 Ineke E. Knot
Ineke E. Knot George D. Zouganelis
George D. Zouganelis Gareth D. Weedall
Gareth D. Weedall Serge A. Wich
Serge A. Wich Robbie Rae
Robbie Rae- 1Institute for Biodiversity and Ecosystem Dynamics, University of Amsterdam, Amsterdam, Netherlands
- 2School of Biological and Environmental Sciences, Liverpool John Moores University, Liverpool, United Kingdom
Many nematode species are parasitic and threaten the health of plants and animals, including humans, on a global scale. Advances in DNA sequencing techniques have allowed for the rapid and accurate identification of many organisms including nematodes. However, the steps taken from sample collection in the field to molecular analysis and identification can take many days and depend on access to both immovable equipment and a specialized laboratory. Here, we present a protocol to genetically identify nematodes using 18S SSU rRNA sequencing using the MinION, a portable third generation sequencer, and proof that it is possible to perform all the molecular preparations on a fully portable molecular biology lab – the Bentolab. We show that both parasitic and free-living nematode species (Anisakis simplex, Panagrellus redivivus, Turbatrix aceti, and Caenorhabditis elegans) can be identified with a 96–100% accuracy compared to Sanger sequencing, requiring only 10–15 min of sequencing. This protocol is an essential first step toward genetically identifying nematodes in the field from complex natural environments (such as feces, soil, or marine sediments). This increased accessibility could in turn improve global information of nematode presence and distribution, aiding near-real-time global biomonitoring.
Introduction
Nematodes are one of the most abundant groups of metazoan organisms (Seesao et al., 2017). It is estimated that less than 4% of nematode species are currently known to science, with global species richness estimated between 106 and 108 (Lambshead, 2004). Many of these species are parasites that threaten the health of plants and animals, including humans. For example, the World Health Organization estimates that worldwide infections with soil-transmitted nematodes cause a human annual disease burden of 3.8 million years lost to disabilities (YLD), a disease burden in the same range as HIV/AIDS (4 million YLD) and twice as high as malaria (1.7 million YLD)1.
Morphological identification is commonly used to identify nematode species, but also has significant drawbacks. For example, easily distinguishable morphological characters are scarce in nematodes, making identification difficult, time-consuming and often unsuccessful to genus or species level (Decraemer and Baujard, 1998; Lawton et al., 1998; Karanastasi et al., 2001; Lambshead, 2004; Hope and Aryuthaka, 2009). As a result, genetic identification is becoming increasingly important in nematology. There have been increased efforts in recent years to resolve the genetic taxonomy of nematodes and barcode nematode species using markers including the 18S small subunit ribosomal RNA gene (18S SSU rRNA), the 28S large subunit ribosomal RNA gene (28S LSU rRNA), the cytochrome oxidase I gene (COI) and the internal transcribed spacer (ITS) regions of the ribosomal RNA locus (Blaxter et al., 1998; Bhadury et al., 2006a, b; Hunt et al., 2016; O’Neil et al., 2017; Seesao et al., 2017; Pafčo et al., 2018).
The majority of these studies used Sanger sequencing but currently there are many sequencing technologies that have become more accessible and affordable for a wide array of applications (Kircher and Kelso, 2010; Van Dijk et al., 2014; Goodwin et al., 2016) such as high-throughput sequencing (HTS) and third generation sequencing (TGS). The latter is defined as single-molecule real-time sequencing (Van Dijk et al., 2014). Massive multiplexing of DNA barcode markers generates a great reduction of per sample sequencing costs and labor time compared to Sanger sequencing (Schuster, 2008; Shokralla et al., 2015). In this paper we explore TGS as an exciting opportunity for novel applications, such as near real-time biomonitoring of parasites, particularly nematodes.
A promising TGS platform is the MinION, introduced in 2014 by Oxford Nanopore Technologies (ONT). The MinION is a portable and compact USB-powered sequencer, generating long reads which can be base called in real-time (Jain et al., 2016). It utilizes a nanopore placed in a biological membrane through which DNA fragments are driven (Deamer et al., 2016), generating a difference in electrical current which can be measured and translated to different DNA bases. More in depth explanation of how the MinION works can be found in reviews by Plesivkova et al. (2019) and Krehenwinkel et al. (2019b). The MinION’s portable nature makes it ideal for field research, proven by sequencing efforts in extreme conditions like the Arctic (Edwards et al., 2016; Goordial et al., 2017), Antarctic (Johnson et al., 2017) and the International Space Station (McIntyre et al., 2016). Shotgun genomic sequencing in a national park in Wales identified closely related plant species (Parker et al., 2017) and DNA barcoding (reviewed in Krehenwinkel et al., 2019b) has allowed for the identification of a variety of vertebrates in a rainforest in Ecuador (Pomerantz et al., 2018) and a rainforest in Tanzania (Menegon et al., 2017), all within hours of collection. Furthermore, the sequencer has successfully been used for real-time detection of Ebola virus during the 2014–2015 Ebola outbreak in West-Africa (Quick et al., 2016), Zika virus in Brazil (Faria et al., 2017; Quick et al., 2017), and the current outbreak of nCoV-20192, which is an important step toward actionable clinical diagnostics. The most popular application of the MinION sequencer so far is the identification of viral or bacterial populations through metagenomics of the 16S rRNA gene (e.g., Greninger et al., 2015; Quick et al., 2015; Benítez-Páez et al., 2016; Schmidt et al., 2017), but metazoan parasites such as nematodes have not yet been examined.
In this paper we highlight the first step toward sequencing nematodes in situ, by genetically identifying parasitic and free-living nematode species with the MinION and testing a portable molecular lab. Specifically, we had four objectives and we sought to: (1) optimize existing 18S SSU rRNA primer sets for MinION sequencing of nematodes; (2) genetically identify nematode species with the MinION; (3) compare the MinION sequencing data to Sanger sequencing data, to assess the quality of MinION data and; (4) test whether we could achieve these results using a portable molecular laboratory, the Bentolab.
Materials and Methods
Barcode Testing With Known Species
We tested DNA barcoding on four different nematode species, Anisakis simplex, Panagrellus redivivus, Turbatrix aceti, and Caenorhabditis elegans. These species represent a subset of parasitic and free-living species with diverse lifestyles.
Anisakis simplex was dissected from fresh mackerel and stored in 70% ethanol, and one individual nematode was selected for DNA extraction. A. simplex is a marine parasite that uses crustaceans as intermediate hosts to infect teleosts and squids (Anderson, 2000). Although humans are accidental hosts for Anisakis spp., there has been a dramatic increase over the last decades in the reported prevalence of anisakiasis, a serious zoonotic disease (Chai et al., 2005).
Panagrellus redivivus was harvested from a fresh culture growing on oatmeal medium and used for DNA extraction. P. redivivus is a free-living nematode that has been used as a model system to study organ development, signal transduction, and toxicology and recently had its full genome and transcriptome sequenced (Srinivasan et al., 2013). The species is amongst others suggested as a comparative model for Strongyloides, as parasitic taxa are typically difficult to culture and analyze independently of their hosts (Blaxter et al., 1998).
Turbatrix aceti was harvested from a fresh culture in an apple cider vinegar medium and used for DNA extraction. The nematodes were washed in distilled water three times before DNA extraction, to mitigate an inhibiting effect of the vinegar medium on the subsequent Polymerase Chain Reaction (PCR). T. aceti is a free-living nematode that is mostly researched in relation to aging phenotypes, that are shared with other free-living nematodes such as Caenorhabditis elegans (Reiss and Rothstein, 1975). It is also used as live food in the larval stages of many fish species (Brüggemann, 2012). It lacks proper genetic studies, making it an interesting representative for the majority of nematode species that are mostly studied morphologically.
Caenorhabditis elegans strain N2 was grown on nematode growth medium (NGM) plates with E. coli OP50 for several days using standard procedures (Brenner, 1974) and subsequently harvested for DNA extraction.
DNA Extraction, PCR, and Sequencing
We extracted the DNA using the GeneJET Genomic DNA Purification Kit (ThermoFisher Scientific Ltd., Paisley, United Kingdom) according to manufacturer’s instructions for mammalian tissue and rodent tail genomic DNA purification (protocol A), except that samples were lysed overnight (step 3) to ensure complete cuticle break down. DNA purity was measured on a NanoDrop 2000 spectrophotometer (software: NanoDrop2000, version 1.4.2; ThermoFisher Scientific Ltd., Paisley, United Kingdom).
We amplified an internal fragment of the 18S SSU rRNA gene from our DNA samples, using the primers and thermocycler protocol optimized by Floyd et al. (2005). This fragment is ∼900 bp in length and widely used for nematode species identification. According to ONT’s instruction we adapted the primers from Floyd et al. (2005) to include an adapter tail at the 5′ end (“MinION tail,” in lowercase), which is compatible with the MinION workflows. This resulted in the following forward primer: Nem_18S_F_MinION: 5′ tttctgttggtgctgatattgcCGCGAA TRGCTCATTACAACAGC 3′ and reverse primer: Nem_18S_ R_MinION: 5′ acttgcctgtcgctctatcttcGGGCGGTATCTGATCGC C 3′. A different primer pair, SSU18A and SSU26R (Floyd et al., 2002), was initially tested with the MinION tails, but resulted in no PCR amplification for these samples. Each 25-μl PCR mix contained 2 μl purified DNA extract, 0.5 μl each forward and reverse primers (10 μM; Sigma-Aldrich/Merck Ltd., Poole, United Kingdom), 9.5 μl nuclease free water (NFW; ThermoFisher Scientific Ltd., Paisley, United Kingdom), and 2X GoTaq Hot Start Colorless Master Mix (Promega, Southampton, United Kingdom). PCR was performed on a Bio-Rad T100 Thermal Cycler (Bio-Rad Laboratories Ltd., Watford, United Kingdom). The PCR protocol remained the same as Floyd et al. (2005): initial denaturation for 5 min at 94°C followed by 35 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 54°C and extension for 1 min at 72°C, all followed by a final extension for 10 min at 72°C and cooling to 12°C.
Successful amplification was confirmed using a 2% agarose gel (Agarose I, Molecular Biology Grade; Thermo Fisher Scientific Ltd., Paisley, United Kingdom) made with 1x TBE buffer (Thermo Fisher Scientific Ltd., Paisley, United Kingdom), using 1 μl of NovelJuice nucleic acid stain (Sigma-Aldrich/Merck Ltd., Poole, United Kingdom) loaded with each sample and the size ladder. PCR products were purified using the GeneJET PCR Purification Kit (Thermo Fisher Scientific Ltd., Paisley, United Kingdom) following manufacturer’s instruction and eluted in 50 μl of Elution Buffer. DNA purity was measured on a NanoDrop 2000 spectrophotometer (software: NanoDrop2000, version 1.4.2; Thermo Fisher Scientific Ltd., Paisley, United Kingdom) and DNA concentration on a Qubit 1.0 (Thermo Fisher Scientific Ltd., Paisley, United Kingdom), using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific Ltd., Paisley, United Kingdom). Both Nanodrop and Qubit measurements were measured twice per sample to confirm accuracy of the measurement.
We prepared the MinION library according to the 1D PCR barcoding amplicons/cDNA (SQK-LSK109) protocol from ONT (version PBAC12_9067_v109_revH_23MAY2018). This protocol incorporates a second PCR to attach ONT barcodes to our first-round PCR products as means of indexing, allowing multiple samples to be run on one flow cell and subsequent demultiplexing in the bioinformatics stage. Briefly, the PCR Barcoding Kit (EXP-PBC001; ONT Ltd., Oxford, United Kingdom) was used to prepare a 100-μl PCR mix containing 2 μl barcode (10 μM; ONT Ltd., Oxford, United Kingdom), 48 μl first-round PCR product, and 50 μl 2X LongAmp Taq Master Mix [New England BioLabs (NEB) Inc., Hitchin, United Kingdom].
We tried to prepare the first-round PCR products in equimolar concentrations for the barcoding PCR, but due to large variations in DNA concentrations between the samples we diluted the first-round PCR product of A. simplex and P. redivivus to between 100 and 150 fmol and used all the first-round PCR product for T. aceti. A. simplex received barcode number 05, P. redivivus barcode 06 and T. aceti barcode 07. PCR was performed on a Bio-Rad T100 Thermal Cycler (Bio-Rad Laboratories Ltd., Watford, United Kingdom). The PCR protocol for an amplicon length of ∼1,000 bp (including primers) was as follows: initial denaturation 3 min @ 95°C; denaturation 15 s at 95°C, annealing 15 s at 62°C, extension 50 s at 65°C (all 15 cycles); final extension 50 s at 65°C; hold at 4°C. The PCR products were cleaned up with 1X Agencourt AMPure XP beads (Beckman Coulter Inc., Indianapolis, IN, United States). Finally, 1 μl per purified second-round PCR product was quantified on the Qubit 1.0 (Thermo Fisher Scientific Ltd., Paisley, United Kingdom) using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific Ltd., Paisley, United Kingdom).
The concentration of A. simplex and P. redivivus DNA was too high for Qubit quantification, so we prepared and quantified a 1/5 dilution in NFW (Thermo Fisher Scientific Ltd., Paisley, United Kingdom) that was taken forward. The second-round PCR products were pooled in roughly equimolar concentrations in 47 μl NFW (Thermo Fisher Scientific Ltd., Paisley, United Kingdom).
Library preparation continued using the reagents from the Ligation Sequencing Kit (SQK-LSK109; ONT Ltd., Oxford, United Kingdom), according to manufacturer’s instructions. Briefly, we prepared 325 ng pooled barcoded library in 47 μl NFW (ThermoFisher Scientific Ltd., Paisley, United Kingdom). Amplified product was end-repaired using NEBNext Ultra II End-Repair/dA-tailing Module (NEB Inc., Hitchin, United Kingdom) for 5 min at 20°C and 5 min at 65°C, after which it was cleaned up with 1X Agencourt AMPure XP beads (Beckman Coulter Inc., Indianapolis, IN, United States). Adapter ligation was performed using NEB Blunt/TA Ligation Master Mix (NEB Inc., Hitchin, United Kingdom) and reagents provided in the SQK-LSK109 kit. Ligation took place for 10 min at room temperature. DNA was eluted in 15 μl Elution Buffer after being purified with 0.4X AMPure XP beads and washed with the Short Fragment Buffer provided in the SQK-LSK109 kit. 1 μl of prepared library was quantified on the Qubit 1.0 (Thermo Fisher Scientific Ltd., Paisley, United Kingdom) using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific Ltd., Paisley, United Kingdom) and gave a measure of 6.36 ng/μl, which equates to a molarity of 102.9 fmol.
The protocol from ONT recommends loading 5-50 fmol of amplicon product onto the flow cell, so we diluted 5.44 μl of prepared library in 6.56 μl Elution Buffer to load 40 fmol of library onto the flow cell. The flow cell was primed for loading by flushing the flow cell with 1 ml priming mix (30 μl of Flush Tether in one tube of Flush Buffer), taking care to avoid the introduction of air bubbles. The library was prepared for loading by mixing 37.5 μl Sequencing Buffer, 25.5 μl Loading Beads and 12 μl diluted DNA library, after which the sample was added to a flow cell, type R9.5.1, through the SpotON sample port. Total library preparation time was estimated to be ∼3 h.
We performed the sequencing run using MinKNOW (version 3.4.5; ONT Ltd., Oxford, United Kingdom) on the MinIT (a small powerful computing unit that eliminates the need for a dedicated laptop; ONT Ltd., Oxford, United Kingdom), indicating the flow cell type and experimental kit used. As a metric of flow cell quality the MinKNOW software assesses flow cell active pore count, in the multiplexer (MUX) scan before each run. Higher active pore counts represent a high flow cell quality, with a maximum of 2,048 and a guaranteed level of 800. Our flow cell had 1,097 pores available for sequencing. The flow cell generated 116,620 reads in 10 min of sequencing, after which the run was stopped. The flow cell was subsequently washed using the Wash Kit (EXP-WSH002; ONT Ltd., Oxford, United Kingdom) with 150 μl Solution A, followed by 500 μl of Storage Solution, and stored in the fridge for re-use.
Portable DNA Extraction, PCR and Sequencing
In preparation for field work we tested whether the developed MinION procedure could also be performed on a fully portable system. We prepared the model organism C. elegans for MinION sequencing using a portable molecular lab, the Bentolab Pro3 (Nature Biotechnology, 2016; Bento Bioworks Ltd., London, United Kingdom) and a multi tool (CMFTLi 10.8V Li-Ion Cordless Multifunction Tool, Clarke International Ltd., Epping, United Kingdom) as a low-cost handheld vortex. Most of the procedures are similar to above, but working on the Bentolab required some essential adaptations to protocols.
We extracted the DNA using the GeneJET Genomic DNA Purification Kit (ThermoFisher Scientific Ltd., Paisley, United Kingdom) according to manufacturer’s instructions for mammalian tissue and rodent tail genomic DNA purification (protocol A). Adaptations to the procedure to make this protocol work on the Bentolab were as follows: In step 3, the sample was divided over two 0.2 ml PCR tubes and briefly spun down using the Bentolab’s centrifuge (Bento Bioworks Ltd., London, United Kingdom). Subsequently, the two PCR tubes were incubated for 18 h at 56°C, using the Bentolab’s thermocycler (Bento Bioworks Ltd., London, United Kingdom) as a heating block. The thermocycler protocol performed 18 cycles of 1 h at 56°C. In step 4, the lysate was then transferred to a 1.5 ml centrifuge, 20 μl RNase A was added and vortexed on the multi tool4. Vortexing on a multi tool can be achieved by attaching the “straight saw blade” to the multi tool. This blade provides enough space for up to four centrifuge tubes at the same time. As a safety measure the sharp end of the saw blade was covered with duck tape. Then the centrifuge tube was added to the blade with duck tape, ensuring a tight fit. The multi tool was turned on at the highest speed (21,000 strokes/minute), creating a similar effect as a lab vortex. The vortexing in step 5 and 6 was also performed using the multi tool. In step 7, the 2 ml collection tube of the GeneJet purification column was replaced by a 1.5 ml centrifuge tube with the cap cut off. The Bentolab’s centrifuge can only handle 1.5 ml tubes; use of 2 ml collection/centrifuge tubes will lead to small plastic particles that can lead to reduced efficiency of the centrifuge lock system. Because of the reduced volume of the collection tube, the lysate was added to the prepared column at a maximum of 350 μl at a time, after which the sample was centrifuged for 1 min at 6,000 × g and the flowthrough discarded (with a total of three repeats necessary to complete step 7). In step 8, 250 μl Wash Buffer I was added at a time and centrifuged for 1 min at 8,000 × g and the flowthrough discarded (with a total of two repeats necessary to complete step 8). In step 9, 250 μl Wash Buffer II was added at a time and centrifuged for 4 min at 8,000 × g and the flowthrough discarded (with a total of two repeats necessary to complete step 9, and increased centrifuge time to compensate for the max 8,000 × g force of the Bentolab’s centrifuge). An additional dry spin of 1 min at 8,000 × g was performed, after which the collection tube was discarded and replaced by a sterile 1.5 ml centrifuge tube. In step 10, 50 μl of Elution Buffer was added to the purification column.
PCR was prepared as described above, but this time performed using the Bentolab’s thermocycler (Bento Bioworks Ltd., London, United Kingdom). Also, aluminum foil was used as a sterile work environment as an alternative for a PCR hood. Aluminum foil was taped to the bench space using masking tape. Bleach (1:10 ratio dilution in water) was sprayed on the surface, letting it sit for 3 min, and wiping the surface with clean paper tissue. This process was repeated twice to decontaminate, after which 70% ethanol was used to remove any residual bleach. The Nem_18S_F/R_MinION primers did not work for C. elegans, so the primers from Floyd et al. (2002) were used with MinION tails (in lowercase). The forward primer: SSU18A_MinION: 5′ tttctgttggtgctgatattgcAAAGATTAAGCCATGCATG 3′ and reverse primer: SSU26R_MinION: 5′ acttgcctgtcgctctatcttcCAT TCTTGGCAAATGCTTTCG 3′. Each 25-μl PCR mix contained 2 μl purified DNA extract, 0.5 μl each forward and reverse primers (10 μM; Sigma-Aldrich/Merck Ltd., Poole, United Kingdom), 9.5 μl nuclease free water (NFW; Thermo Fisher Scientific Ltd., Paisley, United Kingdom), and 2X GoTaq Hot Start Colorless Master Mix (Promega, Southampton, United Kingdom). PCR was performed on the Bentolab (Bento Bioworks Ltd., London, United Kingdom). The PCR protocol was adapted from Floyd et al. (2002): initial denaturation 5 min at 94°C; denaturation 1 min at 94°C, annealing 1.5 min at 60°C, extension 2 min at 72°C (all 35 cycles); final extension 10 min at 72°C; hold at 12°C.
Successful amplification was confirmed using a 2% agarose gel (Agarose I, Molecular Biology Grade; Thermo Fisher Scientific Ltd., Paisley, United Kingdom) made with 1x TBE buffer (Thermo Fisher Scientific Ltd., Paisley, United Kingdom). For the Bentolab’s small gel chamber (Bento Bioworks Ltd., London, United Kingdom) we used 27.5 ml of 1X TBE buffer with 0.5 g agarose. The need for a scale was eliminated by using an Eppendorf tube marked with the needed volume corresponding to 0.5 g agarose. Agarose was melted into the TBE buffer using a traditional coffee pot, which has a typical conical shape, on the hob. We have also found this method to work on a camping stove. The gel was then poured into the chamber and left to set for ∼15 min. The comb and shutters were removed and we added 45 ml 1x TBE buffer for the gel electrophoresis run, 60 min at 60V. We again used 1 μl NovelJuice (Sigma-Aldrich/Merck Ltd., Poole, United Kingdom) for the size ladder and per sample for DNA staining, as this DNA stain is safer to work with than traditional ethidium bromide and works both with UV transilluminators and with the blue LED transilluminator of the Bentolab (Bento Bioworks Ltd., London, United Kingdom).
The PCR product was cleaned up using GeneJET PCR Purification Kit (Thermo Fisher Scientific Ltd., Paisley, United Kingdom) following manufacturer’s instruction for DNA purification using centrifuge (protocol A). Adaptations to the procedure to make this protocol work on the Bentolab were as follows: In step 3, the 2 ml collection tube of the GeneJet purification column was replaced by a 1.5 ml centrifuge tube with the cap cut off (see adaptations to DNA purification protocol for explanation). The solution of step 1 was added to the purification column, centrifuged for 1 min at 8,000 × g and the flowthrough discarded. In step 4, 350 μl Wash Buffer was added at a time and centrifuged for 1 min at 8,000 × g and the flowthrough discarded (with a total of two repeats necessary to complete step 4). In step 5, a dry spin of 1.5 min at 8,000 × g was performed. In step 6, the collection tube was discarded and replaced by a clean 1.5 ml centrifuge tube. 50 μl of Elution Buffer was added to the purification column and centrifuged for 1 min at 8,000 × g.
We prepared the MinION library according to the 1D PCR barcoding amplicons/cDNA (SQK-LSK109) protocol from ONT (version PBAC12_9067_v109_revH_23MAY2018). As mentioned above, this protocol incorporates a second PCR to attach ONT barcodes to our first-round PCR products as means of indexing. This not only allows multiple samples to be run on one flow cell, but also allows for demultiplexing in the bioinformatics stage when a flow cell is reused. Washing a flow cell after a run might leave some remnant DNA from previous runs. Therefore, the ONT barcodes help to identify the current sample in the bioinformatics stage. Briefly, the PCR Barcoding Kit (EXP-PBC001; ONT Ltd., Oxford, United Kingdom) was used to prepare a 100-μl PCR mix containing 2 μl barcode (10 μM; ONT Ltd., Oxford, United Kingdom), 2 μl first-round PCR product, 46 μl NFW (ThermoFisher Scientific Ltd., Paisley, United Kingdom) and 50 μl 2X LongAmp Taq Master Mix [New England BioLabs (NEB) Inc., Hitchin, United Kingdom]. C. elegans received barcode number 10. PCR was performed on the Bentolab thermocycler (Bento Bioworks Ltd., London, United Kingdom). The barcoding PCR protocol was slightly adjusted to accommodate the Bentolab’s inability for setting cycles of 15 s and minimum thermocycler temperature of 10°C: initial denaturation 3 min @ 95°C; denaturation 20 s at 95°C, annealing 20 s at 62°C, extension 60 s at 65°C (all 12 cycles); final extension 50 s at 65°C; hold at 10°C. The PCR products were cleaned up with 1X Agencourt AMPure XP beads (Beckman Coulter Inc., Indianapolis, IN, United States) on a 3D-printed magnetic BOMB microtube rack5 (Oberacker et al., 2019).
Library preparation continued using the reagents from the Ligation Sequencing Kit (SQK-LSK109; ONT Ltd., Oxford, United Kingdom), according to manufacturer’s instructions. Since we wouldn’t have an accurate way of quantifying DNA in the field we based the used volume of DNA on a previous MinION run (Knot, unpublished data), to prepare 33 μl barcoded library in 47 μl NFW (Thermo Fisher Scientific Ltd., Paisley, United Kingdom). Amplified product was end-repaired using NEBNext Ultra II End-Repair/dA-tailing Module (NEB Inc., Hitchin, United Kingdom) for 5 min at 20°C and 5 min at 65°C on the Bentolab thermocycler (Bento Bioworks Ltd., London, United Kingdom). The end-repaired library was cleaned up with 1X Agencourt AMPure XP beads (Beckman Coulter Inc., Indianapolis, IN, United States) on a 3D-printed magnetic BOMB microtube rack (Oberacker et al., 2019). Adapter ligation was performed using NEB Blunt/TA Ligation Master Mix (NEB Inc., Hitchin, United Kingdom) and reagents provided in the SQK-LSK109 kit. Ligation took place for 10 min at room temperature. DNA was eluted in 15 μl Elution Buffer after being purified with 0.4X AMPure XP beads and washed with the Short Fragment Buffer provided in the SQK-LSK109 kit.
The flow cell was primed for loading by flushing the flow cell with 1 ml priming mix (30 μl of Flush Tether in one tube of Flush Buffer), taking care to avoid the introduction of air bubbles. The library was prepared for loading by mixing 37.5 μl Sequencing Buffer, 25.5 μl Loading Beads and 12 μl DNA library, after which the sample was added to a flow cell, type R9.5.1, through the SpotON sample port. Total library preparation time was estimated to be ∼4.5 h.
We performed the sequencing run using MinKNOW (version 3.4.5; ONT Ltd., Oxford, United Kingdom) on the MinIT (a small powerful computing unit that eliminates the need for a dedicated laptop; ONT Ltd., Oxford, United Kingdom), indicating the flow cell type and experimental kit used. To test whether old flow cells can still be useful for sequencing small barcoded amplicon libraries, we used a flow cell that was used twice before, once in a 24 h run and once in a 2.5 h run. When reusing a flow cell the starting voltage has to be adjusted and we adjusted this to -225 V, equivalent to ONT’s recommendation after ∼26 h previous run time. As mentioned above, higher active pore counts represent a high flow cell quality, with a maximum of 2,048 and a guaranteed level of 800 for new flow cells. The MUX scan indicated our flow cell had 43 pores available for sequencing. The flow cell generated 2,632 reads in 14 min of sequencing, after which the run was stopped.
Sanger Sequencing
Each of the samples used for the MinION sequencing was also sent for Sanger sequencing (GATC/Eurofins Genomics). Both forward and reverse strands were sequenced using the amplification primers as sequencing primers. Sanger sequence electropherograms were visually inspected and edited using 4Peaks version 1.8 (Nucleobytes B.V., Aalsmeer, the Netherlands). Edited forward strand and reverse complemented reverse strand sequences were aligned using Seaview version 4.7 (Gouy et al., 2010). Nucleotide mismatches were checked in the original electropherogram and resolved. A consensus sequence was derived for each sample and primer sequences trimmed from each end of it. The resulting sequences were 885 bp (A. simplex), 887 bp (P. redivivus), 832 bp (T. aceti), and 844 bp (C. elegans) long.
Bioinformatic Analyses
The raw fast5 MinION reads were basecalled and demultiplexed using Guppy version 3.2.4 + d9ed22f (ONT Ltd., Oxford, United Kingdom) to produce fastq files for each sample. Reads were classified as pass/fail based on a minimum quality score of 7. The fastq files were merged into one per sample and explored using Nanoplot (version 1.28.06), creating plots displaying log transformed read length (“–loglength”). Barcode and primer trimming was performed using Porechop (version 0.2.47). A second round of demultiplexing requiring barcodes at both ends of the reads (“–require_two_barcodes”) was performed using Porechop. Subsequently, the MinION reads were processed using the default settings of the ONTrack pipeline (version 1.4.28; Maestri et al., 2019). Briefly, Seqtk seq9 was used to create fasta files complementary to the fastq files. Reads were clustered using VSEARCH (Rognes et al., 2016), after which the reads in the most abundant cluster were retained. Then 200 randomly sampled reads were used to produce a draft consensus sequence using Seqtk sample and aligned using MAFFT (Katoh et al., 2002). EMBOSS cons10 was then used to retrieve a draft consensus sequence starting from the MAFFT alignment. Another 200 randomly sampled reads using Seqtk sample, different from the first iteration, were mapped to the draft consensus sequence using Minimap2 (Li, 2018) to polish the obtained consensus sequence. Samtools was used to filter and sort the alignment file and compress it to the bam format (Li et al., 2009). Nanopolish index and nanopolish variants – consensus modules from Nanopolish11 were used to obtain a polished consensus sequence. The ONTrack pipeline was run with three iterations, the standard value of the pipeline. This resulted in three polished consensus sequences which were aligned with MAFFT to select the consensus sequence that was produced in the majority of times. All scripts of the pipeline were run within a virtual machine (as part of the ONTrack pipeline), emulating an Ubuntu v18.04.2 LTS operating system, on a Mac laptop without using any internet connection. All the code used for the bioinformatic analyses and additional files necessary to replicate the analyses can be found on https://github.com/ieknot/MinION-DNA-barcoding-of-nematodes. MinION fastq and Sanger fasta accession numbers are reported in the results.
To assess sequence accuracy, MinION raw reads and consensus reads were aligned to the corresponding Sanger-derived reference sequence using BLASTn (Altschul et al., 1990), with no sequence complexity masking (“-dust no-soft_masking false”). The consensus sequences were aligned to the corresponding Sanger sequence using the MUSCLE algorithm (Edgar, 2004) in Seaview version 4.7 (Gouy et al., 2010).
Results
Sequencing Run Quality and Yield
The first and multiplexed flow cell had 1,097 pores available for sequencing. The flow cell generated 116,620 reads containing 6,033 Mb in 10 min of sequencing, after which the run was stopped. During basecalling 71.9% of these reads passed the minimum quality threshold. The basecalled reads were demultiplexed, producing 42,304 reads for analysis (Table 1). The mean read length was 1,015 bp for A. simplex, 1,011 bp for P. redivivus and 504 bp for T. aceti.
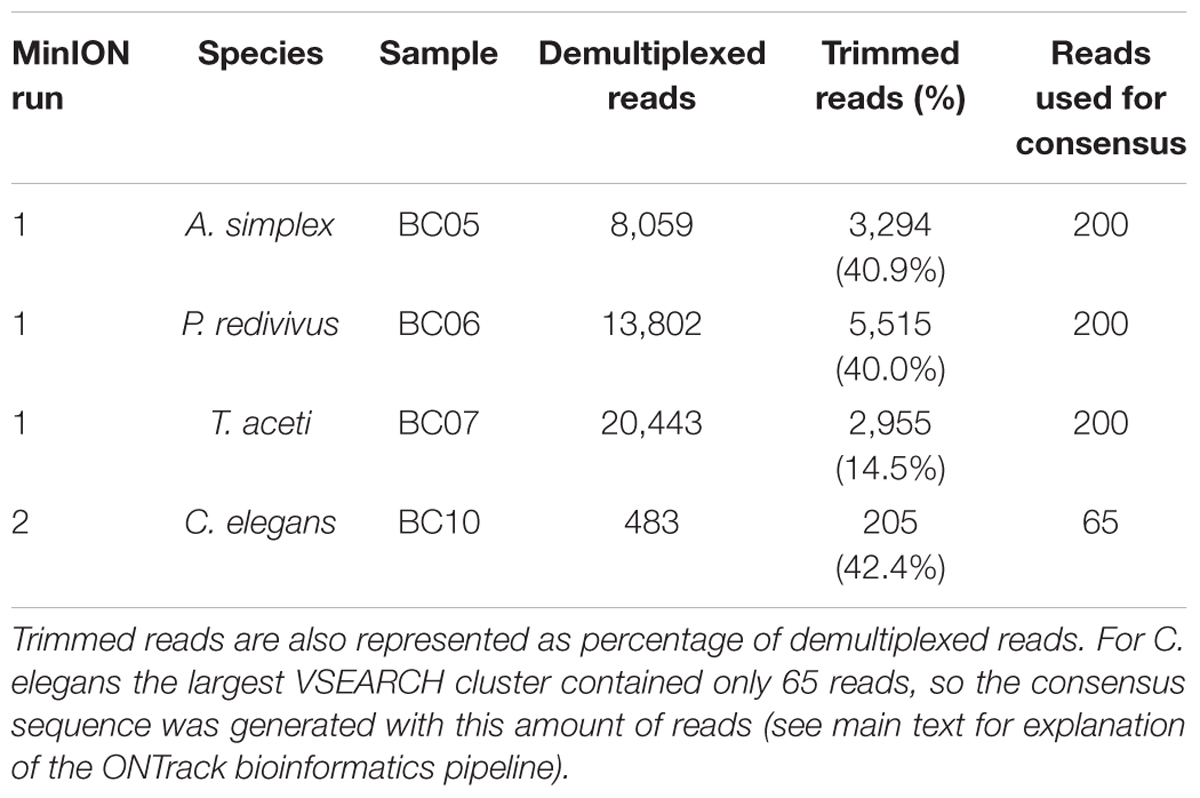
Table 1. Summary of number of reads per sample in subsequent bioinformatics steps.
The second flow cell, used for the library prepared with a fully portable setup, had 43 pores available for sequencing. The flow cell generated 2,632 reads containing 1.94 Mb in 14 min of sequencing, after which the run was stopped. During basecalling 48.9% of these reads passed the minimum quality threshold. The basecalled reads were demultiplexed, producing 205 reads for analysis (Table 1). The mean read length was 833 bp for C. elegans.
To assess the usefulness of the sequence data for taxonomic identification of the samples, the raw reads for each sample were compared to the (Sanger) reference sequences using BLASTn. The distributions of percentage sequence identities for all pairwise read-reference comparisons are shown in Figure 1. The median percent identity was 88.5% for A. simplex, 87.7% for P. redivivus, 89.5% for T. aceti and 82.3% for C. elegans, indicating a ∼11% error rate in sequencing for the first run and a ∼18% error rate in the second run (Figure 1).
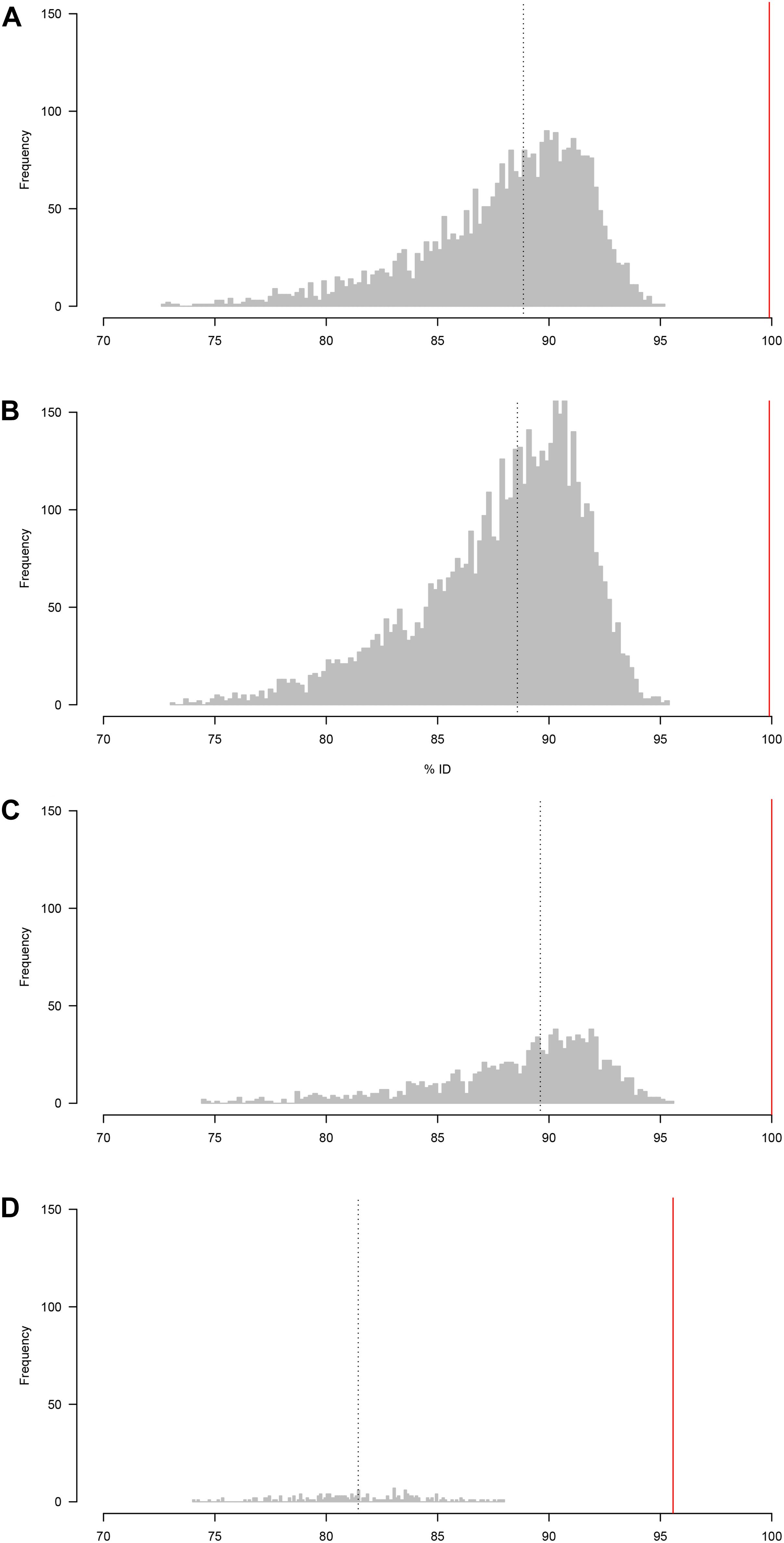
Figure 1. Distribution of percentage sequence identity between raw reads and the (Sanger) reference sequence (A) A. simplex, (B) P. redivivus, (C) T. aceti, and (D) C. elegans. (A–C) were run on a new MinION flow cell, whereas (D) was run on a flow cell that had been used twice before (for a total of 26.5 h, see main text for more details). The median percent identity (indicated by a vertical dotted line) is 88.5% for A. simplex, 87.7% for P. redivivus, 89.5% for T. aceti and 82.3% for C. elegans. Bioinformatic analyses using the ONTrack pipeline generated a consensus sequence for every sample that had a 99.9% (A,B), 100% (C), and 95.6% (D) accuracy compared to their Sanger reads (indicated by a vertical red line).
Bioinformatics Analyses
The second demultiplexing round in Porechop, which included the trimming step to remove primers and ONT barcodes, produced the following number of reads per sample to be taken forward in the ONTrack bioinformatics pipeline (Maestri et al., 2019): 3,294 reads for A. simplex, 5,515 reads for P. redivivus, 2,955 reads for T. aceti and 205 for C. elegans (Table 1). These reads were used in the VSEARCH clustering step of the ONTrack pipeline, where contaminating sequences were removed by only taking the largest cluster of reads forward. T. aceti raw data showed an unexpected short mean read length of 504 bp. The clustering step in the ONTrack pipeline removed contaminating sequences, after which the mean read length of T. aceti improved to 773 bp. From these clusters 200 reads were subsampled per sample for consensus sequence generation. However, for C. elegans the largest VSEARCH cluster contained only 65 reads, so the consensus sequence was generated with this amount of reads (Table 1).
The default setting of the ONTrack pipeline is to run three iterations of the pipeline, generating three consensus sequences per sample. Subsequently, it aligns the consensus sequences generated during each round and selects the final consensus sequence based on the majority rule. Two species, A. simplex and P. redivivus, generated three consensus sequences which were all different. Since they all had the same statistical probability of being correct, the first consensus sequence was randomly selected. T. aceti had a consensus sequence supported by two iterations, and the C. elegans consensus sequence was supported by all three iterations of the pipeline.
Median percent identity between consensus sequences and the reference sequence was significantly improved in all cases (Figure 1). For A. simplex and P. redivivus the accuracy improved to 99.9%, for T. aceti to 100% and for C. elegans to 95.6% (Figure 2). Compared to the raw MinION sequences this is an improvement of 11.4, 12.2, 10.5, and 13.3%, respectively. The MinION datasets generated for this study can be found in the European Nucleotide Archive (ENA) under the project ID PRJEB37489 (samples ERS4397495, ERS4397496, ERS4397497, and ERS4397498). MinION consensus sequences are available in the Supplementary Material.

Figure 2. Species investigated and nucleotide alignments of MinION and Sanger sequences comparing consensus accuracy for (A) A. simplex, (B) P. redivivus, (C) T. aceti, and (D) C. elegans. Sanger sequences have a 100% accuracy. Accuracy shown is the accuracy of the MinION consensus reads. Comparison against accession numbers MF072711.1 (A. simplex), AF083007.1 (P. redivivus), AF202165.2 (T. aceti), and MN519140.1 (C. elegans). The scale bar in the photographs of (A–C) represents 1 mm and in (D) represents 0.5 mm.
Sanger sequence read lengths were 885 bp for A. simplex, 887 bp for P. redivivus, 832 bp for T. aceti and 844 bp for C. elegans. The Sanger reads of all samples matched 100% to a sequence of the correct species on NCBI (Figure 2). Sanger consensus sequences are available at GenBank under the accession numbers MT246663, MT246664, MT246665, and MT246666.
Discussion
We successfully genetically identified four nematode species using 18S SSU rRNA barcoding on the MinION. We also proved that this can be accomplished using a fully portable molecular lab. This was possible by successfully adapting 18S SSU rRNA primers (Floyd et al., 2002, Floyd et al., 2005) with MinION tails. The read lengths of both samples fall within the expected range. Our first run yielded three successful species identifications from MinION reads that have an accuracy of 99.9–100%, when compared to their respective Sanger reads. Our second run yielded a successful species identification from the MinION consensus read that has an accuracy of 95.6%, when compared to Sanger sequencing. The Sanger reads of all samples matched 100% to a sequence of the correct species on NCBI.
The C. elegans data gives a considerably lower accuracy of its MinION consensus sequence than the three other species. We are confident that this is unrelated to the preparation of the sample on a portable setup, as we have applied this setup in a field situation and got MinION consensus sequences that matched closer to the correct species on NCBI (Knot et al., unpublished data). The C. elegans run was set up to test the portable setup before bringing all the equipment out to the field. As such, the primary objective was to prove that everything worked, with data quality being a lower priority. Hence, when the library was loaded and the MinION flow cell MUX scan indicated only 43 working pores, we continued the experiment nonetheless. The MinION generated data for 15 min, after which no active pores remained and the run was stopped. The flow cell that was used twice before, once in a 24 h run and once in a 2.5 h run, multiplexing nine samples in two runs, generating 1,950,657 reads containing 2.11 Gb in total. We therefore feel that the limited accuracy in the C. elegans run has more to do with the limited lifespan of a flow cell, than with the sample preparation or the portable sample preparation. We used the EXP-WSH002 wash kit from ONT, which has now been succeeded by the EXP-WSH003 kit. This latest kit incorporates a nuclease to digest and remove nucleic acid that has been loaded onto a flow cell previously and has proven to be much more efficient in maintaining flow cell quality after a wash than the previous wash kit12. We therefore do not expect future MinION flow cells to deteriorate as much after using the wash kit as was the case for our C. elegans run.
Tackling a large phylum like Nematoda presents challenges that other phyla might be less affected by Kumar et al. (2012). For example, we started exploring the primers developed by Floyd et al. (2005), because these primers are optimized for a wide phylogenetic range of nematodes. However, addition of the MinION tails seems to alter the efficiency of these primers. The tailed primers amplified A. simplex, P. redivivus and T. aceti successfully, but the addition of MinION tails prohibited the primers to amplify C. elegans successfully. We then switched to a primer optimized specifically for soil nematodes (Floyd et al., 2002), and found that this primer with MinION tails amplified C. elegans without problems. Future work will benefit from testing a wider array of nematode primers.
The potential throughput of a MinION R9 chemistry flow cell is ∼20 Gb (Krehenwinkel et al., 2019b), and has been shown to be sufficient to generate new draft genomes of nematodes through shotgun sequencing (Eccles et al., 2018; Fauver et al., 2019). Future barcoding work could focus on maximizing the utility of each flow cell by multiplexing samples in one run or harvesting the long-read potential unique to TGS platforms like PacBio and ONT. For example, Srivathsan et al. (2019) have developed an improved low-cost MinION pipeline where they multiplexed 3,500 samples per flow cell. However, preparing so many samples for sequencing requires significant labor time (Krehenwinkel et al., 2019b; Piper et al., 2019; Srivathsan et al., 2019). Heeger et al. (2018) used PacBio circular consensus sequencing to show the feasibility of long-read metabarcoding of environmental samples using a ∼4,500 bp ribosomal DNA marker that included most of the eukaryote SSU and LSU rRNA genes and the complete ITS region. Krehenwinkel et al. (2019a, b) showed that long-read barcoding using the MinION of a similar ribosomal DNA region, spanning ∼4,000 bp, has great potential for in situ species identification too, although degraded DNA can be a limiting factor in generating long-read barcodes. Small scale projects that do not require such high throughput could alternatively focus on using the newly released Flongle flow cell instead of a traditional MinION flow cell, at a cost of $90 instead of $475-$900 (depending on number of flow cells purchased), respectively. The membrane in this flow cell contains less nanopores to generate a throughput of 1–2 Gb, to accommodate projects with lower throughput demands (Krehenwinkel et al., 2019b). There is a trade-off between flexibility, where a project can sequence samples whenever they want, and cost-effectiveness, where a project can sequence as many samples as possible to get the lowest possible costs per sample. The latter is highly unlikely to be necessary and achievable in very remote regions, given the previously mentioned time restrictions this places on projects.
Conclusion
The use of the MinION opens up exciting possibilities for next-generation biomonitoring. The high efficiency of the MinION consensi compared to the Sanger sequences shows that the MinION can be used to identify diverse nematode species. Extrapolating our results to potential application in a field setting, our results suggest that barcoding with the MinION can generate enough reads for reliable identification within 15 min, assuming good DNA quality and depending on the number of samples that are multiplexed. Our study shows the potential for barcoding eukaryotes and can aid biomonitoring of invertebrate species. Optimizing portable sequencing methods for nematode identification is the first step to sequencing nematode species in the field. One of the challenges ahead for TGS of nematode species lies in the identification of nematodes species from mixed samples from complex natural environments like soil, marine sediments or feces. This challenge could be overcome in several ways. Improvements in the underlying MinION technologies is crucial and will improve accuracy and decrease error rates, just as previous improvements have already shown (Eisenstein, 2019). Further optimization of the bioinformatics analyses is also of high importance. Improved algorithms will lead to higher accuracy in species identification. These improvements will open up possibilities like near real-time genetic identification of nematodes from e.g., soil or feces, which would allow for analyses of soil nematodes as indicator of soil environment disturbance or rapid parasite identification.
Data Availability Statement
The datasets generated for this study are publicly available. The MinION dataset can be found in the European Nucleotide Archive under the project ID PRJEB37489 (samples ERS4397495, ERS4397496, ERS4397497, ERS4397498; and runs ERR4030416, ERR4030415, ERR4030417, ERR4030418, respectively). The Sanger sequences are available at GenBank under the accession numbers MT246663, MT246664, MT246665, and MT246666. All the code used for the bioinformatic analyses and additional files necessary to replicate the analyses, including a detailed explanation of dataset content, can be found on https://github.com/ieknot/MinION-DNA-barcoding-of-nematodes.
Author Contributions
SW, RR, and IK designed the study. IK performed DNA extraction, prepped the Sanger sequencing samples, and performed the nanopore experiments. GZ, RR, and IK optimized the primers. GZ and IK performed PCRs. GW and IK optimized the bioinformatics pipeline. IK, GW, and RR wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was financially supported by the World Wildlife Fund-Netherlands and the University of Amsterdam.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Anne-Sophie Crunchant and Noémie Bonnin for discussions about and feedback on this manuscript, and Yannis Ancele for his help with some of the bioinformatics. We thank Anna de Luis Otero, Prof. Arthur Edison and Prof. Ian Chin-Sang for permission to use the pictures of A. simplex, P. redivivus, and C. elegans, respectively.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00100/full#supplementary-material
The MinION consensus FASTA files correspond to the species as follows: Data Sheet 1 is A. simplex, Data Sheet 2 is P. redivivus, Data Sheet 3 is T. aceti and Data Sheet 4 is C. elegans.
Footnotes
- ^ Global Health Estimates 2016: Disease burden by Cause, Age, Sex, by Country and by Region, 2000–2016. Geneva, World Health Organization; 2018 [online] https://www.who.int/healthinfo/global_burden_disease/estimates/en/index1.html.
- ^ https://nanoporetech.com/about-us/news/novel-coronavirus-ncov-2019-information-and-updates; https://artic.network/ncov-2019
- ^ https://www.bento.bio
- ^ Inspired by Holly Ganz’ use of a multi tool for bead beating: https://youtu.be/Q7PM1xoMjiU.
- ^ https://bomb.bio/protocols/
- ^ https://github.com/wdecoster/NanoPlot
- ^ https://github.com/rrwick/Porechop
- ^ https://github.com/MaestSi/ONTrack
- ^ https://github.com/lh3/seqtk
- ^ http://emboss.open-bio.org/rel/dev/apps/cons.html
- ^ https://github.com/jts/nanopolish
- ^ https://store.nanoporetech.com/flow-cell-wash-kit-r9.html
References
Altschul, S. F., Gish, W., Miller, W., and Myers, E. W. Dj Lipman. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anderson, R. C. (2000). “Nematode parasites of vertebrates,” in Their Development and Transmission, ed. R. C. Anderson (Wallingford: CABI Publishing).
Benítez-Páez, A., Portune, K. J., and Sanz, Y. (2016). species-level resolution of 16S RRNA gene amplicons sequenced through the MinIONTM portable nanopore sequencer. GigaScience 5, 1–9. doi: 10.1186/s13742-016-0111-z
Bhadury, P., Mc Austen, D. T., Bilton, P. J. D., Lambshead, A. D., Rogers, A. D., Smerdon, G. R., et al. (2006a). Development and evaluation of a DNA-barcoding approach for the rapid identification of nematodes. Mar. Ecol. Prog. Ser. 320, 1–9. doi: 10.3354/meps320001
Bhadury, P., Mc Austen, Bilton, D. T., Lambshead, P. J. D., and Rogers, A. D. (2006b). Molecular detection of marine nematodes from environmental samples. overcoming eukaryotic interference. Aqu. Microb. Ecol. 44, 97–103. doi: 10.3354/ame044097
Blaxter, M. L., De Ley, J. R., Garey, L. X., Liu, P., Scheldeman, A., Vierstraete, J. R., et al. (1998). A molecular evolutionary framework for the phylum nematoda. Nature 392, 71–75.
Brenner, S. (1974). The genetics of caenorhabditis elegans. Genetics 7, 71–94. doi: 10.1002/cbic.200300625
Brüggemann, J. (2012). Nematodes as live food in larviculture – a review. J. World Aquacul. Soc. 43, 739–763. doi: 10.1111/j.1749-7345.2012.00608.x
Chai, J. Y., Murrel, K. D., and Lymbery, A. J. (2005). Fish-borne parasitic zoonoses. Status and issues. Int. J. Parasitol. 35, 1233–1254. doi: 10.1016/j.ijpara.2005.07.013
Deamer, D., Akeson, M., and Branton, D. (2016). Three decades of nanopore sequencing. Nat. Biotechnol. 34, 518–524. doi: 10.1038/nbt.3423
Decraemer, W., and Baujard, P. (1998). A polytomous key for the identification of species of the family trichodoridae thorne, 1935 (Nematoda. Triplonchida). Fundam. Appl. Nematol. 21, 37–62.
Eccles, D., Chandler, J., Camberis, M., Henrissat, B., Koren, S., Le Gros, G., et al. (2018). De novo assembly of the complex genome of nippostrongylus brasiliensis using MinION long reads. BMC Biol. 16:6. doi: 10.1186/s12915-017-0473-4
Edgar, R. C. (2004). MUSCLE, multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edwards, A., Ar Debbonaire, S. M., Nicholls, S. M. E., Rassner, B., Sattler, J. M., Cook, T., et al. (2016). In-field metagenome and 16S RRNA gene amplicon nanopore sequencing robustly characterize glacier microbiota. BioRxiv 1–40. [Preprint]. doi: 10.1101/073965
Eisenstein, M. (2019). Playing a long game. Nat. Methods 16, 683–686. doi: 10.1038/s41592-019-0507-7
Faria, R. N., Quick, J., Morales, I., Thézé, J., Jesus, J. G., Giovanetti, M., et al. (2017). Establishment and cryptic transmission of zika virus in Brazil and the Americas. Nature 546, 406–410. doi: 10.1038/nature22401
Fauver, J. R., Martin, J., Weil, G. J., Mitreva, M., and Fischer, P. U. (2019). De novo assembly of the brugia malayi genome using long reads from a single MinION flowcell. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-55908-y
Floyd, R., Abebe, E., Papert, A., and Blaxter, M. (2002). Molecular barcodes for soil nematode identification. Mol. Ecol. 11, 839–850.
Floyd, R. M., and Ad Rogers, P. J. D. Lambshead Cr Smith. (2005). Nematode-specific PCR primers for the 18S small subunit RRNA gene. Mol. Ecol. Notes 5, 611–612. doi: 10.1111/j.1471-8286.2005.01009.x
Goodwin, S., McPherson, JD., and McCombie, WR. (2016). Coming of age, ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Goordial, J. I, Altshuler, K., Hindson, K., Chan-Yam, E., and Marcolefas Whyte, LG. (2017). In situ field sequencing and life detection in remote (79°26’N) canadian high arctic permafrost ice wedge microbial communities. Front. Microbiol. 8:2594. doi: 10.3389/fmicb.2017.02594
Gouy, M., Guindon, S., and Gascuel, O. (2010). Sea view version 4, A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Greninger, A. L., Sn Naccache, S., Federman, G., Yu, P., Mbala, V., Bres, D., et al. (2015). Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 7, 1–13. doi: 10.1186/s13073-015-0220-9
Heeger, F., Yurkov, A., Mazzoni, C. J., Bourne, E. C., Spröer, C., Overmann, J., et al. (2018). Long-read DNA metabarcoding of ribosomal RNA in the analysis of fungi from aquatic environments. Mol. Ecol. Resour. 18, 1500–1514. doi: 10.1111/1755-0998.12937
Hope, W. D., and Aryuthaka, C. (2009). A partial revision of the marine nematode genus elzalia (Monhysterida, Xyalidae) with new characters and descriptions of two new species from khung kraben bay, east thailand. J. Nematol. 41, 64–83.
Hunt, V. L., Ij Tsai, A., Coghlan, A. J., Reid, N., Holroyd, B. J., Foth, A., et al. (2016). The Genomic Basis of Parasitism in the Strongyloides Clade of Nematodes. Nat. Genet. 48, 299–307. doi: 10.1038/ng.3495
Jain, M., Olsen, H. E., Paten, B., and Akeson, M. (2016). The Oxford nanopore MinION, delivery of nanopore sequencing to the genomics community. Genome Biol. 17, 1–11. doi: 10.1186/s13059-016-1103-0l
Johnson, S. S., Zaikova, E., Goerlitz, D. S., Bai, Y., and Tighe, S. W. (2017). Real-Time DNA sequencing in the antarctic dry valleys using the Oxford nanopore sequencer. J. Biomol. Tech. 28, 2–7. doi: 10.7171/jbt.17-2801-009
Karanastasi, E., Decraemer, W., Zheng, J., Martins De Almeida, M. T., and Brown, D. (2001). Interspecific differences in the fine structure of the body cuticle of trichodoridae thorne, 1935 (Nematoda, Diphtherophorina) and review of anchoring structures of the epidermis. Nematology 3, 525–533. doi: 10.1163/156854101753389130
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT, a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kircher, M., and Kelso, J. (2010). High-throughput DNA sequencing – concepts and limitations. BioEssays 32, 524–536. doi: 10.1002/bies.200900181
Krehenwinkel, H., Pomerantz, A., Henderson, J. B., Kennedy, S. R., Lim, J. Y., Swamy, V., et al. (2019a). Nanopore sequencing of long ribosomal DNA amplicons enables portable and simple biodiversity assessments with high phylogenetic resolution across broad taxonomic scale. GigaScience 8, 1–16. doi: 10.1093/gigascience/giz006
Krehenwinkel, H., Pomerantz, A., and Prost, S. (2019b). Genetic biomonitoring and biodiversity assessment using portable sequencing technologies. Curr. Future Dir.. Genes 10, 1–16. doi: 10.3390/genes10110858
Kumar, S., Koutsovoulos, G., Kaur, G., and Blaxter, M. (2012). Toward 959 nematode genomes. Worm 1, 42–50. doi: 10.4161/worm.19046
Lambshead, P. J. D. (2004). “Marine nematode biodiversity,” in Nematology, Advances and Perspectives, eds Z. X. Chen, S. Y. Chen, and D. W. Dickson (Oxford University Press), 1.
Lawton, J. H., De Bignell, B., Bolton, G. F., Bloemers, P., Eggleton, P. M., Hammond, M., et al. (1998). Biodiversity inventories, indicator taxa and effects of habitat modification in tropical forest. Nature 391, 72–76. doi: 10.4103/1658-354x.84113
Li, H. (2018). Minimap2, Pairwise Alignment For Nucleotide Sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Maestri, S., Cosentino, E., Paterno, M., Freitag, H., Garces, J. M., Marcolungo, L., et al. (2019). A rapid and accurate minion-based workflow for tracking species biodiversity in the field. Genes 10:468. doi: 10.3390/genes10060468
McIntyre, A. B. R., Rizzardi, L., Yu, A. M., Alexander, N., Rosen, G. L., Botkin, D. J., et al. (2016). Nanopore sequencing in microgravity. NPJ Microgravity 2, 1–9. doi: 10.1038/npjmgrav.2016.35
Menegon, M., Cantaloni, C., Rodriguez-Prieto, A., Centomo, C., Abdelfattah, A., Rossato, M., et al. (2017). On site DNA barcoding by nanopore sequencing. PLoS One 12:e0184741. doi: 10.1371/journal.pone.0184741
Oberacker, P., Stepper, P., Bond, D. M., Höhn, S., Focken, J., Meyer, V., et al. (2019). Bio-on-magnetic-beads (BOMB), Open platform for high-throughput nucleic acid extraction and manipulation. PLoS Biol. 17:e3000107. doi: 10.1371/journal.pbio.3000107
O’Neil, N. J., Olsen, H. E., Snutch, T. P., Tyson, J. R., Hieter, P., and Jain, M. (2017). MinION-based long-read sequencing and assembly extends the caenorhabditis elegans reference genome. Genome Res. 28, 266–274. doi: 10.1101/gr.221184.117
Pafčo, B., Èížková, D., Kreisinger, J., Hasegawa, H., Vallo, P., Shutt, K., et al. (2018). Metabarcoding analysis of strongylid nematode diversity in two sympatric primate species. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-24126-3
Parker, J., Helmstetter, A. J., Devey, D., Wilkinson, T., and Papadopulos, AST. (2017). Field-based species identification of closely-related plants using real-time nanopore sequencing. Sci. Rep. 7, 1–8. doi: 10.1038/s41598-017-08461-5
Piper, A. M., Batovska, J., Cogan, N. O. I., Weiss, J., Cunningham, J. P., Rodoni, B. C., et al. (2019). Prospects and challenges of implementing DNA metabarcoding for high-throughput insect surveillance. GigaScience 8, 1–22. doi: 10.1093/gigascience/giz092
Plesivkova, D., Richards, R., and Harbison, S. (2019). A Review Of The Potential Of the MinIONTM single-molecule sequencing system for forensic applications. Wiley Interdiscip. Rev. Forensic Sci. 1:e1323. doi: 10.1002/wfs2.1323
Pomerantz, A., Peñafiel, N., Arteaga, A., Bustamante, L., Pichardo, F., Coloma, L. A., et al. (2018). Real-time DNA barcoding in a rainforest using nanopore sequencing, opportunities for rapid biodiversity assessments and local capacity building. GigaScience 7, 1–14. doi: 10.1093/gigascience/giy033
Quick, J., Ashton, P., Calus, S., Chatt, C., Gossain, S., Hawker, J., et al. (2015). Rapid Draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 16, 1–14. doi: 10.1186/s13059-015-0677-2
Quick, J., Nd Grubaugh, S. T., Pullan, I. M., Claro, A. D., Smith, K., Gangavarapu, G., et al. (2017). Multiplex PCR method for MinION and illumina sequencing of zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1266. doi: 10.1038/nprot.2017.066
Quick, J., Nj Loman, S., Duraffour, J. T., Simpson, E., Severi, L., Cowley, J., et al. (2016). Real-Time, portable genome sequencing for ebola surveillance. Nature 530, 228–232. doi: 10.1038/nature16996
Reiss, U., and Rothstein, M. (1975). Age related changes in isocitrate lyase from the free living nematode, turbatrix aceti. J. Biol. Chem. 250, 826–830.
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH, a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Schmidt, K., Mwaigwisya, S., Crossman, L. C., Doumith, M., Munroe, D., Pires, C., et al. (2017). Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J. f Antimicrob. Chemother. 72, 104–114. doi: 10.1093/jac/dkw397
Schuster, S. C. (2008). Next-generation sequencing transforms today’s biology. Nat. Methods 5, 16–18. doi: 10.1038/nmeth1156
Seesao, Y., Gay, M., Merlin, S., Viscogliosi, E., Aliouat-Denis, C. M., and Audebert, C. (2017). A review of methods for nematode identification. J. Microbiol. Methods 138, 37–49. doi: 10.1016/j.mimet.2016.05.030
Shokralla, S., Porter, T. M., Gibson, J. F., Dobosz, R., Janzen, D. H., Hallwachs, W., et al. (2015). Massively parallel multiplex dna sequencing for specimen identification using an illumina MiSeq platform. Sci. Rep. 5, 1–7. doi: 10.1038/srep09687
Srinivasan, J., Ar Dillman, M. G., Macchietto, L., Heikkinen, M., Lakso, K. M., Fracchia, I., et al. (2013). The draft genome and transcriptome of panagrellus redivivus are shaped by the harsh demands of a free-living lifestyle. Genetics 193, 1279–1295. doi: 10.1534/genetics.112.148809
Srivathsan, A., Hartop, E., Puniamoorthy, J., Lee, W. T., Kutty, S. N., Kurina, O., et al. (2019). Rapid, large-scale species discovery in hyperdiverse taxa using 1D MinION sequencing. BMC Biol. 17:96. doi: 10.1186/s12915-019-0706-9
Keywords: MinION, DNA barcoding, biomonitoring, 18S (SSU) rRNA gene, Anisakis simplex, Panagrellus redivivus, Turbatrix aceti, Caenorhabditis elegans
Citation: Knot IE, Zouganelis GD, Weedall GD, Wich SA and Rae R (2020) DNA Barcoding of Nematodes Using the MinION. Front. Ecol. Evol. 8:100. doi: 10.3389/fevo.2020.00100
Received: 07 June 2019; Accepted: 27 March 2020;
Published: 30 April 2020.
Edited by:
David Andrew Bohan, INRA Centre Dijon Bourgogne Franche-Comté, FranceReviewed by:
Virginia León-Règagnon, Institute of Biology, National Autonomous University of Mexico, MexicoHenrik Krehenwinkel, University of Trier, Germany
Stefan Prost, Centre for Translational Biodiversity Genomics (LOEWE-TBG), Germany
Copyright © 2020 Knot, Zouganelis, Weedall, Wich and Rae. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ineke E. Knot, aW5la2VAaW5la2Vrbm90Lm5s