 Niccolò Forin1,2†
Niccolò Forin1,2† Sebastiano Nigris1†
Sebastiano Nigris1† Samuele Voyron3
Samuele Voyron3 Mariangela Girlanda3
Mariangela Girlanda3 Alfredo Vizzini3
Alfredo Vizzini3 Giorgio Casadoro1
Giorgio Casadoro1 Barbara Baldan1,2*
Barbara Baldan1,2*- 1Botanical Garden, University of Padua, Padua, Italy
- 2Department of Biology, University of Padua, Padua, Italy
- 3Department of Life Sciences and System Biology, University of Turin, Turin, Italy
Despite their essential role in the environment, the number of known fungal species is still low compared to the recent estimates of the fungal biodiversity (from 2.2 to 3.8 million species), principally because of their often cryptic or ambiguous morphological traits. Recent studies have reported that only about approx. 35,000 correctly identified fungal species are represented by DNA sequences in public databases. This corresponds to a mere 1% of the estimated total number of species. Thus, the linkage of curated DNA sequence data to properly identified voucher specimens is of fundamental importance to fill the present gap between the different sizes of described and sequenced fungal diversity. For this purpose, mycological herbarium collections are considered an important source for fungal DNA-barcoding, and collection-based sequencing is a relevant priority for the coming decades. Unfortunately, ancient herbarium samples have both time and conservation related DNA damages, besides exogenous DNA contamination, that make nucleic acid extraction and amplification challenging. Here, we present the results of DNA extraction, ITS2 amplification and Illumina MiSeq sequencing of 36 specimens from the Saccardo Mycological Herbarium that were collected in the late XIX century and assigned to the genus Peziza. High-throughput sequencing was chosen as an alternative to the conventional Sanger- and cloning-based methods to overcome the high fragmentation of the ancient DNA and the massive occurrence of non-target DNA from fungal contaminants. Our approach has permitted the assignment of ITS2 sequences to 23 out of the 36 specimens studied in this work, thus providing a univocal DNA sequence for those one century old samples. Furthermore, the ITS2 sequence analysis has permitted a taxonomic study of the samples that has resulted in a re-evaluation of five samples at the species level and 18 samples at genus or higher level. Our results highlight the possibility to apply the technique presented in this work also to the old and more precious type specimens in order to relate a DNA sequence to these important samples, coupling the traditional morphological description of the species with a DNA sequence.
Introduction
In terms of biodiversity fungi are one of the least described groups of multicellular eukaryotes even though they comprise 2.2–3.8 million species (Hawksworth and Lücking, 2017). In fact, despite their important role in ecological systems (Fisher et al., 2012; Martínez-García et al., 2017; Bani et al., 2018), only a small fraction of the estimated species has been formally described (approx. 120,000), and for many of them (approx. 85,000) no DNA sequences have been deposited in public DNA databases so far (Hawksworth and Lücking, 2017). On the other hand, fungal metagenome sequencing is providing scientists with a large number of datasets composed of sequences from environmental/uncultured fungi with no or poor taxonomic annotation. Thus, the linkage of curated DNA sequence data to expertly identified voucher specimens is a fundamental step to fill the present gap between the different sizes of described and sequenced fungal diversity.
In 2012, the Consortium for the Barcode of Life recognized the internal transcribed spacer (ITS) regions of the nuclear ribosomal RNA gene cluster as the primary fungal barcode marker (Schoch et al., 2012). This region includes the highly variable non-coding ITS1 and ITS2 regions separated by the 5.8S coding gene and situated between 18S (small subunit; SSU) and 28S (large subunit; LSU) coding genes. Thanks to the rapid evolutionary rate of the ITS1 and ITS2 non-coding regions their sequences have been used for over 20 years in the identification of fungal species (Nilsson et al., 2008). On the other hand, the highly conserved 18S, 5.8S, and 28S genes have allowed the development of universal primers for PCR amplification and sequencing of either the entire ITS region or the ITS1 and ITS2 regions separately (White et al., 1990; Gardes and Bruns, 1993; Martin and Rygiewicz, 2005; Toju et al., 2012). An additional advantage of this DNA marker is that the fungal genome contains multiple tandemly repeated copies of the ribosomal RNA gene cluster (including the ITS), thus making possible the amplification of this region from small amounts of DNA (Xu, 2016). The sequencing of these marker regions can be easily applied to living organism material, therefore it can be particularly useful also for DNA barcoding of mycological collections (Osmundson et al., 2013).
The fungi collections conserved worldwide in herbaria are considered a great source of genetic material and the collection-based sequencing projects are assuming an important priority in the coming decades because the herbaria can host a large number of unknown or under represented fungal lineages (Brock et al., 2009; Osmundson et al., 2013). In addition, ancient herbarium collections may contain type specimens, in particular holotypes. These specimens have great scientific value because they represent the only certain link to a specific Latin binomial name; therefore, the eventual existence of molecular information from these important samples might help to solve taxonomic controversies (Liimatainen et al., 2014; Mutanen et al., 2014). However, it is well known that obtaining molecular information from very old biological material can be particularly challenging, the main problem being the fragmentation of the ancient DNA (aDNA). The nucleic acids of old biological samples are often degraded into small fragments whose size is generally less than 500 bp (Pääbo et al., 2004; Dabney et al., 2013). Although the DNA of the herbarium specimens naturally degrades in the course of time, it has been demonstrated that the fragmentation degree of the aDNA depends mainly on the methods used to collect, dry and store the specimens rather than on the age of the herbarium material (Erkens et al., 2008). This is especially true for old herbaria where the specimens were never collected and stored for optimal DNA conservation but rather for optimal conservation of the specimens' morphological traits. For example, the adopted drying methods can have different effects on the DNA quality, and the use of some chemicals as pesticides can lead to a high-level degradation of the specimens' DNA (Kigawa et al., 2003; Nagy, 2010; Staats et al., 2011).
Another obstacle that makes DNA extraction and sequencing from herbarium samples challenging is the presence of contaminant material associated to the conserved specimens. Ancient collections are frequently contaminated by other organisms, like airborne fungi or bacteria. Moreover, the specimens are often not properly handled and conserved in sealed containers but rather are stored on paper sheets stacked in herbarium packages. This close contact between different specimens enhances the probability of cross-contamination between the samples in the packages (Yeates et al., 2016). The presence of exogenous fungal DNA is an issue affecting sequencing projects involving ancient mycological collections, since contaminant DNA may outcompete the target DNA during the amplification step. Furthermore, the universal primers adopted in the barcoding procedure cannot discriminate the target DNA from the more recent and intact exogenous DNA, and this can lead to an overrepresentation of non-target sequences with the consequence of either a reduced or no sequencing success.
In addition, the amount of herbarium material usable for DNA extraction is often limited because many specimens are poor in available tissue and because the sampling to yield a sufficient amount of DNA can in some cases cause irreparable damage to the herbarium specimens. Notwithstanding such problems, DNA was successfully extracted and amplified from lichen and plant herbarium samples older than 100 years (Lehtonen and Christenhusz, 2010; Redchenko et al., 2012) and from fungal samples older than 200 years (Larsson and Jacobsson, 2004). These results demonstrate that ancient collections could still represent a source of material for molecular studies.
In this sense, the Pier Andrea Saccardo mycological collection conserved in the herbarium of the Botanical Garden of the University of Padua can represent a rich source of specimens suitable for DNA barcoding. This collection, started by P.A. Saccardo around 1874, contains almost 70,000 fungal specimens encompassing over 18,500 different species which have not been subjected to molecular studies so far. The herbarium was first put together by Saccardo with samples collected by himself around Treviso (his home town) and Padua (Botanical Garden and surroundings); then it was enriched with Italian and foreign specimens sent to him by colleagues and with whole mycological collections presented to him by mycologists from around the world. During his day, Saccardo was recognized as an expert mycologist, as shown by the many specimens that were sent to him from scientists for sample identification. The Saccardo collection has a special scientific importance due to the presence of about 4,500 type specimens (mostly assigned to Ascomycota) that have been used for the first morphological descriptions of new fungal species, and these specimens have long been used by mycologists from all over the world for morphological studies and taxonomic revisions. Unfortunately, for this reason some types of the Saccardo collection have been damaged or lost.
Because of its importance for mycologists, and in order to preserve the scientific value of the Saccardo collection, a long-term DNA barcoding project has been initiated to obtain molecular data from the most important type specimens with the scope of making the results available to the scientific community. To overcome all the above described issues related to ancient DNA analysis, we decided to apply a high throughput sequencing method yielding a large number of sequences per sample. This technique, commonly used for community analysis, has already been used for the sequencing of old insect type specimens, partially demonstrating the feasibility of the sequencing method (Prosser et al., 2016). Preliminary results show that the high degree of DNA fragmentation of the Saccardo samples renders the amplification of the entire ITS sequence (about 600 bp) difficult. Therefore, we focused on the amplification and sequencing of the ITS2 region only. Although ITS1 and ITS2 regions (respectively, ranging from 100 to 280 bp and from 120 to 280 bp; Tedersoo et al., 2015) give similar results in fungal metabarcoding studies (Bazzicalupo et al., 2012; Blaalid et al., 2013), the ITS2 fragment is generally less variable in length compared with ITS1 and lacks the problem of co-amplification of a 5′ SSU intron that is common in many ascomycetes (Lindahl et al., 2013). ITS2 has also a relatively conserved secondary structure among eukaryotes, which potentially enables scientists to perform higher level phylogenetic comparisons (Koetschan et al., 2010), and it is somewhat better represented than ITS1 in databases (Nilsson et al., 2009).
Here we present the results of DNA extraction, ITS2 amplification, sequencing and data analysis of 36 non-type specimens classified by Saccardo as members of the genus Peziza (Ascomycota, Pezizomycetes, Pezizales, Pezizaceae). Given the precious nature of the type specimens present in the collection, non-type samples were selected to set up and test the methods for a future application to the more valuable type specimens. The results obtained in this work appear encouraging since for a large part of the samples it was possible to obtain the target sequence, allowing us to re-evaluate the taxonomic position of each sequenced sample.
Materials and Methods
Sampling
A total of 36 Peziza specimens (28 different species) was sampled from the 323 Peziza specimens (166 different species including 8 types) collected in the Saccardo mycological herbarium, an example of which is shown in Figure 1. The samples for sequencing were selected to cover the size range (from 1 mm to 50 mm) and different conservation conditions (intact or fragmentated ascoma). For the present study no type material was used and for some species multiple specimens were analyzed (Table 1). Care was taken to preserve the overall integrity of each specimen taken from this historical and scientifically important fungal collection, with the permission and supervision of the herbarium curator. The specimens were sampled by removing a tiny piece of dried ascoma tissue using sterilized tweezers and scalpel.

Figure 1. Pictures of some representative Peziza samples investigated in this work and their herbarium label. (A) P. craterium; (B) P. crenata; (C) P. vesiculosa, and (D) P. brunnea. In the last panel, beside the original label a more recent morphological revision is present, and a detail of the fungus is visualized with a 30x enlargement. Except where differently indicated, the black bars represent 10 mm.
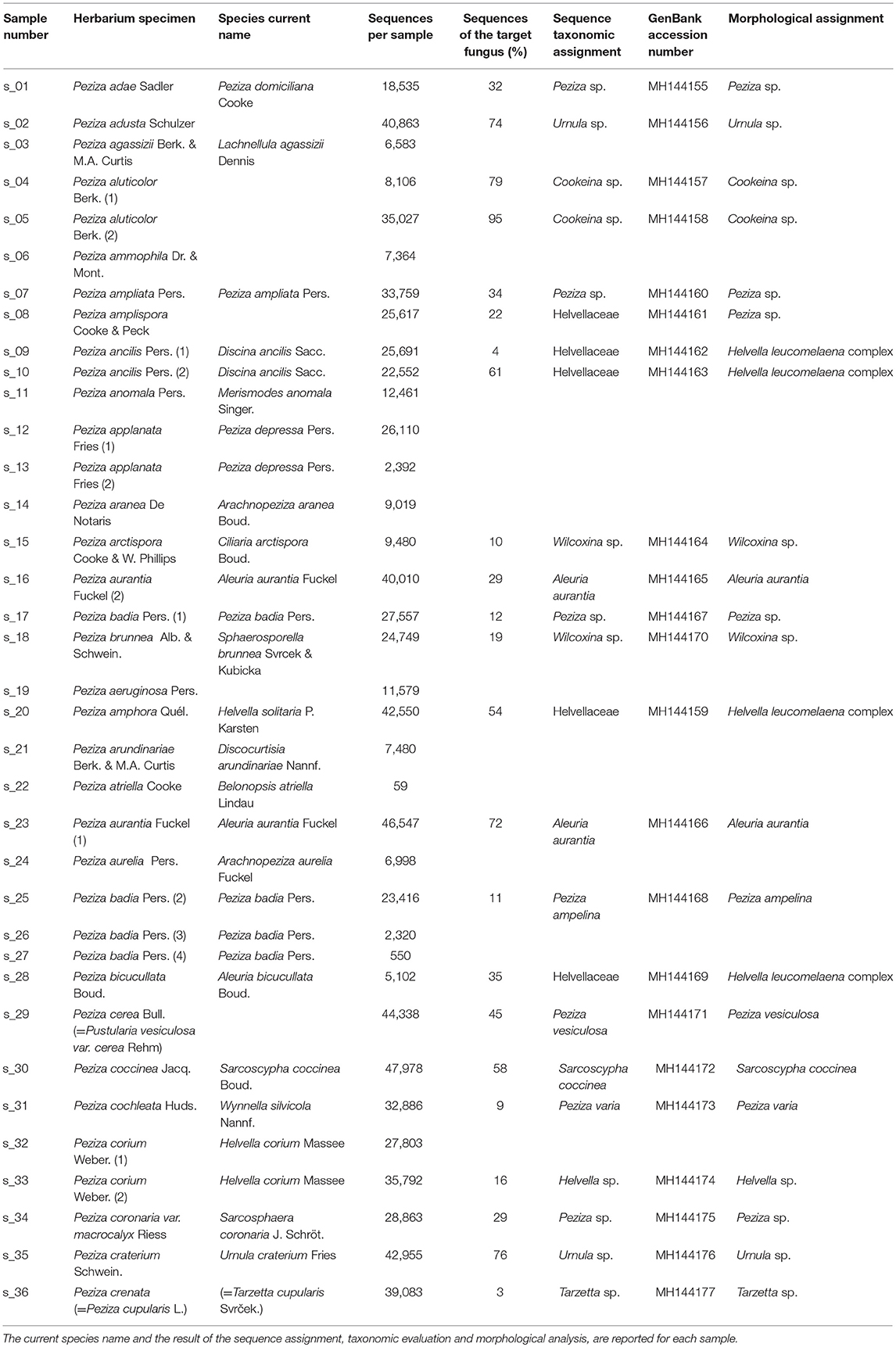
Table 1. List of Peziza specimens involved in this study.
DNA Extraction, PCR Amplification and Library Preparation
DNA was extracted using a CTAB method (Rogers and Bendich, 1985; Doyle and Doyle, 1990) slightly modified in order to obtain DNA with suitable quality from small amounts of material. The fungal tissue was ground into a fine powder in a 1.5 mL microfuge tube by using a pestle with quartz sand and liquid nitrogen. The ground tissue was mixed with 1 mL of prewarmed (65°C) CTAB extraction buffer (2% w/v CTAB; 100 mM Tris-HCl; 20 mM EDTA; 1.4 M NaCl; 2% w/v polyvinylpyrrolidone K30) with the addition of proteinase K (1 mg/mL) and RNase A (10 mg/mL), and then incubated at 65°C overnight. After a centrifugation at max speed for 10 min in a microfuge, the liquid phase was transferred to a new 2 mL microfuge tube and 800 μL of phenol:chloroform:isoamyl alcohol (25:24:1) was added, mixed several times by inversion and centrifuged for 10 min at 10,000 rpm. The upper liquid phase was moved to a new microfuge tube and 600 μL of chloroform:isoamyl alcohol (24:1) was added, mixed several times by inversion and centrifuged for 10 min at 10,000 rpm (this step was repeated twice).
DNA was precipitated by adding ice-cold isopropanol (2/3 of the recovered volume) and incubated at −20°C for 2 h. After a centrifuge at 10,000 rpm for 10 min, the DNA pellet was washed with 300 μL of ice-cold 70% ethanol, resuspended in 40 μL of sterile water and, after an incubation overnight at 4°C, stored at −20°C. DNA concentration and purity (absorbance at 230, 260, and 280 nm) were assessed using both NanoDrop spectrophotometer (Thermo Fisher Scientific) and Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). The extracted DNA from some Peziza samples (P. adae s_01, P. badia s_17 and P. cochleata s_31) was compared with the DNA extracted from a fresh fungus (Agaricus bisporus), running the same amount of DNA samples in agarose gel (0.8% agarose in TAE buffer, stained with Eurosafe DNA dying, Euroclone).
In addition, to have a more precise overview on the integrity of the nucleic acid obtained from the Peziza specimens, the length of the extracted aDNA fragments was determined by microcapillarity electrophoresis using the Agilent Bioanalyzer (Agilent Technology) following the manufacturer's instructions.
Amplification of the nuclear ribosomal ITS2 region for the Illumina sequencing was carried out using a two-step PCR process. In the first PCR the region of interest was amplified using the universal ITS3/ITS4 primers built on the conserved regions flanking the ITS2 (White et al., 1990), while in the second PCR the products of the first amplification were amplified using ITS3mod/ITS4 tagged primers in order to add different 5 bp identifier tags to distinguish sequences from each specimen (Voyron et al., 2016). In this work 18 different tags were used, and the second PCR was done in four replicates for each couple of tagged primers.
The first PCR reaction was carried out in a total volume of 25 μL including 5 μL of 5X Wonder Taq reaction buffer (5 mM dNTPs, 15 mM MgCl2; Euroclone), 0.5 μL of bovine serum albumin (BSA, 10 mg/mL), 0.5 μL each of two primers (10 μM), 0.5 μL of Wonder Taq (5 U/μL), 2 μL of genomic DNA and ddH2O to reach the final volume. The PCR conditions used were: 95°C for 3 min; 35 cycles of 95°C for 30 s, 52°C for 40 s and 72°C for 45 s; 72°C for 5 min. The second PCR reaction was performed similarly to the previous amplification except for the absence of the BSA, the use of 2 μL of the first PCR amplicons as template and the use of the tagged primers. The PCR products were checked in agarose gel (1.2% agarose in TAE buffer, stained with Eurosafe DNA dying, Euroclone), and the four replicates of each sample were pooled together and purified using the PureLink PCR Purification Kit (Invitrogen). After the quantification with Qubit, the purified amplicons were mixed in equimolar amount to prepare two sequencing libraries of 18 samples each. The libraries were sent to IGATech (Udine, Italy) for a paired-end sequencing using the Illumina MiSeq technology (2 × 300 bp).
NGS Data Processing, OTU Identification and Taxonomic Analysis
Raw data of the Illumina sequencing were analyzed using the Illumina pipeline developed for a community analysis of orchid mycorrhizal soil fungi (Voyron et al., 2016) as guideline. Forward and reverse reads from each library were merged using PEAR v0.9.10 (Zhang et al., 2014) with the quality score threshold set at 28, the minimum length of reads after trimming set at 150 bp and the minimum overlap size set at 100 bp. The merged reads were processed using the software package QIIME v1.9.1 (Caporaso et al., 2010). To avoid incorrect assignment, demultiplexing of sequences was performed with a maximum number of errors in the tag sequence of 0. At the same time, the sequences were processed using a minimum sequence length cutoff of 150 bp, minimum quality score of 28, a sliding window test of quality score of 50, a maximum length of homopolymers of 13, a maximum number of ambiguous bases of 0 and a maximum number of mismatches in forward and reverse primers of 3, and then reoriented when necessary to 5′ to 3′.
Sequences were dereplicated with VSEARCH v2.3.4 (Rognes et al., 2016) to form clusters with an identity of 100% and the ITS2 variable region was extracted using ITSx software (http://microbiology.se/software/itsx/; Bengtsson-Palme et al., 2013). The ITSx was used to eliminate the conserved regions where the primers are located as these regions can introduce bias during the subsequent clustering and taxonomic assignment, because they increase similarity among sequences (Bálint et al., 2014). In addition, setting the option -t F (list of organism group - Fungi) all the ITS2 regions not detected as Fungi were eliminated. The picking of the Operational Taxonomic Units (OTUs) was performed with a 98% of similarity clustering using VSEARCH and the clusters with less than 10 sequences were discarded.
Chimera sequences were then removed using a de novo chimera detection with UCHIME algorithm (Edgar et al., 2011) implemented in the VSEARCH pipeline. The last release of the UNITE dataset version 7.2 (https://unite.ut.ee) for QIIME was used as reference for the taxonomic assignment of OTUs (Abarenkov et al., 2010; Kõljalg et al., 2013) and, consequently, for the identification of specimen target sequence. The OTU abundance table was created with VSEARCH, considering a 98% of identity, to determine the number of times an OTU was found in each sample pooled in the libraries. The sum of these numbers for a sample gives the total sequences found for that specific sample. Because a number of OTUs returned no matches compared with the UNITE database, we also compared these sequences with those deposited in NCBI GenBank, excluding uncultured/environmental sample sequences, using the BLASTN algorithm (BLAST-search, http://www.ncbi.nlm.nih.gov/BLAST/; Altschul et al., 1997) and the first 10 hits were evaluated considering consistency between hits, E-value, similarity higher than 96%, query coverage, organism source and publication status of the hits.
The OTU abundance plots were obtained using the R package Phyloseq (McMurdie and Holmes, 2013) that combined the OTU abundance table and the UNITE taxonomic assignment of the OTUs.
Molecular Phylogenetic Analysis
Sequences obtained from the Peziza specimens were further investigated by a phylogenetic analysis in order to complement and confirm or revise the previous taxonomic assignment. The sequences used to infer the phylogenetic analysis were chosen on the bases of BLASTN results isolating a sub-dataset of taxonomically close well annotated sequences, and according to the outcomes of recent phylogenetic studies (Harrington and Potter, 1997; Hansen et al., 2002; Weinstein et al., 2002; Hansen and Pfister, 2006; Perry et al., 2007; Ekanayaka et al., 2016).
ITS2 sequences of each dataset were aligned using the default options in MUSCLE (Edgar, 2004) as implemented in MEGA7 (Kumar et al., 2016). The phylogenetic analysis was conducted in MEGA7 by Maximum Likelihood method based on the General Time Reversible model (Nei and Kumar, 2000) to obtain a more accurate identification of the fungal specimens. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value (1000 bootstrap replicates). A discrete Gamma distribution was used to model evolutionary rate differences among sites. The phylogenetic trees were drawn to scale, with branch lengths measured in the number of substitutions per site and supporting values (MLB, Maximum likelihood bootstrap values) below 50 were generally regarded unreliable and hidden. In this way, six separated phylograms were obtained relatively to the families Pyronemataceae, Pezizaceae, Sarcosomataceae, and Helvellaceae and to the genera Cookeina and Sarcoscypha of the family Sarcoscyphaceae.
Morphological Analysis
Microscopic structures were examined in dried material using a Leitz Diaplan light microscope with 40 × or 100 × (immersion oil) objectives. Dried material was rehydrated in water or 3% KOH. Hymenial elements were studied by teasing apart a piece of hymenium with a scalpel. Structural features of the excipulum were studied using vertical, median sections made by hand. Other chemicals used were Melzer's reagent and Cotton blue in lactic acid. The spore size was obtained by measuring 30 spores judged to be mature.
Results and Discussion
DNA Extraction
The adopted protocol has permitted the extraction of satisfactory amounts of DNA with a concentration ranging from 10 to 200 ng per mg of sample. The purity ratio 260/280 ranged between 1.4 and 2.1 while the ratio 260/230 ranged between 0.7 and 2.1. These values indicate a low level of protein contamination whereas they show the presence of phenolic contaminants and/or other molecules, such as fungal secondary metabolites absorbing at 230 nm. Since the samples used in this study are more than one century old, we expected a certain grade of DNA degradation due to the time and the conservation conditions. The comparison between some Peziza ancient DNA and the DNA from fresh fungal tissue (Agaricus bisporus) extracted using the CTAB method previously described is reported in Figure S1A. The herbarium DNA resulted in highly-degraded fragments shorter than 500 bp (Figure S1A, lanes 2, 3, 4), while the DNA extracted from fresh material appears as a sharp band, the length of which is larger than 10 Kb (Figure S1A, lane 1). The fragmentation level of the extracted aDNA was further investigated using the Bioanalyzer to obtain a more accurate analysis. The resulting electrophoregrams of three representative samples show the fragment length distribution demonstrating a severe DNA degradation (Figures S1B–D). The sample quality analysis and the comparison between aDNA and DNA from fresh tissue demonstrated that the optimized extraction protocol used in this work allows the extraction of DNA from such challenging samples.
NGS Sequencing and Operational Taxonomic Units (OTUs) Analysis
Libraries sequencing generated a total of 4,481,568 raw reads: 2,029,302 reads for library 1 and 2,452,266 reads for library 2. The reads were merged so that 977,339 and 1,138,933 assembled reads were obtained (96% in the library 1 and 93% in the library 2). After demultiplexing and quality filtering the number of reads decreased to 474,221 and 560,754 for library 1 and 2, respectively. The dereplication step produced a number of 22,734 and 25,337 clustered sequences. On these two different datasets, the analysis using the ITSx software recognized 19,427 (library 1) and 21,955 (library 2) fungal ITS2 sequences. These sequences were grouped into OTUs with a 98% identity and chimera-checked, and the result was a total of 106 OTUs for library 1 and 101 OTUs for library 2. The number of fungal sequences in each sample, reported in Table 1, was obtained from the OTU abundance table. The general OTU taxonomic assignment revealed the presence of fungal organisms belonging to Basidiomycota and Ascomycota phyla in all the original herbarium specimens (Figure 2). As expected by considering both the old age and the lack of precautions during the manipulation of the various specimens throughout the herbarium long life, the majority of the contaminants is represented by common airborne molds such as Aspergillus and Penicillium (Ascomycota, Eurotiomycetes, Eurotiales, Aspergillaceae); Epicoccum, Alternaria and Cladosporium (Ascomycota, Dothideomycetes, Pleosporales, Didymellaceae), as well as xerophilic fungi such as Wallemia spp. (Basidiomycota, Wallemiomycetes, Wallemiales, Wallemiaceae; Zalar et al., 2005) (Figure 3). The taxonomic assignment results showed that the herbarium samples present external (airborne mold species) and intra-herbarium DNA contaminations (Figure S2).
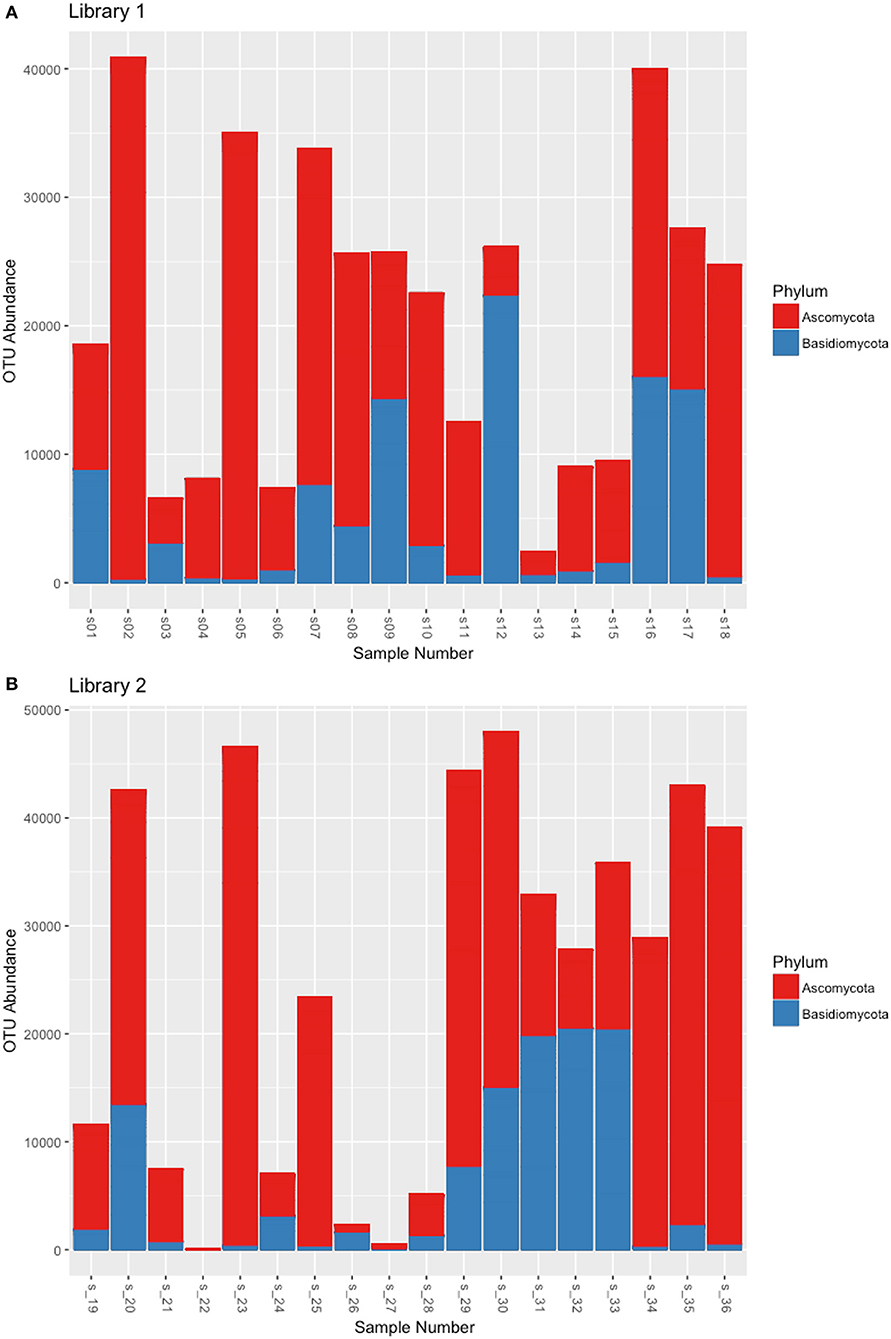
Figure 2. OTU distribution among the Ascomycota and Basidiomycota phyla that resulted from the NGS experiments. (A) OTUs obtained from the reads of Library 1 samples, (B) OTUs obtained from the reads of Library 2 samples.
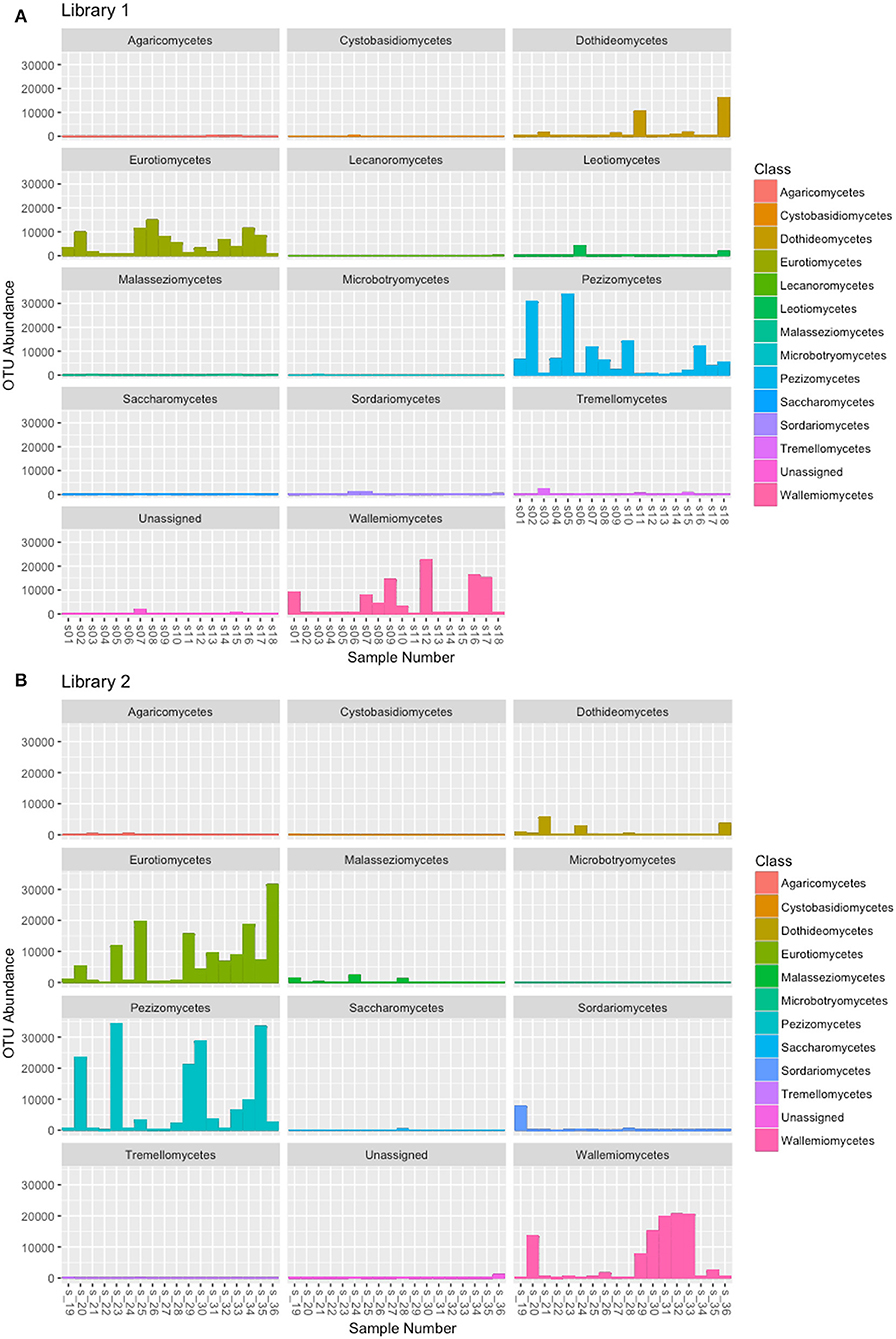
Figure 3. OTU abundance in the fungal classes obtained by the taxonomic assignment of each sample. (A) OTUs obtained from the reads of Library 1 samples, (B) OTUs obtained from the reads of Library 2 samples.
In order to assign the correct sequence to the analyzed Saccardo samples, we evaluated the sequencing results by taking into account the modern taxonomy of the specimens as reported in the Index Fungorum (http://www.indexfungorum.org) and the number of reads per OTUs in order to discriminate between the target sequence and possible contaminations. During the years, many fungal species were moved to other different genera or placed in synonymy with other existing species due to the continuous morphological revisions and the development of molecular techniques for their identification. We have found that the current name of some Peziza species used in this work differs from the taxonomic annotation reported in the Saccardo collection (Table 1). For example, some of the collection species are now placed in synonymy with other Peziza species (e.g., P. applanata = P. depressa); others are considered as members of another family in the Ascomycota order Pezizales (e.g., P. aurantia = Aleuria aurantia, from Pezizaceae to Pyronemataceae family), or of a different order (i.e., P. aranea = Arachnopeziza aranea, from Pezizales to Helotiales order) or even as members of the Basidiomycota phylum (e.g., P. anomala = Merismodes anomala). Therefore, an updated assignment of the ancient samples according to the modern taxonomy can be of great help in identifying the correct sequence of the specimens, in particular when a contamination between herbarium samples is present.
Despite the high contamination level of the examined samples, the NGS sequencing yielded good results because it has allowed us to assign ITS2 sequences to 23 out of the 36 Saccardo herbarium specimens studied in this work (Table 1). In particular, the ITS2 sequence analysis permitted the identification of five samples at the species level and 13 samples only at genus level, while five samples resulted unassigned but were confirmed as belonging to the order Pezizales. For 13 analyzed samples we were unable to assign any sequence.
Taxonomic Assignment of the Analyzed Specimens
The taxonomic assignment has been obtained combining the sequence analysis, phylogenetic analysis and morphological analysis. The results (Table 1) show the different scenarios that can be encountered when sequencing old herbarium specimens. Morphological analysis has always confirmed molecular inference with the exception of P. amplispora (s_08), in which there was a discrepancy between the two methods of analysis used (Table 1 and discussion below).
Samples With an Assigned Sequence and a Taxonomic Evaluation at Species Level
Peziza aurantia
The taxonomic assignment, reported in Table 1, confirmed the identity of P. aurantia (s_16, s_23) specimens giving as result the name of the species reported in the Index fungorum (Aleuria aurantia; Ascomycota, Pezizomycetes, Pezizales, Pyronemataceae). The result was also confirmed by the phylogenetic analysis reported in the Pyronemataceae tree (Figure 4) where the sequences clustered with the Aleuria aurantia sequences in a highly supported clade (MLB = 93%).
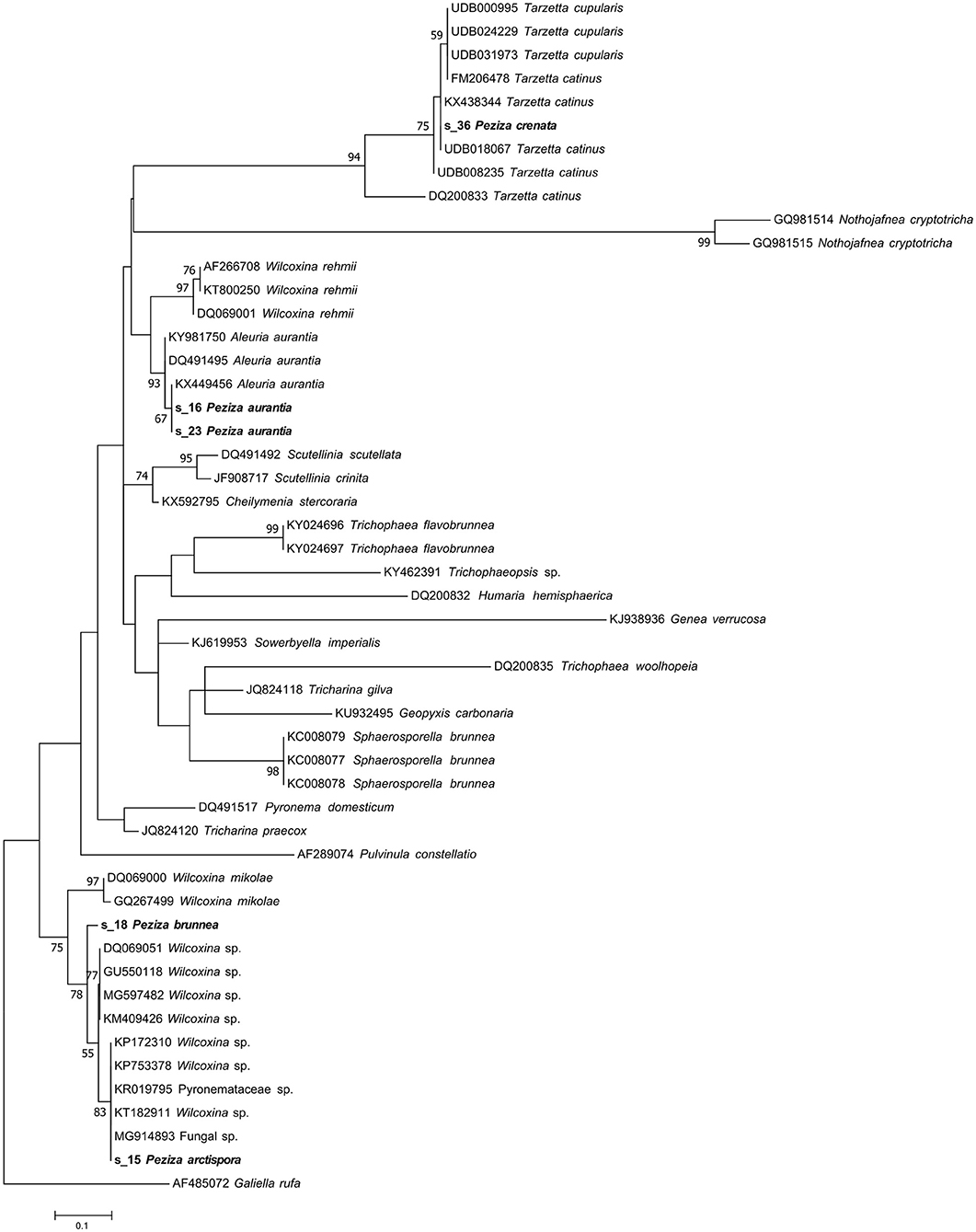
Figure 4. Phylogram generated from maximum likelihood analysis of sequences of the family Pyronemataceae based on ITS2 sequence data. Maximum likelihood bootstrap values ≥50 are given above the nodes. The Accession Number indicates the sequences retrieved from the public databases. The tree was rooted with Galiella rufa (Ascomycota, Pezizomycetes, Pezizales, Sarcosomataceae). The Saccardo specimens are evidenced in bold.
Peziza cerea
In the case of P. cerea (s_29), a deeper study of species synonyms, literature and phylogenetic analysis was necessary to confirm the identity of this herbarium sample. The herbarium specimen placed under the name P. cerea by Saccardo has a papery label where the fungus was previously classified as Pustularia vesiculosa var. cerea (Figure 1C). The name Peziza cerea sensu Bulliard has been suggested as synonym of Peziza vesiculosa by Hansen (Hansen et al., 2002). The NGS sequencing approach provided a sequence that has been recognized as derived from the target fungus and the UNITE taxonomic assignment indicates that the ITS2 sequence belongs to Peziza vesiculosa. The Pezizaceae phylogenetic analysis clearly confirmed this result by placing the specimen sequence in the highly supported (MLB = 97%) Peziza vesiculosa clade (Figure 5).
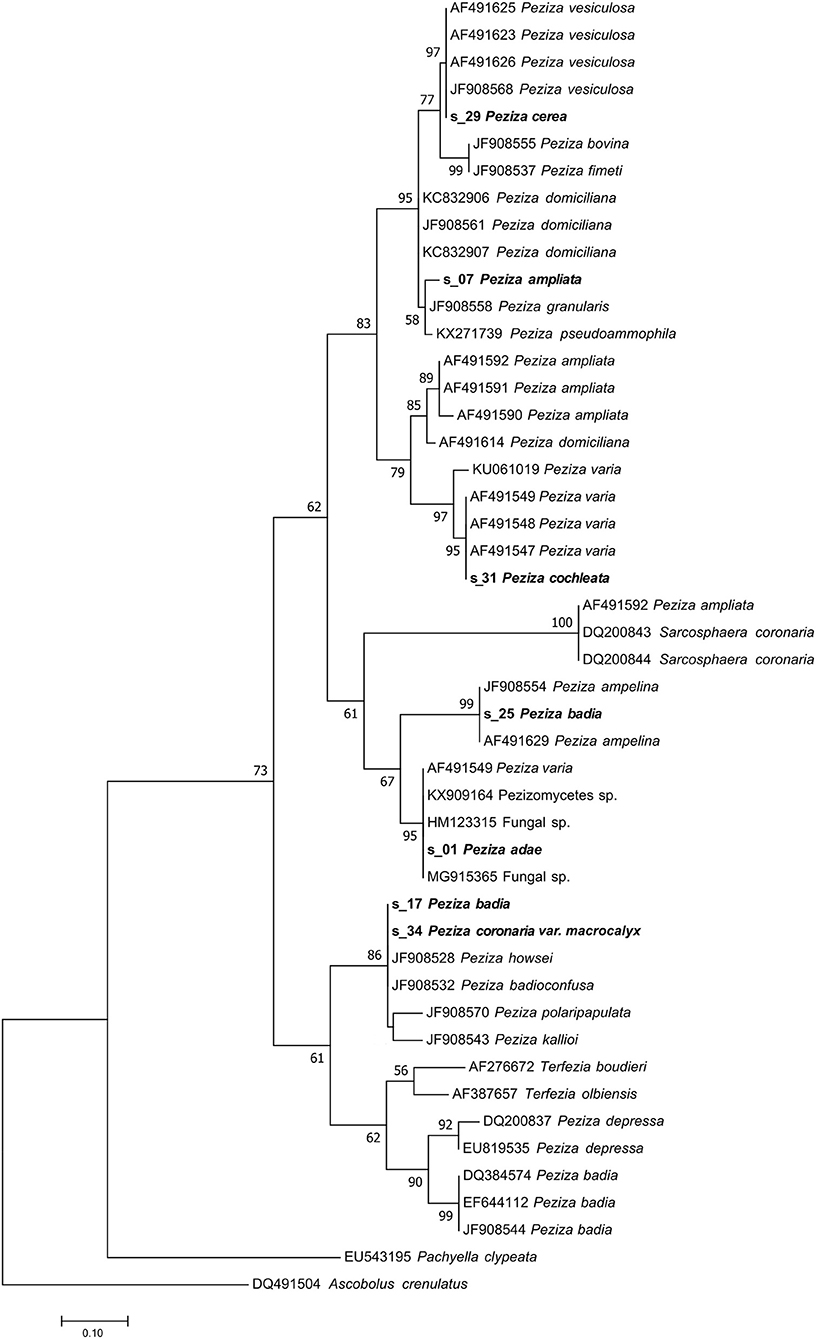
Figure 5. Phylogram generated from maximum likelihood analysis of sequences of the family Pezizaceae based on ITS2 sequence data. Maximum likelihood bootstrap values ≥50 are given above the nodes. The Accession Number indicates the sequences retrieved from the public databases. The tree was rooted with Ascobolus crenulatus (Ascomycota, Pezizomycetes, Pezizales, Ascobolaceae). The Saccardo specimens are evidenced in bold.
Peziza badia (2)
This sample produced a high number of sequences and about 11% of them were recognized as belonging to the target fungus. The modern fungal taxonomy confirms the name of the species and sequences annotated as P. badia are present in GenBank. However, the taxonomy assignment identified the fungus as P. ampelina, a result also confirmed by the phylogenetic analysis that placed the sequence in the highly supported P. ampelina clade (MLB = 99%) (Figure 5).
Peziza coccinea
The taxonomic analysis of the sequence assigned to the target fungus revealed that the specimen is a Sarcoscypha sp., congruent with the current name Sarcoscypha coccinea. Moreover, a more detailed phylogenetic analysis of the sample using selected sequences of the genus Sarcoscypha revealed that all the S. coccinea sequences clustered together and the analyzed specimen sequence appeared among them in the S. coccinea clade (MLB = 63%) (Figure 6). This result underscores that our sample belongs to the species S. coccinea, thus confirming and refining its taxonomical assignment.
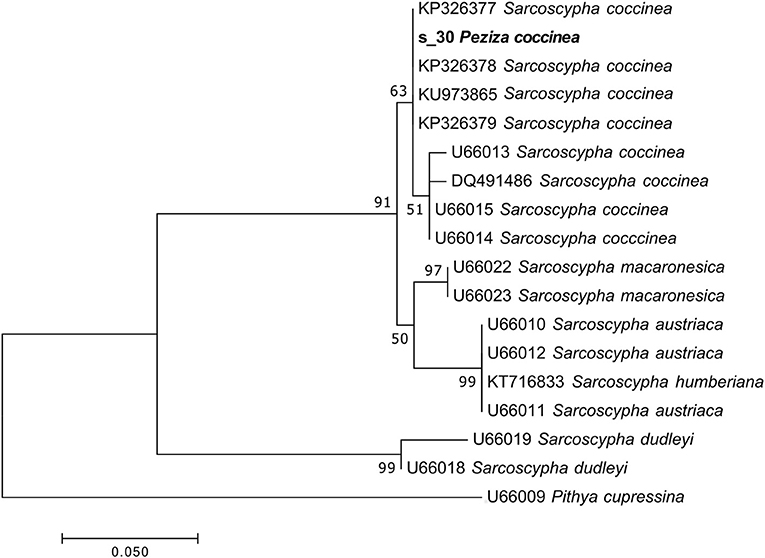
Figure 6. Phylogram generated from maximum likelihood analysis of sequences of the genus Sarcoscypha based on ITS2 sequence data. Maximum likelihood bootstrap values ≥50 are given above the nodes. The Accession Number indicates the sequences retrieved from the public databases. The tree was rooted with Pithya cupressina (Ascomycota, Pezizomycetes, Pezizales, Sarcosyphaceae). The Saccardo specimen is evidenced in bold.
Peziza cochleata
A large number of sequences (32,886) were obtained from P. cochleata but only few of them (9%) were assigned to the target fungus. Both taxonomic and phylogenetic analysis revealed that the target sequence groups with the ITS2 sequences of P. varia in a highly supported clade (MLB = 95%) (Figure 5). This result has demonstrated that the analyzed specimen is not actually a P. cochleata, that the modern taxonomy reports as Wynnella silvicola (Ascomycota, Pezizomycetes, Pezizales, Helvellaceae).
The molecular analysis (UNITE taxonomic assignment and phylogenetic analysis) and the morphological revision of P. badia 2 (s_25) and P. cochleata (s_31) did not confirm the previous classification.
All the above findings represent the best scenario that could be obtained by the molecular analysis of our ancient herbarium specimens, and this was possible because the sequences obtained for the target fungi matched with well annotated sequences in public databases. When the old sample identification is not confirmed by the molecular analysis, also in the presence of correct DNA sequences, a morphological revision is recommended to support the new molecular identification.
Samples With an Assigned Sequence and a Taxonomic Evaluation at Genus Level
P. adae (s_01), P. adusta (s_02), P. aluticolor (s_04, s_05), P. ampliata (s_07), P. arctispora (s_15), P. badia 1 (s_17), P. brunnea (s_18), P. coccinea (s_30), P. coronaria var. macrocalyx (s_34), P. corium 2 (s_33), P. craterium (s_35) and P. crenata (s_36) samples yielded a good number of sequences (Table 1), but, although the identification of the sample-associated sequence was possible, the taxonomic assignment did not allow species identification due to the lack of well annotated reference sequences in online databases.
Peziza adae
Since the taxonomic assignment of the specimen by UNITE comparison was unsuccessful, the sequence of P. adae was compared with the NCBI (GenBank) database. The BLASTN analysis showed that the sequence has the highest similarity value (100%) with some unidentified sequences annotated as Fungal sp. (HM123315, MG915365), Peziza sp. (KY462519) and Pezizomycetes sp. (KX909164). The phylogenetic analysis confirmed the result by placing the sequence in the same clade (MLB = 95%) as the above cited NCBI sequences (Figure 5). This shows that the specimen belongs to the family Pezizaceae and probably to the genus Peziza. So, the sequencing permitted to assign a sequence to our sample, but the taxonomic analysis did not allow us to clearly identify the species of this herbarium specimen.
Peziza ampliata
The sequence of the sample classified as P. ampliata has been identified by the taxonomic assignment as P. domiciliana. However, a BLASTN analysis showed that the similarity value between the sequence found and P. domiciliana sequences in the NCBI database was, in our opinion, not high enough (96%) to allow the annotation of the sample as P. domiciliana. It is also notable that some sequences of P. ampliata are present in the NCBI database. In agreement with our observations, the phylogenetic analysis placed our sample in an unsupported minor clade (MLB = 58%) composed by different Peziza species (P. granularis JF908558, P. pseudoammophila KX271739) which is part of a well-supported major clade (MLB = 95%) consisting of collections of P. domiciliana, P. fimeti, P. bovina and P. vesiculosa (s_29 P. cerea included) (Figure 5). This major clade is sister (MLB = 83%) to a clade formed by the collections of P. ampliata present in the NCBI database and the collections of P. varia (s_31 P. cochleata included). This suggests that the sample of the collection is not P. ampliata but probably a species more related to P. domiciliana without any reference ITS2 sequences in the public databases. For this reason, we can only confirm that this specimen belongs to the genus Peziza, as originally annotated by Saccardo without being able to assign it species name.
Peziza badia (1) and peziza coronaria var. macrocalyx
The analysis of the reads produced by these samples has permitted to assign a sequence to the analyzed fungi. However, the sequences found for the two specimens are identical and they have the same nucleotide identity with the ITS2 sequences of two different Peziza species: P. badioconfusa (JF908532) and P. howsei (JF908528). Probably the two samples classified as two different species in the Saccardo collection are the same species, but we are not able to add details about the species identification, apart from placing them in the Peziza genus. Anyway, the presence of deposited sequences of P. badia and Sarcosphaera coronaria (Ascomycota, Pezizomycetes, Pezizales, Pezizaceae), the current name of P. coronaria var. macrocalyx, excludes the previous identification of the specimens reported in the herbarium samples. This idea is also confirmed by the phylogenetic analysis of the family Pezizaceae where the sequences of the two specimens were grouped together and far from the ITS2 of P. badia and S. coronaria (Figure 5).
Peziza aluticolor (1 and 2)
For both of the samples present in the herbarium we were able to obtain a large number of sequences (79 and 95% for specimen 1 and 2, respectively) of the target fungi. From the Index Fungorum it seems that this species has not changed name although Seaver (1913) considered P. aluticolor as a synonym of Cookeina colensoi (Ascomycota, Pezizomycetes, Pezizales, Sarcoscyphaceae). The taxonomic assignment identified our samples as Cookeina garethjonesii. However, although the phylogenetic analysis using the ITS2 sequences confirmed that both samples actually belong to the genus Cookeina, a species-level identification is impossible because in the phylogram the sequences of C. garethjonesii appear in the same highly supported clade (MLB = 97%) with some sequences of C. speciosa (Figure 7).
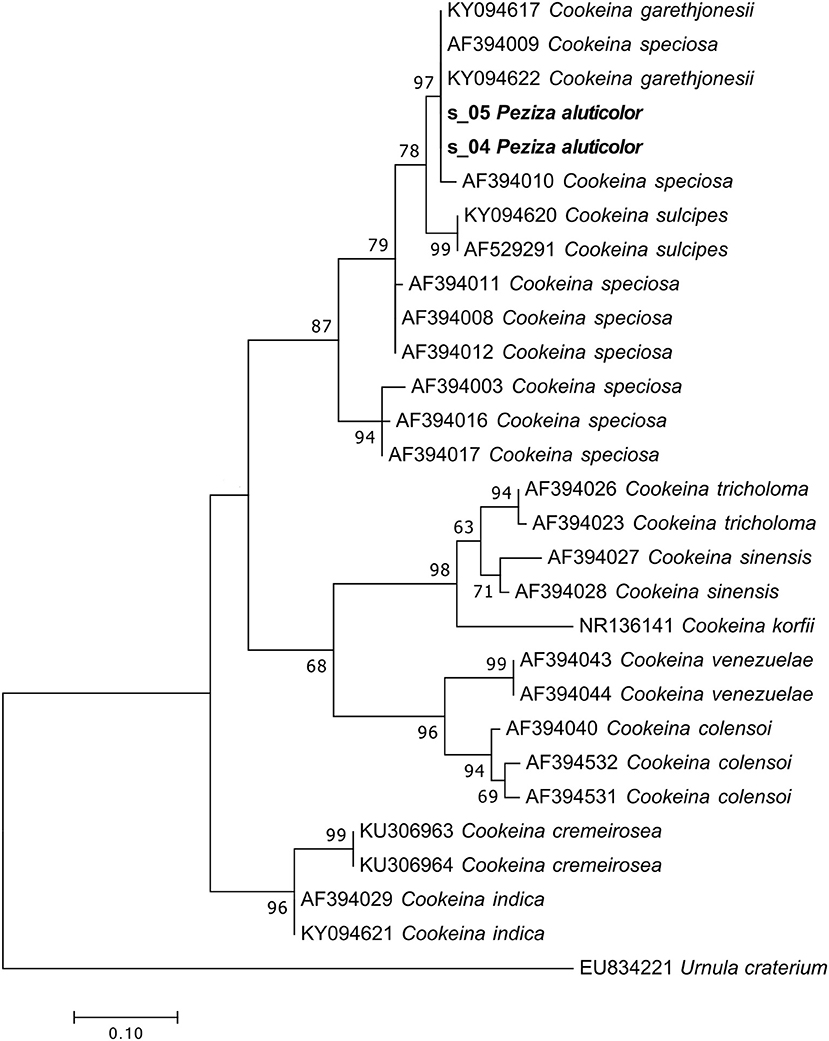
Figure 7. Phylogram generated from maximum likelihood analysis of sequences of the genus Cookeina based on ITS2 sequence data. Maximum likelihood bootstrap values ≥50 are given above the nodes. The Accession Number indicates the sequences retrieved from the public databases. The tree was rooted with Urnula craterium (Ascomycota, Pezizomycetes, Pezizales, Sarcosomataceae). The Saccardo specimens are evidenced in bold.
Peziza adusta and peziza craterium
These specimens were originally annotated as two distinct species, but the herbarium label of P. craterium has an annotation that places it in synonymy with P. adusta (Figure 1A). However, the UNITE taxonomic assignment annotated the P. adusta sequence as Strumella coryneoidea (Ascomycota, Pezizomycetes, Pezizales, Sarcosomataceae) whereas the sequence of the specimen P. craterium (current name Urnula craterium) only as a Sarcosomataceae. Interestingly, U. craterium and S. coryneoidea are two different names for the same species (Davidson, 1950).
Phylogenetic analysis of the specimen sequences and of some representative sequences of the family Sarcosomataceae divided the two S. coryneoidea (the only ones present in GenBank) in two distinct clades, separating our sequences in these two clades (Figure 8). The positions in the tree of the two Strumella sequences support a previous observation about a possible misidentification of one of them (Köpcke et al., 2002); unfortunately, all these considerations make us confident to assign the two herbarium specimens only to the genus Urnula. As reported in Table 1, the morphological analysis has been useful to confirm the genus assignment.
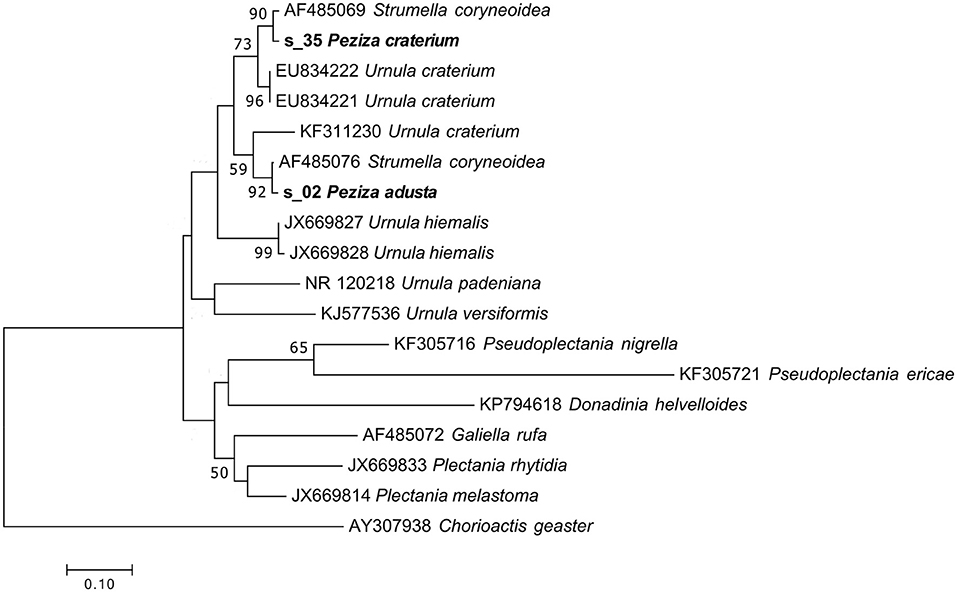
Figure 8. Phylogram generated from maximum likelihood analysis of sequences of the family Sarcosomataceae based on ITS2 sequence data. Maximum likelihood bootstrap values ≥50 are given above the nodes. The Accession Number indicates the sequences retrieved from the public databases. The tree was rooted with Chorioactis geaster (Ascomycota, Pezizomycetes, Pezizales, Chorioactidaceae). The Saccardo specimens are evidenced in bold.
Peziza arctispora and peziza brunnea
Sequences related to the target fungus P. arctispora (approx. 10% of the total of read number) provided as best taxonomic assignment a generic Pyronemataceae (Ascomycota, Pezizomycetes, Pezizales), while the specimen annotated by Saccardo as P. brunnea (Figure 1D; current name Sphaerosporella brunnea; Ascomycota, Pezizomycetes, Pezizales, Pyronemataceae) produced 24,749 sequences and 19% of these were assigned to Trichophaea sp. (Ascomycota, Pezizomycetes, Pezizales, Pyronemataceae).
Phylogenetic analysis of these two samples revealed that both ITS2 sequences grouped within a Wilcoxina cluster, but the tree topology is not clear enough to permit further taxonomical considerations (Figure 4). On the other hand, our ITS2 sequence analysis has not validated a revision from a morphological point of view made in 1974 (H.J. Larsen) that recognized P. brunnea as Nothojafnea cryptotricha (Ascomycota, Pezizomycetes, Pezizales, Pyronemataceae). This is another example that shows how important the role of molecular analysis is in the identification or revision of fungal species.
Peziza corium (2)
NGS sequencing of the P. corium (2) specimen produced 35,792 sequences, 16% assigned to the target fungus. The taxonomic assignment of the sample ITS2 sequence gave as result a generic Pezizales annotation while the phylogenetic analysis placed the sequence in a clade of Helvellaceae between two groups of Helvella species (Figure 9). The position of the sample sequence makes impossible a more detailed identification based on the ITS2, with the result of a Helvella sp. sequence assignment.
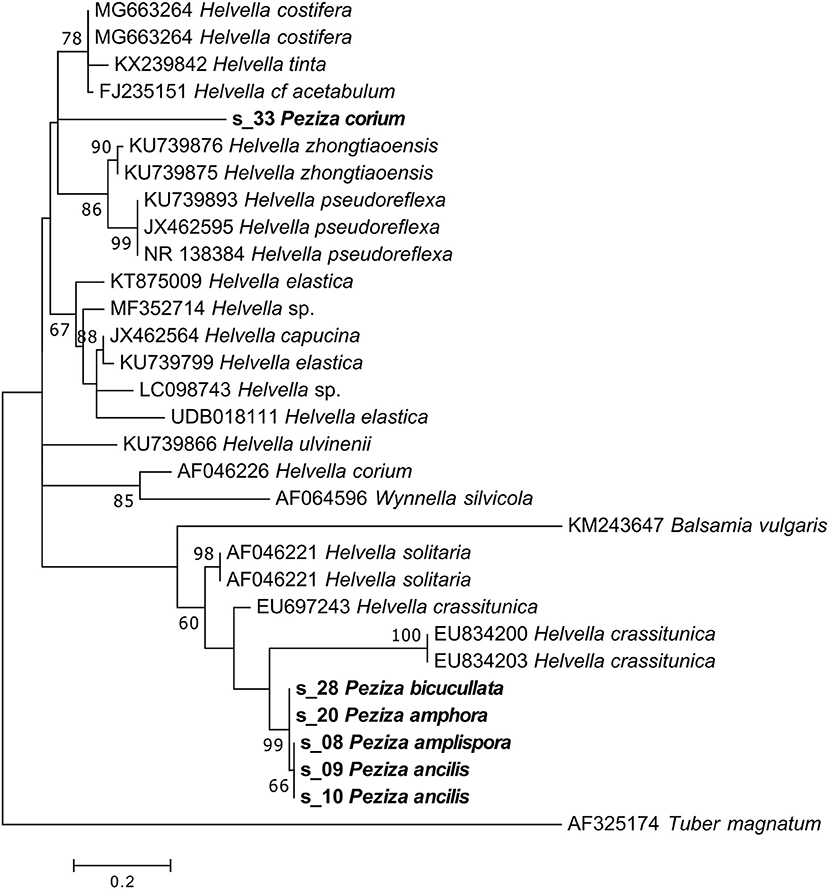
Figure 9. Phylogram generated from maximum likelihood analysis of sequences of the family Helvellaceae based on ITS2 sequence data. Maximum likelihood bootstrap values ≥50 are given above the nodes. The Accession Number indicates the sequences retrieved from the public databases. The tree was rooted with Tuber magnatum (Ascomycota, Pezizomycetes, Pezizales, Tuberaceae). The Saccardo specimens are evidenced in bold.
Peziza crenata
The herbarium label of this specimen shows a handwritten annotation where Saccardo himself posited a morphological similarity with P. cupularis (Figure 1B) and this species is reported in the Index Fungorum as Tarzetta cupularis (Ascomycota, Pezizomycetes, Pezizales, Pyronemataceae). The NGS analysis assigned the samples to the genus Tarzetta, in agreement with the suggestion made by Saccardo. Even though the taxonomic analysis has not provided any information about the species, this result could be still useful for a further analysis of the sample. In fact, the assigned sequence was compared to others of the Pyronemataceae family by phylogenetic analysis in order to obtain a more detailed molecular identification of the specimen. The resulting phylogenetic cladogram (Figure 4) confirmed the genus assignment but not a species-level identification.
Samples With a Recognized Target Sequence but Where a Taxonomic Assignment Was Not Possible
For P. amplispora (s_08), P. ancilis (s_09, s_10), P. amphora (s_20) and P. bicucullata (s_28) specimens we are able to assign a sequence, but the taxonomic identification cannot be done. Although they were determined with four different species names, the samples are probably conspecific, as indicated by the phylogenetic analysis (Figure 9). The phylogenetic analysis of these samples placed their sequences in a highly supported clade (MLB = 99%) separated from the Helvella sequences (Figure 9), suggesting that they could be generically annotated as Helvellaceae. This result probably depends on the fact that only a small fraction of the known fungal species is represented by molecular data in the public databases, and to such a purpose it is worth remembering that no molecular information is presently available not only for many species, but also for entire genera. The morphological analysis of the five specimens highlighted that, based on the presence of a regularly cupulate to planar apothecium with a ± prominent distinct stipe bearing blunt ribs not or scarcely continuing onto the receptacle, a non-amyloid ascus apex (in Melzer's reagent), spores 16–20 × (11) 12–14 μm with a large inner guttula, P. ancilis (s_09, s_10), P. amphora (s_20) and P. bicucullata (s_28) are conspecific and assignable to the Helvella leucomelaena complex (Skrede et al., 2017). By contrast, the specimen of P. amplispora (s_08), characterized by a non-stipitate apothecium, an amyloid ascus apex, large, non-guttulate spores up to 30 × 16 μm, is assignable to the genus Peziza. This is probably a case of cross-contamination. Therefore, the fact that the corresponding sequence is identical to those recovered from the samples belonging to the Helvella leucomelaena complex suggests that it arose by cross-contamination and not through amplification of the genuine P. amplispora (s_08) ITS2.
These examples illustrate the current gap between the number of described morphological fungal species and the sequences available in the public databases. Therefore, an increase of the number of public sequences obtained from herbarium collections appears of paramount importance, although the ancient samples might require time and efforts to optimize the molecular analysis workflow due to high level of DNA degradation and contamination of their specimens.
Samples Where a Target Sequence Was Not Produced
The samples P. agassizii (s_03), P. ammophila (s_06), P. anomala (s_11), P. applanata 1 and 2 (s_12, s_13), P. aranea (s_14), P. aeruginosa (s_19), P. arundinariae (s_21), P. atriella (s_22), P. aurelia (s_24), P. badia 3 and 4 (s_26, s_27) and P. corium 1 (s_32) produced only sequences deriving from contaminating DNA (Table 1). The negative results of the last group of samples might be due to an inefficient amplification of the target DNA in favor of the fungal contaminants. This effect might be explained by considering that the high degradation level of the aDNA (Dabney et al., 2013) leads to a decreased amplification efficiency in the PCR with a consequent lower target ITS2 number in comparison with the contaminant ITS2 sequences.
General Considerations on the NGS Approach for the Study of Herbarium Samples
The ancient fungal collections were created by mycologists principally to have a catalog of fungi as wide as possible, and in those times, it appeared especially important to preserve the specimen's morphological characteristics. Unfortunately, this was accomplished by keeping the samples under conditions that were not the best for the conservation of the nucleic acid integrity. For example, for more than a century in the case of the Saccardo collection, the samples have been treated with chemicals against pests, but these compounds can have a detrimental effect on DNA integrity (Whitten et al., 1999). Moreover, a problem that is much more critical for mycological collection than for plant herbaria is represented by contamination of the samples with airborne environmental fungi, as the generally used universal barcode primers cannot discriminate between the contaminating DNA and the target DNA, thus generating a mix of fungal sequences that enhance the risk of sequencing failure. In fact, the classic DNA extraction followed by PCR amplification and Sanger sequencing has revealed its limitation in the case of old and highly contaminated specimens, as the Saccardo collection turned out to be, and caused poor results, frustration and time and money loss. The method we present here has been designed to overcome the technical issues inherent to ancient herbarium samples. NGS allows one to discard the large amount of non-target sequences and to obtain the ITS2 sequence of the target fungus from a minimal amount specimen biomass, thus protecting the herbarium collection.
In this work, we analyzed 36 samples belonging to Peziza sensu Saccardo, demonstrating that these experimental procedures and data analysis pipeline allowed in many cases a sequence assignment and a taxonomic re-evaluation of samples that are more than 100-years old. Our results underscore that accurate taxonomic assignments require comparing sequences against different databases and phylogenetic analyses. These complementary approaches have allowed us to place the sample sequences in the correct taxon. The NGS platform, which is scalable for a large number of samples, has been applied here as an important test on ancient specimens as a proof of principle for the investigation of no type specimens. The encouraging results obtained in this work suggest that this technique may be used to obtain marker sequences from the approximately 4,500 type specimens conserved in the Saccardo herbarium.
The Saccardo collection could represent a source of genetic material to increase the number of fungal sequences in public databases. Moreover, since we succeeded in the ITS2 amplification and NGS sequencing, the technique might be extended to other molecular markers, focusing on relatively short sequences to overcome the fragmentation of the ancient DNA, thus improving the taxonomic identification and the phylogenetic analysis of the samples. In this sense, next-generation sequencing of important samples can be crucial to support the traditional macro- and microscopic morphological analysis, in particular when the analysis considers poorly sequenced taxa and/or taxa with evanescent morphological traits. In particular, herbarium-derived sequences matching the target taxa only at the genus level provide interesting material and scope for taxonomic revisions based on total (morphological and molecular) evidence.
Data Availability Statement
The ITS2 sequences annotated as obtained by Saccardo's specimens have been deposited in the NCBI database and the Accession Numbers are reported in Table 1. The entire datasets considered for this manuscript are not publicly available because they contain data not involved in this study but currently investigated for other purposes. Data is available upon request, and requests to access the datasets should be directed to Prof. Barbara Baldan, barbara.baldan@unipd.it.
Author Contributions
NF and SN equally made the experimental work, analyzed the results, and co-wrote the manuscript. SV contributed to the setting up of the experimental procedures, helped with the data and phylogenetic analysis, discussed the results and critically read the manuscript. MG discussed the results and critically read the manuscript and AV contributed to the phylogenetic and morphological analysis, discussed the results and critically read the manuscript. GC and BB conceived the research, discussed the results and critically read the manuscript.
Funding
This work was funded through a grant (PANN15T3_00789, Legge 6/2000) by MIUR (Ministero Istruzione Università e Ricerca) Italy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor William Martin (University of Düsseldorf, Germany) for critical comments and useful suggestions and the curator of the Botanical Garden Herbarium Dr. Rossella Marcucci for the continuous support given to us to obtain the samples used in this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2018.00129/full#supplementary-material
References
Abarenkov, K., Nilsson, R. H., Larsson, K. H., Alexander, I. J., Eberhardt, U., Erland, S., et al. (2010). The UNITE database for molecular identification of fungi recent updates and future perspectives. New Phytol. 186, 281–285. doi: 10.1111/j.1469-8137.2009.03160.x
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bálint, M., Schmidt, P. A., Sharma, R., Thines, M., and Schmitt, I. (2014). An illumina metabarcoding pipeline for fungi. Ecol. Evol. 4, 2642–2653. doi: 10.1002/ece3.1107
Bani, A., Pioli, S., Ventura, M., Panzacchi, P., Borruso, L., Tognetti, R., et al. (2018). The role of microbial community in the decomposition of leaf litter and deadwood. Appl. Soil Ecol. 126, 75–84. doi: 10.1016/j.apsoil.2018.02.017
Bazzicalupo, A. L., Bálint, M., and Schmitt, I. (2012). Comparison of ITS1 and ITS2 rDNA in 454 sequencing of hyperdiverse fungal communities. Fungal Ecol. 6, 102–109. doi: 10.1016/j.funeco.2012.09.003
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 25, 914–919. doi: 10.1111/2041-210X.12073
Blaalid, R., Kumar, S., Nilsson, R. H., Abarenkov, K., Kirk, P. M., and Kauserud, H. (2013). ITS1 versus ITS2 as DNA metabarcodes for fungi. Mol. Ecol. Resour. 13, 218–224. doi: 10.1111/1755-0998.12065
Brock, P. M., Döring, H., and Bidartondo, M. I. (2009). How to know unknown fungi: the role of a herbarium. New Phytol. 181, 719–724. doi: 10.1111/j.1469-8137.2008.02703.x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Dabney, J., Meyer, M., and Paabo, S. (2013). Ancient DNA damage. CSH Perspect. Biol. 5:a012567. doi: 10.1101/cshperspect.a012567
Davidson, R. W. (1950). Urnula craterium is possibly the perfect stage of Strumella coryneoidea. Mycologia 42, 735–742. doi: 10.2307/3755615
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Ekanayaka, A. H., Hyde, K. D., and Zhao, Q. (2016). The genus Cookeina. Mycosphere 7, 1399–1413. doi: 10.5943/mycosphere/7/9/13
Erkens, R. H. J., Cross, H., Maas, J. W., Hoenselaar, K., and Chatrou, L. W. (2008). Assessment of age and greenness of herbarium specimens as predictors for successful extraction and amplification of DNA. Blumea 53, 407–428. doi: 10.3767/000651908X608052
Fisher, M. C., Henk, D. A., Briggs, C. J., Brownstein, J. S., Madoff, L. C., McCraw, S. L., et al. (2012). Emerging fungal threats to animal, plant and ecosystem health. Nature 484, 186–194. doi: 10.1038/nature10947
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for Basidiomycetes: application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Hansen, K., Læssøe, T., and Pfister, D. H. (2002). Phylogenetic diversity in the core group of Peziza inferred from ITS sequences and morphology. Mycol. Res. 106, 879–902. doi: 10.1017/S0953756202006287
Hansen, K., and Pfister, D. H. (2006). Systematics of the Pezizomycetes-the operculate discomycetes. Mycologia 98, 1029–1040.
Harrington, F. A., and Potter, D. (1997). Phylogenetic relationships within Sarcoscypha based upon nucleotide sequences of the internal transcribed spacer of nuclear ribosomal DNA. Mycologia 89, 258–267. doi: 10.2307/3761080
Hawksworth, D. L., and Lücking, R. (2017). “Fungal diversity revisited: 2.2 to 3.8 million species,” in The Fungal Kingdom, eds J. Heitman, B. Howlett, P. Crous, E. Stukenbrock, T. James, and N. Gow (Washington, DC: ASM Press), 79–95. doi: 10.1128/microbiolspec.FUNK-0052-2016
Kigawa, R., Nochide, H., Kimura, H., and Miura, S. (2003). Effects of various fumigants, thermal methods and carbon dioxide treatment on DNA extraction and amplification: a case study on freeze-dried mushroom and freeze-dried muscle specimens. Collection Forum 18, 74–85.
Koetschan, C., Förster, F., Keller, A., Schleicher, T., Ruderisch, B., Schwarz, R., et al. (2010). The ITS2 Database III-sequences and structures for phylogeny. Nucleic Acids Res. 38, D275–D279. doi: 10.1093/nar/gkp966
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Köpcke, B., Weber, R. W. S., and Anke, H. (2002). Galiellalactone and its biogenetic precursors as chemotaxonomic markers of the Sarcosomataceae (Ascomycota). Phytochemistry 60, 709–714. doi: 10.1016/S0031-9422(02)00193-0
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Larsson, E., and Jacobsson, S. (2004). Controversy over Hygrophorus cossus settled using ITS sequence data from 200 year-old type material. Mycol. Res. 108, 781–786. doi: 10.1017/S0953756204000310
Lehtonen, S., and Christenhusz, M. (2010). Historical herbarium specimens in plant molecular systematics - an example from the fern genus Lindsaea (Lindsaeaceae). Biologia 65, 204–205. doi: 10.2478/s11756-010-0008-8
Liimatainen, K., Niskanen, T., Dima, B., Kytövuori, I., Ammirati, J. F., and Frøslev, T. G. (2014). The largest type study of Agaricales species to date: bringing identification and nomenclature of Phlegmacium (Cortinarius) into the DNA era. Persoonia 33, 98–140. doi: 10.3767/003158514X684681
Lindahl, B. D., Nilsson, R. H., Tedersoo, L., Abarenkov, K., Carlsen, T., Kjøller, R., et al. (2013). Fungal community analysis by high-throughput sequencing of amplified markers - a user's guide. New Phytol. 199, 288–299. doi: 10.1111/nph.12243
Martínez-García, L. B., De Deyn, G. B., Pugnaire, F. I., Kothamasi, D., and van der Heijden, M. G. A. (2017). Symbiotic soil fungi enhance ecosystem resilience to climate change. Glob. Change Biol. 23, 5228–5236. doi: 10.1111/gcb.13785
Martin, K. J., and Rygiewicz, P. T. (2005). Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol. 5:28. doi: 10.1186/1471-2180-5-28
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Mutanen, M., Kekkonen, M., Prosser, S. W. J., Hebert, P. D. N., and Kaila, L. (2014). One species in eight: DNA barcodes from type specimens resolve a taxonomic quagmire. Mol. Ecol. Resour. 15, 967–984. doi: 10.1111/1755-0998.12361
Nagy, Z. T. (2010). A hands-on overview of tissue preservation methods for molecular genetic analyses. Org. Divers. Evol. 10, 91–105. doi: 10.1007/s13127-010-0012-4
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press.
Nilsson, R. H., Kristiansson, E., Ryberg, M., Hallenberg, N., and Larsson, K. H. (2008). Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 8, 193–201. doi: 10.4137/EBO.S653
Nilsson, R. H., Ryberg, M., Abarenkov, K., Sjökvist, E., and Kristiansson, E. (2009). The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol. Lett. 296, 97–101. doi: 10.1111/j.1574-6968.2009.01618.x
Osmundson, T. W., Robert, V. A., Schoch, C. L., Baker, L. J., Smith, A., Robich, G., et al. (2013). Filling gaps in biodiversity knowledge for macrofungi: contributions and assessment of an herbarium collection DNA barcode sequencing project. PLoS ONE 8:e62419. doi: 10.1371/journal.pone.0062419
Pääbo, S., Poinar, H., Serre, D., Jaenicke-Després, V., Hebler, J., Rohland, N., et al. (2004). Genetic analyses from ancient DNA. Annu. Rev. Genet. 38, 645–679. doi: 10.1146/annurev.genet.37.110801.143214
Perry, B. A., Hansen, K., and Pfister, D. H. (2007). A phylogenetic overview of the family Pyronemataceae (Ascomycota, Pezizales). Mycol. Res. 111, 549–571. doi: 10.1016/j.mycres.2007.03.014
Prosser, S. W. J., Dewaard, J. R., Miller, S. E., and Hebert, P. D. N. (2016). DNA barcodes from century-old type specimens using next-generation sequencing. Mol. Ecol. Resour. 16, 487–497. doi: 10.1111/1755-0998.12474
Redchenko, O., Vondrák, J., and Košnar, J. (2012). The oldest sequenced fungal herbarium sample. Lichenologist 44, 715–718. doi: 10.1017/S002428291200031X
Rogers, S. O., and Bendich, A. J. (1985). Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant. Plant Mol. Biol. 5, 69–76. doi: 10.1007/BF00020088
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584–e2522. doi: 10.7717/peerj.2584
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Skrede, I., Carlsen, T., and Schumacher, T. (2017). A synopsis of the saddle fungi (Helvella: Ascomycota) in Europe – species delimitation, taxonomy and typification. Persoonia 39, 201–253. doi: 10.3767/persoonia.2017.39.09
Staats, M., Cuenca, A., Richardson, J. E., Vrielink-van Ginkel, R., Petersen, G., Seberg, O., et al. (2011). DNA Damage in Plant Herbarium Tissue. PLoS ONE 6:e28448. doi: 10.1371/journal.pone.0028448
Tedersoo, L., Anslan, S., Bahram, M., Põlme, S., Riit, T., Liiv, I., et al. (2015). Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 10, 1–43. doi: 10.3897/mycokeys.10.4852
Toju, H., Tanabe, A. S., Yamamoto, S., and Sato, H. (2012). High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 7:e40863. doi: 10.1371/journal.pone.0040863
Voyron, S., Ercole, E., Ghignone, S., Perotto, S., and Girlanda, M. (2016). Fine-scale spatial distribution of orchid mycorrhizal fungi in the soil of host-rich grasslands. New Phytol. 213, 1428–1439. doi: 10.1111/nph.14286
Weinstein, R. N., Pfister, D. H., and Iturriaga, T. (2002). A phylogenetic study of the genus Cookeina. Mycologia 94, 673–682. doi: 10.1080/15572536.2003.11833195
White, T. J., Bruns, T., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. Sninsky, T. J. White (San Diego, CA: Academic Press), 315–322.
Whitten, W. M., Williams, N. H., and Glover, K. V. (1999). Sulphuryl fluoride fumigation: effect on DNA extraction and amplification from herbarium specimens. Taxon 48, 507–510. doi: 10.2307/1224563
Yeates, D. K., Zwick, A., and Mikheyev, A. S. (2016). Museums are biobanks: unlocking the genetic potential of the three billion specimens in the world's biological collections. Curr. Opin. Insect Sci. 18, 83–88. doi: 10.1016/j.cois.2016.09.009
Zalar, P., Sybren de Hoog, G., Schroers, H.-J., Frank, J. M., and Gunde-Cimerman, N. (2005). Taxonomy and phylogeny of the xerophilic genus Wallemia (Wallemiomycetes and Wallemiales, cl. et ord. nov.). Antonie Van Leeuwenhoek 87, 311–328. doi: 10.1007/s10482-004-6783-x
Keywords: ancient DNA, DNA barcoding, fungi, herbarium collection, NGS sequencing, nuclear ribosomal internal transcribed spacer, Pier Andrea Saccardo
Citation: Forin N, Nigris S, Voyron S, Girlanda M, Vizzini A, Casadoro G and Baldan B (2018) Next Generation Sequencing of Ancient Fungal Specimens: The Case of the Saccardo Mycological Herbarium. Front. Ecol. Evol. 6:129. doi: 10.3389/fevo.2018.00129
Received: 30 March 2018; Accepted: 10 August 2018;
Published: 31 August 2018.
Edited by:
Michael David Martin, Norwegian University of Science and Technology, NorwayReviewed by:
Denis Baurain, University of Liège, BelgiumAndreas Frisch, Norwegian University of Science and Technology, Norway
Copyright © 2018 Forin, Nigris, Voyron, Girlanda, Vizzini, Casadoro and Baldan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Baldan, YmFyYmFyYS5iYWxkYW5AdW5pcGQuaXQ=
† These authors have contributed equally to this work