 Hasdra Elena Sánchez Maya1
Hasdra Elena Sánchez Maya1 Yuridia Mercado-Flores1
Yuridia Mercado-Flores1 Alejandro Téllez-Jurado1
Alejandro Téllez-Jurado1 Juan Pablo Pérez-Camarillo2
Juan Pablo Pérez-Camarillo2 Omar Mejía3
Omar Mejía3 Miguel Angel Anducho-Reyes1*
Miguel Angel Anducho-Reyes1*- 1Laboratorio de Agrobiotecnología, Universidad Politécnica de Pachuca, Zempoala, Mexico
- 2Centro de Investigación y Desarrollo en Agrobiotecnología Alimentaria, Pachuca Ciudad del Conocimiento y la Cultura, San Agustín Tlaxiaca, Mexico
- 3Departamento de Zoología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Miguel Hidalgo, Mexico
Head smut in maize (Zea mays) is a systemic disease caused by the phytopathogenic fungus Sporisorium reilianum. One of the most severely affected regions in Mexico is Valle del Mezquital in the state of Hidalgo, a high production zone with irrigated agriculture that produces over 50% of maize supplies in the state. This study was designed to analyze the genetic diversity of this phytopathogen isolated from different zones of Valle del Mezquital using the following molecular markers: ITS, aspartyl protease, β-tubulin, and the mitochondrial endonuclease LAGLIDADG. Our objective was to estimate the level of genetic variability in a regional space and understand the evolutionary processes to which non-model organisms are subjected. A total of 53 strains of S. reilianum were isolated from the sampling sites and then purified. A Bayesian Analysis of Population Structure (BAPS) analysis of the genetic structuring allowed us to recover three genetic groupings (K = 3), while molecular variance analysis (AMOVA) showed a very low fixation index (FST = 0.0146), which was not statistically significant and indicated little between-group genetic differentiation. Also, the value determined for the coefficient of genetic differentiation (Gst) showed little variation among the genetic groups, while the gene flow number (Nm) was 8.25. The Tajima’s D estimator and haplotype network recently indicated a rapid population expansion. Together, these results suggest that reproduction and migration are key factors in the evolution and virulence of this fungus.
Introduction
Head smut in maize (Zea mays) is a systemic disease caused by the phytopathogenic fungus Sporisorium reilianum f. sp. zeae (Kühn). This process begins during the life cycle of S. reilianum, when temperature and humidity are optimal for the germination of mature uninucleate diploid teliospores in the soil, which produces a structure called promycelium, whose nucleus is divided by meiosis to generate four haploid nuclei that enter lateral cells to constitute four basidiospores of different sexual compatibility. This is considered the saprophytic yeast phase of the fungus that reproduces by gemmation. The infection process occurs during the first 10–20 days after germination via the mesocotyl or root where two sexually compatible basidiospores fuse to form the infective dikaryotic hyphae that penetrate the plant’s epidermal cells via appressoria that develop in the tip of the hyphae. Tissue colonization is progressive around the vascular bundles and the mycelium can be observed in all plant tissues (Ghareeb et al., 2011; Álvarez-Cervantes et al., 2016), though the disease does not become evident until the flowering stage, characterized by carbonaceous masses of spores called sori that take the place of normal pollen and grain formation by completely destroying the cob and/or spike (Matyac and Kommedahl, 1985b; Martínez et al., 2002; Ghareeb et al., 2011; Álvarez-Cervantes et al., 2016).
Kühn wrote the earliest known report in 1875, on a specimen of the original strain found in Egypt by Dr. Reil in 1868 (Halisky, 1963). This disease, however, is now distributed worldwide, especially in areas of extensive maize cultivation, such as Europe, North, Central, and South America, Africa, Asia, Australia, New Zealand, western India, and Palestine. One of the most severely affected region in Mexico since 1960 is Valle del Mezquital in the state of Hidalgo, a high-production zone with irrigated agriculture that supplies over 50% of maize in the state (Little et al., 2012; Quezada et al., 2014; Álvarez-Cervantes et al., 2016). Although there are reports of the use of Bacillus subtilis as a biological control, the methods traditionally utilized to reduce the incidence of this disease in this area are genetic: primarily planting resistant hybrid seeds coupled with chemical control using fungicides to combat the disease and obtain good yields (Mercado-Flores et al., 2014, 2016). Despite efforts to solve this problem, studies have identified strains that are resistant to fungicides and hybrids that, as products of selection processes, have increased disease incidence (Pérez and Bobadilla, 2007; Mercado-Flores et al., 2016; Álvarez-Cervantes et al., 2016).
Knowledge of the distribution of genetic diversity allows us to identify degrees of similarity, detect rare alleles shared by geographically separate populations, and trace gene flows, migration patterns, and recombination phenomena. It also leads to inferences regarding forms of reproduction, and evaluations to determine if populations of phytopathogenic fungi with regular cycles of sexual reproduction present more numerous genotypes and, therefore, higher levels of genetic diversity than populations with asexual reproduction (Chen and McDonald, 1996; Wang, 1997; Lee and Neate, 2007; Ciampi et al., 2008; Meng et al., 2015). Analyzing the genetic structure of populations of phytopathogenic fungi in agricultural ecosystems is thus fundamental for understanding, in time and space, the evolutionary forces that generate genetic variation among, and within, them (McDonald and Linde, 2002; Lumbsch et al., 2008). This information may be useful for developing biological control strategies that go beyond the traditional methods based on resistant vegetable crops, cultural practices, and pesticide applications (McDonald, 1997; Palloix et al., 2009).
This study was conceived to analyze the genetic diversity of this fungal phytopathogen isolated from various zones in Valle del Mezquital using several markers: three nuclear molecular markers Internal Transcribed Spacer (ITS), aspartyl protease (Eap1), and β-tubulin plus a mitochondrial molecular marker that corresponds to a gene fragment that codes for the endonuclease LAGLIDADG. Our aim was to estimate the level of genetic variability in a regional space and understand the evolutionary processes to which populations are subjected in relation to the management of domesticated maize crops, in these non-model organisms.
Materials and Methods
Sampling and Isolation of S. reilianum
Sampling of the biological material was performed during the rainy season in July–October 2016. We gathered 53 samples of spikes and cobs from corn plants that had signs of the characteristic carbonaceous masses of S. reilianum f. sp. zeae (Kühn) infection, in cornfields at 13 sites in 6 municipalities in the study region. The strains of the Cinta Larga population obtained were from sampling conducted between 2012 and 2015 (Figure 1 and Table 1).
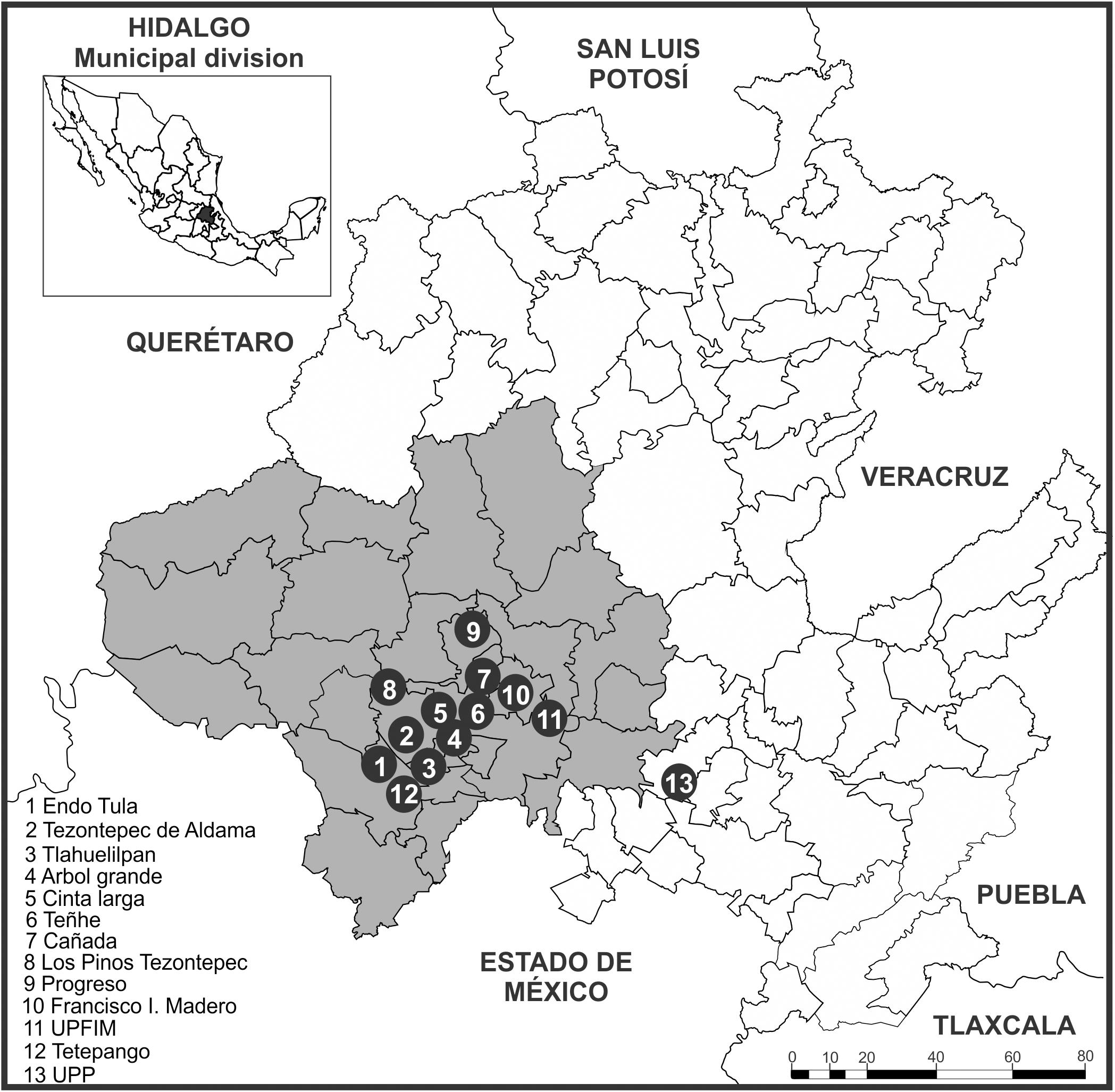
Figure 1. Map of the municipal divisions and geographic location of Valle del Mezquital (gray). Circles with numbers indicate the sites where biological material was collected (INEGI, 2018).
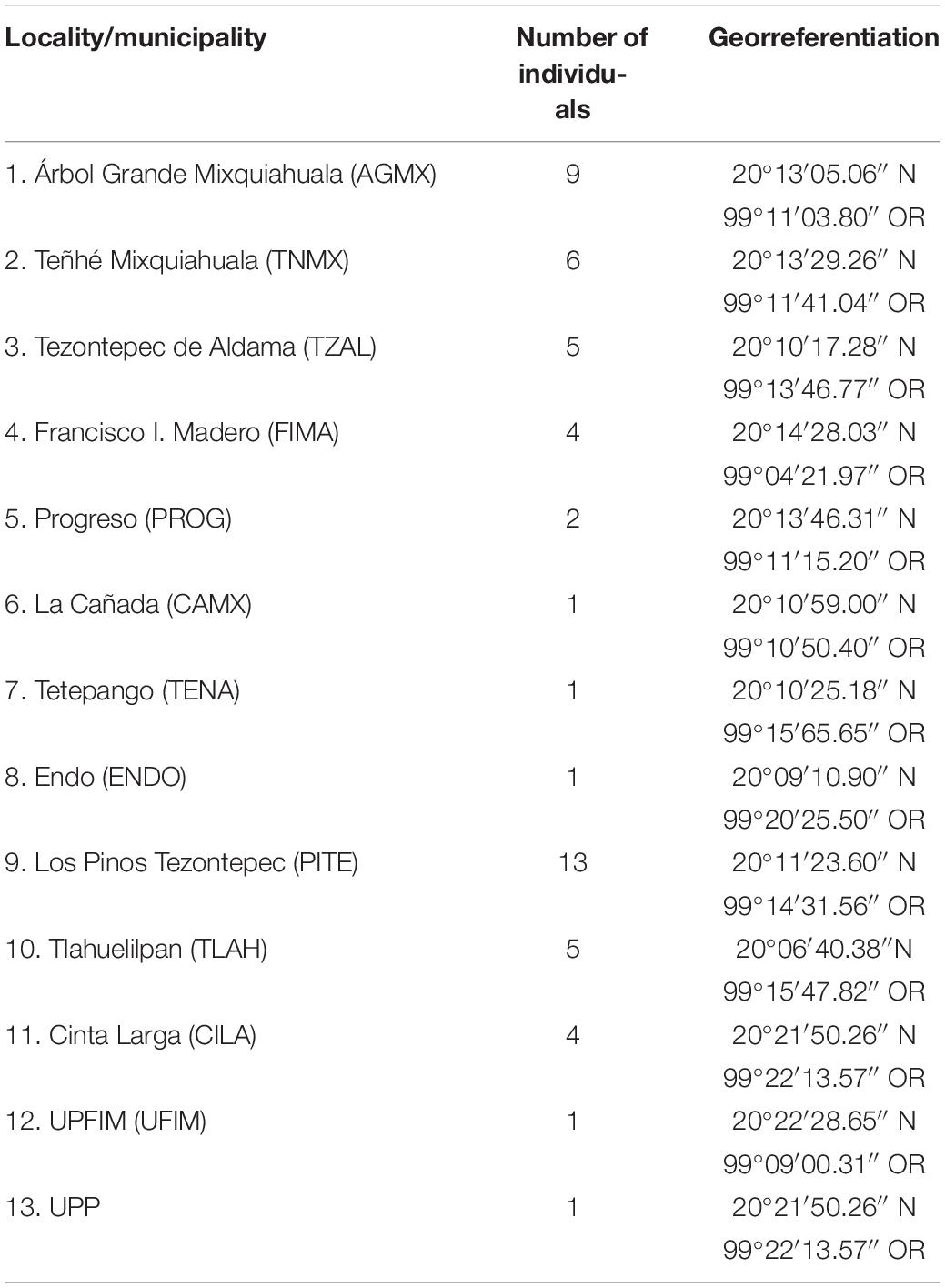
Table 1. Geographic distribution of the samples of S. reilianum.
All the infected vegetable material was placed in paper bags for transport to the laboratory, where samples were dried for 24–30 h at ±2°C. The teliospores were recovered and conserved at 20 ± 2°C until used (Quezada et al., 2014). The monosporic samples were obtained using the technique described by Zhao et al. (2015), that consists in placing 30 mg of spores in a 1% aqueous solution (p/v) of CuSO4 for 24 h, then washing them three times with sterile distilled water, and recovering the teliospores through centrifugation at 14,000 rpm. Once disinfected, all samples were re-suspended in 1 mL of sterile PBS (Na2HPO4 and KH2PO4) (pH 7.0) and serial dilutions were prepared from 10–1 to 10–4. Finally, 100 μL of the 10–4 dilution was inoculated in solid YEPD culture medium [2.0% dextrose (P/V), 2.0% peptone (P/V), 1.0% (P/V) yeast extract, and 2.0% agar (P/V)], supplemented with 0.1 mg/mL of gentamicin, and then incubated at 28°C under darkness for 5 days. Individual, isolated colonies were cultivated in liquid YEPD medium and incubated at 28°C under darkness for 3 days with agitation at 150 rpm. The purity of each culture was verified by microscopy. In the final step, 2 mL of each culture was taken and centrifuged at 12,000 rpm for 10 min. Cells were recovered from the bottom of the tube to genomic DNA extraction.
Genomic DNA Extraction
Genomic DNA was extracted following the method described by Hoffman and Winston (1987). To verify the quality and size of the DNA extracted, electrophoresis was performed in agarose gel at 1% (P/V) in TAE 1× buffer at 90 V for 45 min. The DNA samples in gel were visualized by staining with a solution of ethidium bromide (200 mg/mL). Images were recorded in an ultraviolet light transilluminator (Multidoc-it Digital Imaging System UVP) using Doc-it® LS Image Analysis UVP software and a Canon PC1089 camera.
Polymerase Chain Reaction and Sequencing
Three nuclear loci and one mitochondrial locus from each monosporic culture were amplified by PCR and then sequenced. The three amplified loci included an ITS region, a fragment of the β-tubulin gene, and the gene that codes for an aspartyl protease (Eap1). Amplification of the ITS1–ITS4 region was done with two initiators: ITS1 (5′-TCC GTA GGT GAA CCT GCG G-3′) and ITS4 (5′-TCC TCC GCT TAT TGA TAT GC-3′) (Zhang et al., 2015). PCR conditions consisted of an initial denaturalization step at 94°C for 5 min, followed by 35 cycles of denaturalization at 94°C for 1 min, alignment at 55°C for 1 min, extension at 72°C for 3 min, and a final extension step at 72°C for 2 min. The amplification of the β-tubulin gene fragment was performed with the initiators βtub3 (5′-TGG GCY AAG GGT YAY TAY AC-3′) and βtub4r (5′-GCC TCA GTR AAY TCC ATY TCR TCC AT-3′) (Huang et al., 2009) under the following conditions: an initial denaturalization step at 95°C for 5 min, followed by 35 cycles of denaturalization at 94°C for 1 min, alignment at 56°C for 1 min, extension at 72°C for 1.5 min, and a final extension step of 7 min at 72°C. Amplification of the gene that codes for an aspartyl protease (Eap1) was performed with the initiators Eap1F (5′-ATG CAA CTC AAG CTC TCG TTT GTT GC-3′) and Eap1R (5′-TTA GGC CTT GTT GGC GAA GCC G-3′) (Mandujano et al., 2013), as follows: an initial denaturalization step at 95°C for 5 min, then 35 cycles of denaturalization at 94°C for 1 min, alignment at 52°C for 1 min, extension at 72°C for 1 min, and a final extension step of 5 min at 72°C. The sole mitochondrial locus a fragment of the gene that codes for the putative endonuclease LAGLIDADG was performed with the specific initiators SR1F (5′-CAG GTT ATG TAT GGG CC-3′) and SR1R (5′-TTG AGC GAT GAC CAT TCC-3′) (Xu et al., 1999), under the following PCR program: an initial denaturalization step at 95°C for 5 min, followed by one cycle of denaturalization at 94°C for 4 min, alignment at 56°C for 1 min, extension at 72°C for 1 min, followed by 35 cycles of denaturalization at 94°C for 1 min, alignment at 52°C for 1 min, and extension at 72°C for 2 min, ending with a final extension step of 2 min at 72°C. The reaction mixture of all PCR was carried out with a volume of 50 μL that contained 5.0 μL of the reaction buffer (10×), 5 μL of MgCl2 (25 mM), 1 μL of the forward and reverse primers (10 μM), 1 μL of DNA (100 ng/μL), 0.5 μL of Taq DNA polymerase (5 μ/μL), 1 μL of dNTPs (10 mM), and 35.5 μL of H2O. The endonuclease LAGLIDADG family, specifically the hegII1 gene, was used because the design of the primers that amplify this gene fragment permits differentiating the phytopathogenic fungi S. reilianum from U. maydis. This is important because, although two diseases have very different morphological characteristics, they coexist in cultivated fields (Xu et al., 1999). Visualization of the PCR products was achieved by agarose gel electrophoresis at 0.7% (P/V) in TAE 1× buffer at 90 V for 45 min and the image was captured in an ultraviolet light transilluminator (Multidoc-it Digital Imaging System UVP) using Doc-it® LS Image Analysis UVP software and a Canon PC1089 camera. Finally, the PCR products were purified using Wizard® SV Gel and the PCR Clean-UP System (Promega, Madison, WI, United States) and sequenced using the BigDye terminator v3.1 kit and an 8-capillary automated Model 3500 sequencer (Applied Biosystems®).
Data Analysis
The data generated by the sequencer were visualized with Sequencing Analysis v. 5.3 software (Applied Biosystems®). The electropherograms were transformed into nucleotide sequences in text format, inspected visually and then edited manually using BioEdit sequence alignment editor v. 7.2.6 software1. Once the individual sequences were obtained and edited, they were compared using the BLAST program algorithm2 from the GenBank database of the National Center for Biotechnology Information (NCBI). An ultrametric phylogenetic tree was generated using the BEAST v.2.2.1 program3 (Bouckaert et al., 2014), the substitution model estimated with JModelTest 2.1.4 software4 (Darriba et al., 2012), and the parameters of the relaxed molecular clock with a lognormal distribution and birth–death model. Three independent tests were run, each with 10 million generations. Next, the files generated by the runs were combined by the LogCombiner v.1.8.3 program (Drummond et al., 2012) with a pre-determined burn-in (10%). First, the spatial genetic structure was determined using Bayesian Population Structure Analysis 6.0 software (BAPS)5 (Corander et al., 2004). For this part of the procedure, the program was executed with 20 replicates for each k level (1–6) without origin information, then the “groupings of individuals” function was applied. Once the genetic groups were defined the next step was to determine a set of diversity estimators, such as nucleotidic and haplotype diversity, GC content, polymorphism, the genetic differentiation coefficient between populations (Gst), the gene flow number (Nm), and Tajima’s D of each population, using the DNAsp v. 5.10.1 program6. Likewise, estimates of the genetic structure within, and among, the sampling sites were examined hierarchically by Molecular Variance Analysis (AMOVA) (Excoffier et al., 1992), which meant calculating the paired fixation index, or Fst, the Fu’s Fs test, and the mismatch distributions with 5,000 permutations, utilizing Arlequin 3.5 software7 (Excoffier and Lischer, 2010). The demographic history of these S. reilianum populations was analyzed using the statistical methodology based on coalescence, implementing the Bayesian Skyline Plot routine (BSP) from BEAST v2.5.1. (Bouckaert et al., 2014). The Piecewise-Constant Skyline model was chosen and the relaxed uncorrelated lognormal molecular clock parameter was applied with an estimated mutation rate of 0.69%/MY for S. reilianum (Drake et al., 1998; Kasuga et al., 2002; Lang and Murray, 2008; Farlow et al., 2015). Tracer 1.5 was used to summarize the later estimates of the diverse parameters sampled by the Markov chain, which allowed us to reconstruct the fungus’ demographic history. Results were graphed using a routine established in R-CRAN (R Core Team, 2014). Finally, the construction of the haplotype networks was performed with the Media Joining Network algorithm of the POPART v. 1.7 program8 and then edited in the Inkscape program9.
Results
Sampling and Isolation of S. reilianum
A total of 53 strains of S. reilianum were isolated and purified from the 53 samples of teliospores obtained from the infected spikes and cobs. In most cases, isolation in solid YEPD medium revealed a characteristic morphology with irregular cream-colored colonies of butyrous consistency. Later, inoculation of the isolated colonies in liquid YEPD medium generated slightly elongated, oval-shaped yeasts.
Genetic Groups
The BAPS analysis of genetic structuring using the four concatenated markers revealed three (3) as the most likely grouping number [k = 3; log value (marginal likelihood) = −874.705] after 20 independent runs. The first genetic group included 46 samples and was found at all sites except those in the municipality of Endo. The second group had six individuals from the municipalities of La Cañada, Los Pinos Tezontepec, Teñhé Mixquiahuala, and Tezontepec de Aldama. The third group had only one individual, from the municipality of Los Pinos Tezontepec (Figure 2).
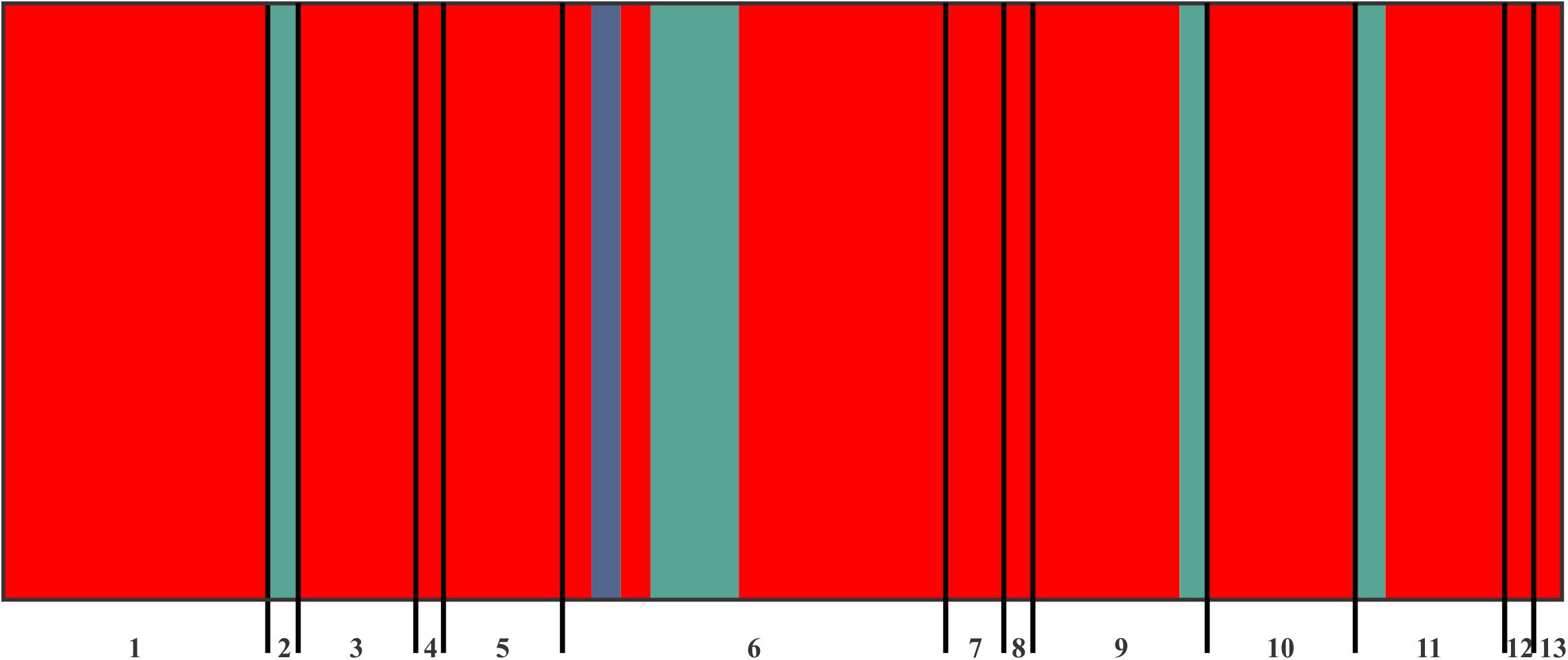
Figure 2. Bayesian groupings (BAPS) for the concatenated ITS, β-tubulin, Eap1, and mitochondrial gene LAGLIDADG of S. reilianum individuals. Vertical lines indicate the posterior probability of each individual belonging to one of the three genetic clusters recovered in this study (red, blue, and green). 1, Árbol Grande Mixquiahuala (AGMX); 2, La Cañada (CAMX); 3, Cinta Larga (CILA); 4, Endo (ENDO); 5, Francisco I. Madero (FIMA); 6, Los Pinos Tezontepec (PITE); 7, Progreso (PROG); 8, Tetepango (TENA); 9, Teñhé Mixquiahuala (TNMX); 10, Tezontepec de Aldama (TZAL); 11, Tlahuelilpan (TLAH); 12, UPFIM (UFIM); 13, UPP.
Analysis of the Molecular Variation of the Nuclear and Mitochondrial Sequences
The similarity analysis of the 53 sequenced fragments corresponding to the nuclear ITS region (ITS1, ITS2, and 5.8S), Eap1, and β-tubulin genes, as well as the mitochondrial gene LAGLIDADG of S. reilianum at the GenBank database of the NCBI, using the algorithm implemented in the BLAST program, showed an identity range of 97–99% with the sequences of the SRZ2 (FQ311469.1) and DT0620 (FJ167357.1) strains of S. reilianum on deposit.
The sequencing of the nuclear ITS showed an average size of 643 pb, except for the PITE07, PROG01, TZAL04, TLAH05, and UPP01 strains, which ranged in size from 642 to 640 pb. Analysis of the ITS1, 5.8S and ITS2 that formed the ITS region revealed that ITS1 had a size of 217 pb for 48 individuals, while the TLAH04, TZAL04, PROG01, and PITE07 strains measured 216 pb, and UPP01 214 pb. The 5.8S gene showed no variation, since all those individuals presented a size of 133 pb with a GC content of 46%. Likewise, ITS2 showed no variation in fragment size, since all individuals measured 293 pb, with GC at 63%.
The nucleotidic analysis of the β-tubulin gene showed a size of 906 bp with an average GC content of 63%, while those of the variable and invariable sites were 58 and 848, respectively. The analysis of the nucleotidic sequence of the Eap1 gene revealed a size of 1182 bp for all individuals. The average number of variable sites was of 23, while for the invariable or monomorphic sites it was 1159. The sequencing of the mitochondrial LAGLIDADG gene for all individuals of S. reilianum had a size of 865 bp and a GC content of 35%. The number of variable sites was just 1, but there were 864 invariable sites.
The concatenated sequences of the ITS, Eap1, β-tub, and mitochondrial LAGLIDADG gene produced a total size of 3596 nucleotides. The number of invariable sites was estimated to be 3508, with 88 variable sites. The estimated number of parsimoniously informative sites was 33, with 92 mutations. The nucleotidic diversity estimated for the four independent regions showed an interval (Pi) of 0.0047–0.0002, while the haplotype diversity presented a range of 0.174–0.852. The estimates of nucleotidic and haplotype diversity for the 53 concatenated sequences from the 13 sites sampled were Pi = 0.0019 ± 3 × 10–4 and Hd = 0.983 ± 7 × 10–5, respectively. The total estimated Tajima’s D value was −2.33761, P < 0.01. Independently of those results, the analysis of nucleotidic and haplotype diversity for the nuclear concatenated genes ITS, β-tubulin, and Eap1 showed a fragment of 2734 nucleotides with h = 38, Hd = 0.977 ± 0.0001, and Pi = 0.0024.
The polymorphic analysis of the genetic groups generated by Bayesian grouping (BAPS) showed a range of nucleotidic diversity of 0.0011–0.0057, while the results of the neutrality tests for Tajima’s D for genetic groups I and II were −1.60440 and −0.89961, respectively, with non-significant probability values (P > 0.05). Because genetic group III had only one individual it was not susceptible to descriptors of diversity or historical demography (Table 2).
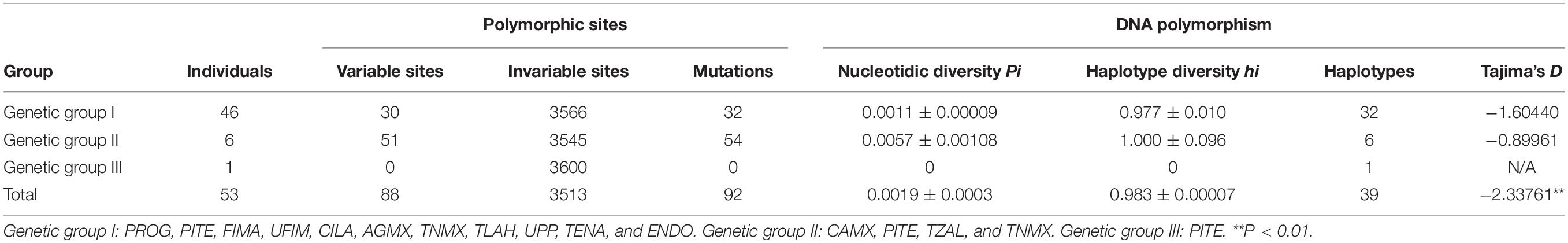
Table 2. Polymorphism rates by genetic group.
The nuclear sequences of the ITS region, β-tubulin, and Eap1 genes and LAGLIDADG mitochondrial sequences are on deposit in the GenBank under access numbers MN049251–MN049462. The vouchers that correspond to S. reilianum are on deposit in the Agrobiotechnology Laboratory of the Universidad Politécnica de Pachuca.
Genealogical Analysis
The haplotype networks generated for genetic groups I and II by the Media Joining Network method using the concatenated ITS region, β-tubulin, and Eap1 nuclear genes, and the LAGLIDADG mitochondrial gene revealed two networks made up of 29 haplotypes for group I, and 6 haplotypes for group II (Figure 3). Both networks showed a star-shaped distribution with no geographic structuring, perhaps indicative of a recent process of population expansion. The large number of mutations in some nodes demonstrates ample genetic variability. For genetic group I, haplotype 4 is considered ancestral with four individuals and is distributed geographically in Francisco I. Madero (FIMA), Tezontepec de Aldama (TZAL), Teñhé Mixquiahuala (TNMX), and Árbol Grande Mixquiahuala (AGMX). The average estimated geographic distance between sampling sites was 9.55 km. Haplotypes 5, 6, 7, 8, 10, 11, 12, 13, 14, 15, 16, 17, 18, 20, 21, 22, 23, 25, 26, 27, 28, 29, 30, 31, and 32 were unique and distributed geographically in Tlahuelilpan (TLAH), Los Pinos Tezontepec (PITE), Francisco I. Madero (FIMA), Cinta Larga (CILA), Tezontepec de Aldama (TZAL), “Teñhé” Mixquiahuala (TNMX), Árbol Grande Mixquiahuala (AGMX), Tetepango (TENA), Endo (ENDO), and UPP. Finally, haplotypes 15, 16, and 18 had the highest number of mutational steps (Figure 3). Genetic group II included six haplotypes, characterized basically by the number of mutational steps in their nodes. We identified haplotype 4 as ancestral, as it is distributed in Los Pinos Tezontepec (PITE) and Tlahuelilpan (TLAH), with five derived haplotypes with high genetic variability distributed in Tlahuelilpan (TLAH), Los Pinos Tezontepec (PITE), “Teñhé” Mixquiahuala (TNMX), and La Cañada, Mixquiahuala (CAMX).
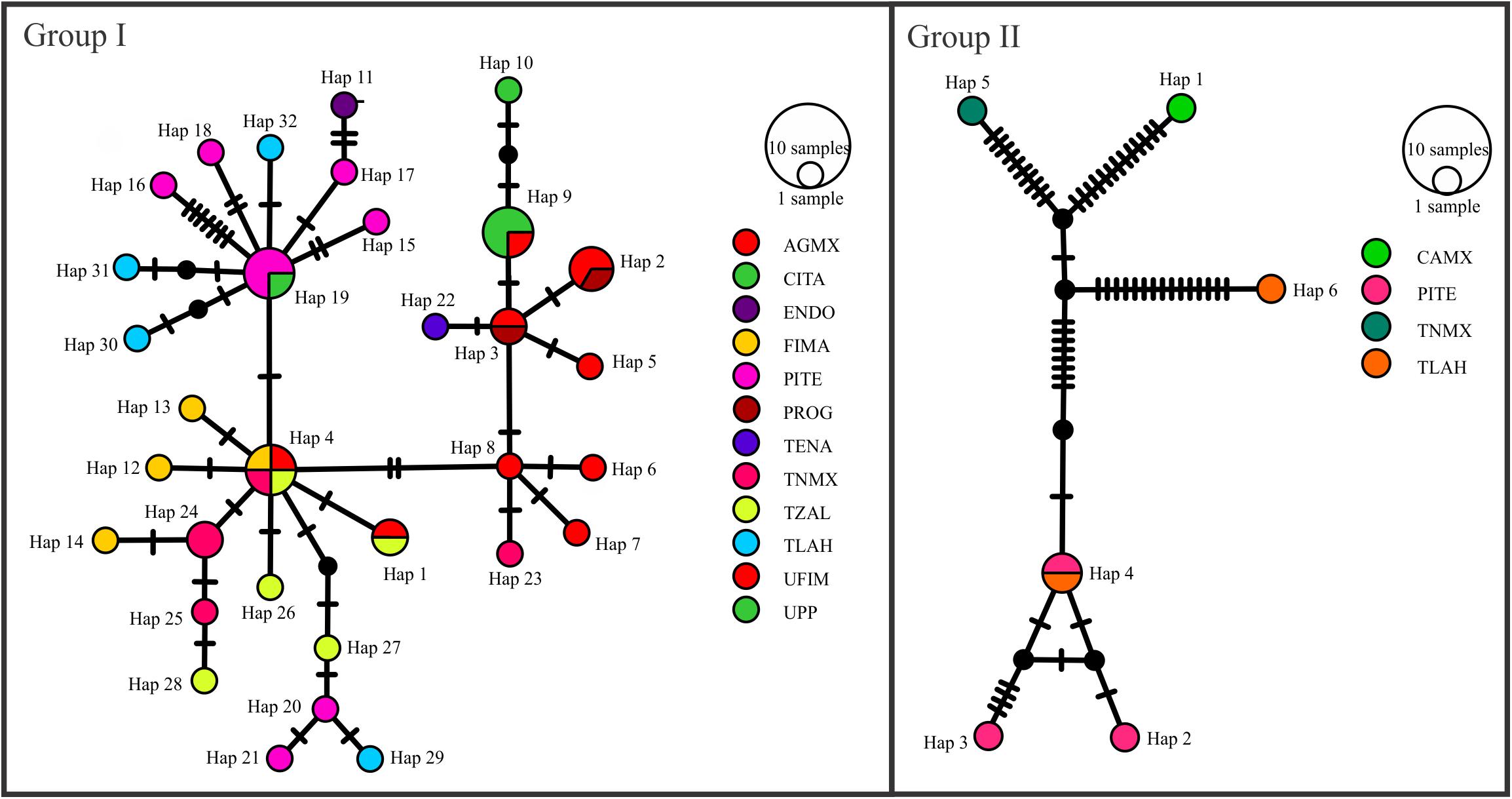
Figure 3. Haplotype networks constructed using the Media Joining Network method for the concatenated ITS, β-tubulin, Eap1 nuclear genes, and mitochondrial gene LAGLIDADG. The networks shown for genetic groups I and II had 27 and 6 haplotypes, respectively, all identified from isolates of S. reilianum collected in cornfields at different sites in Valle del Mezquital. The diagonal lines to the connections between haplotypes represent the number of mutations; colors represent the geographic localities. Circle size is proportional to the frequency of the different haplotypes.
Genetic Structure
After defining the groups using BAPS, we performed a AMOVA of the four concatenated genes, which showed that 98.54% of the genetic diversity was distributed within the groups, with the rest distributed among the three groups. No level of the hierarchy showed significant genetic differentiation (P > 0.05). The fact that the fixation index (FST = 0.0146) was not statistically significant indicates little genetic differentiation among the three groups. However, the estimate of the fixation index (FST = 0.0521) based on the localities showed that some were genetically diverse (P < 0.001) and that together they form a genetically cohesive group. The value of the coefficient of genetic differentiation showed a low variation of just 2.9% among the genetic groups (Gst = 0.0294), while the gene flow number (Nm) was 8.25 which, on a small geographic scale, is indicative of a high level of gene exchange among groups.
Demographic History Analysis
The tests of mutation neutrality using Fu’s Fs test and Tajima’s D estimators suggest processes or events of population expansion in some of the genetic groups generated by BAPS. The estimated values Fu’s Fs values for the four concatenated DNA fragments led us to reject the hypothesis of constant population size only for genetic group I, which reached statistical significance (Fs = −25.93739; P < 0.05), while the Tajima’s D values calculated were negative at −1.47753 (P < 0.05) and −0.57461 (P > 0.05) for genetic groups I and II, respectively. Because this was statistically significant for genetic group I, we were also able to reject a scenario of neutral selection or population equilibrium. The results of the neutrality tests for Fu’s Fs and Tajima’s D for the total population were −25.07692 (P < 0.05) and −2.28043 (P = 0.0006), respectively; negative results that also suggest a process of population expansion.
The histograms of the mismatch distribution of the haplotypes obtained for the four concatenated genes presented a unimodal distribution for genetic group I, suggesting that the populations analyzed had undergone a recent process of demographic or, perhaps, geographic expansion with high levels of migration between neighboring populations. Moreover, the demographic parameters estimated for the rugosity index (r = 0.025, P = 0.4) and the deviation from the sum of the squares (SDD = 0.003, P = 0.3) allow us to confirm and accept the null hypothesis of population expansion (Figure 4). No mismatch analysis was run for genetic groups II and III due to the small sample sizes.
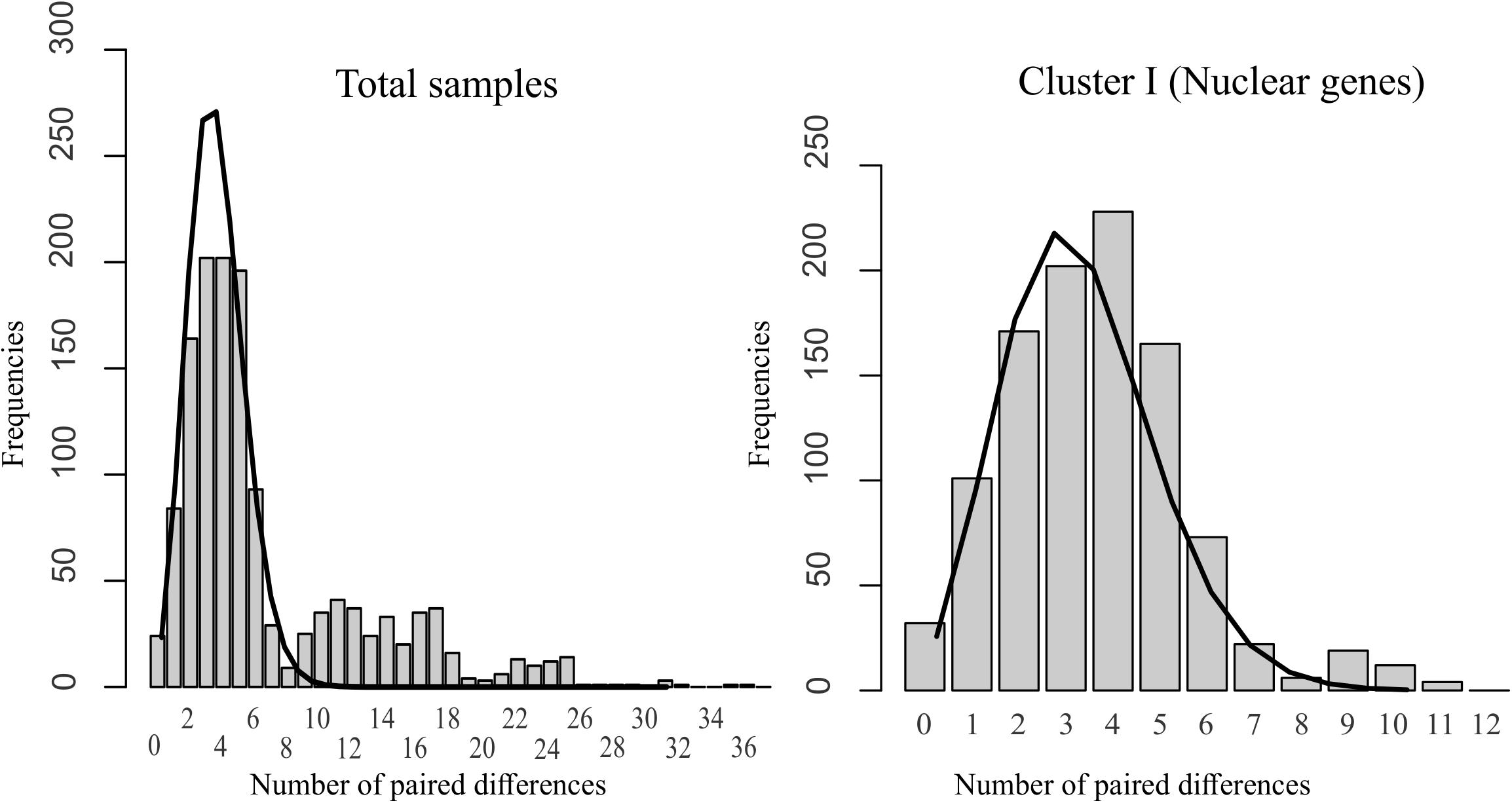
Figure 4. Distribution mismatch for genetic group I determined by BAPS and the total sample of S. reilianum using the ITS region, β-tubulin, and Eap1 nuclear genes. Gray bars represent the observed values; the black line represents the expected distribution under a hypothesis of population expansion.
Group I presented an estimated expansion time of 276.65 years utilizing a mutation rate for the nuclear genes of 2.18% Myr, and of 122.05 years using a mutation rate of 4.94% Myr; calculated on the basis of the value τ = 3.859. The Bayesian methods of skyline plots for genetic group I coincide with the results of the mismatch analysis for this group. BSPs revealed a demographic expansion approximately 20,000–50,000 years ago using a mutation rate of 2.18% and 4.94% Myr for the nuclear genes, respectively (Figure 5).
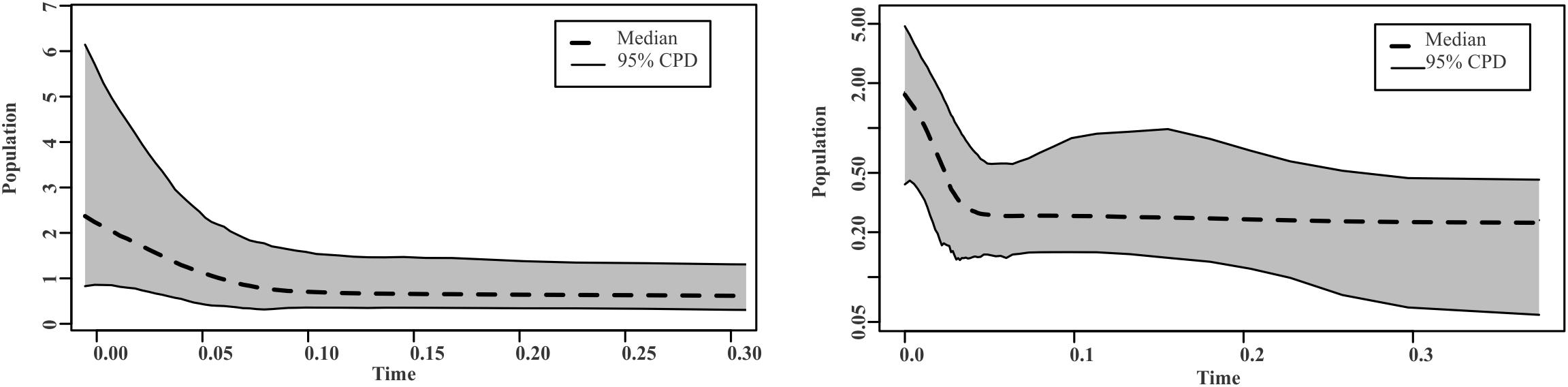
Figure 5. Graphs of Bayesian Skyline Plots for the ITS region, β-tubulin, and Eap1 nuclear genes, showing a process of demographic expansion over time, assuming a divergence index of 2.18 and 4.94% Myr for S. reilianum in the region of Valle del Mezquital.
Discussion
Head smut in maize has been considered a problematic fungal disease since the 1970s due to its high incidence in Mexico, especially in Valle del Mezquital and the Valles Altos of the states of Hidalgo and Mexico (Matyac and Kommedahl, 1985a; Jin et al., 2000; Quezada-Salinas et al., 2017).
In the infection cycle of this pathogen, the most damage becomes visible during flowering, as the mycelia established in the meristem produce sori that replace the maize kernels (female inflorescence) and the anthers. As they mature, the sori release teliospores that fall to the ground (Quezada-Salinas et al., 2017). In this way, the teliospore that results from sexual reproduction is the infectious structure that disseminates is spread by rain, wind, fauna, and human activity (INIFAP-Hidalgo, 2012).
Some information generated demonstrates that the kinds of reproduction of phytopathogenic fungi – asexual, sexual, and para-sexual – profoundly affect epidemiological landscapes, the genetic dynamics of populations, their evolutionary trajectories, and disease control and management (Meng et al., 2015). In this regard, sexual reproduction may offer evolutionary advantages over the asexual form, for it can generate greater genetic variation through intragenic recombination or the reorganization of existing genes, while simultaneously increasing the efficiency of natural selection against undesirable alleles and virulence factors (Meng et al., 2015). In addition, the bodies of sexual fructification are tolerant to stress and can survive for long periods under extreme climatic conditions or in the absence of host. All these factors increase their capacity to evolve rapidly in response to changes in control and management strategies, the use of resistant seeds, and fungicide applications (Chen and McDonald, 1996; Sakai et al., 2001; Parker and Gilbert, 2004; Meng et al., 2015). The sampling, isolation, and culturing of the sexual structures – teliospores – of S. reilianum produced low estimates of nucleotidic diversity (Pi = 0.0019 ± 3 × 10–4) but high values of haplotype diversity (Hd = 0.983 ± 7 × 10–5). This suggests a process of rapid population growth and the accumulation of mutations, given the short spatial–temporal history of these populations; factors that may favor the establishment of resistant, more virulent strains. A similar phenomenon has been described in the fungi R. solani AG-1 IA and Alternaria alternata, allowing to suggest that sexual reproduction plays an important role in the epidemic and evolution of the pathogen in the spite absence of observed teleomorphs (Rosewich et al., 1999; Pascual et al., 2000; Ciampi et al., 2005; Meng et al., 2015).
Our analysis of genetic diversity using an AMOVA with the four concatenated genes in a specific region of the State of Hidalgo demonstrated that 98.54% of diversity is distributed within the groups, while the rest is distributed among groups. Similar findings were reported by Jiménez-Becerril et al. (2016) who analyzed the mitochondrial genetic diversity of Ustilago maydis using samples from 10 states in Mexico. The highest proportion of genetic variation occurred within states (77.46%), while between-state variation was lower (10.37%). The statistical analysis of heterozygosity revealed an average of 2.4 alleles per locus, with an effective number of alleles of 1006. These findings led them to conclude that genetic diversity was greater on small geographic scales as a result of the high sexual recombination of the fungus (Zambino et al., 1997; Munkacsi et al., 2006). Similarly, Valverde et al. (2000) examined the nuclear DNA of 30 isolates from five localities in Mexico with RFLP. They observed that the highest genetic variation was within states (77.46%), while between-state variation was low (10.37%) (Jiménez-Becerril et al., 2016).
We must also consider that the degree of sexual vs. asexual reproduction in natural populations of organisms can deeply affect their genetic variability and population structure. Populations with regular sexual reproduction continually recombine their genes, while organisms with asexual reproduction are limited in terms of recombining their genes, and can generate only a few clones. In this way, sexual recombination can, apparently favor the formation of new combinations of genes or new genotypes, where the selection phenomenon which could include fungicides, resistant plant lines or pesticides would act on suitable individuals that would be clonally increased by asexual reproduction to ensure high frequencies (Wang, 1997).
Two varieties of S. reilianum have been described, based on host specificity; one that infects maize (SRZ) and another that infects sorghum (SRS) (Zuther et al., 2012). In relation to their sexual behavior, reference is made to the mating type locus that is composed of two unlinked genomic regions, a and b, that code for a pheromone, a pheromone receptor and homeodomain proteins. Locus b, exists in at least five different alleles and encodes two subunits of a heterodimeric homeodomain transcription factor, while locus a, can be one of three alleles, each with two pheromone precursors. These different versions, designated a1, a2, and a3, are idiomorphs, each one of which idiomorph encodes different pheromones and pheromone receptors. The pheromones for a particular receptor have an identical sequence even if they exist in a different allele; that is, the pheromones of a1 and a3 that bind to the a2 receptor have the same sequence (Schirawski et al., 2005). However, and despite the narrow range of hosts of the two varieties and the similarity of the genetic material within the mating type loci (Radwan, 2013), SRS can only successfully produce teliospores in sorghum while SRZ can only able to do so in maize. Likewise, in their respective hosts, each variety is able to resist the plants’ attempts to eliminate infection, while, in the other species, the fungus succumbs to the plant’s defense mechanisms. This selection phenomenon, probably does not favor variation within the mating type locus to reduce promiscuity between different species, though some studies sustain that it favors the maintenance of other specific genes that allow proliferation within the host, despite the plants’ attempts to eliminate infection (Wallen and Perlin, 2018).
Another interesting finding of our study was the distribution of clonal haplotypes over small areas, as this may suggest that migration and other mechanisms, such as teliospores or hosts with different levels of resistance, could ensure the dispersion of fungal material. In this context, one problem at the local or regional level in some states of Mexico is the lack of an agroecological culture that seeks to adequately manage certain economically important crops. This means that the dissemination of fungal structures may be fostered through physical media like transport in organic material used as fertilizer, or contaminated seeds, farm machinery, or other agricultural equipment (Jones and Belmar, 1989; Görcsös et al., 2015). The latter factor could be determinant by allowing the movement of contaminating biological material to distant, but contiguous, regions. In contrast, that study found an excess of rare, unique haplotypes, supported by the negative values of the Tajima’s D estimator and the haplotype network. This finding could indicate that these populations recently underwent rapid population expansion or selective sweep.
It is clear that the kind of reproduction of most phytopathogenic fungi and anthropogenic activities are important factors in their evolution and virulence. For this reason, the data generated in this study hypothetically suggest demographic and historical processes of population expansion in a time period of 20,000–50,000 years. In Munkacsi et al. (2007) estimated the dates of divergence among four, monophyletic species of smut fungi Ustilago maydis, U. scitaminea, S. reilianum, S. sorghi, that specifically infect maize, sorghum, sugarcane, and their wild ancestors. In the absence of fossil records, they used a calibrated date of 10,000 years that corresponded to the domestication of maize, and a calibrated date of 50 million years consistent with the origin of the wild ancestors of their domesticated host. Their results showed that the most recent divergence date calculated between S. reilianum and S. fastigiatum was 600,000 years ago. This led to the conclusion that while this divergence occurred well before the moment of domestication, the host range S. reilianum may have been influenced by human activity. In a context with no adequate fossil records or molecular clocks, Bayesian Skyline analysis shows a phenomenon of population expansion behavior in a short time. Our study also reveals that there is no restriction on gene flow among the genetic groups of S. reilianum (Nm = 8.25) because the cultural practices used when planting cornfields in the six municipalities of Valle del Mezquital are inadequate, since they may include infected seeds and/or poor hygiene when handling irrigation systems and machinery. Finally, the presence of rare, low-frequency haplotypes may be explained by the reduced use of hybrid, resistant seed or creole corn, inadequate irrigation water, deficient handling of technological components like fertilizers, and inadequate control of diseases, plagues, and weeds (Pérez-Camarillo et al., 2009).
On this topic, Wang et al. (2015) proposed that the long-term management of diseases caused by phytopathogenic fungi should include tolerant hybrids and the controlled use of fungicides to minimize gene flow by limiting the propagation of reproductive structures through shared irrigation systems and machinery. Preventing the transmission of phytopathogenic fungi through infected seeds could require improved hygienic procedures, moderate fungicide applications in soil and on plants, and restrictions on the distribution and sale of contaminated seeds, especially when growers share them in, and between, communities.
Data Availability Statement
The datasets generated for this study can be found in the GenBank (NCBI) access numbers MN049251–MN049462.
Author Contributions
HS and MA-R conceived the ideas, performed the experimental design, directed the over-all study, and drafted the manuscript. YM-F, OM, JP-C, and AT-J helped perform the analysis of results and revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Jaime Ortega Bernal for his technical assistance, the Comité Estatal de Sanidad Vegetal del Estado de Hidalgo (CESAVEH) for its help in obtaining the samples of teliospores from the infected spikes and cobs in maize crops included in this work, and Paul Kersey for kindly translating and proofreading the manuscript.
Footnotes
- ^ https://bioedit.software.informer.com/7.2/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ https://beast.community/programs
- ^ https://github.com/ddarriba/jmodeltest2
- ^ http://www.helsinki.fi/bsg/software/BAPS/
- ^ http://www.ub.edu/dnasp/
- ^ http://cmpg.unibe.ch/software/arlequin35
- ^ http://popart.otago.ac.nz/index.shtml
- ^ https://inkscape.org/is/
References
Álvarez-Cervantes, J., Hernández-Domínguez, E. M., Tellez-Tellez, M., Mandujano-González, V., Mercado-Flores, Y., and Díaz-Godinez, G. (2016). “Stenocarpella maydis and Sporisorium reilianum: two pathogenic fungi of maize,” in Fungal Pathogenicity, ed. S. Sultan, (Croatia: INTECH), 45–60.
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Chen, R. S., and McDonald, B. A. (1996). Sexual reproduction plays a major role in the genetic structure of populations of the fungus Mycosphaerella graminicola. Genetics 142, 1119–1127.
Ciampi, M. B., Kuramae, E. E., Fenille, R., Meyer, M. C., Souza, N. L., and Ceresini, P. C. (2005). Intraspecific evolution of Rhizoctonia solani AG-1 IA associated with soybean and rice in Brazil based on polymorphisms at the ITS-5.8S rDNA operon. Eur. J. Plant Pathol. 113, 183–196. doi: 10.1007/s10658-005-2231-7
Ciampi, M. B., Meyer, M. C., Costa, M. J. N., Zala, M., McDonald, B. A., and Ceresini, P. C. (2008). Genetic structure of populations of Rhizoctonia solani anastomosis group-1 IA from soybean in Brazil. Phytopathology 98, 932–941. doi: 10.1094/PHYTO-98-8-0932
Corander, J., Waldmann, P., Marttinen, P., and Sillanpää, M. J. (2004). BAPS 2: enhanced possibilities for the analysis of genetic population structure. Bioinformatics 20, 2363–2369. doi: 10.1093/bioinformatics/bth250
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Drake, J. W., Charlesworth, B., Charlesworth, D., and Crow, J. F. (1998). Rates of spontaneous mutation. Genetics 148, 1667–1686.
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under linux and windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 31, 479–491.
Farlow, A., Long, H., Arnoux, S., Sung, W., Doak, T. G., Nordborg, M., et al. (2015). The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201, 737–744. doi: 10.1534/genetics.115.177329
Ghareeb, H., Becker, A., Iven, T., Feussner, I., and Schirawski, J. (2011). Sporisorium reilianum infection changes inflorescence and branching architectures of maize. Plant Physiol. 156, 2037–2052. doi: 10.1104/pp.111.179499
Görcsös, G., Irinyi, L., Radó, L., Tarcali, G., and Sándor, E. (2015). Diversity of Cryphonectria parasítica populations from the Carpathian basin. Acta Microbiol. Immunol. Hungar. 62, 247–266. doi: 10.1556/030.62.2015.3.3
Halisky, P. (1963). Head smut of sorghum, Sudan grass, and corn, caused by Sphacelotheca reiliana (Kühn) Clint. Hilgardia 34, 287–304. doi: 10.3733/hilg.v34n08p287
Hoffman, C., and Winston, F. (1987). A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57, 267–272. doi: 10.1016/0378-1119(87)90131-4
Huang, C. H., Lee, F. L., and Tai, C. J. (2009). The (-tubulin gene as a molecular phylogenetic marker for classification and discrimination of the Saccharomyces sensu stricto complex. Antonie Van Leeuwenhoek 2, 135–142. doi: 10.1007/s10482-008-9296-1
INEGI (Instituto Nacional de Estadística y Geografía) (2018). División Geoestadística Municipal y Municipios con Mayor Población. Available online at: https://www.inegi.org.mx/app/cuadroentidad/AnuarioGeografico/Hgo/2018 (accessed June, 2018).
INIFAP-Hidalgo (2012). INIFAP (Instituto Nacional de Investigaciones Forestales, Agricolas y Pecuarias). New Orleans, LA: Centro de Investigación Regional Centro.
Jiménez-Becerril, M. F., Hernández-Delgado, S., Solís-Oba, M., and González-Prieto, J. M. (2016). Analysis of mitochondrial genetic diversity of Ustilago maydis in Mexico. Mitochondr. DNA 29, 1–8. doi: 10.1080/24701394.2016.1229776
Jin, Q. M., Li, J. P., Zhang, X. W., Wang, G. X., Song, S. Y., Liu, Y. C., et al. (2000). Establishment IPM of system of corn diseases and pest insects in the spring corn belt. J. Maize Sci. 8, 84–88.
Jones, R. K., and Belmar, S. B. (1989). Characterization and pathogenicity of Rhizoctonia spp. isolated from rice, soybean and other crops grown in rotation with rice in Texas. Plant Dis. 73, 1004–1010.
Kasuga, T., White, T., and Taylor, J. (2002). Estimation of nucleotide substitution rates in Eurotiomycete fungi. Mol. Biol. Evol. 19, 2318–2324. doi: 10.1093/oxfordjournals.molbev.a004056
Lang, G. I., and Murray, A. W. (2008). Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 178, 67–82. doi: 10.1534/genetics.107.071506
Lee, S. H., and Neate, S. M. (2007). Population genetic structure of Septoria passerinii in northern great plains barley. Phytopathology 97, 938–944. doi: 10.1094/PHYTO-97-8-0938
Little, C., Perumal, R., Tesso, T., Prom, L., Odvody, G., and Magill, C. (2012). Sorghum pathology and biotechnology. Fungal Dis. Perspect. 6, 10–30.
Lumbsch, H. T., Buchanan, P. K., May, T. W., and Mueller, G. M. (2008). Phylogeography and biogeography of fungi. Mycol. Res. 112, 423–424. doi: 10.1016/j.mycres.2008.02.002
Mandujano, G. V., Arana, C. A., Anducho-Reyes, M. A., Tellez, J. A., Gonzalez, B. A., and Mercado-Flores, Y. (2013). Biochemical study of the extracellular aspartyl protease Eap1 from the phytopathogen fungus Sporisorium reilianum. Protein Expres. Purif. 92, 214–222. doi: 10.1016/j.pep.2013.10.003
Martínez, C., Roux, C., Jauneau, A., and Dargent, R. (2002). The biological cycle of Sporisorium reilianum f.sp. zeae: an overview using microscopy. Mycologia 94, 505–514.
Matyac, C. A., and Kommedahl, T. (1985a). Factors affecting the development of head smut caused by Sphacelotheca reiliana on corn. Phytopathology 75, 577–581.
Matyac, C. A., and Kommedahl, T. (1985b). Occurrence of chlorotic spots on corn seedlings infected with Sphacelotheca reiliana and their use in evaluation of head smut resistance. Plant Dis. 69, 251–254. doi: 10.1094/PD-69-251
McDonald, B. A. (1997). The population genetics of fungi: tools and techniques. Phytopathology 87, 448–453. doi: 10.1094/phyto.1997.87.4.448
McDonald, B. A., and Linde, C. (2002). The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica 124, 163–180.
Meng, J. W., Zhu, W., He, M.-H., Wu, E.-J., Duan, G.-H., Xie, Y.-K., et al. (2015). Population genetic analysis reveals cryptic sex in the phytopathogenic fungus Alternaria alternata. Sci. Rep. 5:18250. doi: 10.1038/srep18250
Mercado-Flores, Y., Cárdenas-Álvarez, I. O., Rojas-Olvera, A. V., Pérez-Camarillo, J. P., Leyva-Mir, S. G., and Anducho-Reyes, M. A. (2014). Application of Bacillus subtilis in the biological control of the phytopathogenic fungus Sporisorium reilianum. Biol. Control. 76, 36–40. doi: 10.1016/j.biocontrol.2014.04.011
Mercado-Flores, Y., Cartagena-Luna, A., Sanchez-Maya, H. E., Pérez Camarillo, J. P., Ortega-Bernal, J., Álvarez-Cervantes, J., et al. (2016). “El carbón de la espiga del maíz en el valle del mezquital,” in Biotecnología y Alimentos en Hidalgo: Transitando a la Bioeconomía, eds L. Batalla-Díaz, C. A. Gómez-Aldapa, J. Castro-Rosas, and A. Téllez-Jurado , (México: Amalgama Arte S.A. Company), 105–116.
Munkacsi, A. B., Kawakami, S., Pan, J. J., Lee, K., Stoxen, S., Hang, J., et al. (2006). Genome-wide assessment of tandem repeat markers for biogeographical analyzes of the corn smut fungus Ustilago maydis. Mol. Ecol. Notes 6, 221–223. doi: 10.1111/j.1471-8286.2005.01199.x
Munkacsi, A. B., Stoxen, S., and May, G. (2007). Domestication of maize, sorghum, and sugarcane did not drive the divergence of their smut pathogens. Evolution 61, 388–403. doi: 10.1111/j.1558-5646.2007.00036.x
Palloix, A., Ayme, V., and Moury, B. (2009). Durability of plant major resistance genes to pathogens depends on the genetic background, experimental evidence and consequences for breeding strategies. New Phytol. 183, 190–199. doi: 10.1111/j.1469-8137.2009.02827.x
Parker, I. M., and Gilbert, G. S. (2004). The evolutionary ecology of novel plant-pathogen interactions. Annu. Rev. Ecol. Evol. 35, 675–700. doi: 10.1146/annurev.ecolsys.34.011802.132339
Pascual, C. B., Toda, T., Raymondo, A. D., and Hyakumachi, M. (2000). Characterization by conventional techniques and PCR of Rhizoctonia solani isolates causing banded leaf sheath blight in maize. Plant Pathol. 49, 108–118. doi: 10.1046/j.1365-3059.2000.00429.x
Pérez, J., and Bobadilla, M. (2007). Carbón de la Espiga Del Maìz, Sìntesis de Resultados Del Ciclo Agrìcola P.V. 2006. Houston, TX: HGO.
Pérez-Camarillo, J. P., Bobadilla-Melendez, M., Martínez-Ruíz, E., Vazquez-Carrillo, G., Garrido-Mora, E., and Mercado-Flores, Y. (2009). Aportaciones de investigación para el manejo de maíz de riego, bajo presión de enfermedades en el Valle del Mezquital, Hidalgo. Innovando Juntos 7, 4–13.
Quezada, A., De León, C., Hernández, A., and Nava, C. (2014). Evaluation of inoculation methods on maize seeds with Sporisorium reilianum f. sp. zeae (Kühn) Langdon & Fullerton. Rev. Mexic. Fitopatol. 31, 80–90.
Quezada-Salinas, A., Moreno-Velázquez, M., De León-García de Alba, C., Nava-Díaz, C., and Solano-Báez, A. R. (2017). Resistencia genética a Sporisorium reilianum f. sp. zeae en líneas seleccionadas de maíz (Zea mays L.) con endospermo blanco y amarillo. R. Mex. Fit. 35:55740.
R Core Team (2014). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Radwan, G. L. H. E. (2013). Molecular Comparison and DNA Fingerprinting of Sporisorium Reilianum and Peronosclerospora Sorghi Relating to Host Specificity and Host Resistance. Doctoral dissertation, Texas A&M University, College Station, TX.
Rosewich, U. L., Pettway, R. E., McDonald, B. A., and Kistler, H. C. (1999). High levels of gene flow and heterozygote excess characterize Rhizoctonia solani AG-1 IA (Thanatephorus cucumeris) from Texas. Fungal Genet. Biol. 28, 148–159. doi: 10.1006/fgbi.1999.1174
Sakai, A. K., Allendorf, F. W., Holt, J. S., Lodge, D. M., Molofsky, J., With, K. A., et al. (2001). The population biology of invasive species. Annu. Rev. Ecol. Syst. 32, 305–332.
Schirawski, J., Heinze, B., Wagenknecht, M., and Kahmann, R. (2005). Mating type loci of Sporisorium reilianum: novel pattern with three a and multiple b specificities. Eukaryot. Cell 4, 1317–1327. doi: 10.1128/ec.4.8.1317-1327.2005
Valverde, M. E., Vandemark, G. J., and Paredes, O. (2000). Genetic diversity of Ustilago maydis strains. World J. Microb. Biotechnol. 16, 49–55.
Wallen, R. M., and Perlin, M. H. (2018). An overview of the function and maintenance of sexual reproduction in dikaryotic Fungi. Front. Microbiol. 9:503. doi: 10.3389/fmicb.2018.00503
Wang, L., Liu, L. M., Hou, Y. X., Li, L., and Huang, S. W. (2015). Pathotypic and genetic diversity in the population of Rhizoctonia solani AG1-IA causing rice sheath blight in China. Plant Pathol. 64, 718–728. doi: 10.1111/ppa.12299
Wang, X. O. (1997). Genetic variability in the canker pathogen fungus, Gremmeniella abietina. Contribution of sexual compared with asexual reproduction. Mycol. Res. 101, 1195–1201. doi: 10.1017/s0953756297003791
Xu, M., Melchinger, A., and Lubberstedt, T. (1999). Species-specific detection of the maize pathogen Sporisorium reiliana and Ustilago maydis by dot blot hybridization and PCR-based assays. Plant Dis. 83, 390–395. doi: 10.1094/pdis.1999.83.4.390
Zambino, P. J., Groth, J. V., Lukens, L., Garton, J. R., and May, G. (1997). Variation at the b mating type locus of Ustilago maydis. Phytopathology 87, 1233–1239. doi: 10.1094/PHYTO.1997.87.12.1233
Zhang, Y. Y., Huang, N., Xiao, X. H., Huang, L., Liu, F., Su, W. H., et al. (2015). Molecular variation of Sporisorium scitamineum in mainland China revealed by internal transcribed spacers. Genet. Mol. Res. 14, 7894–7909. doi: 10.4238/2015.July.14.15
Zhao, X., Ye, J., Wei, L., Zhang, N., Xing, Y., Zuo, W., et al. (2015). Inhibition of the spread of endophytic Sporisorium reilianum render maize resistance to dead smut. Crop J. 3, 87–95. doi: 10.1016/j.cj.2015.02.001
Keywords: head smut, LAGLIDADG, BAPS, maize, phytopathogenic
Citation: Sánchez Maya HE, Mercado-Flores Y, Téllez-Jurado A, Pérez-Camarillo JP, Mejía O and Anducho-Reyes MA (2020) Molecular Variation of the Phytopathogenic Fungus Sporisorium reilianum in Valle del Mezquital, Hidalgo. Front. Ecol. Evol. 8:36. doi: 10.3389/fevo.2020.00036
Received: 07 August 2019; Accepted: 07 February 2020;
Published: 21 February 2020.
Edited by:
Michael David Martin, Norwegian University of Science and Technology, NorwayReviewed by:
Claudia Barros Monteiro Vitorello, University of São Paulo, BrazilYouxiong Que, Fujian Agriculture and Forestry University, China
Copyright © 2020 Sánchez Maya, Mercado-Flores, Téllez-Jurado, Pérez-Camarillo, Mejía and Anducho-Reyes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Angel Anducho-Reyes, YW5kdWNob0B1cHAuZWR1Lm14