 Caitlin Mudge
Caitlin Mudge Rebecca Dallwitz
Rebecca Dallwitz Bastien Llamas
Bastien Llamas Jeremy J. Austin
Jeremy J. Austin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 11 September 2020
Sec. Paleoecology
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.505233
Natural history collections provide a critical temporal view of past biodiversity and are instrumental in the study of extinct populations. However, the value of historical specimens relies on correct species identification, collection date and collection locality. The Australian National Maritime Museum (ANMM) holds an unusual artifact – an electric lamp made from a dried whale penis – with unknown age, species-of-origin and collection locality. We used ancient DNA methods to generate a partial mitochondrial DNA (mtDNA) genome sequence to establish the identity and provenance of the whale, and accelerator mass spectrometry (AMS) radiocarbon dating to determine the approximate year of death. Mitochondrial DNA sequences from the 16S rRNA gene and the control region indicate that the specimen belonged to a sperm whale (Physeter macrocephalus) and a modern radiocarbon age suggests it was collected post-1950s. We were unable to determine the collection locality of the whale due to the very broad geographic distribution of its mtDNA haplotype. Our results suggest the specimen was possibly collected as a souvenir during post-war whaling, where nearly 30,000 male sperm whales were killed annually. This study supports and extends previous research that applies ancient DNA and radiocarbon dating techniques to enhance the value of natural history collections, by identifying the species-of-origin and age of historical specimens.
Natural history museum collections provide a rich source of information for the study of evolution and systematics, and provide a critical temporal view of past biodiversity, adaptation and extinction (Allmon, 1994; Suarez and Tsutsui, 2004). However, the value of museum collections relies on correct species identification, collection date and collection locality (Hall, 1974; Goodwin et al., 2015). Missing or improper labeling and identification is common in specimens collected as trophies or curiosities, or where biological specimens were incorporated into objects such as clothing, furniture and tools. There are ways to recover sample identification and collection date using morphological characteristics (Eyualem and Blaxter, 2003) and radiocarbon dating (Cerling et al., 2016). However, collection locality is generally more difficult to establish. Ancient DNA analysis can be used to identify the species-of-origin and collection locality using phylogeographic analysis of genetic material recovered from the unknown specimen (Hartnup et al., 2011; Bi et al., 2013; Besnard et al., 2015; Thomas et al., 2017; Bastian et al., 2018). Combining specimen genetic data with more traditional sources of evidence, such as archives, specimen labels, and evidence from historical taxidermy can help develop robust predictions of specimen provenance.
Whales have been exploited by humans for thousands of years for their valuable meat, oil and blubber (Berta et al., 2015). Commercial hunting commenced in the tenth century in Europe (Reeves, 2018) and fifth century in Japan (Kasuya, 2018), and spread worldwide in the nineteenth century. Whale hunting became industrialized in the 1860s with the invention of cannon-fired harpoons and steam-powered ships (Hjort, 1932; Reeves, 2018). During the twentieth century whale harvesting increased dramatically, especially for fin (Balaenoptera physalus), blue (Balaenoptera musculus), sei (Balaenoptera borealis), and minke (Balaenoptera spp.) whales. It is estimated that 2.9 million large whales were caught and killed during the twentieth century (Rocha et al., 2015). Some whale species have never been able to fully recover from this and require intense conservation to maintain stable population sizes (Jackson et al., 2016). Estimates show that the number of humpback whales (Megaptera novaeangliae) in the North Atlantic Ocean dwindled from 240,000 to 9,000 during the twentieth century (Roman and Palumbi, 2003). During this time whales were hunted primarily as a source of food and oil. However, baleen, the filter feeding system from baleen whales such as right whales (Eubalaena spp.) and humpback whales, was used in a variety of nineteenth century products, including carriage springs, umbrella ribs, corset stays and fishing rods (Lauffenburger, 1993). Whale bones and teeth were used for art (scrimshaw), tools and utensils (Dyer, 2018). These secondary uses of whale body parts have resulted in a large number of historical artifacts, kept and traded as antiques or stored and displayed in museums as part of social history exhibits associated with whaling (Eastop and Mcewing, 2005).
DNA recovered from historical artifacts made of baleen, whale bone and teeth can provide provenance and other contextual information that enhances an object’s social and historical value (Solazzo et al., 2017). DNA from these artifacts can also aid in population genetic studies to increase our understanding of the effects of whaling, because they provide crucial temporal and geographic sampling (Baker and Clapham, 2004; Eastop and Mcewing, 2005; Sinding et al., 2012, 2016). Pichler et al. (2005) and Sinding et al. (2012) have shown that it is possible to extract sufficient DNA from whale baleen samples to be used for population genetic studies and detail methods to be used for minimally destructive analysis of baleen artifacts. Sinding et al. (2016) successfully developed an XY homolog PCR assay for molecular sexing of historical baleen whale artifacts, allowing for a deeper contextual understanding of the social aspect of whaling. The first example of sequencing mitochondrial genomes from eighteenth century baleen artifacts was reported by Eastop and Mcewing (2005). Mitochondrial DNA extracted from the baleen stiffened stomacher of a deliberately concealed eighteenth century garment was found to be from an extinct, previously undescribed, North Atlantic lineage of right whales (Eubalaena glacialis) (Eastop, 2006). This result demonstrates how deeper knowledge of the history of museum specimens can inform understanding of current diversity.
The Australian National Maritime Museum (ANMM) holds an unusual whale artifact – an electric lamp made from the dried penis of an unidentified whale (Figure 1). The whale penis is mounted on a wooden plinth with a standard light bulb, cord and plug. It is a highly unusual example of the way whale products were souvenired, fetishized and converted into contemporary objects. The lamp, like scrimshaw, represents an important historical artifact that reflects the cultural and social aspects of the whaling industry and whalers themselves. The collection date, species-of-origin and collection locality of the lamp are unknown. We used a combination of radiocarbon dating and ancient DNA analysis to attempt species identification and to determine collection locality and date.

Figure 1. Whale penis lamp (accession number 00042380) held at the Australian National Maritime Museum (ANMM; image used with permission; credit to ANMM).
We obtained a piece (5 mm × 5 mm) of dried tissue from the whale lamp (accession number 00042380) in 2017 and sent it the University of Adelaide for DNA testing.
We controlled for contamination of the historic museum sample with contemporary DNA and previously amplified mitochondrial DNA (mtDNA) PCR products by conducting all pre-PCR work at dedicated ancient DNA facilities at the Australian Centre for Ancient DNA, University of Adelaide. No contemporary whale samples or DNA had ever been present in the pre-PCR laboratory. We conducted all DNA extraction, library preparation and PCR set-up in the pre-PCR laboratory physically separate from post-PCR laboratories and included the use of dead-air glove boxes fitted with internal UV lights, regular decontamination of all work areas and equipment with sodium hypochlorite, PPE including disposable laboratory gown, face mask, shoe covers and double-gloving and strict one-way movement of personnel (shower > freshly laundered clothes > pre-PCR laboratory > post-PCR laboratory).
We extracted DNA from a 3 mm × 3 mm piece of dried tissue that was rehydrated in 1 ml of 0.5 M EDTA for 2 h and then finely minced with a sterile scalpel blade and then digested and extracted using a Qiagen DNeasy Tissue Kit (Qiagen, Valencia, California, United States) as described by Austin et al. (2013), omitting the carrier RNA, and with a final elution volume of 80 μl. We included a negative extraction control during the DNA extraction procedure to monitor for contamination.
We initially used hybridization enrichment and high throughput sequencing to attempt to generate a mitogenome sequence from the sample for species identification and to determine collection locality. Twenty microliters of DNA extract was converted to truncated Illumina libraries, with 7 bp dual internal barcodes, using the method described by Meyer and Kircher (2010). We included a library blank (no DNA) to monitor for contamination. A mtDNA genome hybridization enrichment was performed on 100 ng of the truncated library using biotinylated bison (Bison bison) mtDNA genome baits, following the method described by Richards et al. (2019). We have previously shown that bison mtDNA baits can be used to enrich mtDNA genomes from phylogenetically distant taxa including marsupials and birds (Richards et al., 2019). As members of the placental mammal superorder Cetartiodactyla, the bison mtDNA baits should successfully enrich whale mtDNA, which is the suspected identity of the artifact based on morphology. The bison baits were made in-house following the methods of Richards et al. (2019).
Subsequently, and as a result of low mtDNA genome sequence recovery, we also produced a shotgun sequencing library from 150 ng of the truncated library by amplifying with primers that completed the Illumina adapter sequence as described by Richards et al. (2019). We used the shotgun sequencing data to investigate the cause of the poor mtDNA genome enrichment results. The shotgun library, enriched library and matched library blanks were sequenced on separate Illumina MiSeq runs using a 2 × 150 bp paired end kit at the Australian Genome Research Facility (AGRF, Melbourne).
We demultiplexed raw sequencing data by P7 adapter indexes using bcl2fastq into BaseSpace at AGRF. Reads in the resulting FASTQ files started with 7 bp internal barcodes that we trimmed using fastp v0.20.0 (Chen et al., 2018). Sequencing data were then processed using the PALEOMIX v1.2.14 pipeline (Schubert et al., 2014). Briefly, AdapterRemoval v2.3.1 (Schubert et al., 2016) was used to filter residual 3’ adapter sequences and low-quality ends, merge overlapping mate reads and, discard reads shorter than 25 bp. Read mapping was performed using the backtrack algorithm in BWA v0.7.15 (Li and Durbin, 2010) and the recommended parameter space for ancient DNA (Schubert et al., 2012), duplicates were removed using Picard Tools v2.1.1 (Broadinstitute, 2018) and paleomix rmdup_collapsed, and local realignment around indels was performed with GATK (McKenna et al., 2010). Damage patterns were assessed using MapDamage v2.0 (Jonsson et al., 2013). We mapped sequencing data against one representative of baleen whales (Mysticeti, blue whale, Balaenoptera musculus) and toothed whales (Odontoceti, sperm whale, Physeter macrocephalus) mitogenome sequences (GenBank accessions MF409242 and KU891393, respectively) and genome assemblies (GenBank accessions VNFD00000000.1 and UEMC00000000.1, respectively). For mitogenome results unique mapped reads were visualized in Geneious Prime 2020.0.4 (Biomatters) to generate a consensus sequence using a strict consensus approach with “N” called for bases with <3× read depth.
In order to identify the species of whale we used BLAST searches (nucleotide database, Megablast, with default parameters) within Geneious 20.0.4 using the consensus mtDNA 16S rRNA gene sequence obtained from reads mapped to the sperm whale and blue whale mtDNA genomes. Based on BLAST searches and knowledge of cetacean phylogeny (Mcgowen et al., 2019) we aligned the consensus 16S rRNA sequence to representative mtDNA genomes from sperm whale (Physeter macrocephalus), pygmy sperm whale (Kogia breviceps), dwarf sperm whale (Kogia sima), killer whale (Orcinus orca), Northern right whale (Eubalaena glacialis), and blue whale (Balaeonoptera musculus) to confirm species identity.
Based on results of the initial BLAST search we used the consensus mtDNA sequence from reads mapped to the closest related species, in this case the sperm whale reference mitogenome, to try and identify the collection locality of the whale sample. Morin et al. (2018) identified 80 mitogenome haplotypes from 180 different sperm whales collected from the Pacific Atlantic and Indian Oceans. We aligned the whale consensus mtDNA sequence to these 80 existing mitogenome sequences, trimmed all sites that contained an N or gap, and constructed a Minimum Spanning (Bandelt et al., 1999) haplotype network using PopART (Leigh and Bryant, 2015).
Based on the HTS results, to further confirm species identification and to attempt to refine geographic collection locality we targeted an 89 bp (excluding primers) section of the mtDNA 16S ribosomal RNA gene and a 285 bp (excluding primers) section of the mtDNA control region, using standard PCR and Sanger sequencing. We selected the 16S rRNA region to confirm the hybridization enrichment results. The 285 bp segment of the control region was chosen as it contains 30 of the 31 phylogenetically informative variable sites identified in a 384 bp alignment of global sperm whale mtDNA control region sequences (Alexander et al., 2013) and 30 of 34 variable sites identified in a 619 bp alignment of global sperm whale mtDNA control region sequences by Alexander et al. (2016).
We designed two sets of PCR primers to amplify overlapping fragments of 190 bp and 199 bp for the control region, using an alignment of 41 sperm whale control region sequences from Alexander et al. (2016) (Fragment 1, forward primer: AGATAAATACAAACCCACAGTGCT, reverse primer: TAA TACGAGCTTTCACTGATCG; Fragment 2, forward primer: ACACGCTATGTATAATAGTGCATTCAATT, reverse primer: GTTGCTGGTTTCACGCGGCA). The 16S primers were the generic mammal primers 16S6 and 16S7 from Poinar et al. (2001).
We performed PCRs in 25 μl volumes containing 2 μl of DNA (3.82 ng/μl), 1× High Fidelity PCR Buffer (Invitrogen, California, United States), 250 μM of each dNTP, 400 nM of each primer (IDT), 2 mM MgSO4, 1 μg/μl RSA (Sigma) and 0.5 units of Platinum Taq DNA Polymerase High Fidelity (Invitrogen, California, United States). Thermocycling conditions were: denaturation at 94°C for 2 min, followed by 50 cycles of 94°C for 15 s, annealing at 55°C for 15 s and extension at 68°C for 30 s, with a final extension step at 68°C for 10 min. We included a PCR no template control and the negative extraction control in each PCR attempt. Successful PCR amplifications were sent to AGRF (Adelaide) for bi-directional Sanger sequencing. Sequence chromatograms were edited and assembled using Geneious v11.0.4. We used a BLAST search (nucleotide database, Megablast, with default parameters) to confirm the identity of the 16S rRNA sequence and aligned the control region sequence to the 41 global control region haplotypes (384 bp) from Alexander et al. (2016). We trimmed the control region alignment to 285 bp and constructed a Minimum Spanning (Bandelt et al., 1999) haplotype network using PopART (Leigh and Bryant, 2015). DNA sequences are available on GenBank (accession numbers: MN563138 and MN548398).
We sent a second sample of the whale specimen to the Rafter Radiocarbon Laboratory, GNS Science, New Zealand for radiocarbon dating (sample code R 41180/1) by accelerator mass spectrometry (AMS). A 295.5 mg sub-sample was pretreated by Soxhlet extraction with a standard series of solvents increasing in polarity (hexane, propanol and ethanol), aimed to remove lipids, waxes, resins and preservatives that may have been applied to the sample material. This was followed by repeated washes in dH2O and then heating to 85°C in 0.1 M HCl, aimed to further remove any acid soluble, carbon-bearing compounds, including any atmospheric CO2 absorbed onto the sample surface. δ13C was measured by isotope-ratio mass spectrometry. The AMS date was calibrated to calendar years (BP) using the Post-bomb Marine calibration curve (Reimer et al., 2009) which accounts for radiocarbon fluctuations in the ocean post-World War II.
DNA and radiocarbon dating results suggest that the whale lamp is modern (post-1950) and made from a sperm whale (Physeter macrocephalus). Phylogeographic analysis of the partial mitogenome sequence and the control region sequence was unable to identify a geographic origin for the whale.
Mitochondrial DNA genome enrichment and HTS yielded 2,768,286 retained reads after quality filtering. Only 1414 (0.094%) and 847 (0.067%) unique reads mapped to the sperm whale and blue whale mtDNA reference genomes, respectively. We observed an elevated frequency of purines at the position immediately preceding the start of sequencing reads, as well as an increase of C-to-T and G-to-A substitutions at the start and end of sequencing reads, respectively (Supplementary Figure 1). These patterns are characteristic of degraded DNA (Jonsson et al., 2013). The library blank returned only 1772 retained reads, none of which mapped to the sperm and blue whale mtDNA reference genomes, indicating that the results obtained from the whale sample were not due to contamination during the laboratory process. We focused on the 16S rRNA gene for species identification of the sample. For reads that mapped to the sperm whale or blue whale reference mtDNA genomes we recovered an 898 and 796 bp contiguous consensus sequence, respectively, for the 16S rRNA gene. BLAST searches revealed that both consensus sequences were identical to the sperm whale 16S rRNA gene, and at least 6.2% divergent from all other whale sequences. Among six species of toothed and baleen whale there were 140 variable sites in the 898 bp of aligned mtDNA 16S rRNA sequence (Table 1). The sequence from the whale sample was identical to the sperm whale but 56–85 substitutions different from the pygmy and dwarf sperm whale, killer whale, Northern right whale and blue whale (Table 1). These results indicate that the sample came from a sperm whale.

Table 1. Summary of DNA-based species identification of the whale lamp.
Despite our previous research (Richards et al., 2019) showing that bison mtDNA baits are effective across distantly related taxa, we were unable to recover a complete mtDNA genome from the sample. Only 5111 bp of the sperm whale reference mtDNA genome was covered by three or more reads (average fragment length: 92 bp), so we were only able to call a consensus base for 31% of the mitogenome. The partial whale mitogenome was 1 bp different from sperm whale haplotype 47 (Morin et al., 2018, a single individual from the NW Pacific) and 1 bp different from a second sperm whale haplotype (representing 35 collapsed haplotypes from Morin et al., 2018) from 96 individuals collected in the Atlantic, Indian and Pacific Oceans (Figure 2A). Due to the very broad distribution of this haplotype, which includes >50% of the individuals from Morin et al.’s (2018) study, we were unable to identify the ocean from which the sperm whale was collected.
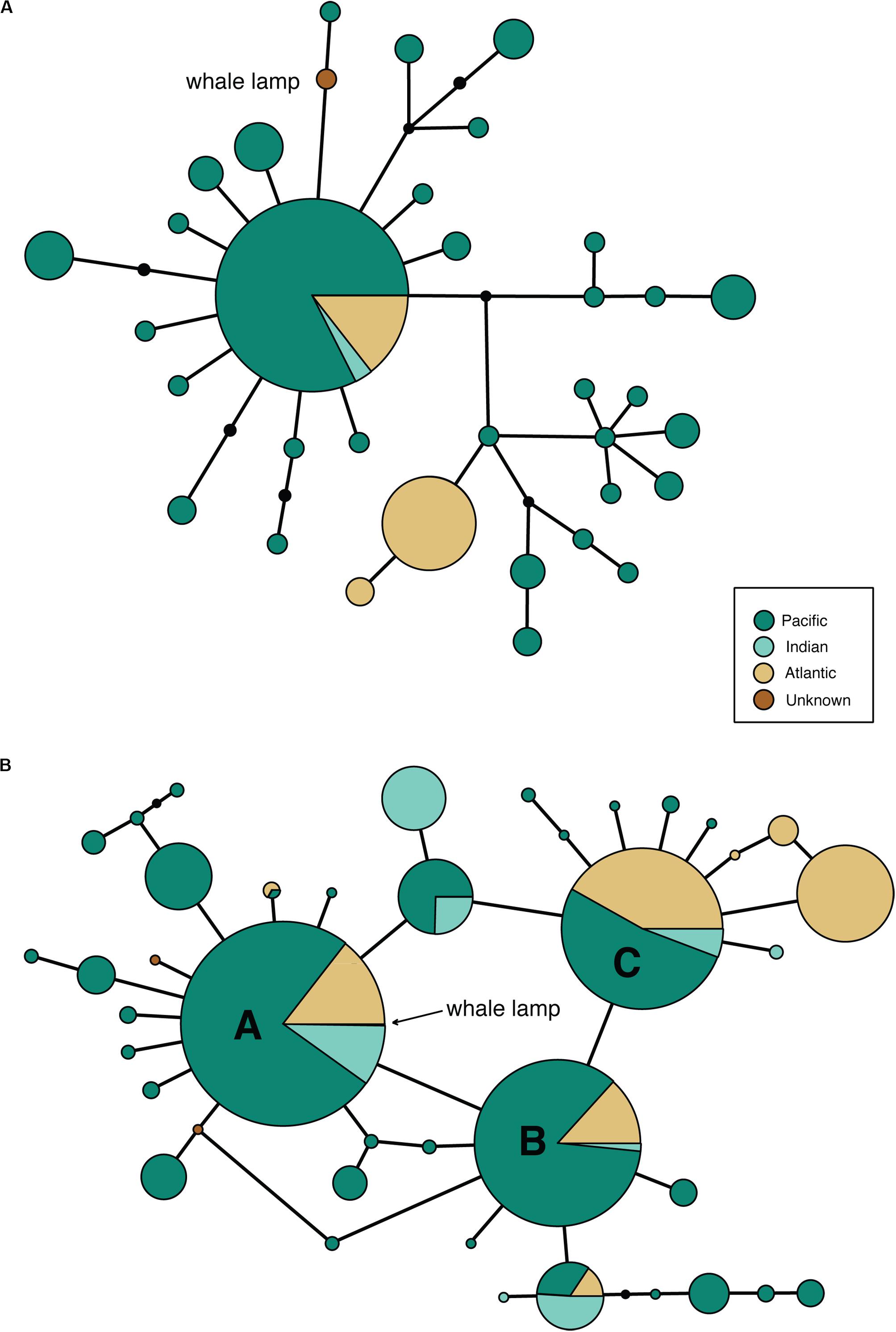
Figure 2. Minimum spanning mtDNA haplotype networks for (A) 35 global sperm whale mtDNA genome haplotypes from 180 individuals, based on an alignment of 5097 bp of sequence obtained from the whale lamp specimen. Black circles are unobserved haplotypes, lines represent a single nucleotide substitution. The whale lamp haplotype is shown in brown. (B) 41 global sperm whale mtDNA control region haplotypes from 1546 individuals, based on an alignment of 285 bp of sequence obtained from the whale lamp specimen. Black circles are unobserved haplotypes, lines represent a single nucleotide substitution. The whale lamp haplotype sits within the most common haplotype A.
We used the shotgun sequencing data to investigate the cause of the poor mtDNA genome results. Shotgun sequencing yielded 2,868,424 retained reads after quality filtering, with only 4909 (0.17%) unique reads mapping to the sperm whale reference genome. The endogenous mtDNA content was very low (0.0016%, 47 unique reads) compared to the endogenous nuclear content (0.17%, 4862 reads). The mtDNA enrichment, via hybridization, was 57-fold (from 0.0016 to 0.094%) which is within the range (2–828) of fold-enrichment previously observed across a range of samples, preservation conditions, taxa and baits used (Templeton et al., 2013; Mohandesan et al., 2017; Bover et al., 2019; Richards et al., 2019). Therefore, the poor mtDNA genome recovery from the sample appears to be related to its very low endogenous mtDNA content.
We successfully amplified and sequenced 89 bp of the mtDNA 16S rRNA gene and 285 bp of the mtDNA control region using traditional PCR and Sanger sequencing. No amplicons were generated from the extraction or PCR negative controls. The 16S rRNA sequence was identical to the sequence obtained via hybridization enrichment and to the sperm whale reference mtDNA genome (KU891373), and at least seven substitutions different from all other available cetacean sequences (Table 1). This result corroborates the findings from the HTS data, confirming that the sample belongs to a sperm whale. Alexander et al. (2016) reported 30 variable sites in the 285 bp of control region sequence among 41 sperm whale mtDNA haplotypes sampled from the Atlantic, Indian and Pacific Oceans. Sperm whales, despite being migratory animals, show moderate mtDNA structure due to female philopatry – 33 of 39 control region haplotypes with known collection locality have only been found in one ocean basin, two haplotypes (J,BB) occur in two oceans and only four haplotypes (A,B,C,N) are found in all three oceans (Table 2 and Figure 2B). The 285 bp control region sequence from the whale lamp was identical to the most common sperm whale haplotype (haplotype A, Table 2 and Figure 2B), which has been found in 475 of 1546 sperm whales (31%) previously sampled across all three oceans by Alexander et al. (2013). Thus, as a consequence of the very broad distribution of haplotype A we are unable to identify the geographic origin of the whale sample to a specific ocean basin.
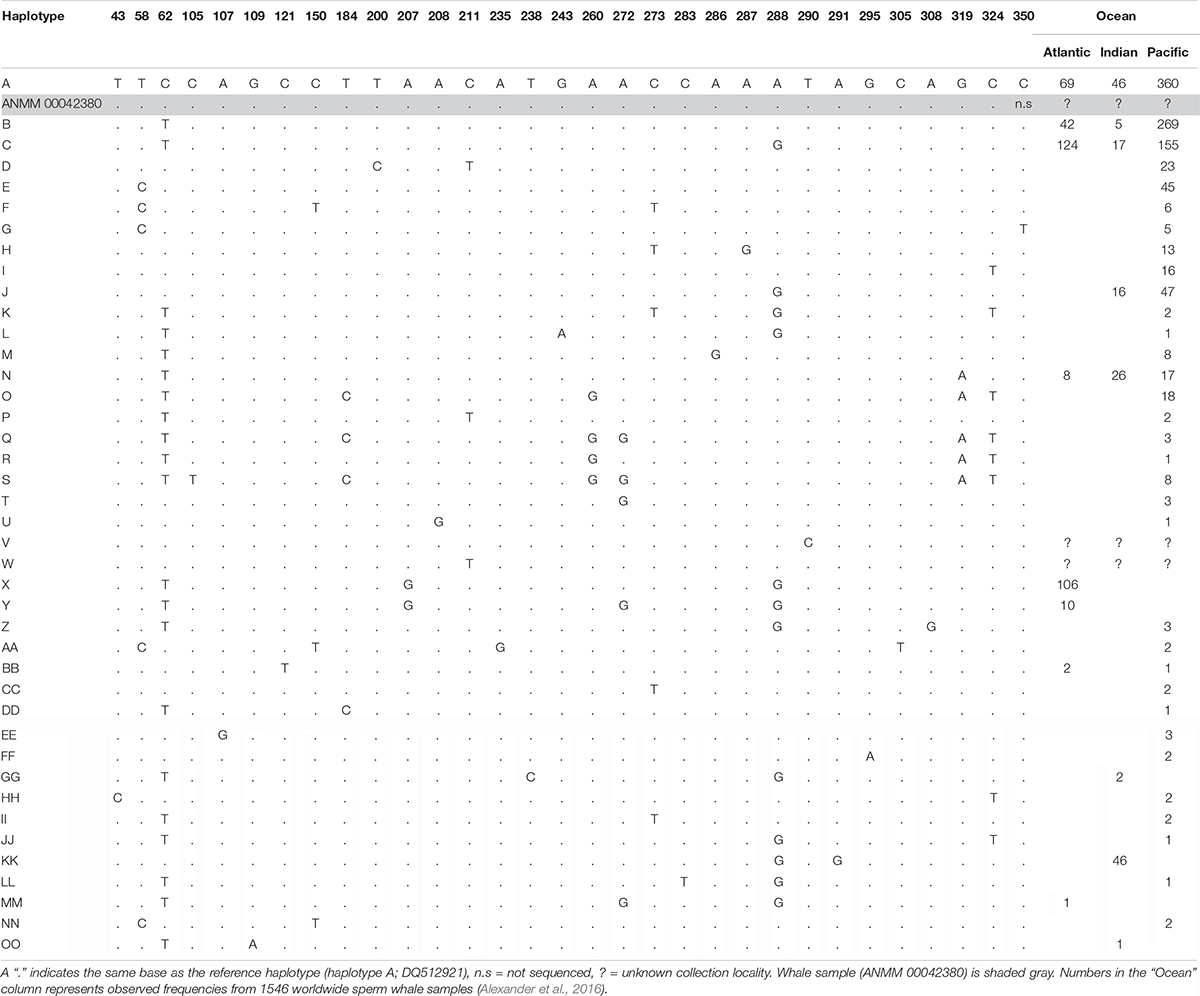
Table 2. Summary of 41 sperm whale mtDNA control region haplotypes and 31 variable sites for a 394 bp alignment from Alexander et al. (2016).
AMS radiocarbon dating of the whale sample yielded a modern date (14C fraction modern = 1.0109 ± 0.0026, δ13C = −13.5 ± 0.2). Radiocarbon in the ocean post-1950s is highly variable making calibration to calendar year (BP) difficult in the case of marine animals. Post-bomb marine calibration indicates a calendar age between 1962 and 1963, only 20 years before whaling was banned.
From two independent DNA analyses of the whale lamp we confirm that the specimen was collected from a sperm whale (Physeter macrocephalus) sometime after the 1950s. Using a partial mitogenome sequence and control region sequences we were unable to identify from which ocean the whale was sourced. Interestingly, while the HTS methods revealed low endogenous content of the sample, traditional PCR and Sanger sequencing yielded results in a fraction of the time and cost, demonstrating that these methods are still useful and appropriate for specific applications.
Morin et al. (2018) described world-wide mitogenome phylogeography of sperm whales from 180 samples and identified 17 different haplotypes that have the same mtDNA control region sequence as the whale lamp. Therefore, additional phylogeographic resolution may be obtained if a complete mitogenome could be generated from the sample. However, this would require significant additional resources, given the very low endogenous content of the sample, and would require sequencing the mtDNA genome via multiple, short, overlapping PCR amplicons or by multiple rounds of hybridization enrichment and increased sequencing effort. An alternative option to resolve collection location of the sperm whale sample would be to investigate markers in the nuclear genome. Hancock-Hanser et al. (2013) propose a way of discovering nuclear loci for population-wide studies in cetaceans using cross-species capture. However, this approach is currently very challenging given the very low endogenous DNA content of the sample and, most importantly, the lack of suitable nuclear single nucleotide polymorphism reference population data for sperm whales.
Hunting of sperm whales increased after WWII and continued until 1988 when the International Whaling Commission introduced a moratorium (Whitehead, 2018). Post-war whaling killed up to 30,000 sperm whales a year (Whitehead, 2018) and the industry targeted large males, thus it is perhaps not surprising that the whale lamp, fitted with an electric light, was made from a male whale killed in the 1950s or 1960s.
As whales are an extremely important part of the marine ecosystem and were hunted for thousands of years, increasing the number of whale samples associated with a collection location would allow an in-depth analysis of genetic and population structure to help our understanding of these creatures both past and present and determine the full impact of whaling. As shown in Eastop and Mcewing (2005) understanding the context of whaling artifacts can provide an insight into historic whale populations that contemporary diversity does not represent.
The datasets generated for this study can be found in GenBank (accession numbers MN563138 and MN548398).
Ethical review and approval was not required for the animal study because only museum specimens were included in the analysis.
CM, RD, and JA contributed to the conception and design of the study. CM and JA performed the genetic laboratory work. CM and BL performed the bioinformatics analysis. CM wrote the first draft of the manuscript. CM, BL, RD, and JA wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
The research was funded by the School of Biological Sciences, University of Adelaide. BL was supported by an Australian Research Council Future Fellowship (FT170100448).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Australian National Maritime Museum for collaborating on this research and supplying tissue samples for destructive testing, the Rafter Radiocarbon Laboratory for performing the radiocarbon dating analysis and past and present members of the Australian Centre for Ancient DNA, University of Adelaide, for providing technical support and comments on previous drafts of the manuscript. We thank Alana Alexander for kindly providing sperm whale mtDNA haplotype frequency data and the two reviewers for constructive comments which have improved the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.505233/full#supplementary-material
Alexander, A., Steel, D., Hoekzema, K., Mesnick, S. L., Engelhaupt, D., Kerr, I., et al. (2016). What influences the worldwide genetic structure of sperm whales (Physeter macrocephalus)? Mol. Ecol. 25, 2754–2772. doi: 10.1111/mec.13638
Alexander, A., Steel, D., Slikas, B., Hoekzema, K., Carraher, C., Parks, M., et al. (2013). Low diversity in the mitogenome of sperm whales revealed by next-generation sequencing. Genome Biol. Evol. 5, 113–129. doi: 10.1093/gbe/evs126
Allmon, W. D. (1994). The value of natural history collections. Curator 37, 83–89. doi: 10.1111/j.2151-6952.1994.tb01011.x
Austin, J. J., Joseph, L., Pedler, L. P., and Black, A. B. (2013). Uncovering cryptic evolutionary diversity in extant and extinct populations of the southern Australian arid zone Western and Thick-billed Grasswrens (Passeriformes: Maluridae: Amytornis). Conserv. Genet. 14, 1173–1184. doi: 10.1007/s10592-013-0504-9
Baker, S. C., and Clapham, P. J. (2004). Modelling the past and future of whales and whaling. TRENDS Ecol. Evol. 19, 365–371. doi: 10.1016/j.tree.2004.05.005
Bandelt, H. J., Forster, P., and Rohl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Bastian, F., Jacot-Des-Combes, C., Hänni, C., and Perrier, M. (2018). Determination of the geographical origin of leather shields from Zanzibar using ancient DNA tools. J. Archaeol. Sci. 19, 323–333. doi: 10.1016/j.jasrep.2018.03.002
Berta, A., Sumich, J., and Kovacs, K. (2015). “Exploitation and conservation,” in Marine Mammals: Evolutionary Biology, Elsevier (Cambridge, MA: Academic Press), 599–655. doi: 10.1016/b978-0-12-397002-2.00015-6
Besnard, G., Bertrand, J. A. M., Delahaie, B., Bourgeois, Y. X. C., Lhuillier, E., and Thébaud, C. (2015). Valuing museum specimens: high-throughput DNA sequencing on historical collections of New Guinea crowned pigeons (Goura). Biol. J. Linn. Soc. 117, 71–82. doi: 10.1111/bij.12494
Bi, K., Linderoth, T., Vanderpool, D., Good, J. M., Nielsen, R., and Moritz, C. (2013). Unlocking the vault: next-generation museum population genomics. Mol. Ecol. 22, 6018–6032. doi: 10.1111/mec.12516
Bover, P., Llamas, B., Mitchell, K. J., Thomson, V. A., Alcover, J. A., Lalueza-Fox, C., et al. (2019). Unraveling the phylogenetic relationships of the extinct bovid Myotragus balearicus Bate 1909 from the Balearic Islands. Q. Sci. Rev. 215, 185–195. doi: 10.1016/j.quascirev.2019.05.005
Broadinstitute (2018). Picard Toolkit. Available online at: http://broadinstitute.github.io/picard/ (accessed January 13, 2020).
Cerling, T. E., Barnette, J. E., Chesson, L. A., Douglas-Hamilton, I., Gobush, K. S., Uno, K. T., et al. (2016). Radiocarbon dating of seized ivory confirms rapid decline in African elephant populations and provides insight into illegal trade. Proc. Natl. Acad. Sci. U.S.A. 113, 13330–13335. doi: 10.1073/pnas.1614938113
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890.
Dyer, M. P. (2018). “Scrimshaw,” in Encyclopedia of Marine Mammals, 3rd Edn, eds B. Wursig, J. Thewissen, and K. Kovacs (Cambridge, MA: Academic Press), 841–845.
Eastop, D. (2006). “Sound recording and text creation: oral history and the deliberately concealed garments project,” in Textiles and Text: Re-Establishing the Links Between Archival and Object-Based Research. Postprints of the Third AHRC Research Centre for Textile Conservation and Textile Studies Conference, eds M. Hayward and E. Kramer (London: Archetype).
Eastop, D., and Mcewing, R. (2005). “Informing textile and wildlife conservation: DNA analysis of baleen from an 18th-century garment found deliberately concealed in a building,” in Scientific Analysis of Ancient and Historic Textiles: Informing Preservation, Display and Interpretation: Postprints of the First Conference of the AHRB Research Centre for Textile Conservation and Textile Studies, eds P. Wyeth and R. Janaway (London: Archetype).
Eyualem, A., and Blaxter, M. (2003). Comparison of biological, molecular, and morphological methods of species identification in a set of cultured panagrolaimus isolates. J. Nematol. 35, 119–128.
Goodwin, Z. A., Harris, D. J., Filer, D., Wood, J. R., and Scotland, R. W. (2015). Widespread mistaken identity in tropical plant collections. Curr. Biol. 25, 1066–1067.
Hall, A. V. (1974). Museum specimen record data storage and retrieval. Taxon 23, 23–28. doi: 10.2307/1218085
Hancock-Hanser, B. L., Frey, A., Leslie, M. S., Dutton, P. H., Archer, F. I., and Morin, P. A. (2013). Targeted multiplex next-generation sequencing: advances in techniques of mitochondrial and nuclear DNA sequencing for population genomics. Mol. Ecol. Resour. 13, 254–268. doi: 10.1111/1755-0998.12059
Hartnup, K., Huynen, L., Te Kanawa, R., Shepherd, L. D., Millar, C. D., and Lambert, D. M. (2011). Ancient DNA recovers the origins of mâori feather cloaks. Mol. Biol. Evol. 28, 2741–2750. doi: 10.1093/molbev/msr107
Jackson, J. A., Carroll, E. L., Smith, T. D., Zerbini, A. N., Patenaude, N. J., and Baker, C. S. (2016). An integrated approach to historical population assessment of the great whales: case of the New Zealand southern right whale. R. Soc. Open Sci. 3, 150669–150669. doi: 10.1098/rsos.150669
Jonsson, H., Ginolhac, A., Schubert, M., Johnson, P. L., and Orlando, L. (2013). mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Kasuya, T. (2018). “Whaling, Japanese,” in Encyclopedia of Marine Mammals (Third Edition), eds B. Würsig, J. G. M. Thewissen, and K. M. Kovacs (Cambridge, MA: Academic Press), 1066–1070. doi: 10.1016/b978-0-12-804327-1.00271-5
Lauffenburger, J. A. (1993). Baleen in museum collections: its sources, uses, and identification. J. Am. Inst. Conserv. 32, 213–230. doi: 10.2307/3179545
Leigh, J. W., and Bryant, D. (2015). POPART: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210x.12410
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Mcgowen, M. R., Tsagkogeorga, G., Álvarez-Carretero, S., Dos Reis, M., Struebig, M., Deaville, R., et al. (2019). Phylogenomic resolution of the cetacean tree of life using target sequence capture. Syst. Biol. 69, 1–24.
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meyer, M., and Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protoc. 2010:db.rot5448.
Mohandesan, E., Speller, C. F., Peters, J., Uerpmann, H.-P., Uerpmann, M., De Cupere, B., et al. (2017). Combined hybridization capture and shotgun sequencing for ancient DNA analysis of extinct wild and domestic dromedary camel. Mol. Ecol. Resour. 17, 300–313. doi: 10.1111/1755-0998.12551
Morin, P. A., Foote, A. D., Baker, C. S., Hancock-Hanser, B. L., Kaschner, K., Mate, B. R., et al. (2018). Demography or selection on linked cultural traits or genes? Investigating the driver of low mtDNA diversity in the sperm whale using complementary mitochondrial and nuclear genome analyses. Mol. Ecol. 27, 2604–2619. doi: 10.1111/mec.14698
Pichler, F. B., Dalebout, M. L., and Baker, C. S. (2005). Nondestructive DNA extraction from sperm whale teeth and scrimshaw. Mol. Ecol. Notes 1, 106–109. doi: 10.1046/j.1471-8278.2001.00027.x
Poinar, H. N., Kuch, M., Sobolik, K. D., Barnes, I., Stankiewicz, A. B., Kuder, T., et al. (2001). A molecular analysis of dietary diversity for three archaic Native Americans. Proc. Natl. Acad. Sci. U.S.A. 98, 4317–4322. doi: 10.1073/pnas.061014798
Reeves, R. R. (2018). “Hunting,” in Encyclopedia of Marine Mammals, 3rd Edn, eds B. Würsig, J. G. M. Thewissen, and K. M. Kovacs (Cambridge, MA: Academic Press), 492–496.
Reimer, P. J., Baillie, M. G. L., Bard, E., Bayliss, A., Beck, J. W., Blackwell, P. G., et al. (2009). IntCal09 and Marine09 radiocarbon age calibration curves, 0–50,000 Years cal BP. Radiocarbon 51, 1111–1150. doi: 10.1017/s0033822200034202
Richards, S. M., Hovhannisyan, N., Gilliham, M., Ingram, J., Skadhauge, B., Heiniger, H., et al. (2019). Low-cost cross-taxon enrichment of mitochondrial DNA using in-house synthesised RNA probes. PLoS One 14:e0209499. doi: 10.1371/journal.pone.0209499
Rocha, R., Clapham, P., and Ivashchenko, Y. (2015). Emptying the Oceans: a summary of industrial whaling catches in the 20th century. Mar. Fish. Rev. 76, 37–48. doi: 10.7755/mfr.76.4.3
Roman, J., and Palumbi, S. R. (2003). Whales before whaling in the North Atlantic. Science 301, 508–510. doi: 10.1126/science.1084524
Schubert, M., Ermini, L., Der Sarkissian, C., Jonsson, H., Ginolhac, A., Schaefer, R., et al. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 9, 1056–1082. doi: 10.1038/nprot.2014.063
Schubert, M., Ginolhac, A., Lindgreen, S., Thompson, J. F., Al-Rasheid, K. A. S., Willerslev, E., et al. (2012). Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13:178. doi: 10.1186/1471-2164-13-178
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Sinding, M.-H., Tervo, O. M., Grønnow, B., Gulløv, H. C., Toft, P. A., Bachmann, L., et al. (2016). Sex determination of baleen whale artefacts: implications for ancient DNA use in zooarchaeology. J. Archaeol. Sci. 10, 345–349. doi: 10.1016/j.jasrep.2016.11.001
Sinding, M.-H., Thomas, M., Gilbert, P., Grønnow, B., Christian Gulløv, H. A., Toft, P., et al. (2012). Minimally destructive DNA extraction from archaeological artefacts made from whale baleen. J. Archaeol. Sci. 39, 3750–3753. doi: 10.1016/j.jas.2012.06.020
Solazzo, C., Fitzhugh, W., Kaplan, S., Potter, C., and Dyer, J. M. (2017). Molecular markers in keratins from Mysticeti whales for species identification of baleen in museum and archaeological collections. PLoS One 12:e0183053. doi: 10.1371/journal.pone.0183053
Suarez, A. V., and Tsutsui, N. D. (2004). the value of museum collections for research and society. BioScience 54, 66–74.
Templeton, J. E. L., Brotherton, P. M., Llamas, B., Soubrier, J., Haak, W., Cooper, A., et al. (2013). DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investigat. Genet. 4, 26. doi: 10.1186/2041-2223-4-26
Thomas, J. E., Carvalho, G. R., Haile, J., Martin, M. D., Castruita, J. A. S., Niemann, J., et al. (2017). An ?Aukward’ tale: a genetic approach to discover the whereabouts of the last great auks. Genes 8:164. doi: 10.3390/genes8060164
Keywords: historic DNA, radiocarbon dating (AMS), whaling, mitochondrial DNA, natural history collection, sperm whale (Physeter macrocephalus), PCR, Sanger and high throughput sequencing
Citation: Mudge C, Dallwitz R, Llamas B and Austin JJ (2020) Using Ancient DNA Analysis and Radiocarbon Dating to Determine the Provenance of an Unusual Whaling Artifact. Front. Ecol. Evol. 8:505233. doi: 10.3389/fevo.2020.505233
Received: 16 October 2019; Accepted: 24 August 2020;
Published: 11 September 2020.
Edited by:
Gael Le Roux, UMR 5245 Laboratoire Ecologie Fonctionnelle et Environnement (ECOLAB), FranceReviewed by:
Nic Rawlence, University of Otago, New ZealandCopyright © 2020 Mudge, Dallwitz, Llamas and Austin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caitlin Mudge, Y2FpdGxpbi5tdWRnZUBhZGVsYWlkZS5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.