 W. Owen McMillan
W. Owen McMillan Luca Livraghi
Luca Livraghi Carolina Concha
Carolina Concha Joseph J. Hanly
Joseph J. Hanly
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ecol. Evol., 24 July 2020
Sec. Evolutionary Developmental Biology
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.00221
This article is part of the Research TopicEvo-Devo of Color Pattern FormationView all 20 articles
Butterfly wing patterns have emerged as exceptional model systems with which to link the developmental and genetic processes that generate morphological variation with the ecological and evolutionary processes that shape this variation in natural populations. Among butterflies, research on species within the genus Heliconius has provided remarkable opportunities to explore how phenotypic diversity is generated within the context of an extraordinary adaptive radiation. Wing pattern diversity among the 48 species and hundreds of intraspecific variants arose within the last 12–14 million years and includes striking pattern convergence between distantly related species, as well as marked pattern divergence between closely related populations and species. Here, we synthesize recent research aimed at gaining a mechanistic understanding of how this variation is generated. This research integrates decades of controlled crossing experiments, and the discovery of major wing patterning genes (optix, aristaless1, WntA and cortex) with recent functional genetic manipulation using CRISPR/Cas9 targeted mutagenesis. The emerging data provides a rich framework with which to explore the repeatability of evolution, particularly within the context of how natural selection acts on divergent gene regulatory networks to generate both highly convergent, as well as highly divergent phenotypes. Overall, the functional data show that the gene regulatory networks underlying pattern variation diverge rapidly in Heliconius; yet these networks retain enough flexibility so that natural selection can drive the evolution of nearly identical patterns from different developmental genetic starting points. Moreover, for the first time this research is starting to illuminate the links between the genetic changes modulating pattern variation and how they influence the larger gene networks that are ultimately responsible for patterning a butterfly wing. There are still large gaps in our understanding, but current research priorities are well laid out and experimental methodologies are in place to address them. The challenge is to synthesize diverse research strategies into a cohesive picture of morphological evolution.
“…on these expanded membranes, nature writes, as on a tablet, the story of the modifications of species.” Henry Walter Bates, in “Naturalist on the river Amazons” (1864).
On the 28th of May 1848, Henry Walter Bates, then only 23 years old, stepped off the boat in Para, Brazil with his friend Alfred Russell Wallace. Bates was to spend the next 11 years exploring the Amazon Basin and his collections, particularly his butterfly collections, provided some of the most visually intuitive examples of evolution by natural selection (Bates, 1864). During his explorations, Bates was struck by the remarkable similarity in the vivid wing patterns of Heliconius and other butterfly and moth species and how these patterns would change rapidly as he traveled across the region, as if “touched by an enchanter’s wand” (Bates, 1879). His insights led to the development of mimicry theory, whereby natural selection drives the evolution of phenotypic similarity (Pasteur, 1982). In the intervening 150 years, research has started to identify the genetic and developmental processes responsible for the evolution of butterfly wing patterns generally, including the mimetic wing patterns of Heliconius. For this reason, the study of butterfly wing patterns continues to speak to research questions at the forefront of biology today, much as it did during that early dawn of evolutionary theory.
Here we review recent research into the genetic and developmental basis of wing pattern change. We focus on the vivid wing patterns of Heliconius, which are used both as aposematic warning signals and as conspecific mating cues (Benson, 1972; Mallet et al., 1990; Jiggins et al., 2001; Kapan, 2001; Chouteau et al., 2017; Merrill et al., 2019). This combination of natural and sexual selection is associated with remarkable variation in wing patterns both within and between species, including numerous cases of parallel evolution of similar wing patterns between distantly related species (Figure 1). Classical crossing work done in the 1950s and 1960s by Beebe, Crane, Sheppard, Turner, and others (Turner, 1972; Sheppard et al., 1985; Beebe, 2005) established that pattern variation within a specific Heliconius species had a relatively simple genetic basis, with most variation explained by a small number of autosomal loci. Later, linkage mapping and positional cloning demonstrated that the same suite of major patterning loci are modulating pattern variation broadly across the genus and that variation around these loci is responsible for both convergent change between species and divergent change within species (Joron et al., 2006; Kapan et al., 2006; Kronforst et al., 2006; Huber et al., 2015). The advent of more powerful genomic technologies culminated in the identification of specific genes that modulate pattern variation across the genus (Reed et al., 2011; Martin et al., 2012; Nadeau et al., 2016; Westerman et al., 2018). These loci join a growing list shown to play a general role in wing pattern variation in butterflies and moths (Table 1).

Figure 1. Evolution’s canvas: Wing pattern mimicry among species in the melpomene/cydno/silvaniform clade (MCS) (above line) and the erato/sapho/sara clade (ESS) (below line). The two lineages diverged from each other about 12 MYA (Kozak et al., 2015), yet have recently evolved a complement of identically patterned wings. Species in the MCS clade are: (A) H. hecale zuleika, (B) H. atthis, (C) H. melpomene ecuadorensis, (D) H. pachinus, (E) H. cydno chioneus, (F) H. m. plesseni, (G) H. m. cythera, (H), H. m. rosina. Species in the ESS clade are: (J) H. sapho sapho, (K) H. erato notabilis, (L) H. e. cyrbia, (M) H. e. demophoon, (N) H. sara sara, (O) H. peruvianus, (P) H. e. etylus, (Q) H. hewitsoni, (R) H. hecalesia formosus. Heliconius doris (I) is from a third clade that falls between the ESS and MCS lineages.

Table 1. Wing genes that have been identified by linkage or association as involved with development of wing color or pattern in butterflies and moths.
The discovery of “wing patterning” genes in Heliconius allows us to make broad general conclusions about how morphological diversity is generated. Foremost, patterning loci identified in Heliconius have also been shown to affect pattern variation broadly across butterflies and even moths. For example, the gene cortex appears to be a major target of evolution across the lepidoptera (Nadeau et al., 2016; van’t Hof et al., 2016). In Heliconius, the locus acts across both wing surfaces and underlies variation in white, yellow, orange, and black pattern elements (Joron et al., 2006). Cortex has also been implicated in pattern variation in other butterflies (VanKuren et al., 2019), the peppered moth Biston betularia and other geometrids (van’t Hof et al., 2016, 2019), as well as the silkmoth Bombyx mori (Ito et al., 2016). Secondly, nearly all the patterning genes thus far identified are highly conserved at the amino acid level and are used extensively throughout embryonic and larval development, and later simply redeployed on a developing wing to affect pattern variation. The transcription factor optix, for example, was first discovered in Drosophila, where ectopic expression of the gene in the developing antennae generates ectopic eyes (Seimiya and Gehring, 2000). It is also expressed in the Drosophila pupal wing and is involved with vein patterning there (Organista et al., 2015). In Heliconius, optix is expressed in the optic lobe (Martin et al., 2014), where it presumably plays a role in neural development. However, in Heliconius, optix has also evolved new expression domains and is expressed in the developing pupal wing from about 60 h after pupation, where its expression perfectly prefigures red pattern variation on Heliconius wings (Reed et al., 2011). Thirdly, pattern variation is produced mostly by regulating when and where these genes are expressed during development. The gene WntA is a member of the Wnt family of signaling ligands and has similarly evolved new patterning roles, with dramatic shifts in WntA expression fueling pattern variation in Heliconius (Martin et al., 2012; Concha et al., 2019) and other nymphalid butterflies (Gallant et al., 2014; Martin and Reed, 2014; Mazo-Vargas et al., 2017).
This review synthesizes recent research aimed at gaining a more mechanistic understanding of how wing color pattern variation in Heliconius is generated. We touch only briefly on the biology and ecology of the group that provides the context for understanding their remarkable pattern evolution. This aspect of Heliconius has been reviewed many times (see Supple et al., 2014; Kronforst and Papa, 2015), culminating in a recent book (Jiggins, 2017). Similarly, we do not review the body of research that led to the identification of the four major patterning genes. This was a sustained effort going back more than 50 years, which, coupled with ideas from evolutionary developmental genetics (Brakefield and French, 1999; Nijhout, 2001; Carroll, 2008), allowed the remarkable progress that has been made over the last 3 years. Rather, we focus on the research that is using the discovery of key patterning loci to begin to unravel the regulatory networks that lay at the heart of pattern diversity. Specifically, we outline how one can cast the origin of pattern diversity on a butterfly wing as a basic question of scale cell fate, and then discuss how recent experiments on Heliconius are beginning to illuminate the role that major patterning loci play in the determination of scale cell fate. Finally, we highlight the research challenges and opportunities moving forward.
The identification of the patterning loci that have been the targets of natural selection in Heliconius is a major research step; however, it is important not to lose sight of the fact that genes do not make phenotypes directly (Gawne et al., 2018). Rather the products of genes interact with each other, and with parts of the genome and the environment to form complex webs of interactions that unfold through development (Rudel and Sommer, 2003). In the case of butterfly wing patterns, phenotype is realized in the patterned differentiation of wing epithelial cells into specific scale cell color types (Figure 2). Understanding how butterfly wing patterns form will require us to dissect how these cell types are specified, how they become spatially arranged, and how they differentiate.

Figure 2. Pattern formation on a butterfly wing: (A) In the late larval stage, the presumptive wing imaginal disk is patterned with lacunae (which will form the veins) indicating that the spatial arrangement of the wing has already been patterned. Aside from this, the wing consists of two opposing sheets of undifferentiated epithelial cells, each attached to the basement membrane. Unlike in Drosophila, this wing disk can be straightforwardly mapped to the adult wing and will not undergo major morphometric rearrangement. The expression domains of several transcription factors and signaling molecules are known at this stage in butterflies (summarized in Hanly et al., 2019), and notably, despite scale cells having not been specified yet, the ligand WntA is already expressed in domains that prefigure adult wing pattern. (B) At pupation, the wing disk expands rapidly. During the first 18h of pupal development, scale precursor cells differentiate from their neighbors by notch-delta mediated lateral inhibition (Reed, 2004). Each scale precursor is surrounded by six non-scale epithelial cells, equating to a 3:1 epithelia to scale ratio in the wing. These scale precursor then undergo a series of divisions and apoptosis events that result in a scale cell, which will form the blade of the scale and a socket cell, which will form a supportive structure to hold the scale in place; this indicates homology between the butterfly scale and the D. melanogaster bristle, which is supported by the expression of achaete-scute in the scale precursor cell. This process also requires the presence of planar cell polarity in the tissue. (C) After the generation of the scale and socket cell, the scale begins to send out a large, actin-driven cytoplasmic protrusion which will form the blade of the scale. In Heliconius, the presumptive Type I scales begin this process earlier than their neighbors, which indicates that there is developmental heterochrony between different scale cell types. The expression of additional genes have by now been patterned to prefigure adult wing pattern; for example the gene optix is expressed specifically in cells that will become Type III red scales in Heliconius. (D) Selector genes such as optix and aristaless1 in turn induce the expression of pigment biosynthesis in specific regions across the developing wing. (E) In mid to late pupal development, scale cell differentiation occurs across the wing, including morphological differentiation and pigment synthesis and deposition. Pigment deposition occurs in a specific temporal manner, with red ommochrome appearing first, followed by melanin and finally deposition of 3-OHK (yellow) in Type I scales. (F) This results in the adult wing pattern at eclosion.
The wing patterns of butterflies and moths are composed of overlapping wing scales, which are homologous to insect bristles (Overton, 1966; Galant et al., 1998; Zhou et al., 2009). Each scale cell generally produces a single pigment and has an elaborate nanostructure. It is the combination of pigment and ultrastructural properties of the scale cells that creates the wing colors we observe (Nijhout, 1991). The fantastic array of complex patterns that we find in nature are generated by varying how different scale cell types are arranged across the wing surface. Importantly, the development of pattern on a butterfly wing is largely devoid of processes such as cell migration which complicate inference, and the developing wing tissue can be mapped accurately onto the corresponding adult phenotype (Nijhout, 1991). We therefore propose that the process of coloring a butterfly wing can be broken into a series of spatially programmed decisions, where progenitor cells acquire specific fates through the interplay of cell signaling and differential gene expression (Figure 2). Tweaks to these developmental decisions lead to the variation in wing patterns seen both within and between the 20,000 species of butterflies (Kristensen et al., 2007).
The process of pattern formation occurs throughout wing development and is linked to the processes underlying scale cell development. In the final instar of butterfly larvae, developing wing disks increase their size through cell proliferation and begin to acquire the shape and venation that is characteristic of the adult wing (Figure 2). At this point the wings are composed of a basal lamina straddled by a single cell epithelial layer on both the ventral and dorsal surfaces (Greenstein, 1972; Ghiradella and Radigan, 1976; Nardi and Magee-Adams, 1986; Dinwiddie et al., 2014). Developing cells at this stage are still composed of undifferentiated epithelia, but the inductive signals required for scale cell identity are being laid out through the expression of transcription factors and signaling molecules (summarized in Hanly et al., 2019). It is during the pupal stage that the morphological development of the wing scales begins. Shortly after pupation, Notch signaling induces scale cell differentiation through lateral inhibition resulting in an array of aligned, parallel cell rows (Reed and Serfas, 2004). Cytological studies have shown that these cells in turn undergo two rounds of cell division promoting a neural lineage and a scale cell lineage (Stossberg, 1938). Neural precursor cells undergo apoptosis, whereas the scale cell precursors undergo a second round of cell division forming a socket cell from which a scale cell arises through actin bundle elongation (Dinwiddie et al., 2014). During pupal development, the scale cells grow dramatically in size and after about 60 h, scale cells fated to become a specific color can often be distinguished (Reed et al., 2008). In our model, during this period all scale cells must acquire a cell identity through the patterned expression of major patterning genes, which in turn will trigger the expression of further differentiation factors or terminal effector genes. Cells must also communicate with each other so that the correct edges and boundaries to delimit pattern elements are appropriately arranged. By 100 h of pupal development, we know that all scale cells are undergoing actin-dependent laminar extension and depositing cuticle, including scale type-specific gene expression of pigment synthesis pathways. By this stage, the wing patterning process is complete.
The extraordinary diversity in wing patterns among the 48 Heliconius species arose within the last 12–14 million years (Kozak et al., 2015) and includes remarkable pattern convergence between distantly related species. Pattern matching is perhaps best exemplified by convergent change in two of the major lineages within the genus - the H. erato/sapho/sara (ESS) and the H. melpomene/cydno/silvaniform (MCS) species groups (see Figure 1). The two lineages diverged from a common ancestor early in the formation of the genus and have been evolving separately for the past 12 million years, diverging in many aspects of their ecology and reproductive biology (Jiggins, 2017). Nonetheless, within the last 2.5–4.5 million years the groups have converged on a similar complement of wing pattern types, including a number of cases of near-perfect wing pattern mimicry. Although hybridization and introgression play a role in the evolution of novel patterns within the ESS and MCS clades (Edelman et al., 2019), there is no evidence for hybridization between the two groups.
Within this evolutionary backdrop, much of the wing pattern variation in Heliconius is created by differences in the distribution of only three major scale cell types. Type I yellow scales contain the ommochrome precursor 3-hydroxykynurenine (3-OHK) (Brown, 1967); whereas Type I white scales lack pigment and the color is the result of the scale cell morphology (i.e., structural) (Gilbert et al., 1988). Type II scale cells are pigmented with melanin and Type III scale cells contain the ommochrome pigments xanthommatin and dihydroxanthommatin (Gilbert et al., 1988). The timing of scale cell enlargement and maturation differs between scale types, with Type I scale cells maturing first, followed by Type III scale cells. Scale cells fated to become melanic (Type II) mature last (Aymone et al., 2013). Pigment development largely follows differences in scale cell maturation. Scales fated to become yellow or white are evident as colorless patches beginning at about 6 days after pupation. At the same developmental time point, red-fated scale cells are orange and progressively darken to red over the next 24 h. After the red pigments have matured, melanic scales develop in a ‘wave’ emanating from the center of the wing; whereas yellow-fated scales remain colorless until a few hours before adult emergence. These changes in scale cell color are associated with upregulation of genes involved in the uptake and synthesis of pigment, some of which show strong pattern specific expression, while others do not (Reed et al., 2008; Ferguson and Jiggins, 2009; Hines et al., 2012).
The process of scale cell differentiation provides a useful context to begin to understand the function of the major patterning loci that have been identified in Heliconius (Figure 3). The patterning genes WntA and cortex are expressed during larval wing development well before scale cells have begun to form (Martin et al., 2012; Nadeau et al., 2016). These genes provide key aspects of the positional information signals to downstream pigment and scale cell maturation pathways. Later in pupal development (about 40 h after pupation), the Type I scale cells begin to undergo laminar extension before their Type II and III neighbors (Gilbert et al., 1988; Reed et al., 2008; Aymone et al., 2013). Type III scales mature next and begin by expressing the transcription factor optix, signaling cells to produce the red ommochrome pigments (Reed et al., 2011). Type II cells contain melanins, which are laid down later in pupal development. The white/yellow difference between Type I scale cells is due to the presence or absence of the ommochrome precursor 3-OHK. This color switch appears to be regulated by the differential expression of aristaless1 (Westerman et al., 2018), with the uptake of 3-OHK from the pupal hemolymph occurring right before adult emergence (Reed et al., 2008).
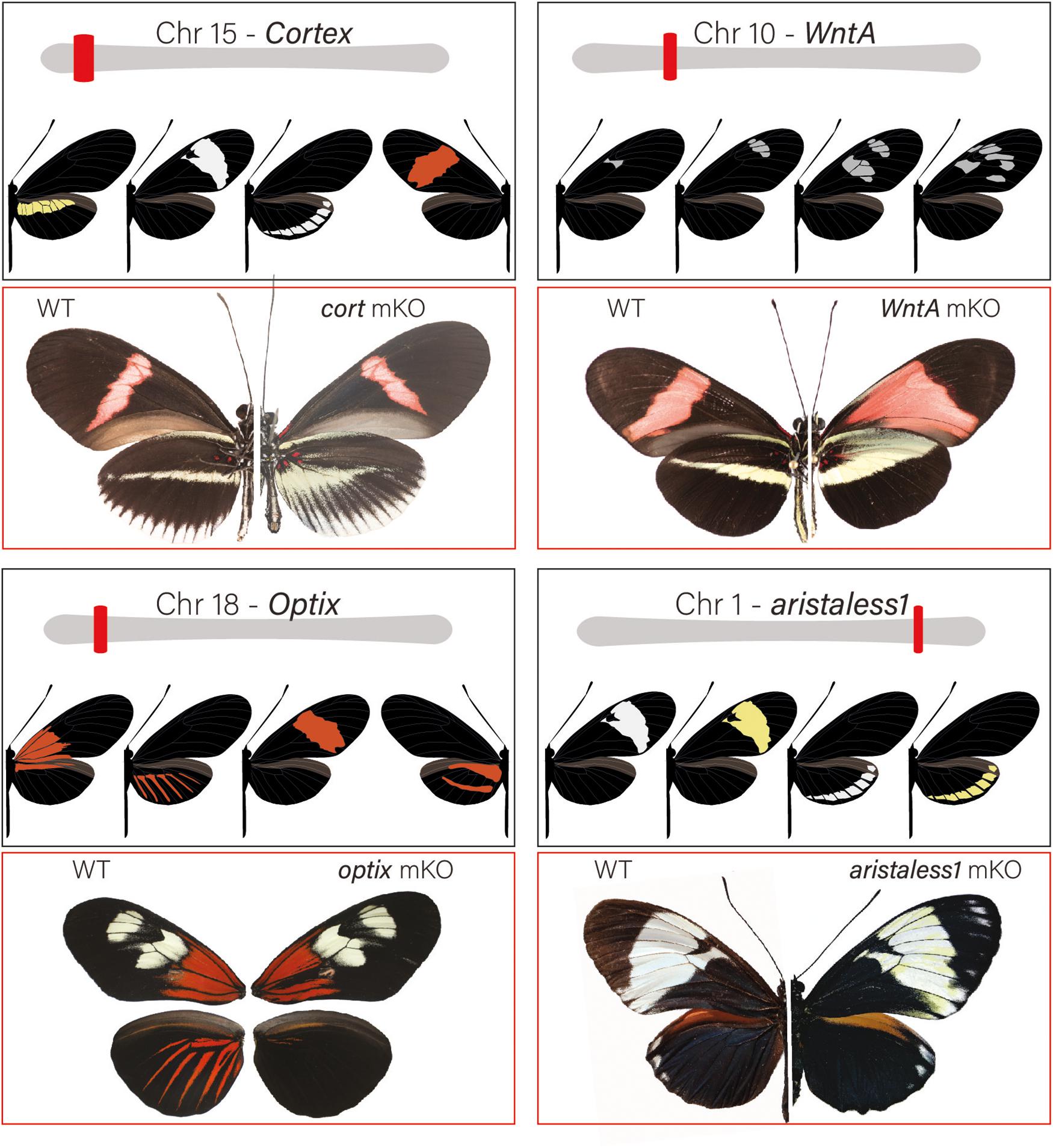
Figure 3. Major patterning switch genes in Heliconius butterflies: Most of the color pattern variation in Heliconius is the result of 4 major effect loci. Loci are unlinked and allelic variation around them is associated with both convergent and divergent change among species. Over the past decade, loci have been positionally cloned and genotype × phenotype, expression and, most recently, KOs with CRISPR/Cas9 have identified specific patterning genes. The gene cortex is located on chromosome 15 and variation around the gene is associated with differences in the yellow hindwing bar, forewing band color, and the white margin and ventral color phenotypes among different geographic morphs of H. erato and H. melpomene. In H. numata, variation around the region that contains cortex controls all variation in wing patterns and is responsible for differences in yellow, orange, and black pattern elements among sympatric color pattern morphs (Nadeau et al., 2016; Saenko et al., 2019). cortex KO results in the appearance of Type I scales across the wing surface of Heliconius butterflies (see Livraghi et al., 2020). The signaling ligand WntA is on chromosome 10 and variation around the locus is associated with differences in forewing band size, shape and position in H. erato and other Heliconius species. KO results in the expansions of color pattern elements in proximal wing regions in H. erato, but generates a wide variety of phenotypic effects across the Heliconius radiation (Concha et al., 2019). The gene optix is on chromosome 18 and variation around this gene is responsible for variation in red color pattern elements. optix KOs result in the transformation of red Type III scales to melanic Type II scales. Finally, variation around the gene aristaless1 on chromosome 1 modulates the presence or absence of yellow pigment in Type I scales. Aristaless1 KOs result in the development of yellow scales suggesting that the gene represses synthesis/deposition of the yellow pigment, 3-hydroxy-L-kynurenine.
Substantial insights into the function that major switch genes play in pattern variation has been gained with the recent development of CRISPR/Cas9 technology in Heliconius (Mazo-Vargas et al., 2017; Zhang et al., 2017b; Westerman et al., 2018; Concha et al., 2019; Lewis et al., 2019) (Figure 3). For the first time, researchers can perform functional assays by inducing targeted mutations in coding regions as well as putative regulatory elements in order to observe the resulting mutant wing pattern phenotypes (reviewed in Livraghi et al., 2018). This research is illuminating the roles that the four major patterning genes play in pattern variation, how they interact during development to influence scale cell fate, and how gene patterning networks evolve within the context of adaptive phenotypic change.
Two major patterning genes, WntA and cortex, are expressed early in wing development and act to establish positional information for the downstream expression of scale cell selector genes. Under a simple model (Figure 4), cortex acts as a “master” regulator of scale cell identity, where scales switch between Type I and Type II/III scales depending on the expression of cortex. A certain threshold of cortex expression may initiate a differentiation cue that sets up a permissive environment for either melanization (optix negative) or red pigment synthesis (optix positive). In contrast, cells below that threshold become either yellow or white depending on the expression of aristaless1, which represses the pathway leading to the synthesis and deposition of 3-OHK pigment (Westerman et al., 2018). Allelic variation in the expression of cortex is responsible for variation in white, yellow, and red/orange patches across the radiation (Figure 3). However, the expression of the gene appears to be more complex than initially envisioned. Although previous expression studies using in situ hybridization have shown a strong correlation between the position of cortex mRNA in wing disks of fifth instar larvae and distal black pattern elements in the adult wings of H. numata and H. melpomene (Nadeau et al., 2016; Saenko et al., 2019), the color switches induced by cortex knockouts (KOs) do not seem to act in a pattern specific way in H. erato and H. melpomene (Livraghi et al., 2020). Instead, cortex KOs result in ectopic switches from melanic (Type II) and red/brown/orange (Type III) into white/yellow (Type I) scale cells across the whole surface of both wings in H. erato and H. melpomene. The switch is associated with both a shift in scale cell color and nanostructure. Thus, the loss of function of cortex causes a wing scale cell to change its identity and fully acquire the identity of a Type I scale cell. The broad effects of cortex across the wings of Heliconius underscore the potential for this gene to generate much wider pattern variation than other major effects genes. This wide range of effects is exemplified by the polymorphic H. numata, where variation in yellow, white, black and orange elements all map to this locus (Joron et al., 2006; Saenko et al., 2010; Nadeau et al., 2016).
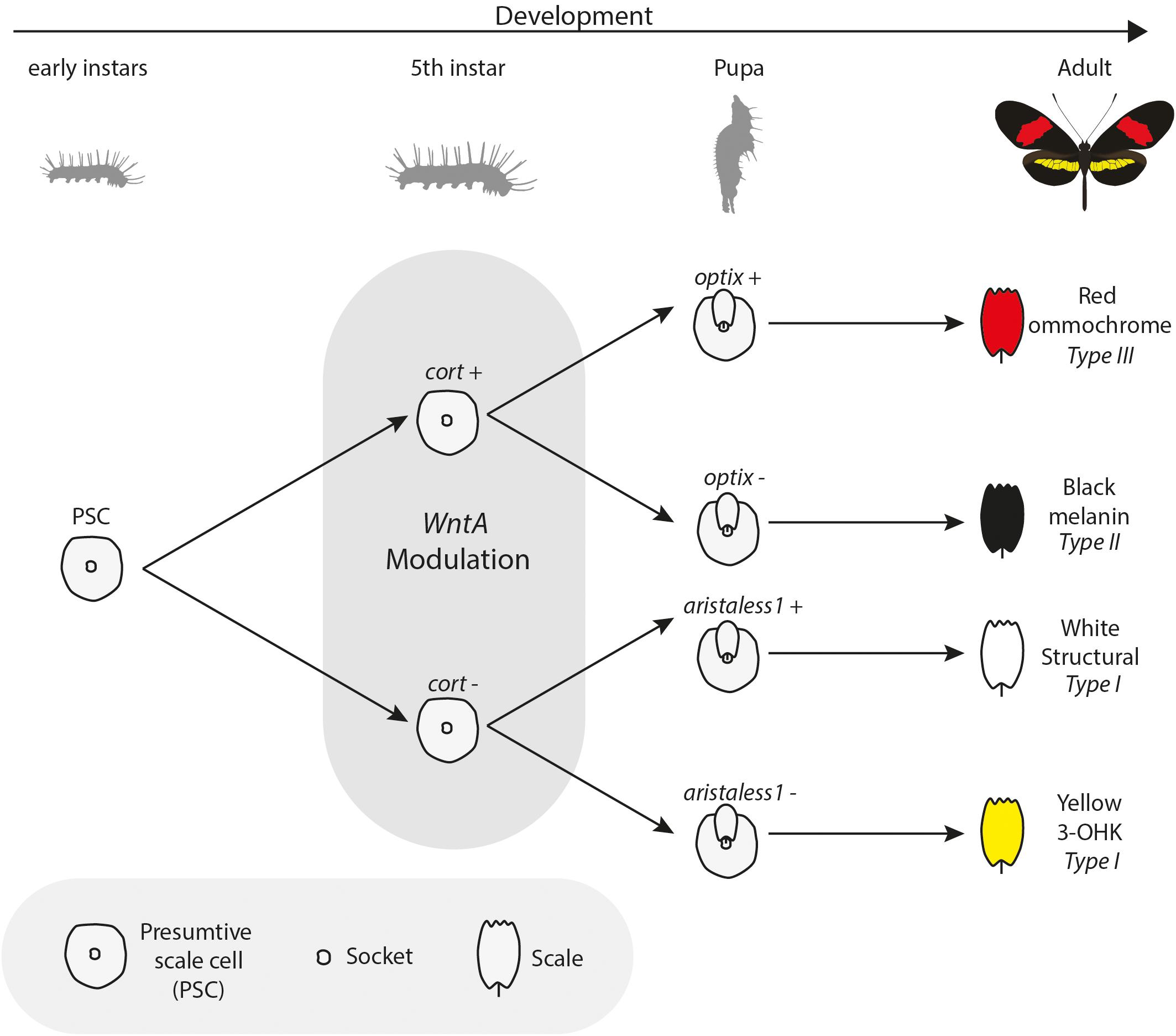
Figure 4. Bifurcating model of interactions among color pattern loci: During wing development, scales express key effector genes that lead to alternate scale cell types. Early presumptive scale cells (PSCs) express cell specification genes such as cortex, which initiate differentiation into Type II (optix−) or Type III (optix +) scales. In the absence of cortex, scale cells differentiate into Type I scales, which differ in pigmentation state based on 3-OHK synthesis controlled by aristaless1 expression. Under this model, WntA acts as a landscape modifier, whereby Wnt signaling modifies the trans environment experienced by differentiating scale cells, in turn delineating the boundaries in which specific differentiation factors can act. Model inspired by Larry Gilbert and collaborators (Gilbert et al., 1988).
If cortex is acting as a “on-off” switch among scale cell types, the signaling ligand WntA might be best conceptualized as modifying the overall regulatory landscape used to provide the positional information for the establishment of boundaries between scale cell types. In contrast to cortex, WntA effects are limited to specific wing pattern elements and the extent of pattern induction across the wing varies both within and between species in the ESS/MCS radiation (Figure 5). The key role of WntA in orchestrating scale cell identity is illustrated by the observation that the shift in scale cell fate happens in every possible direction–from black to red, yellow, and white; as well as from any given color to black, and also from red to yellow scales (Figure 5A). Thus, WntA appears to regulate scale cell fate in a more flexible manner than cortex. Furthermore, unlike cortex where scale cells revert to Type I in cortex KO cells, the exact scale cell phenotype induced by a WntA signal is highly context-dependent (Concha et al., 2019). The complexity of these interactions is best demonstrated in the different responses of optix to WntA KOs in divergent color pattern morphs of H. erato (Figure 5B). Within a given genetic background, WntA KOs result in an upregulation of optix expression and concomitant development of red scales. However, the opposite effect is observed in yellow-banded morphs of H. erato, where the existing yellow patterns expand, even across red scales which are known to express optix in wild-type butterflies.
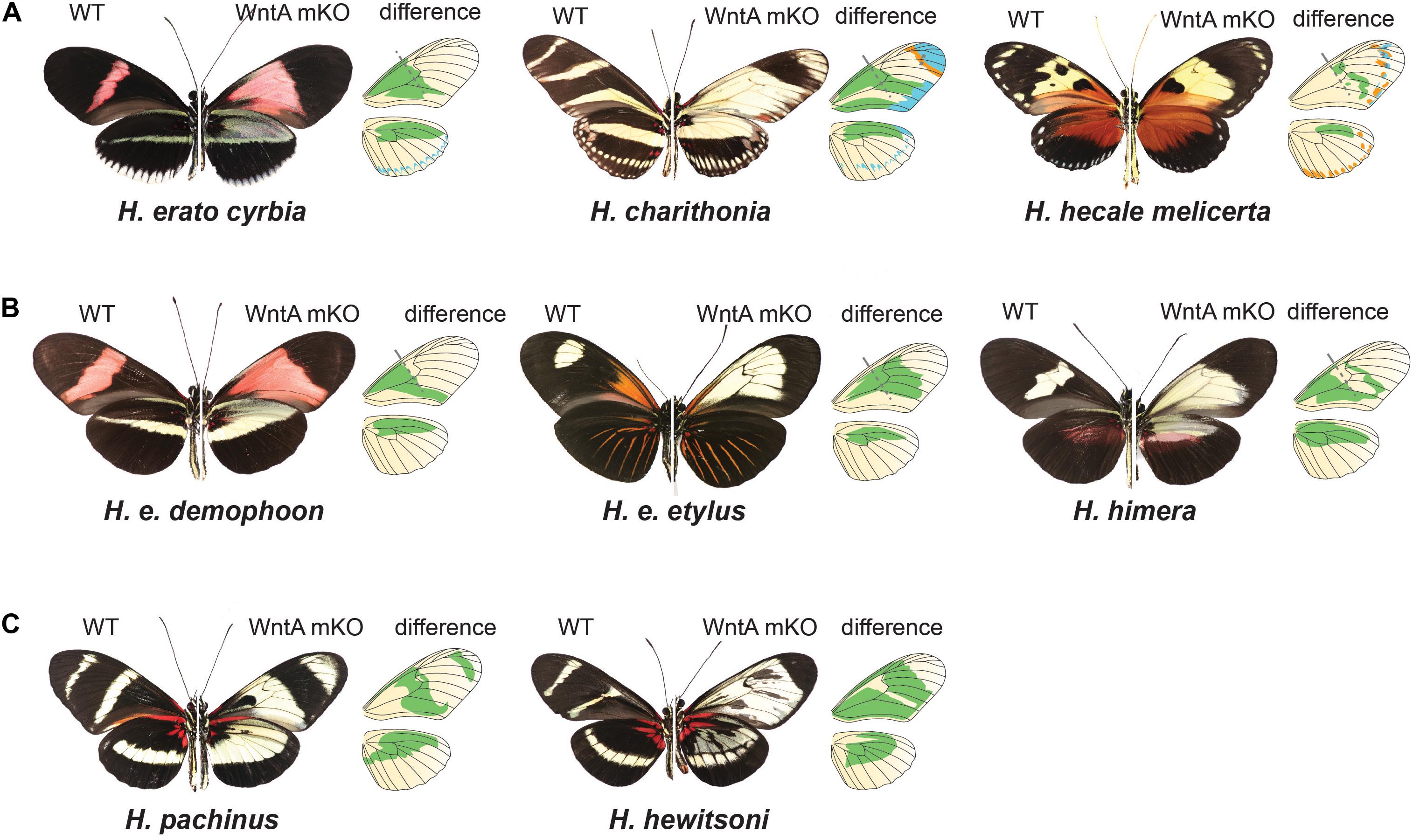
Figure 5. Chance and contingency on a Heliconius wing: CRISPR/Cas9 experiments demonstrate the pervasive role that WntA plays in orchestrating pattern variation and underscores how rapidly the underlying GRNs shaping wing patterns can diverge. (A) WntA seems to be broadly involved in establishing pattern boundaries across the wing and causes shifts in the ultimate scale cell fate in every possible direction. (B) WntA KO effects are highly context specific within H. erato color pattern races. While the same proximal elements are affected, the color switch varies depending on genetic background. (C) The underlying GRNs controlling mimicry have diverged substantially between the MCS/ESS lineages. This is exemplified by the co-mimetic species H. pachinus and H. hewitsoni. WntA KOs in H. hewitsoni affect pattern across most of the forewing, except the most distal regions. By contrast, H. pachinus KO effects extend more distally, and forewing band regions remain unchanged. For each Heliconius wild type and mutant image, we show a drawing of the wings, where green represents the wing color pattern domains that are changed by knocking out the WntA gene. In (A), light blue represents changes in distal patterns and orange represents an extension of black patterns.
The other two major patterning genes identified in Heliconius, optix and aristaless1, are both transcription factors that are expressed later in wing scale cell development in response to signals laid down either directly or indirectly by WntA and cortex. The transcription factor optix is expressed at about 72 h after pupation in Type III scale cells that are fated to become red. Functional knockout of this gene in H. erato results in the loss of red color patterns and is associated with changes in scale cell morphology (Zhang et al., 2017b). Consistent with a role in switching between ommochrome and melanin synthesis, an RNAseq experiment comparing optix mutant and wild type wings in two species of butterflies, Vanessa cardui and Junonia coenia, showed that optix, either directly or indirectly, regulates the expression of many known pigmentation genes involved in ommochrome and melanin synthesis (Zhang et al., 2017b). Lastly, the homeodomain transcription factor aristaless1 causes a change in Type I scale cells and promotes the development of structural white scales by repressing the synthesis or uptake of yellow 3-OHK (Westerman et al., 2018). The aristaless gene is duplicated in Lepidoptera (Martin and Reed, 2010) and in divergent color pattern races of H. cydno there is a strong peak of differentiation associated with the presence/absence of white coloration in a putative cis-regulatory region midway between the two aristaless genes, aristaless1 and aristaless2. Both genes are differentially expressed in the forewings of white and yellow H. cydno morphs, with aristaless1 strongly upregulated late in wing development of the white morph. Consistent with a role in patterning, functional knockout of aristaless1 in white morphs results in the transformation of white scale cells into yellow scale cells (Figure 3 and Westerman et al., 2018).
In Heliconius, there are many replicated cases of mimetic convergence between species within the ESS and MCS clades (see Figure 1 for some examples), providing a powerful context to understand how patterning networks co-evolve to produce variation. One unresolved question is the extent to which similar or distinct mechanisms have generated convergent phenotypes – in other words, are phenotypic repeats based on parallel reiterations of the same process, or do distinct developmental pathways lead to the same outcome? One of the more intriguing conclusions to emerge from comparative studies between species in the two Heliconius clades is how labile the GRNs responsible for patterning a Heliconius wing are (Concha et al., 2019). WntA knockouts in co-mimetic species of Heliconius result in very different phenotypes, demonstrating that the underlying GRNs controlling mimicry have diverged substantially between the two major clades (Concha et al., 2019; Figure 5C). Therefore, while WntA has been the repeated target of selection, its role in establishing pattern elements varies greatly between co-mimics. This fact is perhaps best demonstrated by comparing the KO phenotype between the two co-mimics H. hewitsoni (ESS clade) and H. pachinus (MCS clade). The two co-occur in forests of Panama and Costa Rica and have independently evolved identical wing patterns. However, the WntA KO forewing phenotypes are very different from each other, with WntA affecting pattern from the base of the forewing to nearly the tip in H. hewitsoni, whereas, the KO phenotype in H. pachinus causes a switch from melanic (Type III) to yellow Type I scale cells much more distally. In addition, knocking out WntA in H. pachinus does not affect the black melanic patch across the midsection of the forewing (Figure 5C).
The WntA knockout effects across the radiation highlight rapid divergence in the developmental landscape responsible for pattering a Heliconius wing; including changes in cis-regulatory variation around the locus, divergence in the interpretation of WntA signals by downstream developmental pathways, and variable epistatic interactions between WntA and other patterning loci (Concha et al., 2019). This finding is consistent with a recent study showing considerable differences in the expression patterns of transcription factors and signaling molecules between the two co-mimetic species H. e. demophoon and H. m. rosina (Hanly et al., 2019). Together these findings reinforce a growing appreciation that gene regulatory networks can diverge rapidly (True and Haag, 2001) and create a complex set of historical contingences that we are only beginning to appreciate (see Kittelmann et al., 2018; Sorrells et al., 2018; Ellison and Bachtrog, 2019). On a Heliconius wing, this divergence creates extraordinary flexibility for natural selection to drive the rapid evolution of highly convergent wing patterns in unpredictable ways.
While the functional genomic work on major wing patterning genes is beginning to provide a picture of the molecular events underlying the process of scale cell differentiation, there are many unresolved questions regarding the details of this process.
The major wing patterning loci in Heliconius are “evolutionary hotspots” and have been repeatedly targeted at both micro and macro-evolutionary scales to produce pattern variation. However, wing pattern development is likely to involve the coordinated expression of hundreds of genes. At the most basic level, we currently have little understanding of how WntA or cortex signals are propagated and interpreted by developing scale cells. Indeed, cortex does not conform to the typical paradigm of a wing patterning locus (sensu; Martin and Courtier-Orgogozo, 2017). Unlike the other wing patterning genes (Table 1), cortex is neither a conserved transcription factor nor a component of a known cell signaling pathway. Rather, the gene shows the closest homology to the cdc20/fizzy family of cell-cycle regulators (Nadeau et al., 2016). In Drosophila, Fizzy proteins are known to accumulate at kinetochores and centrosomes and regulate APC/C activity through the degradation of cyclins, which has been shown to prevent mitotic entry (Raff et al., 2002; Meghini et al., 2016). Specifically, expression of fizzy-related in Drosophila follicle cells induces cell endocycling and results in polyploidy, and cortex has been shown to interact with the APC/C and downregulate cyclins during female meiosis (Schaeffer et al., 2004; Shcherbata et al., 2004; Pesin and Orr-Weaver, 2007; Swan and Schüpbach, 2007). The Heliconius ortholog also localizes to the nucleus in developing pupal wings, suggesting it may also be interacting with the APC/C in developing butterfly scales (Livraghi et al., 2020). Other work has shown a correlation between scale cell ploidy and pigmentation state where increased ploidy coincides with darker scales (Cho and Nijhout, 2013; Iwata and Otaki, 2016) and cortex may thus be modulating pigmentation state by varying endocycle rates across the wing. Alternatively, cortex may be acting by modulating the developmental rate of scale cells during wing development. Melanic scales develop slower than the other scale cell types with melanin being synthesized and deposited later in wing development (Gilbert et al., 1988; Reed et al., 2008; Aymone et al., 2013). By controlling the faster or slower development of the scales, cortex may be modulating the final pigmentation state of a scale cell. This hypothesis of “developmental heterochrony” to explain the mechanism of butterfly wing color patterning was proposed 20 years ago (Koch et al., 2000) without gaining much supporting evidence over the years.
The mechanism of how the locus containing cortex affects pattern variation is probably complex. Although cortex is clearly involved in determining scale cell identity, there are other genes that are physically linked to cortex that might also play a role in patterning. Most notably, the genes domeless and washout show pattern specific expression in both H. melpomene and H. numata (Nadeau et al., 2016; Saenko et al., 2019). Domeless encodes a transmembrane receptor of the JAK/STAT signaling pathway (Brown et al., 2001) while washout is involved in actin polymerization through the activation of the Arp2/3 complex (Liu et al., 2009; Verboon et al., 2015), raising the interesting possibility that it may be involved in the formation of adult scales through the control of actin bundle elongation (Dinwiddie et al., 2014). It is therefore possible that multiple linked genes around cortex contribute to wing patterning evolution in Heliconius. Theory predicts that in some cases the linking of such genes might be favored by evolution (Charlesworth and Charlesworth, 1976), leading to the emergence of “supergenes”: tightly linked functional genetic elements that allow a switch between phenotypes to be maintained in a stable local polymorphism (Joron et al., 2011; Le Poul et al., 2014). Indeed, there is some indication that such a system is acting to control polymorphism in H. numata, where all wing pattern variation is controlled by inversions surrounding cortex, domeless and washout (Joron et al., 2011; Nadeau et al., 2016; Saenko et al., 2019). However, we currently have no functional validation for other genes at this locus, nor evidence for interactions between them.
WntA, in contrast, is a classic cell signaling molecule belonging to the family of Wnt signaling ligands. However, we also currently lack an understanding of how WntA signals are transduced into a coordinated scale differentiation program. Wnt signaling may act through the canonical pathway, by which β-catenin is permitted to translocate to the nucleus and act as a transcriptional cofactor (reviewed in Collu et al., 2014), or alternatively it may signal through a non-canonical modality such as the planar cell polarity pathway (PCP) (Lawrence and Casal, 2018), or by a third mechanism such as Wnt-calcium signaling (Agrawal and Hasan, 2015). Understanding the mechanism by which WntA causes patterned scale cell differentiation will be an important first step in unraveling the larger gene regulatory networks that pattern the wings.
One idea to emerge from developmental biology is that loci that have become the nexus of morphological change, so called hotspot genes, are often associated with complex cis-regulatory variation (Stern and Orgogozo, 2009). This is certainly true of the major patterning loci identified in Heliconius. Expression, association, and CRISPR studies have all revealed that cis-regulatory variation is important in driving wing patterning differences. Nonetheless, we still don’t understand how new regulatory regions evolve and what role, if any, ancient regulatory architecture plays in wing pattern variation. Association studies in Heliconius suggest that there is a high degree of modularity with regards to the cis-regulatory elements controlling color pattern variation (Supple et al., 2013; Wallbank et al., 2016; Enciso-Romero et al., 2017; Van Belleghem et al., 2017). For example, different red color pattern elements in H. melpomene and H. erato are associated with discrete genomic intervals in non-coding regions around optix (Wallbank et al., 2016; Van Belleghem et al., 2017). A similar modular architecture is also observed for variation in forewing band shape (WntA) and the presence or absence of a yellow hindwing bar (cortex) within H. erato (Van Belleghem et al., 2017). The idea is that new wing pattern phenotypes can evolve rapidly by simply reshuffling these discrete regulatory regions. In the MCS lineage, for example, the phylogenetic patterns are consistent with at least four instances of between species hybridization that allowed these elements to cross species boundaries, caused pattern change, and probably promoted speciation in the group (Wallbank et al., 2016).
A model where “enhancer shuffling” drives morphological differentiation, however, was not strongly supported by recent work that has more directly probed the relationship between regulatory and phenotypic variation (Lewis et al., 2019). Here, the authors identified and excised a number of putative cis-regulatory regions in genomic intervals associated with red pattern variation in H. erato using a combination of epigenetic profiling and genotype × phenotype analysis. Surprisingly, they found that deletions of a single cis-regulatory element were highly pleiotropic and affected red pattern elements in both the forewing and hindwing. The level of pleiotropy observed suggests that more constraints are acting at color pattern loci than previously hypothesized and this is difficult to reconcile under a model where a single regulatory region affects a single pattern feature. A possible resolution to this apparent conflict between the association and functional data may be explained by the idiosyncratic properties of phenotypes segregating in Heliconius hybrid zones, whereby a single locus can act in a modular fashion in localized population structures; however, because of an ever shifting trans-regulatory landscape, excising a specific enhancer after patterns have become established and genetic backgrounds have diverged is not sufficient to induce the predicted phenotype (see Lewis and Van Belleghem, unpublished, for a discussion of this topic). At this point, we clearly need more functional studies that assess the evidence for and against modularity in driving the evolution of Heliconius wing patterns.
In Heliconius, much of the pattern variation across the genus is driven by variation at homologous genes. However, even in this relatively recent radiation, developmental systems drift appears to be shifting the wiring of the underlying GRNs (Concha et al., 2019). Expanding functional studies across the lepidoptera will be necessary to start addressing questions relating to the evolutionary constraints acting across GRNs as a whole. Moreover, while mapping studies across Heliconius consistently implicate four major patterning genes, variation in other loci across the genome also have quantitative effects on pattern variation (Papa et al., 2013). Indeed, more recent mapping experiments show that structural coloration in Heliconius is not achieved using any of the known patterning loci and that different loci are used to produce structural colors in the two distantly related co-mimics H. erato and H. melpomene (Parnell et al., 2018; Brien et al., 2019). Also, while cortex has been repeatedly mapped in within-species pattern variation, recent studies have begun to identify other genes, not previously identified in Heliconius, as responsible for pattern variation (Table 1). In this light, it will be very interesting to determine if these other conserved components of GRNs underlying wing patterning in other lepidoptera contribute to pattern variation in Heliconius.
While individual components have been identified through evolutionary approaches, very few regulatory connections have been resolved, and we still don’t know the broad applicability of discoveries made in Heliconius to other butterflies or moths. From an evolutionary perspective, Heliconius wings represent a highly derived type of a more ancestral patterning system called the Nymphalid Ground Plan (NGP) (Nijhout and Wray, 1988). In this sense, Heliconius wings may not represent the best system with which to address questions relating to the conservation of GRNs in other lepidoptera, as it is difficult to draw homologies between color pattern elements at larger evolutionary scales. For example, while WntA has been implicated in color pattern evolution across both Heliconius and other nymphalids (Martin et al., 2012; Gallant et al., 2014; Van Belleghem et al., 2017), the pattern elements it affects vary substantially. In species displaying NGP-like patterns, WntA functions as an organizing factor of Central and External Symmetry Systems in at least three distantly related nymphalids (Mazo-Vargas et al., 2017), suggesting a conserved role across large evolutionary scales. On the other hand, it is noteworthy that all of the four major patterning loci discovered in Heliconius affect pattern in other butterflies and moths. In this light, the apparent flexibility in the GRNs that we are observing in Heliconius may reflect the nature of wing patterning networks generally. Thus, while a particular set of conserved loci might have a function in generating pattern variation, how they generate that variation may differ substantially among lineages. For example, the eyespot patterns that are found in many nymphalid butterflies appear to have evolved once; but the gene regulatory networks underlying these patterns have diverged significantly over the past 65 million years such that the eyespots of different species are controlled by different sets of genes (Oliver et al., 2012).
In multicellular organisms, phenotype is ultimately realized in the patterned differentiation of cells into specific functional types. In this respect, butterfly wings have a unique combination of features which make them an ideal system to uncover how changes in a DNA sequence are translated and interpreted through development to generate variation: a relatively simple developmental system, large phenotypic diversity, and experimental tractability.
There is clearly much to be discovered, but the last few years have witnessed remarkable progress in our understanding of pattern formation on butterfly wings. Research priorities are clearly laid out and technical improvements, including techniques for characterizing the developmental landscape of protein–DNA interactions (Park, 2009; Rao et al., 2014; Buenrostro et al., 2015; Lewis et al., 2016, 2019) and the expression profiles of individual cells (Kulkarni et al., 2019), coupled with more efficient functional manipulation provide us with the necessary instruments to further uncover the gene regulatory networks underlying wing pattern variation. In this respect, one of the key technical limitations facing Heliconius is the lack of transgenic tools. Our current use of CRISPR as a functional tool has been limited to the analysis of mosaic loss-of-function mutants, where only a subset of cells carry the desired mutation on the adult wing. While this can be a strength, allowing the analysis of the effects of highly pleiotropic genes and permitting the observation of effects at clone boundaries, in many cases this makes it difficult to determine the extent to which a given induced mutation affects pattern across the wing. These shortcomings could be bypassed by developing technologies that allow researchers to routinely produce gain-of-function mutations; as well as those that would allow better spatiotemporal control over genetic manipulations. For example, by placing reporter constructs under specific inducible elements, the ability of a putative cis-regulatory element to drive expression in the tissue of interest could be directly visualized (Tong et al., 2014). Nonetheless, if we stay on pace we will have come a long way toward fulfilling the prophecy made by Bates (1864) and “the study of butterflies—creatures selected as the types of airiness and frivolity—instead of being despised, will someday be valued as one of the most important branches of Biological science.”
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
WM was supported by the Smithsonian Tropical Research Institute and the Smithsonian Scholarly Studies Program. LL was supported by BBSRC BB/R007500/1. JH was supported by the NSF award IOS-1656553. CC was supported by the Smithsonian Institute Biodiversity Genomics Fellowship and NSF IOS-1656389.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the Butterfly Ecology and Evolution Research group at STRI, as well as the Butterfly Genetics group at the University of Cambridge, the Martin Lab at GWU and the Papa Lab at UPR-RP for constructive conversations that helped to formulate this review. In addition, we thank Marcus Kronforst, Bob Reed, and Steven Van Belleghem for providing images of butterflies, and two reviewers for their helpful feedback on this manuscript.
Agrawal, T., and Hasan, G. (2015). Maturation of a central brain flight circuit in Drosophila requires Fz2/Ca2+ signaling. eLife 4:e07046. doi: 10.7554/eLife.07046.001
Aymone, A. C. B., Valente, V. L. S., and de Araújo, A. M. (2013). Ultrastructure and morphogenesis of the wing scales in Heliconius erato phyllis (Lepidoptera: Nymphalidae): what silvery/brownish surfaces can tell us about the development of color patterning? Arthropod. Struct. Dev. 42, 349–359. doi: 10.1016/j.asd.2013.06.001
Bates, H. W. (1864). The Naturalist on the River Amazons: A Record of Adventures, Habits of Animals, Sketches of Brazilian and Indian Life, and Aspects of Nature under the Equator, During Eleven Years of Travel. London: J Murry. Available online at: https://www.biodiversitylibrary.org/page/13494275#page/9/mode/1up
Beebe, W. (2005). Polymorphism in reared broods of Heliconius butterflies from Surinam and Trinidad. Zoologica 40, 139–143.
Benson, W. W. (1972). Natural selection for Müllerian mimicry in Heliconius erato in Costa Rica. Science 176, 936–939. doi: 10.1126/science.176.4037.936
Brakefield, P. M., and French, V. (1999). Butterfly wings: the evolution of development of colour patterns. Bioessays 21, 391–401. doi: 10.1002/(sici)1521-1878(199905)21:5<391::aid-bies6>3.0.co;2-q
Brien, M. N., Enciso-Romero, J., Parnell, A. J., Salazar, P. A., Morochz, C., Chalá, D., et al. (2019). Phenotypic variation in Heliconius erato crosses shows that iridescent structural colour is sex-linked and controlled by multiple genes. Interface Focus 9:20180047. doi: 10.1098/rsfs.2018.0047
Brown, K. S. (1967). Chemotaxonomy and chemomimicry: the case of 3-hydroxykynurenine. Syst. Zool. 16, 213–216.
Brown, S., Hu, N., and Hombría, J. C. (2001). Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr. Biol. 11, 1700–1705. doi: 10.1016/s0960-9822(01)00524-3
Buenrostro, J. D., Wu, B., Chang, H. Y., and Greenleaf, W. J. (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109:21.29.1-21.29.9. doi: 10.1002/0471142727.mb2129s109
Carroll, S. B. (2008). Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134, 25–36. doi: 10.1016/j.cell.2008.06.030
Charlesworth, D., and Charlesworth, B. (1976). Theoretical genetics of Batesian mimicry II. Evol. Supergenes 55, 305–324. doi: 10.1016/s0022-5193(75)80082-8
Cho, E. H., and Nijhout, H. F. (2013). Development of polyploidy of scale-building cells in the wings of Manduca sexta. Arthropod. Struct. Dev. 42, 37–46. doi: 10.1016/j.asd.2012.09.003
Chouteau, M., Llaurens, V., Piron-Prunier, F., and Joron, M. (2017). Polymorphism at a mimicry supergene maintained by opposing frequency-dependent selection pressures. Proc. Natl. Acad. Sci. 114, 8325–8329. doi: 10.1073/pnas.1702482114
Collu, G. M., Hidalgo-Sastre, A., and Brennan, K. (2014). Wnt-Notch signaling crosstalk in development and disease. Cell. Mol. Life Sci. 71, 3553–3567. doi: 10.1007/s00018-014-1644-x
Concha, C., Wallbank, R. W. R., Hanly, J. J., Fenner, J., Livraghi, L., Rivera, E. S., et al. (2019). Interplay between developmental flexibility and determinism in the evolution of mimetic Heliconius wing patterns. Curr. Biol. 29, 3996.e–4009.e. doi: 10.1016/j.cub.2019.10.010
Cong, Q., Shen, J., Zhang, J., Li, W., Kinch, L. N., Calhoun, J. V., et al. (2019). Genomics reveals the origins of ancient specimens. bioRxiv [Preprint] doi: 10.1101/752121
Dinwiddie, A., Null, R., Pizzano, M., Chuong, L., Leigh Krup, A., Ee Tan, H., et al. (2014). Dynamics of F-actin prefigure the structure of butterfly wing scales. Dev. Biol. 392, 404–418. doi: 10.1016/j.ydbio.2014.06.005
Edelman, N. B., Frandsen, P. B., Miyagi, M., Clavijo, B., Davey, J., Dikow, R. B., et al. (2019). Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599. doi: 10.1126/science.aaw2090
Ellison, C., and Bachtrog, D. (2019). Contingency in the convergent evolution of a regulatory network: dosage compensation in Drosophila. PLoS Biol. 17:e3000094. doi: 10.1371/journal.pbio.3000094
Enciso-Romero, J., Pardo-Diaz, C., Martin, S. H., Arias, C. F., Linares, M., McMillan, W. O., et al. (2017). Evolution of novel mimicry rings facilitated by adaptive introgression in tropical butterflies. Mol. Ecol. 26, 5160–5172. doi: 10.1111/mec.14277
Ferguson, L. C., and Jiggins, C. D. (2009). Shared and divergent expression domains on mimetic Heliconius wings. Evol. Dev. 11, 498–512. doi: 10.1111/j.1525-142X.2009.00358.x
Galant, R., Skeath, J. B., Paddock, S., Lewis, D. L., and Carroll, S. B. (1998). Expression pattern of a butterfly achaete-scute homolog reveals the homology of butterfly wing scales and insect sensory bristles. Curr. Biol. 8, 807–813. doi: 10.1016/s0960-9822(98)70322-7
Gallant, J. R., Imhoff, V. E., Martin, A., Savage, W. K., Chamberlain, N. L., Pote, B. L., et al. (2014). Ancient homology underlies adaptive mimetic diversity across butterflies. Nat. Commun. 5:4817. doi: 10.1038/ncomms5817
Gawne, R., McKenna, K. Z., and Nijhout, H. F. (2018). Unmodern synthesis: developmental hierarchies and the origin of phenotypes. Bioessays 40:1600265. doi: 10.1002/bies.201600265
Ghiradella, H., and Radigan, W. (1976). Development of butterfly scales. II. Struts, lattices and surface tension. J. Morphol. 150, 279–297. doi: 10.1002/jmor.1051500202
Gilbert, L. E., Forrest, H. S., Schultz, T. D., and Harvey, D. J. (1988). Correlations of ultrastructure and pigmentation suggest how genes control development of wing scales of Heliconius butterflies. J. Res. Lepidoptera 26, 141–160.
Greenstein, M. E. (1972). The ultrastructure of developing wings in the giant silkmoth, Hyalophora cecropia. II. Scale-forming and socket-forming cells. J. Morphol. 136, 23–51. doi: 10.1002/jmor.1051360103
Hanly, J. J., Wallbank, R. W. R., McMillan, W. O., and Jiggins, C. D. (2019). Conservation and flexibility in the gene regulatory landscape of heliconiine butterfly wings. Evodevo 10, 1–15. doi: 10.1186/s13227-019-0127-4
Hines, H. M., Papa, R., Ruiz, M., Papanicolaou, A., Wang, C., Nijhout, H. F., et al. (2012). Transcriptome analysis reveals novel patterning and pigmentation genes underlying Heliconius butterfly wing pattern variation. BMC Genomics 13:288. doi: 10.1186/1471-2164-13-288
Huber, B., Whibley, A., Poul, Y. L., Navarro, N., Martin, A., Baxter, S., et al. (2015). Conservatism and novelty in the genetic architecture of adaptation in Heliconius butterflies. Heredity 114, 515–524. doi: 10.1038/hdy.2015.22
Ito, K., Katsuma, S., Kuwazaki, S., Jouraku, A., Fujimoto, T., Sahara, K., et al. (2016). Mapping and recombination analysis of two moth colour mutations, Black moth and Wild wing spot, in the silkworm Bombyx mori. Heredity 116, 52–59. doi: 10.1038/hdy.2015.69
Iwata, M., and Otaki, J. M. (2016). Spatial patterns of correlated scale size and scale color in relation to color pattern elements in butterfly wings. J. Insect Physiol. 85, 32–45. doi: 10.1016/j.jinsphys.2015.11.013
Jiggins, C. D. (2017). The Ecology and Evolution of Heliconius Butterflies. Oxford: Oxford University Press.
Jiggins, C. D., Naisbit, R. E., Coe, R. L., and Mallet, J. (2001). Reproductive isolation caused by colour pattern mimicry. Nature 411, 302–305. doi: 10.1038/35077075
Joron, M., Frezal, L., Jones, R. T., Chamberlain, N. L., Lee, S. F., Haag, C. R., et al. (2011). Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477, 203–206. doi: 10.1038/nature10341
Joron, M., Papa, R., Beltrán, M., Chamberlain, N., Mavárez, J., Baxter, S., et al. (2006). A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol. 4:e303. doi: 10.1371/journal.pbio.0040303
Kapan, D. D. (2001). Three-butterfly system provides a field test of müllerian mimicry. Nature 409, 338–340. doi: 10.1038/35053066
Kapan, D. D., Flanagan, N. S., Tobler, A., Papa, R., Reed, R. D., Gonzalez, J. A., et al. (2006). Localization of Müllerian mimicry genes on a dense linkage map of Heliconius erato. Genetics 173, 735–757. doi: 10.1534/genetics.106.057166
Kittelmann, S., Buffry, A. D., Franke, F. A., Almudi, I., Yoth, M., Sabaris, G., et al. (2018). Gene regulatory network architecture in different developmental contexts influences the genetic basis of morphological evolution. PLoS Genet. 14:e1007375. doi: 10.1371/journal.pgen.1007375
Koch, P. B., Behnecke, B., and ffrench-Constant, R. H. (2000). The molecular basis of melanism and mimicry in a swallowtail butterfly. Curr. Biol. 10, 591–594. doi: 10.1016/s0960-9822(00)00494-2
Kozak, K. M., Wahlberg, N., Neild, A. F. E., Dasmahapatra, K. K., Mallet, J., and Jiggins, C. D. (2015). Multilocus species trees show the recent adaptive radiation of the mimetic Heliconius butterflies. Syst. Biol. 64, 505–524. doi: 10.1093/sysbio/syv007
Kristensen, N. P., Scibke, M. J., and Karsholt, O. (2007). Lepidoptera phylogeny and systematics: the state of inventorying moth and butterfly diversity. Zootaxa 1668, 699–747. doi: 10.11646/zootaxa.1668.1.30
Kronforst, M. R., Kapan, D. D., and Gilbert, L. E. (2006). Parallel genetic architecture of parallel adaptive radiations in mimetic Heliconius butterflies. Genetics 174, 535–539. doi: 10.1534/genetics.106.059527
Kronforst, M. R., and Papa, R. (2015). The functional basis of wing patterning in Heliconius butterflies: the molecules behind mimicry. Genetics 200, 1–19. doi: 10.1534/genetics.114.172387
Kulkarni, A., Anderson, A. G., Merullo, D. P., and Konopka, G. (2019). Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 58, 129–136. doi: 10.1016/j.copbio.2019.03.001
Kunte, K., Zhang, W., Tenger-Trolander, A., Palmer, D. H., Martin, A., Reed, R. D., et al. (2014). doublesex is a mimicry supergene. Nature 507, 229–232. doi: 10.1038/nature13112
Lawrence, P. A., and Casal, J. (2018). Planar cell polarity: two genetic systems use one mechanism to read gradients. Development 145:dev168229. doi: 10.1242/dev.168229
Le Poul, Y., Whibley, A., Chouteau, M., Prunier, F., Llaurens, V., and Joron, M. (2014). Evolution of dominance mechanisms at a butterfly mimicry supergene. Nat. Commun. 5:5644. doi: 10.1038/ncomms6644
Lewis, J. J., Geltman, R. C., Pollak, P. C., Rondem, K. E., Van Belleghem, S. M., Hubisz, M. J., et al. (2019). Parallel evolution of ancient, pleiotropic enhancers underlies butterfly wing pattern mimicry. Proc. Natl. Acad. Sci. 116, 24174–24183. doi: 10.1073/pnas.1907068116
Lewis, J. J., van der Burg, K. R. L., Mazo-Vargas, A., and Reed, R. D. (2016). ChIP-Seq-annotated Heliconius erato genome highlights patterns of cis-regulatory evolution in Lepidoptera. Cell Rep. 16, 2855–2863. doi: 10.1016/j.celrep.2016.08.042
Liu, R., Abreu-Blanco, M. T., Barry, K. C., Linardopoulou, E. V., Osborn, G. E., and Parkhurst, S. M. (2009). Wash functions downstream of Rho and links linear and branched actin nucleation factors. Development 136, 2849–2860. doi: 10.1242/dev.035246
Livraghi, L., Hanly, J. J., Loh, L. S., Ren, A., Warren, I. A., Concha, C., et al. (2020). The gene cortex controls scale colour identity in Heliconius. bioRxiv [Preprint] doi: 10.1101/2020.05.26.116533
Livraghi, L., Martin, A., Gibbs, M., Braak, N., Arif, S., and Joron, M. (2018). CRISPR/Cas9 as the key to unlocking the secrets of butterfly wing pattern development and its evolution. Adv. Insect Physiol. 54, 85–115. doi: 10.1016/bs.aiip.2017.11.001
Mallet, J., Barton, N., Lamas, G., Santisteban, J., Muedas, M., and Eeley, H. (1990). Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in Heliconius hybrid zones. Genetics 124, 921–936.
Martin, A., and Courtier-Orgogozo, V. (2017). “Morphological evolution repeatedly caused by mutation in signaling ligand genes,” in Diversity and Evolution of Butterfly Wing Patterns, eds T. Sekimura and H. F. Nijhout (Singapore: Springer Singapore), 1–54. doi: 10.1007/978-981-10-4956-9
Martin, A., McCulloch, K. J., Patel, N. H., Briscoe, A. D., Gilbert, L. E., and Reed, R. D. (2014). Multiple recent co-options of Optix associated with novel traits in adaptive butterfly wing radiations. Evodevo 5:7. doi: 10.1186/2041-9139-5-7
Martin, A., Papa, R., Nadeau, N. J., Hill, R. I., Counterman, B. A., Halder, G., et al. (2012). Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc. Natl. Acad. Sci. U.S.A. 109, 12632–12637. doi: 10.1073/pnas.1204800109
Martin, A., and Reed, R. D. (2010). wingless and aristaless2 define a developmental ground plan for moth and butterfly wing pattern evolution. Mol. Biol. Evol. 27, 2864–2878. doi: 10.1093/molbev/msq173
Martin, A., and Reed, R. D. (2014). Wnt signaling underlies evolution and development of the butterfly wing pattern symmetry systems. Dev. Biol. 395, 367–378. doi: 10.1016/j.ydbio.2014.08.031
Martin, S. H., Singh, K. S., Gordon, I. J., Omufwoko, K. S., Collins, S., Warren, I. A., et al. (2020). Whole-chromosome hitchhiking driven by a male-killing endosymbiont. PLoS Biol. 18:e3000610. doi: 10.1371/journal.pbio.3000610
Matsuoka, Y., and Monteiro, A. (2018). Melanin pathway genes regulate color and morphology of butterfly wing scales. Cell Rep. 24, 56–65. doi: 10.1016/j.celrep.2018.05.092
Mazo-Vargas, A., Concha, C., Livraghi, L., Massardo, D., Wallbank, R. W. R., Zhang, L., et al. (2017). Macroevolutionary shifts of WntA function potentiate butterfly wing-pattern diversity. Proc. Natl. Acad. Sci. U.S.A. 114, 10701–10706. doi: 10.1073/pnas.1708149114
Meghini, F., Martins, T., Tait, X., Fujimitsu, K., Yamano, H., Glover, D. M., et al. (2016). Targeting of Fzr/Cdh1 for timely activation of the APC/C at the centrosome during mitotic exit. Nat. Commun. 7, 12607–12617. doi: 10.1038/ncomms12607
Merrill, R. M., Rastas, P., Martin, S. H., Melo, M. C., Barker, S., Davey, J., et al. (2019). Genetic dissection of assortative mating behavior. PLoS Biol. 17:e2005902. doi: 10.1371/journal.pbio.2005902
Nadeau, N. J., Pardo-Diaz, C., Whibley, A., Supple, M. A., Saenko, S. V., Wallbank, R. W. R., et al. (2016). The gene cortex controls mimicry and crypsis in butterflies and moths. Nature 534, 106–110. doi: 10.1038/nature17961
Nardi, J. B., and Magee-Adams, S. M. (1986). Formation of scale spacing patterns in a moth wing. Dev. Biol. 116, 278–290. doi: 10.1016/0012-1606(86)90131-4
Nijhout, H. F. (1991). The Development and Evolution of Butterfly Wing Patterns. Washington, DC: Smithsonian Institution Press, doi: 10.18984/yadoriga.1998.177_2
Nijhout, H. F. (2001). Elements of butterfly wing patterns. J. Exp. Zool. 291, 213–225. doi: 10.1002/jez.1099
Nijhout, H. F., and Wray, G. A. (1988). Homologies in the colour patterns of the genus Heliconius (Lepidoptera: Nymphalidae). Biol. J. Linnean Soc. 33, 345–365. doi: 10.1111/j.1095-8312.1988.tb00449.x
Oliver, J. C., Tong, X.-L., Gall, L. F., Piel, W. H., and Monteiro, A. (2012). A single origin for Nymphalid butterfly eyespots followed by widespread loss of associated gene expression. PLoS Genet. 8:e1002893–e1002898. doi: 10.1371/journal.pgen.1002893
Organista, M. F., Martín, M., de Celis, J. M., Barrio, R., López-Varea, A., Esteban, N., et al. (2015). The spalt transcription factors generate the transcriptional landscape of the Drosophila melanogaster wing pouch central region. PLoS Genet. 11:e1005370. doi: 10.1371/journal.pgen.1005370
Overton, J. (1966). Microtubules and microfibrils in morphogenesis of the scale cells of Ephestia kühniella. J. Cell Biol. 29, 293–293.
Papa, R., Kapan, D. D., Counterman, B. A., Maldonado, K., Lindstrom, D. P., Reed, R. D., et al. (2013). Multi-allelic major effect genes interact with minor effect QTLs to control adaptive color pattern variation in Heliconius erato. PLoS One 8:e57033. doi: 10.1371/journal.pone.0057033
Park, P. J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680. doi: 10.1038/nrg2641
Parnell, A. J., Bradford, J. E., Curran, E. V., Washington, A. L., Adams, G., Brien, M. N., et al. (2018). Wing scale ultrastructure underlying convergent and divergent iridescent colours in mimetic Heliconius butterflies. J. R. Soc. Interf, 15:20170948. doi: 10.1098/rsif.2017.0948
Pasteur, G. (1982). A classificatory review of mimicry systems. Ann. Rev. Ecol. Syst. 13, 169–199. doi: 10.1146/annurev.es.13.110182.001125
Pesin, J. A., and Orr-Weaver, T. L. (2007). Developmental role and regulation of cortex, a meiosis-specific anaphase-promoting complex/cyclosome activator. PLoS Genet. 3:e202. doi: 10.1371/journal.pgen.0030202
Raff, J. W., Jeffers, K., and Huang, J.-Y. (2002). The roles of Fzy/Cdc20 and Fzr/Cdh1 in regulating the destruction of cyclin B in space and time. J. Cell Biol. 157, 1139–1149. doi: 10.1083/jcb.200203035
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Reed, R. D. (2004). Evidence for Notch-mediated lateral inhibition in organizing butterfly wing scales. Dev. Genes Evol. 214, 43–46. doi: 10.1007/s00427-003-0366-0
Reed, R. D., McMillan, W. O., and Nagy, L. M. (2008). Gene expression underlying adaptive variation in Heliconius wing patterns: non-modular regulation of overlapping cinnabar and vermilion prepatterns. Proc. Biol. Sci. 275, 37–45. doi: 10.1098/rspb.2007.1115
Reed, R. D., Papa, R., Martin, A., Hines, H. M., Counterman, B. A., Pardo-Diaz, C., et al. (2011). optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333, 1137–1141. doi: 10.1126/science.1208227
Reed, R. D., and Serfas, M. S. (2004). Butterfly wing pattern evolution is associated with changes in a Notch/Distal-less temporal pattern formation process. Curr. Biol. 14, 1159–1166. doi: 10.1016/j.cub.2004.06.046
Rudel, D., and Sommer, R. J. (2003). The evolution of developmental mechanisms. Dev. Biol. 264, 15–37. doi: 10.1016/S0012-1606(03)00353-1
Saenko, S. V., Brakefield, P. M., and Beldade, P. (2010). Single locus affects embryonic segment polarity and multiple aspects of an adult evolutionary novelty. BMC Biol. 8:111. doi: 10.1186/1741-7007-8-111
Saenko, S. V., Chouteau, M., Piron-Prunier, F., Blugeon, C., Joron, M., and Llaurens, V. (2019). Unravelling the genes forming the wing pattern supergene in the polymorphic butterfly Heliconius numata. Evodevo 10, 16–12. doi: 10.1186/s13227-019-0129-2
Schaeffer, V., Althauser, C., Shcherbata, H. R., Deng, W.-M., and Ruohola-Baker, H. (2004). Notch-dependent fizzy-related/Hec1/Cdh1 expression is required for the mitotic-to-endocycle transition in Drosophila follicle cells. Curr. Biol. 14, 630–636. doi: 10.1016/j.cub.2004.03.040
Seimiya, M., and Gehring, W. J. (2000). The Drosophila homeobox gene optix is capable of inducing ectopic eyes by an eyeless-independent mechanism. Development 127, 1879–1886.
Shcherbata, H. R., Althauser, C., Findley, S. D., and Ruohola-Baker, H. (2004). The mitotic-to-endocycle switch in Drosophila follicle cells is executed by Notch-dependent regulation of G1/S, G2/M and M/G1 cell-cycle transitions. Development 131, 3169–3181. doi: 10.1242/dev.01172
Sheppard, P. M., Turner, J. R. G., Brown, K. S. Jr., Benson, W. W., and Singer, M. A. (1985). Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Philos. Trans. R. Soc. Lond. 308, 433–610. doi: 10.1098/rstb.2010.0410
Sorrells, T. R., Johnson, A. N., Howard, C. J., Britton, C. S., Fowler, K. R., Feigerle, J. T., et al. (2018). Intrinsic cooperativity potentiates parallel cis-regulatory evolution. eLife 7:e37563. doi: 10.7554/eLife.37563
Stern, D. L., and Orgogozo, V. (2009). Is genetic evolution predictable? Science 323, 746–751. doi: 10.1126/science.1158997
Stossberg, M. (1938). Die zellvorgänge bei der entwicklung der flügelschuppen von Ephestia kühniella Z. Zeitschrift Morphol. Okol. Tiere 34, 173–206. doi: 10.2307/43261719
Supple, M., Papa, R., Counterman, B., and McMillan, W. O. (2014). The genomics of an adaptive radiation: insights across the Heliconius speciation continuum. Adv. Exp. Med. Biol. 781, 249–271. doi: 10.1007/978-94-007-7347-9_13
Supple, M. A., Hines, H. M., Dasmahapatra, K. K., Lewis, J. J., Nielsen, D. M., Lavoie, C., et al. (2013). Genomic architecture of adaptive color pattern divergence and convergence in Heliconius butterflies. Genome Res. 23, 1248–1257. doi: 10.1101/gr.150615.112
Swan, A., and Schüpbach, T. (2007). The Cdc20 (Fzy)/Cdh1-related protein. Cort, cooperates with Fzy in cyclin destruction and anaphase progression in meiosis I and II in Drosophila. Development 134, 891–899. doi: 10.1242/dev.02784
Tong, X., Hrycaj, S., Podlaha, O., Popadic, A., and Monteiro, A. (2014). Over-expression of Ultrabithorax alters embryonic body plan and wing patterns in the butterfly Bicyclus anynana. Dev. Biol. 394, 357–366. doi: 10.1016/j.ydbio.2014.08.020
True, J. R., and Haag, E. S. (2001). Developmental system drift and flexibility in evolutionary trajectories. Evol. Dev. 3, 109–119. doi: 10.1046/j.1525-142x.2001.003002109.x
Turner, J. R. G. (1972). The genetics of some polymorphic forms of the butterflies Heliconius melpomene (Linnaeus) and H. erato (Linnaeus). II. The hybridization of subspecies of H. melpomene from Surinam and Trinidad. Zoologica 52:125.
Van Belleghem, S. M., Rastas, P., Papanicolaou, A., Martin, S. H., Arias, C. F., Supple, M. A., et al. (2017). Complex modular architecture around a simple toolkit of wing pattern genes. Nat. Ecol. Evol. 1:52. doi: 10.1038/s41559-016-0052
VanKuren, N. W., Massardo, D., Nallu, S., and Kronforst, M. R. (2019). Butterfly mimicry polymorphisms highlight phylogenetic limits of gene reuse in the evolution of diverse adaptations. Mol. Biol. Evol. 36, 2842–2853. doi: 10.1093/molbev/msz194
van’t Hof, A. E., Campagne, P., Rigden, D. J., Yung, C. J., Lingley, J., Quail, M. A., et al. (2016). The industrial melanism mutation in British peppered moths is a transposable element. Nature 534, 102–105. doi: 10.1038/nature17951
van’t Hof, A. E., Reynolds, L. A., Yung, C. J., Cook, L. M., and Saccheri, I. J. (2019). Genetic convergence of industrial melanism in three geometrid moths. Biol. Lett. 15:20190582. doi: 10.1098/rsbl.2019.0582
Verboon, J. M., Rincon-Arano, H., Werwie, T. R., Delrow, J. J., Scalzo, D., Nandakumar, V., et al. (2015). Wash interacts with lamin and affects global nuclear organization. Curr. Biol. 25, 804–810. doi: 10.1016/j.cub.2015.01.052
Wallbank, R. W. R., Baxter, S. W., Pardo-Diaz, C., Hanly, J. J., Martin, S. H., Mallet, J., et al. (2016). Evolutionary novelty in a butterfly wing pattern through enhancer shuffling. PLoS Biol. 14:e1002353. doi: 10.1371/journal.pbio.1002353
Westerman, E. L., VanKuren, N. W., Massardo, D., Tenger-Trolander, A., Zhang, W., Hill, R. I., et al. (2018). Aristaless controls butterfly wing color variation used in mimicry and mate choice. Curr. Biol. 28, 3469.e–3474.e. doi: 10.1016/j.cub.2018.08.051
Woronik, A., Tunström, K., Perry, M. W., Neethiraj, R., Stefanescu, C., de la Paz Celorio-Mancera, M., et al. (2019). A transposable element insertion is associated with an alternative life history strategy. Nat. Commun. 10:5757. doi: 10.1038/s41467-019-13596-13592
Zhang, L., Martin, A., Perry, M. W., van der Burg, K. R. L., Matsuoka, Y., Monteiro, A., et al. (2017a). Genetic basis of melanin pigmentation in butterfly wings. Genetics 205, 1537–1550. doi: 10.1534/genetics.116.196451
Zhang, L., Mazo-Vargas, A., and Reed, R. D. (2017b). Single master regulatory gene coordinates the evolution and development of butterfly color and iridescence. Proc. Natl. Acad. Sci. U.S.A. 114, 10707–10712. doi: 10.1073/pnas.1709058114
Keywords: Heliconius, morphological evolution, CRISPR/Cas9, cell fate, patterning loci
Citation: McMillan WO, Livraghi L, Concha C and Hanly JJ (2020) From Patterning Genes to Process: Unraveling the Gene Regulatory Networks That Pattern Heliconius Wings. Front. Ecol. Evol. 8:221. doi: 10.3389/fevo.2020.00221
Received: 04 March 2020; Accepted: 16 June 2020;
Published: 24 July 2020.
Edited by:
Mark Rebeiz, University of Pittsburgh, United StatesReviewed by:
Sean Mullen, Boston University, United StatesCopyright © 2020 McMillan, Livraghi, Concha and Hanly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. Owen McMillan, bWNtaWxsYW5vQHNpLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.