 Fraser J. Combe
Fraser J. Combe Jonathan S. Ellis
Jonathan S. Ellis Kirsty L. Lloyd
Kirsty L. Lloyd Bradley Cain1
Bradley Cain1 W. Edwin Harris
W. Edwin Harris
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 21 June 2016
Sec. Biogeography and Macroecology
Volume 4 - 2016 | https://doi.org/10.3389/fevo.2016.00072
We used genetic tools to assess phylogeographic structure of the common dormouse (Muscardinus avellenarius) since the end of the last glacial maximum, to identify post-glacial dispersal routes and to describe population units for conservation. Comparative analysis of mitochondrial genes (Cytochrome b, 704 bp, D-loop, 506 bp) and one nuclear gene (Beta-Fibrinogen, 550 bp) was conducted to reconstruct the recent demographic history within and between UK and continental European populations. Our analysis indicated phylogeographic variation in the UK is similar in magnitude to that found in other regions of continental Europe and suggests a recent population expansion. We present evidence which supports a single post-glacial colonization into the UK. Dispersal time calculations, calibrated with geophysical events, are coincident with the start of the Holocene period, 7.5–11 kya, a time when geological evidence suggests temperatures were stable, woodland habitat was prevalent and a land bridge was present to allow the dispersal of small mammals into the UK. We discuss our findings in the context of the extant geographical genetic structure described here and in relation to conservation management of this threatened species.
The phylogeographic structure of many species within the Northern hemisphere is thought to have been shaped by climatic changes and glacial episodes 126–25 kya, during the Late Pleistocene (Webb and Bartlein, 1992; Hewitt, 2004). This period was characterized by extreme climatic fluctuations, which had a major role in shaping contemporary biogeography (Emerson and Hewitt, 2005). There is evidence that when climatic conditions were relatively extreme during the Pleistocene ice ages, temperate species in continental Europe were displaced to southern areas, for example to refugia in the Iberian and Apennine peninsulas, and the Balkan regions (Hewitt, 2004; Teacher et al., 2009). In order to understand how evolutionary processes have shaped biodiversity, it is useful to study how historical range contractions and expansions, potential admixture between lineages, and associated genetic processes have influenced current spatial patterns of this diversity. A modern approach to investigate the evolutionary history of species is to interpret molecular phylogenetic data in the context of climatic and environmental changes (Franks and Hoffmann, 2012).
While the impact of post-glacial colonization routes from refugia are thought to have defined the biogeography of most extant species in temperate regions, for the majority of species the details remain debated or merely unknown (Hewitt, 1996, 2004; Willis and Whittaker, 2000). Phylogeographic studies allow us to make inferences on historical dispersal routes; in the case of many European species, the hypothesis of post-glacial expansion from refugia found in southern areas of continental Europe is generally supported (Hewitt, 2004). Fossil records indicate that many species were found in lower latitudes where climatic conditions were not as extreme (Provan and Bennett, 2008). However, there are several competing hypotheses proposed for the specific details and timing of post-glacial expansion of plant and animal species during the last glacial maximum (LGM), specifically in relation to colonization of the UK. There is evidence that species only dispersed from lower latitudes of continental Europe as glacial ice retreated, at the end of the Pleistocene (e.g., the timing of dispersal of cold-tolerant Lusitanian plants is coincident with this period, Beatty and Provan, 2013, 2014). As such there is consensus that species dispersed into Northern regions and the UK from 23 kya onwards as glacial ice caps retreated and tundra steppes were no longer frozen. The Early Migration Hypothesis (hereafter, EMH) posits dispersal prior to the Younger Dryas period (12.6–11.7 kya; Herman and Searle, 2011). Species migrating at this time must have been more cold tolerant and able to survive the colder conditions of the Younger Dryas period (Stewart et al., 2010; Montgomery et al., 2014). The Late Migration Hypothesis (hereafter, LMH) predicts that species may have not dispersed and colonized Northern regions of Europe until a warmer time period, the Holocene (11.5 kya). Evidence is equivocal for LMH and EMH predominantly affecting modern European biodiversity for species studied so far (Montgomery et al., 2014).
Regarding the UK, there is evidence a land bridge connecting continental Europe and the UK was present during the Holocene period (23 kya to approximately 7.5 kya) until sea levels rose significantly and separated the UK from continental Europe (Sturt et al., 2013). This land bridge is thought to have acted as an important mechanism for the dispersal of different species into the UK (Montgomery et al., 2014). The specific ecological requirements of different species (e.g., food resources and suitable habitat) are thought to have driven the route and timing of expansion into new areas (Lesbarrères, 2009). Range expansion by many modern species has been proposed to have occurred during a single post-glacial event. However, there is not clear evidence supporting a single source of the hypothesized dispersal. For example, there is evidence that some species share a most recent common ancestor with populations in Eastern Europe, such is the case in the pygmy shrew (Sorex minutus; Vega et al., 2010) and the pool frog (Rana lessonae; Zeisset and Beebee, 2001), while other species appear to have originated from Western European populations, such as the common frog (R. temporaria; Teacher et al., 2009). Alternatively, water voles (Arvicola terrestris) show English and Welsh haplotypes nested within Eastern European clades from Iberian refugia and Scottish haplotypes nest within a Western European clade (Piertney et al., 2005), suggesting multiple episodes of colonization. While the timing of these recolonization events are poorly understood, advances in phylogenetic methods allow us to calibrate species or population divergence, in conjunction with historical geological and fossil data, with much greater precision (Hewitt, 2000; Herman et al., 2014). Thus, justifying the fossil record with estimated divergence time of genetic lineages is a powerful approach for understanding modern biogeographical patterns.
Here, we investigate the phylogeographic pattern of dispersal of the common dormouse, Muscardinus avellanarius, into the UK. The common dormouse has been described as a “small, elusive nocturnal mammal” (Bright et al., 2006), which naturally occurs at low population densities and exhibits arboreal behavior (Bright et al., 1994; Juškaitis, 2003). Dormice are thought to have narrow habitat requirements related to woodland habitat complexity, for example, features such as mature shrubs or hazel coppice stands, and food availability such as the presence of hazel, honeysuckle, and bramble (Bright et al., 2006). As such, they are important as a bio indicator of ancient woodland health (Morris, 2003). Because of the empirical observation that dormice have a limited range associated with such habitat requirements, it is thought they have a relatively small ecological window for dispersal (Mortelliti et al., 2010). Therefore, the hazel dormouse is a compelling species to investigate the effects of the LGM on geographical expansion under a regime of climate change.
Phylogeographic data for the common dormouse within continental Europe have suggested that lineages from Western and Eastern populations are relatively divergent, but that within these clades there is low genetic diversity (Mouton et al., 2012). While it is understood that genetic data are sometimes critical to delineate units for conservation (Moritz, 1994; Crandall et al., 2000; Funk et al., 2012), there is no available information available to describe the genetic biogeography of the common dormouse in the UK. Thus, while there is a long-term decline in dormouse populations in the UK precipitating a national monitoring programme, habitat restoration efforts and population reintroductions (Bright et al., 2006), information about regional genetic variation is lacking to inform these efforts or to monitor the effects, if any, of reintroduction projects. Our objectives were to (1) provide phylogeographic coverage of common dormouse populations in the UK relative to its continental European range; (2) assess the geographical and temporal patterns of genetic variation within the UK, and between the UK and continental European populations, and; (3) assess competing post-glacial expansion hypotheses (EMH vs. LMH) by estimating the timing of dispersal in this species.
Non-invasive genetic sampling of hair was conducted during summer nest-box surveys in 2014 and 2015. Samples were stored in sterile tubes at −20°C. A total of 125 samples were collected from 25 populations around the UK (Table 1, Figure 1) and stored under licenses from Natural England and the UK National Trust. This sample size is commensurate with current standards for biogeographical genetic studies with the goal of estimating long-term divergence of lineages (see Gillespie, 2004; Mouton et al., 2012). Study sites were chosen to represent the current natural range of the common dormouse in the UK. Three of our study sites were from reintroduced populations within the UK (Table 1) as part of the dormouse reintroduction programme, to enable us to quantify genetic differentiation, if any, between these and natural populations. We analyzed UK data we produced along with previously published sequences available on GenBank from continental Europe (Mouton et al., 2012) and outgroup taxa.
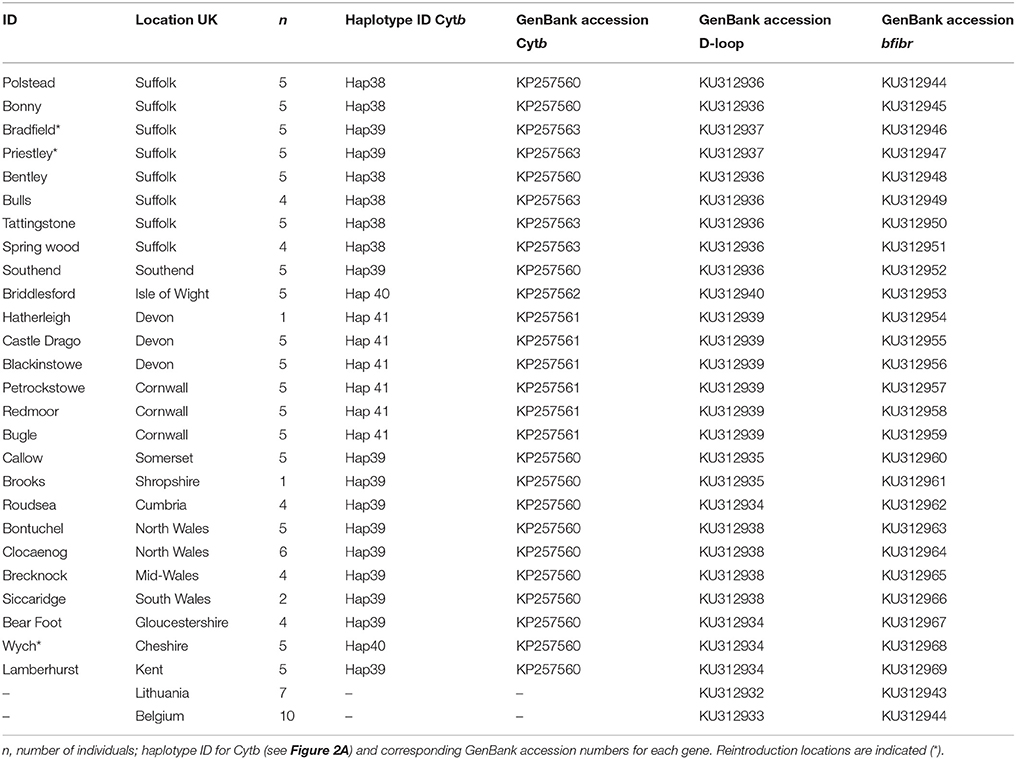
Table 1. Geographic location of sampling sites.
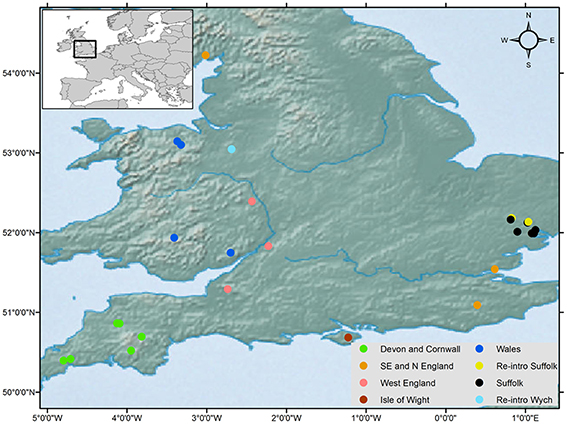
Figure 1. Geographical distribution of the common dormouse, Muscardinus avellinarius, and samples collected around the UK from 25 sites (125 samples) and haplotype distribution for concatenated sequences in Figure 2B. Key indicates the genetic grouping of populations and three reintroduced populations.
Total genomic DNA was extracted from hair roots using a Quick DNA extraction kit (Zymo research, USA) following the manufacturer's protocol with the addition of 20 μl of 1 M dithiothreitol during lysis. A 704 bp fragment of the mitochondrial Cytochrome b (Cytb) gene was amplified and sequenced using primers LMA14255 and RMA15192 (Mouton et al., 2012). DNA degradation was an issue affecting amplification using these primers. Consequently, modified internal primers from Bentz and Montgelard (1999) were used (primers MUSCAR_RINTERN/MUSCAR_LINTERN) to amplify the gene in two fragments of between 300 and 440 bp from which contiguous sequences were generated to give the full 704 bp sequence. Additionally we amplified and sequenced a 506 bp fragment of the D-loop mtDNA (Stacy et al., 1997) using primers (M15997/H16401) and 550 bp of Intron 7 of the nuclear gene Beta-Fibrinogen (bfibr) in the same localities (Table 1) using primers (BFIBR1/BFIBR2) previously used in closely related species (Seddon et al., 2001).
Amplification was performed in 20-μl PCR reactions, containing < 10 ng of lyophilised DNA, 0.2 μM of each primer, 10 μl Bioline 2x PCR biomix (Bioline, UK) and Bovine serum albumin 0.1 μg/μl. PCR amplification was performed using a G-Storm GS1 Thermal Cycler, with the following program: 95°C for 15 min; followed by 35 cycles of 95°C for 30 s, 48°C (Cytb), 60°C (bfibr), or 58°C (D-loop) for 30 s, and 72°C for 45 s; and a final elongation at 72°C for 10 min. Amplified products were cleaned using ZR DNA sequencing clean-up kit (Zymo Research). DNA sequencing was then performed using BigDye v3.1 terminator and run at Manchester University DNA Sequencing Facility on an ABI 3730 48-well capillary DNA Analyser (Applied Biosystems, California, USA).
Sequences were quality checked and aligned using BIOEDIT 7.2.5 (Hall, 1999) and further analyses were undertaken in MEGA 6.0 (Tamura et al., 2013). The mutational model that best fit the data was identified using FINDMODEL (Posada and Crandall, 1998). The Maximum-likelihood (ML) trees were constructed using MEGA 6.0 (Tamura et al., 2013). The robustness of the trees was assessed by 1000 bootstrap replications (Felsenstein, 1985). A Bayesian phylogeny was produced with MRBAYES v3.2.1 (Huelsenbeck and Ronquist, 2001). It was run for 100,000,000 generations and Bayesian posterior probabilities were set at a 50% majority rule consensus of trees sampled every 1000 generations and the consensus tree was generated in FIGTREE v1.3.1 (Rambaut, 2009). We consider nodes to be well supported with bootstrap values >70 (Hillis and Bull, 1993). A minimum spanning haplotype network was constructed using NETWORK 4.6.1.2. Bandelt et al. (1999) to examine genetic structure and geographical distribution of the mtDNA haplotypes for both Cytb and D-loop. Haplotype (h) and nucleotide (π) diversity was estimated using DnaSP 5.10.01 (Librado and Rozas, 2009). Two neutrality test statistics, Tajima's D and Fu's Fs, were also estimated with DnaSP 5.10.01 (Librado and Rozas, 2009). The net distance between groups and average distances within groups were calculated using MEGA 6.0 (Tamura et al., 2013).
To estimate how genetic variation was distributed within and among populations across geographical regions, an analysis of molecular variance (AMOVA) was performed based on pairwise differences using ARLEQUIN 3.0 (Excoffier and Lischer, 2010). AMOVA was conducted at three hierarchical levels of population subdivisions: among groups [UK and CNE (Central Northern Europe) groups]; among populations (sub lineages within the UK); and within each population. F-statistics was estimated ARLEQUIN 3.0 (Excoffier and Lischer, 2010) based on mtDNA sequences (Φ ST) The significance of these parameters was estimated by 10,000 permutations of the distance matrix.
The divergence time of UK populations was calculated using Bayesian sampling in BEAST 2.1.2 (Bouckaert et al., 2014). For this analysis we used DNA sequences only for Cytb, due to sequences being available on GenBank for continental European dormice (Mouton et al., 2012). This also allowed us to estimate the time of divergence for other genetic clades found in continental Europe (Central Northern Europe, Turkey, Balkans and Western Europe, Belgium). Analyses were performed under the GTR+G substitution model parameter (estimated by FINDMODEL), simulated with a substitution rate of 1% per million years, applying a relaxed log-normal molecular clock in BEAST 2.1.2 (Bouckaert et al., 2014) The substitution rate was selected based on previous studies in the common dormouse (Mouton et al., 2012) and previous studies conducted in the dormouse family, Gliridae (Montgelard et al., 2003). The molecular clock was calibrated using prior distributions on the time to most recent common ancestor (TMRCA) of the UK clade. The UK clade was given a normal distribution truncated at lower and upper limits of 7.5 and 18 kya respectively, to coincide with the presence of the land bridge and the beginning of the Devensian time period when most temperate species were thought to disperse into the UK (Montgomery et al., 2014). This time period spans both the EMH and LMH hypotheses and was chosen in order to allow us to resolve them. The analyses were repeated without the prior on the UK clade to test the effect of the priors on the posterior distributions. All other settings were defaults provided by BEAST. Two independent runs were performed with 100,000,000 Markov Chain Monte Carlo (MCMC) samples every 1000th generation. Convergence of chains was visualized using TRACER 1.6 (Rambaut and Drummond, 2013) with a burn-in of the first 10 million generations.
Demographic history was analyzed using mismatch analysis conducted in ARLEQUIN 3.5 (Excoffier and Lischer, 2010) and DNASP 5 (Librado and Rozas, 2009) for the UK and compared to the Central Northern European clade (CNE) (i.e., Mouton et al., 2012). Multimodal distributions are considered to correspond to a condition of demographic stability or multiple colonization, whereas recent sudden population expansions would be observed by unimodal patterns (Slatkin and Hudson, 1991).
A total of 704 nucleotide positions of the target Cytb were resolved in all 125 individuals from around the UK. We found a total of five haplotypes, four of which are unique to known European haplotypes (all sequences have been deposited in GenBank; Table 1, Figure 2A), and all 704 positions were parsimony informative. We found nucleotide frequencies of 31% T, 26% C, 28% A, and 15% G. Among samples from England and Wales nucleotide diversity (π) was 0.00275 ± 0.00023 per site (Table 2), marginally lower than the CNE lineage (0.00337 ± 0.00707). Haplotype diversity we found was similar to that reported in continental Europe (UK 0.727 ± 0.052, EU 0.786 ± 0.096).
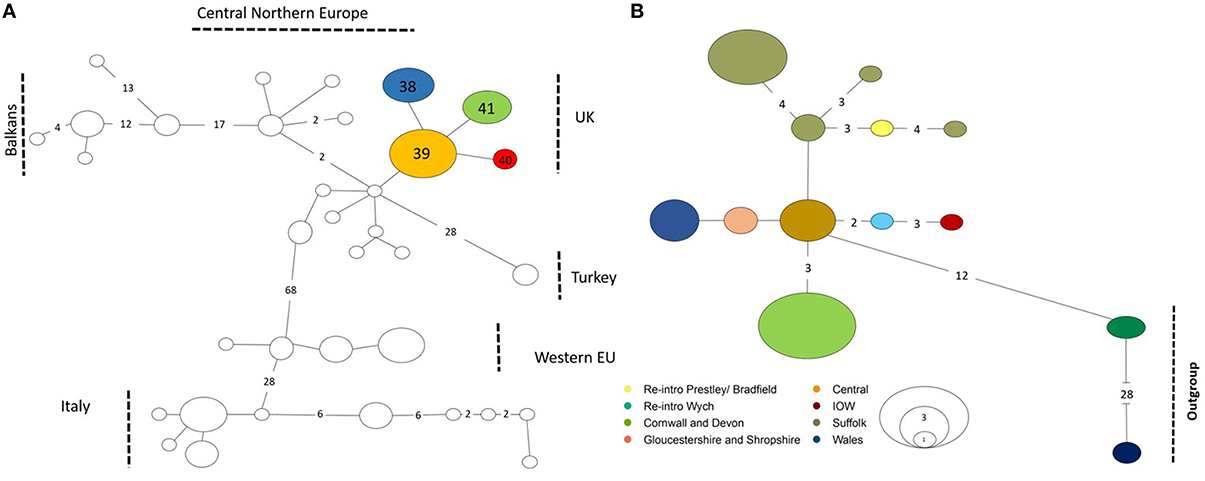
Figure 2. Haplotype map minimum spanning network. Numbers along the branches correspond to the number of mutational steps observed between haplotypes, no number is one mutational step, size of the circles correspond to the number of populations for each haplotype. (A) Number for UK haplotypes given in the circles correspond to Table 1. (B) D-loop haplotypes are colored as found in key. Reintroduction sites in the key relate to * in Table 1 and outgroups are sequences from Central Northern Europe and Western European haplotypes found in Mouton et al. (2012).
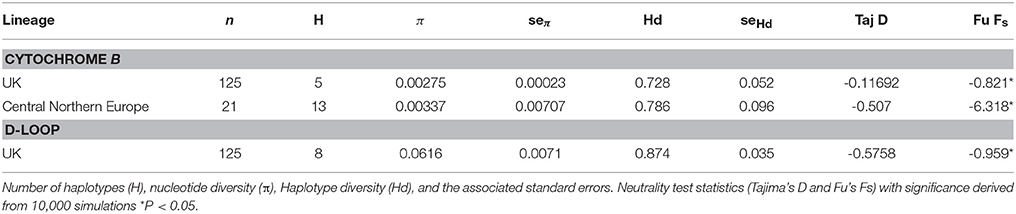
Table 2. Genetic diversity of Cytb for both the UK and Europe and D-loop for the UK.
ML analyses were performed using the GTR + Gamma model suggested for the data by the Akaike Information Criterion (AIC) in FINDMODEL. ML (Figure 3) and Bayesian inference tree showed identical topologies aligning UK dormice as a lineage close to CNE. The median joining network (Figure 2A) showed geographical partitioning of UK and CNE populations. In the cluster of UK haplotypes, only one mutational step was observed between each population forming geographical groupings of a SE England, N England and Wales (hap 39), Isle of Wight (hap 40), SW England (hap 41), and Suffolk (hap 40) (Figures 1, 2A). In total, UK dormice show a 0.3% genetic difference with mainland EU. The Cumbria sample (the most northerly extant population in England) forms a cluster with this central haplotype group. Haplotype 39 (Table 1) was found in 11 populations and is the most frequent group sampled in this study that also form a grouping with CNE clade (Figure 2A).
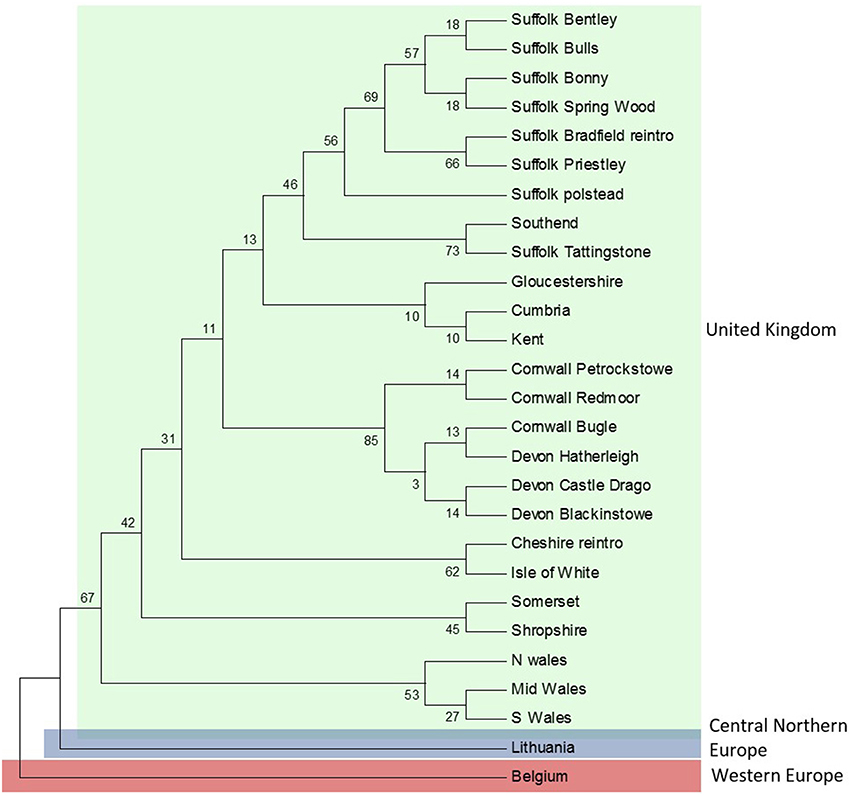
Figure 3. Maximum-likelihood (ML) topology for concatenated sequences (Cytb, D-loop, and bfibr). The numbers on the branches indicate the bootstrap values.
A 506 bp fragment of D-loop was sequenced for 125 individuals (Table 1). Haplotype diversity (0.8738 ± 0.0035) and nucleotide diversity (0.0616 ± 0.0071) were higher than we found in Cytb (Table 2). Tests for departure from neutrality on both Cytb and D-loop (Tajima's D and Fu's Fs) were significant, (both P < 0.05), consistent with a recent demographic expansion. The bfibr (726 bp) nuclear marker was sequenced for all individuals around the UK, Lithuania and France (no other sequences are available on GenBank for common dormice). This gene was found to be monomorphic across all populations in the UK, however there was divergence between the UK and continental European populations. As such bfibr and D-loop sequences were only further analyzed as concatenated sequences along with Cytb.
Concatenated sequence analysis (1760 bp) showed a total of 7 unique haplotypes, and had higher resolution to reveal regional genetic variation than that of Cytb alone (Figure 2B). This consists of additional genetic groups in Wales, North England and South East England, Central England, East England (Suffolk region), South West England (Devon and Cornwall), and the Isle of Wight. Phylogenetic analysis indicates a divergence between the clade containing the UK and CNE samples (Figure 3) with those from Western Europe. Due to a clustering of CNE and UK populations, AMOVA analyses only included these two genetic groups. The AMOVA analysis for Cytb shows most of the variation is within populations (70.72%, p < 0.001) however, relatively high (24.62%, p < 0.001) variation between populations within groups and less variation between groups (4.66%, p < 0.001) was also observed (Table 3). The AMOVA for D-loop (Table 3) shows most variation was shown between populations within populations (75.40, P < 0.001). This indicates population structuring between sampling locations within the UK but no significant variation between UK and CNE.
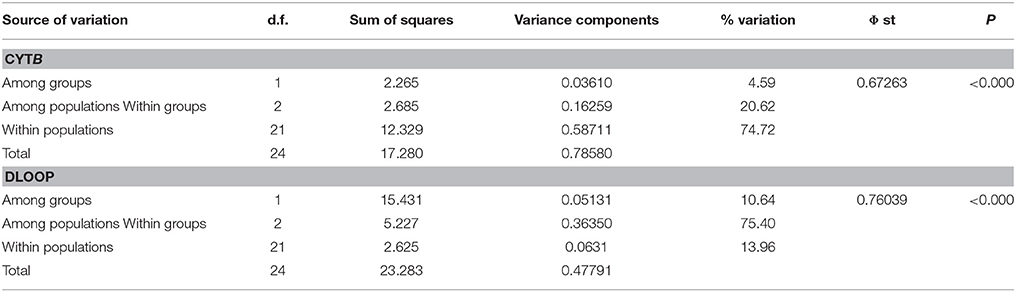
Table 3. Hierarchical distribution of mtDNA using Analysis of Molecular Variance (AMOVA) of Cytb and Dloop (d.f. degrees of freedom, Φ Phist, P = significance value).
In Cytb and D-loop the reintroduced population of Wych in North England shows a close grouping with the Isle of Wight haplotype (Table 1). For Cytb, only one mutation is observed between sequences as seen in both the ML tree and haplotype network (Figures 2, 3), whilst for D-loop there are four mutational steps present (Figure 2B). Samples sequenced from Suffolk in East England show a close genetic grouping with each other, especially in the D-loop haplotype network (Figure 2B). Two populations sampled in Suffolk were part of a national re-introduction programme and clustered separately from natural populations in Suffolk (by a single mutation). Based on our Cytb analysis, these reintroduced populations group with the South East and North England clade (Figure 2A), likely due to the genetic source of these populations from Southern England. Based on our D-loop analysis, one reintroduced Suffolk population (Bradfield) falls into the CE clade and the second reintroduced population groups with another Suffolk population (Bonny; Table 1, Figure 2B). The last population is geographically close (less than 1 km) with available connective habitat to the population in which it forms a haplotype connection with (see Figure 2B), but to assess whether there is evidence of gene flow or whether this is consistent with ancestral polymorphism, additional data and analysis would be required.
Molecular clock analysis was based on the mutation rate for Cytb previously estimated in the Gliridae (1% per Myr) and new calibration points for known periods of time when a land bridge was present between UK and EU between 7,5 and 25 kya at the end of the LGM (Shennan et al., 2000). Based on these priors, TMRCA estimates show the existing UK dormice populations originated from a CNE clade around 10.8 kya (median 95% higher posterior density, HPD), at the end of the LGM (Figure 4). The 95% HPD estimates range from 8.7 to 14.9 kya and all sampled parameters achieved a minimum effective samples size (ESS) average of 314. Test runs with alternative prior distributions did not influence posterior estimates of this parameter. There is some variation in the estimates in the time of origin for this split (Figure 4), however the 95% HPD values fall within the timings of the land bridge being present after the end of the Younger Dryas period. Our TMRCA estimates for the divergence Turkey and CNE clades are 22.7–34 kya (median 95% HPD, 27.9 kya), a time at the end of the LGM as glacial caps were retreating from Northern Europe. Estimates also place a Balkan and Turkey clade divergence at 35.6 kya and a Western European (Belgium) clade divergence at 65.5 kya. Mismatch distribution analysis (Figure 5) showed a unimodal distribution for the UK and CNE indicating a recent, rapid population expansion, which is consistent with an expansion at the end of the LGM when a land bridge facilitated dispersal of dormice to the UK.
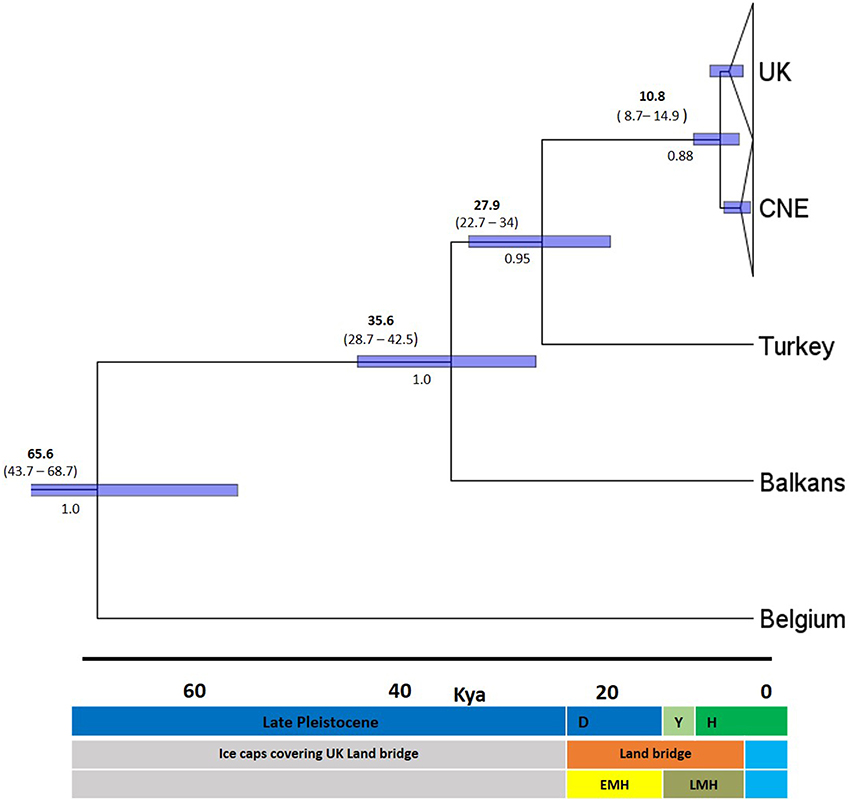
Figure 4. Maximum clade credibility chronogram of the Cytb dataset. Derived from an analysis in BEAST using the GTR+I+G model and a relaxed clock. Numbers above indicate median 95% higher posterior density (HPD) values in bold and age ranges below, Node bars also indicate 95% HPD age ranges. Numbers below branches are Bayesian posterior probabilities (BPPs). Dates in kya, definitions in middle key D-, Devensian period; Y-, Younger Dryas; H-, Holocene period. Blue highlights period when UK was isolated from continental Europe by rising sea levels. The bottom bar indicates the approximate timings of the two colonization hypotheses early migration hypothesis (EMH) and late migration hypothesis (LMH).
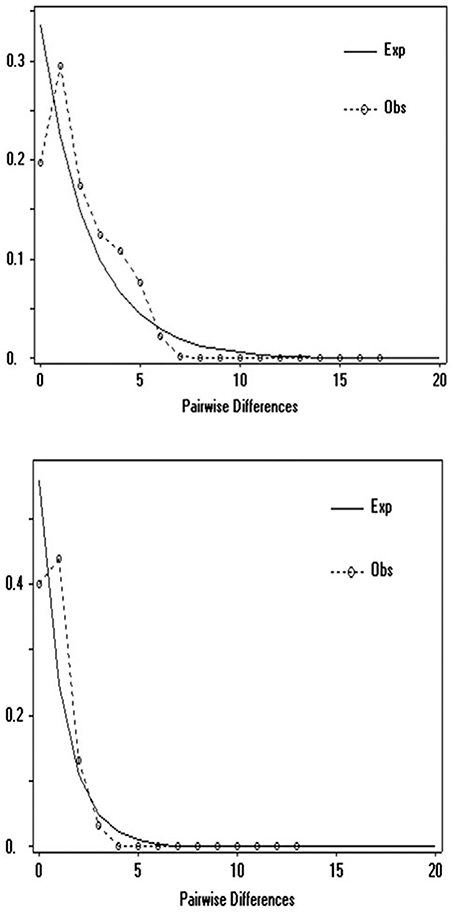
Figure 5. Mismatch analysis of Cytb sequences (704 bp) top, Central Northern European population (n = 25), bottom UK populations (n = 125).
Here, we present the first evidence of genetic divergence between UK common dormouse populations and those in continental Europe. Our sequence analysis of Cytb, in the context of published data from continental Europe (Mouton et al., 2012), suggests significant geographic partitioning of sub-lineages within the UK. Further, it is shown that these UK lineages are most closely related to populations located in CNE (Figure 2) and the within-population variation detected here is similar to that identified within regional subgroups in continental Europe by Mouton et al. (2012). The nuclear gene bfibr, although monomorphic in the UK, shows genetic subdivision from continental Europe with a similar mutational difference to that of Cytb and D-loop studied here. Comparisons of genetic diversity for mitochondrial genes observed in the UK are comparable to that found in the genetic clades of continental Europe which may be a result of recent genetic divergence in the order of magnitude in thousands of years. Because the entire mitochondrial genome is inherited as a unit, sequencing more than one mitochondrial gene, as we have done, whilst offering more resolution on the history of the maternal lineage, still represents a single lineage and thus has some inherent limitations. For this reason the addition of a nuclear gene in our analysis helps to confirm the identification of divergence between continental Europe and the UK. While there is evidence for reciprocal monophyly between continental European and UK dormice, further study of adaptive genetic variation in UK dormice is perhaps needed to inform management of the UK common dormouse as distinct.
We describe the genetic structure of the common dormouse within the UK for the first time. Here, we present evidence of regional genetic clustering of populations around the UK based on mtDNA variation clustering (Figure 3). This regional variation is possibly explained by gross geographical features; the UK has several major rivers and uplands in the North, West, and South of the country which may be important geographical boundaries leading to the further genetic clustering seen in this study. Although, the nuclear gene bfibr was monomorphic in our samples, the allelic sequence variant we report is unique to the UK. Mitochondrial DNA evolves at a much faster rate (5–10 fold) and as such mtDNA is more suitable for resolving contemporary events and defining evolutionary significant units (Wan et al., 2004). While the extent of regional genetic variation we describe is modest, it suggests the possibility of local adaptation and genetic differentiation which warrants further study at a finer geographical scale.
The phylogeographic structure we describe contrasts with that of previous small mammal studies. Other species of small mammal such as water voles, bank voles, field voles, and pygmy shrews show distinct population clusters in Scotland, Wales and Cornwall, or a so-called “Celtic fringe” within the UK (Searle et al., 2009). This is possibly explained by a two-stage colonization where the initial colonists were largely replaced by the second wave, leaving peripheral populations in Northern and Western areas of the UK. A pattern of single colonization was observed in dormice. This is possibly explained by the relatively low dispersal capability of the common dormouse.
Haplotype network analysis shows a clustering between the Isle of Wight and the reintroduced Wych population in North England. Based on the geographical distance between the Isle of Wight and Wych (over 130 km), the relatively close genetic relatedness of these populations is surprising. The Isle of Wight island population is relatively isolated off the south coast of England. However, the Wych population was reintroduced in 2001, with founding individuals originating from the Isle of White (pers. comm. Nida al Faluja, PTES). In Suffolk, the reintroduced populations we studied revealed one population grouping with natural Suffolk and South East England populations, suggesting genetic exchange between native and introduced stock. Thus, our analysis reveals the ability to pick up the genetic signatures of reintroduced populations to identify source populations when records are lacking. The consequences of admixture between lineages must be considered in genetic management to reduce the risk of losing adaptive potential of native populations and potential outbreeding depression in contrast with increasing genetic variation to reduce inbreeding depression (Frankham et al., 2011; Houde et al., 2011; Weeks et al., 2011). Further, research integrating ecological and demographic data and utilizing data from polymorphic functional genetic markers will aid the investigation of the potential consequences and to assess present-day gene flow.
Single post-glacial colonization into the UK through a Central North Western European clade is robustly supported in the haplotype network, Bayesian and ML trees. Molecular clock analyses timed this around the beginning of the Holocene period, supporting a late migration of dormice (LMH) along with several collateral lines of evidence. First, the land bridge “Doggerland” connecting the UK and mainland Europe was present around the beginning of the Holocene period. It is thought that Doggerland landmass had suitable vegetation for dormice dispersal during the early Holocene period (Mix et al., 2001; Spinney, 2008; Steffensen et al., 2008) until the formation of the English Channel approximately 8000 kya (Weninger et al., 2008). A key factor on colonization events would have been a cold period at the very end of the last glaciation, the Younger Dryas (11700–12900 kya) upon which a more temperate period followed (Hewitt, 2004; Steffensen et al., 2008). Thus, the TMRCA estimate after the Younger Dryas period is expected. In addition, the earliest dated dormice fossil remains in the UK from the post ice age period are from 9000 years ago using radio carbon dating (Montgomery et al., 2014) concordant with the estimated ranges in this study. Britain also has a congruent mammal species composition as found in Belgium and the Netherlands (Montgomery et al., 2014). Similarly, the Isle of Wight haplotype in southern UK, was isolated from the mainland around the same period. In total, these lines of evidence are consistent with the suggestion that dormice expanded through this land bridge to what is now the UK after the Younger Dryas period. This study suggests that the younger Dryas period may be more important than once thought in the shaping of the current phylogeographic structure in UK mammalian fauna.
The pattern of regional genetic variation of the common dormouse described here has a direct relevance to conservation management in the UK. Habitat enhancements can be directly related to improving connectivity between populations which were once historically connected. Alternatively, this information can be used to inform the genetic captive management, reintroduction or augmentation of species. The program of reintroduction of common dormice in the UK, in conjunction with the national Biodiversity Action Plan for the species, has a goal of both bolstering the quality and size of extant populations but also restoring additional populations to sites which were once formerly occupied but have gone locally extinct. There have been some important successes in these reintroductions; however the captive born founders for these reintroduced populations come from stock of heterogeneous origin. This is evidenced in the results presented here, where the northerly reintroduced Wych population appears to be genetically discontinuous with geographically close populations and a similar situation can be seen in South East England (Suffolk populations). We further suggest that, because there is no critical overall extinction risk for the common dormouse, preserving the natural pattern of genetic variation observed in natural populations could, and perhaps should, be considered when reintroducing animals back into the wild. A second consideration is to explicitly consider the genetic biodiversity, represented by regional genetic structure, to understand the possible impacts of gene flow between these populations managing the risk of losing adaptive genetic variation via reintroduction itself.
Rising sea levels caused flooding of Doggerland that historically connected the UK and continental Europe around 7.5 kya, dormice have been isolated and under environmental change. Thus, the future persistence of the UK dormouse will be reliant on the phenotypic plasticity or the adaptive genetic variation available. Dormice in the UK have seen population declines of around 50% in the last 25 years (Bright et al., 2006), and have become extinct from many Northern areas of the UK where they were historically present, a pattern not observed in the rest of Northern Europe. What is particularly concerning for threatened species, such as dormice, is climate change and expanding human populations will lead to increased fragmentation and hence further isolation of genetic variants, such as those presented in this study. Variable annual climate fluctuations are known to affect hibernating mammal species, due to food availability upon emergence (Inouye et al., 2000), and given the relatively low dispersal potential of dormice, genetic consequences of these climatic changes may be more severe. As such, if there are further population declines to the UK dormice, due to the relatively close genetic relationship of a CNE genetic clade, this may be a suitable source for future reintroductions.
There has been a considerable improvement in our current knowledge and understanding of the post-glacial expansion of species and the resulting genetic diversity of species on their range boundaries. This knowledge should play an important role in informing policy decisions both at local and national scales with regards to genetic conservation of species. Another consideration is that these peripheral populations on the range boundaries are often referred to as leading-edge populations and considered to easily become extinct (Hampe and Petit, 2005), it is considered that conservation efforts should ensure the survival of these populations as they play a critical role in the future response to climate change, due to their role in historical range movements expanding into new habitats and harbor evolutionary potential to respond to climatic changes (Lesica and Allendorf, 1995). Therefore, we propose that further research is required in common species such as the dormouse which are at risk or susceptible to declines. Finally, we recommend that monitoring programmes take into account not just population demographics but genetic make-up and predict the evolutionary potential of species to firstly, diagnose declines faster and secondly, to prevent further declines in the future.
FC was involved with experimental design, collecting samples, analyzing data, and wrote the manuscript. WH was involved with experimental design, interpretation of analysis and writing the manuscript with FC. JE was involved with Experimental design, data analysis and writing the manuscript. KL collected and analyzed data for the manuscript and contributed to proof of the manuscript. BC was involved with data analysis of genetic data and contributed to writing the manuscript. PW was involved with experimental design and contributed to writing the manuscript.
This project was funded by the People Trust for Endangered Species, UK Mammal Grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study could not be carried out without the help of all those who provided tissue samples. We also thank Nida Al-Fulaij and Ian White from the PTES for their knowledge and help in contacting sites from around the UK and Dr Simone Bullion for her insightful knowledge of dormice within the UK and Suffolk. This work was funded by the People's Trust for Endangered Species, Small Mammal Grant.
Bandelt, H. J., Forster, P., and Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Beatty, G. E., and Provan, J. (2013). Post-glacial dispersal, rather than in situ glacial survival, best explains the disjunct distribution of the Lusitanian plant species Daboecia cantabrica (Ericaceae). J. Biogeogr. 40, 335–344. doi: 10.1111/j.1365-2699.2012.02789.x
Beatty, G. E., and Provan, J. (2014). Phylogeographical analysis of two cold-tolerant plants with disjunct Lusitanian distributions does not support in situ survival during the last glaciation. J. Biogeogr. 41, 2185–2193. doi: 10.1111/jbi.12371
Bentz, S., and Montgelard, C. (1999). Systematic position of the African dormouse graphiurus (Rodentia, Gliridae) assessed from cytochrome b and 12S rRNA mitochondrial genes. J. Mamm. Evol. 6, 67–83. doi: 10.1023/A:1020590430250
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537.
Bright, P., Morris, P., and Mitchell-Jones, T. (2006). The Dormouse Conservation Handbook, Vol. 75. Peterborough, ON: English Nature.
Bright, P. W., Mitchell, P., and Morris, P. A. (1994). Dormouse distribution: survey techniques, insular ecology and selection of sites for conservation. J. Appl. Ecol. 31, 329–339.
Crandall, K. A., Bininda-Emonds, O. R. R., Mace, G. M., and Wayne, R. K. (2000). Considering evolutionary processes in conservation biology. Trends Ecol. Evol. 15, 290–295. doi: 10.1016/S0169-5347(00)01876-0
Emerson, B. C., and Hewitt, G. M. (2005). Phylogeography. Curr. Biol. 15, R367–R371. doi: 10.1016/j.cub.2005.05.016
Excoffier, L., and Lischer, H. E. L. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. doi: 10.1111/j.1755-0998.2010.02847.x
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Frankham, R., Ballou, J. D., Eldridge, M. D. B., Lacy, R. C., Ralls, K., Dudash, M. R., et al. (2011). Predicting the probability of outbreeding depression. Conserv. Biol. 25, 465–475. doi: 10.1111/j.1523-1739.2011.01662.x
Franks, S. J., and Hoffmann, A. A. (2012). Genetics of climate change adaptation. Annu. Rev. Genet. 46, 185–208. doi: 10.1146/annurev-genet-110711-155511
Funk, W. C., McKay, J. K., Hohenlohe, P. A., and Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 27, 489–496. doi: 10.1016/j.tree.2012.05.012
Gillespie, J. H. (2004). Population Genetics. Available online at: http://books.google.se/books?id=xf-ROozfS14C&printsec=frontcover&dq=intitle:Population+Genetics+a+concise&ie=ISO-8859-1&cd=1&source=gbsnpapers2://publication/uuid/C62EF24C-5B60-425C-A02C-0378D2EBB5B9
Hall, T. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hampe, A., and Petit, R. J. (2005). Conserving biodiversity under climate change: the rear edge matters. Ecol. Lett. 8, 461–467. doi: 10.1111/j.1461-0248.2005.00739.x
Houde, A. L. S., Fraser, D. J., O'Reilly, P., and Hutchings, J. A. (2011). Relative risks of inbreeding and outbreeding depression in the wild in endangered salmon. Evol. Appl. 4, 634–647. doi: 10.1111/j.1752-4571.2011.00186.x
Herman, J. L., Challis, C. J., Novák, A., Hein, J., and Schmidler, S. C. (2014). Simultaneous Bayesian estimation of alignment and phylogeny under a joint model of protein sequence and structure. Mol. Biol. Evol. 31, 2251–2266. doi: 10.1093/molbev/msu184
Herman, J. S., and Searle, J. B. (2011). Post-glacial partitioning of mitochondrial genetic variation in the field vole. Proc. Biol. Sci. 278, 3601–3607. doi: 10.1098/rspb.2011.0321
Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. doi: 10.1038/35016000
Hewitt, G. M. (1996). Some genetic consequences of ice ages, and their role, in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276. doi: 10.1006/bijl.1996.0035
Hewitt, G. M. (2004). The structure of biodiversity – insights from molecular phylogeography. Front. Zool. 1:4. doi: 10.1186/1742-9994-1-4
Hillis, D. M., and Bull, J. J. (1993). An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 42, 182–192. doi: 10.1093/sysbio/42.2.182
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogeny. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Inouye, D. W., Barr, B., Armitage, K. B., and Inouye, B. D. (2000). Climate change is affecting altitudinal migrants and hibernating species. Proc. Natl. Acad. Sci. U.S.A. 97, 1630–1633. doi: 10.1073/pnas.97.4.1630
Juškaitis, R. (2003). New data on distribution, habitats and abundance of dormice (Gliridae) in Lithuania. Acta Zool. Acad. Sci. Hung. 49(Suppl. 1), 55–62.
Lesbarrères, D. (2009). Post-glacial phylogeography: New insight into an old story: the post-glacial recolonization of European biota. Heredity (Edinb). 102, 213. doi: 10.1038/hdy.2008.121
Lesica, P., and Allendorf, F. W. (1995). When are peripheral populations valuable for conservation? Conserv. Biol. 9, 753–760. doi: 10.1046/j.1523-1739.1995.09040753.x
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Mix, A. C., Bard, E., and Schneider, R. (2001). Environmental processes of the ice age: land, oceans, glaciers (EPILOG). Q. Sci. Rev. 20, 627–657. doi: 10.1016/S0277-3791(00)00145-1
Montgelard, C., Matthee, C., and A, Robinson, T. J. (2003). Molecular systematics of dormice (Rodentia: Gliridae) and the radiation of Graphiurus in Africa. Proc. Biol. Sci. 270, 1947–1955. doi: 10.1098/rspb.2003.2458
Montgomery, W. I., Provan, J., McCabe, A. M., and Yalden, D. W. (2014). Origin of British and Irish mammals: Disparate post-glacial colonisation and species introductions. Q. Sci. Rev. 98, 144–165. doi: 10.1016/j.quascirev.2014.05.026
Moritz, C. (1994). Defining evolutionarily-significant-units for conservation. Trends Ecol. Evol. 9, 373–375. doi: 10.1016/0169-5347(94)90057-4
Morris, P. A. (2003). A review of research on British dormice (Gliridae) and the effect of increasing public and scientific awareness of these animals. Acta Zool. Acad. Sci. Hung. 49(Suppl. 1), 125–130.
Mortelliti, A., Amori, G., Capizzi, D., Rondinini, C., and Boitani, L. (2010). Experimental design and taxonomic scope of fragmentation studies on European mammals: current status and future priorities. Mamm. Rev. 40, 125–154. doi: 10.1111/j.1365-2907.2009.00157.x
Mouton, A., Grill, A., Sara, M., Kryštufek, B., Randi, E., Amori, G., et al. (2012). Evidence of a complex phylogeographic structure in the common dormouse, Muscardinus avellanarius (Rodentia: Gliridae). Biol. J. Linn. Soc. 105, 648–664. doi: 10.1111/j.1095-8312.2011.01807.x
Piertney, S. B., Stewart, W. A., Lambin, X., Telfer, S., Aars, J., and Dallas, J. F. (2005). Phylogeographic structure and postglacial evolutionary history of water voles (Arvicola terrestris) in the United Kingdom. Mol. Ecol. 14, 1435–1444. doi: 10.1111/j.1365-294X.2005.02496.x
Posada, D., and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Provan, J., and Bennett, K. D. (2008). Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571. doi: 10.1016/j.tree.2008.06.010
Rambaut, A. (2009). FigTree, a Graphical Viewer of Phylogenetic Trees. Institute of Evolutionary Biology at University of Edinburgh. Available online at: http://tree.bio.ed.ac.uk/software/figtree/
Rambaut, A., and Drummond, A. J. (2013). Tracer V1.6. Available online at: http://beast.bio.ed.ac.uk/tracer
Searle, J., Kotlík, P., Rambau, R. V., Markov,á, S., Herman, J. S., and McDevitt, A. D. (2009). The Celtic fringe of Britain: insights from small mammal phylogeography. Proc. Biol. Sci. 276, 4287–4294. doi: 10.1098/rspb.2009.1422
Seddon, J. M., Santucci, F., Reeve, N. J., and Hewitt, G. M. (2001). DNA footprints of European hedgehogs, Erinaceus europaeus and E. concolor: Pleistocene refugia, postglacial expansion and colonization routes. Mol. Ecol. 10, 2187–2198. doi: 10.1046/j.0962-1083.2001.01357.x
Shennan, I., Lambeck, K., Flather, R., Horton, B., McArthur, J., Innes, J., et al. (2000). Modelling Western North Sea palaeogeographies and tidal changes during the Holocene. Geol. Soc. Lond. Spec. Publ. 166, 299–319. doi: 10.1144/GSL.SP.2000.166.01.15
Slatkin, M., and Hudson, R. R. (1991). Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129, 555–562.
Stacy, J. E., Jorde, P. E., Steen, H., Ims, R. A., Purvis, A., and Jakobsen, K. S. (1997). Lack of concordance between mtDNA gene flow and population density fluctuations in the bank vole. Mol. Ecol. 6, 751–759.
Steffensen, J. P., Andersen, K. K., Bigler, M., Clausen, H. B., Dahl-Jensen, D., Fischer, H., et al. (2008). High-resolution Greenland ice core data show abrupt climate change happens in few years. Science 321, 680–684. doi: 10.1126/science.1157707
Stewart, J. R., Lister, A. M., Barnes, I., and Dalén, L. (2010). Refugia revisited: individualistic responses of species in space and time. Proc. Biol. Sci. 277, 661–671. doi: 10.1098/rspb.2009.1272
Sturt, F., Garrow, D., and Bradley, S. (2013). New models of North West European Holocene palaeogeography and inundation. J. Archaeol. Sci. 40, 3963–3976. doi: 10.1016/j.jas.2013.05.023
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Teacher, A. G. F., Garner, T. W. J., and Nichols, R. A. (2009). European phylogeography of the common frog (Rana temporaria): routes of postglacial colonization into the British Isles, and evidence for an Irish glacial refugium. Heredity (Edinb). 102, 490–496. doi: 10.1038/hdy.2008.133
Vega, R., Fløjgaard, C., Lira-Noriega, A., Nakazawa, Y., Svenning, J. C., and Searle, J. B. (2010). Northern glacial refugia for the pygmy shrew sorex minutus in europe revealed by phylogeographic analyses and species distribution modelling. Ecography (Cop.) 33, 260–271. doi: 10.1111/j.1600-0587.2010.06287.x
Wan, Q.-H., Wu, H., Fujihara, T., and Fang, S.-G. (2004). Which genetic marker for which conservation genetics issue? Electrophoresis 25, 2165–2176. doi: 10.1002/elps.200305922
Webb, T., and Bartlein, P. J. (1992). Global changes during the last 3 Million Years: climatic controls and biotic responses. Annu. Rev. Ecol. Syst. 23, 141–173. doi: 10.1146/annurev.es.23.110192.001041
Weeks, A. R., Sgro, C. M., Young, A. G., Frankham, R., Mitchell, N. J., Miller, K. A., et al. (2011). Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol. Appl. 4, 709–725. doi: 10.1111/j.1752-4571.2011.00192.x
Weninger, B., Schulting, R., Bradtmöller, M., Clare, L., Collard, M., Edinborough, K., et al. (2008). The catastrophic final flooding of Doggerland by the Storegga Slide tsunami. Documanta Praehist. XXXV, 1–24. doi: 10.4312/dp.35.1
Willis, K. J., and Whittaker, R. J. (2000). Perspectives: paleoecology. Refug. Debate. Sci. 287, 1406–1407. doi: 10.1111/j.1365-294X.2011.05211.x
Keywords: phylogeography, common dormouse, genetic structure, conservation genetics, postglacial colonization
Citation: Combe FJ, Ellis JS, Lloyd KL, Cain B, Wheater CP and Harris WE (2016) After the Ice Age: The Impact of Post-Glacial Dispersal on the Phylogeography of a Small Mammal, Muscardinus avellanarius. Front. Ecol. Evol. 4:72. doi: 10.3389/fevo.2016.00072
Received: 29 January 2016; Accepted: 06 June 2016;
Published: 21 June 2016.
Edited by:
Angela McGaughran, Australian National University, AustraliaReviewed by:
Jonathan J. Fong, Lingnan University, Hong KongCopyright © 2016 Combe, Ellis, Lloyd, Cain, Wheater and Harris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. Edwin Harris, ZS5oYXJyaXNAbW11LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.