
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Sci. , 06 August 2024
Sec. Freshwater Science
Volume 12 - 2024 | https://doi.org/10.3389/fenvs.2024.1404230
This article is part of the Research Topic Biological Contaminants of Concern in Water and Wastewater: An Environmental Health Perspective View all 15 articles
 Oscar Victor Cardenas-Alegria1,2†
Oscar Victor Cardenas-Alegria1,2† Victor Benedito Costa Ferreira1,2†
Victor Benedito Costa Ferreira1,2† Wylerson Guimarães Noguera3
Wylerson Guimarães Noguera3 David Tavares Martins1,2
David Tavares Martins1,2 Artur Pedro Martins Neto1
Artur Pedro Martins Neto1 Paulo Rógenes Monteiro Pontes4
Paulo Rógenes Monteiro Pontes4 Rosane Barbosa Lopes Cavalcante4Sandy Ingrid Aguiar Alves1,2
Rosane Barbosa Lopes Cavalcante4Sandy Ingrid Aguiar Alves1,2 Artur Luiz da Costa da Silva5Rosilene Gomes Costa6,7
Artur Luiz da Costa da Silva5Rosilene Gomes Costa6,7 Edian Franklin Franco de Los Santos8
Edian Franklin Franco de Los Santos8 Vasco Ariston de Carvalho Azevedo3,9
Vasco Ariston de Carvalho Azevedo3,9 Rommel Thiago Juca Ramos1,2,3*
Rommel Thiago Juca Ramos1,2,3*One of the primary challenges in the spread of infectious diseases is the consumption of poorly or untreated water, which is increasingly being used due to the growth of different human activities and the effect of urbanization on freshwater sources, which are often used for consumption purposes. The determination of pathogenic bacteria in freshwater rivers influenced by anthropogenic activities allows for the assessment of the impact these factors have on water quality. Thus, the purpose of this study was to identify the diversity of pathogenic bacteria and virulence genes in the Uraim River in the northern region of Brazil. For this purpose, surface water was collected from five points with varying degrees of anthropogenic impact along the Uraim River. In situ measurements of physicochemical components were conducted, and metagenomic analysis was used for the identification of pathogenic bacteria and virulence genes. Regarding the physicochemical parameters, variability was observed among the different analysis points, as well as diversity among bacteria and virulence genes. Notably, enterobacteria and the ESKAPE group were highlighted among the bacteria, with significant negative associations found between dissolved oxygen and the diversity of virulence genes and between deforestation and population density with the presence of ESKAPE group bacteria.
Global estimates suggest that 80% of industrial and municipal wastewater is discharged into the environment without prior treatment, leading to adverse effects on human health and ecosystems. This is particularly prevalent in less developed countries due to deficiencies in sanitation and wastewater treatment facilities (Lin et al., 2022).
However, water pollution primarily occurs due to industrialization, agricultural activities, natural factors, inadequate water supply, and inadequate sewage treatment facilities. According to the UNESCO World Water Development Report 2021, approximately 829,000 people die each year from diarrhea caused by the consumption of unsafe water due to lack of basic sanitation, including nearly 300,000 children under the age of five, accounting for 5.3% of all deaths in this age group (Lin et al., 2022).
On the other hand, urbanization worldwide is rapidly progressing. From 1950 to 2020, the global population living in cities increased from 0.8 billion (29.6%) to 4.4 billion (56.2%), and it is projected to reach 6.7 billion (68.4%) by 2050. A key issue is water scarcity, a critical aspect of water security that directly impacts the health and wellbeing of urban residents, urban environmental quality, and socioeconomic development (He et al., 2021; Strokal et al., 2021).
Freshwater is a finite natural resource essential for life on Earth. It is crucial for urban, agricultural, and industrial activities, as well as providing a habitat for a rich diversity of macro and microorganisms. However, anthropogenic activities, climate change, and a growing global population pose threats to its quality and availability worldwide (Pandey et al., 2014; Chopyk et al., 2020).
While the monitoring of bacterial indicator organisms (e.g., Escherichia coli) is widely standardized, waterborne viral and protozoan pathogens remain poorly characterized (Ko et al., 2022). A better understanding of the transport of pathogens from agricultural or urban areas to rivers is a major challenge. Many environmental changes caused by anthropogenic activities have a direct impact on water resource development, climate warming, and interactions between humans and animals, both domestic and wild. These factors promote the dissemination and diversification of pathogenic microorganisms (Pandey et al., 2014). Additionally, the use of various chemicals to ensure production serves as selective components for microbial adaptation, potentially altering ecosystem composition (Schwarzenbach et al., 2010).
In the northern region of Brazil, specifically in the Amazon part, there is significant macro-diversity, and it is made up of various bodies of water. In the state of Pará, the municipality of Paragominas is characterized by two main rivers: the Paragominas River and the Uraim River, the latter belonging to the Northeast Atlantic Western hydrographic region and the Gurupi hydrographic sub-region (Agência Nacional de Águas, 2020), the hydrographic basin is mixed, covering the urban and rural areas of the municipality, has a drainage area of 4,873 km2, a length of the main river of 129.5613 km and a circular shape (Correa, 2017).
Over the past 20 years, these areas have been utilized for diverse agricultural activities, as seen in the municipality of Paragominas. The latter drains the city center towards the northwest and serves as the primary water source for distribution to the population through water acquisition stations (Pereira Júnior et al., 2023a). However, studies carried out in this region are scarce; the few accessible works were on the characterization of physical and chemical characteristics, diatom studies, and geoprocessing analyses (Santos and Picanço, 2008; De Araujo, 2010; Giuliatti et al., 2017; Da Silva et al., 2020), no studies are using metagenomic analysis in the region. In this context, this study aims to identify the diversity of pathogenic bacteria and virulence genotypes in the Uraim River and their relationship with anthropogenic activities.
The samples were obtained from the Uraim River in September 2022, which flows through Paragominas. Five sampling points were selected based on varying levels of anthropogenic impact: P1 (P1A, P1B, P1C), P2 (P2A, P2B, P2C), P3 (P3A, P3B, P3C), P4 (P4A, P4B, P4C), and P5 (P5A, P5B, P5C), were collected 1 m away from the banks and in the center of the river for each point. Three biological replicates were collected at each point, and fifteen samples were obtained from all points.
The point P1 was located in an untouched, pristine site, while the final point (P5) was located downstream after passing through the city. The remaining points were intermediate between these two extremes. The samples were collected at a depth of 20 cm, with a volume of 5L taken at each point in triplicate, with help from a Van Dorn bottle. They were preserved on ice and immediately transported to the laboratory. Upon arrival, the water samples were filtered using a negative pressure filtration system with the assistance of a vacuum pump and 47 mm membranes with a porosity of 0.22 µm. These membranes were preserved in 50 mL falcon tubes containing TSE buffer (Tris HCl 10 mM pH = 8, Sodium Chloride 100 mM, EDTA 1 mM pH = 8) and stored at −20°C.
Physical-chemical component measurements were conducted in situ using a multi-parameter water analyzer (HI 9829, HANNA), considering the parameters of pH, electrical conductivity (EC), dissolved oxygen (DO), and total dissolved solids (TDS).
The methodology for mapping the anthropic signature of land use and land cover in rivers employs the concept of a hydrographic basin and geoprocessing techniques. The deforestation level and population density were evaluated following the methodology of Cavalcante et al. (2023). The deforested area was calculated as the sum of all areas with non-natural land use and land cover based on the classification of the Mapbiomas Brasil project (https://brasil.mapbiomas.org/en/) version six for the year 2020.
The sampling points were chosen following a bioprospecting approach, with consideration given to the gradient of anthropogenic impact and accessibility for sample collection. These points were situated at the outset of the pristine site, specifically at coordinates 3° 3′28.512″S, 47° 25′2.172″W. Following the initial point, the subsequent sampling locations were designated as P2 (3° 0′24.737″S, 47° 22′52.941″W), P3 (2° 59′56.264″S, 47° 22′17.735″W), P4 (2° 58′56.895″S, 47° 21′37.938″W) and extending to the river outlet from the city of Paragominas at P5 (2° 57′54.169″S, 47° 20′15.140″W).
In the laboratory, the membranes containing the samples were first shaken at 250 RPM at room temperature. Subsequently, the liquid containing the suspended particles retained in the membrane was centrifuged at 13200 RPM for 10 min in Eppendorf tubes. Genetic material was then extracted following the manufacturer’s instructions of the Pro-DNeasy PowerSoil kit (Qiagen). The integrity of the obtained genetic material was assessed through horizontal electrophoresis. DNA quantification was conducted using Qubit (Thermo Fisher Scientific), and purity indices were obtained via Nanodrop (Thermo Fisher Scientific).
For the construction of DNA libraries, the samples were processed according to the manufacturer’s protocol for the Nextera XT DNA Library kit (Illumina). Sequencing was performed using the NextSeq 550 System High-Output kit, generating paired-end reads of 150 bp, using the Illumina NovaSeq 500/550 High-Output platform.
The quality of the sequencing reads was initially analyzed using the software FASTQC–Version 0.11.9 (Wingett and Andrew, 2018). Subsequently, the reads were trimmed and filtered with a minimum Phred quality score of 20 using the software Trimmomatic (Bolger et al., 2014). The obtained sequences were submitted to the National Center for Biotechnology Information NCBI database (https://www.ncbi.nlm.nih.gov/), with Submission ID SUB14513086 and BioProject ID PRJNA1122411, accessible with the following link https://www.ncbi.nlm.nih.gov/sra/PRJNA1122411.
The pathogenic species in the collections were identified by comparing the output from Kraken2 with a local database of pathogenic bacteria from various isolates associated with pathogenicity in food, water, and clinical hosts. Analyses were conducted at both the species and genus levels. The local database was created using complete genome records labelled as pathogenic by the NCBI Pathogen Detection Project (https://www.ncbi.nlm.nih.gov/pathogens/). Records were extracted using the NCBI FTP protocol and manipulated using the Python programming language. For information extraction from HTML pages and for the grouping, filtering, and sorting of records, the beautifulSoup and pandas packages were utilized, respectively. The records were saved in the MongoDB database, employing the NoSQL format, hosted in a Docker container https://github.com/labgm/pathogen-detection.git.
For the identification of different taxonomic groups, the previously trimmed reads were used. These reads were then evaluated using the Plants, Fungi, and Protists (PFP) database within the Kraken2 program (Wood et al., 2019). The output format of Kraken2 was converted to be recognized by the MicrobiomeAnalyst program (Chong et al., 2020) for the transformation of abundances using Trimmed Mean of M-values (TMM), then, relative abundances were processed to determine alpha and beta diversity indices and associations between taxa.
For the identification of bacterial virulence factors, the Virulence Factor Database (VFDB) (Liu et al., 2022) was used. This database contains 32 genera of bacterial pathogens, classified into 14 basal categories. The database was indexed using the Bowtie2 software (Langmead and Salzberg, 2012), and then the reads obtained from the sequencing were aligned against the database to capture the presence and abundance of virulence factors in each sample. The alignment result was processed using the Pileup software to identify and quantify the aligned reads at each position of the reference database. The information obtained from Pileup, including the columns of identification and abundance, served as the basis for the subsequent step where they were organized into a tabular structure file containing the identified virulence factors and their respective abundance values for each sample. This facilitated the comparison and interpretation of the data.
The data obtained from the different analyses were evaluated for normality using the Shapiro-Wilk test. Subsequently, they were analyzed using ANOVA or Kruskal–Wallis tests, multiple regression, and principal component analysis (PCA) using the PAST 4.05 software (Hammer et al., 2001). For the preparation of the graphs, the Orange Data Mining program (Demsar et al., 2013), as well as the Seaborn and Vegan-Altair libraries of Python, were used.
The points where samples were collected in the Uraim River are highlighted in yellow in Figure 1, observed through satellite images. In Figure 1A, the population density in inhabitants per square kilometer along the river is emphasized, allowing for the observation of human settlement and agricultural areas near certain areas of the river. In Figure 1B, the percentage of deforestation in the areas along the river is further observed. It is noted that the collection sites showed a deforestation percentage ranging from 70% to 73%, with population densities ranging from 1 to 184 inhabitants per square kilometer.
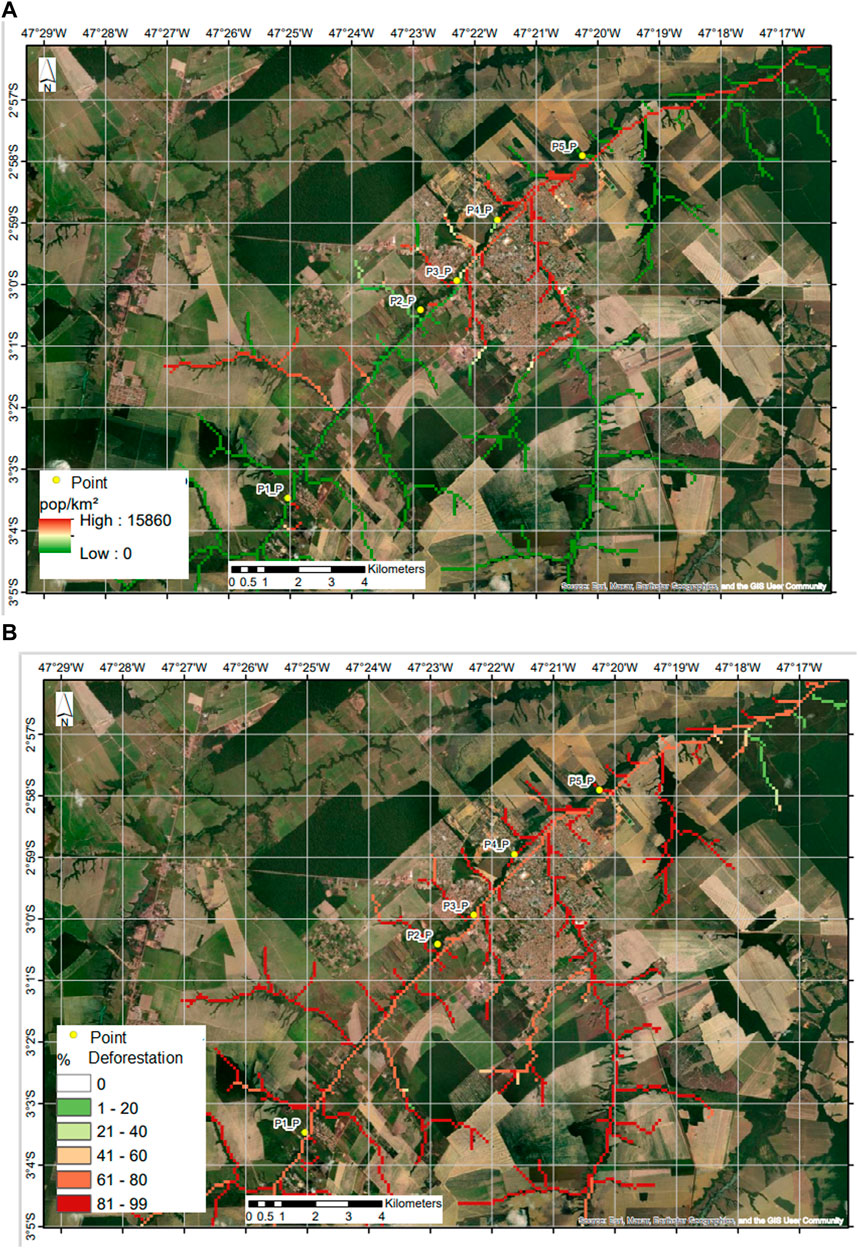
Figure 1. Satellite images of the municipality of Paragominas-PA delineating the path of the Uraim River as it crosses the municipality. (A) Population density in inhabitants per square kilometer along the river. (B) Percentage of deforestation in the areas along the river.
The Uraim River basin exhibits considerable deforestation of its native vegetation, resulting in a 25% reduction in average precipitation during the deforested period compared to years with forest presence, consequently leading to a 22% reduction in channel flow (Sardinha and Ventura, 2022). Different anthropogenic activities have repercussions on various water resources (Pereira Júnior et al., 2023a).
In the municipality of Paragominas-PA, the forest area was 3420.27 km2 in 2000 and decreased to 2784.23 km2 in 2021, representing a forest area loss of 18.6%. Regarding the expansion of the urban area, the development began in 2000 (133156 km2) and 2021 (20.2687 km2), which is linked to population growth: in 2000, the population of Paragominas was 76450 inhabitants, and it increased to 115538 inhabitants in 2021. This implies changes in ecosystems, such as the reduction of the precipitation rate that affects the water recharge of rivers, streams, and creeks, among others (Pereira Júnior et al., 2023a). These characteristics are evident in the images obtained for this study and also in the alterations in chemical and microbiological components.
In the physicochemical analyses, acidic pH was found, ranging between 3.547 and 4.627, with significant differences between the points (ANOVA; F = 23.47; p-value = 0.0001). notably, these differences occurred in the comparison between: points P1 and P2 (Tukey’s test; Q = 9.235; p-value = 0.001); P2 and P3 (Tukey’s test; Q = 6.527; p-value = 0.011); P1 and P4 (Tukey’s test; Q = 7.707; p-value = 0.004); P3 and P4 (Tukey’s test; Q = 4.999; p-value = 0.048); P2 and P5 (Tukey’s test; Q = 2.014; p-value = 0.0003), and P4 between P5 (Tukey’s test; Q = 9.721; p-value = 0.001) (Supplementary Figure S1).
The values of electrical conductivity varied across all points but were not significant. In the case of dissolved oxygen, a decrease was evidenced at P5, with average values of 4.413, showing significant differences (ANOVA; F = 8.148; p-value = 0.006) for this point compared to the others. For total dissolved solids (TDS), despite the variation in values at the collection points, the differences were not significant (Table 1).
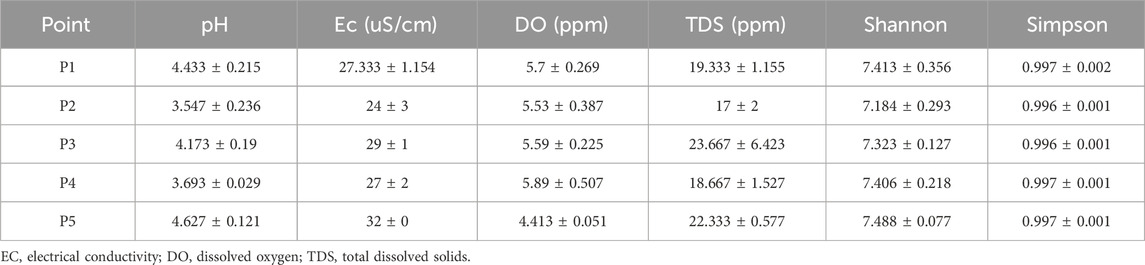
Table 1. Physical-chemical characteristics and total microbial diversity at different sampling points in the Uraim River, Paragominas-PA.
These findings underscore a potential public health concern should the population consume untreated water, including water from the river, typically used for domestic, agricultural, and livestock purposes. Anthropogenic activities stand out as primary factors influencing the quality of surface waters (Tornevi et al., 2014; Vinueza et al., 2021). In the Uram River area, agricultural practices have been observed to increase the concentrations of various chemical compounds in the river. As it flows through urban areas, the river’s natural pH, nitrogen and oxygen concentrations, turbidity, and other parameters can be altered, affecting the standards of freshwater quality intended for human consumption (Pereira Júnior et al., 2023b).
In the sequencing, an average of 35468174.13 reads were obtained from the different sample collection points, of which about 33% were used for taxonomic classification. Bacteria accounted for 30% of the reads, while opportunistic pathogenic bacteria for humans represented 0.34%, Table 1.
Regarding total diversity in these ecosystems, up to 7.190 different species were identified, with Shannon index values above 7.184 and Simpson index values close to 1, indicating no dominance of a single species (Table 1). In the Tama River in Tokyo, Japan, it was observed that bacterial communities exhibited significant differences along the river’s course, showcasing variations in diversity (Mizusawa et al., 2021). This phenomenon was also noted in the Uraim River in Paragominas, Brazil, highlighting the dynamic nature of microbial communities in urban river systems. Interestingly, the Amazon region, home to the Amazon River, lacks comprehensive studies on microbial diversity. Despite being the world’s largest river in terms of discharge, the Amazon River remains relatively understudied, with existing research exhibiting considerable heterogeneity (Li et al., 2021).
In our study of the Uraim River, the most abundant phyla were Pseudomonadota and Actinomycetota, the taxonomic class of Betaproteobacteria was noteworthy, followed by Alphaproteobacteria, Actinomycetes and Gammaproteobacteria. However, in freshwater studies, the dominant classes are usually Alphaproteobacteria, Betaproteobacteria, and Actinobacteria, which complex changes in nutrient composition can alter (Santos-Júnior et al., 2020; Mizusawa et al., 2021).
A higher abundance of pathogenic species was detected at point P5, averaging 226.7 (Figure 2A). The Shannon index showed lower values on average at point P2, with a value of 3.955 ± 0.592, and the highest average value of 4.312 ± 0.102 was found at point P3 (Figure 2B). The Simpson index presented average values between 0.914 and 0.971 at the same aforementioned points (Figure 2C).
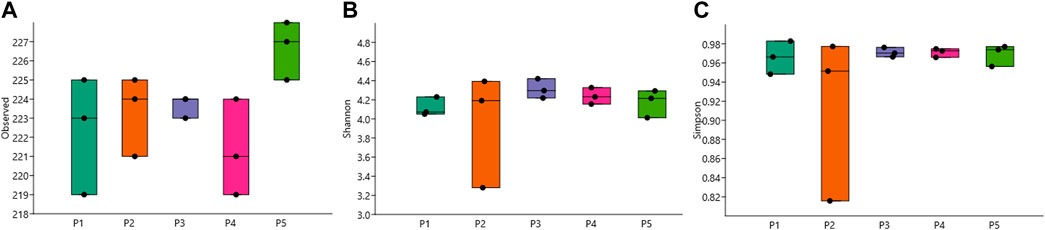
Figure 2. Box plot of alpha diversity values of opportunistic pathogenic bacteria for humans, showing (A) Observed, (B) Shannon, and (C) Simpson indices.
Beta diversity analysis of biological replicates revealed a clustering trend in the bacterial composition corresponding to P5, with R2 values of 0.576 (PERMANOVA F-value = 3.3961; p-value = 0.001), indicating a cumulative variation of 70.6% for the PCoA analysis (Supplementary Figure S2).
Regarding the phylum composition of pathogenic microorganisms identified across all sampling points, Pseudomonadota, Bacillota, and Campylobacterota are highlighted, respectively. Among the eight microorganisms identified, Gammaproteobacteria were predominant, followed by Betaproteobacteria and Bacilli, with the remaining classes taxonomic in smaller proportions (refer to Supplementary Figure S3). Among the 27 identified families, Enterobacteriaceae, Burkholderiaceae, and Pseudomonadaceae exhibited the highest abundance (see Figure 3A). Particularly for Enterobacteriaceae, a substantial increase was observed in points 4 and 5, with statistically significant differences noted between these points (ANOVA; F = 78.96; p-value = 1.59 × 10−7) (see Figure 3B).
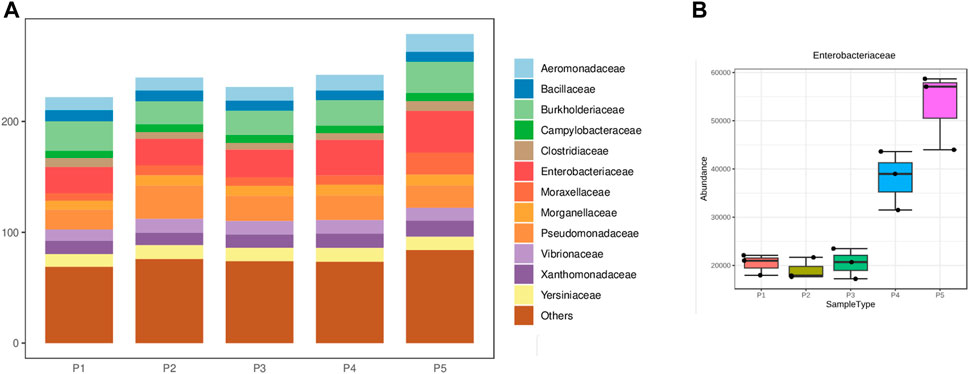
Figure 3. (A) Distribution of the main taxonomic family groupings, (B) Proportion of Enterobacteriaceae at different sampling points.
Metagenomic analysis allows for the simultaneous screening of various potential pathogens and fecal indicators. The river’s microbiome serves as a significant indicator of urban pollution alteration, demonstrating a prevalence of human intestinal bacteria, predominantly pathogenic (Acharya et al., 2022). Among the most abundant genera identified at all detecting points were Burkholderia and Pseudomonas, with a notable presence of Acinetobacter, Escherichia, Klebsiella, and Enterobacter at point 5 (Figure 4A). The most abundant pathogenic species belong to the ESKAPE group. Less frequently detected species include Burkholderia cepacia complex (Bcc), E. coli, and Stenotrophomonas maltophila (Figure 4B). Regarding ESKAPE group species, Enterobacter spp. Exhibited a distribution with minor variations across sampling points (R2 = 0.768; p-value = 1.825 × 10−5), consistent with the observations for Acinetobacter baumanii and Klebsiella pneumoniae, which displayed higher abundance at point 5. Notably, Pseudomonas aeruginosa demonstrated an inverse trend compared to the species above (Figure 4C), which could influence changes environmental characteristics and microbial communities (Philippot et al., 2021), but we could not identify specific facts against these microorganisms due to the complexity of the variables that make up the ecosystems.
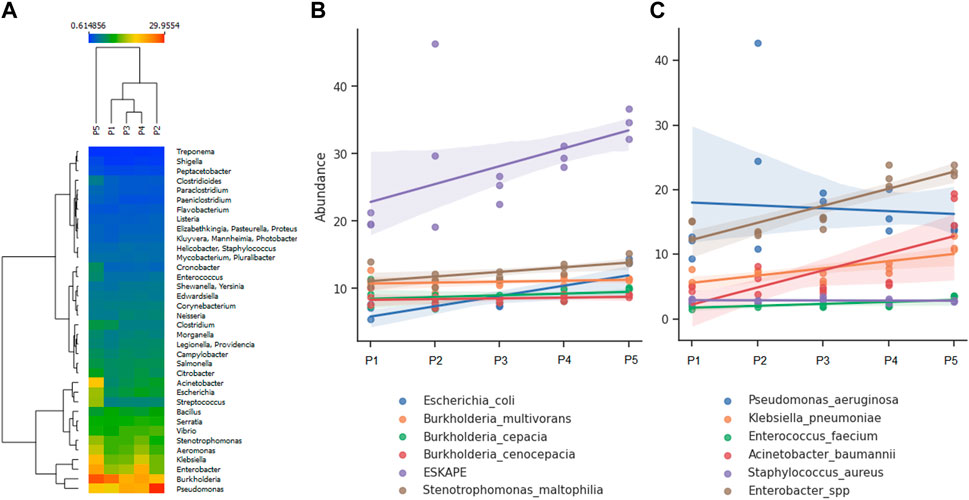
Figure 4. Analysis of abundance according to the identified bacterial genera and species: (A) Most abundant genera, (B) Most abundant species, (C) Species composing the ESKAPE group.
In the correlation analysis with the relative abundances of pathogenic bacterial species, Enterobacter cloacae stands out as significantly associated with the presence of Klebsiella variicola, Citrobacter freundii, Aeromonas veronii and Enterobacter kobei. The latter is correlated with the presence of E. coli and is involved with the occurrence of Acinetobacter nosocomiais, Streptococcus susis, Enterococcus hirae and Enterococcus faecium, which are associated with Streptococcus pyogenes and Streptococcus equi, as indicated by the interaction network presented in Figure 5.
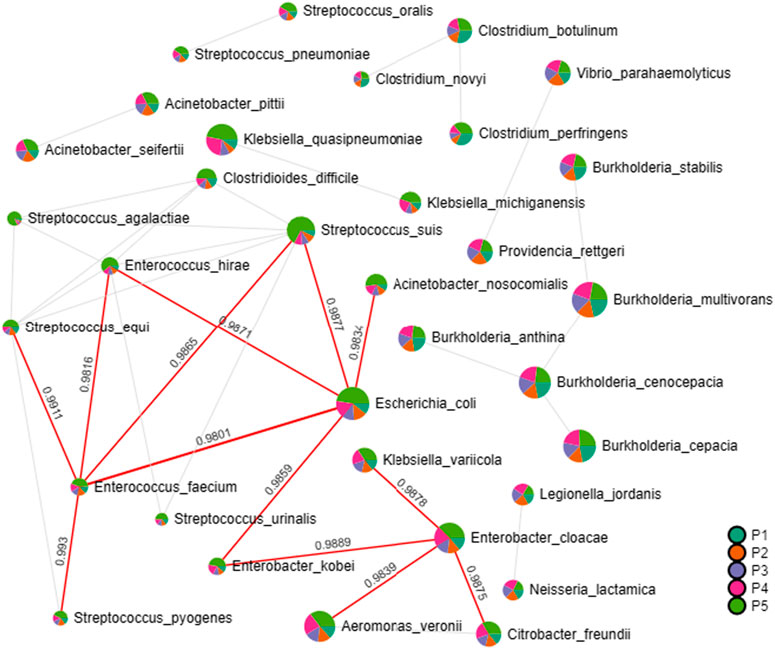
Figure 5. Interaction network revealing associations of presence among pathogenic bacteria. Generated through correlation analysis with Pearson coefficient algorithm. Significant index values are highlighted in red lines.
The substantial presence of human pathogens is notable, constituting approximately 7% of all taxonomically described bacterial species. These pathogenic entities span 24 distinct taxonomic classes, with prominent contributions from Gammaproteobacteria and Actinomycetia in terms of genus and species diversity. The identified pathogen pool encompasses 1513 species categorized into 327 genera, of which 267 genera encompass at least one established pathogenic species (Bartlett et al., 2022). Waterborne illnesses persist as a significant global health concern, prompting numerous regulatory bodies to utilize coliform assessments in water quality testing as an indicator of fecal contamination levels and associated disease risks (Hamner et al., 2019).
The findings presented in this study align with the expected bacterial populations that thrive in sewage-contaminated environments, characterized by a dominance of Proteobacteria and Bacteroidetes (Godoy et al., 2020). Additionally, the prevalence of Actinobacteria in freshwater environments is consistent with previous studies (Mizusawa et al., 2021).
The presence of pathogenic microorganisms allows us to assess the quality of these aquatic systems in terms of environmental impact and anthropogenic activities. In this study, the presence of the ESKAPE group is notable, given its ability to possess and transfer antibiotic resistance genes (Denissen et al., 2022; Aguilar-Salazar et al., 2023). The results highlight a higher abundance, on the bodies of water, of K. pneumoniae, E. cloacae, E. faecium, and E. faecalis. Additionally, the metagenomic analysis reveals the presence of bacteria that serve as bioindicators for chronic diseases in humans, such as Fusobacterium nucleatum associated with colorectal cancer (VanEvery et al., 2023).
Upon examination of the virulence genes identified across the various sampling points, as illustrated in Figure 6, it was noted that point P5 exhibited a greater prevalence of unique genes, not identified in other points. This observation aligns with expectations, given that P5 is situated downstream from the urbanized area of Paragominas, experiencing a range of anthropogenic influences and cumulative pressures from upstream points. Additionally, a total of 315 genes were found to be common among all five sampling locations, indicating a potential presence of native core microbiota unaffected by anthropogenic activities.
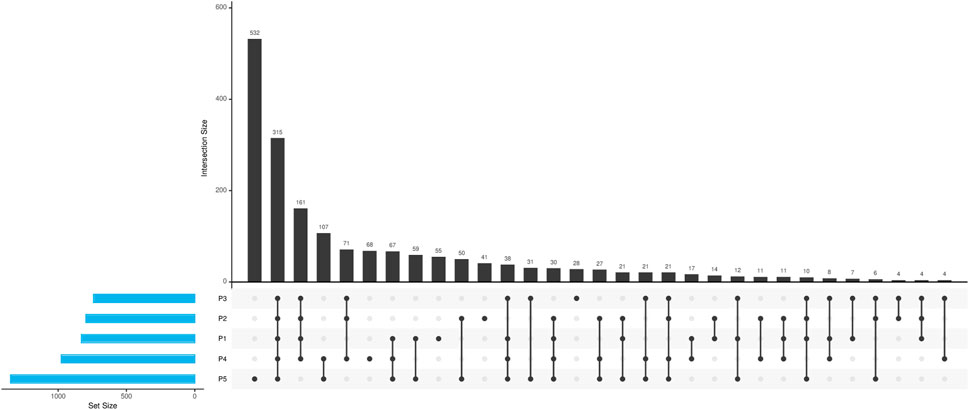
Figure 6. Characterization of virulence genes identified in different points of the Uraim River.
Virulence factors (VFs) can affect a wide range of cellular processes, such as cell-cell signaling, ion secretion, protein synthesis, mitosis, cytoskeleton structure, and mitochondrial function, although their role in natural environments is not yet well understood (Vinueza et al., 2021). Our analyses identified numerous genes related to virulence factors, especially in sites with higher anthropogenic activity (Figure 6), particularly in P5 where there is a greater abundance and diversity of VFs. This site also showed a higher quantity of pathogenic bacteria (Supplementary Figures S4A, B).
The diversity of virulence genes calculated was highest at point P5 with a value of 105.6 for the Fisher alpha diversity index, followed by 77.96 at point P4, values close to 60 at points P1 and P2, and the lowest value of 58 at P3 (Figure 7A). The most relevant virulence mechanism is adhesion, present in all points; points P2 and P5 highlight the effector delivery system mechanism, and in P1, the nutritional/metabolic factor and invasion mechanisms are present (Figure 7B).
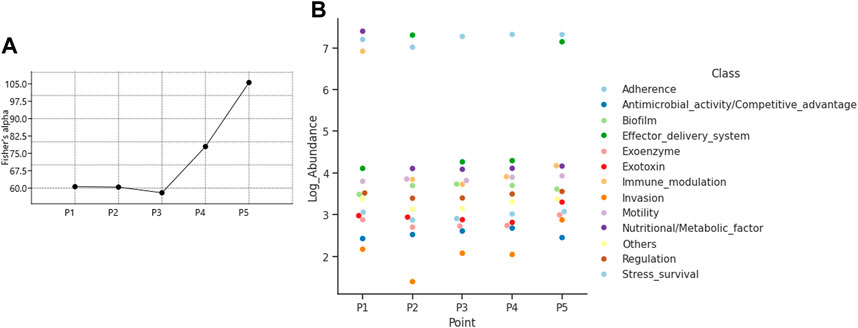
Figure 7. Characterization of virulence genes (A) Fisher alpha diversity index, and (B) Composition of virulence mechanisms.
In studies conducted on rivers traversing urban areas, it has been revealed that urbanization increases environmental risk and the occurrence of resistance and virulence genes compared to those in rural regions. Among the main virulence factors (VFs) are offensive, nonspecific, defensive, and regulatory VFs, which were found in all samples. The dominant subtypes include iron acquisition systems, adhesion, secretion systems, regulation mechanisms, toxins, and antiphagocytosis (Mizusawa et al., 2021; Li et al., 2022). In our samples, secretion systems and adhesion were the most relevant factors.
In all sampling points, a high abundance of the gene fbpC, which is involved in the Secreted fibronectin-binding protein C (VF0311), was observed. These adherence proteins can ensure the adhesion of these pathogens to hosts, with many of these molecules being diverse and not fully described (Aono et al., 2023).
At sampling point P5, there is a notable abundance of genes involved in secretion systems, with the most prominent being the gene vgrG/tssI, which is associated with the synthesis of type VI secretion system tip protein VgrG (VF1337). At P2, the gene vgrG1a, which synthesizes the type VI secretion system substrate VgrG1 (VF0334), stands out (Supplementary Figure S4A).
The identification of genes related to Type III and Type IV secretion systems is essential in bacterial pathogenesis, facilitating direct interactions with host cells. The presence of these genes may reflect regional variations in the bacteria-host dynamics or environmental factors that favor certain pathogenic strategies (Rout et al., 2023). Metagenomic analysis of the Ganges River (India) revealed the widespread distribution of genes associated with flagella, which are related to Type III and Type IV secretion systems, the latter being the most abundant marker identified in different sites. This study highlights the complexity of bacterial virulence and its dependence on environmental context. The detection of flagella-associated genes in various locations implies a widespread reliance on motility and adhesion as critical virulence factors. This suggests a common strategy employed by bacteria to establish infection and colonization across different geographic areas (Godoy et al., 2020; Rout et al., 2023).
In point 1, the gene lic2A synthesizes glycosyltransferase (VF0044), and glnA1 is involved in the production of Probable glutamine synthetase, type I (VF0816) (Supplementary Figure S4A). The glnA1 gene is involved in nitrogen metabolism and is necessary for the development of Mycobacterium tuberculosis and E. coli in the environment (Xu et al., 2023).
The secretion systems identified in this study are characteristic of a variety of Gram-negative bacteria, contributing to the virulence of Type I and Type VI secretion systems, which are commonly described in the genome of P. aeruginosa (Green and Mecsas, 2016), and in members of the genus Pseudomonas, Acinetobacter, Klebsiella, and Enterobacter, which are part of the ESKAPE group and they are the most abundant among the identified pathogenic bacteria identified in our results, which could contribute to the high abundance of this gene.
Another mechanism of adaptation and resistance is the production of biofilms, whose production is induced by the presence of N-acyl-homoserine lactone (Alio et al., 2023), providing significant advantages to bacteria such as resistance to antibiotics and protection against host immune responses. In the environment, biofilms allow bacteria to resist unfavorable conditions (Gomes et al., 2020; Rout et al., 2023), and are present across different sites studied (P1, P2, P3, P4 e P5) in Paragominas.
In the PCA analysis, an accumulated percentage of 88.04% of variation in the variables was found, with 58.8% variation in principal component 1 (PC1), highlighting the following associations: between the physicochemical components of pH, Ec, and TDS with the alpha diversity indices of Shannon, although these were not evident in one analyzed point. In the case of P5, a strong correlation was observed with population occupancy, and Fisher’s alpha diversity of virulence genes (Pearson; ρ = 0.972; p-value = 0.006). Furthermore, a high correlation was identified between the relative abundance of the ESKAPE group and the percentage of deforestation (Pearson; ρ = 0.86; p-value = 0.061); a significant inverse association between dissolved oxygen variables and deforestation percentage (Pearson; ρ = −0.912; p-value = 0.03); and a significant association between virulence gene diversity and the percentage of inhabitants per surface area (Pearson; ρ = 0.972; p-value = 0.005) and with deforestation (Pearson; ρ = 0.907; p-value = 0.033) (Figure 8).
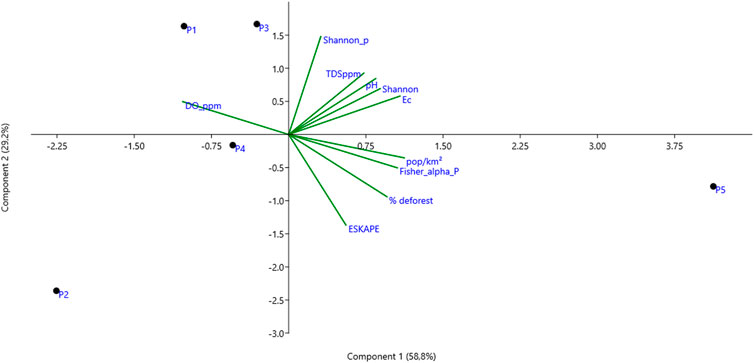
Figure 8. PCA analysis of the different relevant variables in this study for each sampling point in the Uraim River in the city of Paragominas/Brazil. DO ppm: Dissolved Oxygen, Shannon_p: Shannon alpha diversity index of pathogenic bacteria, TDS ppm: Total Dissolved Solids, Ec: Electrical Conductivity, pop/Km2: Number of inhabitants per Km2, deforestation percentage.
The significant associations found between urban and rural population growth and the functioning and biodiversity of the ecosystem by microorganisms responsible for the purification and removal of nutrients in rivers, as well as the nutrient cycle and interactions between plants in the soil (Gomes et al., 2020), are reflected in different analytes. Similarly, biochemical and chemical oxygen demands are influenced by the urbanization process, temperature, dissolved oxygen, ammonia, and phosphorus (Giuliatti et al., 2017).
In this study, the assessed physicochemical properties underwent significant alterations, acting as crucial factors that modify bacterial community structures within these ecosystems and facilitate the acquisition of genetic components associated with their adaptation, including virulence genes for higher organisms (Godoy et al., 2020). Among the identified components, DO is inversely associated with the diversity of virulence genes and the presence of certain pathogens. This association could be attributed to anthropogenic activities, with human presence notably increasing the likelihood of greater contact with these types of organisms.
The metagenomic investigation of the Uraim River, utilizing samples extracted from its surface water, underscores the discernible influence of anthropogenic endeavors on both the diversity and prevalence of pathogenic bacterial strains and their corresponding virulence genes at various sampling locations along the riverine course. Notable among the bacterial taxa are the Enterobacteria and members of the ESKAPE group, with a clear linkage observed to deforestation activities and the density of human habitation in the vicinity of the Uraim River. Similarly, there is an observed trend of increased virulence gene diversity in sites influenced by anthropogenic activities.
However, the limitations identified in the development of this work were the number of points, which could be increased to be more precise regarding the causes of the observed impacts; carrying out studies in different periods (rainy and dry) and several times in each period, to compare the effect in the region, mainly due to agricultural activities that can generate bioaccumulation of chemical products that can later be carried to the river in the period of begin of rainy.
Studies investigating the presence of microorganisms in the water and their impact on the health of human populations and animals consuming river water are necessary to assess potential impacts and guide public policies aimed at minimizing/mitigating anthropogenic impact and its effects in a One Health approach. In this context, it is noteworthy that in 2022, the municipality of Paragominas established an institutional program called “Renascer Uraim” (“Rebirth Uraim”), aimed at implementing preservation actions for the river. These actions range from environmental education to river cleaning and restoration of native vegetation along its banks, which could positively impact the occurrence of pathogenic microorganisms observed due to anthropogenic activities.
The data presented in the study was deposited in the National Center for Biotechnology Information NCBI database (https://www.ncbi.nlm.nih.gov/), accession number ID SUB14513086 and BioProject ID PRJNA1122411.
OC-A: Conceptualization, Formal Analysis, Writing–original draft, Writing–review and editing, VF: Software, Writing–original draft, WN: Data curation, Writing–review and editing. DM: Methodology, Writing–review and editing. AM: Software, Writing–review and editing, PM: Methodology, Writing–review and editing. RL: Methodology, Writing–review and editing. SA: Validation, Writing–review and editing, AL: Resources, Writing–review and editing, RG: Validation, Writing–review and editing. EF: Resources, Validation, Writing–review and editing, VA: Investigation, Resources, Writing–review and editing, RR: Conceptualization, Funding acquisition, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Dean’s Office for Research and Graduate Studies/Federal University of Pará–PROPESP/UFPA (PAPQ), UFPA (Universidade Federal do Pará), CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), UFPA (Universidade Federal do Pará), CNPQ (Conselho Nacional de Desenvolvimento Científico e Tecnológico) project #312316/2022-4, and, SECTET (Secretaria de Estado de Ciência, Tecnologia e Educação Superior, Profissional e Tecnológica).
We thank the PROPESP/UFPA (Pró-Reitoria de Pesquisa e Pós-Graduação/Universidade Federal do Pará for the financial support on this paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2024.1404230/full#supplementary-material
Acharya, K., Blackburn, A., Mohammed, J., Tamiru Haile, A., Mekpnnen Hiruy, A., and Werner, D. (2022). Metagenomic water quality monitoring with a portable laboratory. Water Res. 184, 116112. doi:10.1016/j.watres.2020.116112
Agência Nacional de Águas, (ANA) (2020). Corpos Hídricos Superficiais e Dominialidade. Sist. Nac. Inf. Sobre Recur. Hídricos. Available at: https://www.snirh.gov.br/(Accessed June 3, 2024).
Aguilar-Salazar, A., Martínez-Vázquez, A. V., Aguilera-Arreola, G., De Jesus De Luna-Santillana, E., Cruz-Hernández, M. A., Escobedo-Bonilla, C. M., et al. (2023). Prevalence of ESKAPE bacteria in surface water and wastewater sources: multidrug resistance and molecular characterization, an updated review. Water 15, 3200. doi:10.3390/w15183200
Alio, I., Moll, R., Hoffmann, T., Mamat, U., Schaible, U. E., Pappenfort, K., et al. (2023). Stenotrophomonas maltophilia affects the gene expression profiles of the major pathogens Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro multispecies biofilm model. Microbiol. Spectr. 11, e0085923. doi:10.1128/spectrum.00859-23
Aono, R., Emi, S., Okabe-Watanabe, K., Nariya, H., Matsunaga, N., Hitsumoto, Y., et al. (2023). Autolysin as a fibronectin receptor on the cell surface of Clostridium perfringens. Anaerobe 83, 102769. doi:10.1016/j.anaerobe.2023.102769
Bartlett, A., Padfield, D., Lear, L., Bendall, R., and Vos, M. (2022). A comprehensive list of bacterial pathogens infecting humans. Microbiology 168, 001269. doi:10.1099/mic.0.001269
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi:10.1093/bioinformatics/btu170
Cavalcante, R., Fleischmann, A., and Pontes, P. R. (2023). Looking upstream: analyzing the protection of the drainage area of Amazon rivers. doi:10.31223/X51M4F
Chong, J., Liu, P., Zhou, G., and Xia, J. (2020). Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821. doi:10.1038/s41596-019-0264-1
Chopyk, J., Nasko, D. J., Allard, S., Bui, A., Pop, M., Mongodin, E. F., et al. (2020). Seasonal dynamics in taxonomy and function within bacterial and viral metagenomic assemblages recovered from a freshwater agricultural pond. Environ. Microbiome 15, 18. doi:10.1186/s40793-020-00365-8
Correa, D. L. (2017). Análise da Susceptibilidade a Inundações na Bacia Hidrográfica do Rio Uraim, Paragominas-PA. Belém PA: Universidade Federal do Pará.
Da Silva, A. C. de S., Pereira Junior, A., and Farias, N. (2020). Diatomáceas como indicadoras da qualidade da água em rios urbanos/diatoms as indicators of water quality in urban rivers. Braz. J. Dev. 6, 34616–34643. doi:10.34117/bjdv6n6-125
De Araujo, R. da C. (2010). Efeitos do desmatamento sobre o ciclo hidrológico: uma comparação entre a Bacia do Rio Curua-Una e a Bacia do Rio Uraim. Belem, PA: Universidade Federal do Pará.
Demsar, J., Curk, T., Erjavec, A., Demsar, J., Curk, T., Erjave, A., et al. (2013). Orange: data mining toolbox in Python. J. Mach. Learn. Res. 8, 2349–2353. Available at: https://www.jmlr.org/papers/volume14/demsar13a/demsar13a.pdf
Denissen, J., Reyneke, B., Waso-Reyneke, M., Havenga, B., Barnard, T., Khan, S., et al. (2022). Prevalence of ESKAPE pathogens in the environment: antibiotic resistance status, community-acquired infection and risk to human health. Int. J. Hyg. Environ. Health 244, 114006. doi:10.1016/j.ijheh.2022.114006
Giuliatti, N. M., Rodrigues, A. B. M., Brito, V. L., Silva, I. A. V. D., and Pereira Junior, A. (2017). “Demanda bioquímica e química de oxigênio no rio uraim e o processo de urbanização no município de paragominas-pa.” in Blucher engineering proceedings (Belo Horizonte, Brasil: Editora Blucher), 938–948. doi:10.5151/xveneeamb-094
Godoy, R. G., Marcondes, M. A., Pessôa, R., Nascimento, A., Victor, J. R., Duarte, A. J. D. S., et al. (2020). Bacterial community composition and potential pathogens along the Pinheiros River in the southeast of Brazil. Sci. Rep. 10, 9331. doi:10.1038/s41598-020-66386-y
Gomes, I. B., Maillard, J.-Y., Simões, L. C., and Simões, M. (2020). Emerging contaminants affect the microbiome of water systems—strategies for their mitigation. Npj Clean. Water 3, 39. doi:10.1038/s41545-020-00086-y
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems: an overview. Microbiol. Spectr. 4 (1), 13. doi:10.1128/microbiolspec.VMBF-0012-2015
Hammer, O., Harper, D. A. T., and Ryan, P. D. (2001). PAST: paleontological statistics software package for education and data analysis. Palentologica Electron 4 1, 1–9. Available at: http://palaeo-electronica.org.
Hamner, S., Brown, B., Hasan, N., Franklin, M., Doyle, J., Eggers, M., et al. (2019). Metagenomic profiling of microbial pathogens in the little bighorn river, Montana. Int. J. Environ. Res. Public. Health 16, 1097. doi:10.3390/ijerph16071097
He, C., Liu, Z., Wu, J., Pan, X., Fang, Z., Li, J., et al. (2021). Future global urban water scarcity and potential solutions. Nat. Commun. 12, 4667. doi:10.1038/s41467-021-25026-3
Ko, K. K. K., Chng, K. R., and Nagarajan, N. (2022). Metagenomics-enabled microbial surveillance. Nat. Microbiol. 7, 486–496. doi:10.1038/s41564-022-01089-w
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi:10.1038/nmeth.1923
Li, K., Hu, J., Li, T., Liu, F., Tao, J., Liu, J., et al. (2021). Microbial abundance and diversity investigations along rivers: current knowledge and future directions. WIREs Water 8, e1547. doi:10.1002/wat2.1547
Li, R., Zhu, L., Wang, Y., and Zhu, Y.-G. (2022). Metagenomic insights into environmental risk of field microplastics in an urban river. Water Res. 223, 119018. doi:10.1016/j.watres.2022.119018
Lin, L., Yang, H., and Xu, X. (2022). Effects of water pollution on human health and disease heterogeneity: a review. Front. Environ. Sci. 10, 880246. doi:10.3389/fenvs.2022.880246
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi:10.1093/nar/gkab1107
Mizusawa, N., Reza, Md. S., Oikawa, C., Kuga, S., Iijima, M., Kobiyama, A., et al. (2021). Diversity and functions of bacterial communities in water and sediment from the watershed of the Tama River flowing a highly urbanized area. Fish. Sci. 87, 697–715. doi:10.1007/s12562-021-01543-4
Pandey, P. K., Kass, P. H., Soupir, Mi. L., Biswas, S., and Singh, V. (2014). Contamination of water resources by pathogenic bacteria. Amb. Express 4, 51. doi:10.1186/s13568-014-0051-x
Pereira Júnior, A., Morales, G. P., Beltrão, N. E. S., Barbosa, A. J. S. D. S., Gutierrez, L. A. C. L., Jesus, E. D. S., et al. (2023a). Geospatial analysis of deforestation, land use and impacts of water resources in paragominas, Pará. DELOS Desarro. LOCAL Sosten. 16, 3399–3433. doi:10.55905/rdelosv16.n48-026
Pereira Júnior, A., Morales, G. P., Beltrão, N. E. S., Silva, A. P. D. S., and Pereira, L. C. (2023b). Multitemporal analysis of land use and land cover in the Uraim river microbasin, Paragominas–Pará: Análise multitemporal do uso e cobertura da terra na microbacia do rio Uraim, Paragominas, Pará. Concilium 23, 383–404. doi:10.53660/CLM-1093-23D20A
Philippot, L., Griffiths, B. S., and Langenheder, S. (2021). Microbial community resilience across ecosystems and multiple disturbances. Microbiol. Mol. Biol. Rev. 85, e00026-20. doi:10.1128/MMBR.00026-20
Rout, A. K., Tripathy, P. S., Dixit, S., Behera, D. U., Behera, B., Das, B. K., et al. (2023). Unveiling the microbiome landscape: a metagenomic study of bacterial diversity, antibiotic resistance, and virulence factors in the sediments of the river ganga, India. Antibiotics 12, 1735. doi:10.3390/antibiotics12121735
Santos, E. M., and Picanço, O. A. S. (2008). Recomposição do ecossistema florestal da nascente do rio uraim localizado no município de paragominas/pa. Belem, PA: Universidade Federal do Pará.
Santos-Júnior, C. D., Sarmento, H., Pellon de Miranda, F., Henrique-Silva, F., and Logares, R. (2020). Uncovering the genomic potential of the Amazon River microbiome to degrade rainforest organic matter. Microbiome 8, 151. doi:10.1186/s40168-020-00930-w
Sardinha, A. S., and Ventura, K. S. (2022). “Análise preliminar dos riscos ambientais da Bacia do Rio Uraim, em um Município da Amazônia Oriental,” in Proceedings of (Brazil: Universidade Estadual de Campinas), 61–64. doi:10.20396/iwisdw.n1.2022.4790
Schwarzenbach, R. P., Egli, T., Hofstetter, T. B., Von Gunten, U., and Wehrli, B. (2010). Global water pollution and human health. Annu. Rev. Environ. Resour. 35, 109–136. doi:10.1146/annurev-environ-100809-125342
Strokal, M., Bai, Z., Franssen, W., Hofstra, N., Koelmans, A. A., Ludwig, F., et al. (2021). Urbanization: an increasing source of multiple pollutants to rivers in the 21st century. Npj Urban Sustain 1, 24. doi:10.1038/s42949-021-00026-w
Tornevi, A., Bergstedt, O., and Forsberg, B. (2014). Precipitation effects on microbial pollution in a river: lag structures and seasonal effect modification. PLoS ONE 9, e98546. doi:10.1371/journal.pone.0098546
VanEvery, H., Franzosa, E. A., Nguyen, L. H., and Huttenhower, C. (2023). Microbiome epidemiology and association studies in human health. Nat. Rev. Genet. 24, 109–124. doi:10.1038/s41576-022-00529-x
Vinueza, D., Ochoa-Herrera, V., Maurice, L., Tamayo, E., Mejía, L., Tejera, E., et al. (2021). Determining the microbial and chemical contamination in Ecuador’s main rivers. Sci. Rep. 11, 17640. doi:10.1038/s41598-021-96926-z
Wingett, S., and Andrew, S. (2018). FastQ Screen: a tool for multi-genome mapping and quality control. F1000Research 7, 1338. doi:10.12688/f1000research.15931.2
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. doi:10.1186/s13059-019-1891-0
Keywords: anthropogenic activity, microbial diversity, bacterial pathogens, virulence genes, Uraim River
Citation: Cardenas-Alegria OV, Ferreira VBC, Noguera WG, Martins DT, Martins Neto AP, Monteiro Pontes PR, Lopes Cavalcante RB, Aguiar Alves SI, Luiz da Costa da Silva A, Gomes Costa R, Franco de Los Santos EF, Azevedo VAdC and Ramos RTJ (2024) Microbiome analyses of the Uraim River in the Amazon and georeferencing analyses to establish correlation with anthropogenic impacts of land use. Front. Environ. Sci. 12:1404230. doi: 10.3389/fenvs.2024.1404230
Received: 20 March 2024; Accepted: 15 July 2024;
Published: 06 August 2024.
Edited by:
Visva Bharati Barua, University of North Carolina at Charlotte, United StatesReviewed by:
Sol Park, FAMU-FSU College of Engineering, United StatesCopyright © 2024 Cardenas-Alegria, Ferreira, Noguera, Martins, Martins Neto, Monteiro Pontes, Lopes Cavalcante, Aguiar Alves, Luiz da Costa da Silva, Gomes Costa, Franco de Los Santos, Azevedo and Ramos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rommel Thiago Juca Ramos, cm9tbWVscmFtb3NAdWZwYS5icg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.