 Anat M. Belasen
Anat M. Belasen Molly C. Bletz
Molly C. Bletz Domingos da Silva Leite
Domingos da Silva Leite Luís Felipe Toledo
Luís Felipe Toledo Timothy Y. James
Timothy Y. James- 1Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, MI, United States
- 2Department of Biology, University of Massachusetts Boston, Boston, MA, United States
- 3Instituto de Biologia, Universidade Estadual de Campinas, Campinas, Brazil
- 4Laboratório de História Natural de Anfíbios Brasileiros, Departamento de Biologia Animal, Instituto de Biologia, Universidade Estadual de Campinas, Campinas, Brazil
Habitat fragmentation and wildlife disease are two widespread drivers of biodiversity loss, yet few empirical studies have explored their interactions. In this study, we utilized a naturally fragmented island system to examine the impacts of fragmentation on genetic diversity and amphibian infection dynamics. We determined the impacts of fragmentation on genetic diversity at the immunity locus MHC IIB, a hypothesized predictor of disease susceptibility. Contrary to the expectation that MHC diversity would remain high due to balancing selection, island populations lost genetic diversity at this locus while simultaneously experiencing positive selection at MHC IIB. We then used Next-Generation Sequencing to identify a variety of potential eukaryotic parasites from amphibian skin swabs. Island populations exhibited higher potential parasite richness (proportion of eukaryotic microbe operational taxonomic units or OTUs from parasitic taxa) relative to mainland populations. MHC homozygotes hosted a lower diversity of potential parasites, and population-level MHC diversity was negatively associated with parasite richness. Our results show that genetic erosion can occur at the MHC IIB locus following fragmentation, which may contribute to increased susceptibility to parasites.
Introduction
Habitat loss and fragmentation are arguably the most widespread drivers of biodiversity loss (Perfecto and Vandermeer, 2010). Habitat fragmentation can significantly impact population resilience against stressors including emerging infectious diseases (Dobson and Foufopoulos, 2001). These effects include the loss of genetic diversity over time due to genetic drift compounded by inbreeding (Newmark, 1995; Zuidema et al., 2017), which in turn may increase disease susceptibility (Altizer et al., 2003). Fragmentation may also reduce host contact rates and disease transmission, which could alter selection on disease resistance alleles. This could also result in the loss of genetic diversity if there was strong selection for resistance to a few remaining local pathogens.
Genetically eroded hosts have exhibited increased disease susceptibility both in laboratory experiments (Arkush et al., 2002; Spielman et al., 2004; Ilmonen et al., 2008; Ellison et al., 2012) and in studies of natural populations (Pearman and Garner, 2005; Roca et al., 2010; Ellison et al., 2011). Low genetic diversity has also been associated with increased levels of parasitism, which suggests that there is a relationship between immune function and genetic diversity (Macdougall-Shackleton et al., 2005) and that genetically eroded individuals may be more susceptible to opportunistic infections (Anaissie, 1992). These associations between inbreeding and disease may be related to loss of resistance alleles at immunogenetic loci—genetic regions that are important in pathogen recognition and immune response.
Immunogenetic studies in vertebrates often focus on the major histocompatibility complex (MHC), a gene family composed of two major classes (Bernatchez and Landry, 2003). Molecules encoded by the MHC bind with antigenic peptides, which they present on the outside of cells in infected tissue to signal T-cells to mount an immune response (Richmond et al., 2009). MHC heterosis (heterozygote advantage) is believed to be common, as heterozygosity is associated with a broader repertoire of MHC molecules and therefore enhanced immunity (Doherty and Zinkernagel, 1975; McClelland et al., 2003; Whitehorn et al., 2011). Indeed, MHC heterozygosity can be exceedingly high relative to other loci (Gaudieri et al., 2000; Landry et al., 2001; Hambuch and Lacey, 2002), even in otherwise genetically impoverished populations (Aguilar et al., 2004). In some cases, however, specific MHC genotypes have been more strongly associated with disease resistance than heterozygosity (Schwensow et al., 2007; Deter et al., 2008).
MHC genotype determines the structure of the peptide binding region of MHC molecules. This structure in turn determines the types of antigen peptides, and essentially the types of pathogens, that can be recognized (Piertney and Oliver, 2006). Alleles encoding MHC molecules that can recognize endemic pathogens are most likely to be common in the local host gene pool. For example, a human HLA (MHC analog) haplotype associated with malaria resistance is common in West Africa, where malaria is endemic, but not elsewhere (Hill et al., 1991). Introduced pathogens are more likely to cause severe outbreaks and declines, which may be because in naïve host populations, particular MHC alleles associated with an effective pathogen-specific immune response are likely to be relatively uncommon. Indeed, long-term parasite release has been hypothesized as a mechanism driving the susceptibility of island endemics to introduced pathogens and parasites (reviewed in Wikelski et al., 2004).
Amphibians serve as models for understanding the interactions of fragmentation, genetics, and disease. Habitat fragmentation is considered one of the most important drivers of amphibian declines globally (Kiesecker et al., 2001; Cushman, 2006). While this may be due to direct effects of fragmentation on amphibian dispersal, amphibians also experience indirect effects including loss of genetic diversity. Many amphibians are especially sensitive to this genetic impoverishment, in part due to naturally small effective population sizes (Allentoft and O'Brien, 2010). Reduced genetic diversity in amphibians has been correlated with decreased growth, survival, and population resilience (Lesbarrères et al., 2005; Arens et al., 2007). Although few studies have tested for correlations between genetic diversity and disease susceptibility (but see Pearman and Garner, 2005), increased appreciation of the widely variable disease susceptibility within and among amphibian species (Briggs et al., 2010; Gahl et al., 2012; Bielby et al., 2015; Gervasi et al., 2015) has led to increased focus on the relationship between amphibian genetics and disease. Notably, the MHC IIB locus has been shown to modulate susceptibility to chytridiomycosis caused by the fungal skin pathogen Batrachochytrium dendrobatidis (Bd), with studies suggesting both that specific alleles and heterozygosity can provide an advantage against Bd (Savage and Zamudio, 2011; Bataille et al., 2015; Kosch et al., 2016).
The majority of recent amphibian disease research focuses on only a few pathogens, despite widespread evidence that amphibians are vulnerable to a diversity of pathogens (Kiesecker et al., 2001; Briggs et al., 2010). Multiple pathogens and parasites have been implicated in amphibian declines, notably the chytrid fungi Bd and B. salamandrivorans, the zygomycete fungus Mucor amphibiorum, oomycete parasites including Saprolegnia spp., Ranaviruses, and metazoans including trematodes (Speare et al., 1994; Martel et al., 2013; Gleason et al., 2014). Amphibians are also known to experience infection by alveolates including Perkinsea spp. (Chambouvet et al., 2015), the apicomplexan Hepatozoon (Desser et al., 1995), the ichthyosporean Anurofeca richardsi (Rowley et al., 2013), the ectoparasitic ciliate Epistylus sp. (Pritchett and Sanders, 2007), and a variety of helminth parasites (Dyer, 1991; Yildirimhan et al., 2006). Much attention has focused on the role of Bd as an important disease agent in amphibians worldwide, while fewer disease surveys have explored other parasitic taxa.
In this study, we examined the effects of fragmentation on amphibian immunogenetic diversity at the MHC IIB locus and potential susceptibility to a diversity of parasites. The effects of fragmentation are often subject to time lags whereby the impacts of current anthropogenic fragmentation may not be fully realized for several generations (Tilman et al., 1994; Arenas et al., 2014; Mona et al., 2014). Therefore, to examine the long-term impacts of fragmentation on immunogenetic diversity, we utilized a natural laboratory in the Brazilian Atlantic Forest as a model system. The Atlantic Forest contains dozens of land-bridge islands, which were connected to the mainland prior to the Pleistocene (Suguio et al., 2005). These islands are essentially forest patches that were naturally isolated by marine incursions 12,000–20,000 years ago; thus they represent very old habitat fragments. Remaining frog populations were once part of contiguous mainland populations, and are now isolated to the islands (Bell et al., 2012; Duryea et al., 2015).
We sampled populations of a single frog species (Cycloramphidae: Thoropa taophora) that is widespread across the Atlantic Forest coast. We collected tissue and skin swab samples to respectively analyze frog MHC IIB diversity and parasite diversity using DNA sequencing. These data allowed us to test the following predictions: (i) immunogenetic diversity at the MHC IIB locus is maintained despite overall losses of neutral genetic diversity in fragmented (island) populations; (ii) genetically eroded populations exhibit higher parasitism; and (iii) MHC IIB genotype is associated with differences in parasite richness.
Material and Methods
Study System and Field Sampling
The study area spans seven land-bridge islands located off of the north coast of São Paulo state in Southeastern Brazil, and four sites found on the nearby coastal mainland (Figure 1A). The island and mainland sites are characterized by typical Atlantic rainforest vegetation bordered by rocky coastal areas and exhibit similarly low levels of human perturbation. On the Atlantic Forest land-bridge islands, a small number of amphibian species have maintained stable populations since island formation (Bell et al., 2012). The best-studied of these is Thoropa taophora, a Cycloramphid frog that inhabits wet rocks and is widespread along the São Paulo coast (Sazima, 1971; Bokermann, 1974; Giaretta and Facure, 2004). The island T. taophora populations were recently shown to possess reduced genetic diversity relative to mainland populations (evaluated with microsatellite loci), with estimated divergence times approximately consistent with island age estimates (ranging from 4,000 to 28,000 years for most island populations), little significant migration among populations, and no effect of island size or distance from mainland on migration or population genetic diversity (Duryea et al., 2015).
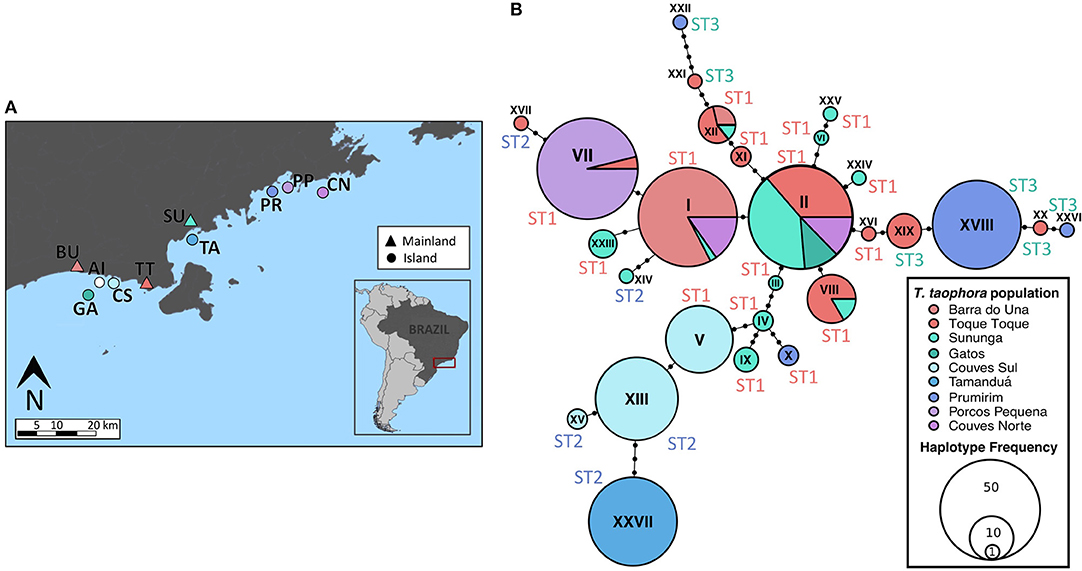
Figure 1. Locations of Thoropa taophora frog populations and MHC IIB Haplotype network. Sample sizes of adult frogs captured and sampled are listed in Table 1. (A) Map of populations sampled in January-February 2015. Inset map shows the location of the study area in southeastern Brazil. Mainland sites are represented with triangles and islands with circles. Colors of markers correspond to colors of populations in haplotype network. (B) Haplotype network showing the MHC IIB locus amplified from Thoropa taophora. Black dots on branches indicate the number of mutations between haplotypes.
Adult T. taophora frogs (n = 4–30 per site, 179 total) were individually sampled from each of the 10 study populations between January and March 2015 (SISBio collection permit 27745-13). Frogs were washed with sterile water and then swabbed using a standard sampling protocol (Hyatt et al., 2007). Toe tissue was collected from each individual with sterilized dissection scissors for analysis of host genetics. DNA was extracted from swab and tissue samples using the Qiagen DNeasy Blood and Tissue kit.
Immunogenetic Analyses
A ~120 bp fragment of the MHC IIB locus was PCR-amplified from tissue DNA extracts (May and Beebee, 2009; Supplementary Material) and sequenced at the University of Michigan Sequencing Core. Haplotypes were predicted using Phase (Version 2.1.1) (Stephens et al., 2001) to obtain allele frequencies, which were used to calculate allelic richness (NA) and observed and expected heterozygosity (Ho and He). Segregating sites (S) and nucleotide diversity (π) were calculated in DnaSP (Version 5; Librado and Rozas, 2009). T-tests were used to evaluate differences in S and π among populations, and simple linear regression was used to compare MHC NA and heterozygosity with microsatellite NA and heterozygosity (extracted from Duryea et al., 2015) in R (Version 3.3.0; Lighten et al., 2014).
The ratio of non-synonymous to synonymous nucleotide changes (dN/dS) across the MHC alignment and associated p-values for departure from the neutral expectation (dN/dS = 1, no selection) were calculated in MEGA (vrs. 6.06-mac; Tamura et al., 2013). dN/dS was calculated across the entire dataset, and also for each population and for each habitat type (island vs. mainland). Tajima's D and the p-value associated with its departure from a neutral expectation (D = 0, random evolutionary processes; Tajima, 1989) were calculated in DnaSP. To examine evolutionary and geographic patterns across MHC IIB alleles, an MHC haplotype network was visualized and the fixation index (FST; a measure of population differentiation) was calculated using the pegas package in R (Paradis, 2010). Mean FST was calculated for nine microsatellite markers from the same populations (extracted from Duryea et al., 2015). A partial Mantel correlation was used to evaluate associations between MHC FST and geographic distance among populations while accounting for neutral (microsatellite) FST. MHC FST was compared with mean microsatellite FST to determine whether similar evolutionary processes were impacting the MHC.
MHC haplotypes were condensed into functionally different genotypes, known as “supertypes.” Supertyping is used to partition haplotypes into groups based on amino acid properties that are assumed to affect antigen binding capability (Reche and Reinherz, 2003). Supertype clusters were predicted using Discriminant Analysis of Principal Components (DAPC) implemented with the adegenet package in R (vrs. 3.3.0; Jombart et al., 2010).
Detection of Potential Parasites
Swab samples were assayed for Bd using duplicate quantitative polymerase chain reaction (qPCR; Boyle et al., 2004). To examine broader parasite diversity, DNA extracts were PCR-amplified with dual-indexed, barcoded broad eukaryote primers, and then sequenced on the Illumina MiSeq platform (Stoeck et al., 2010). Sequences were quality-filtered and processed using Quantitative Insights into Microbial Ecology (QIIME; Caporaso et al., 2010) and UCHIME2 (Edgar, 2016) and assigned taxonomy using Silva 97 and Genbank reference databases (Supplementary Material). Briefly, sequences that passed quality filtering were grouped into operational taxonomic unit (OTU) clusters at the standard microbial threshold of 97% similarity. Taxonomy was assigned to OTUs by comparing a representative sequence from each sequence cluster against the Silva 97 reference database using the BLAST search algorithm to find the closest match. Because this database has limited taxonomic resolution for certain groups (e.g., some Fungi contain few or no representative sequences in Silva 97), the representative sequence set was then compared against GenBank sequence database using the BLASTn search algorithm to confirm OTU identity.
OTUs were categorized as either potentially parasitic or non-parasitic using a standardized Google Scholar search. The search was performed with the parameters “[taxon name] AND parasite OR pathogen.” Resulting articles were examined manually to determine whether published accounts of the taxon as a parasite in either amphibians or other animal hosts existed.
Potential parasite OTUs (PP OTUs) were rarefied to 200 sequences (determined via visual examination of the minimum asymptotic point of read/sample histograms). Datasets with and without opportunistic pathogens/parasites were analyzed separately to provide liberal (i.e., all PP OTUs, including likely parasites and opportunistic PP OTUs) and conservative (i.e., only likely parasites, excluding opportunistic PP OTUs) estimates of potential parasite richness. The fraction of PP OTUs that were likely to be opportunistic OTUs (only opportunistic PP OTUs) were also analyzed to test whether loss of genetic diversity is associated with increased opportunistic infections. For all three PP OTU fractions, alpha diversity metrics (Observed OTUs, Chao 1 index, and phylogenetic diversity) as well as beta diversity distance matrices were computed in QIIME and analyzed across groups in R.
Independent samples t-tests were used to examine associations between alpha diversity and site type (island vs. mainland) or MHC genotype (homozygous vs. heterozygous, supertype). Associations between beta diversity and genetic or geographic distance of host populations were tested with Mantel correlations to test the null hypothesis that parasite community structure is a function of geography rather than host properties. Simple Linear Regressions were used to determine the relationship between population-level MHC genetic diversity and log10 mean parasite richness.
Results
Neutral Genetic and MHC Diversity
Twenty-seven MHC IIB haplotypes that condensed into three functionally divergent supertypes were recovered from the T. taophora study populations (Figure 1B). At most two alleles were recovered from each individual frog. MHC IIB diversity was significantly lower in island populations in terms of nucleotide diversity [π, independent samples t-test, t(8) = 3.209, p < 0.012; Table 1]. Island populations also showed trends toward lower MHC class IIB diversity in terms of allelic richness (NA; Mann-Whitney Test, U = 2, p = 0.067) and segregating sites S [t-test, t(8) = 1.926, p = 0.09; Table 1]. MHC IIB allelic richness was positively associated with microsatellite allelic richness across all populations (Linear Regression weighted by sample size, one-tailed p = 0.04, R2 = 0.42). There were no differences in observed or expected MHC IIB heterozygosity between island and mainland populations (HO and HE, independent samples t-tests, p > 0.05 for each metric).
When all data were pooled and analyzed, there was a statistically significant signature of positive selection (dN/dS > 1) at the MHC IIB locus (dN/dS = 2.811, p = 0.003). In addition, comparison of island vs. mainland populations also revealed signatures of positive selection for all groups (all coastal populations: dN/dS = 2.292, p = 0.012; all island populations: dN/dS = 2.843, p = 0.003). However, when populations were analyzed individually, 2/3 mainland populations showed signatures of positive selection while only 1/6 islands exhibited a dN/dS significantly >1 (Table 1). This is likely due to low allele number in island populations (only sites with four or more alleles had dN/dS ratios significantly >1).
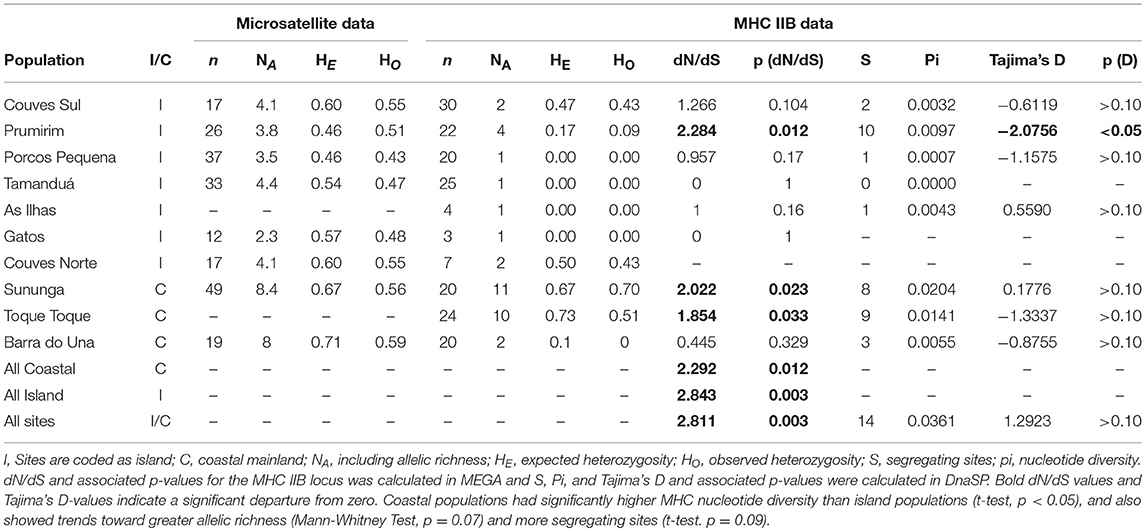
Table 1. Summary statistics for microsatellite data (extracted from Duryea et al., 2015) and MHC IIB data.
Geographic distance showed no significant association with MHC IIB FST (partial Mantel test,—r = −0.0179, p > 0.5). To further examine whether neutral processes or selection were more likely influencing MHC class IIB diversity (see Radwan et al., 2009), we compared the MHC FST across all the T. taophora study populations with the overall FST averaged across all microsatellite loci sequenced by Duryea et al. (2015). The overall MHC FST value was 0.729, which falls above 2SE of the mean microsatellite FST (0.496 ± 0.082 SE).
Bd Infection Prevalence and Load Among Study Populations
All study populations exhibited low Bd loads (range 1–100 ZE with the majority ~10 ZE). Mean Bd infection load and prevalence did not significantly differ among populations (ANOVA, p = 0.2 for infection load among all Bd positive populations; t-test, p = 0.6 for prevalence between pooled mainland and pooled island populations; Figure S1). There was also no significant association between Bd load and neutral genetic diversity (p > 0.05 in Simple Linear Regression for both Ho and NA of microsatellites).
Potential Parasite Identification
After quality and chimera filtering, there were 845 total eukaryotic OTUs found across all samples. Eukaryotic OTUs that were determined through the standardized literature search to be potentially parasitic made up ~11% (97/845) of the total OTUs. The prevalence of coinfections was extremely high: 150/168 individuals with potential parasites hosted two or more potentially parasitic OTUs (hereafter PP OTUs), and a maximum of 18 PP OTUs was recovered from one individual (Figure 2A). PP OTUs were taxonomically diverse (Figure 2B), with 37 likely parasites and 60 likely opportunistic pathogens (Table S1).
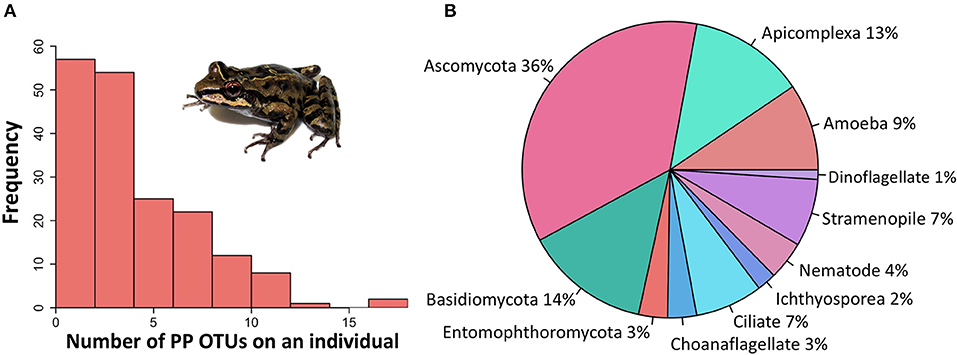
Figure 2. Frequency and diversity of potentially parasitic eukaryote OTUs identified from Thoropa taophora skin swab samples. Sequences were recovered by NGS of the 18S v4 region from individual frog skin swab extracts. OTUs were clustered at 97% similarity and taxonomically assigned via BLAST search of the Silva 97 database and GenBank. A standardized literature search was used to categorize all OTUs as potentially parasitic or not. (A) Frequency distribution of potentially parasitic OTUs across individual swab samples. (B) Taxonomic distribution of the 97 potentially parasitic micro-eukaryotic OTUs.
Many of the potential parasites were identified as endoparasites, such as Apicomplexa which are known to infect blood cells. Tissue and swab samples from eight individual frogs for which apicomplexan sequences were recovered in our 18S dataset were further analyzed to validate the amplification of internal parasites from skin swabs. In all eight of these samples, PCR-amplification with apicomplexan primers resulted in bands in sample pairs (tissue and swab samples from the same individual frog) for at least one of the two primer sets, with 4/8 sample pairs showing strong amplification with both primer sets. Clean DNA sequences were obtained from sample pairs from 6/8 individual frogs. When the sequences were searched against GenBank using BLASTn, all six sample pairs matched the same apicomplexan identity: five of the tissue/swab sample pairs matched Hepatozoon sp., and the remaining pair matched Adelina sp.
Potential Parasite Diversity and Distribution
Beta diversity of PP OTUs did not correlate with geographic or genetic distance (Mantel correlation, p > 0.05). Overall, alpha diversity of PP OTUs did not vary by site type (t-tests for observed OTUs, PD, and Chao1, p > 0.05). However, when opportunistic PP OTUs were excluded from analyses, PP OTU alpha diversity was lower in island populations (observed OTUs: 0.42 average PP OTUs in islands vs. 1.10 in mainland, Mann-Whitney test, U = 2,883.5, p < 0.01; PD: 0.25 island vs. 0.64 mainland, Mann-Whitney test, U = 2,841, p < 0.01; Chao 1: 0.42 island vs. 1.10 mainland, Mann-Whitney test, U = 2,883.5, p < 0.01). Site type did not contribute to differences in alpha diversity of opportunistic PP OTUs (t-tests for observed OTUs, PD, and Chao1, p > 0.05).
Richness varied by MHC IIB genotype for the overall PP OTU dataset and for the PP OTU dataset with opportunistics excluded, with fewer PP OTUs in MHC IIB homozygotes for both fractions (all PP OTUs: observed OTUs: 4.05 mean PP OTUs in homozygotes vs. 5.59 in heterozygotes, Mann-Whitney test, U = 3,889.5, p < 0.05; PD: 1.04 in homozygotes vs. 1.51 in heterozygotes, Mann-Whitney test, U = 3,697.5, p < 0.001; Chao 1: n.s., t-test, p > 0.05; PP OTUs with no opportunistics: observed OTUs: 0.45 mean PP OTUs in homozygotes vs. 1.3 in heterozygotes, Mann-Whitney test, U = 3,711.5, p < 0.001; PD: n.s., t-test, p > 0.05; Chao 1: 0.46 in homozygotes vs. 1.28 in heterozygotes, Mann-Whitney test, U = 3,500.5, p < 0.05). When only opportunistic PP OTUs were analyzed, richness did not vary by MHC IIB genotype (t-tests, p > 0.05 for all alpha diversity metrics).
MHC IIB Supertype had no effect on alpha diversity in the total PP OTU dataset. However, when opportunistics were excluded, PP OTU richness was significantly lower in frogs possessing Supertype 3 (observed OTUs: n.s., t-test, p > 0.05; PD: 0.18 in ST3+ vs. 0.48 in ST3-, Mann-Whitney test, U = 1485.5, p < 0.05; Chao1: 0.37 in ST3+ vs. 0.74 in ST3-, Mann-Whitney test, U = 1,486, p < 0.05). When only opportunistic PP OTUs were analyzed, frogs possessing MHC IIB Supertype 2 hosted a greater number of PP OTUs (observed OTUs: 5.04 in ST2+ vs. 3.33 in ST2-, Mann-Whitney test, U = 3,619.5, p < 0.01; PD: n.s., t-test, p > 0.05; Chao1: 5.26 in ST2+ vs. 3.36 in ST2-, Mann-Whitney test, U = 3,653, p < 0.01).
On average, fewer total eukaryotic OTUs were recovered from island frogs than mainland frogs (Mann-Whitney test, U = 2,787, p < 0.01). To evaluate relative parasite pressure among populations while accounting for differences in baseline eukaryotic diversity, the proportion of PP OTUs to total eukaryotic OTUs on each individual was calculated for further statistical analyses. Relative to mainland frogs, island frogs hosted proportionately more PP OTUs overall (0.29 mean proportion PP OTUs:total OTUs in islands vs. 0.20 in mainland, Mann-Whitney test, U = 4,963, p < 0.001) and proportionally more opportunistic PP OTUs (0.26 mean opportunistic PP OTUs:total OTUs in islands vs. 0.17 in mainland, Mann-Whitney test, U = 5,072, p < 0.001; Figure 3A), but similar levels of only non-opportunistic PP OTUs (t-test, p > 0.05). On the population level, MHC class IIB diversity was negatively associated with a greater proportion of PP OTUs for all three fractions of PP OTUs (all PP OTUs: SLR, β = −0.025, p < 0.001, R2 = 0.16 for NA, β = −0.247, p < 0.001, R2 = 0.07 for HO; only non-opportunistic PP OTUs: SLR, β = −0.015, p < 0.05, R2 = 0.08 for NA, β = −0.145 but n.s. with p > 0.05 for HO; only opportunistic PP OTUs: β = −0.031, p < 0.001, R2 = 0.21 for NA, β = −0.338, p < 0.001, R2 = 0.12 for HO; Figure 3B).
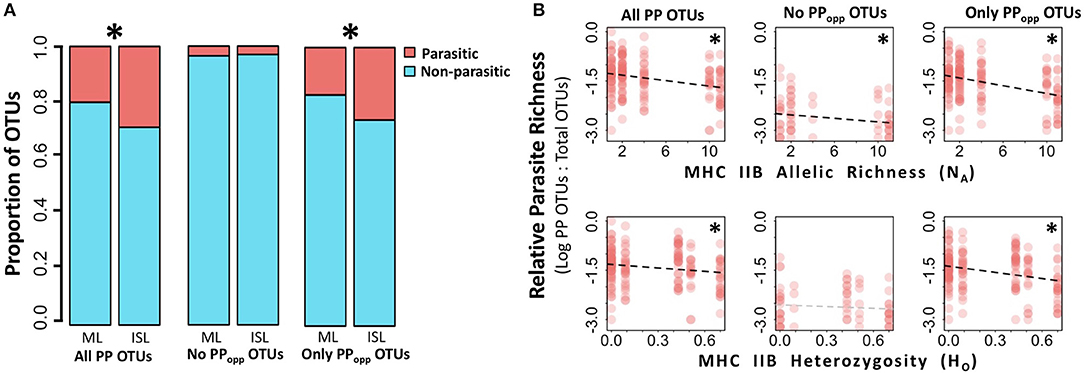
Figure 3. Site type (mainland vs. island) and MHC IIB influence the proportion of OTUs that are potentially parasitic. For analyses, potentially parasitic OTUs (PP OTUs) were divided into three fractions: (1) “All PP OTUs” includes all OTUs considered potentially parasitic; (2) “No PPopp OTUs” excludes opportunistic parasites; and (3) “Only PPopp OTUs” includes only opportunistic parasites. Significant results with alpha = 0.05 are denoted by an asterisk. (A) Proportion of PP OTUs (top/red) vs. non-parasites (bottom/blue) from Thoropa taophora skin swab extracts were significantly higher (Mann-Whitney U-tests, p < 0.05) in island (ISL) populations relative to mainland (ML) populations for PP OTUs fractions 1 and 3. (B) Relative parasite richness decreases as population-level MHC IIB diversity decreases (SLRs, p < 0.05) for PP OTU fractions 1 and 3. PP OTU fraction 1 is shown in the top left and bottom left panels, fraction 2 is shown in the middle two panels, and fraction 3 is shown in the top right and bottom right panels. Relative Parasite Richness was calculated as the proportion of total eukaryotic OTUs that were considered potential parasites (i.e., PP OTUs/total OTUs) and is plotted on a log10 scale. Two measures of population-level MHC IIB diversity were analyzed: allelic richness (NA, top three panels), and observed heterozygosity (HO, bottom three panels).
To distinguish between effects of site type and MHC IIB diversity on proportional diversity of PP OTUs, these analyses were repeated for only mainland populations. The negative association between proportion of PP OTUs remained present and significant for all PP OTUs (SLR, β = −0.02, p < 0.05, R2 = 0.10 for NA, β = −0.27, p < 0.01, R2 = 0.11 for HO) and for only opportunistic PP OTUs (SLR, β = −0.026, p < 0.01, R2 = 0.15 for NA; β = −0.357, p < 0.01, R2 = 0.16 for HO). While only non-opportunistic PP OTUs showed negative associations (negative regression slopes) with the two metrics of MHC IIB population-level diversity, these relationships were no longer significant when only mainland populations were analyzed (SLR, p > 0.05).
Discussion
The ecological and evolutionary impacts of long-term fragmentation on land-bridge island fauna has been studied in a number of systems (Karr, 1982; Aguilar et al., 2004; Hurston et al., 2009; Estrada-Villegas et al., 2010; Duryea et al., 2015; Belasen et al., 2016; Santonastaso et al., 2017). In a previous study examining immunogenetic diversity in island vertebrates, MHC diversity remained high while overall diversity dropped (Aguilar et al., 2004). This led us to predict that island T. taophora populations would display a similar pattern. Contrary to our expectations, we found that MHC class IIB diversity is reduced in island populations relative to their mainland counterparts (Table 1) and appears to have eroded at a rate similar to the loss of neutral diversity. This suggests a relatively strong effect of genetic drift on the MHC IIB locus. However, the larger overall dN/dS, the high level of MHC genetic differentiation (FST) relative to neutral loci, as well as a lack of MHC geographic structure support the action of positive selection. These patterns may have been generated by divergent selection, and suggest that the divergence observed in MHC IIB is not solely a product of genetic isolation but rather that selection may be leading to acceleration of differences among populations.
Although the idea that high MHC diversity is common remains prevalent in the literature, MHC diversity has been shown to correlate with overall diversity in a number of species, suggesting that demographic processes that affect neutral loci can extend to the MHC (reviewed in Radwan et al., 2009). Low MHC diversity is not necessarily an indication of imminent extinction; very low MHC diversity has been demonstrated in stable and viable wildlife populations (Ellegren et al., 1993) and entire species (Slade, 1992). Nevertheless, reduced diversity at immunogenetic loci could reduce resistance to infections: hosts would be less likely to possess matched resistance alleles to novel or invading pathogens, while pathogens could adapt more quickly or escape immune cell recognition in less immunogenetically variable hosts (Radwan et al., 2009; Siddle et al., 2010).
To examine whether fragmentation and genetics influence infections in T. taophora, we first conducted a standard assay for the fungal amphibian pathogen, Bd. The presence of Bd in almost all study populations but at very low loads (Figure S1) suggests Bd resistance in this species, enzootic dynamics of Bd in the study area, and/or unsuitable environmental conditions. T. taophora inhabits lowland coastal areas and can tolerate some contact with seawater. The salinity of the habitat (Stockwell et al., 2015) as well as high and variable temperatures may negatively impact the growth of Bd. Experimental studies are needed to distinguish between Bd resistance in T. taophora and low environmental suitability for Bd.
To gain a clearer picture of infections in T. taophora, we screened for a diversity of micro-eukaryotes in skin swab samples. Our Next-Generation Sequencing (NGS) analysis combined with a standardized literature review recovered 97 potentially parasitic (PP) micro-eukaryote OTUs, and up to 18 unique PP OTUs found on a single individual (Figure 2A). These results demonstrate a high frequency and diversity of coinfections, and underscore the limitations of using targeted assays for only one or few known pathogens. Although there were no differences among populations in Bd prevalence or load, there were significant patterns when we examined the PP OTUs identified with NGS. As we hypothesized, island populations exhibited higher parasitism, i.e., proportionately more PP OTUs than mainland populations (Figure 3A and Table 2), despite higher PP OTU diversity at mainland sites. This was only true for the total PP OTU dataset and the dataset only including likely opportunistic PP OTUs, but not for the more conservative PP OTU dataset that excluded opportunistics. This implies that opportunistic PP OTUs are driving this trend, although this result could also be due to a lack of statistical power in the very small conservative PP OTU dataset (n = 37 OTUs). A higher diversity of PP OTUs on islands could be attributed to higher contact between hosts and/or hosts and parasites in the limited island geographic area. However, island frogs hosted significantly fewer total eukaryotic OTUs than mainland frogs, implying that the microbiome of island frogs may be depauperate despite potentially higher contact rates.
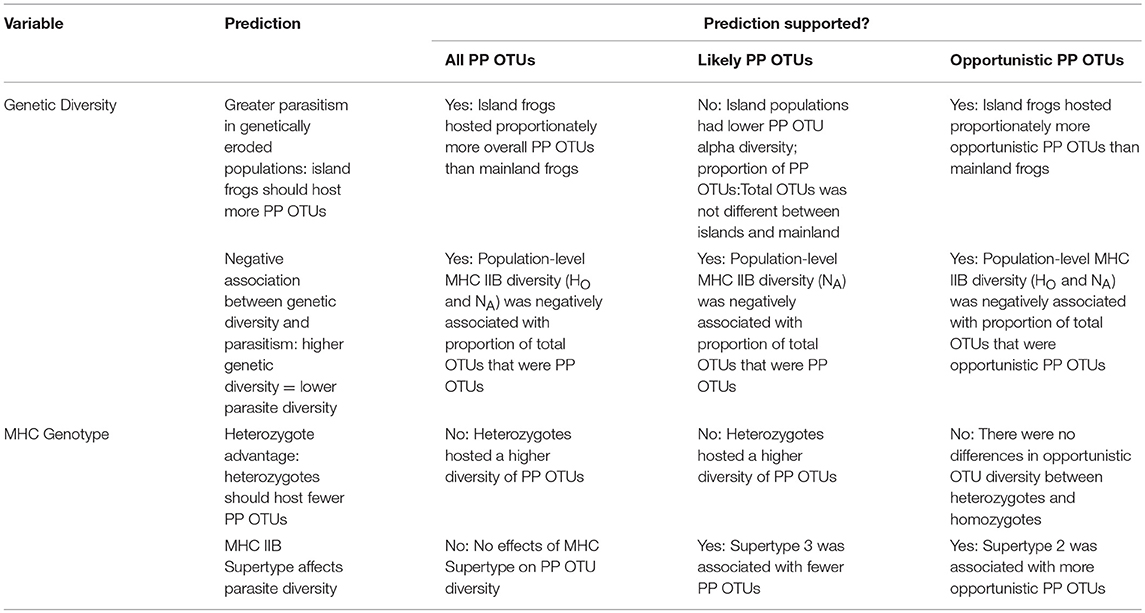
Table 2. Summary of predicted outcomes and observed study results.
Our results also indicate significant associations between PP OTUs and MHC class IIB. On the individual level, PP OTU alpha diversity was higher in MHC IIB heterozygotes both when the entire PP OTU dataset was analyzed and when opportunistic PP OTUs were excluded (Table 2). It remains unclear whether this relationship is driven by balancing selection on the MHC in the presence of a higher diversity of pathogens and parasites, or whether heterozygotes exhibit higher tolerance for pathogens and parasites, resulting in higher apparent infections. MHC class IIB supertype was also associated with differences in PP OTU richness. Supertype 3 was associated with fewer non-opportunistic PP OTUs, whereas Supertype 2 was associated with greater numbers of opportunistic PP OTUs than those possessing either Supertype 1 or Supertype 3. These results suggest that MHC IIB genotype can influence infection dynamics, and specifically that in T. thoropa Supertype 3 may be associated with the greatest resistance against likely parasites. More research is needed to determine the relationship between parasite richness and disease outcomes, as well as the evolutionary consequences of elevated parasite diversity on host immunogenes.
On the population level, reduced MHC class IIB diversity corresponded with proportionally more PP OTUs (PP OTUs: total eukaryotic OTUs; Figure 3B and Table 2). To address the possibility that these relationships were confounded by genetic structure or other factors covarying with site type (such as environmental differences between sites), we excluded island populations and re-analyzed the data, and recovered the same negative associations between MHC IIB diversity and proportional richness PP OTUs. The largest negative slopes and highest R2-values were observed for the dataset only including opportunistic PP OTUs (across all populations: β = −0.031, R2 = 0.21 for NA, β = −0.338, R2 = 0.12 for HO). This relatively strong negative relationship would be expected if population-level MHC IIB diversity reflects the immunocompetence of individuals, as immunocompromised individuals tend to be more susceptible to opportunistic infections (Anaissie, 1992).
The negative associations of all three PP OTU fractions with MHC IIB diversity suggest that populations that lose allelic diversity at the MHC class IIB locus may be subject to increased parasitism. Epidemics select for rare alleles in the surviving population, a mechanism known as negative frequency-dependent selection (Lively and Dybdahl, 2000). Loss of rare alleles through the loss of immunogenetic diversity may thus increase population-level susceptibility to new pathogen invaders (Berngruber et al., 2013). This dovetails with the finding that rare immunogenetic types have been associated with increased disease resistance in natural populations (Schwensow et al., 2007). Due to limited sample sizes, we did not possess the statistical power to determine whether rare alleles (haplotypes) were associated with lower parasite richness in T. taophora. Nonetheless, the association of population-level MHC IIB diversity with lower PP OTU richness suggests that populations with more rare alleles may experience fewer infections.
Our approach for identifying a broad diversity of parasites does not come without caveats. First, the specificity of the taxonomic match reported for each OTU is dependent upon whether data from closely related taxa occur in the reference database. Nearly half (39/97) of our potentially parasitic OTUs had a closest match from GenBank of <97% (Table S1), which is the commonly accepted (and arguably conservative) sequence similarity cutoff below which microbial organisms are considered distinct species. For example, one OTU in our dataset (“New.CleanUp.ReferenceOTU912”) matched Perkinsus qugwadi, a species which is known to parasitize mollusks (Blackbourn et al., 1998). However, this represents a match of low identity (83%), indicating that our OTU is likely a related organism. Lower on the list of closest GenBank matches for our OTU is an unidentified alveolate parasite of the frog Rana sphenocephala (Davis et al., 2007). This further supports that our OTU represents an unidentified but likely parasitic eukaryote.
A second caveat of our approach is that we categorized some OTUs as potentially parasitic based on known parasitism in vertebrate taxa other than amphibians. We argue that it is reasonable to treat these as potential parasites because (1) many of these OTUs are present on a number of individuals from a number of populations (Table S1), which reduces the likelihood that these are incidental or transient; and (2) they represent poorly-studied taxa, particularly in non-human animals. A number of the PP OTUs that are non-specifically listed as “animal parasites” are classified within the Apicomplexa, which is an entirely parasitic phylum.
Third, the presence of these PP OTUs in a sample does not necessarily imply disease: many parasitic microbes can be commensal in healthy individuals or at low loads. However, the presence of parasite organisms is necessary for disease, implies a less than healthy microbiome (Willing et al., 2010), and is likely to activate host immune responses even in the absence of symptomatic disease. While interpretations of our dataset are limited by a lack of demonstrated pathology, we present a preliminary study that advances understanding of the diversity and distribution of potentially parasitic microbes inhabiting the skin of non-human animal hosts. Complementary approaches including microscopy and/or experimental infections would be useful in future studies to confirm parasitism by PP OTUs identified with a similar NGS-based parasite screening approach.
We note that we are unable to exclude other factors that may contribute to infection susceptibility, including contributions by other genetic loci (including other immunogenetic loci), effects of individual-level genome-wide diversity or heterozygosity (Acevedo-Whitehouse and Cunningham, 2006), aspects of innate immunity (Richmond et al., 2009), or environmental influences on immunity and/or disease. In addition, because the MHC region we have amplified is relatively short (~120 bp), our analysis may underestimate the number of haplotypes among populations. However, despite some level of ascertainment bias, we have recovered a genetic pattern that is significantly associated with variation in potential parasite diversity.
Lastly, we note that the remaining ~89% of eukaryotes that were amplified from the skin samples could be ecologically important as well. Eukaryotes in the amphibian skin microbiome have only recently come under study (Kueneman et al., 2016), though bacteria found in the amphibian skin microbiome are known to play significant roles in amphibian host health. In particular, some bacteria are known to inhibit Bd (Myers et al., 2012) and may contribute to disease resistance (Harris et al., 2009). Preliminary research on eukaryotes suggests that fungi isolated from frog skin may in reality contribute more to Bd resistance than bacteria (Kearns et al., 2017). Symbiotic micro-eukaryotes have the potential to serve as competitors or hyperparasites of harmful microbes in the host-associated microbiome. Our method of combining NGS with a survey of available literature can be applied in future research to tie host-associated microbiota data with demonstrated ecological functions of the microbiome.
The results of our study have several implications. First, the effects of fragmentation on immunogenetics can be strong, although these may be dependent on the focal taxon and the conditions under study. Second, using targeted assays to study specific pathogens may underestimate infections, to the extent that analyses may be completely blind to significant ecological patterns. Third, fragmentation may increase disease susceptibility by altering host genetics. Our results support prioritizing genetic diversity in vulnerable wildlife populations to improve survival and resilience. Future studies of field infections should bear in mind that diverse coinfections in amphibians may be highly prevalent, and thus should consider a broad diversity of pathogens and parasites if infection status is a variable of interest.
Data Availability Statement
The supporting data and sequences for this study can be found in the GenBank (MK348627 - MK348653 and MK350361 - MK351204).
Ethics Statement
All methods were approved by the Institutional Animal Care and Use Committee of the University of Michigan (protocols PRO00005605 and PRO00007691).
Author Contributions
AB, TJ, and LT conceived of the study. AB, TJ, LT, and DL coordinated the field and lab work. AB, TJ, and LT collected field samples. AB performed the labwork. AB and MB performed the data analysis. All authors provided contributions to the writing and gave final approval before submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Paula Morão and Luis Moreno for assistance in field logistics, field work, and labwork; Vinicius Hansser, Amanda Piffer, and Cesar Alexandre for assistance in the field; Carlos Almeida for providing site coordinates; Rebecca Clemons and Nicholas Farrugia for assistance with labwork; Alisha Quandt, William Davis, Thibaut Jombart, and Anna Savage for assistance with bioinformatics and data analysis; and Kelly Zamudio and Celio Haddad for assistance with fieldwork planning. We also thank two reviewers for providing feedback on earlier versions of the manuscript. We wish to acknowledge funding from NSF (OISE-1159513), CNPq (300980/2014-0), FAPESP (#2016/25358-3), and a Block Grant from the U. Michigan Dept. of Ecology and Evolutionary Biology.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2018.00236/full#supplementary-material
References
Acevedo-Whitehouse, K., and Cunningham, A. A. (2006). Is MHC enough for understanding wildlife immunogenetics? Trends Ecol. Evol. 21, 433–438. doi: 10.1016/j.tree.2006.05.010
Aguilar, A., Roemer, G., Debenham, S., Binns, M., Garcelon, D., and Wayne, R. K. (2004). High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc. Natl. Acad. Sci. U.S.A. 101, 3490–3494. doi: 10.1073/pnas.0306582101
Allentoft, M. E., and O'Brien, J. (2010). Global amphibian declines, loss of genetic diversity and fitness: a review. Diversity 2, 47–71. doi: 10.3390/d2010047
Altizer, S., Harvell, D., and Friedle, E. (2003). Rapid evolutionary dynamics and disease threats to biodiversity. Trends Ecol. Evol. 18, 589–596. doi: 10.1016/j.tree.2003.08.013
Anaissie, E. J. (1992). Opportunistic mycosis in the immunocompromissed host: experience at a cancer center and review. Clin. Infect. Dis. 14, 43–53. doi: 10.1093/clinids/14.Supplement_1.S43
Arenas, M., Mona, S., Trochet, A., Sramkova Hanulova, A., Currat, M., Ray, N., et al. (2014). “The scaling of genetic diversity in a changing and fragmented world,” in Scaling in Ecology and Biodiversity Conservation, eds K. Henle, S. G. Potts, W. E. Kunin, Y. G. Matsinos, J. Similä, J. D. Pantis, et al. (Sofia: Pensoft Publishers), 55–60.
Arens, P., Van Der Sluis, T., Van't Westende, W. P. C., Vosman, B., Vos, C. C., and Smulders, M. J. M. (2007). Genetic population differentiation and connectivity among fragmented Moor frog (Rana arvalis) populations in the Netherlands. Landsc. Ecol. 22, 1489–1500. doi: 10.1007/s10980-007-9132-4
Arkush, K. D., Giese, A. R., Mendonca, H. L., Mcbride, A. M., Marty, G. D., and Hedrick, P. W. (2002). Resistance to three pathogens in the endangered winter-run chinook salmon (Oncorhynchus tshawytscha): effects of inbreeding and major histocompatibility complex genotypes. Can. J. Fish. Aquat. Sci. 59, 966–975. doi: 10.1139/F02-066
Bataille, A., Cashins, S. D., Grogan, L., Skerratt, L. F., Hunter, D., McFadden, M., et al. (2015). Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc. Biol. Sci. 282:20143127. doi: 10.1098/rspb.2014.3127
Belasen, A., Brock, K., Li, B., Chremou, D., Valakos, E., Pafilis, P., et al. (2016). Fine with heat, problems with water: microclimate alters water loss in a thermally adapted insular lizard. Oikos 126, 447–457. doi: 10.1111/oik.03712
Bell, R. C., Brasileiro, C. A., Haddad, C. F. B., and Zamudio, K. R. (2012). Evolutionary history of Scinax treefrogs on land-bridge islands in south-eastern Brazil. J. Biogeogr. 39, 1733–1742. doi: 10.1111/j.1365-2699.2012.02708.x
Bernatchez, L., and Landry, C. (2003). MHC studies in non-model vertebrates: what have we learned about natural selection in 15 years? J. Evol. Biol. 16, 363–377. doi: 10.1046/j.1420-9101.2003.00531.x
Berngruber, T. W., Froissart, R., Choisy, M., and Gandon, S. (2013). Evolution of virulence in emerging epidemics. PLoS Pathog. 9:e1003209. doi: 10.1371/journal.ppat.1003209
Bielby, J., Fisher, M. C., Clare, F. C., Rosa, G. M., and Garner, T. W. J. (2015). Host species vary in infection probability, sub-lethal effects, and costs of immune response when exposed to an amphibian parasite. Sci. Rep. 5:10828. doi: 10.1038/srep10828
Blackbourn, J., Bower, S. M., and Meyer, G. R. (1998). Perkinsus qugwadi sp.nov. (incertae sedis), a pathogenic protozoan parasite of Japanese scallops, Patinopecten yessoensis, cultured in British Columbia, Canada. Can. J. Zool. 76, 942–953. doi: 10.1139/z98-015
Bokermann, W. (1974). Notas sobre as especies de Thoropa Fitinger (Amphibia, Leptodactylidae). An. Acad. Bras. Cienc. 37, 525–537. doi: 10.2307/302397
Boyle, D. G., Boyle, D. B., Olsen, V., Morgan, J. A. T., and Hyatt, A. D. (2004). Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Organ. 60, 141–148. doi: 10.3354/dao060141
Briggs, C. J., Knapp, R. A., and Vredenburg, V. T. (2010). Enzootic and epizootic dynamics of the chytrid fungal pathogen of amphibians. Proc. Natl. Acad. Sci. U.S.A. 107, 9695–9700. doi: 10.1073/pnas.0912886107
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chambouvet, A., Gower, D. J., Jirk,u, M., Yabsley, M. J., Davis, A. K., Leonard, G., et al. (2015). Cryptic infection of a broad taxonomic and geographic diversity of tadpoles by Perkinsea protists. Proc. Natl. Acad. Sci. U.S.A. 112, E4743–E4751. doi: 10.1073/pnas.1500163112
Cushman, S. A. (2006). Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol. Conserv. 128, 231–240. doi: 10.1016/j.biocon.2005.09.031
Davis, A. K., Yabsley, M. J., Kevin Keel, M., and Maerz, J. C. (2007). Discovery of a novel alveolate pathogen affecting southern leopard frogs in Georgia: description of the disease and host effects. Ecohealth 4, 310–317. doi: 10.1007/s10393-007-0115-3
Desser, S. S., Hong, H., and Martin, D. S. (1995). The life history, ultrastructure, and experimental transmission of Hepatozoon catesbianae n. comb., an apicomplexan parasite of the bullfrog, Rana catesbeiana and the mosquito, Culex territans in algonquin park, Ontario. J. Parasitol. 81, 212–22. doi: 10.2307/3283922
Deter, J., Bryja, J., Chaval, Y., Galan, M., Henttonen, H., Laakkonen, J., et al. (2008). Association between the DQA MHC class II gene and Puumala virus infection in Myodes glareolus, the bank vole. Infect. Genet. Evol. 8, 450–458. doi: 10.1016/j.meegid.2007.07.003
Dobson, A., and Foufopoulos, J. (2001). Emerging infectious pathogens of wildlife. Philos. Trans. R. Soc. B Biol. Sci. 356, 1001–1012. doi: 10.1098/rstb.2001.0900
Doherty, P. C., and Zinkernagel, R. M. (1975). Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256, 50–52. doi: 10.1038/256050a0
Duryea, M. C., Zamudio, K. R., and Brasileiro, C. A. (2015). Vicariance and marine migration in continental island populations of a frog endemic to the Atlantic Coastal forest. Heredity 115, 225–234. doi: 10.1038/hdy.2015.31
Dyer, W. G. (1991). Helminth parasites of amphibians from Illinois and adjacent midwestern states. Trans. Illinois State Acad. Sci. 84, 125–143.
Edgar, R. C. (2016). UCHIME2: improved chimera prediction for amplicon sequencing. bioRxiv [Preprint]. doi: 10.1101/074252
Ellegren, H., Hartman, G., Johansson, M., and Andersson, L. (1993). Major histocompatibility complex monomorphism and low levels of DNA fingerprinting variability in a reintroduced and rapidly expanding population of beavers. Proc. Natl. Acad. Sci. U.S.A. 90, 8150–8153. doi: 10.1073/pnas.90.17.8150
Ellison, A., Allainguillaume, J., Girdwood, S., Pachebat, J., Peat, K. M., Wright, P., et al. (2012). Maintaining functional major histocompatibility complex diversity under inbreeding: the case of a selfing vertebrate. Proc. R. Soc. B Biol. Sci. 279, 5004–5013. doi: 10.1098/rspb.2012.1929
Ellison, A., Cable, J., and Consuegra, S. (2011). Best of both worlds? Association between outcrossing and parasite loads in a selfing fish. Evolution 65, 3021–3026. doi: 10.1111/j.1558-5646.2011.01354.x
Estrada-Villegas, S., Meyer, C. F. J., and Kalko, E. K. V. (2010). Effects of tropical forest fragmentation on aerial insectivorous bats in a land-bridge island system. Biol. Conserv. 143, 597–608. doi: 10.1016/j.biocon.2009.11.009
Gahl, M. K., Longcore, J. E., and Houlahan, J. E. (2012). Varying responses of Northeastern North American amphibians to the chytrid pathogen Batrachochytrium dendrobatidis. Conserv. Biol. 26, 135–141. doi: 10.1111/j.1523-1739.2011.01801.x
Gaudieri, S., Dawkins, R. L., Habara, K., Kulski, J. K., and Gojobori, T. (2000). SNP profile within the human major histocompatibility complex reveals an extreme and interrupted level of nucleotide diversity. Genome Res. 10, 1579–1586. doi: 10.1101/gr.127200
Gervasi, S. S., Civitello, D. J., Kilvitis, H. J., and Martin, L. B. (2015). The context of host competence: a role for plasticity in host-parasite dynamics. Trends Parasitol. 31, 419–425. doi: 10.1016/j.pt.2015.05.002
Giaretta, A. A., and Facure, K. G. (2004). Reproductive ecology and behavior of Thoropa miliaris (Spix, 1824) (Anura, Leptodactyidae, Telmatobiinae). Biota Neotrop. 4, 1–10. doi: 10.1590/S1676-06032004000200008
Gleason, F. H., Chambouvet, A., Sullivan, B. K., Lilje, O., and Rowley, J. J. L. (2014). Multiple zoosporic parasites pose a significant threat to amphibian populations. Fungal Ecol. 11, 181–192. doi: 10.1016/j.funeco.2014.04.001
Hambuch, T. M., and Lacey, E. A. (2002). Enhanced selection for MHC diversity in social tuco-tucos. Evolution 56, 841–845. doi: 10.1111/j.0014-3820.2002.tb01395.x
Harris, R. N., Lauer, A., Simon, M. A., Banning, J. L., and Alford, R. A. (2009). Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis. Aquat. Organ. 83, 11–16. doi: 10.3354/dao02004
Hill, A. V. S., Allsopp, C. E. M., Kwiatkowski, D., Anstey, N. M., Twumasi, P., Rowe, P. A., et al. (1991). Common West African HLA antigens are associated with protection from severe malaria. Nature 352, 595–600. doi: 10.1038/352595a0
Hurston, H., Voith, L., Bonanno, J., Foufopoulos, J., Pafilis, P., Valakos, E., et al. (2009). Effects of fragmentation on genetic diversity in island populations of the Aegean wall lizard Podarcis erhardii (Lacertidae, Reptilia). Mol. Phylogenet. Evol. 52, 395–405. doi: 10.1016/j.ympev.2009.03.028
Hyatt, A. D., Boyle, D. G., Olsen, V., Boyle, D. B., Berger, L., Obendorf, D., et al. (2007). Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis. Aquat. Organ. 73, 175–192. doi: 10.3354/dao073175
Ilmonen, P., Penn, D. J., Damjanovich, K., Clarke, J., Lamborn, D., Morrison, L., et al. (2008). Experimental infection magnifies inbreeding depression in house mice. J. Evol. Biol. 21, 834–841. doi: 10.1111/j.1420-9101.2008.01510.x
Jombart, T., Devillard, S., Balloux, F., Falush, D., Stephens, M., Pritchard, J., et al. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94. doi: 10.1186/1471-2156-11-94
Karr, J. R. (1982). Population variability and extinction in the avifauna of a tropical land bridge Island. Ecology 63, 1975–1978. doi: 10.2307/1940137
Kearns, P. J., Fischer, S., Fernandez-Beaskoetxea, S., Gabor, C. R., Bosch, J., Bowen, J. L., et al. (2017). Fighting fungi with fungi: the mycobiome contribution to emerging disease in amphibians. Peerj Prepr. 5:e3017v1. doi: 10.7287/peerj.preprints.3017v1
Kiesecker, J. M., Blaustein, A. R., and Belden, L. K. (2001). Complex causes of amphibian population declines. Nature 410, 681–684. doi: 10.1038/35070552
Kosch, T. A., Bataille, A., Didinger, C., Eimes, J. A., Rodríguez-Brenes, S., Ryan, M. J., et al. (2016). Major histocompatibility complex selection dynamics in pathogen-infected túngara frog (Physalaemus pustulosus) populations. Biol. Lett. 12:345. doi: 10.1098/rsbl.2016.0345
Kueneman, J. G., Woodhams, D. C., Treuren, W., and Van Archer, H. M. (2016). Inhibitory bacteria reduce fungi on early life stages of endangered Colorado boreal toads (Anaxyrus boreas). Int. Soc. Microb. Ecol. 10, 934–944. doi: 10.1038/ismej.2015.168
Landry, C., Garant, D., Duchesne, P., and Bernatchez, L. (2001). “Good genes as heterozygosity”: the major histocompatibility complex and mate choice in Atlantic salmon (Salmo salar). Proc. R. Soc. B Biol. Sci. 268, 1279–1285. doi: 10.1098/rspb.2001.1659
Lesbarrères, D., Primmer, C. R., Laurila, A., and Meril,ä, J. (2005). Environmental and population dependency of genetic variability-fitness correlations in Rana temporaria. Mol. Ecol. 14, 311–323. doi: 10.1111/j.1365-294X.2004.02394.x
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lighten, J., Van Oosterhout, C., and Bentzen, P. (2014). Critical review of NGS analyses for de novo genotyping multigene families. Mol. Ecol. 23, 3957–3972. doi: 10.1111/mec.12843
Lively, C. M., and Dybdahl, M. F. (2000). Parasite adaptation to locally common host genotypes. Nature 405, 679–681. doi: 10.1038/35015069
Macdougall-Shackleton, E. A., Derryberry, E. P., Foufopoulos, J., Dobson, A. P., and Hahn, T. P. (2005). Parasite-mediated heterozygote advantage in an outbred songbird population. Biol. Lett 1, 105–107. doi: 10.1098/rsbl.2004.0264
Martel, A., Spitzen-van der Sluijs, A., Blooi, M., Bert, W., Ducatelle, R., Fisher, M. C., et al. (2013). Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc. Natl. Acad. Sci. U.S.A. 110, 15325–15329. doi: 10.1073/pnas.1307356110
May, S., and Beebee, T. J. C. (2009). Characterisation of major histocompatibility complex class II alleles in the natterjack toad, Bufo calamita. Conserv. Genet. Resour. 1, 415–417. doi: 10.1007/s12686-009-9096-6
McClelland, E. E., Penn, D. J., and Potts, W. K. (2003). Major histocompatibility complex heterozygote superiority during coinfection. Infect. Immun. 71, 2079–2086. doi: 10.1128/IAI.71.4.2079-2086.2003
Mona, S., Ray, N., Arenas, M., and Excoffier, L. (2014). Genetic consequences of habitat fragmentation during a range expansion. Heredity 112, 291–299. doi: 10.1038/hdy.2013.105
Myers, J. M., Ramsey, J. P., Blackman, A. L., Nichols, A. E., Minbiole, K. P. C., and Harris, R. N. (2012). Synergistic inhibition of the lethal fungal pathogen Batrachochytrium dendrobatidis: the combined effect of symbiotic bacterial metabolites and antimicrobial peptides of the frog Rana muscosa. J. Chem. Ecol. 38, 958–965. doi: 10.1007/s10886-012-0170-2
Newmark, W. D. (1995). Extinction of mammal populations in Western North American National Parks. Conserv. Biol. 9, 512–526. doi: 10.1046/j.1523-1739.1995.09030512.x
Paradis, E. (2010). pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26, 419–420. doi: 10.1093/bioinformatics/btp696
Pearman, P. B., and Garner, T. W. J. (2005). Susceptibility of Italian agile frog populations to an emerging strain of Ranavirus parallels population genetic diversity. Ecol. Lett. 8, 401–408. doi: 10.1111/j.1461-0248.2005.00735.x
Perfecto, I., and Vandermeer, J. (2010). The agroecological matrix as alternative to the land-sparing/agriculture intensification model. Proc. Natl. Acad. Sci. U.S.A. 107, 5786–5791. doi: 10.1073/pnas.0905455107
Piertney, S. B., and Oliver, M. K. (2006). The evolutionary ecology of the major histocompatibility complex. Heredity (Edinb) 96, 7–21. doi: 10.1038/sj.hdy.6800724
Pritchett, K. R., and Sanders, G. E. (2007). Epistylididae ectoparasites in a colony of African clawed frogs (Xenopus laevis). J. Am. Assoc. Lab. Anim. Sci. 46, 86–91.
Radwan, J., Biedrzycka, A., and Babik, W. (2009). Does reduced MHC diversity decrease viability of vertebrate populations? Biol. Conserv. 143, 537–544. doi: 10.1016/j.biocon.2009.07.026
Reche, P. A., and Reinherz, E. L. (2003). Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J. Mol. Biol. 331, 623–641. doi: 10.1016/S0022-2836(03)00750-2
Richmond, J. Q., Savage, A. E., Zamudio, K. R., and Rosenblum, E. B. (2009). Toward immunogenetic studies of amphibian chytridiomycosis: linking innate and acquired immunity. Bioscience 59, 311–320. doi: 10.1525/bio.2009.59.4.9
Roca, V., Foufopoulos, J., Valakos, E., and Pafilis, P. (2010). Parasitic infracommunities of the Aegean wall lizard Podarcis erhardii (Lacertidae, Sauria): isolation and impoverishment in small island populations. Amphib. reptil. 30, 493–503. doi: 10.1163/156853809789647176
Rowley, J. J. L., Gleason, F. H., Andreou, D., Marshall, W. L., Lilje, O., and Gozlan, R. (2013). Impacts of mesomycetozoean parasites on amphibian and freshwater fish populations. Fungal Biol. Rev. 27, 100–111. doi: 10.1016/j.fbr.2013.09.002
Santonastaso, T., Lighten, J., van Oosterhout, C., Jones, K. L., Foufopoulos, J., and Anthony, N. M. (2017). The effects of historical fragmentation on major histocompatibility complex class II β and microsatellite variation in the Aegean island reptile, Podarcis erhardii. Ecol. Evol. 7, 4568–4581. doi: 10.1002/ece3.3022
Savage, A. E., and Zamudio, K. R. (2011). MHC genotypes associate with resistance to a frog-killing fungus. Proc. Natl. Acad. Sci. U.S.A. 108, 16705–16710. doi: 10.1073/pnas.1106893108
Sazima, I. (1971). The occurrence of marine invertebrates in the stomach contents of the frog Thoropa miliaris. Cienc. Cult. 23, 647–648. doi: 10.1163/_q3_SIM_00374
Schwensow, N., Fietz, J., Dausmann, K. H., and Sommer, S. (2007). Neutral versus adaptive genetic variation in parasite resistance: importance of major histocompatibility complex supertypes in a free-ranging primate. Heredity 99, 265–277. doi: 10.1038/sj.hdy.6800993
Siddle, H. V., Marzec, J., Cheng, Y., Jones, M., and Belov, K. (2010). MHC gene copy number variation in Tasmanian devils: implications for the spread of a contagious cancer. Proc. R. Soc. B Biol. Sci. 277, 2001–2006. doi: 10.1098/rspb.2009.2362
Slade, R. W. (1992). Limited MHC polymorphism in the southern elphant seal. Proc. Biol. Sci. 249, 163–171. doi: 10.1098/rspb.1992.0099
Speare, R., Shea, P. O., Shipton, W. A., and Thomas, A. D. (1994). Mucor amphibiorum in the toad, Bufo marinus, in Australia. J. Wildl. Dis. 30, 399–407. doi: 10.7589/0090-3558-30.3.399
Spielman, D., Brook, B. W., Briscoe, D. A., and Frankham, R. (2004). Does inbreeding and loss of genetic diversity decrease disease resistance? Conserv. Genet. 5, 439–448. doi: 10.1023/B:COGE.0000041030.76598.cd
Stephens, M., Smith, N. J., and Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989. doi: 10.1086/319501
Stockwell, M. P., Clulow, J., and Mahony, M. J. (2015). Evidence of a salt refuge: chytrid infection loads are suppressed in hosts exposed to salt. Oecologia 177, 901–910. doi: 10.1007/s00442-014-3157-6
Stoeck, T., Bass, D., Nebel, M., Christen, R., Jones, M. D. M., Breiner, H. W., et al. (2010). Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 19, 21–31. doi: 10.1111/j.1365-294X.2009.04480.x
Suguio, K., Angulo, R. J., Carvalho, A. M., Corrêa, I. C. S., Tomazeli, L. J., Willwock, J. A., et al. (2005). “Paleoníveis do mar e paleolinhas da costa,” in Quaternário do Brasil, eds C. R. G. Souza, K. Suguio, A. M. S. Oliveira, and P. E. Oliveira (São Paulo: Associação Brasileira de Estudos do Quaternário), 378.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tilman, D., May, R. M., Lehman, C. L., and Nowak, M. A. (1994). Habitat destruction and the extinction debt. Nature 371, 65–66. doi: 10.1038/371065a0
Whitehorn, P. R., Tinsley, M. C., Brown, M. J. F., Darvill, B., and Goulson, D. (2011). Genetic diversity, parasite prevalence and immunity in wild bumblebees. Proc. R. Soc. B Biol. Sci. 278, 1195–1202. doi: 10.1098/rspb.2010.1550
Wikelski, M., Foufopoulos, J., Vargas, H., and Snell, H. (2004). Galápagos birds and diseases: invasive pathogens as threats for island species. Ecol. Soc. 9:5. doi: 10.5751/ES-00605-090105
Willing, B. P., Dicksved, J., Halfvarson, J., Andersson, A. F., Lucio, M., Zheng, Z., et al. (2010). A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139, 1844–1854.e1. doi: 10.1053/j.gastro.2010.08.049
Yildirimhan, H. S., Bursey, C. R., and Goldberg, S. R. (2006). Helminth parasites of the taurus frog, Rana holtzi, and the uludag frog, Rana macrocnemis, with remarks on the helminth community of turkish anurans. Comp. Parasitol. 73, 237–248. doi: 10.1654/4191.1
Keywords: habitat fragmentation, amphibians, parasites, immunogenetics, Brazilian Atlantic Forest
Citation: Belasen AM, Bletz MC, Leite DS, Toledo LF and James TY (2019) Long-Term Habitat Fragmentation Is Associated With Reduced MHC IIB Diversity and Increased Infections in Amphibian Hosts. Front. Ecol. Evol. 6:236. doi: 10.3389/fevo.2018.00236
Received: 30 August 2018; Accepted: 19 December 2018;
Published: 10 January 2019.
Edited by:
Miguel Arenas, University of Vigo, SpainReviewed by:
Ryan Calsbeek, Dartmouth College, United StatesMatthew Fisher, Imperial College London, United Kingdom
Yessica Rico, Instituto de Ecología (INECOL), Mexico
Copyright © 2019 Belasen, Bletz, Leite, Toledo and James. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anat M. Belasen, YWJlbGFzZW5AdW1pY2guZWR1