
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 03 March 2020
Sec. Paleoecology
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.00037
This article is part of the Research Topic Applied Uses of Ancient DNA View all 14 articles
 Clio Der Sarkissian1*
Clio Der Sarkissian1* Per Möller2
Per Möller2 Courtney A. Hofman3,4
Courtney A. Hofman3,4 Peter Ilsøe5
Peter Ilsøe5 Torben C. Rick6Tom Schiøtte7
Torben C. Rick6Tom Schiøtte7 Martin Vinther Sørensen7
Martin Vinther Sørensen7 Love Dalén8,9
Love Dalén8,9 Ludovic Orlando1,5
Ludovic Orlando1,5The shells of marine mollusks represent promising metagenomic archives of the past, adding to bones, teeth, hairs, and environmental samples most commonly examined in ancient DNA research. Seminal work has established that DNA recovery from marine mollusk shells depends on their microstructure, preservation and disease state, and that authentic ancient DNA could be retrieved from specimens as old as 7,000 years. Here, we significantly push the temporal limit for shell DNA recovery to ≥100,000 years with the successful genetic characterization of one Portlandia arctica and one Mytilus mussel sample collected within a dated permafrost layer from the Taimyr Peninsula, Russia. We expand the analysis of ancient DNA in carbonate shells to a larger number of genera (Arctica, Cernuella, Crassostrea, Dreissena, Haliotis, Lymnaea, Margaritifera, Pecten, Ruditapes, Venerupis) from marine, freshwater and terrestrial environments. We demonstrate that DNA from ancient shells can provide sufficient resolution for taxonomic, phylogenetic and/or population assignment. Our results confirm mollusk shells as long-term DNA reservoirs, opening new avenues for the investigation of environmental changes, commercial species management, biological invasion, and extinction. This is especially timely in light of modern threats to biodiversity and ecosystems.
Applications of ancient DNA (aDNA) to ecological studies are plentiful, especially now that high-throughput DNA sequencing (HTS) technologies make it possible to generate genome-wide data at the scale of populations for the last million years (Orlando et al., 2013; Der Sarkissian et al., 2015; Leonardi et al., 2016; Nielsen et al., 2017; Fages et al., 2019; Narasimhan et al., 2019). By allowing high-resolution genomic history reconstructions, aDNA has the power to reveal how changes in the environment (e.g., climate, pollution levels), or interactions with other species and populations (e.g., hybridization, admixture, invasions), have potentially affected past organisms or populations through adaptation, distribution shifts, or extinction (e.g., Green et al., 2010; Lorenzen et al., 2011; Orlando and Cooper, 2014; Palkopoulou et al., 2015). Although most ancient DNA studies have focused on mammal bones and teeth (Green and Speller, 2017), new knowledge in ecology and evolution could be gained by applying ancient genomics to shell-producing mollusks (Coutellec, 2017). These animals indeed grow their carbonate shells in an incremental fashion, simultaneously recording biological and environmental data (Fortunato, 2015; Steinhardt et al., 2016; Butler et al., 2019), which can be tracked through a multitude of proxies, including morphology, isotopes (e.g., ∂18Oshell, ∂13Cshell), trace elements and metal composition (e.g., Ba, Cd, Cu, Mo, Pb, U, Zn). The rich fossil record of mollusk shells, thus, provides an equally rich environmental archive of seawater paleo-temperature and salinity (Surge et al., 2003; Chauvaud et al., 2005; Hiebenthal et al., 2012; Vokhshoori and McCarthy, 2014; Reynolds et al., 2016; Black et al., 2017), pollution levels (Vander Putten et al., 2000; Liehr et al., 2005; Pérez-Mayol et al., 2014), stress (Hiebenthal et al., 2012; Trivellini et al., 2018), infection history (Paillard et al., 2004; Trinkler et al., 2010), as well as food availability and primary productivity (Lartaud et al., 2010; Sadler et al., 2012).
Despite recent advances in ancient genomics and the ecological relevance of mollusks, no mollusk genomic time transect has ever been produced. The potential of applying HTS to ancient marine mollusk shells has, however, been assessed for the first time in a recent study that revealed their metagenomic content (Der Sarkissian et al., 2017). This study identified species with aragonitic shell microstructures, such as Mytilus mussels, Haliotis abalones, Arctica islandica quahogs, and Venerupis/Ruditapes clams, as good candidates for mollusk DNA content and/or preservation, with the longest DNA survival observed for a Mytilus specimen dated to ∼7,000 years Before Present (year BP; Der Sarkissian et al., 2017). This timeframe allows for the examination of many important questions relative to ecological changes and the genomic responses of mollusks, as it is characterized by several climatic transitions (Marcott et al., 2013), e.g., the Little Ice Age in the North Hemisphere (∼14th century to ∼1850 C.E.) or post-industrial global warming (Mann et al., 1999, 2009). During this period of intensifying human impact, mollusks have been exposed to increasing pollution levels that have resulted from rising human population densities, industrial activities, extensive farming, fishing and transportation (Jackson, 2001). They have also been commercially exploited as resources for food, nacre or pearls (Fortunato, 2015), and have been displaced over long distances intentionally for aquaculture purposes, or unintentionally (e.g., through ship transportation) (Carlton, 1999). Although successful DNA recovery may prove challenging, investigating mollusk shell DNA preservation beyond the 7,000 year BP limit holds the potential to provide valuable information about the impact of earlier climatic shifts in the Quaternary, such as the Pleistocene glacial/interglacial/interstadial oscillations, the transition into the Holocene, and subsequent increased human environmental impacts (Stephens et al., 2019). Other important areas of research include the applicability of ancient shell genomics to freshwater and terrestrial mollusks (Lydeard et al., 2004) and determining if ancient mollusk shell DNA enclose useful phylogenetic and population structure information.
To further unlock the potential of ancient mollusk shell DNA for ecological studies, we apply an aDNA HTS approach to a range of samples that extends previous assessments to significantly older specimens, to a larger number of species, as well as to mollusks of the freshwater and terrestrial niches. We then illustrate the importance of ancient mollusk DNA for understanding the impact of environmental change, extinction and biological invasion. We also explore the limitations of such investigations and evaluate the research potential of each genus examined, including genera of commercial interest.
For aDNA analyses, we selected three fossils identified as mollusk shells, sampled in a river-cut sediment succession along the lower reaches of the Bol’shaya Balaknya River (site BBR17 in Möller et al., 2019b, with close-by subsites 17A and 17B), situated in the southern portion of the Taimyr Peninsula, Russian Federation (N73° 37,084′; E105° 38,178′; Supplementary Table S1). This location is south of the limits of repeated Kara Sea-based glaciations during the last glacial cycle (the Zyryanka, Marine Isotope Stage (MIS) 5d-2) (Svendsen et al., 2004; Möller et al., 2015, 2019b). The samples were all found in a distinct marine silty clay designated sediment unit A, overlaid by fluvial sand and gravel (sediment unit B) (see sediment log in Möller et al., 2019b). The marine unit A sediment is rich in bivalve and gastropod shells (Supplementary Table S2), including subarctic species such as Buccinum undatum, Mytilus edulis, and Macoma balthica, not present today in the Kara Sea (to the north) and the Laptev Sea (to the east), thus suggesting interglacial conditions at sediment deposition (Möller et al., 2019b). Three shells (Portlandia arctica) from the unit A sediments were dated by Electron-Spin Resonance (ESR) to 101–105 kyr (± 9–12 kyr; averaged here to ≥100 kyr). The above-lying unit B fluvial sediments were dated to their deposition by Optically Stimulated Luminescence to 42–43 kyr (± 3–4 kyr), which is within the Middle Zyryanka (MIS 3), while redeposited mollusks (P. arctica) within the unit B sediments, eroded from underlying marine unit A, gave ESR ages of 122 and 123 kyr (± 15 kyr). Möller et al., 2019b thus concluded that the unit A marine sediment can be placed with high confidence to their deposition within the Karginsky interglacial (MIS 5e), equivalent to the North West European Eemian. Site BBR 17 is located ∼50 km from the ocean today, but paleoenvironmental reconstructions showed marine inundation of the Taimyr lowlands in front of the retreating ice margin at the transition between the Taz glaciation (MIS 6) and into the Karginsky interglacial (MIS 5e), with sea level at its maximum reaching levels in excess of 80 m above present sea level in this area (Möller et al., 2019b). The shells subjected to aDNA analyses were sampled from frozen marine unit A sediment (permafrost) at site BBR 17A and have been kept frozen since the time of collection in 2010 to optimize DNA preservation post-excavation.
An additional 39 mollusk shell specimens from nine genera (Arctica, Cernuella, Crassostrea, Dreissena, Haliotis, Lottia, Lymnaea, Margaritifera, Mytilus) were obtained from museum and laboratory collections. When available, genus- or species-level identification based on morphological examination, as well as collection location and date were retrieved from the information curated together with the specimens (Supplementary Table S1). Samples were selected to represent genera from marine, freshwater and terrestrial niches, with particular relevance to important ecological questions, such as those related to economical exploitation, environmental changes, biological invasions and extinctions, as well as to optimize comparison with available genomic resources.
We carried out shell DNA extraction, shotgun DNA library construction and amplification setup following (Der Sarkissian et al., 2017) in conditions strictly limiting DNA contamination at the ancient DNA facilities of the Centre for GeoGenetics, University of Copenhagen, Denmark. In order to monitor DNA contamination during pre-PCR laboratory work, every step was simultaneously performed on non-template Extraction Blank Controls (EBCs; Supplementary Table S1).
Fragments of the shells’ ventral margin were first decontaminated for 10 min under agitation in one volume of 1% sodium hypochlorite, before being rinsed three times in three volumes of distilled water, air-dried, and reduced to powder with a mortar and pestle (Der Sarkissian et al., 2017). We then performed DNA extraction based on the method developed in Yang et al., 1998; Gamba et al., 2014, 2016. Shell powder (112–2,990 mg) was incubated overnight at 37°C in 15 mL of digestion buffer (0.45 mM EDTA, 0.5% N-laurylsarcosyl, 0.25 mg/mL proteinase K) under constant mixing. Centrifugation at 3,000 RPM for 2 min allowed separating remaining solids from the liquid phase, which was then concentrated to a volume of ∼200 μL using an Amicon Ultra-15 30 kDa centrifugal filter unit (Merck Millipore) at 3,000 RPM for 50–60 min. The retentate was subjected to the MinElute PCR Purification kit (Qiagen), and purified DNA was obtained in a final elution volume of 60 μL EB buffer in presence of Tween (0.05% final concentration, hereafter, EB-Tween).
Double-indexed blunt-ended DNA libraries were constructed following a protocol modified from Orlando et al., 2013; Seguin-Orlando et al., 2013, where adapters carried an identifying 7 bp-index and were paired in a combination unique to each library (Rohland et al., 2015). We used the NEBNext Quick DNA Library Prep Master Mix Set for 454 (New England Biolabs) with a starting volume of 42.6 μL shell DNA extract in reaction volumes of 50 μL for end-repair (12°C for 20 min then 37°C for 15 min) and ligation (20°C for 20 min; 0.5 μM Illumina adapter final concentration), and 25 μL for fill-in (37°C for 20 min then 80°C for 20 min). After end-repair and ligation, the reaction mixes were purified using the MinElute kit with elution volumes of 30 and 20 μL EB-Tween, respectively.
For each library, we used real-time PCR (qPCR) to estimate the optimal number of amplification cycles to obtain DNA amounts compatible with Illumina sequencing, while minimizing library clonality. We performed two independent qPCRs per library in 20 μL of the following reaction mix: 1 μL 1/20 diluted DNA library (in EB buffer), 2.5 units AccuPrime DNA polymerase, 1× AccuPrime mix (Thermo Fisher Scientific), 1 mg/mL BSA, 0.2 μM primer inPE1.0 (5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCC TACACGACGCTCTTCCGATCT-3′) and 0.2 μM of an Illumina 6 bp-indexed (“I”) primer (5′-CAAGCAGAAGACGGCA TACGAGATIIIIIIGTGACTGGAGTTCAGACGTGTGCTCTTC CG-3′), and 0.8 μL of 1:4:2000 ROX:SybR:DMSO DNA dye mix (Thermo Fisher Scientific). qPCRs were performed on a LightCycler 480 Real-Time PCR System instrument (Roche Applied Science) with the following conditions: activation at 95°C for 5 min; 40 cycles of: denaturation at 95°C for 15 s, annealing at 60°C for 30 s, elongation at 68°C for 30 s; final elongation at 68°C for 5 min.
We analyzed qPCR data with the LightCycler Software 4.0 and the second derivative maximum method to estimate Ct values used as a relative measure of DNA concentration for each library. We found that libraries built from EBCs differed in Ct values by 4–6 additional cycles from those constructed from ancient shells. As the corresponding 16–64-fold increase in the amount of DNA in ancient samples relative to EBCs was not large enough to confidently rule out a significant impact of laboratory-derived DNA contamination, we subjected EBC libraries to the same PCR amplification and sequencing protocols as ancient shell libraries (see below).
Each double-indexed library was PCR-amplified using one Illumina IS4 primer and one unique 6 bp-indexed primer, thus, resulting in the introduction of a third, external index. The same conditions as for qPCR were applied, apart from non-diluted DNA and total reaction volumes of 5 and 25 μL, respectively, and from the absence of ROX:SybR:DMSO. We derived the optimal number of cycles to apply from the qPCR results on a per-library basis (14–29 for ancient samples and 34–35 for EBCs; Supplementary Table S1). In order to limit clonality, all libraries were first amplified for 12 cycles, before MinElute-purified reaction mixes (25 μL EB elution volume) were re-amplified as above for the required remaining number of cycles and in four independent reaction volumes (25 μL each) per library. Amplified libraries were purified using the Agencourt AMPure XP system (Beckman Coulter) and 25 μL of bead solution. Libraries eluted in 25 μL EB were quantified on a 2200 TapeStation Instrument (High Sensitivity D1000 Screen Tape; Agilent).
All triple-indexed libraries were pooled together in equimolar proportions and sequenced in paired-end mode on an Illumina HiSeq4000 platform at the Danish National High Throughput DNA Sequencing Centre. Raw sequencing data were deposited on the European Nucleotide Archive (ENA) public database (project PRJEB35671). Previously published ancient mollusk DNA data from Der Sarkissian et al. (2017) were retrieved from ENA (project PRJEB20113) and reanalyzed here.
Post-sequencing, we de-multiplexed DNA reads and only retained those displaying the expected, unique combination of indexes, i.e., unlikely to represent chimeric sequences. We used PALEOMIX v1.2.13 (Schubert et al., 2014) to trim adapters and collapse overlapping pair-end mates with AdapterRemoval2 v2.3.0 (Schubert et al., 2016) as described in Fages et al. (2019), i.e., applying default parameters and quality/length filters –minlength 25 –trimns –trimqualities –minquality 2.
Initial molecular taxonomic assignment was performed by mapping shotgun DNA reads to 145,457 reference sequences of the mitochondrial cytochrome c oxidase subunit I gene 5′-extremity barcode region (COI-5P) compiled for the Mollusca phylum from the Barcode of Life Database (BOLD1). Building on the results, reads were then mapped to reference sequences for mitochondrial complete genomes, nuclear genome or transcript assemblies, when available for the identified genus/species (Supplementary Table S3). Some of the genera studied show Doubly Uniparental Inheritance, where the mitochondrial genome of the mother (or F-genome) is transmitted to both the somatic and gonadic cells of the female offspring, but only to the somatic cells of the male offspring, the gonadic cells of which get their mitochondrial genome from the father (M-genome). As a consequence, we used both F- and M-genomes as reference sequences for mapping (Supplementary Table S3). The end of the circular reference mitochondrial genome sequences was extended using the first 30 bp of the sequence in order to take the circularity of the mitochondrial genomes into account. We used the BWA v0.5.9 aln alignment command (Li and Durbin, 2009) in PALEOMIX (Schubert et al., 2014) with default parameters and disabled seeding as recommended in Schubert et al., 2012. We kept all reads mapping to the BOLD Mollusca database, whereas only reads showing mapping qualities ≥30 against the mitochondrial and nuclear DNA reference sequences were retained. We removed duplicated non-collapsed reads with MarkDuplicates in Picard Tools version 1.1192 and duplicated collapsed reads with the PALEOMIX FilterUniqueBAM Python script (Schubert et al., 2014). Average depth-of-coverage (coverage) was calculated using PALEOMIX and considering only unique high-quality reads (after quality filtering and duplicate removal). When multiple reference sequences were available for a given genus, we retained the one yielding the highest coverage for each sample.
For samples showing maximal coverage ≥3× when mapping against complete mitochondrial genome reference sequences, we built complete mitochondrial genome consensus sequences from which we constructed maximum-likelihood phylogenies. This was achieved using the perl script wrapper.pl (for circular genomes) as well as the C + + programs bam2prof, endoCaller and log2fasta within the schmutzi pipeline (Renaud et al., 2015), taking into consideration rates of cytosine deamination at the 5′- and 3′- read ends, and only retaining bases with quality ≥30. For each sample, we called a first consensus from the rescaled BAM file, then re-mapped all shotgun reads to the newly built consensus with PALEOMIX as described above, and called a final consensus from the resulting rescaled BAM with schmutzi.
We then constructed alignments from the consensus sequences for ancient samples obtained here and in Der Sarkissian et al. (2017), as well as all sequences previously published for the genus/species of interest. We extracted the sequences of 12 genes (CYTB, COX2, NADH1, NADH4, COX3, NADH2, NADH3, COX1, ATP6, NADH4L, NADH5, NADH6) and aligned them independently with PRANK (Löytynoja and Goldman, 2005). For the analyses of Mytilus sp. sequences, we added the sequences of the 12S ribosomal RNA gene (12S rRNA), and 23 transfer RNAs (tRNA). Maximum-likelihood trees were reconstructed using the program PhyML 3.0 (Guindon et al., 2010) with an approximate likelihood-ratio test (aLRT) and a Shimodaira-Hasegawa-like procedure for branch support estimation. Using the SMS heuristic procedure for model selection implemented in PhyML 3.0 (Lefort et al., 2017), we assessed the best substitution models for the concatenated alignments: the Generalized Time-Reversible model, with invariant sites and 4 gamma site-rate categories for the Mytilus DNA alignment, and the Variable Time substitution model with invariant sites, a gamma site-rate distribution and alignment-based estimation of amino-acid frequency equilibrium for the mollusk protein alignment.
For all samples positively identified through comparison with the BOLD marker database and showing an average coverage ≥3×, a COI-5P consensus sequence was built as described for complete mitochondrial genomes. The obtained consensus was then compared to all the sequences available in the BOLD database for the corresponding genus/species using Median Joining Network (epsilon = 0) in POPART v1.7 (Leigh and Bryant, 2015) and the vouchers’ geographical origin as curated in the BOLD database metadata. When available, published mitochondrial sequences from modern populations for species of interest were retrieved for comparison to ancient sequences using the same procedure. These comprised Mytilus trossulus (Breton et al., 2006; Smietanka et al., 2010, 2013; Zbawicka et al., 2014b; Śmietanka and Burzyński, 2017), A. islandica (Glöckner et al., 2013), Ruditapes decussatus (Cordero et al., 2014; Sanna et al., 2017), R. philippinarum (Cordero et al., 2017), and Crassostrea angulata (Grade et al., 2016).
The ≥100 kyr Tx101A Mytilus sample from the Taimyr Peninsula could be compared to modern Mytilus populations for which genomic data are publicly available (Fraïsse et al., 2016, 2017). This comparative dataset was previously obtained through capture-based enrichment sequencing targeting 4.3 Mb DNA sequences assembled from M. edulis BAC clones and M. galloprovincialis cDNAs in 1,269 contigs (Fraïsse et al., 2016, 2017). It is composed of 80 individuals from ten populations: M. trossulus from the Baltic Sea (N = 8) and the North American Atlantic coast (N = 7), M. edulis from the North American Atlantic coast (N = 11), as well as from the Wadden Sea (N = 8) and the Bay of Biscay (N = 8; respectively, outside and inside the Atlantic hybrid zone with M. galloprovincialis), M. galloprovincialis from Portugal (N = 6) and Brittany (N = 8; respectively, outside and inside the Atlantic hybrid zone with M. edulis), as well from the Occidental (N = 8) and Oriental Mediterranean basins (N = 8), and M. platensis from the Kerguelen Islands (N = 8). In order to avoid computational analysis biases, the raw data published in Fraïsse et al. (2016, 2017) were re-mapped to the contig reference sequence at the same time as raw data for the Tx101A ancient sample and EBCs. The same BWA mapping parameters were used as described above for both ancient and modern samples, except that the options for mismatch penalty (-M 2), gap open penalty (-O 3) and minimum seed length (-k 10) were added as in Fraïsse et al. (2016, 2017), and that seeding was disabled for Tx101A and EBCs.
Low depth-of-coverage for Tx101A when mapped against the Mytilus contigs precluding accurate genotype calling, subsequent analyses where carried out using genotype likelihood in ANGSD v.0.929 (Korneliussen et al., 2014). Genotype likelihoods were estimated with the following options and filters: -doMajorMinor 1 -doMaf 1 -remove_bads 1 -uniqueOnly 1 -minMapQ 30 -C 50 -minQ 30 -baq 1 -skipTriallelic 1 -SNP_pval 1e-6 -doHWE 1 -maxHWEpval 0.05. All analyses were performed either considering all substitution types, or excluding transitions (-rmTrans 1). We used PCAngsd v.0.98 (Meisner and Albrechtsen, 2018) and the option -minMaf 0.05 to perform Principal Component Analysis (PCA) and determine the optimal number of principal components to describe the Mytilus population structure using the minimum average partial (MAP) test. Admixture proportions were estimated using NgsAdmix (Skotte et al., 2013) for a number of clusters equal to the previously determined optimal number of principal components +1 (Meisner and Albrechtsen, 2018), i.e., four, here.
We used mapDamage v2.0.1 (Jónsson et al., 2013) to characterize ancient shell DNA in terms of fragmentation patterns, nucleotide mis-incorporation and fragment size distributions. On the basis of 100,000 Markov Chains Monte Carlo iterations, posterior distributions were obtained for the following damage parameters: rates of deamination in double strands (δD) and single strands (δS), probability of reads not terminating in overhangs (λ, transformed into 1/λ–1, a proxy for the length of overhanging regions). Quality scores of bases likely to represent damaged read positions were then rescaled using default parameters.
We provided a summarizing visualization of the potential for ecological studies of each mollusk genus investigated. In genus-specific radar graphs, we represented: their molecular preservation potential (oldest authenticated mollusk DNA data, DNA recovery rate), the availability or unavailability (encoded as 1 and 0, respectively) of comparative datasets (BOLD barcodes, complete mitochondrial genomes, nuclear transcripts and genome assemblies), and their relevance to ecological questions with regards to the research themes “Environment,” “Commercial” exploitation, and biological “Invasion” (assessed on the basis of bibliography and encoded as 1, relevant and 0, not relevant), and “Extinction.” An “extinction score” was given for each vulnerability category defined by The IUCN Red List of Threatened Species (2019): i.e., 1 = “least concerned,” 2 = “near threatened,” 3 = “vulnerable,” 4 = “endangered,” 5 = “critically endangered,” 6 = “extinct in the wild,” 7 = “extinct,” and a genus extinction score was calculated as the average score across species listed for each genus.
We tested for long-term preservation of aDNA in marine mollusk shells by constructing shotgun DNA libraries using extracts from three mollusk shell samples (Tx100, Tx101A, Tx103) dated to ≥100 kyr. For the three libraries, qPCR showed copy-numbers compatible with HTS, which yielded 653,177–19,314,607 collapsed and non-collapsed pair-end mate reads after trimming and quality filtering (Table 1 and Supplementary Table S1).
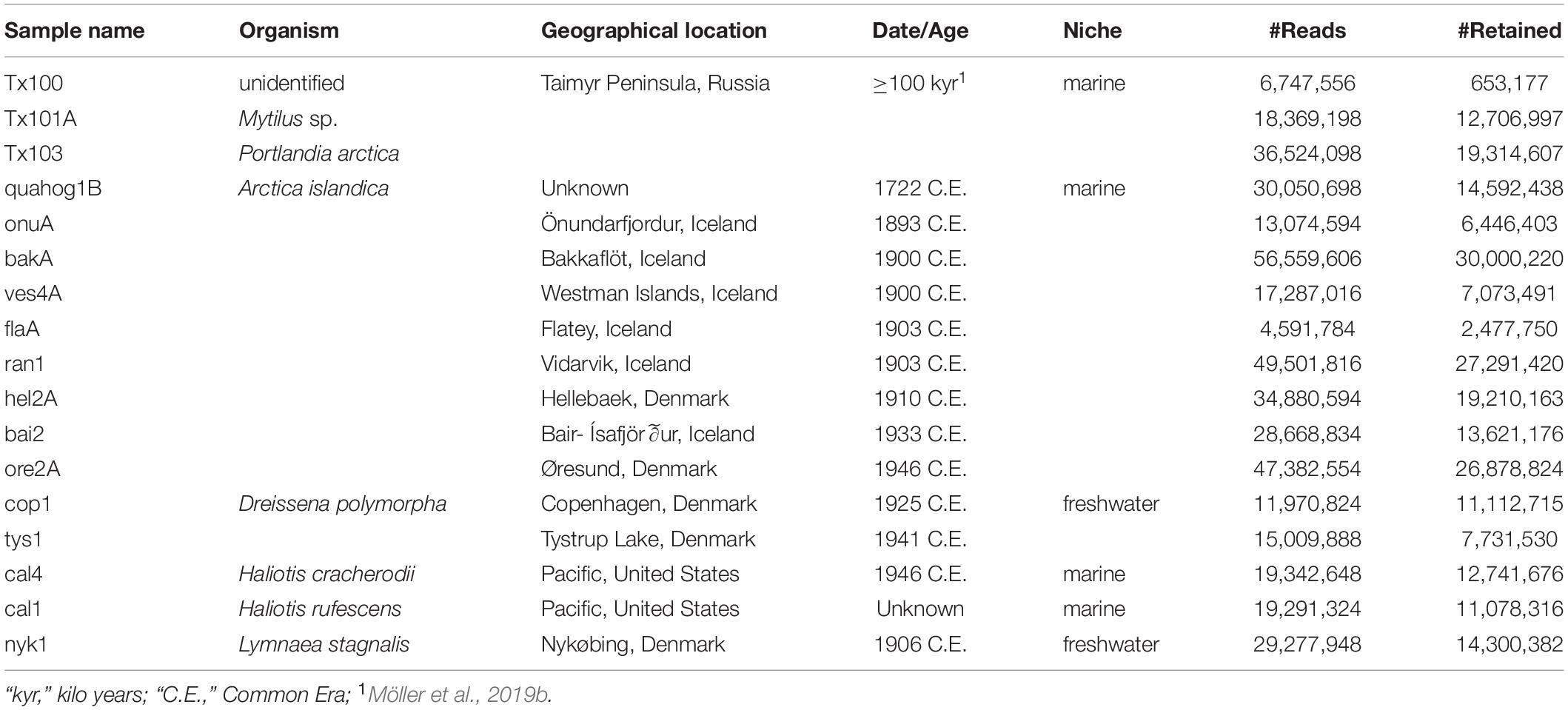
Table 1. Sample information and sequencing statistics.
Taxonomic identification was achieved by mapping against the BOLD Mollusca mitochondrial COI-5P marker reference sequences. No hit was obtained for Tx100, precluding any classification for this specimen. On the basis of mitochondrial DNA, Tx101A could be identified as M. trossulus since 98.6% of the mapped nucleotides aligned to markers of this species, plus 0.9 and 0.5% aligning to markers of the Mytilus genus and the Mytilida order (Supplementary Table S4). Tx101A had been previously identified as M. edulis on the basis of shell morphology in Möller et al., (2019a,b), but M. trossulus and M. edulis can be difficult to distinguish due to their similarity in shape and size (Innes and Bates, 1999; Riginos and Cunningham, 2005). The morphological assignment of the Tx103 sample to P. arctica could be confirmed as 27.3% of the mapped nucleotides align to markers of this species, and 72.7% to the Nuculanida order (Supplementary Table S4).
We attempted to gain more confidence in our taxonomic assignment, and retrieve more specific information about genomic affinities of Tx101A and Tx103 with modern individuals (e.g., at the population level), by comparing their COI-5P sequences with M. trossulus and P. arctica sequences available in the BOLD database. Median Joining Network analyses, however, proved uninformative due to the limited phylogenetic resolution of the COI-5P marker, the scarcity of geographic information provided for BOLD vouchers (Tx101A), and the lack of comparative P. arctica entries (Tx103) (Supplementary Figure S1).
In order to achieve a more robust identification and a characterization of the Tx101A specimen with more resolution, we performed phylogenetic and population structure analyses based on HTS read alignments to all complete mitochondrial genomes published to date, as well as to genome-wide contigs for Mytilus. In absence of such public reference resources for P. arctica, or any close relative taxon, further genomic characterization of the Tx103 specimen was not achievable. The analyses provided further evidence for the mitochondrial identification of Tx101A as M. trossulus. The maximal coverage of the mitochondrial genome was, indeed, obtained when aligning HTS reads to the sequences of M. trossulus F-genomes or recently masculinized F-genomes (24.7–25.7× maximum for GenBank accession number GU936625; Supplementary Table S5). Other Mytilus mitochondrial genomes led to 10–9,267-fold lower coverage (Supplementary Table S5). This pattern was robust to relaxing or strengthening mapping stringency (-n, BWA edit distance option) (Supplementary Figure S2).
A Maximum-likelihood tree reconstructed from concatenated mitochondrial genome sequence partitions confirmed the phylogenetic placement of Tx101A within the clade of M. trossulus (recently masculinized) F-genomes, where it occupies a basal position (Figure 1A). Patterns of coverage are consistent with the phylogenetic position of Tx101A, i.e., the more distant to M. trossulus the mapping reference genome, the lower the coverage (Figure 1B and Supplementary Table S5). When only a 1,171 bp concatenated sequence partition of mitochondrial genes and transfer RNAs from Tx101A was compared to data from 187 present-day M. trossulus individuals (Breton et al., 2006; Smietanka et al., 2010, 2013; Zbawicka et al., 2014b; Śmietanka and Burzyński, 2017), the outlying position of Tx101A was confirmed. Closest relatives equidistant to the Tx101A haplotype were found in the broadly distributed present-day populations of the Baltic, the eastern and western coasts of the North Atlantic and the North Pacific (Figure 1C).
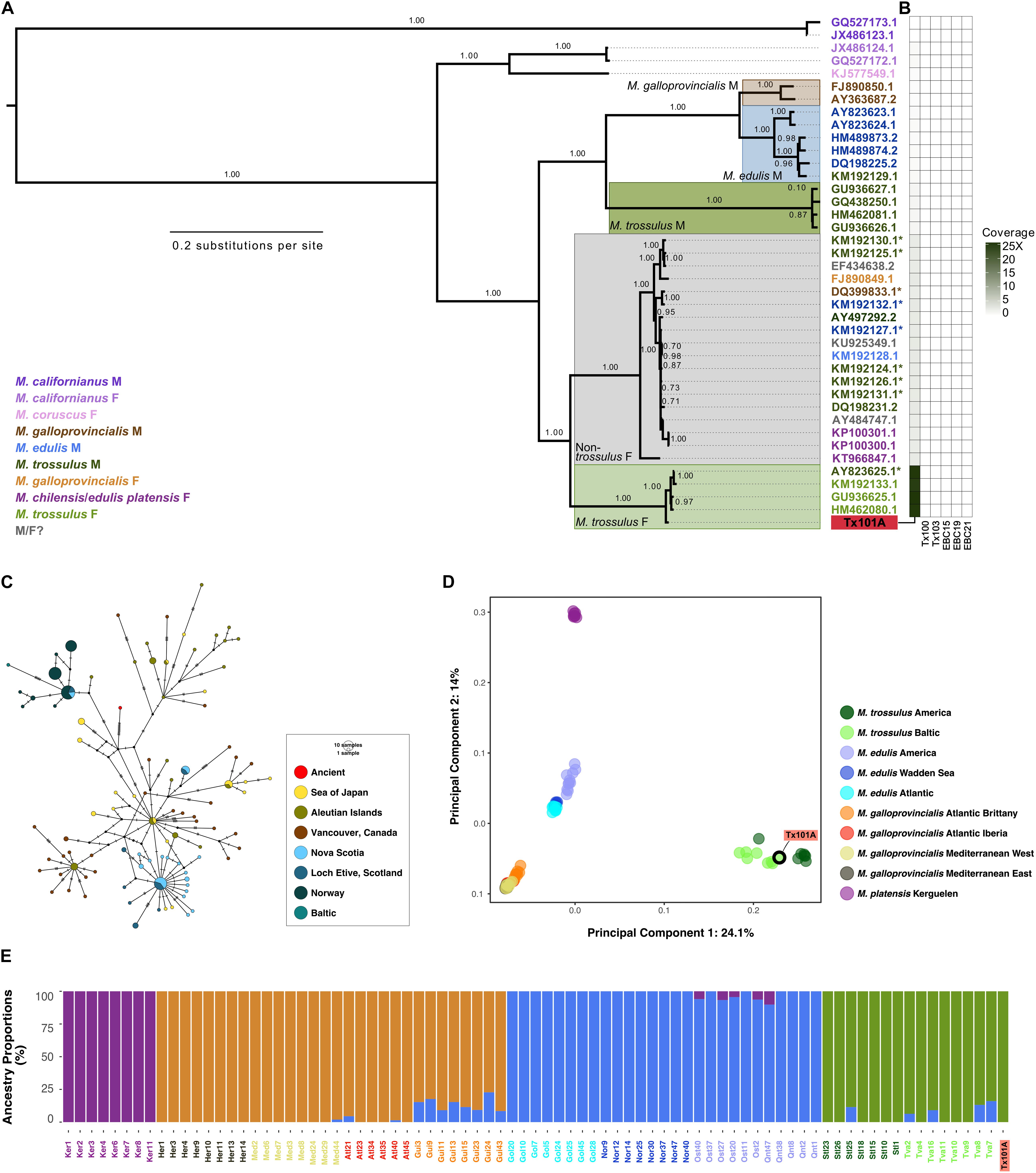
Figure 1. Phylogenetic and population affinities of Tx101A within present-day Mytilus. (A) Maximum likelihood phylogenetic tree built from a concatenated alignment of Mytilus mitochondrial sequences for 12 protein-coding genes, the 12S rRNA gene and 23 tRNAs (total length 14,277 bp). The sex and species of modern individuals were determined based on morphology and nuclear DNA markers. Branch support ≥0.70 is shown as Shimodaira-Hasegawa approximate Likelihood Ratio Test. *Recently masculinized mitochondrial genomes. (B) Average depth-of-coverage when mapping against each Mytilus mitochondrial reference genome considering unique high-quality reads (≥30). (C) Median-Joining network built from a 1,171 bp concatenated alignment of mitochondrial genes (COX3, ND2) and transfer RNA (Ser, Met) from 187 present-day M. trossulus individuals. (D) Principal Component Analyses of the top two principal components in a dataset comprising 11,882 variable sites (considering transversions only) for 80 individuals. The proportion of the variance explained by each component is indicated for each axis. (E) Ancestry proportions estimated considering four ancestral populations (transversions only). Individual color code is the same as in (D).
As hybridization can occur amongst members of the M. edulis complex (M. trossulus, M. edulis, M. galloprovincialis), we examined the nuclear genomic background of Tx101A in order to validate its taxonomic identification and/or estimate potential admixture proportions. PCA of 11,882 variable sites (considering transversions only; 29,440 variable sites considering all substitutions in Supplementary Figure S3) in 80 individuals and along the optimal number of three components (as determined by the MAP test in PCAngsd) confirmed that Tx101A belongs to the M. trossulus population cluster, clearly segregating from the other clusters of the M. edulis complex and of the M. platensis outgroup (Figure 1D and Supplementary Figure S3). This result was confirmed by clustering analyses considering four ancestries, in which a genome-wide affinity of Tx101A for M. trossulus modern populations and no admixture with another M. edulis complex or outgroup population were detected (considering transitions alone; Figure 1E and Supplementary Figure S4).
Considering that the age of the Tx101A and Tx103 samples dramatically pushes back the limit of marine mollusk shell DNA preservation from ∼7,000 years to ≥100 kyr, particular attention was paid to the line of evidence supporting data authenticity: (1) Tx101A and Tx103 were excavated at the same site, but yielded DNA reads that were identified as representative of different Bivalvia sub-classes; (2) our molecular classification confirms previous morphological identification of Tx101A as Mytilus and Tx103 as P. arctica (Möller et al., 2019b, a); (3) the detection of M. trossulus and P. arctica at the BBR 17A site was ecologically coherent with these being considered as (sub-) arctic species of the littoral or deeper marine environments (Möller et al., 2019b); (4) none of the HTS datasets obtained for the extraction blank controls (EBCs) led to the identification of M. trossulus or P. arctica DNA sequences. Compared to those observed for Tx101A and Tx103, significantly lower coverage were attained when mapping EBC reads against reference sequences for: M. trossulus/P. arctica BOLD COI-5P (no hit), Mytilus complete mitochondrial genomes (≥2,335-fold coverage decrease), the M. galloprovincialis genome assembly (≥16,563-fold coverage decrease), Mytilus sp. genome-wide contigs (≥5,704-fold coverage decrease); (5) for both Tx101A and Tx103, high-quality HTS reads aligned to the various reference sequences displayed molecular signatures characteristic of post-mortem DNA fragmentation and cytosine deamination-induced mis-incorporation. When considering the COI-5P alignment, Tx103 showed reads of ∼66.6 bp average length and a 9.4% C-to-T mis-incorporation rate at the 5′-read extremities. As for Tx101A, when considering mitochondrial genome/nuclear contig alignments, HTS reads exhibited ∼82.8/78.5 bp reads, maximum 3.9/6.4% C-to-T mis-incorporation rates, post-mortem deamination rates in overhangs 4.9/5.2-larger than in double stranded DNA, and overhanging ends of 3.0/2.0 bp (1/λ – 1), as expected. In addition, a 10–bp periodicity in the size distribution was observed in the nuclear DNA of Tx101A, but not in its mitochondrial DNA, a specific pattern that was previously proposed to reflect nucleosome protection in authentic and degraded ancient nuclear DNA (Pedersen et al., 2014; Figure 2 and Supplementary Table S6); and (6) all phylogenetic and population structure analyses of mitochondrial and nuclear DNA were consistent with Tx101A being part of M. trossulus. Combined, these results provide compelling evidence that the DNA obtained from ≥100 kyr mollusk shells is authentic.
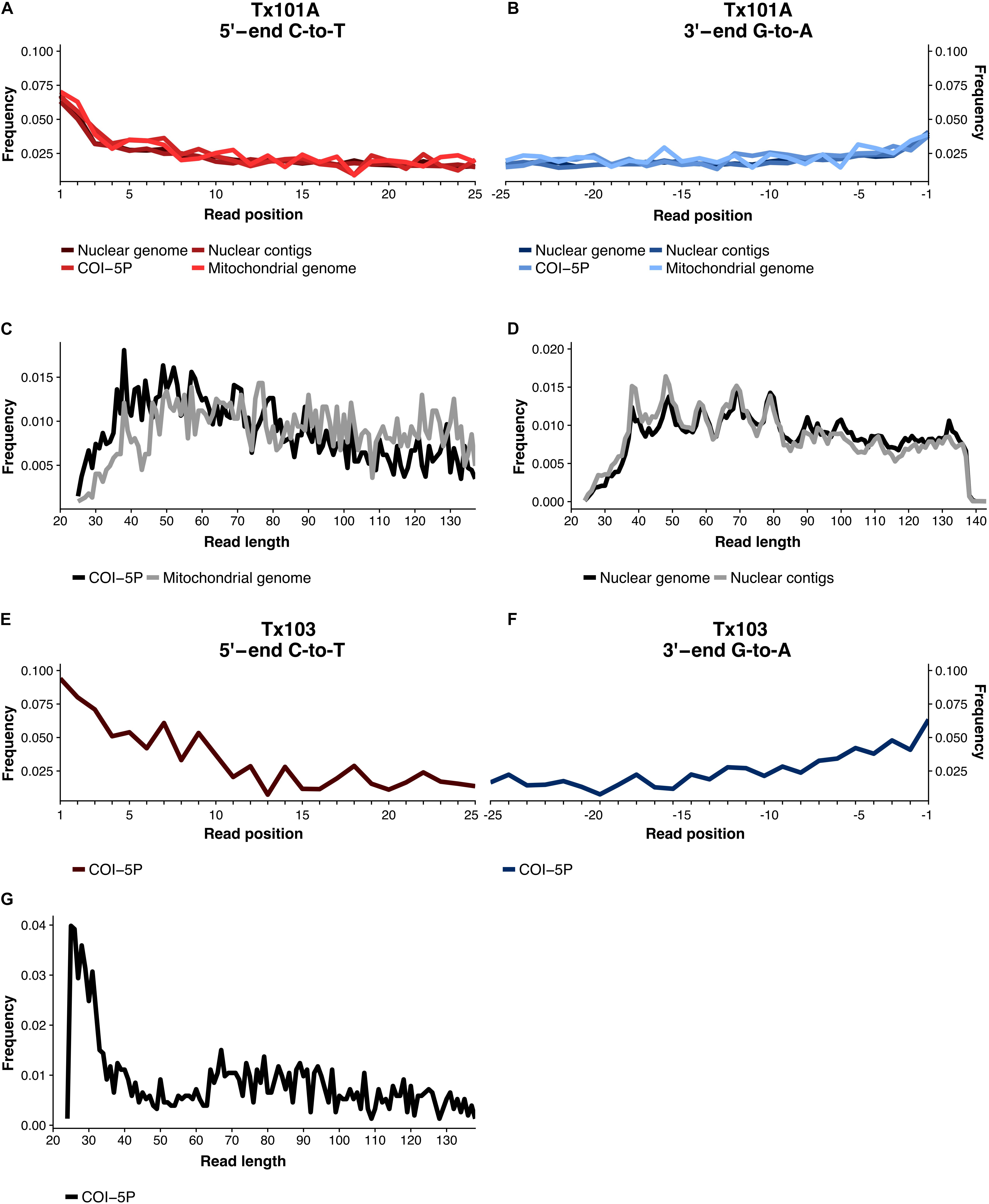
Figure 2. Post-mortem DNA damage patterns. (A) For Tx101A, rates of C-to-T nucleotide mis-incorporation at 5′-read ends, (B) rates of G-to-A at 3′ read-ends. (C) Size distribution of reads mapping to mitochondrial, and (D) nuclear reference sequences. (E) For Tx103, rates of C-to-T nucleotide mis-incorporation at 5′-read ends, (F) Rates of G-to-A at 3′ read-ends. (G) Size distribution of reads mapping to mitochondrial reference sequences. Only unique high-quality (MQ≥30) reads were considered and mapDamage v.2 was run on the full alignment (100,000 iterations).
We next extended the assessment of DNA recovery from ancient mollusk shells to 39 new specimens, 14 of which led to shotgun HTS datasets containing 2,477,750–30,000,220 quality-filtered collapsed and non-collapsed pair-end mate DNA reads (Table 1 and Supplementary Table S1). In subsequent analyses, these were added to 32 previously published ancient shell data (Der Sarkissian et al., 2017) and the two ≥100 kyr Taimyr samples described above to build a dataset comprising 48 specimens from 13 species and 9 genera, with 73% of the samples dated to the last 300 years of the post-industrial era (Figure 3 and Supplementary Table S1). Success rates were variable across genera, with the highest rates observed for Ruditapes/Venerupis sp. (100%, N = 20), Haliotis sp. (70%, N = 7) and A. islandica (57%, N = 12). No successful DNA recovery was achieved for the Lottia gigantea (N = 1), Margaritifera margaritifera (N = 3), and the terrestrial Cernuella virgata specimens (N = 2), thus limiting investigations to marine (N = 13) and freshwater individuals (N = 3) in this study. We found no indication that the overall success rate depends on the amount of shell powder analyzed, as specimens yielding successful and unsuccessful DNA recovery did not show significant differences in input shell median weight (Wilcoxon signed-rank test, p-value = 0.3).
Figure 3. Geographical location and age of the shell samples screened for DNA. (A) Map showing the number and average age of specimens by geographical location. (B) Chronology by genus (Supplementary Table S1). “kyr,” kilo years.
Mapping to the Mollusca database of BOLD COI-5P markers provided sufficient read alignments for taxonomic identification, as all 48 samples, but the LEFB Pecten sp. shell, could be classified at least to the genus level (Figure 4 and Supplementary Table S4). A. islandica, C. angulata, Haliotis tuberculata, R. decussatus, R. philippinarum, and Dreissena polymorpha specimens were unambiguously identified at the species level, and even subspecies level for H. tuberculata coccinea and H. tuberculata tuberculata. We noted, however, 6–30% false positive rates for H. rufescens, V. corrugata, and Lymnaea stagnalis at the genus level. Through resequencing, we also assembled 29 additional complete mitochondrial genomes with coverage ≥2.8-fold that were used to reconstruct a maximum-likelihood tree of mitochondrial genes. The L. stagnalis and V. corrugata specimens could not be included in this analysis as no reference sequences are available for these species. The obtained tree retraces known phylogenetic relationships within gastropods and bivalves, but also confirms the taxonomic identification based on COI-5P sequences (Figure 5). Interestingly, with five new R. decussatus mitochondrial genomes assembled here from ancient specimens, we could confirm a previous hypothesis based on one modern R. decussatus mitochondrial genome that R. decussatus is more closely related to the Paphia sp. than to R. philippinarum despite being named as members of the same genus (Ghiselli et al., 2017).
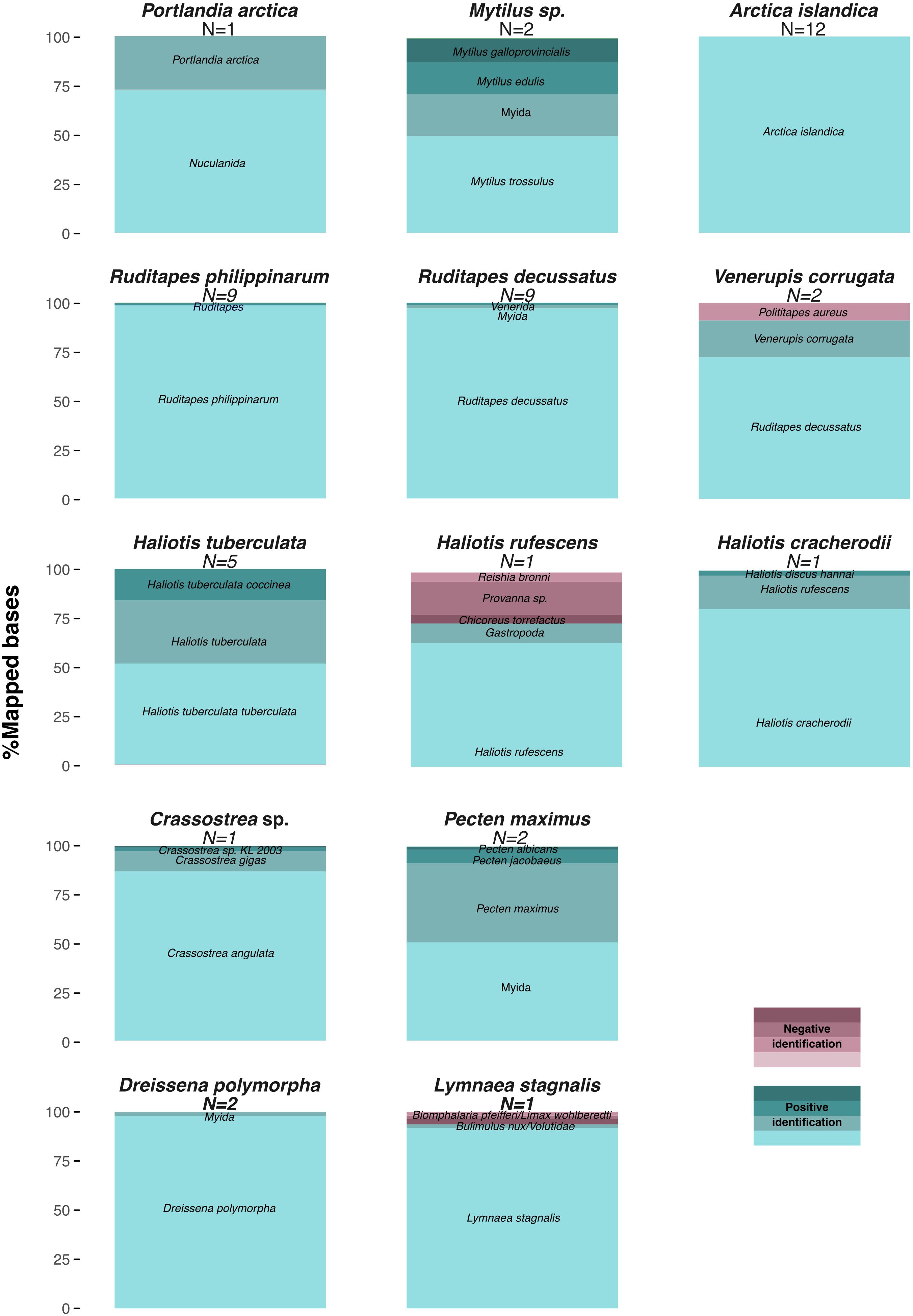
Figure 4. COI-5P barcode-based identification of Mollusca taxa from shell DNA. Proportion of total nucleotides mapped to each reference marker combining all datasets for samples of the same genus and considering unique high quality (≥30) reads and reference markers covered by ≥150 bp (Supplementary Table S4).
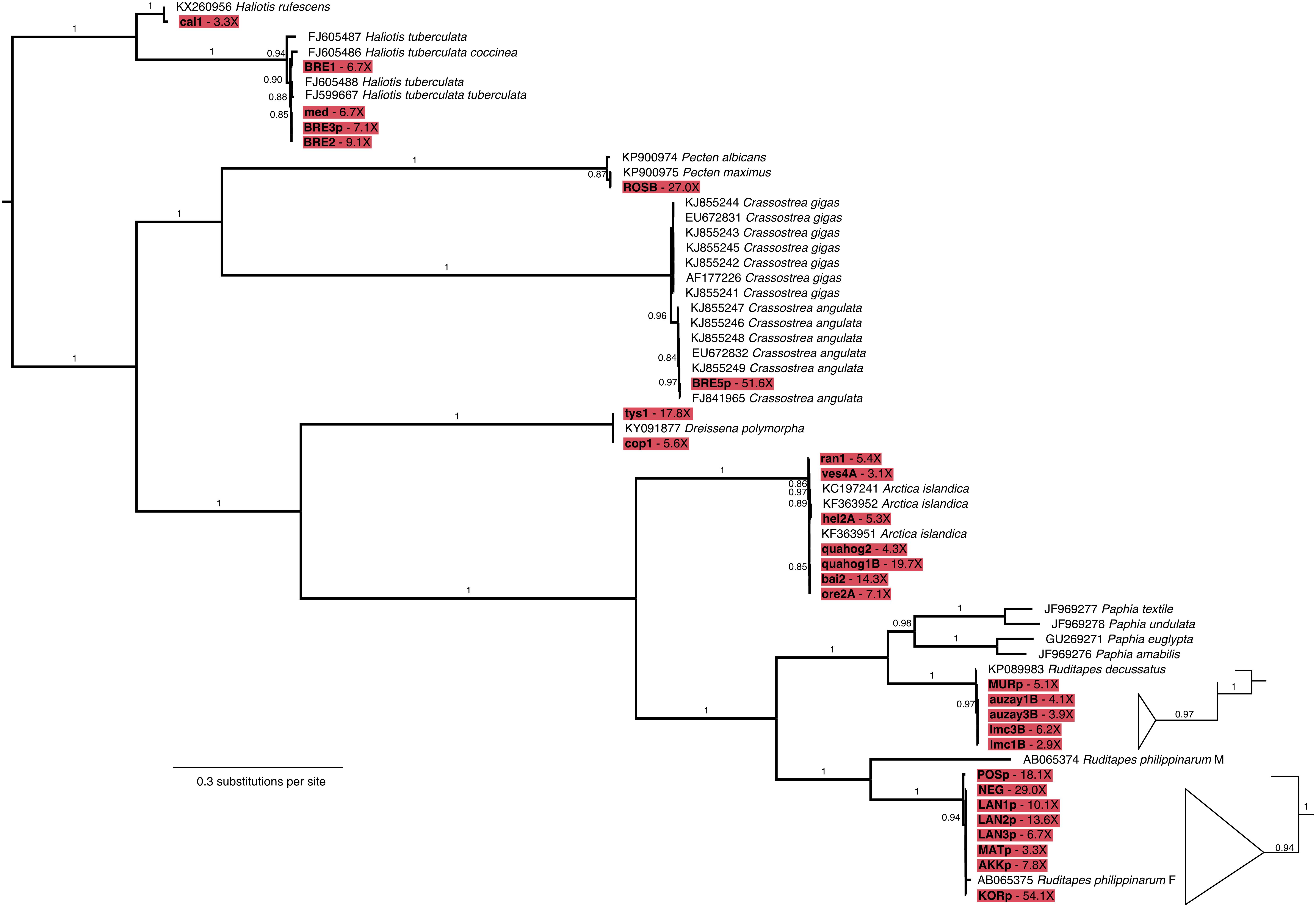
Figure 5. Maximum likelihood phylogenetic tree of mollusk mitochondrial genes. Tree built from a 60-individual alignment of 12 concatenated gene sequences. Sequences assembled from shell DNA in this study are highlighted in red and their coverage is indicated in the tip label, GenBank accession numbers are provided for previously published sequences. Branch support ≥0.70 is shown as Shimodaira-Hasegawa approximate Likelihood Ratio Test. The subtrees for the R. decussatus and R. philippinarum clades were enlarged and branches collapsed in order make branching order more apparent.
Little information about affinities between ancient and modern populations could be retrieved from the phylogenetic tree owing to the limited number of published complete mitochondrial genomes in mollusks. Although more mitochondrial COI-5P sequences are available, analyses proved poorly informative for most genera due to the paucity of sequenced present-day individuals (Pecten maximus) and corresponding geographical metadata (P. maximus, L. stagnalis). Additionally, many ancient haplotypes were found similar to the most abundant and widely distributed haplotypes in modern populations partly due to the limited resolution of the COI-5P marker (C. angulata, D. polymorpha, R. decussatus; Supplementary Figure S1). The mitochondrial phylogenetic tree could, however, show a segregation within R. decussatus between the ∼30 year BP Adriatic Sea MURp individual from the 400–5,500 year BP Atlantic Ocean auzay1B, auzay3B, lmc1B, and lmc3B individuals (Figure 5). Within the other clam species R. philippinarum, both the mitochondrial gene tree and the COI-5P network showed a differentiation between the 1988 C.E. POSp French specimen and the 1983–2012 C.E. LAN1p, LAN2p, LAN3p, NEG, MATp, AKKp and KORp samples from France, Japan and Korea (Figure 5 and Supplementary Figure S1). For A. islandica quahogs, we compared our 73–297 year-old dataset to 24 COI-5P sequences from the BOLD database (Supplementary Figure S1) as well as to 3,029-bp long mitochondrial sequences for 20 modern individuals (Glöckner et al., 2013). Modern quahogs belong to two main clades: one comprising individuals from Iceland, the Baltic and the North Seas, and one composed of individuals from Iceland and the Baltic Sea, with the Icelandic and the Baltic Sea populations displaying a larger diversity than the North Sea population (Glöckner et al., 2013). We found the same pattern in the past as our sequences from ancient quahogs fell within the two clades described previously (Supplementary Figure S5). More present-day data in the form of complete mitochondrial genome sequences are needed to rule out possible sampling biases in both the number and geographical origin of the sequenced individuals.
Authenticity of the aDNA retrieved from the mollusk shells was supported by: (1) congruent molecular and morphological identification, at least at the genus level; (2) increased coverage when mapping against the complete mitochondrial genome reference sequence for the identified species (Supplementary Figure S6); (3) phylogenetic placement of the COI-5P barcodes and mitochondrial genes (Figures 4, 5 and Supplementary Figure S1); and (4) the presence of typical molecular signatures of mitochondrial DNA post-mortem damage in the form of short fragment sizes (45.1–95.2 bp), increased rates of C-to-T mis-incorporation rates at read 5′-ends (0.0–32.2%), increased mis-incorporation rates in single- versus double-strand contexts ∂S/∂D (2.3–70.3), and decreased values for the overhang-length proxy 1/λ – 1 (0.1–4.7). Similar patterns were observed in reads mapping against nuclear reference sequences: DNA fragmentation (41.9–107.8 bp), C-to-T mis-incorporation rates at read 5′-ends (0.0–17.4%), ∂S/∂D (0.1–184.6), and 1/λ – 1 (0.2–16.3) (Supplementary Table S6).
In this work, we broaden the assessment of mollusk DNA recovery from ancient shells in terms of time scale, species, and applications for ecological studies. The successful retrieval of aDNA from ≥100 kyr shells significantly extends the temporal scale of mollusk aDNA analyses, as the oldest specimens that had previously yielded aDNA were ∼7,000 year-old Mytilus shells collected from shell middens in Denmark (Der Sarkissian et al., 2017). The constantly frozen Siberian permafrost marine sediments from which the ≥100 kyr shells were retrieved (Möller et al., 2019b, a) may have provided exceptionally favorable DNA preservation conditions minimizing damage induced by both water and microbial activity (as reviewed in Pedersen et al., 2015). As examples of long-term DNA preservation in similar conditions, the oldest remains for which animal aDNA could be recovered in Siberia were >63.5 kyr and ∼50 kyr mammoth hair, and tooth/femur (Gilbert et al., 2008; Pečnerová et al., 2017), and, outside Siberia, a >560 kyr horse bone from the Yukon Territories, Canada (Orlando et al., 2013). Although earlier studies demonstrated permafrost as one of the best environments for long-term preservation of animal DNA in keratin- and calcium phosphate-based remains, we show here for the first time that permafrost also preserves calcium carbonate-based remains over significant timescales. Past marine diversity was formerly recovered from <43 kyr sediments that yielded DNA from protists, including haptophytes and foraminifera, which are other organisms producing calcium carbonate exoskeletons surviving or dissolving within sediments (Boere et al., 2009; Coolen et al., 2013; Lejzerowicz et al., 2013; Pawłowska et al., 2014; Morard et al., 2017; More et al., 2018; Armbrecht et al., 2019).
Compared to the previous assessment of eight marine species (Der Sarkissian et al., 2017), we demonstrate here that aDNA can be recovered from an extra five species (P. arctica, H. rufescens, H. cracherodii, D. polymorpha, and L. stagnalis), including two freshwater species. Considering the relevance of mollusks to ecological questions about the terrestrial niche (Lydeard et al., 2004), aDNA content of terrestrial mollusk shells is worth further investigation. In the summarizing visualization of the potential of each mollusk genus investigated for ecological studies, radar-plot maximal surface areas indicate Mytilus, Haliotis, and Arctica as the three genera with the most potential for ecological studies based on aDNA (Figure 6). Overall, we observed a DNA recovery success rate of 61.25% for the 80 shells examined (Figure 6), and endogenous contents of 0.09–33.26% for those genera with available reference nuclear genome assemblies (Crassostrea, Dreissena, Haliotis, Lymnaea, Mytilus), and we estimated that no less than 71,000 sequencing reads were required here to obtain 1× coverage of 16,358–18,653 bp mitochondrial genomes. In line with these results suggesting low numbers of shotgun-sequenceable DNA templates in ancient mollusk shell extracts, we were not successful at recovering DNA from three 42–191 year BP M. margaritifera shells, although positive nuclear Small Tandem Repeat (STR) PCR amplification could be achieved from fresh dry shells of the same species by Geist et al. (2008). Our negative results could be explained by the particular storage conditions of the specimens, extensive post-mortem DNA degradation and/or concentrations of recoverable DNA below the sensitivity threshold of our methodology. Target-enrichment protocols may help circumventing these limitations in the future. We, however, caution that such methods will result in the loss of the vast majority of off-target DNA molecules. Their benefits in terms of information gain should be weighed against the risk of heritage loss for each (rare) ancient specimen considered. In addition, the type of capture should carefully be selected and designed to maximize the overlap with datasets generated from comparative modern or ancient populations. Importantly, neither capture nor shotgun HTS aDNA data can, at this stage, provide much information about population dynamics in cases where modern population data available for comparison are nuclear STR genotypes. These indeed typically encompass >100 bp regions (e.g., Knott et al., 2003; Cruz et al., 2005; Gardeström et al., 2008; Feldheim et al., 2011; Van Wormhoudt et al., 2011; Beldade et al., 2012; Morvezen et al., 2013; Borrell et al., 2014; d’Auriac et al., 2017; Jiang et al., 2018) that cannot reliably be typed from aDNA extracts due to their characteristic high fragmentation (here, 41.9–107.8 bp).
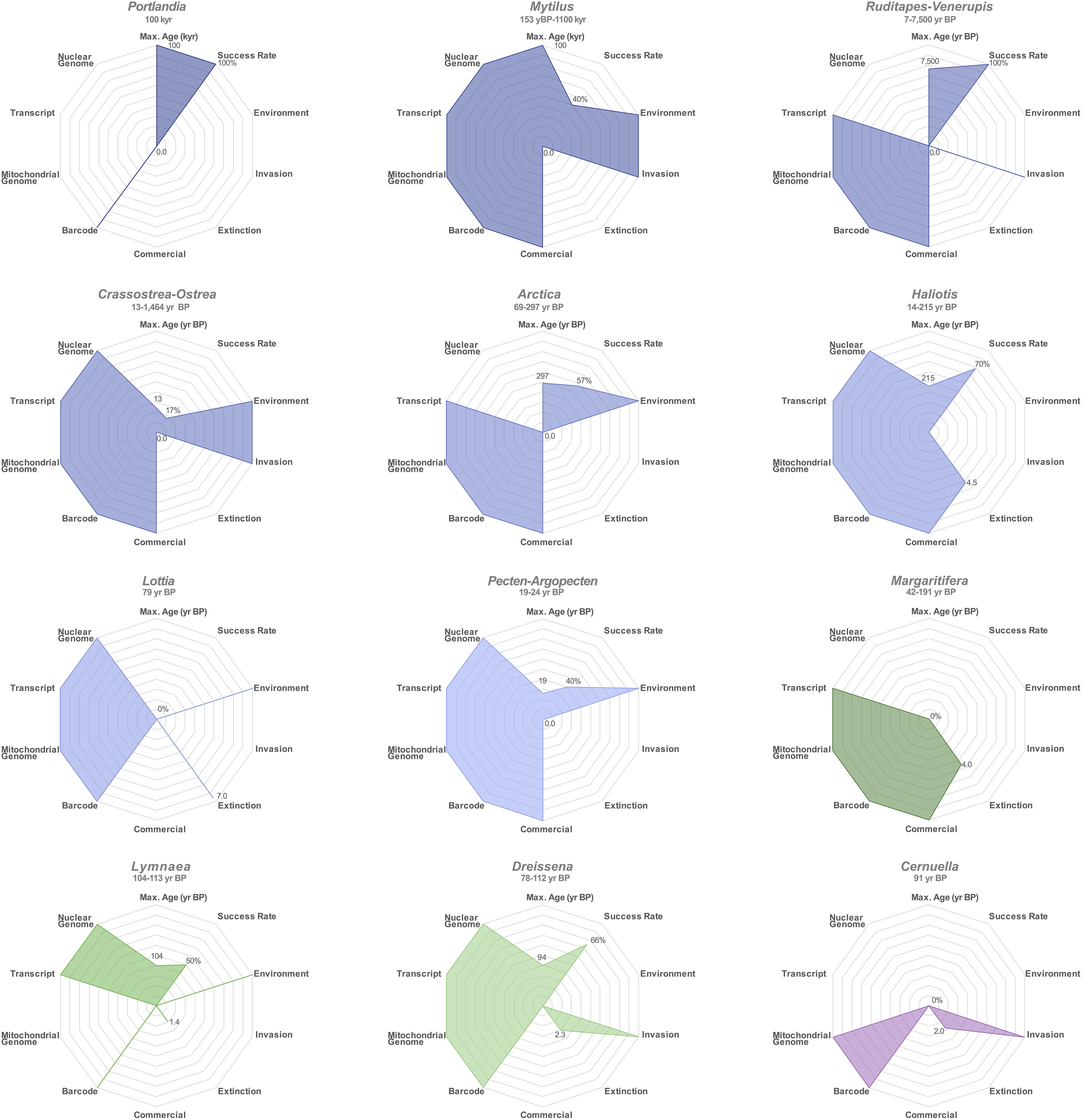
Figure 6. Radar plot summarizing application potential of ancient shell DNA for mollusk genera. Marine genera are represented in blue shades, freshwater genera in green shades and the terrestrial genus Cernuella in purple. “Max. Age” refers the age of the oldest sample for which DNA could successfully be recovered; “kyr,” kilo years; “year BP,” years Before Present.
We observed a substantial heterogeneity among the examined genera in the availability of comparative mitochondrial sequences, complete mitochondrial genomes, nuclear transcripts and genomes (Figure 6). As expected for mostly non-model species, the present study highlights a general paucity of published mitochondrial and nuclear genome assemblies, population-scale data, as well as geographical metadata submitted to barcode databases (BOLD, GenBank), with some mollusk populations better described at the population genomic level, i.e., Crassostrea sp., Mytilus sp., Haliotis sp., and Pecten sp. (Zbawicka et al., 2014a, 2018; Fraïsse et al., 2016; Mathiesen et al., 2016; Wenne et al., 2016; Gutierrez et al., 2017; Harney et al., 2018; Wilson et al., 2018; El Ayari et al., 2019; Masonbrink et al., 2019; Paterno et al., 2019; Vendrami et al., 2019a, b). It is worth noting that the phylogenetically robust data reported here significantly increases the number of released complete mitochondrial genome sequences by 6-fold for R. decussatus, five-fold for R. philippinarum, three-fold for A. islandica, and D. polymorpha, and finally two-fold for H. rufescens, H. tuberculata, and P. maximus. Although mitochondrial DNA is very informative for genus-level classification and investigating the evolution of organellar genomes, it should be stressed that species-level taxonomic identification and population affinity inference should be confirmed by nuclear sequences for those mollusks in which interspecies hybridization is observed, e.g., Crassostrea and Mytilus. Importantly, as genomic data production is expected to increase significantly in the near future, it is crucial that appropriate metadata are made easily accessible to the research community, so that published results can be reproduced and future analyses can incorporate ever-growing datasets. Despite limitations associated with the degraded nature of mollusk shell aDNA and the occasional scarceness of comparable modern population data, phylogenetic and population structure signals could nevertheless be recovered, thus emphasizing the promising potential of mollusk shell aDNA for ecological studies.
Our results directly hint at taxonomic identification based on mitochondrial barcoding, which could reveal helpful in cases where morphological examination of fragmentary or undiagnostic shell remains preclude classification. Mollusk aDNA analyses are compatible with shotgun- or mitochondrial metabarcoding-based (Bush et al., 2019) reconstructions of past communities from bulk samples (e.g., Murray et al., 2013; Grealy et al., 2015; Seersholm et al., 2018) or sediment cores (e.g., Giguet-Covex et al., 2014; Pedersen et al., 2016), which could be of relevance to the study of shell middens. Temporal changes in ecosystems detected through such approaches could reveal interesting insights into the timing and impact of environmental changes, biological invasions and extinctions.
Time series DNA data from mollusk shells hold the potential to illuminate patterns of species or population distribution and diversity. These can be compared to past global and local paleo-environments reconstructed through stable isotope, metal or trace element analyses of the very same shells in order to characterize the temporal and spatial scales of biological responses to climatic events, in particular prior to access to instrumental data or historical records (reviewed in Jones et al., 2009). Marine bivalves such as A. islandica and P. maximus, for which we reported successful aDNA recovery in this study, have previously allowed sclerochronological reconstructions of past seawater temperature and salinity changes at millennial-length annual and seasonal resolutions, respectively (Surge et al., 2003; Chauvaud et al., 2012; Vokhshoori and McCarthy, 2014; Reynolds et al., 2016; Black et al., 2017). At a more local scale, the freshwater L. stagnalis, for which we report shell aDNA recovery here for the first time, is a model commonly used in ecotoxicology (Amorim et al., 2019). Its abundant and widespread distribution in temperate limnic systems and its low dispersal ability can be leveraged to investigate the impact of pollutants in freshwater systems, in particular pesticides, molluscicides, algaecides or industrial (petro-) chemicals pollutants (Amorim et al., 2019). Adding temporally sampled genomic data would allow for controlling the genomic background of L. stagnalis population subjected to toxic stress and provide baselines prior to the use of pesticides and chemicals (Coutellec et al., 2013; Bouétard et al., 2014).
Integrating aDNA and environmental information provides new perspectives to better understand mollusk responses to global and local changes. These are expected to be complex, heterogeneous through time, space and across species. Many gaps still remain in our knowledge of the principles and mechanisms driving these responses; for example, the relative contribution of migration (routes and speed) and in situ tolerance in the form of phenotypic plasticity (morphology, behavior, ecophysiology, reproductive strategies) or evolutionary adaptation (speciation, selection) to the survival of species facing environmental changes (e.g., climate, pollution) of varying magnitude and rate.
Mytilus, Haliotis, Crassostrea, and Ruditapes species represented 73% (8,816,367 tons) of the world marine mollusk production in 2017 (FAO, 2017). Their production can be managed in large-scale hatcheries and has resulted in massive translocation across large geographical scales. For example, Haliotis were commercially fished in Europe in the 19th century, which led to drastic wild stock depletions and the implementation of management plans and regulations for fishing and cultivation (Huchette and Clavier, 2004). European Crassostrea has had a distinct history, as the Portuguese oyster C. angulata was first introduced unintentionally from Asia, probably in the 16th century, and cultivated until it underwent high mortalities associated with iridovirus outbreaks in the 1960–1970s (Grizel and Héral, 1991). It was then substituted by the more resistant Pacific cupped oyster sister species C. gigas with distributions in Asia overlapping those of native C. angulata (Grizel and Héral, 1991). Haliotis and Crassostrea in North America have similar histories of overfishing and stock depletion across the 19 and 20th centuries (Braje et al., 2009, 2016; Rick et al., 2016). As for the Manila clam R. philippinarum, it has a natural distribution on the West coast of the Pacific from the Philippines to Russia. According to historical records, R. philippinarum was first unintentionally introduced from Japan to North America through oyster “hitchhiking” in 1936 C.E. (Quayle, 1964), and then imported from there into Europe partly to overcome the over-exploitation, irregular yields, recruitment failures, and outbreak of bacterial infections in the endemic R. decussatus in 1972–1974 C.E. (Flassch and Leborgne, 1992; Sanna et al., 2017). Human-driven displacements, intense harvest, over-exploitation, and controlled reproduction in hatchery are expected to have impacted exploited mollusk species, in particular with regards to genomic patterns of diversity (e.g., inbreeding) and/or admixture (Astorga, 2014), as well as allele frequencies for loci associated with economically important phenotypic traits (e.g., survival, growth rate, body weight, and yield) (Gutierrez et al., 2018). These could be tracked through time using the same mollusk shell aDNA approach as applied here, which could also potentially reveal the existence of undocumented transfers.
Additionally, as human-mediated transfers may subject farmed mollusks to new pathogens, mollusk shell aDNA could also help track and monitor the spread of infections. For example, in R. philippinarum, high mortalities were observed in 1987 C.E. in Brittany from the so-called “Brown Ring Disease” (BRD) for which the bacteria Vibrio tapetis was identified as the etiological agent (Paillard et al., 1989, 2008; Borrego et al., 1996; Allam et al., 2000). DNA recovered from the shells of R. philippinarum showing substantial brown conchiolin deposits diagnostic of acute BRD (shells labeled as POSp and KORp in the present study; Supplementary Figure S1) was previously found in Der Sarkissian et al., 2017 to show highest affinity for the virulent V. tapetis strain RP2-3, whereas DNA from shells displaying weak BRD infection (LAN1p, LAN2p, and LAN3p) showed highest affinity for the less virulent V. tapetis strain HH6087 (Der Sarkissian et al., 2017; Dias et al., 2018). Here, phylogenetic analyses of the R. philippinarum COI mitochondrial gene in specimens from Landéda, Brittany, reveal a segregation between the 1988 C.E. acute BRD specimen POSp falling within the mainly Asian diversity (China, Japan), and the 1983–1988 C.E. asymptomatic or weak BRD specimens NEG, LAN1p, LAN2p, and LAN3p closely related to mainly European and American individuals (Supplementary Figure S1). These results are in line with a tentative scenario, in which lower virulence V. tapetis strains may have already been present in Brittany before the 1987 C.E. virulent BRD outbreak, which might have been triggered by a second introduction of R. philippinarum stocks, possibly of more recent Asian origin, containing highly virulent strains. The presence of the 2003 C.E. KORp shell in Korea showing acute BRD and a COI sequence closely related to European and American clams could possibly reflect a later transfer from Europe to Korea, where BRD was first identified by molecular methods in 2006 C.E. (Park et al., 2006), and where stocks have underwent severe reductions due to overexploitation and coastal pollution (Cordero et al., 2017).
In a context of global trade, human activities greatly facilitate rapid species displacements over large geographical distances, whether it be intentionally (e.g., for cultivation in hatcheries or direct commercial purposes) or unintentionally (e.g., through waterways, ship and seaplane hull fouling, ballast waters, fishing) (Carlton, 1999). Thanks to a competitive advantage (e.g., long larval phases, rapid growth, early sexual maturity, high fecundity, broad salinity, and temperature tolerance), newly introduced species can proliferate from a point of introduction into new ecosystems in which they become dominant as invasive species (Valéry et al., 2008). Biological invasions are recognized as the second most important threat to biodiversity after habitat destruction due to their substantial negative ecological (e.g., food web alteration and extirpation of native species) and economic impacts [e.g., biofouling, blocking of water pipes (McCarthy et al., 1997)].
Belonging to the mollusk genera examined here, C. gigas and R. philippinarum threaten the European native species of Ostrea edulis and R. decussatus, respectively, and M. galloprovincialis has been listed as one of the “100 world’s worst invasive alien species” (Lowe et al., 2004). D. polymorpha is another well-identified invasive species for which shell aDNA could be retrieved. Today, D. polymorpha has a worldwide distribution and its invasive status is a recognized threat (Lowe et al., 2004). It initially spread from the Ponto-Caspian Basin to Europe in the mid 1800s (or before), and then reached North America from Europe in the mid 1980s (Kinzelbach, 1986; Carlton, 1999). Although it is one of the most studied invasive species with regards to ecological, toxicological and physiological aspects, population genomic data are currently scarce. Early detection of invasive species is of foremost importance in efforts to eradicate them, or at least slow their spread down (Williams et al., 2017). Designing management plans for the control of invasions can greatly benefit from the reconstruction of their history: geographical origin, number, and timing of the introduction(s), as well as dispersal and connectivity patterns. This can be informed by phylogeography in both the native and invaded areas. The timing of invasions, however, can be difficult to estimate with precision. One reason for this is that some mollusk (cryptic) invasive and native species show similar shell morphology due to their close relationships and phenotypic plasticity (Morais and Reichard, 2018). Some of the local species may also be impacted by hybridization with invasive sister species in semi-reproductive isolation (Harrison and Larson, 2014), which is facilitated by high fecundity and dispersal potential in broadcast-spawning aquatic mollusks Dreissena, Crassostrea, and Mytilus species (Voroshilova et al., 2010; Fraïsse et al., 2016; Gagnaire et al., 2018). Mollusk shell DNA is a useful tool to assess the presence and genomic landscape of invaders at given points in time and space. Considering the dynamic and stochastic nature of biological invasions, shell aDNA could help evaluate their impact on native populations and ecosystems, thus showing potential for future studies in ecology and conservation.
A total of 1,526 species of gastropods and bivalves are classified as threatened by The IUCN Red List of Threatened Species (2019). Among them, two species of Haliotis abalones of the North American Pacific coast have been classed as “endangered,” i.e., H. kamtschatkana, and “critically endangered,” i.e., H. cracherodii or black abalone. The latter was shown here to be amenable to aDNA analyses, similarly to another Pacific abalone species, H. rufescens or red abalone. Both species were found in shell middens and in artifacts at prehistoric Native American sites (Erlandson and Rick, 2008; Braje et al., 2009). H. cracherodii experienced critical population declines following intensive fishing in the 1850-1900s and then again after the peak in abalone farming of the 1950–1960s, which severely affected all abalone populations in the Pacific, including H. rufescens (Rogers-Bennett et al., 2002). Abalone population declines were aggravated by the recovery of sea otters (Enhydra lutris nereis and E. l. kenyoni) in the 20th century, abalone predators that had nearly gone extinct following overexploitation by the historical fur trade (Lee et al., 2016). High mortalities due to infections by the bacterium Candidatus xenohaliotis responsible for the Withering syndrome devastated abalone populations (Crosson et al., 2014) and contributed to the low genetic diversities and population differentiations observed in H. cracherodii and H. rufescens today (Gaffney et al., 1996; Burton and Tegner, 2000; Hamm and Burton, 2000). Conservation efforts are focused in restoring and protecting abalones in the Pacific: shore picking of H. rufescens is allowed only under strict rules, and fishing of H. cracherodii (commercial or recreational) have been suspended (Haas et al., 2019). Management and conservation plans would greatly benefit from shell aDNA analyses that could help better understand the dynamics of genomic diversity and distribution for the two species, the effects of exploitation, infection and predator recovery, and complement archeological studies aimed at helping in abalone restoration (Braje et al., 2009, 2016; Hofman et al., 2015).
In this study, we illustrate the potential of ancient mollusk shell mitochondrial and nuclear DNA analyses using geological, archeological and museum specimens from 13 species, spanning ≥100 kyr. Ancient mollusk DNA is of growing relevance for ecological studies as present-day societies are increasingly concerned by on-going global environmental changes (e.g., warming, pollution, sea level rise, ocean acidification, eutrophication) and associated biodiversity loss (Cardinale et al., 2012) continue to increase at an alarming pace. Some environmental stressors are expected to influence directly shell-producing mollusks in their ability to calcify, and hence, their integrity and vulnerability to predators (Kroeker et al., 2013), with some of the affected species playing the role of keystone species with large impacts on ecosystems. Our work highlights the importance of mollusk shell aDNA as a powerful and novel tool for ecological studies.
The datasets generated for this study can be found in the European Nucleotide Archive public database (project PRJEB35671).
Ethical review and approval was not required for this study because no living animals were collected or examined.
CD and LO conceived and coordinated the study. CD performed molecular and computational analyses, with input from LO. PM, CH, PI, TR, MS, TS, LD, and LO provided samples and reagents. CD wrote the article with input from PM, LD, and LO.
This work was supported by the Danish Council for Independent Research, the Natural Sciences (FNU, 4002-00152B), the Danish National Research Foundation (DNRF94), and the Initiative d’Excellence Chaires d’attractivité, Université de Toulouse (OURASI). LO has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programe (Grant agreement no. 681605-PEGASUS). PM and LD received funding from the Swedish Natural Science Research Council (VR) (Grant agreements nos. 621-2008-3759 and 621-2008-4896, respectively), and expedition logistics, including for the Taimyr 2010 expedition, from the Swedish Polar Research Secretariat (SPRS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Martin Sikora at The GLOBE Institute, University of Copenhagen, for transporting samples. We also thank the PALEOMIX group at the University of Copenhagen and the AGES group at the Laboratory AMIS UMR 5288 (CNRS, University Toulouse 3), as well as the staff of the Danish National High-Throughput DNA Sequencing Centre and of AMIS for support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00037/full#supplementary-material
Allam, B., Paillard, C., Howard, A., and Le Pennec, M. (2000). Isolation of the pathogen Vibrio tapetis and defense parameters in brown ring diseased Manila clams Ruditapes philippinarum cultivated in England. Dis. Aquat. Organ. 41, 105–113. doi: 10.3354/dao041105
Amorim, J., Abreu, I., Rodrigues, P., Peixoto, D., Pinheiro, C., Saraiva, A., et al. (2019). Lymnaea stagnalis as a freshwater model invertebrate for ecotoxicological studies. Sci. Total Environ. 669, 11–28. doi: 10.1016/j.scitotenv.2019.03.035
Armbrecht, L. H., Coolen, M. J. L., Lejzerowicz, F., George, S. C., Negandhi, K., Suzuki, Y., et al. (2019). Ancient DNA from marine sediments: precautions and considerations for seafloor coring, sample handling and data generation. Earth Sci. Rev. 196:102887. doi: 10.1016/j.earscirev.2019.102887
Astorga, M. P. (2014). Genetic considerations for mollusk production in aquaculture: current state of knowledge. Front. Genet. 5:435. doi: 10.3389/fgene.2014.00435
Beldade, R., Bell, C. A., Raimondi, P. T., George, M. K., Miner, C. M., and Bernardi, G. (2012). Isolation and characterization of 8 novel microsatellites for the black abalone, Haliotis cracherodii, a marine gastropod decimated by the withering disease. Conserv. Genet. Resour. 4, 1071–1073. doi: 10.1007/s12686-012-9709-3
Black, H. D., Andrus, C. F. T., Lambert, W. J., Rick, T. C., and Gillikin, D. P. (2017). δ15N values in Crassostrea virginica shells provides early direct evidence for nitrogen loading to Chesapeake bay. Sci. Rep. 7:44241. doi: 10.1038/srep44241
Boere, A. C., Abbas, B., Rijpstra, W. I. C., Versteegh, G. J. M., Volkman, J. K., Damsté, J. S. S., et al. (2009). Late-Holocene succession of dinoflagellates in an Antarctic fjord using a multi-proxy approach: paleoenvironmental genomics, lipid biomarkers and palynomorphs. Geobiology 7, 265–281. doi: 10.1111/j.1472-4669.2009.00202.x
Borrego, J. J., Castro, D., Luque, A., Paillard, C., Maes, P., Garcia, M. T., et al. (1996). Vibrio tapetis sp. nov., the causative agent of the brown ring disease affecting cultured clams. Int. J. Syst. Evol. Microbiol. 46, 480–484. doi: 10.1099/00207713-46-2-480
Borrell, Y. J., Arias-Pérez, A., Freire, R., Valdés, A., Sánchez, J. A., Méndez, J., et al. (2014). Microsatellites and multiplex PCRs for assessing aquaculture practices of the grooved carpet shell Ruditapes decussatus in Spain. Aquaculture 42, 49–59. doi: 10.1016/j.aquaculture.2014.01.010
Bouétard, A., Côte, J., Besnard, A.-L., Collinet, M., and Coutellec, M.-A. (2014). Environmental versus anthropogenic effects on population adaptive divergence in the freshwater snail Lymnaea stagnalis. PLoS One 9:e106670. doi: 10.1371/journal.pone.0106670
Braje, T. J., Erlandson, J. M., Rick, T. C., Dayton, P. K., and Hatch, M. B. A. (2009). Fishing from past to present: continuity and resilience of red abalone fisheries on the Channel Islands, California. Ecol. Appl. 19, 906–919. doi: 10.1890/08-0135.1
Braje, T. J., Rick, T. C., Erlandson, J. M., Rogers-Bennett, L., and Catton, C. A. (2016). Historical ecology can inform restoration site selection: the case of black abalone (Haliotis cracherodii) along California’s Channel Islands. Aquat. Conserv. Mar. Freshw. Ecosyst 26, 470–481. doi: 10.1002/aqc.2561
Breton, S., Burger, G., Stewart, D. T., and Blier, P. U. (2006). Comparative analysis of gender-associated complete mitochondrial genomes in marine mussels (Mytilus spp.). Genetics 172, 1107–1119. doi: 10.1534/genetics.105.047159
Burton, R. S., and Tegner, M. J. (2000). Enhancement of red abalone Haliotis rufescens stocks at San Miguel Island: reassessing a success story. Mar. Ecol. Prog. Ser. 202, 303–308. doi: 10.3354/meps202303
Bush, A., Compson, Z. G., Monk, W. A., Porter, T. M., Steeves, R., Emilson, E., et al. (2019). Studying ecosystems with DNA metabarcoding: lessons from biomonitoring of aquatic macroinvertebrates. Front. Ecol. Evol. 7:434. doi: 10.3389/fevo.2019.00434
Butler, P. G., Freitas, P. S., Burchell, M., and Chauvaud, L. (2019). “Archaeology and sclerochronology of marine bivalves,” in Goods and Services of Marine Bivalves, eds A. C. Smaal, J. G. Ferreira, J. Grant, J. K. Petersen, and Ø. Strand , (Cham: Springer), 413–444. doi: 10.1007/978-3-319-96776-9_21
Cardinale, B. J., Duffy, J. E., Gonzalez, A., Hooper, D. U., Perrings, C., Venail, P., et al. (2012). Biodiversity loss and its impact on humanity. Nature 486, 59–67. doi: 10.1038/nature11148
Carlton, J. (1999). Molluscan invasions in marine and estuarine communities. Malacologia 41, 439–454.
Chauvaud, L., Lorrain, A., Dunbar, R. B., Paulet, Y.-M., Thouzeau, G., Jean, F., et al. (2005). Shell of the great scallop Pecten maximus as a high-frequency archive of paleoenvironmental changes. Geochem. Geophys. Geosyst. 6:Q08001. doi: 10.1029/2004GC000890
Chauvaud, L., Patry, Y., Jolivet, A., Cam, E., Goff, C. L., Strand, Ø., et al. (2012). Variation in size and growth of the great scallop Pecten maximus along a latitudinal gradient. PLoS One 7:e37717. doi: 10.1371/journal.pone.0037717
Coolen, M. J. L., Orsi, W. D., Balkema, C., Quince, C., Harris, K., Sylva, S. P., et al. (2013). Evolution of the plankton paleome in the Black Sea from the deglacial to anthropocene. Proc. Natl. Acad. Sci. U.S.A. 110, 8609–8614. doi: 10.1073/pnas.1219283110
Cordero, D., Delgado, M., Liu, B., Ruesink, J., and Saavedra, C. (2017). Population genetics of the Manila clam (Ruditapes philippinarum) introduced in North America and Europe. Sci. Rep. 7:39745. doi: 10.1038/srep39745
Cordero, D., Peña, J. B., and Saavedra, C. (2014). Phylogeographic analysis of introns and mitochondrial DNA in the clam Ruditapes decussatus uncovers the effects of Pleistocene glaciations and endogenous barriers to gene flow. Mol. Phylogenet. Evol. 71, 274–287. doi: 10.1016/j.ympev.2013.11.003
Coutellec, M.-A. (2017). Mollusc shells as metagenomic archives: The true treasure is the chest itself. Mol. Ecol. Resour. 17, 854–857. doi: 10.1111/1755-0998.12716
Coutellec, M.-A., Besnard, A.-L., and Caquet, T. (2013). Population genetics of Lymnaea stagnalis experimentally exposed to cocktails of pesticides. Ecotoxicology 22, 879–888. doi: 10.1007/s10646-013-1082-9
Crosson, L. M., Wight, N., VanBlaricom, G. R., Kiryu, I., Moore, J. D., and Friedman, C. S. (2014). Abalone withering syndrome: distribution, impacts, current diagnostic methods and new findings. Dis. Aquat. Organ. 108, 261–270. doi: 10.3354/dao02713
Cruz, P., Ibarra, A. M., Fiore-Amaral, G., Galindo-Sánchez, C. E., and Mendoza-Carrión, G. (2005). Isolation of microsatellite loci in green abalone (Haliotis fulgens) and cross-species amplification in two other North American red (Haliotis rufescens) and pink (Haliotis corrugata) abalones. Mol. Ecol. Notes 5, 857–859. doi: 10.1111/j.1471-8286.2005.01088.x
d’Auriac, M. B. A., Rinde, E., Norling, P., Lapègue, S., Staalstrøm, A., Hjermann, Ø., et al. (2017). Rapid expansion of the invasive oyster Crassostrea gigas at its northern distribution limit in Europe: Naturally dispersed or introduced? PLoS One 12:e0177481. doi: 10.1371/journal.pone.0177481
Der Sarkissian, C., Allentoft, M. E., Ávila-Arcos, M. C., Barnett, R., Campos, P. F., Cappellini, E., et al. (2015). Ancient genomics. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 370:20130387. doi: 10.1098/rstb.2013.0387
Der Sarkissian, C., Pichereau, V., Dupont, C., Ilsøe, P. C., Perrigault, M., Butler, P., et al. (2017). Ancient DNA analysis identifies marine mollusc shells as new metagenomic archives of the past. Mol. Ecol. Resour. 17, 835–853. doi: 10.1111/1755-0998.12679
Dias, G. M., Bidault, A., Le Chevalier, P., Choquet, G., Der Sarkissian, C., Orlando, L., et al. (2018). Vibrio tapetis displays an original type IV secretion system in strains pathogenic for bivalve molluscs. Front. Microbiol. 9:227. doi: 10.3389/fmicb.2018.00227
El Ayari, T., Trigui El Menif, N., Hamer, B., Cahill, A. E., and Bierne, N. (2019). The hidden side of a major marine biogeographic boundary: a wide mosaic hybrid zone at the Atlantic-Mediterranean divide reveals the complex interaction between natural and genetic barriers in mussels. Heredity 122, 770–784. doi: 10.1038/s41437-018-0174-y
Erlandson, J. M., and Rick, T. C. (2008). “Archaeology, marine ecology, and human impacts on marine environments,” in Human Impacts on Ancient Marine Ecosystems: A Global Perspective, eds T. C. Rick, and J. M. Erlandson, (Berkeley, CA: University of California Press), 1–19.
Fages, A., Hanghøj, K., Khan, N., Gaunitz, C., Seguin-Orlando, A., Leonardi, M., et al. (2019). Tracking five millennia of horse management with extensive ancient genome time series. Cell 177, 1419–1435.e31. doi: 10.1016/j.cell.2019.03.049
FAO (2017). Fishery and Aquaculture Statistics. Aquacultre Production. Available at: http://www.fao.org/fishery/static/Yearbook/YB2017_USBcard/root/aquaculture/yearbook_aquaculture.pdf (accessed February 17, 2020).
Feldheim, K. A., Brown, J. E., Murphy, D. J., and Stepien, C. A. (2011). Microsatellite loci for dreissenid mussels (Mollusca: Bivalvia: Dreissenidae) and relatives: markers for assessing exotic and native populations. Mol. Ecol. Resour. 11, 725–732. doi: 10.1111/j.1755-0998.2011.03012.x
Flassch, J.-P., and Leborgne, Y. (1992). Introduction in Europe, from 1972 to 1980, of the Japanese Manila clam (Tapes philippinarum) and the effects on aquaculture production and natural settlement. ICES Mar. Sci. Symp. 194, 92–96.
Fortunato, H. (2015). Mollusks: tools in environmental and climate research. Am. Malacol. Bull. 33, 310–324. doi: 10.4003/006.033.0208
Fraïsse, C., Belkhir, K., Welch, J. J., and Bierne, N. (2016). Local interspecies introgression is the main cause of extreme levels of intraspecific differentiation in mussels. Mol. Ecol. 25, 269–286. doi: 10.1111/mec.13299
Fraïsse, C., Haguenauer, A., Gérard, K., Weber, A. A.-T., Bierne, N., and Chenuil, A. (2017). Fine-grained genetic-environment association in an admixed population of mussels in the small isolated Kerguelen island. bioRxiv [Preprint]. doi: 10.1101/239244
Gaffney, P. M., Rubin, V. P., Hedgecock, D., Powers, D. A., Morris, G., and Hereford, L. (1996). Genetic effects of artificial propagation: signals from wild and hatchery populations of red abalone in California. Aquaculture 143, 257–266. doi: 10.1016/0044-8486(96)01278-1
Gagnaire, P.-A., Lamy, J.-B., Cornette, F., Heurtebise, S., Dégremont, L., Flahauw, E., et al. (2018). Analysis of genome-wide differentiation between native and introduced populations of the cupped oysters Crassostrea gigas and Crassostrea angulata. Genome Biol. Evol. 10, 2518–2534. doi: 10.1093/gbe/evy194
Gamba, C., Hanghøj, K., Gaunitz, C., Alfarhan, A. H., Alquraishi, S. A., Al-Rasheid, K. A. S., et al. (2016). Comparing the performance of three ancient DNA extraction methods for high-throughput sequencing. Mol. Ecol. Resour. 16, 459–469. doi: 10.1111/1755-0998.12470
Gamba, C., Jones, E. R., Teasdale, M. D., McLaughlin, R. L., Gonzalez-Fortes, G., Mattiangeli, V., et al. (2014). Genome flux and stasis in a five millennium transect of European prehistory. Nat. Commun. 5:5257. doi: 10.1038/ncomms6257
Gardeström, J., Pereyra, R. T., and André, C. (2008). Characterization of six microsatellite loci in the Baltic blue mussel Mytilus trossulus and cross-species amplification in North Sea Mytilus edulis. Conserv. Genet. 9, 1003–1005. doi: 10.1007/s10592-007-9432-x
Geist, J., Wunderlich, H., and Kuehn, R. (2008). Use of mollusc shells for DNA-based molecular analyses. J. Molluscan Stud. 74, 337–343. doi: 10.1093/mollus/eyn025
Ghiselli, F., Milani, L., Iannello, M., Procopio, E., Chang, P. L., Nuzhdin, S. V., et al. (2017). The complete mitochondrial genome of the grooved carpet shell, Ruditapes decussatus (Bivalvia, Veneridae). PeerJ 5:e3692. doi: 10.7717/peerj.3692
Giguet-Covex, C., Pansu, J., Arnaud, F., Rey, P.-J., Griggo, C., Gielly, L., et al. (2014). Long livestock farming history and human landscape shaping revealed by lake sediment DNA. Nat. Commun. 5:3211. doi: 10.1038/ncomms4211
Gilbert, M. T. P., Drautz, D. I., Lesk, A. M., Ho, S. Y. W., Qi, J., Ratan, A., et al. (2008). Intraspecific phylogenetic analysis of Siberian woolly mammoths using complete mitochondrial genomes. Proc. Natl. Acad. Sci. U.S.A. 105, 8327–8332. doi: 10.1073/pnas.0802315105
Glöckner, G., Heinze, I., Platzer, M., Held, C., and Abele, D. (2013). The mitochondrial genome of Arctica islandica; phylogeny and variation. PLoS One 8:e82857. doi: 10.1371/journal.pone.0082857
Grade, A., Chairi, H., Lallias, D., Power, D. M., Ruano, F., Leitão, A., et al. (2016). New insights about the introduction of the Portuguese oyster, Crassostrea angulata, into the North East Atlantic from Asia based on a highly polymorphic mitochondrial region. Aquat. Living Resour. 29:404. doi: 10.1051/alr/2016035
Grealy, A. C., McDowell, M. C., Scofield, P., Murray, D. C., Fusco, D. A., Haile, J., et al. (2015). A critical evaluation of how ancient DNA bulk bone metabarcoding complements traditional morphological analysis of fossil assemblages. Quat. Sci. Rev. 128, 37–47. doi: 10.1016/j.quascirev.2015.09.014
Green, E. J., and Speller, C. F. (2017). Novel substrates as sources of ancient DNA: prospects and hurdles. Genes 8:180. doi: 10.3390/genes8070180
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., et al. (2010). A draft sequence of the Neandertal genome. Science 328, 710–722. doi: 10.1126/science.1188021
Grizel, H., and Héral, M. (1991). Introduction into France of the Japanese oyster (Crassostrea gigas). ICES J. Mar. Sci. 47, 399–403. doi: 10.1093/icesjms/47.3.399
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gutierrez, A. P., Matika, O., Bean, T. P., and Houston, R. D. (2018). Genomic selection for growth traits in pacific oyster (Crassostrea gigas): potential of low-density marker panels for breeding value prediction. Front. Genet. 9:391. doi: 10.3389/fgene.2018.00391
Gutierrez, A. P., Turner, F., Gharbi, K., Talbot, R., Lowe, N. R., Peñaloza, C., et al. (2017). Development of a medium density combined-species SNP array for pacific and European oysters (Crassostrea gigas and Ostrea edulis). G3 7, 2209–2218. doi: 10.1534/g3.117.041780
Haas, H., Braje, T. J., Edwards, M. S., Erlandson, J. M., and Whitaker, S. G. (2019). Black abalone (Haliotis cracherodii) population structure shifts through deep time: management implications for southern California’s northern Channel Islands. Ecol. Evol. 9, 4720–4732. doi: 10.1002/ece3.5075
Hamm, D. E., and Burton, R. S. (2000). Population genetics of black abalone, Haliotis cracherodii, along the central California coast. J. Exp. Mar. Biol. Ecol. 254, 235–247. doi: 10.1016/S0022-0981(00)00283-5
Harney, E., Lachambre, S., Roussel, S., Huchette, S., Enez, F., Morvezen, R., et al. (2018). Transcriptome based SNP discovery and validation for parentage assignment in hatchery progeny of the European abalone Haliotis tuberculata. Aquaculture 491, 105–113. doi: 10.1016/j.aquaculture.2018.03.006
Harrison, R. G., and Larson, E. L. (2014). Hybridization, introgression, and the nature of species boundaries. J. Hered. 105, 795–809. doi: 10.1093/jhered/esu033
Hiebenthal, C., Philipp, E. E. R., Eisenhauer, A., and Wahl, M. (2012). Interactive effects of temperature and salinity on shell formation and general condition in Baltic Sea Mytilus edulis and Arctica islandica. Aquat. Biol. 14, 289–298. doi: 10.3354/ab00405
Hofman, C. A., Rick, T. C., Fleischer, R. C., and Maldonado, J. E. (2015). Conservation archaeogenomics: ancient DNA and biodiversity in the anthropocene. Trends Ecol. Evol. 30, 540–549. doi: 10.1016/j.tree.2015.06.008
Huchette, S. M. H., and Clavier, J. (2004). Status of the ormer (Haliotis tuberculata L.) industry in Europe. J. Shellfish Res. 23, 951–956.
Innes, D. J., and Bates, J. A. (1999). Morphological variation of Mytilus edulis and Mytilus trossulus in eastern Newfoundland. Mar. Biol. 133, 691–699. doi: 10.1007/s002270050510
Jackson, J. B. C. (2001). What was natural in the coastal oceans? Proc. Natl. Acad. Sci. U.S.A. 98, 5411–5418. doi: 10.1073/pnas.091092898
Jiang, L., Nie, H., Li, C., Li, D., Huo, Z., and Yan, X. (2018). The genetic diversity of wild and cultivated Manila clam (Ruditapes philippinarum) revealed by 29 novel microsatellite markers. Electron. J. Biotechnol. 34, 17–21. doi: 10.1016/j.ejbt.2018.05.003
Jones, P. D., Briffa, K. R., Osborn, T. J., Lough, J. M., van Ommen, T. D., Vinther, B. M., et al. (2009). High-resolution palaeoclimatology of the last millennium: a review of current status and future prospects. The Holocene 19, 3–49. doi: 10.1177/0959683608098952
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F., and Orlando, L. (2013). mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Kinzelbach, R. (1986). The recent distribution of the Zebra Mussel, Dreissena polymorpha, in the Aegean Region and in Anatolia. Zool. Middle East 1, 132–138. doi: 10.1080/09397140.1986.10637537
Knott, K. E., Puurtinen, M., and Kaitala, V. (2003). Primers for nine microsatellite loci in the hermaphroditic snail Lymnaea stagnalis. Mol. Ecol. Notes 3, 333–335. doi: 10.1046/j.1471-8286.2003.00444.x
Korneliussen, T. S., Albrechtsen, A., and Nielsen, R. (2014). ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356. doi: 10.1186/s12859-014-0356-4
Kroeker, K. J., Kordas, R. L., Crim, R., Hendriks, I. E., Ramajo, L., Singh, G. S., et al. (2013). Impacts of ocean acidification on marine organisms: quantifying sensitivities and interaction with warming. Glob. Change Biol. 19, 1884–1896. doi: 10.1111/gcb.12179
Lartaud, F., Emmanuel, L., de Rafaelis, M., Pouvreau, S., and Renard, M. (2010). Influence of food supply on the δ13C signature of mollusc shells: implications for palaeoenvironmental reconstitutions. Geomar. Lett. 30, 23–34. doi: 10.1007/s00367-009-0148-4
Lee, L. C., Watson, J. C., Trebilco, R., and Salomon, A. K. (2016). Indirect effects and prey behavior mediate interactions between an endangered prey and recovering predator. Ecosphere 7:e01604. doi: 10.1002/ecs2.1604
Lefort, V., Longueville, J.-E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Leigh, J. W., and Bryant, D. (2015). popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Lejzerowicz, F., Esling, P., Majewski, W., Szczuciński, W., Decelle, J., Obadia, C., et al. (2013). Ancient DNA complements microfossil record in deep-sea subsurface sediments. Biol. Lett. 9:20130283. doi: 10.1098/rsbl.2013.0283
Leonardi, M., Librado, P., Der Sarkissian, C., Schubert, M., Alfarhan, A. H., Alquraishi, S. A., et al. (2016). Evolutionary patterns and processes: lessons from ancient DNA. Syst. Biol. 66, e1–e29. doi: 10.1093/sysbio/syw059
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Liehr, G. A., Zettler, M. L., Leipe, T., and Witt, G. (2005). The ocean quahog Arctica islandica L.: a bioindicator for contaminated sediments. Mar. Biol. 147, 671–679. doi: 10.1007/s00227-005-1612-y
Lorenzen, E. D., Nogués-Bravo, D., Orlando, L., Weinstock, J., Binladen, J., Marske, K. A., et al. (2011). Species-specific responses of Late Quaternary megafauna to climate and humans. Nature 479, 359–364. doi: 10.1038/nature10574
Lowe, S., Browne, M., Boudjelas, S., De Poorter, M., and The Invasive Species Specialist Group, (2004). 100 of the World’s Worst Invasive Alien Species: A Selection from the Global Invasive Species Database. The Invasive Species Specialist Group.
Löytynoja, A., and Goldman, N. (2005). An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. U.S.A. 102, 10557–10562. doi: 10.1073/pnas.0409137102
Lydeard, C., Cowie, R. H., Ponder, W. F., Bogan, A. E., Bouchet, P., Clark, S. A., et al. (2004). The global decline of nonmarine mollusks. Bioscience 54, 321–330.
Mann, M. E., Bradley, R. S., and Hughes, M. K. (1999). Northern hemisphere temperatures during the past millennium: inferences, uncertainties, and limitations. Geophys. Res. Lett. 26, 759–762. doi: 10.1029/1999GL900070
Mann, M. E., Zhang, Z., Rutherford, S., Bradley, R. S., Hughes, M. K., Shindell, D., et al. (2009). Global signatures and dynamical origins of the little ice age and medieval climate anomaly. Science 326, 1256–1260. doi: 10.1126/science.1177303
Marcott, S. A., Shakun, J. D., Clark, P. U., and Mix, A. C. (2013). A reconstruction of regional and global temperature for the past 11,300 years. Science 339, 1198–1201. doi: 10.1126/science.1228026
Masonbrink, R. E., Purcell, C. M., Boles, S. E., Whitehead, A., Hyde, J. R., Seetharam, A. S., et al. (2019). An Annotated Genome for Haliotis rufescens (Red Abalone) and resequenced green, pink, pinto, black, and white abalone species. Genome Biol. Evol. 11, 431–438. doi: 10.1093/gbe/evz006
Mathiesen, S. S., Thyrring, J., Hemmer-Hansen, J., Berge, J., Sukhotin, A., Leopold, P., et al. (2016). Genetic diversity and connectivity within Mytilus spp. in the subarctic and Arctic. Evol. Appl. 10, 39–55. doi: 10.1111/eva.12415
McCarthy, T. K., Fitzgerald, J., and O’Connor, W. (1997). The occurrence of the Zebra mussel Dreissena polymorpha (Pallas, 1771), an introduced biofouling freshwater bivalve in Ireland. Ir. Nat. J. 25, 413–416.
Meisner, J., and Albrechtsen, A. (2018). Inferring population structure and admixture proportions in low-depth NGS data. Genetics 210, 719–731. doi: 10.1534/genetics.118.301336
Möller, P., Alexanderson, H., Funder, S., and Hjort, C. (2015). The Taimyr Peninsula and the Severnaya Zemlya archipelago, Arctic Russia: a synthesis of glacial history and palaeo-environmental change during the Last Glacial cycle (MIS 5e–2). Quat. Sci. Rev. 107, 149–181. doi: 10.1016/j.quascirev.2014.10.018
Möller, P., Benediktsson, Í. Ö., Anjar, J., Bennike, O., Bernhardson, M., Funder, S., et al. (2019a). Data set on sedimentology, palaeoecology and chronology of middle to late pleistocene deposits on the Taimyr Peninsula, Arctic Russia. Data Brief 25:104267. doi: 10.1016/j.dib.2019.104267
Möller, P., Benediktsson, Í. Ö., Anjar, J., Bennike, O., Bernhardson, M., Funder, S., et al. (2019b). Glacial history and palaeo-environmental change of southern Taimyr Peninsula, Arctic Russia, during the Middle and Late Pleistocene. Earth Sci. Rev. 196:102832. doi: 10.1016/j.earscirev.2019.04.004
Morais, P., and Reichard, M. (2018). Cryptic invasions: a review. Sci. Total Environ. 613–614, 1438–1448. doi: 10.1016/j.scitotenv.2017.06.133
Morard, R., Lejzerowicz, F., Darling, K. F., Lecroq-Bennet, B., Winther Pedersen, M., Orlando, L., et al. (2017). Planktonic foraminifera-derived environmental DNA extracted from abyssal sediments preserves patterns of plankton macroecology. Biogeosciences 14, 2741–2754. doi: 10.5194/bg-14-2741-2017
More, K. D., Orsi, W. D., Galy, V., Giosan, L., He, L., Grice, K., et al. (2018). A 43 kyr record of protist communities and their response to oxygen minimum zone variability in the Northeastern Arabian Sea. Earth Planet. Sci. Lett. 496, 248–256. doi: 10.1016/j.epsl.2018.05.045
Morvezen, R., Cornette, F., Charrier, G., Guinand, B., Lapegue, S., Boudry, P., et al. (2013). Multiplex PCR sets of novel microsatellite loci for the great scallop Pecten maximus and their application in parentage assignment. Aquat. Living Resour. 26, 207–213. doi: 10.1051/alr/2013052
Murray, D. C., Haile, J., Dortch, J., White, N. E., Haouchar, D., Bellgard, M. I., et al. (2013). Scrapheap Challenge: A novel bulk-bone metabarcoding method to investigate ancient DNA in faunal assemblages. Sci. Rep. 3:3371. doi: 10.1038/srep03371
Narasimhan, V. M., Patterson, N., Moorjani, P., Rohland, N., Bernardos, R., Mallick, S., et al. (2019). The formation of human populations in South and Central Asia. Science 365:eaat7487. doi: 10.1126/science.aat7487
Nielsen, R., Akey, J. M., Jakobsson, M., Pritchard, J. K., Tishkoff, S., and Willerslev, E. (2017). Tracing the peopling of the world through genomics. Nature 541, 302–310. doi: 10.1038/nature21347
Orlando, L., and Cooper, A. (2014). Using ancient DNA to understand evolutionary and ecological processes. Annu. Rev. Ecol. Evol. Syst. 45, 573–598. doi: 10.1146/annurev-ecolsys-120213-091712
Orlando, L., Ginolhac, A., Zhang, G., Froese, D., Albrechtsen, A., Stiller, M., et al. (2013). Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78. doi: 10.1038/nature12323
Paillard, C., Korsnes, K., Le Chevalier, P., Le Boulay, C., Harkestad, L., Eriksen, A. G., et al. (2008). Vibrio tapetis-like strain isolated from introduced Manila clams Ruditapes philippinarum showing symptoms of brown ring disease in Norway. Dis. Aquat. Organ. 81, 153–161. doi: 10.3354/dao01950
Paillard, C., Percelay, L., Le Pennec, M., and Le Picard, D. (1989). Origine pathogène de l’”anneau brun” chez Tapes philippinarum (Mollusque, bivalve). C. R. Acad. Sci. 309, 235–241.
Paillard, C., Roux, F. L., and Borrego, J. J. (2004). Bacterial disease in marine bivalves, a review of recent studies: trends and evolution. Aquat. Living Resour. 17, 477–498. doi: 10.1051/alr:2004054
Palkopoulou, E., Mallick, S., Skoglund, P., Enk, J., Rohland, N., Li, H., et al. (2015). Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth. Curr. Biol. 25, 1395–1400. doi: 10.1016/j.cub.2015.04.007
Park, K.-I., Paillard, C., Le Chevalier, P., and Choi, K.-S. (2006). Report on the occurrence of brown ring disease (BRD) in Manila clam, Ruditapes philippinarum, on the west coast of Korea. Aquaculture 255, 610–613. doi: 10.1016/j.aquaculture.2005.12.011
Paterno, M., Bat, L., Souissi, J. B., Boscari, E., Chassanite, A., Congiu, L., et al. (2019). A genome-wide approach to the phylogeography of the mussel Mytilus galloprovincialis in the Adriatic and the Black Seas. Front. Mar. Sci. 6:566. doi: 10.3389/fmars.2019.00566
Pawłowska, J., Lejzerowicz, F., Esling, P., Szczuciński, W., Zajączkowski, M., and Pawlowski, J. (2014). Ancient DNA sheds new light on the Svalbard foraminiferal fossil record of the last millennium. Geobiology 12, 277–288. doi: 10.1111/gbi.12087
Pečnerová, P., Díez-del-Molino, D., Dussex, N., Feuerborn, T., von Seth, J., van der Plicht, J., et al. (2017). Genome-based sexing provides clues about behavior and social structure in the woolly mammoth. Curr. Biol. 27, 3505–3510.e3. doi: 10.1016/j.cub.2017.09.064
Pedersen, J. S., Valen, E., Velazquez, A. M. V., Parker, B. J., Rasmussen, M., Lindgreen, S., et al. (2014). Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 24, 454–466. doi: 10.1101/gr.163592.113
Pedersen, M. W., Overballe-Petersen, S., Ermini, L., Sarkissian, C. D., Haile, J., Hellstrom, M., et al. (2015). Ancient and modern environmental DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370:20130383. doi: 10.1098/rstb.2013.0383
Pedersen, M. W., Ruter, A., Schweger, C., Friebe, H., Staff, R. A., Kjeldsen, K. K., et al. (2016). Postglacial viability and colonization in North America’s ice-free corridor. Nature 537, 45–49. doi: 10.1038/nature19085
Pérez-Mayol, S., Blasco, J., Tornero, V., Morales-Nin, B., Massanet, A., and Tovar-Sánchez, A. (2014). Are the shells of Scrobicularia plana useful for monitoring trace metal pollution events? J. Environ. Biol. Acad. Environ. Biol. India 35, 9–17.
Quayle, D. B. (1964). Distribution of introduced marine Mollusca in British Columbia waters. J. Fish. Res. Board Can. 21, 1155–1181. doi: 10.1139/f64-102
Renaud, G., Slon, V., Duggan, A. T., and Kelso, J. (2015). Schmutzi: estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol. 16:224. doi: 10.1186/s13059-015-0776-0
Reynolds, D. J., Scourse, J. D., Halloran, P. R., Nederbragt, A. J., Wanamaker, A. D., Butler, P. G., et al. (2016). Annually resolved North Atlantic marine climate over the last millennium. Nat. Commun. 7:13502. doi: 10.1038/ncomms13502
Rick, T. C., Reeder-Myers, L. A., Hofman, C. A., Breitburg, D., Lockwood, R., Henkes, G., et al. (2016). Millennial-scale sustainability of the Chesapeake Bay Native American oyster fishery. Proc. Natl. Acad. Sci. U.S.A. 113, 6568–6573. doi: 10.1073/pnas.1600019113
Riginos, C., and Cunningham, C. W. (2005). INVITED REVIEW: Local adaptation and species segregation in two mussel (Mytilus edulis × Mytilus trossulus) hybrid zones. Mol. Ecol. 14, 381–400. doi: 10.1111/j.1365-294X.2004.02379.x
Rogers-Bennett, L., Haaker, P. L., Huff, T. O., Dayton, P. K., and Hall, R. (2002). Estimating baseline abundances of abalone in California for restoration. CalCOFI Rep. 43, 97–111.
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S., and Reich, D. (2015). Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370:20130624. doi: 10.1098/rstb.2013.0624
Sadler, J., Carré, M., Azzoug, M., Schauer, A. J., Ledesma, J., Cardenas, F., et al. (2012). Reconstructing past upwelling intensity and the seasonal dynamics of primary productivity along the Peruvian coastline from mollusk shell stable isotopes. Geochem. Geophys. Geosyst. 13:Q01015. doi: 10.1029/2011GC003595
Sanna, D., Lai, T., Cossu, P., Scarpa, F., Dedola, G. L., Cristo, B., et al. (2017). Cytochrome c oxidase subunit I variability in Ruditapes decussatus (Veneridae) from the western Mediterranean. Eur. Zool. J. 84, 554–565. doi: 10.1080/24750263.2017.1395914
Schubert, M., Ermini, L., Der Sarkissian, C., Jónsson, H., Ginolhac, A., Schaefer, R., et al. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 9, 1056–1082. doi: 10.1038/nprot.2014.063
Schubert, M., Ginolhac, A., Lindgreen, S., Thompson, J. F., Al-Rasheid, K. A., Willerslev, E., et al. (2012). Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13:178. doi: 10.1186/1471-2164-13-178
Schubert, M., Lindgreen, S., and Orlando, L. (2016). AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 9:88. doi: 10.1186/s13104-016-1900-2
Seersholm, F. V., Cole, T. L., Grealy, A., Rawlence, N. J., Greig, K., Knapp, M., et al. (2018). Subsistence practices, past biodiversity, and anthropogenic impacts revealed by New Zealand-wide ancient DNA survey. Proc. Natl. Acad. Sci. U.S.A. 115, 7771–7776. doi: 10.1073/pnas.1803573115
Seguin-Orlando, A., Schubert, M., Clary, J., Stagegaard, J., Alberdi, M. T., Prado, J. L., et al. (2013). Ligation bias in illumina next-generation DNA libraries: implications for sequencing ancient genomes. PLoS One 8:e78575. doi: 10.1371/journal.pone.0078575
Skotte, L., Korneliussen, T. S., and Albrechtsen, A. (2013). Estimating individual admixture proportions from next generation sequencing data. Genetics 195, 693–702. doi: 10.1534/genetics.113.154138
Śmietanka, B., and Burzyński, A. (2017). Disruption of doubly uniparental inheritance of mitochondrial DNA associated with hybridization area of European Mytilus edulis and Mytilus trossulus in Norway. Mar. Biol. 164:209. doi: 10.1007/s00227-017-3235-5
Smietanka, B., Burzyński, A., and Wenne, R. (2010). Comparative genomics of marine mussels (Mytilus spp.) gender associated mtDNA: rapidly evolving atp8. J. Mol. Evol. 71, 385–400. doi: 10.1007/s00239-010-9393-4
Smietanka, B., Zbawicka, M., Sańko, T., Wenne, R., and Burzyński, A. (2013). Molecular population genetics of male and female mitochondrial genomes in subarctic Mytilus trossulus. Mar. Biol. 160, 1709–1721. doi: 10.1007/s00227-013-2223-7
Steinhardt, J., Butler, P. G., Carroll, M. L., and Hartley, J. (2016). The application of long-lived bivalve sclerochronology in environmental baseline monitoring. Front. Mar. Sci. 3:176. doi: 10.3389/fmars.2016.00176
Stephens, L., Fuller, D., Boivin, N., Rick, T., Gauthier, N., Kay, A., et al. (2019). Archaeological assessment reveals Earth’s early transformation through land use. Science 365, 897–902. doi: 10.1126/science.aax1192
Surge, D. M., Lohmann, K. C., and Goodfriend, G. A. (2003). Reconstructing estuarine conditions: oyster shells as recorders of environmental change, Southwest Florida. Estuar. Coast. Shelf Sci. 57, 737–756. doi: 10.1016/S0272-7714(02)00370-0
Svendsen, J. I., Alexanderson, H., Astakhov, V. I., Demidov, I., Dowdeswell, J. A., Funder, S., et al. (2004). Late Quaternary ice sheet history of northern Eurasia. Quat. Sci. Rev. 23, 1229–1271. doi: 10.1016/j.quascirev.2003.12.008
The IUCN Red List of Threatened Species (2019). IUCN Red List Threat, Species. Available at: https://www.iucnredlist.org/en (accessed October 13, 2019).
Trinkler, N., Labonne, M., Marin, F., Jolivet, A., Bohn, M., Poulain, C., et al. (2010). Clam shell repair from the brown ring disease: a study of the organic matrix using confocal Raman micro-spectrometry and WDS microprobe. Anal. Bioanal. Chem. 396, 555–567. doi: 10.1007/s00216-009-3114-0
Trivellini, M. M., Van der Molen, S. V., and Márquez, F. (2018). Fluctuating asymmetry in the shell shape of the Atlantic Patagonian mussel Mytilus platensis generated by habitat-specific constraints. Hydrobiologia 822, 189–201. doi: 10.1007/s10750-018-3679-8
Valéry, L., Fritz, H., Lefeuvre, J.-C., and Simberloff, D. (2008). In search of a real definition of the biological invasion phenomenon itself. Biol. Invasions 10, 1345–1351. doi: 10.1007/s10530-007-9209-7
Van Wormhoudt, A., Roussel, V., Courtois, G., and Huchette, S. (2011). Mitochondrial DNA introgression in the European abalone Haliotis tuberculata tuberculata: evidence for experimental mtDNA paternal inheritance and a natural hybrid sequence. Mar. Biotechnol. 13, 563–574. doi: 10.1007/s10126-010-9327-6
Vander Putten, E., Dehairs, F., Keppens, E., and Baeyens, W. (2000). High resolution distribution of trace elements in the calcite shell layer of modern Mytilus edulis: environmental and biological controls. Geochim. Cosmochim. Acta 64, 997–1011. doi: 10.1016/S0016-7037(99)00380-4
Vendrami, D. L. J., Houston, R. D., Gharbi, K., Telesca, L., Gutierrez, A. P., Gurney-Smith, H., et al. (2019a). Detailed insights into pan-European population structure and inbreeding in wild and hatchery Pacific oysters (Crassostrea gigas) revealed by genome-wide SNP data. Evol. Appl. 12, 519–534. doi: 10.1111/eva.12736
Vendrami, D. L. J., Noia, M. D., Telesca, L., Handal, W., Charrier, G., Boudry, P., et al. (2019b). RAD sequencing sheds new light on the genetic structure and local adaptation of European scallops and resolves their demographic histories. Sci. Rep. 9:7455. doi: 10.1038/s41598-019-43939-4
Vokhshoori, N. L., and McCarthy, M. D. (2014). Compound-Specific δ15N amino acid measurements in littoral mussels in the California upwelling ecosystem: a new approach to generating baseline δ15N Isoscapes for coastal ecosystems. PLoS One 9:e98087. doi: 10.1371/journal.pone.0098087
Voroshilova, I. S., Artamonova, V. S., Makhrov, A. A., and Slyn’ko, Y. V. (2010). Natural hybridization of two mussel species Dreissena polymorpha (Pallas, 1771) and Dreissena bugensis (Andrusov, 1897). Biol. Bull. 37, 542–547. doi: 10.1134/S1062359010050158
Wenne, R., Bach, L., Zbawicka, M., Strand, J., and McDonald, J. H. (2016). A first report on coexistence and hybridization of Mytilus trossulus and M. edulis mussels in Greenland. Polar Biol. 39, 343–355. doi: 10.1007/s00300-015-1785-x
Williams, M. R., Stedtfeld, R. D., Engle, C., Salach, P., Fakher, U., Stedtfeld, T., et al. (2017). Isothermal amplification of environmental DNA (eDNA) for direct field-based monitoring and laboratory confirmation of Dreissena sp. PLoS One 12:e0186462. doi: 10.1371/journal.pone.0186462
Wilson, J., Matejusova, I., McIntosh, R. E., Carboni, S., and Bekaert, M. (2018). New diagnostic SNP molecular markers for the Mytilus species complex. PLoS One 13:e0200654. doi: 10.1371/journal.pone.0200654
Yang, D. Y., Eng, B., Waye, J. S., Dudar, J. C., and Saunders, S. R. (1998). Improved DNA extraction from ancient bones using silica-based spin columns. Am. J. Phys. Anthropol. 105, 539–543. doi: 10.1002/(sici)1096-8644(199804)105:4<539::aid-ajpa10>3.0.co;2-1
Zbawicka, M., Sańko, T., Strand, J., and Wenne, R. (2014a). New SNP markers reveal largely concordant clinal variation across the hybrid zone between Mytilus spp. in the Baltic Sea. Aquat. Biol. 21, 25–36. doi: 10.3354/ab00566
Zbawicka, M., Trucco, M. I., and Wenne, R. (2018). Single nucleotide polymorphisms in native South American Atlantic coast populations of smooth shelled mussels: hybridization with invasive European Mytilus galloprovincialis. Genet. Sel. Evol. 50:5. doi: 10.1186/s12711-018-0376-z
Keywords: ancient DNA, mollusk shell, high-throughput DNA sequencing, taxonomic assignment, climate change, invasion, extinction
Citation: Der Sarkissian C, Möller P, Hofman CA, Ilsøe P, Rick TC, Schiøtte T, Sørensen MV, Dalén L and Orlando L (2020) Unveiling the Ecological Applications of Ancient DNA From Mollusk Shells. Front. Ecol. Evol. 8:37. doi: 10.3389/fevo.2020.00037
Received: 03 December 2019; Accepted: 07 February 2020;
Published: 03 March 2020.
Edited by:
Nic Rawlence, University of Otago, New ZealandReviewed by:
Kieren James Mitchell, The University of Adelaide, AustraliaCopyright © 2020 Der Sarkissian, Möller, Hofman, Ilsøe, Rick, Schiøtte, Sørensen, Dalén and Orlando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clio Der Sarkissian, Y2xpby5kZXJzYXJraXNzaWFuQHVuaXYtdGxzZTMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.