative of the rates by which combinatorial microbial variants are generated.
There are several types of horizontal gene transfer mediated by bacteriophage through either specialized (Morse et al., 1956) or generalized (Zinder and Lederberg, 1952) transduction, conjugation (Lederberg and Tatum, 1946) (Lederberg et al., 1952; Cavalli et al., 1953), DNA-mediated transformation (Avery et al., 1944), or cell fusion (Gratia and Thiry, 2003; Gratia, 2005). New DNA entering a cell may either replicate independently or integrate into the host chromosome, or both. Integration into the host chromosome may involve illegitimate (Scwacha and Kleckner, 1994), site specific (Campbell, 1965) or homologous (Clark and Margulies, 1965) recombination. The degree of sequence similarity required to support homologous recombination is modulated by the mismatch repair and SOS systems which themselves are composed of genes and genetic networks subject to mutation and other genetic and physiological changes (Rayssiguier et al., 1989; Thaler, 1994; Magnasco and Thaler, 1996; Moxon and Thaler, 1997; Field et al., 1999). Sequence studies have inferred many of these processes independently (Cohan, 2017, 2019). Thus we know from experimental work that horizontal gene transfer can happen, with some detail as to molecular mechanisms, and from sequencing studies that these processes occur frequently in nature. Approaches need to be developed that directly assay mechanisms that mediate horizontal gene transfer.
Important niches for microbial diversity occur in highly specific associations with EMDOs, such as the gut microbiota in animals and nitrogen-fixing nodules on the roots of legumes. These specialized microbial communities probably cease to exist along with the extinction of their associated animals and plants. On the other hand, much of microbial life is understood only in the context of other microbes. For microbes, Darwin’s “tangled bank” of “elaborately constructed forms, so different from each other, and so dependent upon each other” may consist mostly of other microbes.
A “species” may be considered to be a cluster in nucleotide sequence space. The number of different animal and plant species, although unknown, is the subject of reasonable estimation (Mora et al., 2011). The number of 16S eubacterial and archaeal sequence clusters has also been estimated (Louca et al., 2019) albeit with the caveat that estimates based on 16S necessarily underestimate total sequence diversity (Shevchenko et al., 2019). The difficulty in estimating sequence diversity of viruses inclusive of bacteriophage and archaeaphage lies partly in the plausibility of strains appearing and disappearing more rapidly than a comprehensive survey at any one moment could catch (Hadfield et al., 2018). What seems most likely is that the biosphere’s sequence diversity at present is probably dominated by microbes, including viruses. A possible qualification to the statement of microbial dominance of biospheric sequence space is if somatic immune-system diversity is counted (Lin et al., 2020; Roskin et al., 2020) and turns out to be sufficient to shift the accounting of total diversity in favor of EMDOs. A different type of possible exception concerns heritable biological variation that is not based in polynucleotide sequence. DNA sequence could, in principle, account for all hereditary information. However, the fact that extant cells come only from other cells leaves open the possibility that DNA sequence alone is not sufficient for biological continuity (Thaler, 2009).
The classical way to measure the rate of change in biodiversity is to measure diversity at different times and divide the difference by the amount of time that has passed. For example, if the number of species decreases by half over the course of a year, the rate of loss is 0.5/yr. This classical approach of counting species and seeing how the counts change with time can be applied in specialized contexts, such as monitoring changes in microbial diversity within the intestinal microbiome (Magro et al., 2019). But there are two related problems making this approach difficult and perhaps impossible to generalize.
The first problem is that the extent of current microbial biodiversity is unknown (Dance, 2020), possibly with a large fraction in hard-to-access, rare, or extreme environments (Sogin et al., 2006; Amaral-Zettler et al., 2010). The deep hot biosphere may contain the majority of our planet’s microbial biodiversity (Magnabosco et al., 2019). The second problem is a possible “chicken and egg” paradox that would preclude direct measurement of the rate of microbial biodiversification through a series of independent timepoints: Suppose that the rate of generation of new microbial diversity is very fast. If so, a baseline sequence library may never be finished because new diversity is generated more rapidly than it can be measured. It might require 20 years before there is sufficient understanding of the deep biosphere and other hard-to-access environments to have an adequate baseline library. However, if microbial–including viral–evolution is as rapid as it might be, establishing a complete baseline inventory may prove impossible. These two problems motivate the pursuit of approaches with potential for directly measuring the first derivative of microbial biodiversity, in order to gauge instantaneous directions and rates of change.
Species, in common practice of mitochondrial barcode sequence analysis, are usually assigned on the basis of their closeness to a consensus sequence. Counts taken at successive times determine if consensus sequences are missing and if new ones have appeared. Census counts of a species are enumerated as the number of individuals that are “close enough” to the consensus sequence. Microbiome analysis often enumerates not “species” but the diversity of “operational taxonomic units” (OTUs) in the context of SSU-encoding genes (usually 16S). OTUs are clusters of sequences separated by more than 2% (for fungi) or more than 3% (for eubacteria and archaea) from the nearest cluster.
“Quasispecies” are clouds in a sequence space formed by populations possessing mutable genomes (Eigen, 1993; Bull et al., 2005; Domingo and Perales, 2019; Lu et al., 2020). Modeling suggests that favored quasispecies allow many neutral mutations. Selectively favored mutations migrate the quasispecies cloud or establish a new one. Consider the case when a founder clone seeds a new quasispecies. The distribution of the quasispecies in sequence space is a function of the number of generations elapsed since clonal founding as well as the mutation rate and spectrum. The greater the number of generations and the higher the mutation rate, the larger the quasispecies sequence cloud will be. The size of a quasispecies cloud can be accessed through the positively correlated statistic APD among individuals within the cloud (Dridi et al., 2015; de Azevedo et al., 2017). Consider the case of a population that starts from a single sequence, a founder and its clonal descendants. A larger APD and cloud are functions of the number of generations and the mutation rate per generation. The quasispecies concept is related to the molecular clock, neutral evolution, and Luria-Delbrück mutation.
Zuckerkandl and Pauling in 1965 hypothesized a molecular clock based on the rate of amino acid substitutions in hemoglobin compared with fossil evidence giving an independent measure of time of divergence (Zuckerkandl and Pauling, 1965). Kimura in 1968 proposed that most sequence changes are selectively neutral or nearly neutral and the accumulation of variation in a population follows from the mutation rate, the number of generations, and the chance-driven loss or gain of variants (Kimura, 1968). The Luria-Delbrück interpretation of mutant clone sizes in a growing bacterial population implicitly includes the assumption that the accumulating variants are neutral during an exponential growth phase before selection (Luria and Delbrück, 1943; Stewart et al., 1990).
Phylogeography of mitochondrial genome coalescence, in combination with archeology and paleontology dates the origin of the modern human mitochondrial sequence in the range of 150,000 to 200,000 years ago (Mellars, 2006). Most mitochondrial sequence variation appears to be neutral (Richards et al., 2000; Tishkoff and Williams, 2002; Forster, 2004; Kivisild et al., 2006), but for a different view see Mishmar et al. (2003). The APD of mitochondrial sequences within animal species tends toward the same low value and most variation is probably neutral in divergent phyla such as insects, birds, mammals and, fish (Stoeckle and Thaler, 2018). Humans are an average animal species when analyzed in this way; the human APD is 0.1% and the majority of animal species are in the range of 0.1% to 0.2% (Thaler and Stoeckle, 2016). Outgrowth from a clonal sequence in a similar way over a similar time frame (for different species this varies from a range of ca. “last week” to half a million years ago, with a median at 100,000 to 200,000 years ago) is a plausible way to account for the similarity of mitochondrial sequence clustering within different species across the animal kingdom (Stoeckle and Thaler, 2018). The variance within clusters measured by APD is a prototype of an independent way to obtain the derivative of biodiversity–the pace of change in the sense of population replacement. Successive clonal replacement is very much the manner of evolution in the microbial realm (Tibayrenc et al., 2015; Baym et al., 2016). Baym and colleagues have produced a superb YouTube video demonstrating in an experimental setting the evolution of bacteria to survive in increasingly high concentrations of an antibiotic (Link in Figure 2 caption). It seems reasonable that a method akin to that used to estimate the age since expansion from a clonal sequence of mitochondria within an extant population, i.e., the variance of individuals around a consensus sequence as measured by APD, can also be used to estimate the time or the number of generations that have elapsed since a clonal origin of cellular microbial and viral populations.
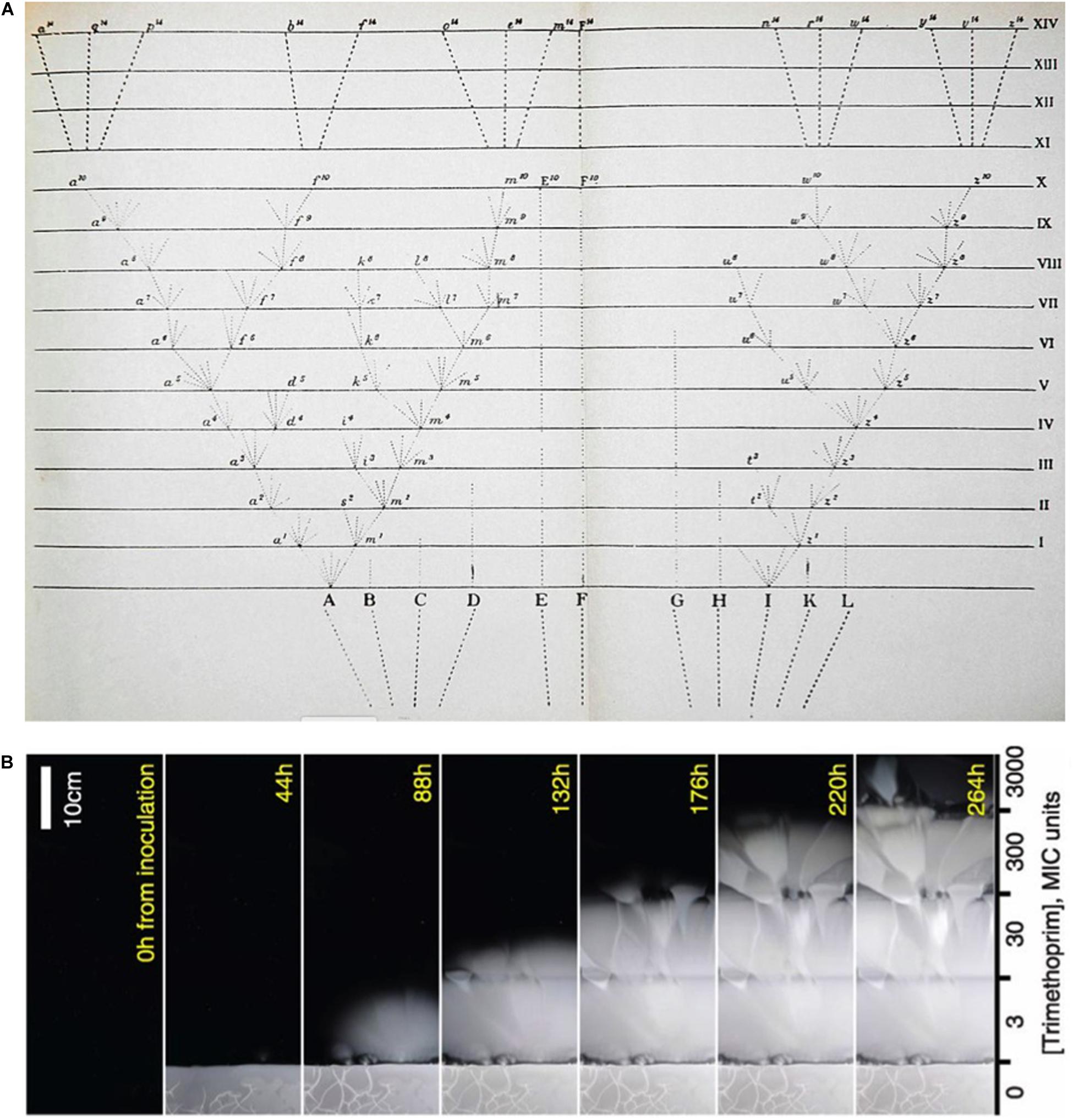
Figure 2. (A) Darwin’s “Tree of Life” diagram from his Origin of Species (Darwin, 1860) juxtaposed with (B) a time-lapse study of bacterial growth across a step-wise concentration gradient of antibiotic (Baym et al., 2016). A video of this marvellous experiment may be accessed here: https://www.youtube.com/watch?v=plVk4NVIUh8.
In the eubacterial and archael realms of microbiome metagenomic sequence many analyses use OTUs rather than the word “species.” OTUs are clusters separated by 2% or 3% in sequence space. The lack of further definitive knowledge follows in some cases because the organisms harboring them have never been cultured. Another part of the ignorance is simply that there are so many different sequence clusters. A related aspect is the semi-ignorance of sequence classification in the absence of experiment. A sequence may be somewhat similar to one in another organism that has been cultured, or to an enzyme for which a reaction has been characterized. There may be a temptation to overestimate the certainty when a new sequence is assigned to an old role. A less presumptuous way to state the level of certainty is that it is a generator of hypotheses whose degree of certainty and testability differs by specific case. OTUs are often based on SSUs (small subunit RNA, 16S in eubacteria and archaea) although with longer reads and better total assembly, more extensive chromosomal contiguous segments are available and horizontal evolution can more easily be taken into account (Nguyen et al., 2016; Palmer et al., 2019). A saving grace of eubacterial, archaea, and eukaryotic cellular sequencing is that there is an overall tree of life on which to organize sequences (Woese et al., 1990; York, 2020). Such an overarching organizing principle is not available for viruses of either prokaryotes or eukaryotes.
The sequences of viruses of eukaryotes and prokaryotes (bacteriophage, archaeaphage) are in some ways easier and in others harder to characterize. Easier, because viral genomes are smaller and tend to be less potentially confusing than others, making it straightforward to assemble full genomes even from short read lengths (e.g., ∼200 bp from Illumina). There are differences in sample preparation to optimize for viruses, and they may have been missed in some studies. Phylogenetically, there are lineages and groups of related viruses, but the trail of relatedness “runs cold” far short of the all-encompassing tree of cellular life (Mavrich and Hatfull, 2017; Low et al., 2019). It seems likely that groups of viruses evolved independently, i.e., viruses of bats did not evolve from viruses of bacteria. Instead, it seems likely that viruses have branched off from cellular life many independent times and may continue to do so. There is recombination creating functional combinatorial diversity among viruses but most of this is between viruses that were related to begin with (Botstein, 1980; Brown et al., 2016).
Consider the distribution of life in sequence space as similar to that of stars in the universe. In this analogy, individuals are stars and species are galaxies. Galaxies correspond to quasispecies in the Eigen formulation. The sequence space outside quasispecies is unused and, at least in the neighborhood of quasispecies-galaxies, largely neutral with regard to selection. Higher taxa might approximate galaxy clusters or other cosmological structures in this planetarium-inspired representation. The enumeration of higher taxa in eukaryotes is proposed as a way to estimate the number of species and characterize their phylogenetic distribution (Mora et al., 2011). It will be of interest to learn if this approach can be usefully extended to microbial life, prokaryotic and eukaryotic.
It is instructive to compare the famous tree diagram from Darwin’s Origin of Species (Darwin, 1860) with a more recent time series of bacterial evolution upward against a gradient of increasing concentrations of an antibiotic (Baym et al., 2016). Darwin’s stick figure diagram of the origin of new species by descent is essentially unchanged in contemporary phylogenetic diagrams and their conceptualization (Figure 2A). When compared with a time-lapse visualization of bacteria evolving against an antibiotic gradient (Figure 2B), what we see are not stick figures but fans of selected clones followed by expansion, apparent neutrality being manifest in the symmetry of each fan’s outgrowth. The accumulation of neutral diversity in the outgrowth is invisible to the eye, but certainty of its occurrence follows from the very property that Darwin precisely articulated in the final paragraph of his Origin of Species as inheritance with variability. Evidence of accumulated invisible diversity during clonal outgrowth is revealed when the expanding edge of growth reaches a higher concentration of antibiotic. At the point where a more resistant clone has occurred, it is selected at the border and the outgrowth scenario iterates.
NGS approaches to microbial metagenomics where each molecule is sequenced separately are amenable to direct measurement of variation in microbial and viral populations, thereby giving precise instantiation to Eigen’s quasispecies. Most sequence variation is likely to be neutral, as Kimura first proposed. A series of sequences across a horizontal line in the bacterial experiment would presumably manifest as a cloud in the manner of Eigen’s quasispecies. Virus outbreak sequences such as HIV and SARS-CoV-2, could also be interpreted as quasispecies as well as the more usual approach as phylogenetic trees, which are optimal for epidemiological tracing (Hadfield et al., 2018).
A gedanken experiment follows. Consider two sets of metagenomic microbiome sequences, i.e., every bit of DNA and RNA sequence that can be obtained from an environmental or medical sample. Map each metagenome in sequence space. They will form a universe of galaxies. Compare the two universes that came from applying similar sequencing and analysis methods to different samples. Suppose that the galaxies coincide. The maps are congruent with respect to the center of all galaxies. However, suppose in one case the galaxies are bigger and more diffuse. What would this mean? The more diffuse galaxies imply more neutral accumulation, which implies that the population has existed longer without going through conditions that enforce sequence uniformity. The compactness of galaxies would be a measure of how recently populations have been through conditions that enforce sequence uniformity. There will be further information if the variance within galaxies that are OTUs within a sample tend to be similar and whether they tend to be similar among different samples. This would be the microbial and viral analog of the analysis that led to the conclusion that the extant population of most animal species is within an order of magnitude similar to humans in terms of age or number of generations since their mitochondria passed though conditions that led to sequence uniformity.
Sequence uniformity in a population can come about via different mechanisms including: (a) Clonal bottlenecks (b) Selective Sweeps or (c) Sorting. These mechanisms sometimes overlap and they cannot be distinguished within the sequence-clustering context discussed here [A more extensive discussion can be found in the section “Conditions that favor clonal uniformity are frequent in biology” of Stoeckle and Thaler (2018)].
Learning the direction and pace of microbial change in biodiversity is predicted to be a key parameter for better understanding all evolution and better thinking about human futures. Both of the approaches proposed here are indirect and imperfect. However, they correlate with the first derivative of microbial biodiversity as it changes over time. The first approach follows from the reasoning of Kimura and Luria and Delbrück to be the integral of the mutation rate and the number of generations since a population originated from a single sequence. This is a measurement of vertical evolution. The second is to inventory elements of horizontal gene transfer, e.g., the origin of transfer for promiscuous conjugative plasmids and transducing bacteriophage that harbor eubacterial or archaeal sequences in their capsids.
A key shortcoming of the proposed approaches, because they try to extract all their information from only a single time point, is that they are insensitive to the loss or gain of an entire group. If an entire group has become extinct, then it is by definition impossible to assay the variation within it. Conversely, when a group is within the sample, it is impossible to say whether or not it was not in the sampled ecosystem previously, e.g., a year ago. This insensitivity to large-scale changes appears (at this moment) to be an insurmountable weakness of any method based on sampling only a single moment in time. In situations where it is possible to take multiple measurements at different times, it will be worthwhile to inquire if and under what specific circumstances measurements indicative of the rate of change within existing populations correlate with the loss or gain of entire groups. The hygiene hypothesis and its intellectual descendant, the disappearing microbiota hypothesis, assert that over years and generations the taxonomic diversity of human microbiomes has been on a downward trajectory that negatively impacts health (Finlay et al., 2021).
These questions, framed here in the context of microbial diversity, are familiar concepts and controversies regarding macroscopic plants, animals, and the relationships between population and evolutionary processes.
Microbes are sometimes thought of as living fossils (Schopf et al., 2015). Some “big history” views imply that microbial evolution must be finished or insignificant because eukaryotes culminating in humans with culture and science now cut the edges of evolution (Chardin, 1959; Marchetti, 1983). Concepts of hierarchical evolution may be biologically inaccurate and lead to flawed interpretations and predictions.
A complication to global thinking is that the derivative of microbial biodiversity is not the same everywhere. On the contrary, trends in microbial biodiversity are situation and context specific. Human gut bacterial diversity is reported to be taxonomically decreasing (Finlay et al., 2021). On the other hand, the diversity of SARS-CoV-2 is increasing, although understanding the phylogeny and significance of variants is difficult (Morel et al., 2020; Mascola et al., 2021). Approaches for analyzing microbial biodiversity are context specific, and new efforts will be needed to synthesize them.
Consider a scenario in which global microbial biodiversity increases while plant and animal diversity decreases (a quite plausible condition for the world in which we live). Does it matter? Might there be consequences for possible human futures? Most variation in evolution is neutral and has no selective consequence. Nevertheless, variation is the raw material from which new evolutionary possibilities are made. Microbial novelty might alter macroscopic ecosystems. It is worth considering how this change might occur. New human pathogens might arise. Prediction is not perfect, but possibilities merit thought. The cyberneticist Ashby framed a context between competitors each of which is considered to be or to possess an array of variety (Ashby, 1952). Each element of variety in one competitor’s array is countered by an element in the other competitor’s array. The advantage in Ashby’s game goes to the competitor with the most variety. As Ashby famously put it, “Only variety can destroy/absorb variety.” Serious thought should be given to consideration of whether Ashby’s metaphor aptly describes human interactions with the microbial world.
If Ashby’s game theory is apt, then it would benefit our side of the competition, the EMDO team, to identify and enhance our own relevant variety. The metaphor encourages the proposal that human needs might be well taken care of while leaving much of our biosphere in a wild, and biologically more complex, state (Waggoner et al., 1996; Wilson, 2016).
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
DT conceived and wrote the manuscript.
This work was supported by the Program for the Human Environment, Rockefeller University.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Thanks to Jesse Ausubel, Fiona Doetsch, Wandrille Duchemin, Jackie Faherty, Geoffrey Fucile, Mark Stoeckle, and Iddo Wernick for discussion and encouragement. The submitted manuscript has been significantly improved through insightful anonymous as well as editorial review and through language polishing by Dale Langford. I believe Socrates described “profound ignorance” as a dangerous lack of awareness of what one does not know. This essay is dedicated to my three sisters: Susanna Drogsvold, Joan Dobbie, and Ellen Beeler. I am a guest associate editor for a special research topics section in Frontiers of Ecology and Evolution: DNA Barcodes: Controversies, Mechanisms and Future Applications.
Amaral-Zettler, L., Artigas, L., Baross, J., Bharathi, P., Boetius, A., Chandramohan, D., et al. (2010). “A global census of marine microbes,” in Life in the World’s Oceans: Diversity, Distribution, and Abundance, ed. A. D. McIntyre (Hoboken, NJ: Wiley Blackwell Publishing Ltd), 221–245.
Avery, O., Macleod, C., and McCarty, M. (1944). Studies on the chemical nature of the substance inducing transformation of pneumococcal types. J. Expt. Med. 79, 137–158.
Baym, M., Lieberman, T. D., Kelsic, E. D., Chait, R., Gross, R., Yelin, I., et al. (2016). Spatiotemporal microbial evolution on antibiotic landscapes. Science 353, 1147–1151. doi: 10.1126/science.aag0822
Botstein, D. (1980). A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 354, 484–490. doi: 10.1111/j.1749-6632.1980.tb27987.x
Brown, P. A., Touzain, F., Briand, F. X., Gouilh, A. M., Courtillon, C., Allee, C., et al. (2016). First complete genome sequence of European turkey coronavirus suggests complex recombination history related with US turkey and guinea fowl coronaviruses. J. Gen. Virol. 97, 110–120. doi: 10.1099/jgv.0.000338
Bull, J. J., Meyers, L. A., and Lachmann, M. (2005). Quasispecies made simple. PLoS Comput. Biol. 1:e61. doi: 10.1371/journal.pcbi.0010061
Campbell, A. (1965). The steric effect in lysogenization by bacteriophage lambda. I. Lysogenization of a partially diploid strain of Escherichia coli K-12. Virology 27, 329–339. doi: 10.1016/0042-6822(65)90112-1
Cavalli, L. L., Lederberg, J., and Lederberg, E. M. (1953). An infective factor controlling sex compatibility in Bacterium coli. J. Gen. Microbiol. 8, 89–103. doi: 10.1099/00221287-8-1-89
Clark, A. J., and Margulies, A. D. (1965). Isolation and characterization of recombination-deficient mutants of Escherichia coli K-12. Proc. Natl. Acad. Sci. U.S.A. 53, 451–459.
Cohan, F. M. (2017). Transmission in the origins of bacterial diversity, from ecotypes to phyla. Microbiol. Spectr. 5:MTB-0014-2016. doi: 10.1128/microbiolspec.MTBP-0014-2016
Cohan, F. M. (2019). Systematics: the cohesive nature of bacterial species taxa. Curr. Biol. 29, R169–R172. doi: 10.1016/j.cub.2019.01.033
Dance, A. (2020). The search for microbial dark matter. Nature 582, 301–303. doi: 10.1038/d41586-020-01684-z
Darwin, C. (1860). On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. New York, NY: D. Appleton and Company.
de Azevedo, S. S. D., Caetano, D. G., Cortes, F. H., Teixeira, S. L. M., Dos Santos Silva, K., Hoagland, B., et al. (2017). Highly divergent patterns of genetic diversity and evolution in proviral quasispecies from HIV controllers. Retrovirology 14:29. doi: 10.1186/s12977-017-0354-5
Di Marco, M., Ferrier, S., Harwood, T. D., Hoskins, A. J., and Watson, J. E. M. (2019). Wilderness areas halve the extinction risk of terrestrial biodiversity. Nature 573, 582–585. doi: 10.1038/s41586-019-1567-7
Dobzhansky, T. (1937). Genetics and the Origin of Species, 3rd Edn. New York, NY: Columbia University Press.
Domingo, E., and Perales, C. (2019). Viral quasispecies. PLoS Genet. 15:e1008271. doi: 10.1371/journal.pgen.1008271
Dridi, M., Rosseel, T., Orton, R., Johnson, P., Lecollinet, S., Muylkens, B., et al. (2015). Next-generation sequencing shows West Nile virus quasispecies diversification after a single passage in a carrion crow (Corvus corone) in vivo infection model. J. Gen. Virol. 96, 2999–3009. doi: 10.1099/jgv.0.000231
Field, D., Magnasco, M. O., Moxon, E. R., Metzgar, D., Tanaka, M. M., Wills, C., et al. (1999). Contingency loci, mutator alleles, and their interactions. Synergistic strategies for microbial evolution and adaptation in pathogenesis. Ann. N. Y. Acad. Sci. 870, 378–382.
Finlay, B. B., Amato, K. R., Azad, M., Blaser, M. J., Bosch, T. C. G., Chu, H., et al. (2021). The hygiene hypothesis, the COVID pandemic, and consequences for the human microbiome. Proc. Natl. Acad. Sci. U.S.A. 118:e2010217118. doi: 10.1073/pnas.2010217118
Forster, P. (2004). Ice Ages and the mitochondrial DNA chronology of human dispersals: a review. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 255–264. doi: 10.1098/rstb.2003.1394
Frazao, N., Sousa, A., Lassig, M., and Gordo, I. (2019). Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc. Natl. Acad. Sci. U.S.A. 116, 17906–17915. doi: 10.1073/pnas.1906958116
Gratia, J. P. (2005). Noncomplementing diploidy resulting from spontaneous zygogenesis in Escherichia coli. Microbiology 151(Pt 9), 2947–2959.
Gratia, J. P., and Thiry, M. (2003). Spontaneous zygogenesis in Escherichia coli, a form of true sexuality in prokaryotes. Microbiology 149(Pt 9), 2571–2584.
Hadfield, J., Megill, C., Bell, S. M., Huddleston, J., Potter, B., Callender, C., et al. (2018). Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123. doi: 10.1093/bioinformatics/bty407
Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (2020). Assessing the State of Knowledge on Biodiversity. Available online at: https://ipbes.net/assessing-knowledge [Accessed April 25 2020].
Kimura, M. (1986). DNA and the neutral theory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 312, 343–354.
Kivisild, T., Shen, P., Wall, D. P., Do, B., Sung, R., Davis, K., et al. (2006). The role of selection in the evolution of human mitochondrial genomes. Genetics 172, 373–387. doi: 10.1534/genetics.105.043901
Lederberg, J., Cavalli, L. L., and Lederberg, E. M. (1952). Sex compatibility in Escherichia Coli. Genetics 37, 720–730.
Lederberg, J., and Tatum, E. L. (1946). Gene recombination in Escherichia coli. Nature 158:558. doi: 10.1038/158558a0
Lin, H., Peng, Y., Chen, X., Liang, Y., Tian, G., and Yang, J. (2020). T cell receptor repertoire sequencing. Methods Mol. Biol. 2204, 3–12. doi: 10.1007/978-1-0716-0904-0_1
Louca, S., Mazel, F., Doebeli, M., and Parfrey, L. W. (2019). A census-based estimate of Earth’s bacterial and archaeal diversity. PLoS Biol. 17:e3000106. doi: 10.1371/journal.pbio.3000106
Low, S. J., Dzunkova, M., Chaumeil, P. A., Parks, D. H., and Hugenholtz, P. (2019). Evaluation of a concatenated protein phylogeny for classification of tailed double-stranded DNA viruses belonging to the order Caudovirales. Nat. Microbiol. 4, 1306–1315. doi: 10.1038/s41564-019-0448-z
Lu, I. N., Muller, C. P., and He, F. Q. (2020). Applying next-generation sequencing to unravel the mutational landscape in viral quasispecies. Virus Res. 283:197963. doi: 10.1016/j.virusres.2020.197963
Luria, S. E., and Delbrück, M. (1943). Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 490–510.
Magnabosco, C., Biddle, J., Cockell, C., Jungbluth, S., and Twing, K. (2019). “Biogeography, ecology, and evolution of deep life,” in Deep Carbon: Past to Present, eds B. Orcutt, I. Daniel, and R. Dasgupta (Cambridge: Cambridge University Press), 524–555.
Magro, D. O., Santos, A., Guadagnini, D., de Godoy, F. M., Silva, S. H. M., Lemos, W. J. F., et al. (2019). Remission in Crohn’s disease is accompanied by alterations in the gut microbiota and mucins production. Sci. Rep. 9:13263. doi: 10.1038/s41598-019-49893-5
Marchetti, C. (1983). On the role of science in the postindustrial society “Logos” - the empire builder. Technol. Forecast. Soc. Change 24, 197–206.
Mascola, J. R., Graham, B. S., and Fauci, A. S. (2021). SARS-CoV-2 viral variants-tackling a moving target. JAMA 11:2776542. doi: 10.1001/jama.2021.2088
Mavrich, T. N., and Hatfull, G. F. (2017). Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2:17112. doi: 10.1038/nmicrobiol.2017.112
Mellars, P. (2006). Why did modern human populations disperse from Africa ca. 60,000 years ago? A new model. Proc. Natl. Acad. Sci. U.S.A. 103, 9381–9386. doi: 10.1073/pnas.0510792103
Mishmar, D., Ruiz-Pesini, E., Golik, P., Macaulay, V., Clark, A. G., Hosseini, S., et al. (2003). Natural selection shaped regional mtDNA variation in humans. Proc. Natl. Acad. Sci. U.S.A. 100, 171–176. doi: 10.1073/pnas.0136972100
Mora, C., Tittensor, D. P., Adl, S., Simpson, A. G., and Worm, B. (2011). How many species are there on Earth and in the ocean? PLoS Biol. 9:e1001127. doi: 10.1371/journal.pbio.1001127
Morel, B., Barbera, P., Czech, L., Bettisworth, B., Hubner, L., Lutteropp, S., et al. (2020). Phylogenetic analysis of SARS-CoV-2 data is difficult. Mol Biol Evol. 15:msaa314. doi: 10.1093/molbev/msaa314
Morse, M. L., Lederberg, E. M., and Lederberg, J. (1956). Transduction in Escherichia Coli K-12. Genetics 41, 142–156.
Murray, A. E., Freudenstein, J., Gribaldo, S., Hatzenpichler, R., Hugenholtz, P., Kampfer, P., et al. (2020). Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol. 5, 987–994. doi: 10.1038/s41564-020-0733-x
Nguyen, N. P., Warnow, T., Pop, M., and White, B. (2016). A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2:16004. doi: 10.1038/npjbiofilms.2016.4
Palmer, M., Venter, S. N., Coetzee, M. P. A., and Steenkamp, E. T. (2019). Prokaryotic species are sui generis evolutionary units. Syst. Appl. Microbiol. 42, 145–158. doi: 10.1016/j.syapm.2018.10.002
Rayssiguier, C., Thaler, D. S., and Radman, M. (1989). The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342, 396–401. doi: 10.1038/342396a0
Richards, M., Macaulay, V., Hickey, E., Vega, E., Sykes, B., Guida, V., et al. (2000). Tracing European founder lineages in the Near Eastern mtDNA pool. Am. J. Hum. Genet. 67, 1251–1276.
Rodriguez, R. L., Gunturu, S., Harvey, W. T., Rossello-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The microbial genomes atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Roskin, K. M., Jackson, K. J. L., Lee, J. Y., Hoh, R. A., Joshi, S. A., Hwang, K. K., et al. (2020). Aberrant B cell repertoire selection associated with HIV neutralizing antibody breadth. Nat. Immunol. 21, 199–209. doi: 10.1038/s41590-019-0581-580
Schopf, J. W., Kudryavtsev, A. B., Walter, M. R., Van Kranendonk, M. J., Williford, K. H., Kozdon, R., et al. (2015). Sulfur-cycling fossil bacteria from the 1.8-Ga Duck Creek Formation provide promising evidence of evolution’s null hypothesis. Proc. Natl. Acad. Sci. U.S.A. 112, 2087–2092. doi: 10.1073/pnas.1419241112
Scwacha, A., and Kleckner, N. (1994). Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell 76, 51–63.
Shevchenko, S. G., Radey, M., Tchesnokova, V., Kisiela, D., and Sokurenko, E. V. (2019). Escherichia coli clonobiome: assessing the strain diversity in feces and urine by deep amplicon sequencing. Appl. Environ. Microbiol. 85:e01866-19. doi: 10.1128/AEM.01866-19
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120.
Stewart, F. M., Gordon, D. M., and Levin, B. R. (1990). Fluctuation analysis: the probability distribution of the number of mutants under different conditions. Genetics 124, 175–185.
Thaler, D. S. (2009). The cytoplasmic structure hypothesis for ribosome assembly, vertical inheritance, and phylogeny. Bioessays 31, 774–783. doi: 10.1002/bies.200800190
Thaler, D. S., and Stoeckle, M. Y. (2016). Bridging two scholarly islands enriches both: COI DNA barcodes for species identification versus human mitochondrial variation for the study of migrations and pathologies. Ecol. Evol. 6, 6824–6835. doi: 10.1002/ece3.2394
Tibayrenc, M., Avise, J. C., and Ayala, F. J. (2015). In the light of evolution IX: clonal reproduction: alternatives to sex. Proc. Natl. Acad. Sci. U.S.A. 112, 8824–8826. doi: 10.1073/pnas.1508087112
Tishkoff, S. A., and Williams, S. M. (2002). Genetic analysis of African populations: human evolution and complex disease. Nat. Rev. Genet. 3, 611–621. doi: 10.1038/nrg865
Trisos, C. H., Merow, C., and Pigot, A. L. (2020). The projected timing of abrupt ecological disruption from climate change. Nature 580, 496–501. doi: 10.1038/s41586-020-2189-9
Waggoner, P., Ausubel, J., and Wernick, I. (1996). Lightening the tread of population on the land: American examples. Popul. Dev. Rev. 22, 531–545.
Williams, T. A., Cox, C. J., Foster, P. G., Szollosi, G. J., and Embley, T. M. (2020). Phylogenomics provides robust support for a two-domains tree of life. Nat. Ecol. Evol. 4, 138–147. doi: 10.1038/s41559-019-1040-x
Woese, C. R., and Fox, G. E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc. Natl. Acad. Sci. U.S.A. 74, 5088–5090.
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains of archea, bacteria, and eukarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579.
York, A. (2020). New data for the tree of life. Nat. Rev. Microbiol. 18:63. doi: 10.1038/s41579-019-0317-z
Zhu, Q., Mai, U., Pfeiffer, W., Janssen, S., Asnicar, F., Sanders, J. G., et al. (2019). Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 10:5477. doi: 10.1038/s41467-019-13443-4
Keywords: biodiversity, microbial diversity, extinction rate, generation of diversity, speciation, bottleneck, DNA bar coding, 16S/18S ribosomal RNA gene analysis
Citation: Thaler DS (2021) Is Global Microbial Biodiversity Increasing, Decreasing, or Staying the Same? Front. Ecol. Evol. 9:565649. doi: 10.3389/fevo.2021.565649
Received: 25 May 2020; Accepted: 16 March 2021;
Published: 19 April 2021.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Frederick M. Cohan, Wesleyan University, United StatesCopyright © 2021 Thaler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S. Thaler, ZGF2aWRzdGhhbGVyQGdtYWlsLmNvbQ==; ZGF2aWQudGhhbGVyQHVuaWJhcy5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.