
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 26 November 2019
Sec. Evolutionary and Population Genetics
Volume 7 - 2019 | https://doi.org/10.3389/fevo.2019.00439
This article is part of the Research TopicHerbarium collection-based plant evolutionary genetics and genomics View all 8 articles
 Laura L. Forrest1
Laura L. Forrest1 Michelle L. Hart1
Michelle L. Hart1 Mark Hughes1
Mark Hughes1 Hannah P. Wilson1,2Kuo-Fang Chung3
Hannah P. Wilson1,2Kuo-Fang Chung3 Yu-Hsin Tseng3
Yu-Hsin Tseng3 Catherine A. Kidner1,4*
Catherine A. Kidner1,4*Over the past 300 years of plant collecting for herbaria, the basic method of preservation has remained remarkably consistent—a plant press with absorbent paper. However, the difficulty of drying plant specimens in the humid tropics has led to a variety of additions to this basic technique, primarily to prevent the specimens succumbing to fungal and bacterial breakdown. These additions include drying gently in tents over low fires, soaking the specimens in alcohol before pressing and, more recently, drying the specimens with forced heat from hair dryers. The process of drying is, however, known to cause breakage and damage to the plant's DNA, as well as providing time for non-plant organisms (bacteria and fungi) to multiply in the tissue, potentially “swamping” the plant specimen's own DNA. Contemporary plant collectors therefore usually collect a separate sample for DNA work, usually rapidly dried in silica gel desiccant; historical collections, however, may have been treated with alcohol and/or heat. We have recently shown that Hyb-Seq provides a reliable method for retrieving robust sequence data from even very old and degraded herbarium specimens. In this study we have used a panel of specimens preserved using a variety of methods to assess whether Hyb-Seq is capable of retrieving informative amounts of sequence data from duplicate specimens preserved using a range of specimen preservation methods. We present data on the amount and quality of DNA and of sequence data retrieved, the variation in error types between preservation techniques and the utility of the data for phylogenetic analysis.
Over the c. 3 centuries that plant collections have been deposited in herbaria, a wide variety of plant preservation techniques have been used (Smith, 1971; Bridson and Forman, 1999; RBGE, 2017), including air-drying, heat, and soaking specimens in alcohol prior to drying (the Schweinfurt method; Schrenk, 1888). The main aim of these methods is to preserve the morphology of the specimens, in particular their reproductive structures, for identification purposes and for taxonomic research, but recent technological developments have opened up their use for genetic studies (Rowe et al., 2011; Nachman, 2013; Bieker and Martin, 2018). However, the reported effect of different collecting methodologies on the preservation of genetic material varies widely. Harris (1993) field-tested three plant drying methods, finding little difference in DNA quality between specimens dried in paper, with heat, or in silica gel. Särkinen et al. (2012) showed, on the other hand, that the ability of PCR to amplify longer loci drops for samples preserved using heat or alcohol.
Previously we have shown that Hyb-Seq allows recovery of hundreds of kilobases of nuclear genomic sequence from herbarium specimens collected in tropical rainforests over 170 years ago (Hart et al., 2016), although unfortunately we do not have records of the preservation methods that were used by the nineteenth century collectors of the specimens sequenced. In order to see how widely applicable Hyb-Seq will be as a method for recovering genetic information from herbarium specimens, in this paper we examine the effect of different preservation methods on DNA quantity and quality and sequence analysis, including library quality, library capture success, sequencing, read assembly and consensus calling, for a range of common plant collection methodologies.
We used Begonia L., one of the largest plant genera, as a model for this study as it is the focus of ongoing phylogenetic and genomic research at Royal Botanic Garden Edinburgh (RBGE) and Academia Sinica and represents many of the challenges faced when working with plants from the wet tropics. Begonia leaves can be fleshy, with a water-filled hypodermis providing structural support, allowing isolated leaves to remain hydrated for many days. Drying Begonia leaves quickly enough to preserve good specimens in the field can thus be difficult given the areas of high humidity where the plants are typically found. Our test data set comprises three species from RBGE's living collection of Southeast Asian Begonia: Begonia bipinnatifida J.J.Sm. (section Petermannia), B. serratipetala Irmsch. (section Petermannia), and B. trichopoda Miq. (section Reichenheimia) (Figure 1A).
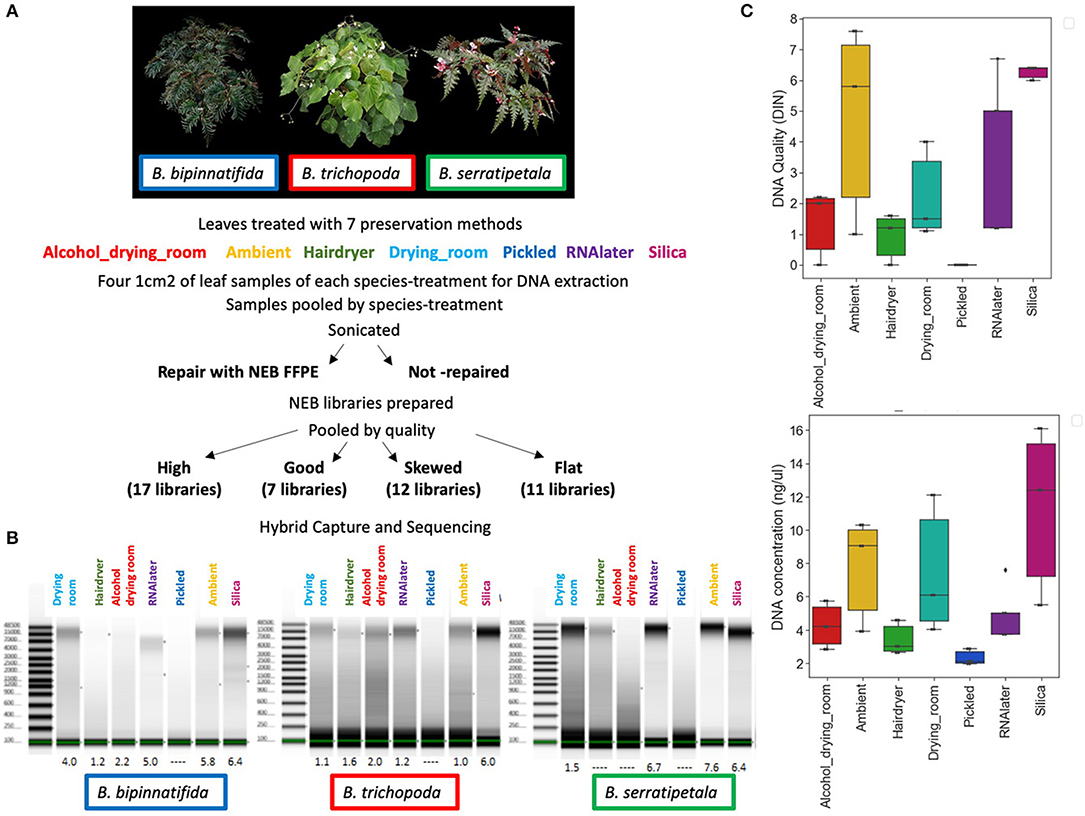
Figure 1. DNA quality and quantity. (A) Individuals sampled: B. bipinnatifida (RBGE living accession 20090801), B. trichopoda (RBGE living accession 20161284), B. serratipetala (RBGE living accession 19681637) and treatment scheme (see section Materials and Methods). (B) Tapestation images of DNA from 4 pooled extractions, prior to splitting the extractions into FFPE repair/not-repair aliquots, with DIN values listed at base. “No DIN assignable” is represented as a dash. (C) Effect of drying method on DNA quality and concentration by treatment (N = 3, one for each species). “No DIN assignable” is represented numerically as zero.
The preservation methods we chose to test reflect those commonly in use at the herbarium of the Royal Botanic Garden Edinburgh. Since the 1990s, most collectors have collected plant tissue into silica gel specifically for genetic analysis (Chase and Hills, 1991; Wilkie et al., 2013); some now also collect into RNAlater™ to preserve the plant's RNA for possible transcriptomic analysis. The vast majority of RBGE's herbarium collections are dried, and some have associated pickled reproductive structures. Many of the tropical collections have been treated with alcohol immediately after collection, to prevent fungal/bacterial growth prior to the drying process. It is also advantageous, in terms of good color preservation, to help the drying along with gentle heat. This can be provided by a permanent or temporary drying room (such as a tent built over a stove) or by using forced heat, in our case via a hair dryer. Drying, alcohol and heat are, however, known to damage plant genomic DNA through fragmentation and base changes (Staats et al., 2011). In addition, fragmentation caused by depurination-driven breakage occurs exponentially through time in storage (Weiß et al., 2016). The accumulation of thymine bases due to the deamination of cytosine also increases with time, leading to an excess of C to T substitutions toward both ends of the DNA fragments. This can be used as a signal of authenticity for sequences derived from ancient DNA (Briggs et al., 2007; Krause et al., 2010; Mouttham et al., 2015; Linderholm, 2016), but could cause convergence of sequences between samples over time, leading to similarity due to age related damage rather than phylogenetic distance. Much of the observed DNA damage in herbarium specimens appears to occur during the initial preservation process (Staats et al., 2011), with further gradual damage during long term storage. However, Staats et al. (2011), sampling herbarium and fresh material of the same individuals separated by 40–120 years, did not find the types of nucleotide misincorporation that are associated with ancient DNA in the herbarium material they studied.
We present results using Hyb-Seq protocols established by Hart et al. (2016) on seven commonly used herbarium specimen preservation methods: silica-gel dried tissue, RNA-later™, ambient drying in newspaper flimsies, drying at a slightly elevated temperature (in a drying room), drying after an alcohol wash (the Schweinfurt method), drying at high temperature using forced heat (the hair dryer method), and pickling in alcohol (Figure 1A). For each of the preservation treatments we compare the DNA quantity and DNA Integrity Number (DIN; a fragmentation parameter as assessed by the Agilent TapeStation; Gassmann and McHoull, 2015).
Increasing use of preserved material for DNA extraction has led companies to produce enzyme mixtures to repair damage commonly caused by preservation in Formalin-fixed, paraffin-embedded (FFPE) blocks. However, these mixtures are advertised as being able to repair many different types of basic DNA damage, and have accordingly been used in studies where despite samples not being preserved in FFPE blocks, the DNA extracted is of a low quality (Gorden et al., 2018). We tested the effect of this repair protocol on preserved plant material by splitting each DNA extraction and using NEB FFPE repair (NEB #M6630S) on one aliquot from each treatment before library preparation.
To capture sequences from the prepared libraries we used a Begonia-specific RNA bait set known to work well with silica-gel preserved tissue samples (Yu-Hsin Tseng and Kuo-Fang Chung unpublished results). We sequenced the captured libraries and compare number of paired-end reads, the efficiency of capture by the bait set, and our ability to call SNPs correctly from the captured reads. In the light of these results we assess the potential for Hyb-Seq to provide useful data for phylogenetic and genomic research using DNA extracted from material preserved using a variety of methods.
We sampled from three Begonia accessions in the living collection at RBGE (Figure 1A; Table 1). Material from individual plants was split between seven different material preservation treatments: the alcohol and drying room method, the ambient method, the hair dryer method, the RBGE drying room, pickling in 70% alcohol, collecting tissue into Invitrogen™ RNAlater™ and subsequent storage at −20°C, and collecting tissue directly into silica gel desiccant.

Table 1. Study material.
1. Alcohol + Drying Room (Schweinfurt method)
Each specimen was immediately placed in a folded piece of dry newspaper. These were stacked to make a pile, then wrapped with four extra sheets of newspaper to create a bundle which was secured with string before being placed in a strong plastic bag. Alcohol (50% ethanol) was added to the bag, which was then sealed securely before shaking to ensure the whole bundle was evenly wet. Specimens were stored in this way for 3 days before being transferred to dry newspapers. The specimens were then dried in the RBGE drying room as described below.
2. Ambient
Each specimen was immediately placed in a folded piece of dry newspaper and added to a field press (not in a plastic bag), along with material from the two other test plants. Blotting cards were placed between specimens. The damp newspaper was replaced daily with dry newspaper until the specimen was completely dry, following the method that is used when there is no forced air or heat available in the field.
3. Hair dryer method
Specimens were placed in dry newspaper, surrounded by blotting card, and then sandwiched between corrugated cardboard, with the corrugations in each layer running parallel to the short sides of the press to allow air flow. A cowling of polythene sheeting was wrapped around the press and secured tightly with gaffer tape, with one long end of the press left open, and the other taped firmly around the end of a 1,600 watt hair dryer. The specimens were dried for 18 h. This method is frequently used by RBGE expeditions to produce specimens with well-preserved color from difficult genera such as Begonia.
4. Drying room
Each specimen was placed in a folded piece of dry newspaper, and then placed between blotting cards, and then between steel corrugated boards to allow air to circulate. Bundles of specimens were placed in a wooden slat herbarium press, with 2 kg of weights on top, and left in the drying room for 2 weeks. The drying room was kept at 30–35°C and 25–30% relative humidity using an electric fan heater and dehumidifier.
5. Pickled
Leaves were torn into sections of c. 1cm2 and placed in 30 ml of 70% ethanol in 50 ml falcon tubes, then stored at room temperature (c. 18°C) for 3 weeks prior to DNA extraction.
6. RNAlater
At least 1.5 ml of Invitrogen™ RNAlater™ was placed in a 2 ml Eppendorf tube. The lid of the tube was then used to punch out 6 circular pieces of tissue from leaves still attached to the living plant. These were then stored at −20°C for 2 weeks before DNA extraction.
7. Silica gel collection
At the point of collection a tissue sample was placed in an open teabag, which was folded over and then stored in 0.2–0.5 mm granular silica gel desiccant in an airtight container.
The RBGE herbarium's integrated pest management strategy requires all incoming specimens to be frozen at −30°C for at least 5 days, so all the herbarium samples (i.e., those preserved using the alcohol and drying room method, the ambient method, the hair dryer method and in the RBGE drying room) were stored at −30°C for 5 days after drying, then transferred to the herbarium and stored in herbarium cabinets (c. 20°C, 50% relative humidity) for 3 days prior to tissue being removed for DNA extraction. The pickled samples and the silica gel dried tissue samples were not given this additional freezing step, as material would not be frozen coming into our spirit or dried tissue collections.
Four separate DNA extractions were made per sample, each using leaf tissue equivalent to c. 1 cm2. Dry plant tissue was placed in 2 ml Eppendorf tubes with two 5 mm tungsten beads and frozen overnight before homogenization using a Qiagen TissueLyser; wet plant tissue was removed from solution, air dried for 30 s−1 min, and then homogenized using mini-pestles and a pinch of acid washed sand. Extractions used standard Qiagen Plant DNeasy mini-columns, and followed the manufacturer's protocol except that the initial incubation at 65°C was increased to 1 h, and with the four extractions combined onto a single DNeasy mini-spin column prior to elution, in order to produce a more concentrated DNA elution. The DNA was eluted in 90 μl of Qiagen AE buffer.
The DNA was quantified using Qubit dsDNA HS chemistry on a DeNovix DS-11, with duplicate reads for all samples. Relative sample purity was assessed using the spectrophotometry option on a DeNovix, and the test set of 23 DNA extractions was also run on Agilent TapeStation Genomic DNA screen tapes to visualize the integrity of the DNA and to obtain DIN scores where possible (Figure 1B).
The DNA extractions were normalized to 1.9 ng/μl (or as close as possible) in resuspension buffer, and re-quantified using Qubit dsDNA HS chemistry on the DeNovix DS-11. The normalized DNA samples were split into two 53 μl aliquots in 1.5 ml tubes and sonicated, for between 2 and 8 cycles of 30 s on, 90 s off, using a Diagenode Bioruptor sonicator. The number of sonication cycles was chosen based on the TapeStation images of the genomic DNA extractions, with fewer cycles required for more degraded DNA. The sonicated DNA was bead-cleaned with 80 μl of beads, and the two aliquots were recombined, run on a Bioanalyzer High Sensitivity DNA chip, and quantified using the DeNovix dsDNA ultra-high Sensitivity assay kit and the DeNovix DS-11. For the pickled plant DNA extractions, no sonication or bead clean was used, as the DNA was already highly degraded and in very low quantity. Each 106 μl aliquot of normalized DNA was split into two equal aliquots, with NEB FFPE repair carried out on one aliquot from each treatment. This was followed by end repair and adaptor ligation, following the NebNext Ultra II DNA Library Prep Kit for Illumina protocol.
Samples that contained over 50 ng of DNA were size-selected using NEBNext Sample Purification beads at c. 0.4X (keeping the supernatant) and 0.2X (keeping the beads), to generate a library of c. 300 bp fragments. Samples with <50 ng of DNA were instead bead-cleaned with 0.9X NEBNext Sample Purification beads. The samples were then quality checked using a Bioanalyzer High Sensitivity DNA chip, and the DeNovix dsDNA ultra-high Sensitivity assay kit, prior to amplification of 7–12 cycles (depending on DNA quantity), for PCR enrichment. Following enrichment, the samples were again quantified using the DeNovix Qubit dsDNA HS assay and run on a Bioanalyzer High Sensitivity DNA chip, in order to normalize each library to 10 nM.
Libraries were scored for quality based on the fragment size profiles of their Bioanalyzer traces. Trace shape most heavily influenced these groupings, although data such as peak fragment length, average fragment length, peak fluorescence (FU), and quantity were also considered at this stage. Pool 1 comprised the 17 highest quality libraries whose traces showed narrow, symmetrical peaks, the 7 libraries in pool 2 had a clear peak at 250–350 bp, but overall the trace showed a left-skewed distribution, pool 3 contained 12 libraries that were profoundly left skewed and pool 4 contained 11 libraries that had a near-flat line on the bioanalyzer (see results).
The volume of each library added to the pools was chosen so that each final pool contained over 500 ng of DNA. Pools were concentrated in a spin-vac down to a final volume of 7 μl.
The bait set was designed using a draft genome of the American species Begonia conchifolia A.Dietr. (section Gireoudia) as a reference (C. Kidner and A. Bombarely, unpublished data) and a transcriptome from vegetative buds and male flowers of B. luzhaiensis T.C.Ku (section Coelocentrum, K-F Chung, SRA: PRJNA378679) to give SE Asian clade orthologs. Bait sequences comprised B. luzhaiensis sequences which had a single unique hit in the B. conchifolia genome and a single unique hit in the proteome of the cucurbit Cucumis sativus L. (1114 sequences). We also added the B. luzhaiensis orthologs for 130 SNP sequences previously placed on the Begonia conchifolia genetic map of Brennan et al. (2012) and B. luzhaiensis sequences of 94 shade-growth associated and key developmental genes. When duplicates were removed, this resulted in 1,239 unique sequences (Supplementary Table 1—Excel list of baits, %ID across Begonia, Annotation).
Hybridization of the libraries followed Nicholls et al. (2015) with some modifications. Reaction volumes were halved, and we used a 14 h hybridization. Some of the hybridizations were repeated at a 5°C lower temperature, after the post-capture PCR did not result in enough DNA for sequencing. The number of post-capture PCR cycles depended on the pool, varying from 13 to 20 cycles. Each pool was then quantified using the Qubit dsDNA HS assay on a DeNovix DS-11, run on a Bioanalyzer High Sensitivity DNA chip, then diluted using 0.1X TE to 15 nM. Pools 3 and 4 were bead-cleaned to remove relatively high adaptor peaks.
Two final pools were made for sequencing, the first by combining the two pools of higher quality libraries (Pools 1 and 2; 24 libraries in total), and the second by combining the two pools of lower quality libraries (Pools 3 and 4; 23 libraries in total) into 40 μl volumes. The 15 nM pools were combined using [(number of libraries per pool)/(total number of libraries that will be run on the lane) × 40] μl of each pool. These two final pools were quantified using the Qubit ds DNA HS kit on a DeNovix DS-11, and run on a Bioanalyzer High Sensitivity DNA chip. A total of 35 μl of each final pool was then sent to the Earlham Institute, with each final pool run on a separate miSEQ lane, with 250 bp paired end reads.
Assembly and further analysis were done on the Cyverse Atmosphere servers (National Science Foundation under Award Numbers DBI-0735191, DBI-1265383, and DBI-1743442. URL: www.cyverse.org) using scripts collected at https://github.com/ckidner/Basic_Hyb_Seq_Assembly. Raw reads were trimmed of adapters and quality trimmed using Trimmomatic-0.36 and Cutadapt. FastQC was used to confirm quality pre-and post-trimming. The capture success was assessed by mapping the trimmed reads to the bait sequences and to the complete plastid genome of B. bipinnatifida (provided by L. Campos Dominguez) using Bowtie2 with default parameters (Langmead and Salzberg, 2012). BWA-MEM was used to map trimmed reads to the reference bait set. We used samtools to remove duplicate reads, and bcftools to generate VCF files with quality filtering of QUAL > 20 and DP > 10 (Li et al., 2009). Consensus sequences were derived from the vcf files using vcfutils_fasta.pl, part of the vcftools package (vcftools.github).
Consensus sequences were concatenated by sample using amas-0.93 and trimmed using trimAl v1.2 (Capella-Gutiérrez et al., 2009). Sequences were aligned using MAFFT (Katoh et al., 2009), with FastTree 2.1 (Price et al., 2010) used to produce a maximum likelihood phylogram for the sequenced libraries.
To assess the errors due to different treatments, firstly a conservative consensus sequence was produced for each species as follows: Mapping stringency in BWA was optimized to a conservative level, setting the “-T” parameter to 100 to minimize mis-matches due to paralog mapping. The stringently mapped reads from the silica samples (both repaired and non-repaired libraries) were used to call variants and the resulting VCF files were quality filtered before generation of consensus sequences per bait per species. These consensus sequences were then used as a reference to map reads from all the treatments for that species. VCF files from the silica reads and the other treatments were compared, to identify SNPs found in both (alleles) and SNPs specific to the non-silica sample (likely errors).
Metrics of the numbers, types and average quality of variants for each treatment which were specific to the treated sample and shared with the silica sample were gathered from the VCF files using gather_vcf_stats.sh (https://github.com/ckidner/Basic_Hyb_Seq_Assembly).
DNA preparations from the samples ranged from excellent quality and quantity (silica dried for B. bipinnatifida and B. serratipetala) to barely any detectable product (pickled B. trichopoda). Both quantity and quality of the DNA were significantly affected by treatment, being best from silica dried material and worst from pickled and hair dryer processed specimens (Figure 1C). One way ANOVA indicated that quantity was only marginally significant at F(6, 16) = 2.204, p < 0.0968, R = 0.453 and quality at F(6, 16) = 3.618, p < 0.0267, R = 0.568. Tukey's HSD test (a = 0.05) supported a difference between silica and pickled treatments. The limitation of sample number prevents us being able to fully statistically assess all the factors which vary between treatments. There was considerable variation in quality, in terms of size distribution and quantity, between libraries produced from these DNA preparations (Figure 2). To prevent the higher-quality libraries swamping the lower-quality ones, samples were pooled by quality for capture, into four pools (Table 2).
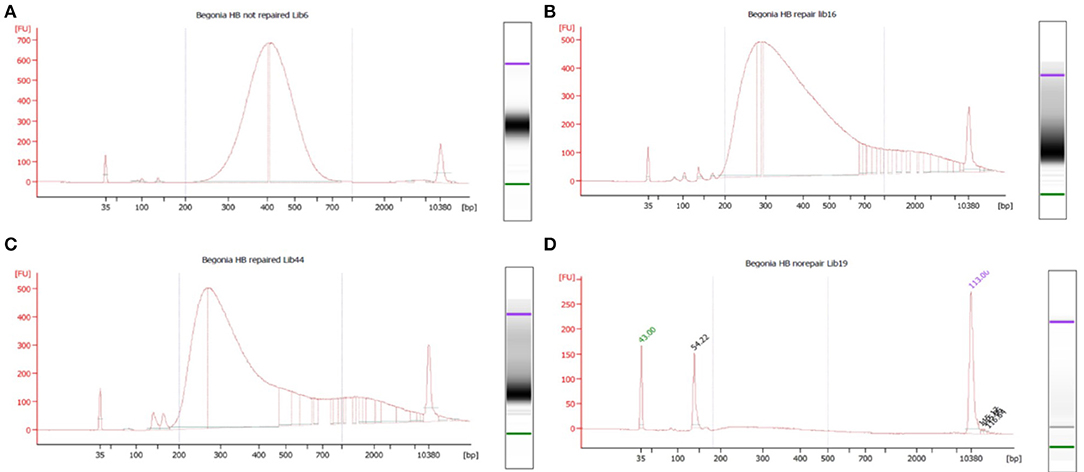
Figure 2. Bioanalyser High Sensitivity DNA assays for representative libraries from each of the four capture pools, showing fragment size distribution. Base pairs (x-axis) and fluorescence units (y-axis) are shown on the graphs. The upper and lower size markers are represented in purple and green, respectively, on the gel image to the right of each graph. (A) B. bipinnatifida, Ambient, not repaired (pool 1—high quality). (B) B. trichopoda, Hair dryer, repaired (pool 2—left skewed). (C) B. serratipetala, Hair dryer, repaired (pool 3—profoundly left skewed). (D) B. trichopoda, Pickled, not repaired (pool 4—flat). A single adaptor peak is clearly visible on the graph and the gel image.
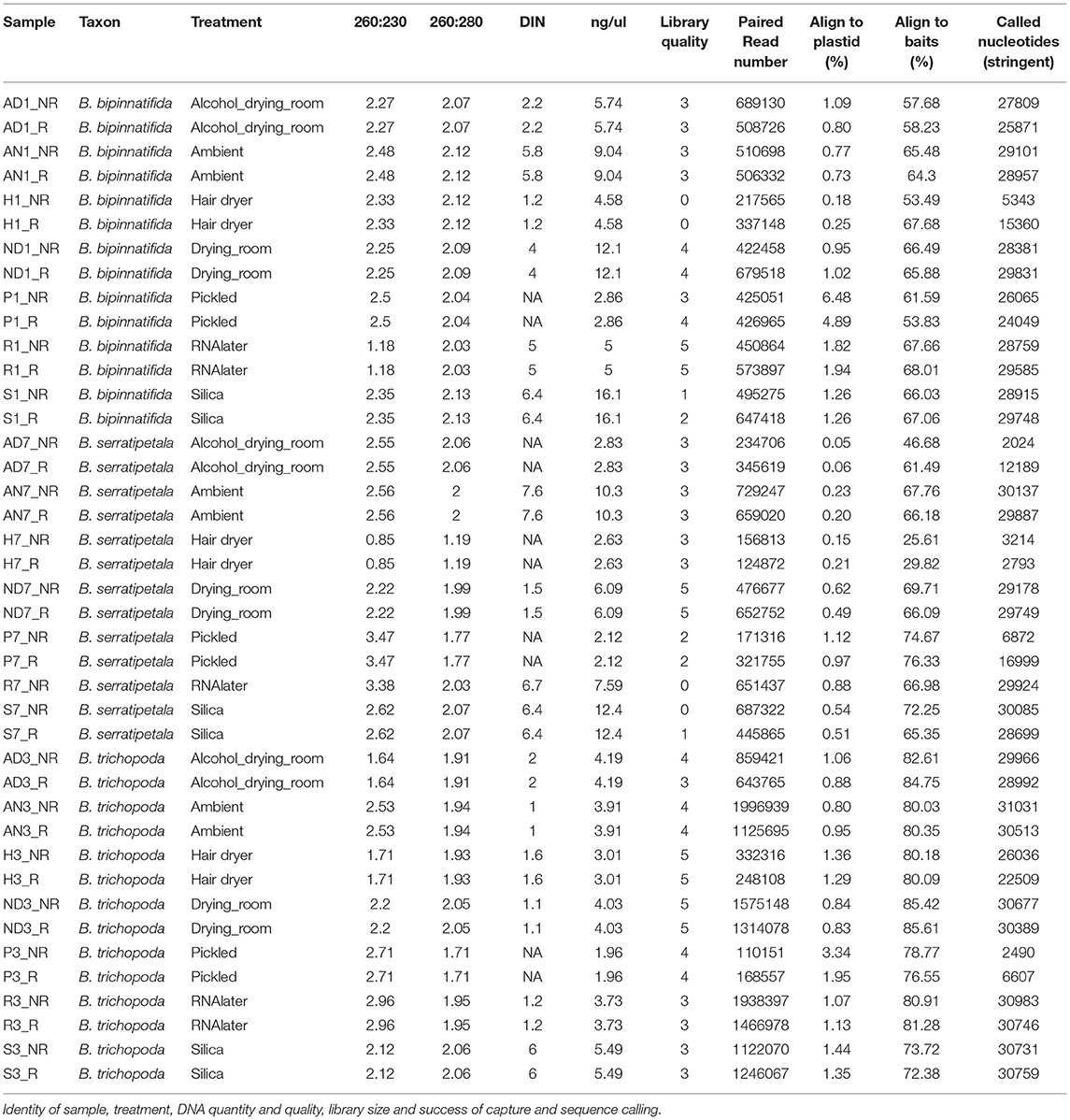
Table 2. Analysis metrics.
There was no strong correlation between number of reads generated and the concentration or quality of the initial DNA (Figure 3, R squared 0.009); though very low amounts (<3 ng/μl) produced low numbers of reads, some low quality/quantity samples from B. trichopoda produced very high numbers of reads.
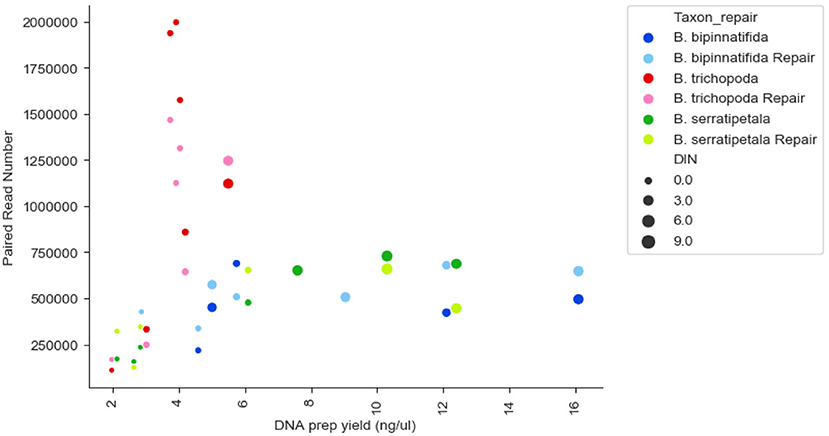
Figure 3. Reads generated per sample by DNA prep yield (ng/μl), DNA quality (DIN), and species. Each species has 14 datapoints: one FFPE repaired and one un-repaired for each of the 7 treatments. DIN is represented by size of marker, species by color of marker, repair by shade of marker.
Capture was excellent across most samples (the exceptions being hair dryer treated B. serratipetala). A small amount of plastid DNA was present in each library, the largest amounts being seen in the pickled samples (Figure 4A). However, this was not sufficient to allow reconstruction of plastid genomes from individual treatments.
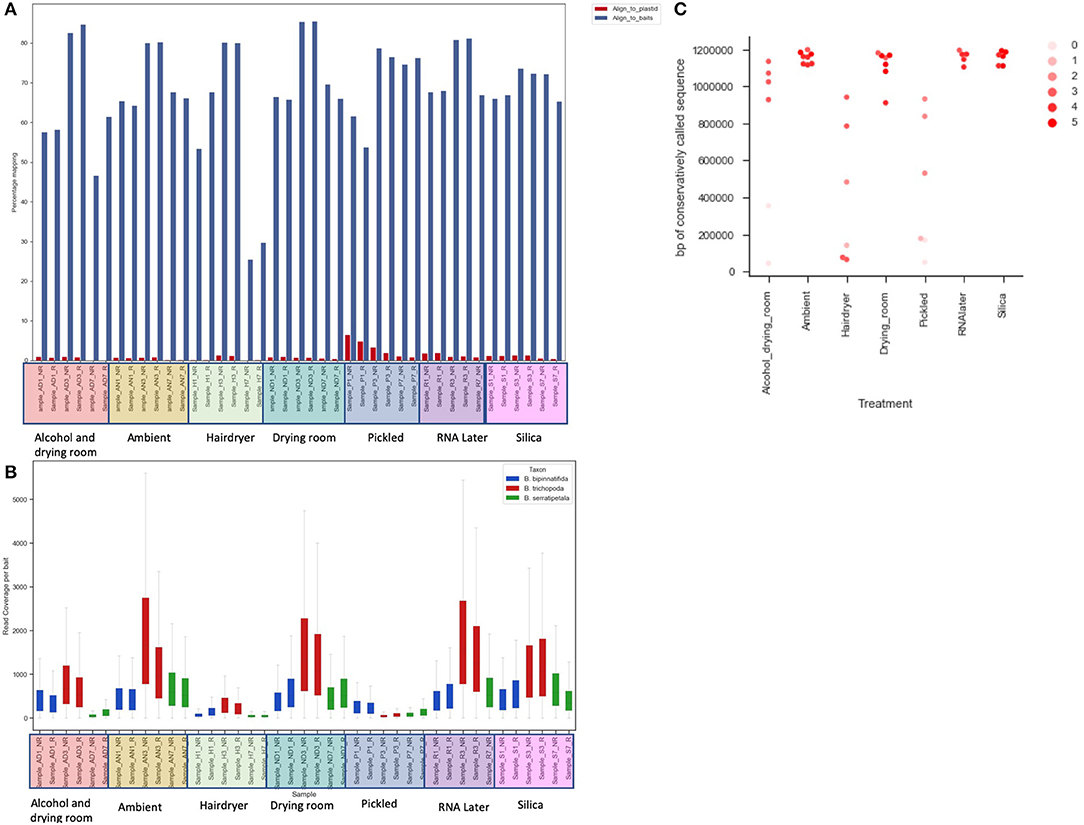
Figure 4. The effect of treatment on recovery of bait sequence, coverage of baits and length of consensus sequence called. (A) Effect of treatment on percentage of reads mapping to baits (blue) and to plastid sequence (red) using BWA. (B) Read coverage per bait by treatment (x-axis) and by species (color of bar). (C) Length of sequence retrieved from each sample using conservative BWA mapping by treatment (x-axis) and library quality (arbitrary scale—see text; shade of marker).
Read coverage per bait was good, but showed a strong variance between species, which may be due to the genetic distance of each species from B. luzhaiensis, the source of the bait sequences. The treatments “Pickled” and “Hair dryer” performed worst. The mapped reads were used to call variants and the VCF files quality filtered before generation of consensus sequences per bait per sample. The sequences for each bait were aligned, trimmed and the alignments concatenated. A Maximum Likelihood (ML) phylogram based on this alignment is shown in Figure 5A. The samples from each individual plant are monophyletic, but there is considerable distance between some (Figure 5B). To some extent this can be explained by high levels of missing data for some of the samples, as shown in Figure 4C.
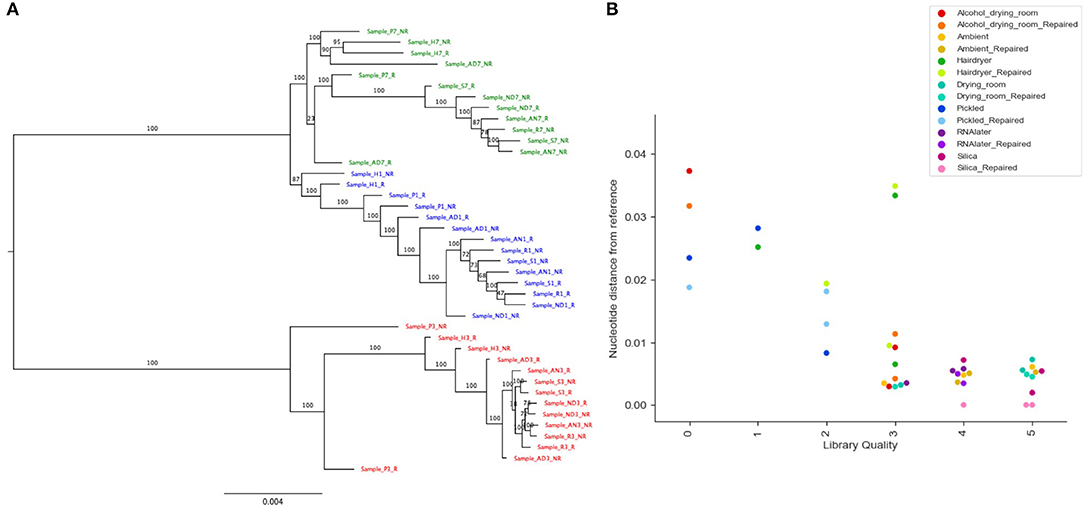
Figure 5. Phylogenetic analysis of recovered sequence. (A) Phylogram of consensus sequences from each sample. Produced using RAxML from a trimmed Mafft alignment of a concatenation of all consensus bait sequences. Samples from B. bipinnatifida are colored in blue, B. serratipetala in green and from B. trichopoda in red. B. trichopoda was designated outgroup. Numbers on the branches represent ML Bootstrap supports. Sample codes represent the tissue preservation treatments, and whether the DNA was FFPE repaired or not. (B) Genetic distance of sample from reference (silica dried repaired consensus) by library quality (arbitrary scale, x-axis), by treatment (color of marker), by repair (shade of marker). Calculated from Mafft alignment using PAUP.
Figure 6 shows the numbers of each type of SNP for each treatment. There is a clear effect of preservation treatment on SNP presence, and also variation between species. For B. bipinnatifida the hair dryer-treated samples showed most errors, due to unique SNPs, and also due to SNPs that are missing from the hair dryer samples but present in the silica-dried reference for this species. For B. serratipetala it was the pickling process that caused the most errors, and in B. trichopoda it was both alcohol and the hair dryer treatment.
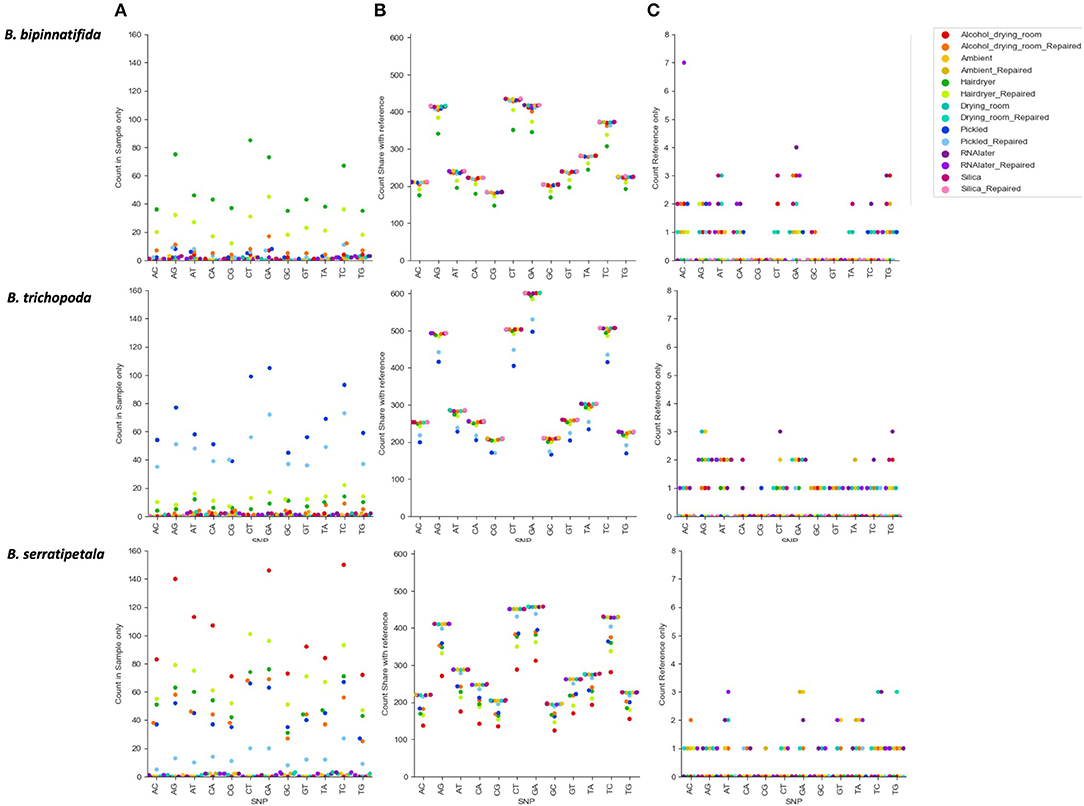
Figure 6. Count of SNP types by species and by treatment. SNPs unique or shared between reference sample (silica dried repaired consensus) and different treatments (colored markers). (A) SNPs found in treatment sample only. These represent likely nucelotide changes due to treatment, or errors due to difficulty calling with low coverage. (B) SNPs shared between treatment sample and reference (true SNPS). (C) SNPs found in reference only (erroneously lost from treatment sample).
The SNPs shared between the reference and the treated sample (SNPs due to heterozygosity in the sample) clearly show a higher proportion of transitions vs. transversions (Figure 6B), but there is no clear pattern in the types of SNPs erroneously called (Figure 6A). For some samples there is a higher proportion of CT and GA SNPs in the errors than in the true SNPs, but numbers are small and we do not detect them at as high a level as found in Staats et al. (2011), perhaps due to the short duration of our treatments. A MapDamage analysis failed to find any signal typical of ancient DNA (Jónsson et al., 2013). Generally the effect of making libraries from repaired vs. unrepaired DNA is small; however, for the hair dryer (B. bipinnatifida, B. serratipetala, and B. trichopoda), pickled (B. serratipetala and B. trichopoda) and alcohol (B. trichopoda) treatments, the libraries from the unrepaired DNA consistently had higher numbers of erroneous SNPs of all types (Figure 6A).
Overall, the percentage of erroneous SNPs ranged as high as 50% (B. trichopoda, alcohol treatment), but in most cases it was below 10% (Figure 7A). The number of erroneous SNPs could be controlled by modifying the filtering options. Distribution of depth and quality for erroneous and correct SNPs shows that the majority of erroneous SNPS had low scores for both, and could be excluded by increasing quality filtering parameters (Figures 7B,C). The distributions shown in Figure 7C suggest that increasing filtering on quality would be the best way to exclude erroneous calls without loss of too much data.
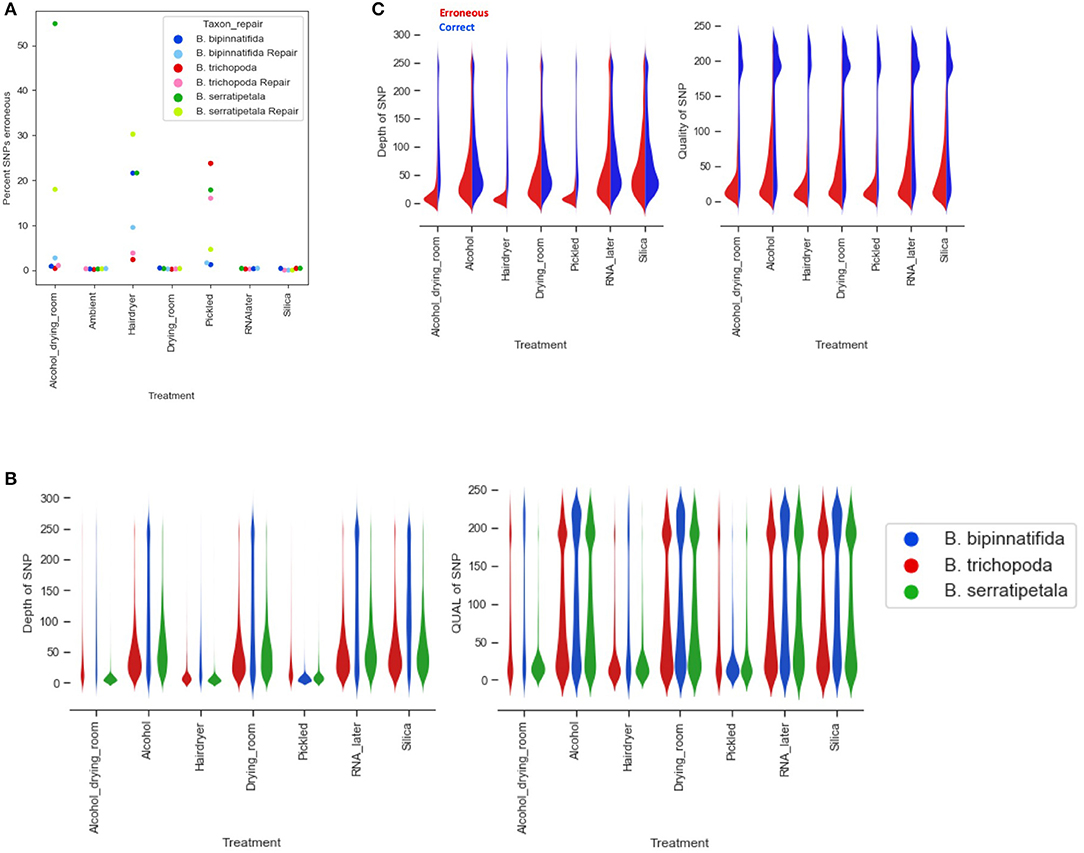
Figure 7. Erroneous SNPs by species and treatment. (A) Percentage of erroneous SNPs by treatment (x axis) and by species (marker color). (B) Depth and quality of all SNPs called by treatment (x axis) and by species (plot color). (C) Depth and quality of SNPS called erroneously (red) and correctly (blue) by treatment.
Re-running the analysis using a quality cut-off which limits the number of erroneous calls likely to be included (QUAL > 160), based on Figure 7C, results in a large decrease in the number of bases called and loci recovered (Figures 8A,B). The stricter settings reduce the distance seen between samples from the same individual but also fail to resolve the two more closely related species, B. bipinnatifida and B. serratipetala (Figure 8C). Some, but not all, of the heat and alcohol treated specimens and the pickled samples from B. trichopoda produce very little sequence using this quality cut-off, resulting in large amounts of missing data in the alignment.
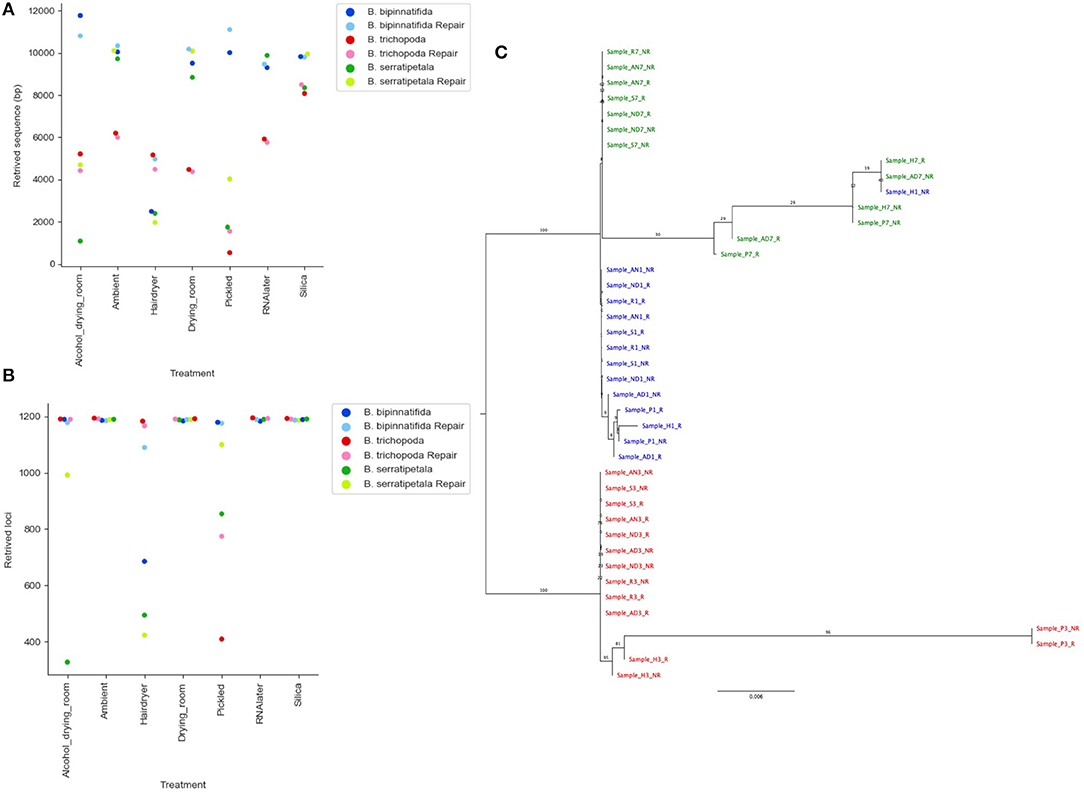
Figure 8. Effect of stricter quality filtering on sequence recovery and phylogenetic analysis. (A) Length of sequence recovered using strict quality filters (QUAL > 160). (B) Number of loci recovered using strict quality filters (QUAL > 160). (C) Phylogram of consensus sequences from each sample. Produced using RAxML from a trimmed Mafft alignment of a concatenation of all consensus bait sequences using strict quality filters (QUAL > 160). Samples from B. bipinnatifida are colored in blue, B. serratipetala in green and from B. trichopoda in red. B. trichopoda was designated outgroup. Numbers on the branches represent ML Bootstrap supports. In (A,B), lighter shades are used for repaired libraries.
The richness of herbarium collections and their value for research and conservation is well-acknowledged (Soltis, 2017). A major limitation in their use for genetic analysis is the poor quality and potential contamination of DNA extracted from herbarium specimens (Drábková et al., 2002; Staats et al., 2011; Weiß et al., 2016; Bakker, 2018). Approaches to access the potential of herbarium specimen DNA whilst overcoming the limitations differ. Genome skimming sequences all the DNA available, making the widest survey of the genetic information present. However, the analysis of this data is complex, as the DNA from the plant of interest may be a small proportion of what is sequenced and genome coverage is patchy (Staats et al., 2013). Analysis is greatly helped by the possession of a reference for the species or a close relative, which is not common for lineages that do not include crops or model plants. Much of higher plant genomes is repetitive DNA and sequences from these regions are generally not useful for genetic analysis (but see Dodsworth et al., 2015). Assembly of plastid genomes is easier, due to higher copy number and better stability of some regions (Bakker et al., 2016). However, plastid DNA can lack sufficient signal to resolve some radiations (e.g., Turner et al., 2016) and by providing information only on the maternal lineage, can obscure hybridization events (Hughes et al., 2018). Hybrid capture, or hybrid baits (Gnirke et al., 2009), by focusing sequencing effort on phylogenetically useful plant sequences, gives much higher coverage and less contamination. The baits themselves provide a reference for assembly, simplifying the bioinformatic work. We have previously shown that this method allows sequence retrieval from some very old herbarium specimens (Hart et al., 2016), and in this paper we trial the method on a range of different plant preservation methods to provide guidance on using this approach to obtain useful sequence data from any herbarium specimen.
With the seven different tissue preservation treatments tested in this study, only a few weeks (from harvesting from the RBGE living collection on the 9th February 2018 to DNA extractions on 6th March 2018) saw a dramatic difference in the quantity and quality of DNA obtained from the different preserved plant tissues. Of the preservation methods used, silica gel proved consistently reliable, while treatments involving heat or alcohol lead to recovery of less DNA, and of more degraded DNA. This is in line with previous reports (Staats et al., 2011; Särkinen et al., 2012; Bressan et al., 2014; Weiß et al., 2016). Contemporary field collections for molecular work are usually silica gel-dried, or less frequently, collected into high salt buffers or RNAlater; however, it is not always possible to work with recent plant material. Loss of habitats, plant rarity or extinction, political unrest in key localities, or simply the high costs involved with field collecting can leave sampling from herbarium collections as the only feasible option. Also, nomenclatural difficulties are best resolved by sequencing from type material.
Our investigation has focused on using Hyb-Seq methods to compare how genetic data gathered from the same plant, treated in different ways, performs in terms of quality, coverage and capturing allelic variation. We were able to retrieve sequence information even from the very poorest samples, with most samples giving thousands of base pairs of reliable sequence. Above a minimal threshold we saw no strong correlation between the amount or quality of the DNA that was extracted from the samples, and the number of sequenced reads generated. In this small sample, the strongest predictor of number of sequenced reads was species (Figure 3). Silica preserved specimens usually gave the most DNA, of the highest quality, and produced the largest amounts of sequence data under conservative and very strict bioinformatic settings. We would thus recommend using tissue preserved in silica gel wherever possible. However, for many studies this is not feasible, and herbarium material preserved in a poor or unknown fashion is all that is available.
The worst performing samples in this data set were: pickled B. trichopoda (both repaired and non-repaired), hair dryer-treated B. serratipetala (both repaired and non-repaired), alcohol-treated and pickled B. serratipetala (non-repaired) and hair dryer-treated B. bipinnatifida (non-repaired). All these samples produced under 4 kb of sequence under the strictest bioinformatics settings. Whether such a return is worth the work and expense will depend on the value of the sample, but we would not have been able to discount these libraries from capture or sequencing based on their poor quality. It is notable that the worst performing samples were not all from plant specimens that had experienced freezing. Freeze-thaw cycles on extracted DNA are known to degrade the DNA molecules through physical stress (Shao et al., 2016; Klingström et al., 2018), but the freezing involved in standard herbarium curation does not prevent the recovery of sequence data.
Much of the DNA we were working with was suboptimal in terms of quantity and quality, and would likely have been rejected by commercial sequencing facilities. The quality of the individual libraries, as shown by Bioanalyzer library traces, were very poor for some of the libraries with low sequence recovery (hair dryer-treated B. bipinnatifida, alcohol-treated and pickled B. serratipetala, pickled B. trichopoda; Supplementary Material). However, the libraries generated from hair dryer-treated B. bipinnatifida were not notably poor but still produced little useable sequence data. This suggests that even if the library trace data is poor, if no better quality material is available, it is worth proceeding with capture and sequencing even for suboptimal libraries. Based on our data, it is difficult to suggest a hard and fast metric that suggests a sample is not worth sequencing. Most of the very poorest performing samples had DNA extractions with low DIN scores, but some samples with low DIN scores produced large amounts of useful data (e.g., B. bipinnatifida pickled, repaired).
The number and type of SNPs recovered varies by species as well as by treatment (Figure 7). We would expect such variation to be even greater when comparisons are made across genera rather than just within Begonia. Some of this variation could be linked to phylogenetic distance between the sequences used for the baits (from B. luzhaiensis) and the sampled species. B. bipinnatifida and B. serratipetala are both in section Petermannia, whereas B. trichopoda is in section Reichenheimia. B. luzhaiensis is section Coelocentrum, more closely related to Petermannia than to Reichenheimia. B. trichopoda however, had the higher capture rates across treatments, despite low DNA concentrations (Figure 4A). This could be related to sequencing success, as more reads were obtained for B. trichopoda samples (Figure 3), but the explanation for the higher number of reads for samples of this species is unclear.
There is considerable variation between species in their response to the different preservation treatments (Figure 4). It is possible that with increased experience and sampling, a picture will emerge of which treatments result in viable samples for different clades of Begonia, or for plants with different functional traits (e.g., hirsuitness, fleshiness, cuticle thickness, stomatal types). However, as most studies using herbarium material will be very limited in choice of samples and it is not possible to change historical collection methods, it still seems that the best option remains “try it and see.”
Our ability to call SNPs is dependent on sufficient read depth to support each call. The advantage that Hyb-Seq has over genome skims is its ability to enrich sequencing libraries sufficiently to allow SNP calling even from poor samples (Enk et al., 2014; Beck and Semple, 2015). High read coverage is essential in confidently calling SNPs and allowing population-level analysis of preserved specimens (Bi et al., 2013). However, it is noteable that not all our SNP calls will be accurate; some SNPs are seen only in the data from the silica-dried reference sample. That they appear in none of the other treatments suggests a miscall can occur even with the best quality DNA we have available. Increasing the filtering to exclude any doubtful SNPs can reduce the data to the extent that species resolution is lost (Figure 8C). A balance must be struck between harvesting what data is possible and ensuring reliability of that data.
Calling SNPs has been possible with all but the very lowest quality samples. However, we would not recommend exclusion of any sample from analyses until a wide range of mapping parameters has been trialed, as even in our worst samples many alleles were correctly called (Table 2). The high percentage of erroneously called SNPs in poor samples could be due to DNA damage (though we do not detect the classical signal of this), or too poor capture resulting in low coverage and loss of the ability to confidently distinguish between alleles and paralogs. One trade-off for phylogenetic reconstructions is increasing taxon sampling by the inclusion of poorly performing samples, vs. difficulties due to missing data. We suggest here that there is a great benefit to sequencing replicates. We also recommend great caution when using herbarium samples for population genetic studies; only the best supported SNPs should be used for such work.
In our study, we have the advantage of being able to compare the sequence data from low quality libraries with that from good libraries produced from the same individual plant, a luxury that will not be available in herbarium-based studies. We conclude that much valuable information can be obtained even from poor samples, if quality filtering is high enough to provide confidence in the consensus sequence generated.
We believe Hyb-Seq provides the best current method to acquire specific nuclear sequence from any herbarium specimen due to its ability to capture highly degraded DNA, filter out contaminants and produce high read coverage. The retrieval of identical sequences across all the samples makes assembly and analysis easier, producing phylogenies with higher confidence (Jones and Good, 2015). It is also possible to include un-captured library in the sequencing reaction to provide a genome skim with sufficient coverage for plastid genome assembly (e.g., Schmickl et al., 2016; Villaverde et al., 2018), though we did not attempt that here. Using these methods gives us access to the genetic information in millions of unique, curated specimens collected over the last 300 years, and greatly expands the questions we can ask and therefore our depth of understanding of plant biodiversity.
The datasets generated for this study can be found in the PRJNA552424 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA552424).
MHu, MHa, LF, HW, and CK designed the experimental structure. MHu and HW prepared the samples. MHa, LF, and HW performed the lab work. K-FC provided the sequences for bait design. CK and Y-HT performed the analyses. MHu, MHa, LF, HW, and CK wrote the manuscript.
The Royal Botanic Garden Edinburgh is supported by the Scottish Government's Rural and Environment Science and Analytical Services Division. Y-HT and K-FC are supported by Ministry of Science and Technology, Taiwan (107-2917-I-564-026).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Ruth Hollands, Dr. James Nicholls and Dr. Flavia Pezzini for assistance in the molecular laboratory, also RBGE-hort for the fantastic job they do maintaining the living collections and RBGE-herbarium for curating the dead collection. Thanks also to Dr. George Argent for discussion of Paddy Wood's collecting methods. Thanks to Dr. Aurealiano Bombarely for the assembly of the B. conchifolia genome, and Lucia Campos Dominguez for the B. bipinnatifida plastid genome. Lastly, thanks to Dr. Michael David Martin and Dr. Freek T. Bakker for the inducement for this study, via an invitation to submit to an article collection on Herbarium collection-based plant evolutionary genetics and genomics.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00439/full#supplementary-material
Supplementary Table 1. Begonia bait set. Sequence identity, annotation, sequence divergence from orthologs of loci chosen as baits.
Supplementary Figure 1. Distribution of true and erroneous GA and CT SNPs. The scales on the y-axes vary between charts. (A) The proportion of true and erroneous SNPS for the dinucleotide GA by species and treatment. (B) The proportion of true and erroneous SNPS for the dinucleotide CT by species and treatment.
Bakker, F. T. (2018). “Herbarium genomics: plant archival DNA explored,” in Paleogenomics, eds C. Lindqvist and O. P. Rajora (Cham: Springer), 205–224. doi: 10.1007/13836_2018_40
Bakker, F. T., Lei, D., Yu, J., Mohammadin, S., Wei, Z., Van de Kerke, S., et al. (2016). Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly (IOGA) pipeline. Biol. J. Linn. Soc. 117, 33–43. doi: 10.1111/bij.12642
Beck, J. B., and Semple, J. C. (2015). Next-generation sampling: pairing genomics with herbarium specimens provides species-level signal in Solidago (Asteraceae). Appl. Plant Sci. 3:1500014. doi: 10.3732/apps.1500014
Bi, K., Linderoth, T., Vanderpool, D., Good, J. M., Nielsen, R., and Moritz, C. (2013). Unlocking the vault: next-generation museum population genomics. Mol. Ecol. 22, 6018–6032. doi: 10.1111/mec.12516
Bieker, V. C., and Martin, M. D. (2018). Implications and future prospects for evolutionary analyses of DNA in historical herbarium collections, Bot. Lett. 165, 409–418. doi: 10.1080/23818107.2018.1458651
Brennan, A., Bridgett, S., Shaukat Ali, M., Harrison, N., Mathews, A., Pellicer, J., et al. (2012). Genomic resources for evolutionary studies in the large, diverse tropical genus Begonia. Trop. Plant Biol. 5, 261–276. doi: 10.1007/s12042-012-9109-6
Bressan, E. A., Rossi, M. L., Gerald, L. T. S., and Figueira, A. (2014). Extraction of high-quality DNA from ethanol-preserved tropical plant tissues. BMC Res. Notes 7:268. doi: 10.1186/1756-0500-7-268
Bridson, D., and Forman, L. (1999). The Herbarium Handbook, 3rd Edn. Richmond, VA: Royal Botanic Gardens Kew.
Briggs, A. W., Stenzel, U., Johnson, P. L. F., Green, R. E., Kelso, J., Prufer, K., et al. (2007). Patterns of damage in genomic DNA sequences from a Neanderthal. Proc. Natl. Acad. Sci. U.S.A. 104, 14616–14621. doi: 10.1073/pnas.0704665104
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Chase, M. W., and Hills, H. H. (1991). Silica gel: an ideal material for field preservation of leaf samples for DNA studies. Taxon 40, 215–220. doi: 10.2307/1222975
Dodsworth, S., Chase, M. W., Kelly, L. J., Leitch, I. J., Macas, J., Novák, P., et al. (2015). Genomic repeat abundances contain phylogenetic signal. Syst. Biol. 64, 112–126. doi: 10.1093/sysbio/syu080
Drábková, L., Kirschnew, J., and Vlcek, C. (2002). Comparison of seven DNA extraction and amplification protocols in historical herbarium specimens of Juncaceae. Plant Mol. Biol. Rep. 20, 161–175. doi: 10.1007/BF02799431
Enk, J. M., Devault, A. M., Kuch, M., Murgha, Y. E., Rouillard, J. M., and Poinar, H. N. (2014). Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 31, 1292–1294. doi: 10.1093/molbev/msu074
Gassmann, M., and McHoull, B. (2015). DNA Integrity Number (DIN) with the Agilent 2200 TapeStation System and the Agilent Genomic DNA ScreenTape Assay: Technical overview. Agilent Technologies. Available online at: http://www.agilent.com/cs/library/applications/5991-5258EN.pdf
Gnirke, A., Melnikov, A., Maguire, J., Rogov, P., LeProust, E. M., Brockman, W., et al. (2009). Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189. doi: 10.1038/nbt.1523
Gorden, E. M., Sturk-Andreaggi, K., and Marshall, C. (2018). Repair of DNA damage caused by cytosine deamination in mitochondrial DNA of forensic case samples. Forensic Sci. Int. Genet. 34, 257–264. doi: 10.1016/j.fsigen.2018.02.015
Harris, S. A. (1993). DNA analysis of tropical plant species: an assessment of different drying methods. Plant Syst. Evol. 188, 57–64. doi: 10.1007/BF00937835
Hart, M. L., Forrest, L. L., Nicholls, J. A., and Kidner, C. A. (2016). Retrieval of hundreds of nuclear loci from herbarium specimens. Taxon 65, 1081–1092. doi: 10.12705/655.9
Hughes, M., Peng, C. I., Lin, C. W., Rubite, R. R., Blanc, P., and Chung, K. F. (2018). Chloroplast and nuclear DNA exchanges among Begonia sect. Baryandra species (Begoniaceae) from Palawan Island, Philippines, and descriptions of five new species. PLoS ONE 13:e0194877. doi: 10.1371/journal.pone.0194877
Jones, M. R., and Good, J. M. (2015). Targeted capture in evolutionary and ecological genomics. Mol. Ecol. 25, 185–202. doi: 10.1111/mec.13304
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L., and Orlando, L. (2013). mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Katoh, K., Asimenos, G., and Toh, H. (2009). “Multiple alignment of DNA sequences with MAFFT,” in Bioinformatics for DNA Sequence Analysis, ed D. Posada (New York, NY: Humana Press), 39–64. doi: 10.1007/978-1-59745-251-9_3
Klingström, T., Bongcam-Rudloff, E., and Pettersson, O. V. (2018). A comprehensive model of DNA fragmentation for the preservation of High Molecular Weight DNA. bioRxiv [Preprint]. doi: 10.1101/254276
Krause, J., Briggs, A. W., Kircher, M., Maricic, T., Zwyns, N., Derevianko, A., et al. (2010). A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr. Biol. 20, 231–236. doi: 10.1016/j.cub.2009.11.068
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Linderholm, A. (2016). Ancient DNA: The next generation—chapter and verse. Biol. J. Linn. Soc. 117, 150–160. doi: 10.1111/bij.12616
Mouttham, N., Klunk, J., Kuch, M., Fourney, R., and Poinar, H. (2015). Surveying the repair of ancient DNA from bones via high-throughput sequencing. Biotechniques 59, 19–25. doi: 10.2144/000114307
Nachman, M. W. (2013). Genomics and museum specimens. Mol. Ecol. 22, 5966–5968. doi: 10.1111/mec.12563
Nicholls, J. A., Pennington, R. T., Koenen, E. J. M., Hughes, C. E., Hearn, J., Bunnefeld, L., et al. (2015). Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae). Front. Plant Sci. 6:710. doi: 10.3389/fpls.2015.00710
Price, M. N., Paramvir, S. D., and Arkin, A. P. (2010). FastTree 2 – Approximately Maximum-Likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Rowe, K. C., Singhal, S., Macmanes, M. D., Ayroles, J. F., Morelli, T. L., Rubidge, E. M., et al. (2011). Museum genomics: low-cost and high-accuracy genetic data from historical specimens. Mol. Ecol. Resour. 11, 1082–1092. doi: 10.1111/j.1755-0998.2011.03052.x
Särkinen, T., Staats, M., Richardson, J. E., Cowan, R. S., and Bakker, F. T. (2012). How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS ONE 7:e43808. doi: 10.1371/journal.pone.0043808
Schmickl, R., Liston, A., Zeisek, V., Oberlander, K., Weitemier, K., Straub, S. C. K., et al. (2016). Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae). Mol. Ecol. Resour. 16, 1124–1135. doi: 10.1111/1755-0998.12487
Schrenk, J. (1888). Schweinfurth's method of preserving plants for herbaria. Bull. Torrey Bot. Club. 15, 292–293. doi: 10.2307/2477483
Shao, W., Khin, S., and Kopp, W. C. (2016). Characterization of effect of repeated freeze and thaw cycles on stability of genomic DNA using pulsed field gel electrophoresis. Biopreserv. Biobank. 10, 4–11. doi: 10.1089/bio.2011.0016
Smith, E. E. Jr. (1971). Preparing Herbarium Specimens of Vascular Plants. Washington, DC: USDA Agricultural Information Bulletin, 348.
Soltis, P. S. (2017). Digitization of herbaria enables novel research. Am. J. Bot. 104, 1–4. doi: 10.3732/ajb.1700281
Staats, M., Cuenca, A., Richardson, J. E., Vrielink-van Ginkel, R., Petersen, G., Seberg, O., et al. (2011). DNA damage in plant herbarium tissue. PLoS ONE 6:e28448. doi: 10.1371/journal.pone.0028448
Staats, M., Erkens, R. H. J., Van de Vossenberg, B., Wieringa, J. J., Kraaijeveld, K., Stielow, B., et al. (2013). Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS ONE 8:e69189. doi: 10.1371/journal.pone.0069189
Turner, B., Paun, O., Munzinger, J., Chase, M. W., and Samuel, R. (2016). Sequencing of whole plastid genomes and nuclear ribosomal DNA of Diospyros species (Ebenaceae) endemic to New Caledonia: many species, little divergence. Ann. Bot. 117, 1175–1185. doi: 10.1093/aob/mcw060
Villaverde, T., Pokorny, L., Olsson, S., Rincón-Barrado, M., Johnson, M. G., Gardner, E. M., et al. (2018). Bridging the micro- and macroevolutionary levels in phylogenomics: Hyb-Seq solves relationships from populations to species and above. N. Phytol. 220, 636–650. doi: 10.1111/nph.15312
Weiß, C. L., Schuenemann, V. J., Devos, J., Shirsekar, G., Reiter, E., Gould, B. A., et al. (2016). Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. R. Soc. Open Sci. 3:160239. doi: 10.1098/rsos.160239
Wilkie, P., Poulson, A. D., Harris, D., and Forrest, L. L. (2013). The collection and storage of plant material for DNA extraction: the teabag method. Gard. Bull. Sing. 65, 231–234. Available online at: https://www.nparks.gov.sg/sbg/research/publications/gardens-bulletin-singapore/listing-of-publications?page=3
Keywords: alcohol damage, Begonia, degraded DNA, herbarium specimens, hybrid baits, preservation method, target capture
Citation: Forrest LL, Hart ML, Hughes M, Wilson HP, Chung K-F, Tseng Y-H and Kidner CA (2019) The Limits of Hyb-Seq for Herbarium Specimens: Impact of Preservation Techniques. Front. Ecol. Evol. 7:439. doi: 10.3389/fevo.2019.00439
Received: 04 July 2019; Accepted: 30 October 2019;
Published: 26 November 2019.
Edited by:
Michael David Martin, Norwegian University of Science and Technology, NorwayReviewed by:
Rafal Marek Gutaker, New York University, United StatesCopyright © 2019 Forrest, Hart, Hughes, Wilson, Chung, Tseng and Kidner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine A. Kidner, Y2tpZG5lckByYmdlLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.